Embed Size (px)
Citation preview

Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 3
Inflammatory Reactionsin Peritonitis and Malignant Obstructive Jaundice
JOHANNA ÖSTERBERG
Clinical and Experimental Studies with Special Emphasis on the Cellular Immune Response
ISSN 1651-6206ISBN 91-554-6138-7urn:nbn:se:uu:diva-4767
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2005


Det finns mitt i skogen en oväntad glänta
som bara kan hittas av den som gått vilse.
Tomas Tranströmer
To my family


Torrnålsgravyr, Lena Sundberg 2005.

List of papers
This thesis is based on the following papers, which will be referred to in the text by their Roman numerals (I-V):
I Alteration in mucosal immune cell distribution in septic rats. Österberg J, Johnsson C, Gannedahl G, Westlund A-M, Haglund U. Shock, 7(3): 182-185, 1997.
II Effect of Linomide on gut immune cell distribution and on TNF- in plasma and ascites. An experimental study in the septic rat.
Österberg J, Haglund U.Shock, 18(5): 471-5, 2002.
III Influence of cyclooxygenase inhibitors on gut immune cell distribution and apoptosis rate in experimental sepsis. Österberg J, Ljungdahl M, Haglund U. Manuscript
IV Microbial translocation and inflammatory response in patients with acute peritonitis.Österberg J, Ljungdahl M, Lundholm M, Engstrand L, Ulf Haglund. Scand J Gastroenterol, 39(7): 657-64, 2004
V Inflammatory response in patients with malignant obstructive jaundice.Ljungdahl M, Österberg J, Ransjö U, Engstrand L, Haglund U. Submitted
Reprints have been made with the permission of the publishers.

Contents
Introduction...................................................................................................11Peritonitis .................................................................................................11Obstructive jaundice.................................................................................13Innate and adaptive immunity ..................................................................13Lymphocytes and the lymphatic system ..................................................15Mononuclear phagocytes..........................................................................15Apoptosis..................................................................................................15Gut defence and mucosal immunology ....................................................16Mediators of the inflammatory response..................................................17Endotoxins................................................................................................17Cytokines..................................................................................................18Prostaglandins ..........................................................................................19SIRS, sepsis and septic shock ..................................................................19The gut in sepsis and shock......................................................................20
Aims of the present investigation..................................................................22Experimental studies I-III: .......................................................................22Clinical studies IV-V:...............................................................................22
Material and methods....................................................................................23Experimental studies (I, II and III) ...........................................................23
Animals and anaesthesia......................................................................23Experimental model.............................................................................23Pilot series............................................................................................24Experimental groups............................................................................24Drug treatments ...................................................................................25
Clinical studies in humans with peritonitis (IV) and malign obstructive jaundice (V)..............................................................................................26Study groups.............................................................................................27Sampling and analyses (I-V) ....................................................................29
Sampling of ascitic fluid, blood, small- bowel biopsies and MLNs...29Bacterial cultures .................................................................................29Pulsed-field gel electrophoresis (PFGE)..............................................29Cytokine measurement ........................................................................30Endotoxin measurement ......................................................................30Monoclonal antibodies.........................................................................30

Immunohistochemistry and microscopic evaluation ............................30Staining for apoptosis ..........................................................................31Colour based image analysis ...............................................................31Statistics...............................................................................................31
Results and comments...................................................................................33Clinical appearance (I-III) and mortality after CLP (Pilot series A)........33Pilot series B.............................................................................................34Pilot series C.............................................................................................35Mucosal morphology (I-III) .....................................................................35Distribution of immune cells in the mucosa (I-III) and in the MLNs (III).............................................................................................37Immune cell distribution in the MLNs (IV-V) .........................................39Apoptosis in the mucosa (III) and MLNs (III and V) ..............................41Cytokine analysis: TNF- (II-III) and IL-6 (III)......................................42Cytokine analysis: TNF- , IL-6 and IL-10 (IV-V)..................................43The IL-10/TNF- ratio (IV-V).................................................................45Bacterial cultures (IV-V)..........................................................................46Endotoxin concentrations (IV-V).............................................................47
General discussion and clinical considerations............................................50
Conclusions...................................................................................................53
Summary in Swedish (sammanfattning på svenska)....................................54
Acknowledgements.......................................................................................57
References.....................................................................................................59

Abbreviations
Ab(s) Antibody (Antibodies) Ag(s) Antigen (Antigens) ALAT Alanine aminotransferase ALP Alkaline phosphatase ANOVA Analysis of variance APC Antigen presenting cells ASAT Aspartate aminotransferase BT Bacterial translocationCLP Cecal ligation and puncture CD Cluster of differentiation CD4+ T cell Helper T lymphocyte CD8+ T cell Cytotoxic T lymphocyte COX-1 Cyclooxygenase-1 COX-2 Cyclooxygenase-2 CRP C-reactive protein E.coli Escherishia coli ELISA Enzyme linked immunosorbent assay EpL Epithelial layer GALT Gut associated lymphoid tissue HPB Hepatic-pancreatic-biliary HPB+ HPB tumor and obstructive jaundice HPB- HPB tumor without obstructive jaundice ICU Intensive care unit IL-6 Interleukin-6 IL-10 Interleukin-10 i.p. Intraperitoneal LAL Limulus amoebocyte lysate LP Lamina propria LPS Lipopolysaccaride (endotoxin) mAb Monoclonal antibody MALT Mucosa associated lymphoid tissue MHC Major histocompatibility complex MLN(s) Mesenteric lymph node(s) MODS Multiple organ dysfunction syndrome PAP Peroxidase-antiperoxidase PCR Polymerase chain reaction

PFGE Pulsed-field gel electrophoresis p.o. Per oral SIRS Systemic inflammatory response syndrome TCR T cell receptor TLR Toll-like receptor TNF- Tumour necrosis factor-alphaWBC White blood cell

11
Introduction
Peritonitis Peritonitis or intra-abdominal sepsis is an inflammation of the peritoneum from any cause. Reports on surgical treatment of peritonitis were available early in the surgical literature (Mikulicz J, 1881), at that time with a mortal-ity rate between 80 – 90 % (116). In 1926 Kirschner reported a decrease in mortality to an average of 50 %, with the development of new operative techniques, the introduction of antibacterial chemotherapy and intensive care (81). After the discovery of penicillin by Fleming, there was no great improvement in treatment results. Only since the mid 1970s have we been able to register further improvement down to about 30 % mortality, due to broadspectrum antibiotics, improvement of ICU monitoring and a more aggressive and optimised surgical approach. Still no definitive therapy exists that can successfully treat sepsis and its complications. The diagnosis is supported by clinical signs, e.g., abdominal pain and tenderness, nausea, vomiting, diminished intestine sounds, fever, shock, radiographic and microbiologic evidence (43). Clinically peritonitis is often classified either as local or as diffuse. Local peritonitis refers to loculi of infection, usually walled-off or contained by adjacent organs (peritoneal defence system = omentum majus and fibrin formation), whereas diffuse peritonitis is synonymous with generalized disease, i.e spread to the entire cavity. The bacteria then reach the general circulation via subperitoneal lymphatic lacunaes, through diaphragmatic lymphatic valves into ductus thoracicus (2). Since intra-abdominal infection takes many forms, there are a number of classifications (Table 1). To compare treatment results from different clinics using different therapy modalities, various scoring systems have developed. APACHE II scoring is one of the most commonly used systems and is pro-posed for grading the severity of the infection and for stratification of patient risk of mortality (123). The normal human intestinal flora contains most 1012
colony forming units per gram of faeces and at least 400 species of bacteria are present in the gut (184, 57). Especially the anaerobic microflora, acts to resist colonization. The pathogens normally detected in peritonitis are the same that commonly are known to translocate: the Gram-negative bacteria as Escherishia coli (E.coli), Klebsiella, and Pseudomonas (13). When peritoni-tis persists, Pseudomonas aeruginosa, Enterobacter, Enterococci and Can-dida albicans may be isolated (168).

12
When the inflammatory response gets out of control, multi organ failure will ensue and surgery can no longer limit the immune response, emphasizing the need for timely operation in suspected peritonitis (Table 2). Intraabdominal infections are found to occur in 25 % of patients with multiple organ failure in surgical ICU.
Table 2. Therapeutic strategy in intra-abdominal sepsis.
1) Primary, refers to spontaneous bacterial invasion of the peritoneal cavity. This mainly occurs in infancy and early childhood, in cirrhotic patients and immunocompromised hosts.
2a) Secondary dependent, describes: I) The acute perforation of a hollow viscous II) Postoperative due to leakageIII) Posttraumatic
2b) Secondary independent, when intra-abdominal organ sustain an insult from splanchnic hypoperfusion e.g. bowel wall necrosis and transloca-tion of enteric bacteria from the gut.
3) Tertiary, is characterized by persistent or recurrent infections with organisms of low intrinsic virulence or with predisposition for the immunocompromised patient. It usually follows operative attempts to treat secondary peritonitis and is almost always associated with a sys-temic inflammatory response.
(17, 76, 101, 106, 107)
Table 1. Peritonitis classified according to etiology.
1) Elimination the source of infection. 2) Removal of infective materials, necroses, and toxins.
Surgery, Percutaneous drainage
3) Optimization of defence mechanisms against infection.
Antibiotics,Immunotherapies
4) Treatment of the consequences of infection. Intensive-care treatment
(33,107,108)

13
Obstructive jaundiceIncreased levels of specific liver variables in serum as bilirubin, alkaline phosphatase (ALP) and alanine aminotransferase (ALAT) are indicators for liver damage in obstructive jaundice (118). Surgery in patients with obstruc-tive jaundice is frequently associated with postoperative, mainly septic, complications (88, 165). These complications have recently been associated with a proinflammatory state resulting from portal and systemic endotoxemia, bacterial translocation, and subsequent activation of the inflammatory cascadeleading to the sepsis syndrome (30, 39, 77, 129, 155). Increased levels of TNF- and interleukin IL-6 and depressed cell mediated immunity have been demonstrated experimentally (12, 174), as well as clinically (4). A reduction in liver reticuloendothelial system function leading to a diminished clearance of endotoxin by Kupffer cells (35, 176) have been correlated to acute renal failure and sepsis (38). Furthermore, it is very likely that the normal clearance of endotoxin to the bile will be decreased in biliary obstruction. Systemic endotoxemia, and the additional inflammatory response caused by the operative trauma itself, have been associated withpostoperative morbidity. Studies of the intestinal mucosa in a jaundice rat model with bile duct ligation, have demonstrated an atrophic mucosa with decreased villous density and thickness, and an increased apoptosis rate most marked in the distal ileum (133).
The absence of bile within the gastrointestinal tract allows intestinal overgrowth and the combination of bacterial overgrowth and mucosal injury appears to promote bacterial translocation (31). It remains uncertain whether preoperative drainage by endoscopic insertion of an endoprosthesis influ-ences the inflammatory response. Recent data from the literature showconflicting results (65, 135, 151, 158). Even with internal drainage, restora-tion of normal immune parameters takes weeks or months (155).
Innate and adaptive immunity The immune system is seen as a guardian of tissue integrity evolved to protect us from invasion by various pathogens. The invasion of host tissues by microorganisms evokes a complex and highly conserved response that serves to recruit inflammatory cells to the site of challenge and to optimise local defences to kill the invading bacteria, minimize microbial dissemina-tion beyond the site of infection, and initiate the processes of tissue repair. Immunity to pathogens links two effector arms of the immune response: the 'frontline' defences of innate (natural or native) immunity with that of adaptive (acquired) immunity.
The innate immunity is particularly important in warding off bacterial and viral infections presenting at the mucosal cell surface (68, 195).

14
The components of innate immunity are: physical and chemical barriers (epithelia, antimicrobial chemicals), natural killer cells and phagocytes (macrophages, neutrophils), blood proteins (complement) and cytokines. The effective development of the overall immune response depends on careful interplay and regulation between innate and adaptive immunity (130). Epithelial- and immune cells of the innate immune defence system can recognize pathogen-associated molecular patterns, such as those that occur in the bacterial cell wall, through their Toll-like receptors (TLRs) (92, 127), and then elicit pathogen-specific cellular immune responses.
Adaptive immune responses develop later and consist of activation of lymphocytes and their products; antibodies (Abs) and cytokines (190). The adaptive immune system is necessary for protection from polymicrobial sepsis and plays a significant role in regulating the inflammatory response to injury (160). The adaptive immune system can be divided into humoral and cell-mediated responses. In humoral immunity, B lymphocytes secrete Abs that eliminate extracellular microbes, this process requires the help of activated T cells. In cell-mediated immunity, T lymphocytes are subdivided due to their membrane-associated glycoproteins into helper (h) T cells (CD4+) and cytotoxic T cells (CD8+). T lymphocytes either activate macrophages to kill phagocytosed microbes (CD4+ T cells) or destroy infected cells (CD8+ T cells). CD4+ T helper lymphocytes may differentiate further into Th1 and T h2 subsets of effector cells, producing distinct sets of cytokines (149). Th1cells favour the phagocyte-cell mediated functions, producing interleukin (IL)-2 and interferon gamma (IFN ), thus activating CD8+ cells. Conversely, T h2 cells secrete IL-4, IL-5, IL-6 and IL-10, activate B cells, stimulate immunoglobulin E (IgE) reactions and downregulate Th1 responses. Unlike B cells, which recognize Ag in its native form, T cells recognize the Ag after it has been processed and attached to specific protein complexes, major histocompatibility complex (MHC) class I or II molecules, on the surfaces of Ag presenting cells (APCs) (19). MHC is the gene region coding for “transplantation Ags”, in humans called the “human leukocyte Ag” (HLA). The contact with the Ag by the specific T cell receptor (TCR) triggers the immune response, through release of cytokines from the activated T cells. T lymphocytes expressing the surface marker CD8 recognize foreign Ag in association with MHC class I, and CD4+ cells recognize Ag in association with MHC class II. MHC class I Ags are expressed on most nucleated cells (58) and MHC class II Ags are expressed on B lymphocytes, APCs, macro-phages, activated T lymphocytes, but also on endothelial and epithelial cells (28).

15
Lymphocytes and the lymphatic systemLymphocytes arise from stem cells in bone marrow, and differentiate in the central lymphoid organs, B cells in bone marrow and T cells in the thymus. They migrate from these tissues and are carried in the bloodstream to the peripheral or secondary lymphoid organs, the lymph nodes, the spleen, and lymphoid tissues associated with the gut mucosa. The peripheral lymphoid organs are the sites of lymphocyte activation by Ag, and naive T lympho-cytes recirculate between the blood and these organs until they encounter Ag. Effector T- and memory cells are often recruited to peripheral sites of inflammation, where microbial Ags are located. Adhesion molecules govern the recirculation process of lymphocytes. The process, by which particular subsets of lymphocytes selectively enter some tissues, but not others, is called “lymphocyte homing”(21, 188). The lymph nodes consist of an outer cortex where the follicles, B cell rich areas, are situated. Germinal centres are follicles, which develop in response to Ag stimulation. The inner para-cortex is the T cell rich zone; approximately 70 % of these T cells are CD4+
cells. Beneath the paracortex is the medulla, consisting of medullary cords and sinus. These cords are populated by macrophages and plasma cells. In rats the mesenteric lymph nodes (MLNs) are located close to the cecal wall and drain the small intestine together with the first part of colon (60).
Mononuclear phagocytesMononuclear phagocytes develop from hematopoietic stem cells in the bone marrow, circulate in the blood as monocytes, and are resident as macrophages in all tissues of the body. They have important function in both innate and adaptive immunity (89). Their effector functions in innate immunity are to phagocytose microbes and to produce cytokines that recruit and activate other inflammatory cells. In the adaptive phase Ag stimulated T cells activate macrophages to destroy phagocytosed microbes. They also have an assessory function as APC, which are to display Ag on their surface that can be recognized by CD4+ T lymphocytes.
ApoptosisApoptosis is a highly regulated DNA-dependent cell suicide mechanism, which occurs under physiological and pathological conditions (194). It is the main method of the body to in a controlled manner get rid of cells, which are in excess, damaged, or no longer needed. Most tissues, and especially the skin, gut, and immune system, depend on well-ordered apoptosis and cell replacement. In contrast to active cellular necrosis that occurs after

16
destruction of the cell membrane, apoptosis refers to the morphological findings characterized by cellular shrink, nuclear and cytoplasmic condensa-tion, membrane blebbing, and fragmentation of this into apoptotic bodies. The apoptotic cell is quickly recognized and phagocytosed by macrophages and/or dendritic cells. Recent evidence indicates that uptake of apoptotic cells impair the immune function of surviving cells and contributes to immunosuppression (172). During the septic syndrome, lymphocyte apoptosis can be triggered by the absence of interleukin-2 (IL-2) or by the release of glucocorticoids, granzymes (granules of cytolytic lymphocytes, natural killer and cytotoxic T cells), or the so-called “death”activators: tumour necrosis factor alpha (TNF- ) or Fas ligand. Apoptosis proceeds via auto-activation of cytosolic and/or mitochondrial caspases (intracellur cysteine proteases), which can be influenced by the pro- and anti-apoptotic proteins of the Bcl-2 family (62). It is now clear that caspase activation is a hallmark of almost all apoptotic systems. Activated caspase-3, is a main effector caspase of the apoptotic cascade within cells (52). Excessive apoptosis is associated with ischaemic heart disease, stroke, neurodegenera-tive disease, sepsis and multiple organ dysfunction syndrome.
Death by apoptosis is essential for function, growth and differentiation of T lymphocytes. In addition, apoptosis is also involved in cytotoxic killing of target cells and the regulation of lymphocyte homeostasis during immune responses. Thus, apoptosis has advantages and disadvantages for the host depending on the specificity of the lymphocyte population affected (11). Sepsis demonstrates a marked dysregulation of the immune system in its ability to fight infection. Studies in experimental animals and critically ill patients have demonstrated that increased apoptosis in lymphoid organs and some parenchymal tissues contributes to immune suppression, anergy, unresponsiveness to treatment, thus leading to organ system dysfunction. In experimental animals, treatment with inhibitors of lymphoid cell apoptosis improves outcome. This new understanding of sepsis may lead to novel therapeutic approaches including pharmacological agents that block apoptosis (72, 100, 125).
Gut defence and mucosal immunologyThe non-immunologic mechanical defence mechanisms consist of the gut barrier and intestinal motility. The intrinsic barriers contains the epithelium itself, composed mostly of enterocytes with their microvilli and covered by a layer of mucus and the stabilizing influence of a normal intestinal microflora (114, 153). The extrinsic barriers exert their effect within the lumen as gastric acid, bile salts and proteolytic enzymes (31, 99). The immunologic mechanisms include secretory IgA and cell mediated immunity. The mucosa-associated lymphoid tissues (MALT) lining the gut are known as a

17
part of the gut-associated lymphoid tissue (GALT) (1, 196). The mucosal surface is colonized by lymphocytes and accessory cells in three main regions: within the epithelial layer (EpL), the lamina propria (LP) and in organized collections in LP such as Peyers’patches (PPs) in order to respond optimally to ingested Ag. GALT also include mesenteric lymph nodes. The majority of intraepithelial lymphocytes are CD8+ cells. The LP, a loose layer of connective tissue in the intestinal mucosa that comprises the villous structure with nerves, blood and lymphatic vessels, contains a large number of activated T cells, mostly CD4+ cells. It also contains populations of macrophages, activated B- lymphocytes, plasma cells, dendritic cells, polymorphonuclear leucocytes, eosinophils and mast cells. PPs are B cell rich areas that also contain small number of CD4+ T cells. The intestinal epithelium that line mucosal surfaces, serves as one of human's primary interfaces with the outside world, and plays a very active role in innate immunity particularly by secreting chemokines and other immune mediators in response to pathogenic microbes. The GALT must constantly distinguish harmless Ags that are present in food or on commensal bacteria from patho-genic assaults by microbes. It is perhaps not surprising, then, that the GALT contains more lymphocytes and macrophages than all of the secondary lymphoid organs combined.
Mediators of the inflammatory responseThe inflammatory response includes complex cascade processes character-ized by disturbances in the microcirculation and up-regulation of inflamma-tory mediators, which activate and recruit inflammatory cells to the site of inflammation. The mediators have a dual role in the inflammatory response; small amounts can have beneficial effects on homeostasis, on the other hand, large amounts can be detrimental to the host by an overwhelming activation.
EndotoxinsEndotoxin- the lipopolysaccaride (LPS) component of the outer membrane of the bacterial cell wall- is the major activator in Gram-negative sepsis. LPS will be released as a consequence of bacteria death and lysis but also as a consequence of growth by cell division (74). Many studies have also shown increased endotoxin release after exposure to different antibiotics (51). Endotoxemia occurs following major abdominal surgery, severe burns, trauma, sepsis and shock, but also secondary to intestinal hypoperfusion and gut ischemia (22, 40). Several studies have showed that the gut is the source of endotoxins released in shock (66). LPS forms a complex with a serum protein (LPS-binding protein; LBP), which binds to the macrophage receptor

18
and stimulates the microbicidal activities of these cells and the secretion of cytokines, above all TNF- (112, 193). LBP may be suitable as a marker for infection and also for monitoring the course of the infectious process in critically ill patients. Endotoxin induces local and systemic inflammation, and many of the features of tissue injury observed in infection by Gram-negative bacteria can be mimicked by the administration of LPS parenteral (115). In addition, endotoxin has been described as superantigen for T cells.
CytokinesCytokines are small molecular weight proteins produced by the cells of both innate and adaptive immunity. They act in networks leading to a change in cell proliferation, differentiation and function (46). Basal production of cytokines is usually low or absent (97), detection in the circulation reflects excessive production at the tissue level. However, concentrations may vary significantly depending on, for example, the origin of the insult (179). Most cytokines are redundant, meaning that several types of cytokines are capable of fulfilling each other tasks (37). Thus, loss or pharmacological blockade of only one type of cytokine is rarely enough to eliminate the unwanted effect of cytokine action. Cytokines are part of the normal systemic response to trauma, burn and sepsis, it is only when the cascade of cytokine release becomes disproportionate, that the systemic inflammatory response syndrome (SIRS) evolves into a pathological reality (3, 18). Cytokines have been implicated as both direct mediators of tissue injury (181) as well as predictors of clinical outcome (144). Some cytokines induce and promote inflammation, and have been named pro-inflammatory cytokines, whereas anti-inflammatory cytokines act to limit and down-regulate the inflammatory response. The balance of these two groups of cytokines defines the overall immune response (179).
TNF- is one of the main proinflammatory cytokines and an early cental mediator of sepsis. It is produced primarily by macrophages, but also by lymphocytes, natural killer cells and endothelial cells. Endotoxins are the most powerful stimulus for release (85). TNF- is pleiotropic and induces cellular effects ranging from proliferation to apoptosis. The principal func-tion is to stimulate the recruitment of neutrophils and monocytes to sites of infection and to activate these cells to eradicate microbes. TNF- increases the neutrophil aggregation and adherence, procoagulant activity, the release of IL-1, IL-6 and IL-8 and leukotrienes, enhance the activity of leucocytes, adhesion molecules on endothelial cells, the phagocytosis and reduces pro-tein C activation (97, 171).
IL-6 has been demonstrated to have most pro- but also anti-inflammatory effects (147). Mononuclear phagocytes from both innate and adaptive immunity mainly produce IL-6. It is a prime mediator of SIRS, mediates B

19
and T-cell differentiation and induction of hepatic acute phase proteins (C-reactive protein: CRP, and fibrinogen), stimulate macrophage activation and inhibition of albumin synthesis (61). Unlike most cytokines, IL-6 is more readily detected systemically, and thus serves more of a classical endocrine function. IL-6 may be used as a marker of tissue injury and to identify patients with systemic inflammation.
IL-10 is an important anti-inflammatory cytokine produced mainly by activated macrophages, and because it inhibits macrophage functions, it is an example of a negative feedback regulator. This cytokine down regulates the expression of TNF- and other inflammatory cytokines and is, thus, involved in the homeostatic control of innate immune reactions and the cell-mediated immunity (102, 103).
ProstaglandinsProstaglandins (PGs) are potent immunomodulators in states of inflamma-tion and sepsis and are produced largely in the LP of the intestine by macro-phages and leukocytes (198). It is one of three members of the eicosanoid family; the other two are thromboxanes and leucotrienes. PGs are arachdoni-cacid metabolites generated through the enzyme COX-1 or –2. These enzymes differ in their inducibility and degree of inhibition by nonsteroidal anti-inflammatory drugs (NSAIDs). COX-1 is constitutively expressed in almost all tissues and is important for the maintenance of a homeostatic function. COX-2 is upregulated in macrophages and endothelial cells during inflammatory conditions such as SIRS and sepsis by proinflammatory cytokines as TNF- (27, 64).
SIRS, sepsis and septic shock The term “systemic inflammatory response syndrome” (SIRS) describes the general systemic inflammatory process in the body independent of cause. Sepsis is defined as a condition with fever, tachycardia, tachypnoe and change in leukocyte count caused by an infection (Table 3). Sepsis is frequently associated with hypoperfusion followed by tissue injury and organ failure. There are three recognized stages in the hierarchy of the inflammatory response, with progressively increased risk of end-organ failure and death: sepsis, severe sepsis, and septic shock. Patients with infection plus two or more elements of the systemic inflammatory response syndrome (SIRS) meet the criteria for sepsis; those who have end-organfailure are considered to have severe sepsis; and those who also have refractory hypotension are considered to be in septic shock (16). Septic

20
shock can be characterized hemodynamically by either an early hyperdynamic or a late hypodynamic phase.
Multiple organ dysfunction syndrome (MODS) represents progressive deterioration of function in several organ systems, in sequence or simultane-ously. The gastrointestinal tract has been suggested as the “motor” of MODS (23). The activation of monocytes/macrophages and neutrophils, with the consecutive release of pro-inflammatory mediators and activation of the coagulation cascade, seems to play a key role in the pathogenesis of sepsis.(182). The early hyperinflammatory phase caused by production of TNF- ,IL-1, IL-6, and interferon gamma especially, is invariably followed by a prolonged period of an anti-inflammatory response(e.g. IL-10, IL-4), which may lead to a hypoinflammatory state. In addition, a patient may oscillate between a pro- and anti-inflammatory state repeatedly (the mixed antagonis-tic response syndrome).
The successful treatment of patients with sepsis syndrome will probably require multi-modal therapies aimed at several of the immunological and physiological disturbances, which are occurring simultaneously – an inten-sive care unit (ICU) package (84, 189). The new staging system of sepsis (PIRO) facilitates a more accurate characterization of the disease and the associated risks and prognosis. PIRO is based on predisposition (especially to genetic factors), the insult infection (the type of infection, source) the response of the host system (SIRS, septic shock, the inflammatory markers) and organ dysfunction.
Table 3. Definition of SIRS and sepsis.
The gut in sepsis and shock There is an increased demand for oxygen in the gut during sepsis (29). The splanchnic oxygenation is affected by reduced oxygen delivery secondary to
1. Temperature > 38 C or < 36 C2. Heart rate > 90 beats/minute 3. Respiratory rate > 20 breaths/minute or PaCO2 of < 4.3 kPa 4. WBC count > 12.0 or < 4.0 cells x 109 /l, or > 10 % immature neutrophils
For the diagnosis of SIRS and sepsis, at least two of these criteria have to be fulfilled. Definition of sepsis requires also a detectable infection.
(Adapted from Bone RC et al. 1992)

21
reduced intestinal blood flow and cardiac output (8, 140). This, together with tissue oedema and microthrombosis leads to lowered oxygen uptake, anaerobic metabolism and metabolic acidosis. The septic condition also causes a reduced oxygen extraction and diminishes the capacity to utilize oxygen in the mucosa, because of a shunting mechanism in the villi. An early indicator of injury to the gut mucosa without histological evidence is an increase in permeability (55, 45) and a low gastrointestinal intramucosal pH (119). Before microscopic evidence of a superficial mucosal injury (54), the increase in mucosal permeability may lead to translocation of toxic factors, for example, bacteria and endotoxin from the intestinal lumen to the MLNs, and from there to distant sites (5, 13).
Bacterial translocation is frequently demonstrated in experimental studies of rodents but is much less frequent, if it occurs at all, in man (120, 156). Certain bacterial species/strains disseminate from the intestinal tract more easily than others and, therefore, the composition of the intestinal flora can affect intestinal permeability (95, 122). The first morphological sign of mucosal injury can be detected using electron microscopy, but commonly the mucosal injury is evaluated by light microscopy. The most frequently used system for grading mucosal injury is the one originally proposed by Chiu and co-workers (24), and later adapted to rats (53) (Table 4). Additional grades (6-8) including: crypt layer-, transmucosal- and transmural infarction have been demonstrated after complete vascular occlusion (131). The injury degree is dependent on the severity of the applied ischaemia /shock and its duration, and the reperfusion component (55, 56).
Table 4. Microscopic criteria in the grading of mucosal injury.
Grade Description
01
Normal mucosa Subepithelial space at villous tip
23
More extended subepithelial space Epithelial lifting along villous sides
45
Denuded villi Loss of villous tissue
normal
moderatete
severe

22
Aims of the present investigation
This study was undertaken with the following aims:
Experimental studies I-III: to investigate changes of the various components of the cell mediated immunity within the intestinal mucosa during experimental sepsis.
to evaluate if the previously described changes of immune cell distribution in the small intestinal mucosa were paralleled by changes in TNF- concentrations in plasma and/or ascites and, furthermore, if pre-treatment with the immunomodulator Linomide influenced the mucosal immune cell distribution and the grade of mucosal damage during sepsis, and /or the TNF- levels.
to determine if pre-treatment with cyclooxygenase (COX) inhibitors in a septic model influences the grade of mucosal injury, the immune response and the rate of apoptosis, as well as if there was any difference in this respect between a selective COX-2 inhibitor as compared to a nonselective COX-1 and -2 inhibitor.
Clinical studies IV-V: to investigate whether translocation of enteric bacteria occurs and whether it is a frequent or rare event in localized or general peritonitis in man. In addition, we sought to study the association between translocation on the one hand and the immune cell response to peritonitis in MLNs as well as the plasma concentrations of TNF- , IL-6, IL-10 on the other.
to investigate the inflammatory response, measured as the concentrations of cytokines (TNF- , IL-6, IL-10) as well as changes in the population of T lymphocytes and macrophages in MLNs, in patients with malignant obstructive jaundice undergoing surgery. Furthermore, we wanted to study the possible association between changes in the amount of immune cells and the number of apoptotic cells.

23
Material and methods
Experimental studies (I, II and III) Animals and anaesthesia The experiments were performed using laboratory rats, because their repro-ducibility in sepsis models are well established and sampling methods for immunological and microbiological analyses exists. Rats with the same sex and age 12-16 weeks (correspond to young adults) were used because rodents appear to have age and gender-based differences in responses to infection and sepsis (6, 63, 150, 178). Male Wistar rats, weighing 220-300 g obtained from Möllegaard (Skensved, Denmark) were acclimated for at least one week in a standard animal care facility before start of the experiments. The rats were maintained according to the recommendations of the National Institutes of Health guidelines (121) for care and use of laboratory animals and the regional research animal ethical committee approved the experiments. The animals were kept in controlled temperature (22 ), humidity and 12-hour light/dark cycles and allowed food and water ad libitum. Anaesthesia was induced by intraperitoneal injection of Eqviticin, 0.432 ml/100 g b.w. (a mixture of chloralhydrate 18.2 mg/100 g b.w. and pentobarbital 4.2 mg/100 g b.w.) (Apoteket AB Production and Laboratories, Umeå, Sweden). Each rat were resuscitated with 37°C saline (2 ml/100 g b.w.) subcutaneously immediately after surgery. At harvesting cardiac arrest arose in anaesthetised animals after blood samples were taken from the portal vein and the aorta.
Experimental model The cecal ligation and puncture (CLP) model of sepsis was developed by Wichterman, Baue, and Chaundry and has been used as a standard model for sepsis in more than 3 decades (187). CLP leads to polymicrobial peritonitis, bacteraemia and cytokine responses caused by leakage of faecal material, and mimics the human scenario of a perforated bowel. Unlike peritoneal inoculation models, there is no need to quantify the amount of bacteria. Standardizing the size of the needle used for puncture, the number of punctures and the length of cecum ligated leads to predictable mortality (164). Aged mice have worse survival than younger mice after CLP and antibiotic treatment in younger mice leads to improved survival but is not

24
beneficial in older mice (178). The CLP model has many advantages compared to Lipopolysaccharide (LPS) injections or bacterial infusion models, which react more severely and have an increased serum cytokine responses (143), much higher than in human sepsis. Immunotherapy for sepsis based on cytokine production after LPS challenge is misdirected because the LPS model does not accurately reproduce the cytokine profile of sepsis. It is conceivable that rapid endotoxin administration may result in death due to the effects of the intense inflammatory response on cardiovas-cular and pulmonary physiology.
In contrast, death at later time points following CLP may be due to multi organ failure, as in most septic humans. The validity of CLP has been reported i.e. in publications showing that the blockade of TNF- failed to prevent death, results similar to that observed in human clinical trials (142). The accelerated expression pattern of inflammatory cytokines in severe sepsis supports the notion that early and aggressive therapy is important in treating multi organ injury syndromes (180). Furthermore, the CLP model can be manipulated to give either lethal or nonlethal sepsis.
Pilot series A. Evaluation of the mortality 48h following CLP (n=5) in the experimental
model.
B. Mucosal grading in lamina propria (LP) at different time points (6, 12, 24 and 48 h) following CLP or sham operation (10 cm from caecum, n=5).
C. Evaluation of TNF- levels (pg/mL) in rat plasma at different time points (1, 2, 3, 6 and 24 h) following CLP (n=4).
Experimental groups All animals in the separate studies were handled in an identical way and divided into different groups (Table 5).
CLP (I-III) group. The abdomen was opened with a 2.5 cm midline incision through the linea alba. The cecum was isolated and ligated just below the ileocecal valve with 4-0 silk. Thereafter both cecal walls were punctured twice with a 23-gauge needle. The bowel was repositioned into the abdomen and the incision was closed.
Sham (I-III) group (controls). Sham-treated rats underwent midline laparo-tomy as CLP- animals with cecal manipulations but without ligation and puncture.
Non operated (I) group. They were only subjected to anaesthesia before harvest.

25
Linomide (II) groups. Linomide in a dose of 160 mg/kg b.w. was adminis-tered via a gastric feeding catheter (p.o.) or intra peritoneally (i.p.) 24 hours before sham operation or CLP.
SC-236 (III) groups. 1 mg/kg of SC-236 was given p.o. 21 h and 1 h before operation to respectively sham and CLP group.
Indometacin (III) groups. 3 mg/kg of indometacin was administered orally 21 h and 1 h before operation to respectively sham and CLP group.
Table 5. Experimental design and analysis (I-III).
Study Groups n n Sample / Method / Analysis
I Sham
CLP
Non.op
8
11
4
Mucosal / HE-staining / Morphology
Mucosal / Immunohistochemistry / Immune cell distribution (mAbs: OX8, W3/25, OX6, R73, ED1)
II
Sham
Sham + Linomide p.o.
Sham + Linomide i.p.
CLP
CLP + Linomide p.o.
CLP + Linomide i.p.
A (24 h)
8
10
9
8
8
8
B (3 h)
8
8
8
12
7
7
Mucosal / HE-staining / Morphology (A)
Mucosal / Immunohistochemistry (A) / Immune cell distribution (mAbs : OX8, W3/25, OX6, R73, ED1)
Plasma / ELISA / Cytokine; TNF- (B)
Ascitic fluid / ELISA / Cytokine; TNF- (A)
III
Sham
CLP
Sham + SC-236
CLP + SC-236
Sham + ind.
CLP + ind.
A (6 h)
8
11
10
10
9
9
B (24 h)
8
11
9
15
11
10
Mucosal / HE-staining / Morphology (A, B)
Mucosa, MLN / Immunohistochemistry (B) / Immune cell distribution (mAbs: OX8, W3/25, OX6, R73, ED1) Apoptosis (caspase-3)
Plasma / ELISA / Cytokines; TNF- ,IL-6 (A, B)
Ascitic fluid / Cytokines; TNF- , IL-6 (A, B)
n = number of animals in the different study groups. A and B present time at harvest. CLP = cecal ligation and puncture, ind. = indometacin, HE= hematoxylin-eosin, MLNs = mesenterial lymph nodes, mAbs = monoclonal antibodies, TNF- = tumour necrosis factor- , IL-6 = interleukin-6, ELISA = enzyme-linked immunosorbent assay.

26
Drug treatments Linomide; LS2616, a synthetic quinoline derivative (Pharmacia-Upjohn, Inc., Stockholm, Sweden), is an immunomodulatory drug that has displayed beneficial effects in several models of inflammation. Linomide has been demonstrated to have a life-preserving effect in sepsis (49). Gonzalo et al have shown an anti-apoptotic effect on peripheral T cells and partial inhibi-tion of the secretion of TNF- after injection of bacterial to mice (49, 50). In vivo, Linomide has been documented to suppress the up-regulation of TNF- and concomitantly increase the expression of anti-inflammatory cytokines as IL-10 (34), and also decrease leukocyte- recruitment and extravasation (82, 197). Li et al. have demonstrated that the protective effect of Linomide in endotoxemic mice induced liver injury was mediated via upregulation of IL-10 (91).
SC-236 and indometacin are both nonsteroidal anti-inflammatory drugs (NSAIDs). SC-236 is a specific COX-2 inhibitor (Searle; Skokie, Illinois, USA) and indometacin is a nonselective COX inhibitor (Confortid Alpharma AB, Stockholm, Sweden). COX-1 is constitutively expressed in most tissues, whereas COX-2 is induced by cytokines, mitogens and endotoxins in inflammatory cells, is responsible for the elevated production of PGs during inflammation (94, 111). Inhibition of COX-2 accounts for the anti-inflammatory and analgesic action of NSAIDs; however, concurrent inhibition of COX-1 inhibits PG-dependent mechanisms such as gastroduo-denal mucosal defence, and platelet aggregation (15, 59). NSAIDs blocks an important negative feedback mechanism sustained by PG and consequently increases TNF production.
Clinical studies in humans with peritonitis (IV) and malign obstructive jaundice (V) The Research Ethics Committee at Uppsala University approved the studies IV and V and all patients gave informed consent before entering the study.
The immune response was studied in patients accepted for surgery due to peritonitis or malign obstructive jaundice (Table 6).

27
IV
During the period February 1997 to December 1999, 20 patients with local peritonitis (LP) and 15 patients with general peritonitis (GP) submitted for an acute operation were investigated. The main inclusion criteria were clinical signs of peritonitis complemented by a typical history and inflamma-tory blood parameters. Eighty percent of the patients with general and 35% with local peritonitis met the criteria for SIRS/sepsis.
V
Forty-five patients were recruited during the period February 1997 to May 2000. Entry criteria included patients with clinical and radiological findings consistent with a tumour in the hepatic-pancreatic-biliary (HPB) region and accepted for surgery. Patients with obstructive jaundice (HPB+ group) were compared to those without jaundice (HPB- group). Nine out of 18 patients in the HPB+ group had persistent jaundice despite previous endoscopic or percutaneous biliary drainage efforts.
Study groups Controls (C group) (IV and V). Twelve patients, 3 men and 9 women with a mean age of 49 (range, 23-70) years, had elective open surgery for gastric disorders or repair of incisional hernias.
Local peritonitis (LP group). Twenty patient (mean age 35 years, range 20–74; 17 men) presented with local peritonitis and clinical suspicion of acute appendicitis. Eighteen of the patients were found to have acute appendicitis.
General peritonitis (GP group). Fifteen patients, 8 men and 7 women with a mean age of 62 (range, 21-84) years were operated on for different gastroin-testinal disorders leading to general peritonitis.
HPB- group. Fifteen patients (mean age, 69 years, range 47-89; 9 men) were investigated; the majority of the patients had primary or secondary (metastasis) liver tumours.
HPB+ group. Thirteen men and five women with a mean age of 58 (range, 24-78) years were included, the diagnosis pancreatic cancer were most common in the group.

28
Table 6. Experimental design and Analysis (IV–V).
Study Groups n Sample / Method / Analysis
IV LP Local peritonitis
GP General peritonitis
C * Control
20
15
12
MLNs, Faecal / Culturing; Aerobic, anaerobic bacteria, PFGE typing
MLNs / Immunohistochemistry / Immune cell distribution (mAbs: CD4, CD8, CD68)
Blood / WBC, CRP
Blood, Ascitic fluid / Culturing; Aerobic, anaerobic bacteria
Plasma / LAL / Endotoxin
Plasma / ELISA / Cytokines; TNF- , IL-6, IL-10
V HPB –
HPB +
15
18
MLNs / Culturing; Aerobic, anaerobic bacteria
MLNs / Immunohistochemistry / Immune cells distribution (mAbs: CD4, CD8, CD68) Apoptosis (caspase-3)
Blood, Ascitic fluid / Culturing; Aerobic, anaerobic bacteria
Blood / WBC, CRP, Bilirubin, ALP, ALAT, ASAT
Plasma / LAL / Endotoxin
Plasma / ELISA / Cytokines; TNF- , IL-6, IL-10
n = number of patients in the different study groups. HPB- = hepato-pancreatic-biliary (HPB) tumour disease without obstructive jaundice; HPB+ = HPB tumour with jaundice. MLNs = mesenterial lymph nodes, mabs = monoclonal antibodies, TNF- = tumour necrosis factor- , IL-6/-10 = interleukin-6/-10, WBC = white blood cell count, CRP = C-reactive protein, ALP = alkaline phosphatase, ALAT = alanine aminotransferase, ASAT = aspartate aminotransferase, LAL = limulus amoebocyte lysate, ELISA = enzyme-linked immunosorbent assay, PFGE = pulsed-field gel electrophoresis. * These patients were also used as controls in study V

29
Sampling and analyses (I-V) Sampling of ascitic fluid, blood, small- bowel biopsies and MLNs Immediately after opening the abdomen ascitic fluid was obtained (II-V). Blood samples were taken from the portal vein and the aorta, respectively (II-III) and in the clinical studies from the brachial vein. The samples were separated by centrifugation at 2500 g for 10 minutes at 4°C, and the plasma stored at -70°C until assayed (Table 5-6).
Two specimens, each 8-15 mm long, from the distal third of the small bowel (I-III) and two lymph nodes from the ileocolic mesentery (MLNs) (II-V) were excised. One of the specimens was immediately frozen in liquid nitrogen and stored at -700C for future immunohistochemical staining, while the other was fixed in buffered 4% formalin and routinely processed and stained with haematoxylin- eosin. In studies IV-V one of the MLNs was cleaned in 70% ethanol and then stored at - 700 C for later bacterial cultures.
Bacterial culturesSamples of venous blood were injected into aerobic and anaerobic blood culture media, respectively, three times at 5-min intervals and incubated at 35 C. If there was any bacterial growth, the sample was spread onto appropriate agar medium and the bacteria were identified using standard bacteriological techniques. Peritoneal exudate was analysed for bacterial growth using standard methods (IV-V). The MLN was thawed, homogenized and then spread onto 5% blood agar and J-agar (for anaerobic bacteria), respectively, and incubated at 35 C for 2 days (IV-V). Faecal samples from the rectum were obtained before surgery and frozen for subsequent culture (IV).
Pulsed-field gel electrophoresis (PFGE)Gel electrophoresis is a technique used for the separation of nucleic acids and proteins, for identification of particular DNA molecules by the band patterns they yield in the gel after being cut with various restriction enzymes. Separation of large (macro) molecules depends upon charge and mass, as well as the frictional force of the gel, which acts as a "molecular sieve". PFGE is a technique in which the direction of current flow in the electropho-resis chamber is periodically altered. This allows fractionation of pieces of DNA ranging from 50,000 to 5 million base pairs, which is much larger than can be resolved on standard gels (26). When adequate migration has occurred, DNA fragments are visualized in an ultraviolet transilluminator after staining with a fluorescent dye, ethidium bromide.

30
In patients with growth of Gram-negative bacteria in the MLN, PFGE typing was performed on representative subsets of E. coli isolates from the MLN and the faecal sample (IV). Bacterial isolates showing indistinguish-able or closely related DNA patterns (< 6 band differences) were regarded as clonally related indicating translocation (173).
Cytokine measurement A sandwich-type enzyme-linked immunosorbent assay (ELISA) is a method that determines cytokines on the basis of their reactivity with specific antibodies. TNF- V IL-6 (III-V) and IL-10 (IV-V) levels in plasma and ascites fluid were determined by ELISA technique according to protocol pro-vided by the manufactures.
Endotoxin measurement The analysis was performed using a chromogenic limulus amoebocyte lysate (LAL) assay for 0.06–0.6 endotoxin units (EU)/mL (IV-V). For calculation purposes, non-measurable levels were assigned a value of 0.001 and the standard curve was used to extrapolate values up to 0.7.
Monoclonal antibodies For the immunohistochemical staining in the experimental studies (I-III), the following monoclonal antibodies (mAbs) were used: OX8 reactive with CD8 antigen on cytotoxic T lymphocytes and natural killer cells; W3/25 reactive with the CD4 antigen on T helper cells and peritoneal macrophages (136, 186); OX6 reactive with MHC class II antigens (113); R73 reactive with the
/ antigen receptor on T lymphocytes (73, 105) and ED1 reactive with monocytes, macrophages and dendritic cells (36, 166). The mAbs used in the clinical studies (IV-V) are specific for CD4, CD8 and CD68, mainly staining CD4+ helper T lymphocytes, CD8+ cytotoxic T lymphocytes and macrophages, respectively (137, 152).
Immunohistochemistry and microscopic evaluation Cryostat sections, 6 m, from the small bowel (I-III) and from MLNs (III-V) were placed on slides before fixation in 100% acetone for 10 minutes. The sections were stained using a peroxidase-antiperoxidase (PAP) technique (170). In brief, they were incubated in 0.3% H2O2 in phosphate buffered saline to inhibit endogenous peroxidase, with normal goat serum to prevent non-specific background staining and finally with the primary mabs. In the next step a secondary antibody, goat anti-mouse IgG1 was applied.

31
The last incubation was performed with horseradish peroxidase-mouse antiperoxidase. The slides were processed with H2O2 and 3-amino-9-ethyl-carbazole (AEC). Finally, the sections were counterstained with Mayer's haematoxylin. The slides were coded and the microscopic evaluation performed in a blinded manner by one examiner. The numbers of positive cells within the intestinal mucosa (I-III) and MLN (III-V) were graded arbi-trarily for every single antibody on a scale from 0 to +++. In addition, all slides were ranked manually according to the number of positive cells. The statistical analysis was based on this ranking.
Staining for apoptosisBriefly, paraffin specimens from mucosa (III) and MLNs (III, V) were deparaffinized in Xylen. The antigen retrieval was done in 10 mM EDTA (pH 8.0). The tissues were incubated in 3 % H2O2 in phosphate buffered saline and normal goat serum, before adding the primary antibody rabbit anti-active caspase-3 (Cell Signaling Technology, Inc., Beverly, MA, USA). After incubation with a secondary goat anti-rabbit antibody and washing, an immunoperoxidase system, ABC-Elite (Vector Laboratories, Inc., Burlingame, CA, USA), was applied. Slides were rinsed and developed with 3.3-diaminobenzidine, DAB (Sigma-Aldrich Inc., St Louis, USA). Finally, the sections were counterstained with Harris’s haematoxylin.
Failure to detect caspases could be due to low levels of enzyme expression, test applied within a very brief time period only, or to a caspase-independent mechanism of apoptotic cell death. We only evaluated one specimen from each tissue, which possibly can influence the outcome of the apoptosis rate. The numbers of positive cells were graded from 0 to +++ and ranked (III, V).
Colour based image analysisAn automatic computerised method for quantification of images of immuno-histochemically stained cells was used in study II (138, 139). This method was developed in the Centre for Image Analysis at Uppsala University, Sweden. For every group stained with the same monoclonal antibody a classifier was created. Stained cells in lamina propria are classified as positive pixels. The positivity in percentage was expressed for all stained cells within eight villi of each specimen.
StatisticsA probability (P) of < 0.05 was considered statistically significant. We assumed the distribution of data to be non-parametric. Continuous variables were expressed as median (range) (I-V). The Wilcoxon rank-sum test was used to test differences in morphology and immunohistochemical grading and

32
ranking between the groups (I-V). A two-way analysis of variance (ANOVA) with repeated measures was used to test differences in cytokine concentrations (II-V) and other inflammatory blood parameters (IV-V) between groups. The Wilcoxon matched-pairs signed-rank test was used to evaluate pre- and postoperative differences in cytokine concentrations within the groups (IV).The Mann-Whitney U test was used to compare physiological and chemical parameters between subgroups of patients or animals (III-V). The Pearson correlation coefficient was used to evaluate the relationship between the concentrations of cytokines and endotoxins (IV). Fisher’s exact test was used to compare the apoptosis rate between the groups (III, V).

33
Results and comments
Below is given a summary of the results considered most important followed by comments. The individual papers (I-V) are to be consulted for details.
Clinical appearance (I-III) and mortality after CLP (Pilot series A)
Twenty-four hours following CLP all animals demonstrated signs associ-ated with late sepsis; tachypnoe, hypothermia, impaired skin circulation, piloerection, weakness, conjunctivitis, anorexia and lethargy.
Macroscopic acute diffuse fibrinopurulent peritonitis existed in all CLP animals at harvest 24 h following induction.
The fibrin deposits in septic rats were unchanged after pre-treatment with Linomide or COX-inhibitors.
In the pilot series A, was the mortality 60% (3/5) 48 h after CLP.
CommentsIn this CLP model cecum was punctured twice with a 23-gauge needle (intermediate diameter), which gives peritonitis and a slowly (1-2 days) de-veloping sepsis with a low initial mortality (126). This type of model is use-ful for studying more long-term effects of the septic states, including immu-nological changes. Twenty-four hours after CLP, we noted fibrin formations around the cecum. This is the result of an important step in the host defence, stimulated by TNF-(42). During the early phase of peritonitis endogenous TNF stimulate nonlym-phoid cells such as granulocytes and macrophages to ingest bacteria, and platelets and fibroblasts to localize the inflammation. We were not able to see any macroscopic differences in the fibrin spreading in animals pre-treated with Linomide or COX inhibitors. The initial production of TNF- must have been enough for the induction of the fibrin production, despite decreased TNF- level after Linomide treatment. In contrast COX inhibitors increased the TNF-
levels in ascites 24 h following CLP, but this was not apparent when inspect-ing the fibrin clots.
34
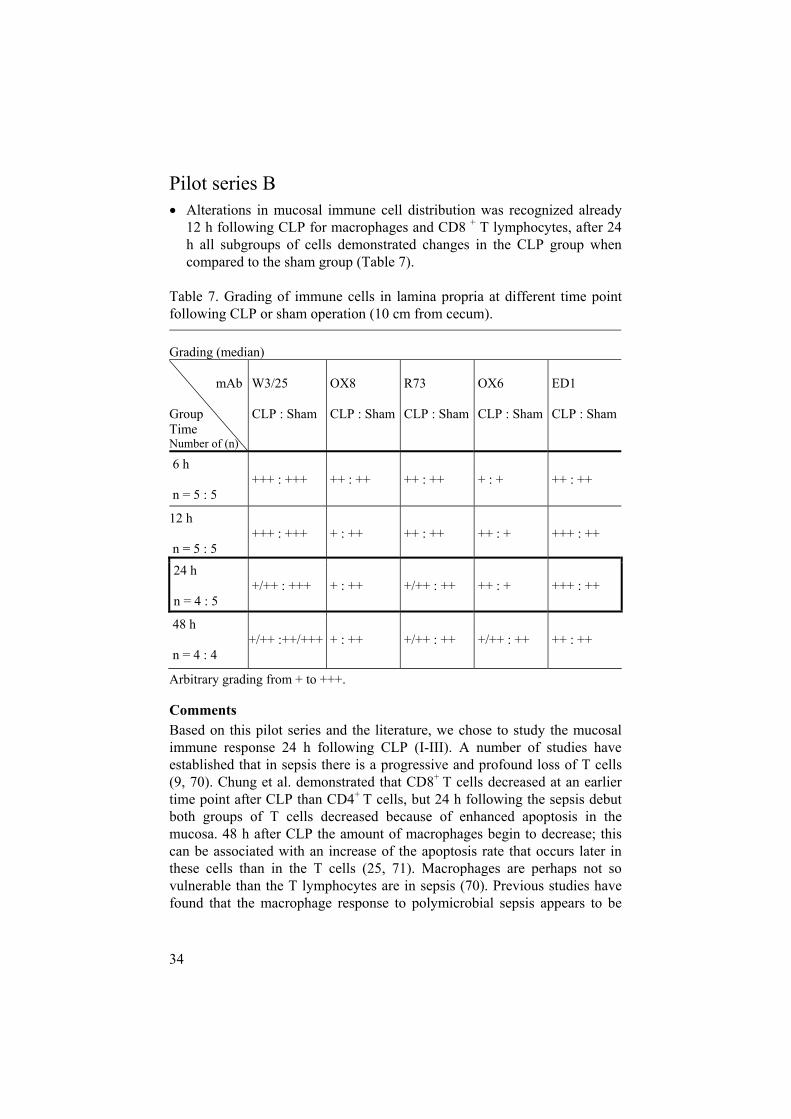
Pilot series B Alterations in mucosal immune cell distribution was recognized already 12 h following CLP for macrophages and CD8 + T lymphocytes, after 24 h all subgroups of cells demonstrated changes in the CLP group when compared to the sham group (Table 7).
Table 7. Grading of immune cells in lamina propria at different time point following CLP or sham operation (10 cm from cecum).
Grading (median) mAb Group Time Number of (n)
W3/25
CLP : Sham
OX8
CLP : Sham
R73
CLP : Sham
OX6
CLP : Sham
ED1
CLP : Sham
6 h
n = 5 : 5 +++ : +++ ++ : ++ ++ : ++ + : + ++ : ++
12 h
n = 5 : 5 +++ : +++ + : ++ ++ : ++ ++ : + +++ : ++
24 h
n = 4 : 5 +/++ : +++ + : ++ +/++ : ++ ++ : + +++ : ++
48 h
n = 4 : 4 +/++ :++/+++ + : ++ +/++ : ++ +/++ : ++ ++ : ++
Arbitrary grading from + to +++.
CommentsBased on this pilot series and the literature, we chose to study the mucosal immune response 24 h following CLP (I-III). A number of studies have established that in sepsis there is a progressive and profound loss of T cells (9, 70). Chung et al. demonstrated that CD8+ T cells decreased at an earlier time point after CLP than CD4+ T cells, but 24 h following the sepsis debut both groups of T cells decreased because of enhanced apoptosis in the mucosa. 48 h after CLP the amount of macrophages begin to decrease; this can be associated with an increase of the apoptosis rate that occurs later in these cells than in the T cells (25, 71). Macrophages are perhaps not so vulnerable than the T lymphocytes are in sepsis (70). Previous studies have found that the macrophage response to polymicrobial sepsis appears to be

35
tissue-specific, i.e. the rise in the apoptotic macrophage count peaked earlier in the liver (6 h) than in the lungs (48 h) (163).
Pilot series C
Fig 1. TNF- changes in plasma following CLP. Mean values ± SEM.
CommentsThe plasma level of TNF- was demonstrated to peak at 3 h following CLP (* P < 0.05 vs. 1h and 24h, Anova). This result determined the experimental model in study II and III.
Mucosal morphology (I-III) Microscopic examination of haematoxylin-eosin stained tissue specimens revealed normal appearance of the mucosa (range 0-1) in the sham operated animals (I-III) as well as in the non-operated animals (I). In all the studies there were a significant difference between untreated peritonitis (CLP) and control (sham) animals.
When Linomide was given intraperitoneally (II) before CLP, a lesser degree of mucosal damage was seen as compared to untreated peritonitis. This was not found after peroral distribution.

36
The median grade of mucosal damage was higher 24 h following CLP (III, series B) compared to the animals harvested 6 h after CLP (III, series A). CLP-animals pre-treated with SC-236 had a lesser degree of mucosal damage as compared to rats with untreated peritonitis or indometacin treated septic rats (III, in both series A and B).
Fig 2. Grade of mucosal injury (Wilcoxon rank-sum test). * p<0.05 vs. Sham, ** p<0.01 vs. Sham, # p<0.05 vs. CLP, ¤ p<0.05 vs. SC+CLP.
CommentsStudies I-III verifies that sepsis causes superficial injury to the villous part of the intestinal mucosa (192). Overall, only a few animals showed a deeper injury in the villi ( grade 4), which made it possible to grade the cell mediated immune response immunohistochemically. Deep mucosal injury has not been seen in the present or in similar experimental models (44). The septic animals in study I had a median grade 1 injury and in the other studies (II and III, series B) they had a median grade 3 injury. The difference could be due to the technique of evaluation, in study I the highest grade of injury given the mucosa was depending on the predominant findings. In the two latter studies the highest grade of mucosal injury was used. This grade did not have to be present in a majority of the observed villi.
The pathogenesis of this injury involves hypoxia due to decreased oxygen delivery, uptake and utilization (95). Increased release of proinflammatory cytokines such TNF- may contribute to this effect. Animals injected with TNF- intravenously have showed a superficial injury to the villi in other series (141).
When Linomide was given intraperitoneally before CLP, a less degree of mucosal damage was seen as compared to untreated peritonitis. This could
Morphology
0
1
2
3
4
5
I II III A III B
Study
Gra
de o
f muc
osal
inju
ry
(med
ian)
Sham
CLP
Lpo+CLP
Lip+CLP
SC+CLP
Ind+CLP
*
**
** ¤
** ¤
** ¤
** ¤
#
* **

37
possibly be an effect of inhibited TNF- secretion by macrophages in the lamina propria and in the peritoneal cavity or an anti-apoptotic effect on small bowel epithelium by Linomide (49, 50). It was demonstrated that the grade of mucosal injury increased with longer duration of the septic period for all the CLP groups (III; series A vs. series B) (192). An increased muco-sal damage impairs the mucosal barrier and leads to enhance risk for toxins and bacterial translocation (14, 31). We could demonstrate that pre-treatment with a selective COX-2 inhibitor (SC-236) attenuated the mucosal damage as compared to untreated peritonitis as well as in the CLP-group pre-treated with the nonselective COX inhibitor (indometacin) (177).
Distribution of immune cells in the mucosa (I-III)and in the MLNs (III)
In the lamina propria of the untreated septic rats there was a significant reduction in the number of CD4+ and CD8+ T lymphocytes as compared to sham. In contrast, 24 hours after induction of peritonitis the number of MHC class II positive cells and macrophages increased compared to sham (I-III).
Linomide pre-treatment, regardless route of administration, significant attenuated the number of macrophages and enhanced the amount of T lymphocytes observed following CLP (II).
Regardless pre-treatment with COX inhibitors there was a significant reduction of CD8+ cells in the EpL for all the CLP groups compared to their sham. Also both pre-treated sham groups had a reduced amount of CD8+ cells compared to untreated sham. COX inhibitors significantly attenuated the increase of macrophages in the LP and MLN seen in the untreated CLP group. Indometacin given before induction of CLP decreased the amount of CD4+ cells further in LP compared to untreated CLP animals, and it also decreased both subgroups of T lymphocytes in MLN compared to the other CLP groups (III).
CommentsNon-operated animals (I) showed an immunohistochemical pattern closely similar to that of the sham group. The increased number of macrophages in the septic group could be explained by recruitment of monocytes from the circulation, by activation of resident macrophages, or by a combination of both. Several different explanations are possible for the observed reduction of T lymphocytes in both EpL and LP. Evidence for increased T cells apoptosis in the mucosa following sepsis and shock has been put forward by

38
many authors (25, 69). Another reason could be that activated T cells have already left the LP for the intra-abdominal cavity or been homed to other parts of the GALT, but no increase of T lymphocytes was seen in the MLN following CLP. The increase in mucosa macrophages means also increased release of their cytokines, which could mediate the cellular immune system of the gut and influence other organ systems in the body leading to MODS (32).
After Linomide treatment the effects of peritonitis on immune cell distri-bution were attenuated. This could be a benefit for the cell-mediated immune response in sepsis. Other studies had demonstrated that Linomide inhibited programmed cell death in peripheral T and B-lymphocytes (50), which could explain our results regarding the T cells but not for the macrophages.
Both COX inhibitors attenuated the increase of macrophages observed in the mucosa and MLN after CLP. These effects could be associated with an increased rise in the apoptotic cell rate after COX inhibitors. Enhanced loss of macrophages can at a later stage of sepsis limit macrophage activity and impair a proper inflammatory response. On the other hand, it can be benefi-cial to reduce harmful cells excreting cytotoxic enzymes or cytokines. All CLP groups demonstrated a reduced amount of cytotoxic T lymphocytes in the EpL regardless the pre-treatment. This reduction can lead to a deterio-rated defence mechanism against MHC class I Ag. CLP + indometacin demonstrated a further decrease of CD4+ cells in LP and MLN compared to the other CLP groups. This might be an effect of the enhanced level of TNF- after indometacin treatment inducing and trigging lymphocyte apoptosis.

39
Immune cell distribution in the MLNs (IV-V) In the LP group, more CD8+ and CD68+ cells were found than in the control group. The GP group had a decreased number of CD4+ and CD8+
T lymphocytes compared with the LP group.
The two patients with proven translocations had lower scores for CD4+
and CD8+ cells than the average of their groups, while the number of CD68+ cells did not differ.
In the HPB+ group, more CD68+ cells were found than in groups HPB-
and C, while the number of CD4+ cells was decreased in the HPB+ group compared to the HPB- group and to controls.
Table 8. Distribution of immune cells in the MLNs. Scoring of positive cells for the antibodies CD4, CD8, and CD68 in the entire lymph node.
Median score Group n mAb: CD4 mAb: CD8 mAb: CD68 C 9/12 ++ IV +++ V ++ + LP 13/20 +++ +++ * ++ * GP 11/15 ++ # ++ # ++ HPB- 11/15 +++ ++ ++ HPB+ 12/18 ++ * † ++ ++/+++ * †
The figures represent the median score of positive cells for each single antibody on an ascending scale from + to +++ in the different groups.
C = controls; LP = local peritonitis; GP = general peritonitis; HPB- = hepato-pancreatic-biliary (HPB) tumour without obstructive jaundice; HPB+ = HPB tumour with jaundice. CD4 = T helper lymphocytes, CD8 = T cytotoxic lymphocytes, CD68 = macrophages. n = number of patients with investigated lymph nodes in the group / total number of patients in that group. * P <0.05 vs. group C, # P <0.05 GP group vs. LP group † P <0.05 group HPB+ vs. group HPB- (Wilcoxon rank-sum test).

40
Within the LP group, the reaction of the different immune cells in relation to the grade of appendicitis was evaluated. The number of CD4+ cells was significantly decreased in gangrenous appendicitis compared to phleg-monous type, while there were no significant differences in the number of CD8+ and CD68+ cells (Table 9).
Table 9. Differences in staining of the antibody CD4 in MLNs in relation to the grade of appendicitis.
MLNs = mesenteric lymph nodes, LP = local peritonitis. The distribution of CD4+
cells was studied in 11 of the patients in the LP group. *P < 0.01 (Wilcoxon rank-sum test).
CommentsIn the human studies we were limited to performing immunohistochemical investigations on MLNs excised from the ileocolic region. The localization of cellular components of the immune system within the MLN is more diffi-cult to grade and rank compared to the intestinal mucosa because of the larger number of lymphocytes. The reason why the GP group had a reduced
Grading score CD4 Grade of appendicitis
+++ Phlegmonous Phlegmonous Phlegmonous Phlegmonous Phlegmonous Phlegmonous * ++ Gangrenous Gangrenous
+ Perforated Perforated Gangrenous

41
amount of T lymphocytes compared to LP group is not fully understood. One explanation could be that in patients with general peritonitis the T cells leave the MLNs to enter inflamed peripheral tissues (20). Hotchkiss et al. reported that clinical abdominal sepsis induced lymphocyte apoptosis and caused depletion of CD4+ T lymphocytes but not depletion of macrophages in the spleen (71). The pattern we found when the distribution of subsets of immune cells was related to the grade of appendicitis indicates a similar chain of events.
Peritonitis patients with proven bacterial translocation had lower scores for T cells in MLNs than the average of their groups. It has previously been demonstrated that translocation of E. coli occurs to a greater extent in mice with T cell depletion in MLNs, lamina propria and intestinal epithelium than in normal mice (47). In patients with obstructive jaundice we demonstrated an attenuation of T helper cells compared to patients without jaundice and controls. An elevated bilirubin concentration could stimulate cytotoxic responses, and, thus, lead to an increased apoptosis (157, 159).
Apoptosis in the mucosa (III) and MLNs (III and V) The apoptosis rate in the small intestinal mucosa was significantly higher in the CLP group compared to the sham group, and the grading of apoptosis was enhanced in EpL and MLNs compared to the LP. However, no significant difference was demonstrated between the CLP groups. Sloughed cells positive for apoptosis were observed in the intestinal lumen, and in a higher number in the CLP groups compared to sham.
The apoptosis rate enhanced in MLN and in epithelial cells, but not in the LP of the sham animals when COX inhibitors were given. Indometacin pre-treated sham animals had a significantly increased degree of apoptosis compared to untreated sham group in MLNs.
In study V apoptosis occurred in all groups but a significantly higher frequency of apoptosis was found in group HPB+ compared to the other groups. The majority of positive stained cells were located in the paracor-tex area of the MLNs (Table 10).

42
Table 10. Immunohistochemical staining for apoptosis (active caspase-3) in mesenteric lymph nodes.
The figures represent the score for stained cells positive for apoptosis on an ascend-ing scale from - to ++ in the different groups. C = controls; HPB- = hepato-pancreatic-biliary (HPB) tumour disease without obstructive jaundice; HPB+ = HPB with obstructive jaundice. P < 0.05 vs. C group; † P < 0.05 HPB+ group versus HPB- group (Wilcoxon rank-sum test, Fisher’s exact test).
CommentsIn the normal villous, active caspase-3 expression was found infrequently, but following CLP a significant increase was observed in both EpL and LP.
An up-regulated apoptosis of mononuclear cells has been observed in animal models of sepsis as well as in patients suffering from severe sepsis (69). NSAIDs promote apoptosis through mechanisms that are both COX-dependent and -independent. In sham animals the nonselective COX inhibi-tor seem to enhance the apoptosis rate more than the selective one.
An increased apoptosis in HPB+ group could contribute to a reduced cellular defence and, thus, be a further explanation why jaundice is associ-ated with an elevated risk for septic complications.
Cytokine analysis: TNF- (II-III) and IL-6 (III) The levels of TNF- and IL-6 were significant higher in ascitic fluid than in circulating blood for all the groups. CLP groups had significant higher IL-6 concentrations in ascitic fluid in series A compared with series B (III).
Linomide p.o. or i.p. before CLP led to significant decreased TNF- levels in ascitic fluid and Linomide i.p. gave also a reduction in portal samples compared to untreated septic animals (II).
Sham animals, pre-treated with COX inhibitors, had a slight increase of the TNF- in ascitic fluid, and the level was higher after treatment with indometacin than with SC-236 in both series in study III. Indometacin
Group - + ++ median score % pos. staining C 6 2 0 - 25 (2/8) HPB- 8 1 2 - 27 (3/11) HPB+ 4 3 6 + * † 69 (9/13) * †

43
given before CLP caused significantly increased TNF- levels in ascitic fluid compared to the other CLP groups (III, series A). CLP animals pre-treated with indometacin had significantly enhanced IL-6 levels in aortic and portal vein samples compared to sham and to the SC-236 treated septic animals. On the other hand only SC-236 pretreat-ment significantly attenuated the IL-6 concentration in plasma and in ascitic fluid compared to untreated septic animals (III, series A).
CommentsThe exaggerated cytokine responses in ascitic fluid compared to blood may indicate more activity at the local site of inflammation and infection (perito-neum) than systemically. These findings are in agreement with previous reports, experimental as well as clinical (41, 67, 110). After Linomide treat-ment we noted a decrease in TNF- level in ascites, which is likely to be the effect of inhibited TNF- secretion by the peritoneal macrophages (49). An altered cytokine release after COX inhibitors could be explained by the fact that administration of NSAID interrupts a negative feedback loop exerted by PGs, allowing mononuclear cells to increase their production of proinflam-matory cytokines (86, 104). Treatment with a nonselective COX-2 inhibitor enhanced the proinflammatory cytokine release after CLP compared to a selective inhibitor, indicating an effect on the balance between the pro- and anti-inflammatory states.
Cytokine analysis: TNF- , IL-6 and IL-10 (IV-V) Preoperatively, the concentrations of TNF- , IL-6 and IL-10 were increased in the GP group compared with the other groups. In the LP group, only the IL-6 level differed significantly from controls. After 24 h, the cytokine plasma concentrations in the two peritonitis groups decreased, although the GP group still had significantly higher IL-6 and IL-10 levels than the other groups (IV).
IL-6, IL-10 but not TNF- , were higher in septic patients compared with nonseptic patients (IV).
Within the HPB+ group, there were no differences in cytokine concentra-tions between patients with or without previous biliary drainage procedures.
Twenty-four hours after start of operation, the cytokine levels, except those of TNF- , increased in all groups. The IL-6 concentration in the HPB+ group demonstrated the highest value and differed significantly to the other groups at this point of time (V).

44
Table 11. Inflammatory blood parameters obtained preoperatively and 24 h after start of the operation. Cytokine Group Preoperative 24 h postoperative
C 3.5 (0.8-17.9) 4.6 (0.5-17.0) TNF- HPB- 2.8 (1.4-13.5) 4.8 (0.7-14.7) (pg/mL) HPB+ 6.2 (0.9-13.7) 4.5 (2.3-15.0) LP 5.5 (1.1-21.7) 4.4 (1.1-17.0) GP 9.2 (2.4-118.7) * † 11.3 (4.9-19.3) C 3.4 (0.6-14.4) 83.5 (29.2-256.5) ‡ IL-6 HPB- 3.9 (0.4-98.2) 103.5 (7.9-563.5) ‡ (pg/mL) HPB+ 11.9 (2.8-69.2) 266.9 (24.1-551.9) * † ‡ LP 46.4 (5.7-422.1) * 31.7 (6.4-398.7) GP 390.6 (176.7-522.3) * † 268.6 (36.7-467.6) * † ‡ C 3.5 (1.4-23.8) 15.1 (9.5-44.1) ‡ IL-10 HPB- 5.6 (1.6-21.8) 16.6 (7.0-88.1) ‡ (pg/mL) HPB+ 9.0 (2.5-18.3) 24.8 (9.4-344.6) ‡ LP 25.7 (2.9-174.7) 12.7 (4.9-90.0) GP 42.6 (25.6-852.8) * † 34.9 (9.6-415.0) * †
C = controls; HPB- = hepato-pancreatic-biliary (HPB) tumour without obstructive jaundice; HPB+ = HPB tumour with jaundice; LP = local peritonitis; GP = general peritonitis; TNF- =tumour necrosis factor alpha; IL = interleukin. Data are presented as median value (range). * P < 0.05 vs. C group; † P < 0.05 HPB+ vs. HPB- or LP vs. GP (Anova); ‡ P < 0.05 for 24h post- versus preoperative values (Mann-Whitney U-test).
CommentsThe operative interventions led to a pronounced inflammatory response, demonstrated by significantly increased concentrations of IL-6 and IL-10 24 hours after the start of surgery. Eighty percent of patients with GP and 35% with LP met the criteria for SIRS/sepsis. Clinical sepsis was diagnosed at time of surgery using previously defined criteria for sepsis (16). Septic patients had higher CRP and IL-6 values compared to non-septic patients. The two patients with proven translocation demonstrated cytokine concen-trations above the median. IL-6 is rapidly released and has a short plasma half-life but is readily measured, in contrast to TNF- . For this reason, and because IL-6 levels change more quickly than the CRP levels, this cytokine is clinically best defined. It may have a prognostic value in patients with peritonitis, detecting early complications after surgery, and may also be useful for the control of therapy. However, no significant difference in IL-6 levels was seen in the subgroups of appendicitis. Other studies have demon-strated that elevated concentrations of TNF- and IL-6 are correlated with mortality (109, 134, 144), and high concentrations of IL-10 have been asso-

45
ciated with an increased risk of developing infectious complications (191). There was no 30- days mortality in any of the groups in IV-V, but these studies are to small for making any statistical analysis of correlations between cytokines levels and morbidity or mortality. Increased concentra-tions of IL-10 were found in the GP and HPB+ group postoperative. IL-10 is an anti-inflammatory cytokine that is activated later in the inflammatory process (10, 78). Increased levels of IL-6 have been demonstrated in patients with obstructive jaundice (80) while increased TNF- levels are more infrequently reported (78).
The IL-10/TNF- ratio (IV-V) The IL-10/TNF- ratio was increased preoperatively in both peritonitis groups compared with controls. The ratio was higher in septic patients compared with nonseptic patients.
The two patients with proven translocation of bacteria to MLNs had pre-operatively IL-10/TNF- ratio values above the median in their group.
IL-10/TNF- ratio was higher in the HPB+ group preoperatively com-pared to the others (V), but the difference did not read statistical signifi-cance. Both HPB groups had higher ratio postoperatively, than preopera-
tively and higher ratio than the peritonitis groups.
Fig 3. The IL-10/TNF- ratio (IV-V). C = controls; HPB- = hepato-pancreatic-biliary (HPB) tumour without obstructive jaundice; HPB+ = HPB tumour with jaundice; LP = local peritonitis; GP = general peritonitis.

46
Comments
The IL-10/TNF- ratio may be of value for evaluating the inflammatory response and the balance between pro- and anti-inflammatory cytokine activation after injury, burn and sepsis. IL-10 inhibits many of the proinflammatory effects of TNF- and a high ratio of IL-10 /TNF- may be linked to coinfections. An elevated IL-10 level can indicate a state of immune paralysis.
Bacterial cultures (IV-V) Positive MLN cultures were obtained in both peritonitis groups but not in controls. In the GP group, 2 out of 12 MLN cultures were positive. The corresponding figure in the LP group was 2/19. Ten of 15 GP patients had a positive bacterial culture in peritoneal fluid. Blood cultures were positive in 3/15 patients in the GP group but in none of the other groups.
Two patients (one from the LP and one from the GP group) had culture-positive MLNs and sterile peritoneal fluid. PFGE typing was performed on subsets of E. coli isolates from MLNs and the faecal samples in these two patients. Identical DNA restriction patterns and a clonal relationship were found from faecal colonies and the MLN in both patients, proving translocation of E. coli from the intestinal bacterial flora to MLNs.
In study V no patient demonstrated enteric bacteria in MLN cultures. In blood cultures, two patients in the HPB+ group demonstrated Gram-negative bacteria (E. coli and pyogenes), while all other blood cultures were sterile. Peritoneal fluid was also sterile in all but one patient.
CommentsIn the present study translocation of bacteria was unequivocally proven to occur in 2 out of 35 (6%) patients with local or general peritonitis. However, this proportion is much less than reported from animal experiments, especially from experiments on rodents in which translocation is often reported to occur in 100% of cases (31, 183). It is believed that the impair-ment of the intestinal mucosal barrier occurring in the critically ill facilitates translocation of bacteria. However, it has been demonstrated experimentally that the quality of the intestinal bacterial flora is more important than gut mucosal injury in determining whether or not translocation will take place, e.g. during sepsis (184, 96). Moreover, bacterial translocation has been dem-onstrated to be associated with a higher incidence of postoperative septic morbidity in patients undergoing laparotomy (124, 156).

47
In the present study, translocation of E. coli to MLNs was found in 4 peritonitis patients (2 in groups LP and GP, respectively) but not in controls. However, two of the patients with positive MLN cultures (one from each group) also had E. coli in samples from peritoneal fluid. We cannot prove that the bacteria found in the MLNs and in the peritoneal fluid were identical in these two patients and therefore we cannot rule out that the growth in the MLNs could have been caused by other mechanism than translocation. Thus, translocation of E. coli from the intestinal bacterial flora to MLNs was clearly shown in two patients by PFGE analyses.
Experimentally bacterial translocation has been demonstrated fairly frequently in obstructive jaundice (162, 185), but more rarely in the clinical situation (77, 87). In the present study, translocation of enteric bacteria to MLNs was not demonstrated in any patient. There might be several explanations to this:
1. Bacterial translocation does not occur to any measurable extent in patients with malignant obstructive jaundice and is of minor importance in the development of septic complications in such patients.
2. Identification of translocating bacteria to MLNs was only made on one occasion. One can only speculate if repeated sampling would give other results.
3. In the present study, conventional culture methods and not PCR techniques were used. The latter may find higher rates of bacterial translocation compared with culture methods, but the timing of the translocating event cannot be decided with PCR (154). Therefore, the obvious risk of contamination and false positive results must be taken into consideration when using sensitive DNA-based techniques such as PCR in this context.
4. The use of preoperative antibiotics may have influenced the number of negative culture results in the present studies. All included patients had antibiotics given 1-3 hours prior to harvesting of MLNs, in accordance with the guidelines for surgical treatment of these diseases in our department.
Endotoxin concentrations (IV-V) The GP group demonstrated the highest concentrations of endotoxin in plasma with a median value of 0.068 EU/mL (0.005–0.680). The corresponding figures in the LP and C groups were 0.041 (0.004–0.150) and 0.030 (0.005–0.100), respectively.

48
The GP patient with proven bacterial translocation had an endotoxin concentration of 0.400 EU/mL and the translocating LP patient 0.040 EU/mL. Moreover, endotoxin concentrations were higher in septic peritonitis patients than in non-septic patients. The differences were not statistically significant.
HPB+-patients demonstrated a median endotoxin plasma concentration of 0.120 EU/mL (0.015-0.310), which differed significantly to the HPB-
0.030 (0.005-0.165) and C groups 0.030 (0.005-0.100).
Fig. 4. Preoperative median concentrations of plasma endotoxins in the different groups.
C = controls; HPB- = hepato-pancreatic-biliary (HPB) tumour without obstructive jaundice; HPB+ = HPB tumour with jaundice; LP = local peritonitis; GP = general peritonitis. * P< 0.05 HPB+ versus HPB- and C groups (Anova).
CommentsGP caused higher concentrations of endotoxins than LP, while the concentra-tion in LP did not differ compared to controls. Moreover, peritonitis patients who met the criteria for sepsis had higher levels of endotoxins than non-septic patients. Although there is no threshold to define when endotoxaemia is harmless or becomes harmful, we find it reasonable to support the assumption that the higher the concentration, the higher the biological effects (22, 48). Patients with obstructive jaundice had higher concentration of endotoxins than those of the GP group. Consequence of impaired gut barrier function in patients with biliary obstruction may be increased exposure to endotoxins (10, 132). Endotoxemia, and the additional
*
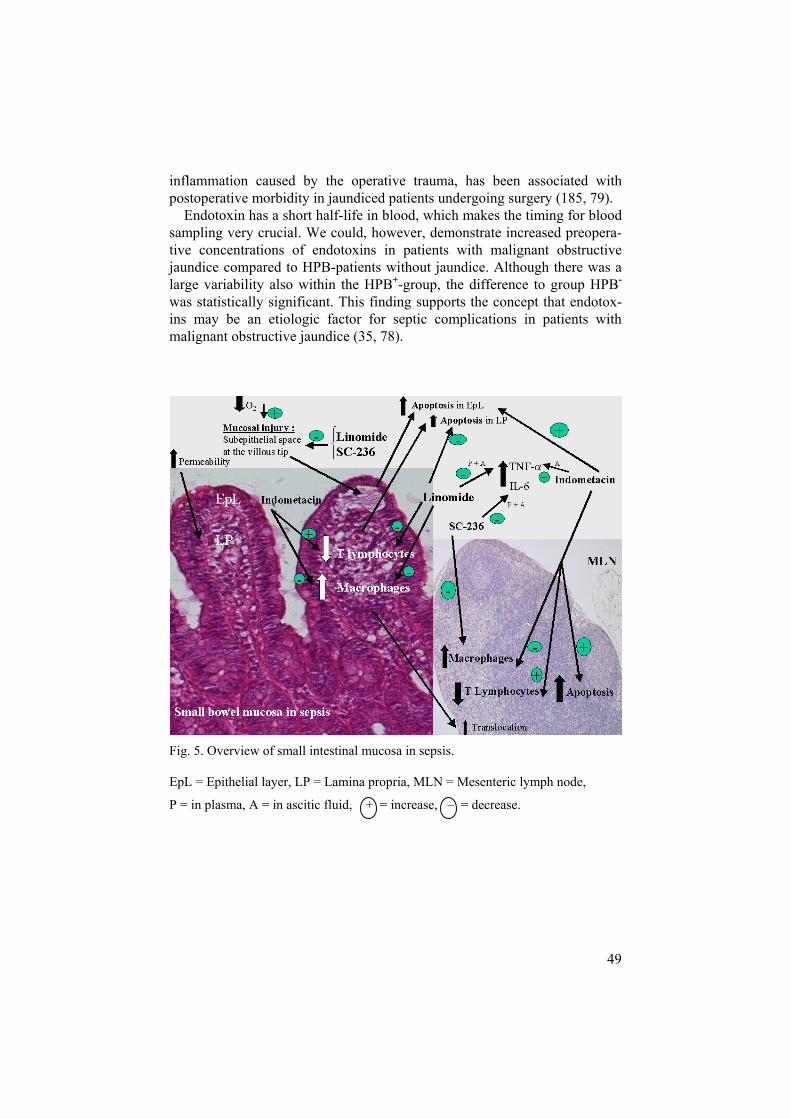
49
inflammation caused by the operative trauma, has been associated with postoperative morbidity in jaundiced patients undergoing surgery (185, 79). Endotoxin has a short half-life in blood, which makes the timing for blood sampling very crucial. We could, however, demonstrate increased preopera-tive concentrations of endotoxins in patients with malignant obstructive jaundice compared to HPB-patients without jaundice. Although there was a large variability also within the HPB+-group, the difference to group HPB-
was statistically significant. This finding supports the concept that endotox-ins may be an etiologic factor for septic complications in patients with malignant obstructive jaundice (35, 78).
Fig. 5. Overview of small intestinal mucosa in sepsis.
EpL = Epithelial layer, LP = Lamina propria, MLN = Mesenteric lymph node,
P = in plasma, A = in ascitic fluid, + = increase, – = decrease.

50
General discussion and clinicalconsiderations
Sepsis and septic shock are still major causes of mortality worldwide, claim-ing millions of lives each year. They are also the leading causes of death in surgical intensive care units in developed countries despite advances in critical care medicine (7, 199). Unfortunately, no definitive therapy yet exists that can successfully treat sepsis and its complications. Recent pro-spective trials have found anti-cytokine and anti-inflammatory therapies to be ineffective. The reasons for such a failure may be: 1) the physiological response in the stressed patient is complex. A single intervention cannot prevent an entire clinical response; 2) the critically-ill patient population is heterogenous and difficult to score, researchers may used various scoring systems for classification; 3) the presence of genetic polymorphisms within the general population has identified subsets of individuals who may have different physiological responses to similar stresses; 4) the lack of precise immunological monitoring. This means that the timing of the therapeutic intervention can be difficult to standardize among patients, which may explain how the same action can cause so different results. A single magic bullet therapy is thus unlikely to work (90, 117, 145).
One should be careful when transferring results obtained in experimental septic shock studies to clinical conditions. One major reason is the fact that the experimental design certainly cannot in all respects mimic the clinical situation. In all our experimental studies, we have used the CLP technique to induce peritonitis and sepsis. We find CLP clinically more relevant compared to e.g. bacterial or LPS injections as it mimics a condition similar to that caused by a perforated gut in man (143).
An intact gastrointestinal mucosal barrier and a normal distribution of T lymfocytes in the mucosa are important factors for the gut immune defence function. Our present data demonstrated that sepsis causes a superficial injury in the epithelial layer and a decrease in the number of cytotoxic T lymfocytes, the latter probably secondary to an increased rate of apoptosis. As a clinical effect this may lead to a deteriorated defence mechanism against MHC class I antigens. We also observed an enhanced apoptosis and a depletion of T lymphocytes, mostly T helper cells, in the lamina propria of the villi. This may impair phagocyte-cell mediated functions, due to a decrease in the activation of macrophages and cytotoxic T cells.

51
T helper cells are also essential for induction of the antibody production.Reduced capacity of innate and cell mediated immune responses, thus promotes translocation of bacteria and endotoxins. This suggests that strate-gies to prevent mucosal damage and lymphocyte apoptosis could represent a potentially important line of therapy.
In the second study we evaluated the effect of pre-treatment with the immunomodulator Linomide on immune cell distribution in the gut follow-ing sepsis. Linomide attenuated the changes in the number of T lymphocytes observed following CLP. This is likely to reflect the anti-apoptotic effect of Linomide (50). Other experimental studies have shown that Linomide suppresses pro-inflammatory cytokines and inhibit leukocyte recruitment in endotoxemia (34, 82). We noted a reduced TNF- level in plasma and ascites and a lesser degree of mucosal damage when Linomide was given intraperitoneally. Peroral administration was not equally effective. In studies II-III a higher cytokine concentration was measured at the local site of inflammation and infection (in peritoneal fluid) than systemically, suggest-ing that in peritonitis-induced sepsis, immunotherapy should target the peri-toneal compartment and suppress the over-activated peritoneal macrophages. Another source for cytokine release is the increased number of macrophages seen in the mucosa (I-III) and in the MLNs (III-V).
Levels of cytokines in the exudate of peritonitis may be used to better stratify the severity of peritonitis and, in future, to guide local therapy (75). Intraperitoneal microdialysis are alredy developed for detection and monitor-ing of peritoneal cytokine production and intestinal ischemia (148, 167).
Early monitoring of the immune responses in critically ill patients may help us to understand pathophysiological aspects of inflammation and infec-tion, as well as provide the basis for interventions directed towards the immune defence system. In addition, monitoring the immune response is a prerequisite for assessment of objective measures of therapeutic success. Cytokine gene polymorphisms may provide a method for determining which patients are at high risk of complications (93).
Our data showed that the plasma concentrations of IL-6 and IL-10, but not TNF- were higher in septic patients compared with nonseptic. TNF-concentrations were low in all patients pre- and postoperatively, and cannot easily be used as prognostic parameters because of the short half-life time. In contrast, IL-6 appears to be a helpful tool for early assessment of SIRS or sepsis in patients with peritonitis, and to grade the severity of peritonitis preoperatively. It may also be a more useful marker of postoperative infec-tive complications than CRP. A normal postoperative course is characterized by decreasing levels of IL-6 (146); CRP is raised a longer period following surgery than IL-6 (as seen in the local peritonitis group, IV). Surgical inter-ventions performed to clear septic foci in the two peritonitis groups, decreased cytokine plasma concentrations postoperatively, although the general peritonitis group still had significantly higher IL-6 and IL-10 levels

52
than the local peritonitis group. Patients, who undergo major surgery for cancer, and patients with obstructive jaundice are at high risk of postopera-tive infection. The HPB+ group demonstrated higher IL-6 levels postopera-tively than the HPB- group (V). A rapid and reliable interleukin-6 test kit is today available for clinical use and can be a helpful tool to get stratified pa-tients with elevated interleukin-6 levels into clinical trials (128). Micro ar-rays for quantitfying different cytokines at the same time, provides a more complete picture of the cytokine pattern and can help to indicate if a pro- or anti-inflammatory state outweighs or to follow the course of events (83, 109). Immune monitoring using a combination of IL-6 and LPS- binding protein (LBP) or procalcitonin (PCT) can discriminate sepsis from SIRS.
So far, most efforts to intervene in the immunopathogenesis of sepsis have been directed at the pro-inflammatory response. None of these inter-ventions has been shown to improve the prognosis of sepsis, possibly because many patients were already in a state in which anti-inflammatory responses dominated. The IL-10/TNF- ratio may be of value for evaluating the inflammatory response and the balance between pro- and anti-inflammatory cytokine activation after sepsis, injury, and burns. Preopera-tively, IL-10/TNF- was increased in both peritonitis groups compared with controls, and higher in septic patients compared with nonseptic. The two patients with proven translocation of bacteria to MLNs had IL-10/TNF-ratio values above the median in their group (IV).
Extensive work on bacterial translocation has been performed in animal models and occurs notably in sepsis, obstructive jaundice, haemorrhagic shock, thermal injury, protein malnutrition, trauma, and intestinal obstruc-tion. In the clinical situation bacterial translocation has been more rarely described, and the clinical significance is not clear and still under debate (169). In study IV, we were able to prove occurrence of bacterial transloca-tion in 2 patients (6%) with peritonitis by the use of pulsed-field gel electro-phoresis (PFGE). Although proven, one can find the rate of translocation low. The use of modern antibiotics regimes preoperatively in major surgery could possibly decrease the rate of bacterial translocation to blood and MLNs. In our septic patients, only a few demonstrated positive blood cultures. No patients in the malignant obstructive jaundice group (V) were found to have enteric bacteria in blood or MLN cultures. In contrast the plasma endotoxin concentrations were elevated in the above mention groups.
It is hypothesized that sepsis and MODS is more probably triggered by the combination of tissue damage and systemic endotoxaemia, than bacterial translocation, which is perhaps a later phenomenon. Future therapeutic strategies could therefore focus more on binding endotoxin in the gut before the triggering event, for example before major surgery. Such a strategy could be combined with the start of early enteral feeding, which has been shown in both clinical and in animal studies to have a beneficial effect on intestinal mucosal barrier function (98, 153, 161, 175).

53
Conclusions
Twenty-four hours after the induction of peritonitis, there is an altered pattern of immunocompetent cells within the intestinal mucosa. Depletion of T lymphocytes in the mucosa may impair the gut immune defence.
Increased concentrations of TNF- were demonstrated in portal blood and in ascitic fluid following CLP induction. Linomide decreased the TNF- level and gave a reduced grade of mucosal injury, it attenuated the decrease of T lymphocytes and the increase of macrophages observed following CLP.
A selective COX-2 inhibitor results in a lesser degree of mucosal injury and a reduced proinflammatory cytokine release in sepsis compared to treatment with a non-selective COX inhibitor. COX inhibitors did not significantly change the already increased apoptosis rate in the mucosa and MLNs, but we observed T lym-phocyte depletion in the mucosa and MLNs more often in the CLP group pre-treated with a nonselective COX inhibitor.
Bacterial translocation occurs during acute peritonitis but seems to be a fairly infrequent phenomenom. Peritonitis causes significant inflammatory cellular reactions as increased cytokine plasma levels and an altered immune cell distribution in MLNs.
Concentrations of endotoxins and cytokines were increased in the HPB+ group as compared to HPB- and controls. A higher rate of apoptosis in MLNs, an increase in the number of macrophages and a decrease of CD4+ T lymphocytes was observed in the HPB+
group. Compared to HPB- group and controls, no evidence of bac-terial translocation was demonstrated. Lymphocyte depletion may be a result of the increased apoptosis rate, which could reduce the ability of the jaundice patient to eradicate infection.

54
Summary in Swedish(sammanfattning på svenska)
Tarmen och dess lymfatiska vävnad (gut associated lymphatic tissue, GALT) är det största och kanske viktigaste immunologiska organet i kroppen. Det initiala inflammatoriska svaret i tarmen vid ”systemic inflammatory response syndrome”(SIRS) eller sepsis inkluderar makrofager och andra fagocyter, påverkan på diverse kaskadsystem samt aktivering av tarmens epithel och endotelceller. Det cellulära immunsvaret uppstår något senare och medieras av CD4+ och CD8+ T lymfocyter. En nedsatt funktion eller ökad programmerad celldöd (apoptos) av dessa celler i tarmens slemhinna (mucosa) och de mesenteriella lymfkörtlarna (MLNs) leder till nedsatt försvar mot mikrober. Sepsis är det systeminflammatoriska svaret på en infektion. Om detta tillstånd leder till endera nedsatt blodtryck (hypoten-sion), nedsatt cirkulation (hypoperfusion) eller organdysfunktion har man en svår sepsis. Septisk chock föreligger om hypotensionen ej svarar adekvat på vätsketillförsel och patienten samtidigt har organdysfunktion eller hypoper-fusion. Peritonit (intra-abdominal sepsis) kan utlösas av svår infektionssjuk-dom, efter intra-abdominala skador eller vara en komplikation till bukkirur-gi. Peritonit leder till septisk chock och multipel organsvikt i relativt hög frekvens. En ytlig mucosaskada i tunntarmen uppstår tidigt vid chock på grund av försämrad tillförsel av syrgas, samtidigt som behovet av syrgas är ökat, vilket innebär att barriären mot tarmlumen försämras. Därmed ökar tarmbar-riärens genomsläpplighet (permeabilitet), vilket underlättar passage för bakterier (vanligen gramnegativa) eller delar av dess vägg (lipopolysaccha-rid, LPS, sk endotoxiner) genom slemhinnan till blodcirkulationen och lymf-systemet, s k translokation. Bakterier och LPS stimulerar det lokala immun-systemet bl.a. makrofager att frisätta signalsubstanser som cytokiner, vilka i sin tur påverkar det inflammatoriska svaret. Obalans mellan det pro- och det anti-inflammatoriska svaret vid sepsis är svårbehandlat och leder till hög mortalitet trots optimerad intensiv vård. Gallvägsstas (ikterus) anses ge ett nedsatt immunförsvar i tarmen, vilket ökar risken för transloka-tion av endotoxiner och tarmbakterier.
Målsättningen har varit att i både djurexperimentella studier samt i klinis-ka studier skapa en ökad kunskap kring tarmens cellulära immunförsvar i samband med peritonit, sepsis och malign gallvägsstas. I de tre experimen-

55
tella arbetena har en standardiserad fekal peritonit framkallats på råtta (cecal ligation and puncture; CLP). Det inflammatoriska svaret (koncentration av cytokinerna; TNF- , IL-6 och IL-10, förändringar i populationer av T-lymfocyter och makrofager i tarmslemhinnan och i MLNs, förekomst och frekvens av apoptos i dessa områden) har studerats. Mekanismerna bakom de aktuella reaktionerna har vidare studerats med hjälp av tillförsel av en syntetisk anti-inflammatorisk substans (Linomid) och genom tillförsel av en ospecifik cyklo-oxygenas (COX) hämmare (indometacin) respektive en specifik COX-2-hämmare (SC-236). Här följer en del av resultaten.
Delarbete I: T-lymfocyterna uttrycktes i betydligt mindre omfattning i peritonitdjurens mucosa jämfört med kontroller medan däremot makrofager-na ökade 24 tim efter CLP. Laparotomi utan CLP hade ingen effekt på immuncellsdistributionen.
Delarbete II: Ökad koncentration av TNF- påvisades i blod och buk-vätska (ascites) efter CLP. Nivåerna av TNF- var alltid högre i ascites än i blod. Linomid administrerat i bukhålan (intraperitonealt; i.p.) före CLP, sänkte TNF- nivåerna signifikant i portablod och i ascites, samt ledde till en lägre grad av mucosa skada jämfört med den icke behandlade CLP grup-pen. Linomid givet peroralt (p.o.) var inte lika effektivt. Linomide givet i.p. eller p.o. före CLP mildrade de förändringar man tidigare (I) sett vad beträf-far immuncellsdistributionen i de septiska djurens mucosa.
Delarbete III: SC-236 administrerat p.o. innan CLP gav en lägre grad av mucosaskada jämfört med den obehandlade CLP gruppen och den grupp som erhållit indometacin före CLP. Nivåerna av TNF- och IL-6 var alltid högre i ascites än i blodet och högre i ascites efter 6 tim än efter 24 tim. Indometacin givet före CLP medförde signifikant högre TNF- koncentra-tion i ascites efter 6 tim jämfört med övriga CLP grupperna. När SC-236 gavs noterades lägre nivåer av IL-6 i ascites och i blodet. I alla CLP grupperna minskade antalet T lymfocyter i mucosan, en ytterligare minsk-ning sågs om djuren hade fått förbehandling med indometacin. Samtidigt sågs både i mucosan och i MLNs en högre frekvens av apoptos 24 tim. efter CLP jämfört med den obehandlade kontrollgruppen. Ökningen av antalet makrofager som även tidigare noterats efter CLP, i mucosa och i MLN minskades till kontrollgruppens nivå efter behandling med COX-hämmare, oavsett selektivitet.
I de två kliniska arbetena (IV-V) har vi undersökt förekomst av endotoxi-nemi och bakteriell translokation. Vi har också studerat om de inflammato-riska parametrarna som analyserades i de experimentella studierna (I-III), påverkades hos patienter med generell peritonit eller lokal peritonit (appendicit) (delarbete IV) respektive hos patienter med tumör i gallvägar eller pankreas med eller utan ikterus (delarbeteV) innan och efter operation. Bakteriell translokation förekom i få fall vid akut peritonit. Förhöjda endo-toxinkoncentrationer förelåg endast hos patienterna med generell peritonit. Både lokal och generell peritonit orsakade stegrade cytokinnivåer jämfört

56
med kontrollgruppen. De högsta koncentrationerna sågs hos patienter med generell peritonit preoperativt. Endast lokal peritonit orsakade signifikanta förändringar av immuncellsdistributionen i MLNs jämfört med kontrollerna.De ikteriska patienterna hade högre endotoxin- och cytokinnivåer i blodet, en högre grad av apoptos i MLN, en ökad mängd makrofager och en mins-kad mängd CD4+ T lymfocyter i sina MLNs jämfört med kontroll gruppen och de patienterna med tumör i gallvägar eller pankreas utan ikterus. Ingen av grupperna uppvisade bakteriell translokation. Minskningen av CD4+ T celler kan vara associerad med den ökade graden av apoptos som sågs hos de ikteriska patienterna. Reducerat antal T lymphocyter i tarmens slemhinna och dess lymfkörtlar kan vara en förklaring till att patienter med peritonit och ikterus har svårare att erradikera bakterier och då lättare får infektiösa komplikationer som leder till sepsis efter kirurgi.
Immunoterapi med substanser som minskar apoptos och stärker det cellu-lära immunförsvaret kan möjligen i framtiden vara ett komplement till de övriga medicinska behandlingar och den avancerade intensivvård som dessa patienter kräver. Immun-monitorering med analys av cytokiner som TNF- ,IL-10 men framförallt IL-6 i kombination med andra biokemiska markörer (t ex. prokalcitonin, lipopolysaccharide binding protein) kommer att spela en allt större roll för diagnos, utvärdering av terapier samt prognos och riskstratifiering av septiska patienter.

57
Acknowledgements
I wish to express my sincere gratitude and appreciation to:
Ulf Haglund, my tutor, for sharing his extensive knowledge in the field of shock research, for his support and constructive criticism and for stimulating to independent work.
Mikael Ljungdahl, my co-tutor, for your enthusiasm and friendship, invalu-able help and support, which have been of the greatest value in the comple-tion of this thesis.
David Bergqvist and Lars Wiklund, former and present Head of the Department of Surgical Sciences, for providing the funding for my research.
Annemarie Westlund, one of my co-authors, for excellent technical assistance, and enthusiasm during this study. For always being so pleasant and helpful, and for offer me a nice cup of tea when needed.
Mari-Anne Carlsson, for excellent technical assistance, invaluable help and support throughout the last period of my work.
My other co-authors, for valuable help: Cecilia Johnsson, Göran Gannedahl, Lars Engstrand, Ulrika Ransjö and the late Monika Lundholm.
Astrid Asklin, Janet Lynn Cunningham, Margareta Ericson, Eva Tanoand Ulla Zettersten, for technical assistance.
Rolf Heuman, for teaching me general surgical principles, for introducing me into the field of science, and for his encouragement, support and friend-ship.
Friends and colleagues at the Department of Surgery, Uppsala and Mora, for sharing your knowledge and valuable support. Especially my col-leagues in Mora who made it possible for me to take time off from my clini-cal work to complete this thesis.

58
Rune Sanbu, for teaching me laparoscopic Nissen fundoplication and for discussions about science and life in general.
Michael Dahlberg and Fredrik Wärnberg, colleagues and room mates during my Uppsala period, for support and gossip about life.
All my friends in MIK committe, for your positive attitude and stimulating advices.
Lena Sundberg, for creating the interesting dry-point engraving on the cover of this book.
Catherine Williams, for skilful linguistic revision.
Göran Engemar, for help with the electronic publishing.
My parents, Karin och Ture Wikström, for giving me a good start in life,for their never ending love and concern. Without your help this would not have been possible!
My brother Magnus and his wife Anna, for love, help and technical assistance.
Oscar and Ebba, constant sources of joy and energy, who kept their mother aware that there are much more in life.
and,
Most of all, Asbjörn, Head of the Department of Surgery at Mora Hospital, my husband, colleague and very best friend. For unlimited love, understanding, humour and mental support when I needed it most!
Finally, I would like to thank all my friends and relatives, for their constant encouragement and support!
This research has been supported by grants from the Swedish Medical Research Council (project no 4502); the Medical Faculty, Uppsala University and the Centre for Clinical research (CKF) Dalarna, Sweden.

59
References
1. Abreu-Martin MT, Targan SR: Regulation of immune responses of the intesti-nal mucosa. Crit Rev Immunol 16:277-309, 1996.
2. Abu-Hijleh MF, Habbal OA, Moqattash ST: The role of the diaphragm in lym-phatic absorption from the peritoneal cavity. J Anat 186:453-67, 1995.
3. Adrie C, Pinsky MR: The inflammatory balance in human sepsis. Intensive Care Med 26:364-75, 2000.
4. Akiyama T, Hasegawa T, Sejima T, Sahara H et al: Serum and bile interleukin 6 after percutaneous transhepatic cholangio–drainage. Hepatogastroenterology 45:665-71, 1998.
5. Alexander JW, Boyce ST, Babcock GF, Gianotti L et al: The process of mi-crobial translocation. Ann Surg 212:496-510, 1990.
6. Angele MK, Schwacha MG, Ayala A, Chaudry IH: Effect of gender and sex hormones on immune responses following shock. Shock 14:81-90, 2000.
7. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G et al: Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associ-ated costs of care. Crit Care Med 29:1303-10, 2001.
8. Arvidsson D, Rasmussen I, Almqvist P, Niklasson F, Haglund U: Splanchnic oxygen consumption in septic and hemorrhagic shock. Surgery 109:190-7, 1991.
9. Ayala A, Chung CS, Xu YX, Evans TA et al: Increased inducible apoptosis in CD4+ T lymphocytes during polymicrobial sepsis is mediated by Fas ligand and not endotoxin. Immunology 97:45-55,1999.
10. Ballinger AB, Woolley JA, Ahmed M, Mulcahy H et al: Persistent systemic inflammatory response after stent insertion in patients with malignant bile duct obstruction: Gut 42:555-9, 1998.
11. Baumann S, Krueger A, Kirchhoff S, Krammer PH: Regulation of T cell apoptosis during the immune response. Curr Mol Med 2:257-72, 2002.
12. Bemelmans MH, Gouma DJ, Greve JW, Buurman WA: Cytokines tumor necrosis factor and interleukin-6 in experimental biliary obstruction in mice. Hepatology 15:1132-6,1992.
13. Berg RD, Garlington AW: Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect Immun 23:403-11, 1979
14. Berg RD: Bacterial translocation from the gastrointestinal tract. Adv Exp Med Biol 473:11-30, 1999.
15. Bernard GR, Wheeler AP, Russell JA, Schein R et al: The effects of ibupro-fen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med 336:912-8, 1997.
16. Bone RC, Balk RA, Cerra FB, Dellinger RP et al: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 101:1644–55, 1992.

60
17. Bosscha K, van Vroonhoven TJ, van der Werken C: Surgical management of severe secondary peritonitis. Br J Surg 86:1371-7, 1999.
18. Bown MJ, Nicholson ML, Bell PR, Sayers RD: Cytokines and inflammatory pathways in the pathogenesis of multiple organ failure following abdominal aortic aneurysm repair. Eur J Vasc Endovasc Surg 22:485-95, 2001.
19. Braciale TJ, Braciale VL: Antigen presentation: structural themes and func-tional variations. Immunol Today 12:124-9, 1991.
20. Bradley LM, Watson SR: Lymphocyte migration into tissue: the paradigm derived from CD4 subsets. Curr Opin Immunol 8:312–20,1996.
21. Butcher EC, Picker LJ: Lymphocyte homing and homeostasis. Science 272:60-6, 1996.
22. Buttenschoen K, Buttenschoen DC, Berger D, Vasilescu C et al: Endotoxe-mia and acute-phase proteins in major abdominal surgery. Am J Surg 181:36-43, 2001.
23. Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV: Multiple-organ-failure syndrome. Arch Surg 121:196-208, 1986.
24. Chiu CJ, McArdle AH, Brown R, Scott HJ, Gurd FN: Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic re-appraisal. Arch Surg 101:478-83, 1970.
25. Chung CS, Xu YX, Chaudry IH, Ayala A: Sepsis induces increased apoptosis in lamina propria mononuclear cells, which is associated with altered cytokine gene expression. J Surg Res 77:63-70, 1998.
26. Clark SM, Lai E, Birren BW, Hood L: "A novel instrument for separating large DNA molecules with pulsed homogenous electric fields." Science241:1203-1205, 1988.
27. Crofford LJ, Lipsky PE, Brooks P, Abramson SB et al: Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum43: 4-13, 2000.
28. Daar AS, Fuggle SV, Fabre JW, Ting A, Morris PJ: The detailed distribution of MHC Class II antigens in normal human organs. Transplantation 38:293-8, 1984.
29. Dahn MS, Lange P, Lobdell K, Hans B et al: Splanchnic and total body oxy-gen consumption differences in septic and injured patients. Surgery 101:69-80, 1987.
30. Deitch EA, Sittig K, Li M, Berg R, Specian RD: Obstructive jaundice pro-motes bacterial translocation from the gut. Am J Surg 159:79-84,1990.
31. Deitch EA: Bacterial translocation of the gut flora. J Trauma 30:184-189, 1990. 32. Deitch EA: Multiple organ failure: Pathophysiology and potential future ther-
apy. Ann Surg 216:117-134, 1992. 33. Dellinger RP, Carlet JM, Masur H, Gerlach H et al: Surviving Sepsis Cam-
paign Management Guidelines Committee. Surviving Sepsis Campaign guide-lines for management of severe sepsis and septic shock. Crit Care Med32:858-73, 2004.
34. Diab A, Michael L, Wahren B, Deng GM et al: Linomide suppresses acute experimental autoimmune encephalomyelitis in Lewis rats by counter-acting the imbalance of pro-inflammatory versus anti-inflammatory cytokines. JNeuroimmunol 85:146-54, 1998.
35. Diamond T, Rowlands BJ: Endotoxaemia in obstructive jaundice. HPB Surg4:81-94, 1991.
36. Dijkstra CD, Döpp EA, Joling PJ, Kraal G: The heterogenity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat

61
recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 54:589–599, 1985.
37. Dinarello CA: Proinflammatory cytokines. Chest 118:503-8, 2000. 38. Ding JW, Andersson R, Stenram U, Lunderquist A, Bengmark S: Effect of
biliary decompression on reticuloendothelial function in jaundiced rats. Br J Surg 79:648-52, 1992.
39. Ding JW, Andersson R, Soltesz V, Willen R, Bengmark S: Obstructive jaun-dice impairs reticuloendothelial function and promotes bacterial translocation in the rat. J Surg Res 57:238-45, 1994.
40. Drewe J, Beglinger C, Fricker G: Effect of ischemia on intestinal permeability of lipopolysaccharides. Eur J Clin Invest 31:138-44, 2001.
41. Ebong S, Call D, Nemzek J, Bolgos G et al: Immunopathologic alterations in murine models of sepsis of increasing severity. Infection and immunity 67: 6603-6610,1999.
42. Echtenacher B, Hultner L, Mänel DN: Cellular and molecular mechanisms of TNF protection in septic peritonitis. J Inflamm 47:85–89, 1996.
43. Evans HL, Raymond DP, Pelletier SJ, Crabtree TD et al: Diagnosis of intra-abdominal infection in the critically ill patient. Curr Opin Crit Care 7:117-21, 2001.
44. Falk A, Redfors S, Myrvold H, Haglund U: Small intestine mucosal lesions in feline septic shock: A study on the pathogenesis. Circ Shock 17:327-337, 1985.
45. Fink MP, Antonsson JB, Wang HL, Rothschild HR: Increased intestinal permeability in endotoxic pigs. Mesenteric hypoperfusion as an etiologic fac-tor. Arch Surg 126:211-8, 1991.
46. Fong Y, Moldawer LL, Shires GT, Lowry SF: The biologic characteristics of cytokines and their implication in surgical injury. Surg Gynecol Obstet170:363-78, 1990.
47. Gautreaux MD, Deitch EA, Berg RD: T lymphocytes in host defence against bacterial translocation from the gastrointestinal tract. Infect Immun 62:2874–84,1994.
48. Glauser MP, Zanetti G, Baumgartner JD, Cohen J: Septic shock: pathogene-sis. Lancet 338:732–6, 1991.
49. Gonzalo JA, Gonza´les-Garcia A, Kalland T, Hedlund G: Linomide, a novel immunomodulator that prevents death in four models of septic shock. Eur J Immunol 23:2372–2374, 1993.
50. Gonzalo JA, Gonza´lez-Garcia A, Kalland T, Hedlund G: Linomide inhibits programmed cell death of peripheral T cells in vivo. Eur J Immunol 24:48–52, 1994.
51. Goscinski G, Lipcsey M, Eriksson M, Larsson A et al: Endotoxin neutraliza-tion and anti-inflammatory effects of tobramycin and ceftazidime in porcine endotoxin shock. Crit Care 8:35-41, 2004.
52. Green DR: Apoptotic pathways: paper wraps stone blunts scissors. Cell 102:1-4, 2000.
53. Haglind E, Haglund U, Lundgren O, Romanus M, Schersten T: Graded intestinal vascular obstruction: I. Description of an experimental shock model in the rat. Circ Shock 7:83-91, 1980.
54. Haglund U, Hulten L, Ahren C, Lundgren O: Mucosal lesions in the human small intestine in shock. Gut 16:979-84, 1975.
55. Haglund U, Bulkley GB, Granger DN: On the pathophysiology of intestinal ischemic injury. Clinical review. Acta Chir Scand 153:321-4, 1987.

62
56. Haglund U, Österberg J: Local consequences of reperfusion in the gut. In Grace PA, Mathie RT (eds.): Ischaemia-reperfusion injury. Blackwell Science Ltd. 1999, 65-70.
57. Hao WL, Lee YK: Microflora of the gastrointestinal tract: a review. Methods Mol Biol 268:491-502, 2004.
58. Harris HW, Gill TJ: Expression of class I transplantation antigens. Transplan-tation 42:109-17, 1986.
59. Hawkey CJ: COX-2 inhibitors. Lancet 353:307-14, 1999. 60. Hebel R, Stromberg MW: Anatomy of the laboratory rat. 1976 Williams &
Wilkins company, Baltimore. 61. Heinrich PC, Castell JV, Andus T: Interleukin-6 and the acute phase response.
Biochem J 265:621-36,1990. 62. Hengartner MO: The biochemistry of apoptosis. Nature 407:770-6, 2000. 63. Hildebrand F, Pape HC, Hoevel P, Krettek C, van Griensven M: The impor-
tance of systemic cytokines in the pathogenesis of polymicrobial sepsis and dehydroepiandrosterone treatment in a rodent model. Shock 20:338-46, 2003.
64. Hinz B, Brune K: Cyclooxygenase-2 – 10 years later. J Pharmacology300:367-375, 2002.
65. Hodul P, Creech S, Pickleman J, Aranha GV: The effect of preoperative biliary stenting on postoperative complications after pancreaticoduodenec-tomy. Am J Surg 186:420-5, 2003.
66. Hoffman WD, Natanson C: Endotoxin in septic shock. Anesth Analg 77:613-24, 1993.
67. Holzheimer RG, Schein M, Wittmann DH: Inflammatory response in perito-neal exudate and plasma of patients undergoing planned relaparotomy for se-vere secondary peritonitis. Arch Surg 130:1314-9, 1995.
68. Horner C, Bouchon A, Bierhaus A, Nawroth PP et al: Role of the innate immune response in sepsis. Anaesthesist 53:10-28, 2004.
69. Hotchkiss, RS, Schmieg RE Jr, Swanson PE, Freeman BD et al: Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med 28: 3207-3217, 2000.
70. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr et al: Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 166:6952-63, 2001.
71. Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH et al: Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol168:2493–500, 2002.
72. Hotchkiss RS, Tinsley KW, Karl IE: Role of apoptotic cell death in sepsis. Scand J Infect Dis 35:585-92, 2003.
73. Hünig T, Wallny HJ, Hartley JK, Lawetsky A, Tiefenthaler G: A mono-clonal antibody to a constant determinant of the rat T cell antigen receptor that induces T cell activation. J Exp Med 169:73–86, 1989.
74. Ishiguro EE, Vanderwel D, Kusser W: Control of lipopolysaccharide biosyn-thesis and release by Escherichia coli and Salmonella typhimurium. J Bacteriol168:328-33, 1986.
75. Jansson K, Redler B, Truedsson L, Magnuson A et al: Intraperitoneal cyto-kine response after major surgery: higher postoperative intraperitoneal versus systemic cytokine levels suggest the gastrointestinal tract as the major source of the postoperative inflammatory reaction. Am J Surg 187:372-7, 2004.
76. Kimber CP, Hutson JM: Primary peritonitis in children. Aust N Z J Surg66:169-70, 1996.

63
77. Kimmings AN, van Deventer SJ, Obertop H, Rauws EA, Gouma DJ: In-flammatory and immunologic effects of obstructive jaundice: pathogenesis and treatment. J Am Coll Surg 181:567-81, 1995.
78. Kimmings AN, van Deventer SJ, Obertop H, Rauws EA et al: Endotoxin, cytokines, and endotoxin binding proteins in obstructive jaundice and after preoperative biliary drainage. Gut 46:725-31, 2000.
79. Kimmings AN, Sewnath ME, Mairuhu WM, Van Zanten AP et al: The ab-normal lipid spectrum in malignant obstructive jaundice in relation to en-dotoxin sensitivity and the result of preoperative biliary drainage. Surgery 129:282-91, 2001.
80. Kimura F, Miyazaki M, Suwa T, Sugiura T et al: Anti-inflammatory response in patients with obstructive jaundice caused by biliary malignancy. J Gastroen-terol Hepatol 16:467-72, 2001.
81. Kirschner M: Die Behandlung der akuten eitrigen freien Bauchfellentzündung. Langenbecks, Arch Klein Chir 142:53-267, 1926.
82. Klintman D, Hedlund G, Thorlacius H: Protective effect of Linomide on TNF-alpha-induced hepatic injury. J Hepatol 36:226-32, 2002.
83. Knight PR, Sreekumar A, Siddiqui J, Laxman B et al: Development of a sensitive microarray immunoassay and comparison with standard enzyme-linked immunoassay for cytokine analysis. Shock 21:26-30, 2004.
84. Kox WJ, Volk T, Kox SN, Volk HD: Immunomodulatory therapies in sepsis. Intensive Care Med 26 Suppl 1:124-8, 2000.
85. Ksontini R, MacKay SL, Moldawer LL: Revisiting the role of tumor necrosis factor alpha and the response to surgical injury and inflammation. Arch Surg133:558-67, 1998.
86. Kunkel SL, Wiggins RC, Chensue SW, Larrick J: Regulation of macrophage tumor necrosis factor production by prostaglandin E2. Biochem Biophys Res Commun 137: 404-409, 1986.
87. Kuzu MA, Kale IT, Col C, Tekeli A et al: Obstructive jaundice promotes bac-terial translocation in humans. Hepatogastroenterology 46:2159-64, 1999.
88. Lacaine F, Fourtanier G, Fingerhut A, Hay JM: Surgical mortality and mor-bidity in malignant obstructive jaundice: a prospective multivariate analysis. Eur J Surg 161:729-34, 1995.
89. Langermans JA, Hazenbos WL, van Furth R: Antimicrobial functions of mononuclear phagocytes. J Immunol Methods 174:185-94, 1994.
90. le Roux P: New concepts in treatment of sepsis. SADJ 59:208-9, 2004. 91. Li X, Klintman D, Sato T, Hedlund G et al: Interleukin-10 mediates the pro-
tective effect of Linomide by reducing CXC chemokine production in en-dotoxin-induced liver injury. Br J Pharmacol 143:865-71, 2004.
92. Lien E, Ingalls RR: Toll-like receptors. Crit Care Med 30:1-11, 2002. 93. Lin MT, Albertson TE: Genomic polymorphisms in sepsis. Crit Care Med
32:569-79, 2004. 94. Lipsky PE: Defining COX-2 inhibitors. J Rheumatol Suppl 60:13-6, 2000. 95. Ljungdahl M, Rasmussen I, Haglund U: Intestinal blood flow and intramuco-
sal pH in experimental peritonitis. Shock 11:44–50, 1999. 96. Ljungdahl M, Lundholm M, Katouli M, Rasmussen I et al: Bacterial translo-
cation in experimental shock is dependent on the strains in the intestinal flora. Scand J Gastroenterol 35:389-97, 2000.
97. Lowry SF: Cytokine mediators of immunity and inflammation. Arch Surg128:1235-41, 1993.

64
98. Luyer MD, Jacobs JA, Vreugdenhil AC, Hadfoune M et al: Enteral admini-stration of high-fat nutrition before and directly after hemorrhagic shock re-duces endotoxemia and bacterial translocation. Ann Surg 239:257-64, 2004.
99. Madara JL: Warner-Lambert/Parke-Davis Award lecture. Pathobiology of the intestinal epithelial barrier. Am J Pathol 137:1273-81, 1990.
100. Mahidhara R, Billiar TR: Apoptosis in sepsis. Crit Care Med 28:105-13, 2000.
101. Malangoni MA: Evaluation and management of tertiary peritonitis. Am Surg66:157-61, 2000.
102. Marchant A, Bruyns C, Vandenabeele P, Ducarme M et al: Interleukin-10 controls interferon-gamma and tumour necrosis factor production during ex-perimental endotoxemia. Eur J Immunol 24:1167-71, 1994.
103. Marchant A, Deviere J, Byl B, De Groote D et al: Interleukin-10 production during septicaemia. Lancet 343:707-8, 1994.
104. Marcinkiewicz J: In vitro cytokine release by activated murine peritoneal macrophages: role of prostaglandins in the differential regulation of tumour necrosis factor alpha, interleukin 1, and interleukin 6. Cytokine 3:327-32, 1991.
105. Marrack P, Kappler J: The T-cell receptor. Science 238:1073–1078, 1987. 106. Marshall JC, Christou NV, Meakins JL: The gastrointestinal tract. The
"undrained abscess" of multiple organ failure. Ann Surg 218:111-9, 1993. 107. Marshall JC, Innes M: Intensive care unit management of intra-abdominal
infection. Crit Care Med 31:2228-37, 2003. 108. Marshall JC, Maier RV, Jimenez M, Dellinger EP: Source control in the
management of severe sepsis and septic shock: an evidence-based review. Crit Care Med 32:513-26, 2004.
109. Martin C, Boisson C, Haccoun M, Thomachot L, Mege JL: Patterns of cytokine evolution (tumor necroris factor-alpha and interleukin-6) after septic shock, hemorrhagic shock, and severe trauma. Crit Care Med 25:1813–9, 1997.
110. Martineau L, Shek PN: Peritoneal cytokine concentrations and survival out-come in an experimental bacterial infusion model of peritonitis. Crit Care Med28:788–794, 2000.
111. Masferrer JL, Zweifel BS, Colburn SM, Ornberg RL: The Role of Cyclooxygenase-2 in Inflammation. Am J Ther 2:607-610, 1995.
112. Mathison JC, Tobias PS, Wolfson E, Ulevitch RJ: Plasma lipopolysaccha-ride (LPS)-binding protein. A key component in macrophage recognition of gram-negative LPS. J Immunol 149:200-6, 1992.
113. McMaster WR, Williams AF: Monoclonal antibodies to Ia antigens from rat thymus: cross reactions with mouse and human and use in purification of rat Ia glycoproteins. Immunological Rev 47:117–137, 1979.
114. McNabb PC, Tomasi TB: Host defense mechanisms at mucosal surfaces. Annu Rev Microbiol 35:477-96, 1981.
115. Michie HR: The value of animal models in the development of new drugs for the treatment of the sepsis syndrome. J Antimicrob Chemother 41:47-9, 1998.
116. Mikulicz J: Über die Anwendung der Antisepsis bei Laparotomien mit beson-derer Rücksicht auf die Drainage der Bauchhöhle. Arch Klein Chir 26:111-117, 1881.
117. Minnich DJ, Moldawer LL: Anti-cytokine and anti-inflammatory therapies for the treatment of severe sepsis: progress and pitfalls. Proc Nutr Soc 63:437-41, 2004.

65
118. Mishra JP, Singh LN: Changes in serum protease inhibitors and liver specific enzymes in experimental jaundice. Indian J Med Sci 50:221-7,1996.
119. Montgomery A, Almqvist P, Arvidsson D, Lindgren S, Haglund U: Early detection of gastrointestinal mucosal ischemia in porcine E. coli sepsis. Acta Chir Scand 156:613-20, 1990.
120. Moore FA, Moore EE, Poggetti R, McAnena OJ et al: Gut bacterial translo-cation via the portal vein: a clinical perspective with major torso trauma. JTrauma 31:629-36, 1991.
121. National Institutes of Health: Guide for the care and use of laboratory ani-mals. Betsheda, MA: National Institutes of Health, 1985, p 86-123.
122. Nettelbladt CG, Katouli M, Bark T, Svenberg T et al: Orally inoculated Escherichia coli strains colonize the gut and increase bacterial translocation af-ter stress in rats. Shock 20:251-6, 2003.
123. Nystrom PO, Bax R, Dellinger EP, Dominioni L et al: Proposed definitions for diagnosis, severity scoring, stratification, and outcome for trials on intraab-dominal infection. Joint Working Party of SIS North America and Europe. World J Surg 14:148-58, 1990.
124. O`Boyle CJ, MacFie J, Mitchell CJ, Jonstone P et al: Microbiology of bac-terial translocation in humans. Gut 42:29-35, 1998.
125. Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL: Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J 15:879-92, 2001.
126. Otero-Anton E, Gonzalez-Quintela A, Lopez-Soto A, Lopez-Ben S et al:Cecal ligation and puncture as a model of sepsis in the rat: influence of the puncture size on mortality, bacteremia, endotoxemia and tumour necrosis fac-tor alpha levels. Eur Surg Res 33:77-79, 2001.
127. Otte JM, Kiehne K, Herzig KH: Antimicrobial peptides in innate immunity of the human intestine. J Gastroenterol 38:717-26, 2003.
128. Panacek EA, Marshall JC, Albertson TE, Johnson DH et al: Monoclonal Anti-TNF: A Randomized Controlled Sepsis Study Investigators. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab')2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med 32:2173-2182, 2004.
129. Papakostas C, Bezirtzoglou E, Pitiakoudis M, Polychronidis A, Simopoulos C: Endotoxinemia in the portal and the systemic circulation in obstructive jaundice. Clin Exp Med 3:124-8, 2003.
130. Parish CR, O'Neill ER: Dependence of the adaptive immune response on innate immunity: some questions answered but new paradoxes emerge. Immu-nol Cell Biol 75:523-7,1997.
131. Park PO, Haglund U, Bulkley GB, Falt K: The sequence of development of intestinal tissue injury after strangulation ischemia and reperfusion. Surgery107:574-80, 1990.
132. Parks RW, Clements WDB, Smye MG, Pope C et al: Intestinal barrier dys-function in clinical and experimental obstructive jaundice and its reversal by internal biliary drainage. Br J Surg 83:1345-9, 1996.
133. Parks RW, Stuart Cameron CH, Gannon CD, Pope C et al: Changes in gastrointestinal morphology associated with obstructive jaundice. J Pathol192:526-32, 2000.
134. Patel RT, Deen KI, Youngs D, Warwick J, Keighley MR: Interleukin-6 as a prognostic indicator of outcome in severe intraabdominal sepsis. Br J Surg81:1306–8, 1994.

66
135. Povoski SP, Karpeh MS Jr, Conlon KC, Blumgart LH, Brennan MF: As-sociation of preoperative biliary drainage with postoperative outcome follow-ing pancreaticoduodenectomy: Ann Surg 230:131-42, 1999.
136. Pulford KA, Carter PB, McMaster WR, Mason DW, Williams AF: Two subsets of rat lymphocytes defined with monoclonal antibodies. Eur J Immunol10: 609–615, 1980.
137. Pulford KA, Rigney EM, Micklem KJ, Jones M et al: KPI (CD68): a new monoclonal antibody that detects a monocyte/macrophage associated antigen in routinely processed tissue sections. J Clin Pathol 42:414–21, 1989.
138. Ranefall P, Egevad L, Nordin B, Bengtsson E: A new method for segmenta-tion of colour images applied to immunohistochemically stained cell nuclei. Anal Cell Pathol 15:145–156, 1997.
139. Ranefall P, Wester K, Bengtsson E: Automatic quantification of immunohis-tochemically stained cell nuclei using unsupervised image analysis. Anal Cell Pathol 16:29–43, 1998.
140. Rasmussen I, Haglund U: Early gut ischemia in experimental fecal peritonitis. Circ Shock 38:22-8, 1992.
141. Remick DG, Kunkel SL: Pathophysiologic alterations induced by tumour necrosis factor. Int Rev Exp Pathol 34:7–25, 1993.
142. Remick DG, Manohar P, Bolgos G, Rodriguez J et al: Blockade of tumour necrosis factor reduces lipopolysaccharide lethality, but not the lethality of ce-cal ligation and puncture. Shock 4:89-95, 1995.
143. Remick DG, Newcomb DE, Bolgos GL, Call DR: Comparison of the mortal-ity and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock 13:110-6, 2000.
144. Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA: Six at six: inter-leukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17:463-7, 2002.
145. Remick DG: Cytokine therapeutics for the treatment of sepsis: why has noth-ing worked? Curr Pharm Des 9:75-82, 2003.
146. Riche FC, Cholley BP, Panis YH, Laisne MJ et al: Inflammatory cytokine response in patients with septic shock secondary to generalized peritonitis. Crit Care Med 28:433-7, 2000.
147. Riedemann NC, Neff TA, Guo RF, Bernacki KD et al: Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Im-munol 170:503-7, 2003.
148. Riese J, Boecker S, Hohenberger W, Klein P, Haupt W: Microdialysis: a new technique to monitor perioperative human peritoneal mediator production. Surg Infect 4:11-5, 2003.
149. Romagnani S: Type 1 T helper and type 2 T helper cells: functions, regulation and role in protection and disease. Int J Clin Lab Res 21:152-8,1991.
150. Saito H, Sherwood ER, Varma TK, Evers BM: Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev 124:1047-58, 2003.
151. Sano T, Ajiki T, Takeyama Y, Kuroda Y: Internal biliary drainage improves decreased number of gut mucosal T lymphocytes and MAdCAM-1 expression in jaundiced rats. Surgery 136:693-9, 2004.
152. Schlossman S, Bloumsell L, Gilks W, Harlan JM et al: Leukocyte typing V: white cell differentiation antigens. New York: Oxford University Press;1995.
153. Schmidt H, Martindale R: The gastrointestinal tract in critical illness. CurrOpin Clin Nutr Metab Care 4:547-51, 2001.

67
154. Schoeffel U, Pelz K, Häring RU, Amberg R et al: Inflammatory conse-quences of the translocation of bacteria and endotoxin to mesenteric lymph nodes. Am J Surg 180:65-72, 2000.
155. Scott-Conner CE, Grogan JB: The pathophysiology of biliary obstruction and its effect on phagocytic and immune function. J Surg Res 57:316-36,1994.
156. Sedman PC, Macfie J, Sagar P, Mitchell CJ et al: The prevalence of gut translocation in humans. Gastroenterology 107:643-9, 1994.
157. Seubert JM, Darmon AJ, El-Kadi AO, D'Souza SJ, Bend JR: Apoptosis in murine hepatoma hepa 1c1c7 wild-type, C12, and C4 cells mediated by bilirubin. Mol Pharmacol 62:257-64, 2002.
158. Sewnath ME, Birjmohun RS, Rauws EA, Huibregtse K et al: The effect of preoperative biliary drainage on postoperative complications after pancreati-coduodenectomy. J Am Coll Surg 192:726-34, 2001.
159. Sheen-Chen SM, Hung KS, Ho HT, Chen WJ, Eng HL: Effect of glutamine and bile acid on hepatocyte apoptosis after bile duct ligation in the rat. World J Surg 28:457-60, 2004.
160. Shelley O, Murphy T, Paterson H, Mannick JA, Lederer JA: Interaction between the innate and adaptive immune systems is required to survive sepsis and control inflammation after injury. Shock 20:123-9, 2003.
161. Sigalet DL, Mackenzie SL, Hameed SM: Enteral nutrition and mucosal im-munity: implications for feeding strategies in surgery and trauma. Can J Surg47:109-16, 2004.
162. Sileri P, Morini S, Sica GS, Schena S et al: Bacterial translocation and intes-tinal morphological findings in jaundiced rats. Dig Dis Sci 47:929-34, 2002.
163. Simovart HE, Poldoja E, Kokk K, Tapfer H et al: Changes of activated macrophages and apoptotic cell count in the organs of rats during experimental sepsis. Medicina 39:932-9, 2003.
164. Singleton KD, Wischmeyer PE: Distance of cecum ligated influences mortal-ity, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Eur Surg Res 35:486-91, 2003.
165. Sitzmann JV, Greene PS: Perioperative predictors of morbidity following hepatic resection for neoplasm. A multivariate analysis of a single surgeon ex-perience with 105 patients. Ann Surg 219:13-7, 1994.
166. Sminia T, Jeurissen SHM: Macrophages in the gastrointestinal tract. Im-munobiology 172:72, 1986.
167. Sommer T, Larsen JF: Intraperitoneal and intraluminal microdialysis in the detection of experimental regional intestinal ischaemia. Br J Surg 91:855-61, 2004.
168. Stafford RE, Weigelt JA: Surgical infections in the critically ill. Curr Opin Crit Care 8:449-52, 2002.
169. Steinberg SM: Bacterial translocation: what it is and what it is not. Am J Surg 186:301-5, 2003.
170. Sternberger LA: Immunocytochemistry. New York: Wiley Medical Publica-tions,1979.
171. Strieter RM, Kunkel SL, Bone RC: Role of tumour necrosis factor-alpha in disease states and inflammation. Crit Care Med 21:447-63, 1993.
172. Sun EW, Shi YF: Apoptosis: the quiet death silences the immune system. Pharmacol Ther 92:135-45, 2001.
173. Tenover FC, Arbeit RD, Goering RV, Mickelsen PA et al: Interpreting chromosomal DNA restriction patterns produced by pulse-field gelelectropho-resis: criteria for bacterial typing. J Clin Microbiol 33:2233–9, 1995.

68
174. Thompson RL, Hoper M, Diamond T, Rowlands BJ: Development and reversibility of T lymphocyte dysfunction in experimental obstructive jaun-dice. Br J Surg 77:1229-32, 1990.
175. Thorlacius H, Nobaek S, Wang XD, Andersson R et al: Lactobacilli attenu-ate bacteremia and endotoxemia associated with severe intra-abdominal infec-tion. Surgery 134:467-73, 2003.
176. Tomioka M, Iinuma H, Okinaga K: Impaired Kupffer cell function and effect of immunotherapy in obstructive jaundice. J Surg Res 92:276-82, 2000.
177. Trevethick MA, Oakley I, Clayton NM, Strong P: Non-steroidal anti-inflammatory drug-induced gastric damage in experimental animals: underly-ing pathological mechanisms. Gen. Pharmacol 26:1455–1459, 1995.
178. Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS et al: Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock 19:310-3, 2003.
179. Wakefield CH, Barclay GR, Fearon KC, Goldie AS et al: Proinflammatory mediator activity, endogenous antagonists and the systemic inflammatory re-sponse in intra-abdominal sepsis. Scottish Sepsis Intervention Group. Br J Surg 85:818-25, 1998.
180. Walley KR, Lukacs NW, Standiford TJ, Strieter RM , Kunkel SL: Balanceof inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun 64:4733-8, 1996.
181. Van der Poll T, Lowry SF: Tumour necrosis factor in sepsis: mediator of multiple organ failure or essential part of host defense? Shock 3:1-12, 1995.
182. Weigand MA, Horner C, Bardenheuer HJ, Bouchon A: The systemic in-flammatory response syndrome. Best Pract Res Clin Anaesthesiol 18:455-75, 2004.
183. Wells CL, Rotstein OR, Pruett TL, Simmons RL: Intestinal bacteria translo-cate into experimental intra-abdominal abscesses. Arch Surg 121:102–7, 1986.
184. Wells CL, Maddaus MA, Simmons RL: Proposed mechanisms for the trans-location of intestinal bacteria. Rev Infect Dis 10:958-79,1988.
185. Welsh FK, Ramsden CW, MacLennan K, Sheridan MB et al: Increased intestinal permeability and altered mucosal immunity in cholestatic jaundice. Ann Surg 227:265-72, 1998.
186. White RAH, Mason DW, Williams AF, Galfre G, Milstein C: T-lymphocyte heterogenity in the rat: separation of the functional subpopulations using monoclonal antibody. J Exp Med 148:664–673, 1978.
187. Wichterman KA, Baue AE, Chaudry IH: Sepsis and septic shock--a review of laboratory models and a proposal. J Surg Res 29:189-201,1980.
188. Wiedle G, Dunon D, Imhof BA: Current concepts in lymphocyte homing and recirculation. Crit Rev Clin Lab Sci 38:1-31, 2001.
189. Vincent JL, Abraham E, Annane D, Bernard G et al: Reducing mortality in sepsis: new directions. Crit Care 6 Suppl 3:1-186, 2002.
190. Vitetta ES, Berton MT, Burger C, Kepron M et al: Memory B and T cells. Annu Rev Immunol 9:193-217,1991.
191. Volk H-D, Reinke P, Docke WD: Immunological monitoring of the inflamma-tory process: which variables? When to assess? Eur J Surg Suppl 584:70–2, 1999.
192. Wollert S, Antonsson J, Gerdin B, Lundberg C et al: Intestinal mucosal injury during porcine faecal peritonitis. Eur J Surg 161:741–750, 1995.
193. Wright SD, Tobias PS, Ulevitch RJ, Ramos RA: Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel re-ceptor on macrophages. J Exp Med 170:1231-41, 1989.

69
194. Wyllie AH: Apoptosis and the regulation of cell numbers in normal and neo-plastic tissues: an overview. Cancer Metastasis Rev 11:95-103, 1992.
195. Yuan Q, Walker WA: Innate immunity of the gut: mucosal defense in health and disease. J Pediatr Gastroenterol Nutr 38:463-73, 2004.
196. Zarzaur BL, Kudsk KA: The mucosa-associated lymphoid tissue structure, function, and derangements. Shock 15:411-20, 2001.
197. Zhang XW, Hedlund G, Borgstrom P, Arfors KE, Thorlacius H: Linomide abolishes leukocyte adhesion and extravascular recruitment induced by tumor necrosis factor alpha in vivo. J Leukoc Biol 68:621-6, 2000.
198. Zifroni A, Treves AJ, Sachar DB, Rachmilewitz D: Prostanoid synthesis by cultured intestinal epithelial and mononuclear cells in inflammatory bowel dis-ease. Gut 24:659-64, 1983.
199. Zimmerman JE, Knaus WA, Wagner DP, Sun X et al: Hakim RB, Nystrom PO. A comparison of risks and outcomes for patients with organ system fail-ure: 1982-1990 Crit Care Med 24:1633-41, 1996.

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 3
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, Uppsala University, is usually a summary of a number of papers. A few copies of the complete dissertation are kept at major Swedish research libraries, while the summary alone is distributed internationally through the series Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine. (Prior to January, 2005, the series was published under the title "Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine".)
Distribution: publications.uu.seurn:nbn:se:uu:diva-4767
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2005





























