Embed Size (px)
DESCRIPTION
Although Rat-l fibroblasts expressing c-myc constitutivelyare unable to arrest growth in low serum, theirnumbers do not increase in culture because of substantialcell death. We show this cell death to be dependentupon expression of c-myc protein and to occur byapoptosis. Regions of the c-myc protein required forinduction of apoptosis overlap with regions necessaryfor cotransformation, autoregulation, and inhibition ofdifferentiation, suggesting that the apoptotic functionof c-myc protein is related to its other functions. Moreover,cells with higher levels of c-myc protein are moreprone to cell death upon serum deprivation. Finally,we demonstrate that deregulated c-myc expression inducesapoptosis in cells growth arrested by a varietyof means and at various points in the cell cycle.
Citation preview

Cell. Vol. 69, 119-128, April 3, 1992. Copyright 0 1992 by Cell Press
Induction of Apoptosis in Fibroblasts by c-myc Protein
Gerard I. Evan,’ Andrew H. Wyllie,t Christopher S. Gilbert,* Trevor D. Littlewood,’ Hartmut Land,’ Mary Brooks,* Catherine M. Waters,’ Linda 2. Penn,’ and David C. Hancock* *Imperial Cancer Research Fund Laboratories 44 Lincoln’s Inn Fields London WC2A 3PX England tDepartment of Pathology University Medical School Teviot Place Edinburgh EH8 9AG Scotland
Summary
Although Rat-l fibroblasts expressing c-myc constitu- tively are unable to arrest growth in low serum, their numbers do not increase in culture because of sub- stantial cell death. We show this cell death to be depen- dent upon expression of c-myc protein and to occur by apoptosis. Regions of the c-myc protein required for induction of apoptosis overlap with regions necessary for cotransformation, autoregulation, and inhibition of differentiation, suggesting that the apoptotic function of c-myc protein is related to its other functions. More- over, cells with higher levels of c-myc protein are more prone to cell death upon serum deprivation. Finally, we demonstrate that deregulated c-myc expression in- duces apoptosis in cells growth arrested by a variety of means and at various points in the cell cycle.
Introduction
The c-myc gene is the cellular homolog of the viral onco- genev-myc, which is found in a number of avian and feline retroviruses that induce leukemias and carcinomas. Re- cent evidence strongly suggests that the c-myc protein (Myc) is a transcription factor (Blackwood and Eisenman, 1991). It possesses a number of functional domains found in other proteins modulating transcription, specifically the leucine zipper characteristic of the Fos-Jun-CREB tran- scription factor families (Landschulz et al., 1988) and the basic-helix-loop-helix motif found in, for example, the MyoD and E box enhancer-binding proteins (Murre et al., 1989). Recently, both a heterodimeric partner (Blackwood and Eisenman, 1991) and a consensus DNA-binding se- quence for Myc (Blackwell et al., 1990; Prendergast and Ziff, 1991) have been identified. However, it is still un- known precisely which genes are regulated by Myc or to what biological end.
The c-myc oncogene has been implicated in the control of normal cell proliferation by many studies. In particular, it is one of the immediate early growth response genes
that are rapidly induced in quiescent cells upon mitogenic induction, suggesting that it plays some role in mediating the transition from quiescence to proliferation. However, unlike the majority of immediate early growth response genes, expression of c-myc is not confined to a brief period during the Go/G1 transition. Although a peak of c-myc ex- pression in fibroblasts is observed some 3 hr after mito- genie stimulation, both c-myc mRNAand protein are con- tinuously present at an appreciable level throughout the cell cycle in proliferating cells. As both c-myc mRNA and protein have very short half-lives in fibroblasts (Moore et al., 1987; Persson et al., 1986; Waters et al., 1991), this sustained presence of Myc protein can only result from continuous synthesis. Ectopic induction of Myc activity is sufficient to drive quiescent growth factor-deprived fibro- blasts into the cell cycle (Eilers et al., 1991). This argues that Myc regulates genes mediating the mitogenic re- sponse, an idea consistent with the protein’s rapid induc- tion by mitogens in quiescent cells. In addition, sustained expression of Myc can block both growth arrest and cell differentiation programs, suggesting a role for Myc also in regulating genes mediating both of these processes.
Untransformed fibroblasts respond to serum or mitogen deprivation by growth arrest in a Gl-like state often termed Go and can remain viable in this arrested state for extended periods. Mitogen withdrawal is accompanied by rapid down regulation of c-myc expression at both the mRNA and protein levels, irrespective of position within the cell cycle (Dean et al., 1986; Waters et al., 1991). Because cells deprived of growth factors eventually become quies- cent, it has been suggested that Myc down regulation is a requirement or even a signal for growth arrest (Freytag, 1988; Waters et al., 1991).
In tumor cells, elevated or deregulated expression of c-myc (and occasionally other myc genes) is so wide- spread as to suggest a critical role for myc gene activation in multi-stage carcinogenesis (Field and Spandidos, 1990; Spencer and Groudine, 1991). Although it is unclear whether it is deregulation or overexpression of c-myc that makes up the major determinant in c-myc oncogene acti- vation, it is nonetheless evident that c-myc activation dis- rupts the growth regulation of cells. For this reason, we have investigated the consequences of deregulated and elevated expression on the behavior of rodent fibroblasts. In this report we show that deregulated c-myc expression is a potent inducer of programmed cell death (apoptosis) when combined with a block to cell proliferation. We dis- cuss the implications of these findings in terms of the bio- logical role of c-myc in normal and tumor cells.
Results
Constitutive c-myc Expression Prevents Growth Arrest in Serum-Deprived Rat-l Fibroblasts and Induces Cell Death Growth arrest is accompanied by rapid down regulation of
Cell 120
Rat- 1 control
10% FCS
0.5% FCS 0.05% FCS
1 Rat-l/A106-143 T 10% FCS
0.5% FCS
0.05% FCS
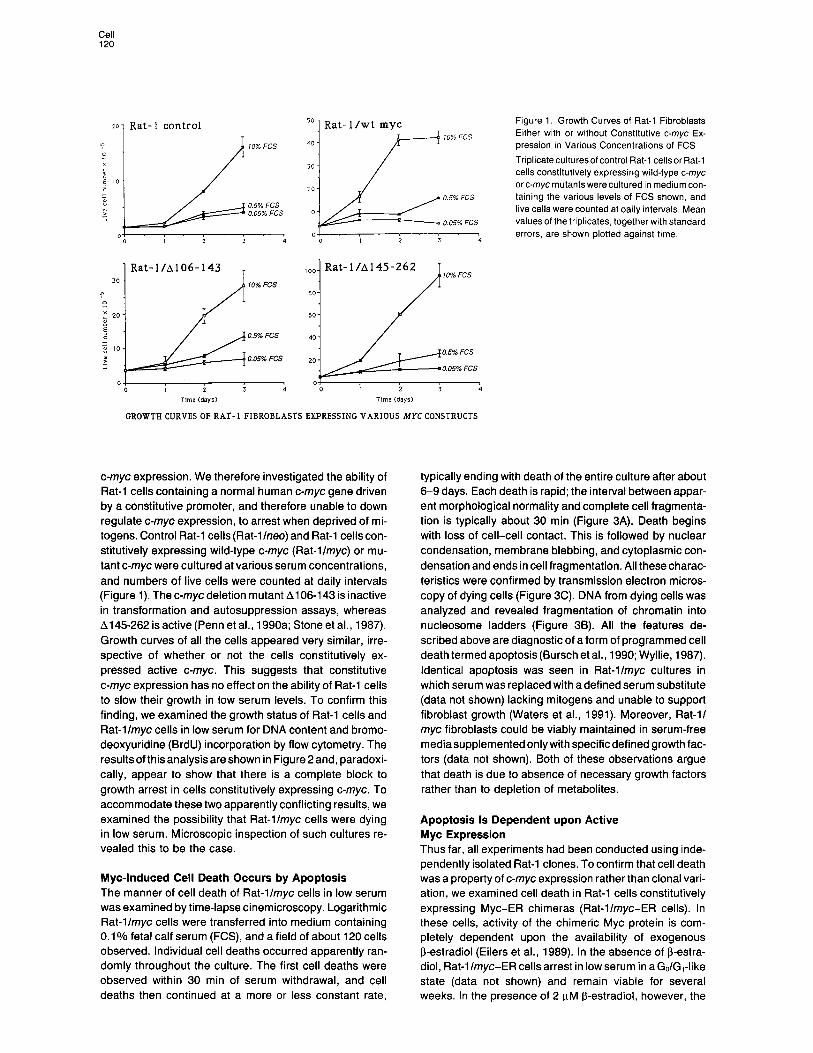
Rat- 1 /wt myc Figure 1. Growth Curves of Rat-l Fibroblasts Either with or without Constitutive c-myc Ex- pression in Various Concentrations of FCS
Tnplicateculturesofcontrol Rat-l cellsor Rat-i cells constitutively expressing wild-type c-myc arc-mycmutantswereculturedin mediumcon- taining the various levels of FCS shown, and live cells were counted at daily intervals, Mean values of the triplicates, together with standard errors, are shown plotted against time.
10% FCS
0.5% FCS
0.05% FCS
GROWTH CURVES OF RAT- 1 FIBROBLASTS EXPRESSING VARIOUS MYC CONSTRUCTS
c-myc expression. We therefore investigated the ability of Rat-l cells containing a normal human c-myc gene driven by a constitutive promoter, and therefore unable to down regulate c-myc expression, to arrest when deprived of mi- togens. Control Rat-l cells (Rat-l Ineo) and Rat-l cells con- stitutively expressing wild-type c-myc (Rat-llmyc) or mu- tant c-myc were cultured at various serum concentrations, and numbers of live cells were counted at daily intervals (Figure 1). The c-myc deletion mutant Al 06-l 43 is inactive in transformation and autosuppression assays, whereas A145262 is active (Penn et al., 1990a; Stone et al., 1987). Growth curves of all the cells appeared very similar, irre- spective of whether or not the cells constitutively ex- pressed active c-myc. This suggests that constitutive c-myc expression has no effect on the ability of Rat-l cells to slow their growth in low serum levels. To confirm this finding, we examined the growth status of Rat-l cells and Rat-llmyc cells in low serum for DNA content and bromo- deoxyuridine (BrdU) incorporation by flow cytometry. The results of this analysis are shown in Figure 2 and, paradoxi- cally, appear to show that there is a complete block to growth arrest in cells constitutively expressing c-myc. To accommodate these two apparently conflicting results, we examined the possibility that Rat-llmyc cells were dying in low serum. Microscopic inspection of such cultures re- vealed this to be the case.
Myc-Induced Cell Death Occurs by Apoptosis The manner of cell death of Rat-llmyc cells in low serum was examined by time-lapse cinemicroscopy. Logarithmic Rat-llmyc cells were transferred into medium containing 0.1% fetal calf serum (FCS), and a field of about 120 cells observed. Individual cell deaths occurred apparently ran- domly throughout the culture. The first cell deaths were observed within 30 min of serum withdrawal, and cell deaths then continued at a more or less constant rate,
typically ending with death of the entire culture after about 6-9 days. Each death is rapid; the interval between appar- ent morphological normality and complete cell fragmenta- tion is typically about 30 min (Figure 3A). Death begins with loss of cell-cell contact. This is followed by nuclear condensation, membrane blebbing, and cytoplasmic con- densation and ends in cell fragmentation. All these charac- teristics were confirmed by transmission electron micros- copy of dying cells (Figure 3C). DNA from dying cells was analyzed and revealed fragmentation of chromatin into nucleosome ladders (Figure 38). All the features de- scribed above are diagnostic of a form of programmed cell death termed apoptosis (Bursch et al., 1990; Wyllie, 1987). Identical apoptosis was seen in Rat-llmyc cultures in which serum was replaced with adefined serum substitute (data not shown) lacking mitogens and unable to support fibroblast growth (Waters et al., 1991). Moreover, Rat-l/ myc fibroblasts could be viably maintained in serum-free mediasupplemented onlywith specificdefined growth fac- tors (data not shown). Both of these observations argue that death is due to absence of necessary growth factors rather than to depletion of metabolites.
Apoptosis Is Dependent upon Active Myc Expression Thus far, all experiments had been conducted using inde- pendently isolated Rat-l clones. To confirm that cell death was a property of c-myc expression rather than clonal vari- ation, we examined cell death in Rat-l cells constitutively expressing Myc-ER chimeras (Rat-l/myc-ER cells). In these cells, activity of the chimeric Myc protein is com- pletely dependent upon the availability of exogenous 6-estradiol (Eilers et al., 1989). In the absence of 8-estra- diol, Rat-l/myc-ER cells arrest in low serum in aGo/G1-like state (data not shown) and remain viable for several weeks. In the presence of 2 PM f3-estradiol, however, the

induction of Apoptosis by c-myc Protein 121
A Rat-llneo control Rat-l/my c
0.1% serum for 48 hrs 0.1% serum for 48 hrs
10% ser”m far 48 hrs 10% serum for 48 hrs
ONA content -
Figure 2. Constituttve c-myc Expression Prevents Growth Arrest in Serum-Deprived Rat-l Cells
Triplicate logarithmicculturesof Rat-llneocontrol and Rat-llmyccells were transferred into medium containing either 10% or 0.1% FCS. After 49 hr, cells were labeled for 1 hr with 2 mM BudR, trypsinized, fixed in ethanol, and stained with propidium iodide and appropriately confugated anti-BudR antibody as described (Ormerod, 1990). Flow cytometric analysis was carried out on a Beckton-Dickinson FACSstar plus. (A) DNA histograms of Rat-l cells.(B) Percentage of cellsstaining with anh-BrdU antibody (i.e., traversing S phase) over a 1 hr perrod.
rapid onset of apoptosis is observed in low serum. In con- trast, Rat-l fibroblasts expressing the transformation- defective mutant of Myc, A106-143 (Penn et al., 1990a; Stone et al., 1987) fused to estrogen receptor (ER) (Rat-l/ A106-143-ER cells) arrest in low serum and exhibit no apoptosis irrespective of the presence of j3-estradiol (Fig- ure 4). Thus, apoptosis of Rat-l/myc-ER depends upon the presence of active Myc protein and is not a trivial result of the addition of j3-estradiol to the culture.
Deregulated Expression of Myc Induces Apoptosis in Serum-Deprived Rat Embryo Fibroblasts Rat-l cells are an immortalized and established cell line. We were therefore interested to determine how general was Myc-induced apoptosis; in particular, whether it oc- curred in a nonestablished primary fibroblast culture. Ac- cordingly, primary rat embryo fibroblasts (REFs) constitu- tively expressing Myc were subjected to serum deprivation and monitored microscopically over a 72 hr period. As with
Rat-llmyc cells, such REFlmyc fibroblasts fail to arrest growth in low serum as determined by flow cytometry(data not shown). As can be seen in Figure 5, substantial apoptosis occurs within 24 hr of transfer into low serum.
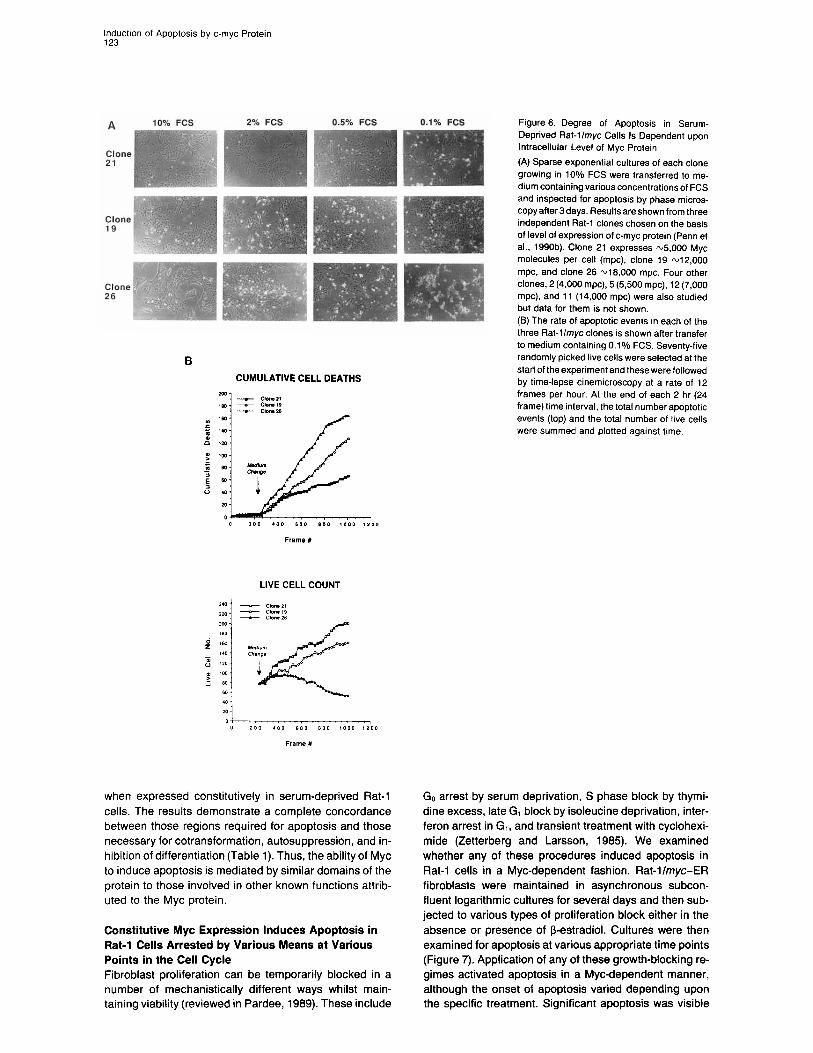
Extent of Myc-Induced Apoptosis Is Related to the Levels of Myc Protein in Cells We next investigated whether there was any correlation between intracellular Myc protein level and susceptibility to Myc-induced apoptosis in Rat-llmyc cells. Various Rat-llmyc cell clones were selected, each of which ex- presses a different steady-state level of Myc protein (Penn et al., 1990b). Each clone was then assayed for apoptosis by two independent assays. First, cells were cultured in various concentrations of FCS, and the degree of cell death was assessed by microscopic examination after 3 days. Results are shown in Figure 6A for the three Rat-l/ myc clones 21, 19, and 26, which representatively span the range of Myc protein levels investigated. Clone 26 expresses most Myc and exhibits significant apoptosis even in serum levels as high as 2%. Apoptosis is even more evident at lower serum levels. In contrast, clone 21, which expresses a level of Myc protein similar to that found in normal fibroblasts (Waters et al., 1991; Moore et al., 1987), exhibits apoptosis only at the lowest serum levels. Clone 19, with an intermediate Myc protein level, exhibits an intermediate phenotype. Results obtained for four other clones (2, 5, 12, and 1 l), each expressing a different Myc level, were consistent with this trend (data not shown). Second, the various Rat-llmyc clones were transferred into 0.1% FCS, and fields containing identical numbers of cells monitored for apoptosis by time-lapse cinemicros- copy. This type of analysis is complicated by the fact that cell number is continuously varying because of both cell division and cell death. For this reason, results were pooled within each 2 hr time interval, and both cumulative cell deaths and live cell numbers at the end of each 2 hr interval were plotted against time. Data obtained using clones 21, 19, and 26 is shown in Figure 6B. The rate of apoptosis is highest in clone 26, intermediate in clone 19, and lowest in clone 21. Again, results obtained with the four other tested clones confirmed this positive correlation between higher Myc levels and higher apoptotic rate (data not shown).
Thus, we conclude that the sensitivity of Rat-l lmyc cells to induction of apoptosis upon serum depletion and its rate both depend upon the level of Myc protein expressed. Even the low levels of Myc protein observed in normal Rat-l fibroblasts are, however, sufficient to induce apop- tosis in serum-deprived cells if Myc expression is deregu- lated.
Regions of the Myc Protein Required for Apoptosis Certain regions of the Myc protein are absolutely required for its known activities in cotransformation, autosuppres- sion, and inhibition of differentiation (Freytag et al., 1990; Penn et al., 1990a; Stone et al., 1987). These regions include the basic-helix-loop-helix-leucine zipper at the C terminus and part of the N-terminal region. We examined the ability of a range of Myc mutants to induce apoptosis

Cell 122
Figure 3. Myc Induces Death by Apoptosis in Serum-Deprived Rat-l Cells
(A) Rat-l/c-myc cells were transferred to me- dium containing 0.1% FCS and observed by time-lapse cinemicroscopy at a rate of one frame every 30 seconds. Representative frames from a typical apoptotic event are shown with the time in minutes given from the last frame when the cell appeared normal. (6) Cell death is accompanied by nucleosome laddering. Rat-l cells constitutively expressing either active A145-262 (lanes 2, 4, and 6) or
2 inactive A106-144 (lanes 3, 5, and 7) c-myc mutants were transferred to medium con- taining 0.1% FCS. Dying cells were harvested at various times after transfer, by virtue of their reduced adherence. In cultures with no dying
1 cells, very few cells were harvested by this method. DNA was isolated and fractionated on a 1.5% agarose gel. Lane 1, standards; Lanes 2 and 3, 0 hr; Lanes 4 and 5, 30 hr; Lanes 6 and 7, 40 hr; Lane 8, dexamethasone-treated
Y thymocytes. (C) Electron microscopic analysis of individual Rat-llmyc cells undergoing apoptosis in low
serum. Frame 1 shows a normal viable Rat-llmyc cell. The nucleus is marked N. Frames 2 to 4 are representative electron micrographs of Rat-11 myc cells at progressively more advanced stages of apoptosis and exhibiting cytoplasmic and nuclear vesicularization.
Rat-l /pMVG/wt c-myc-E R Rat-llpMV6/A106-143 c-myc-E R Figure 4. Apoptosis in Serum-Deprived Rat-II myc Cells Is Dependent upon Active Myc Protein
Log-phase Rat-l cells constitutively express- ing either full length c-myc protein or the dele- tion mutant A106-143 fused to ER were trans- ferred to medium containing 0.1% FCS either with or without 2 KM 8-estradiol. After 3 days, cultures were examined by phase microscopy.
Figure 5. Deregulated Myc Expression In- duces Apoptosis in Serum-Deprived Primary Cells
REFs constitutively expressing c-myc were cul- tured in medium containing 0.1% FCS for 72 hr. At various times, the culture was inspected microscopically. A, 0 hr; B, 24 hr; C, 48 hr; D. 72 hr.
inductron of Apoptosis by c-myc Protein 123
three Rat-llmyc clones is shown after transfer to medium containing 0.1% FCS. Seventy-five
B randomly picked live cells were selected at the
CUMULATIVE CELL DEATHS start of the experiment and these were followed by time-lapse cinemicroscopy at a rate of 12 frames per hour. At the end of each 2 hr (24 frame) time interval, the total number apoptotic events (top) and the total number of live cells were summed and plotted against time.
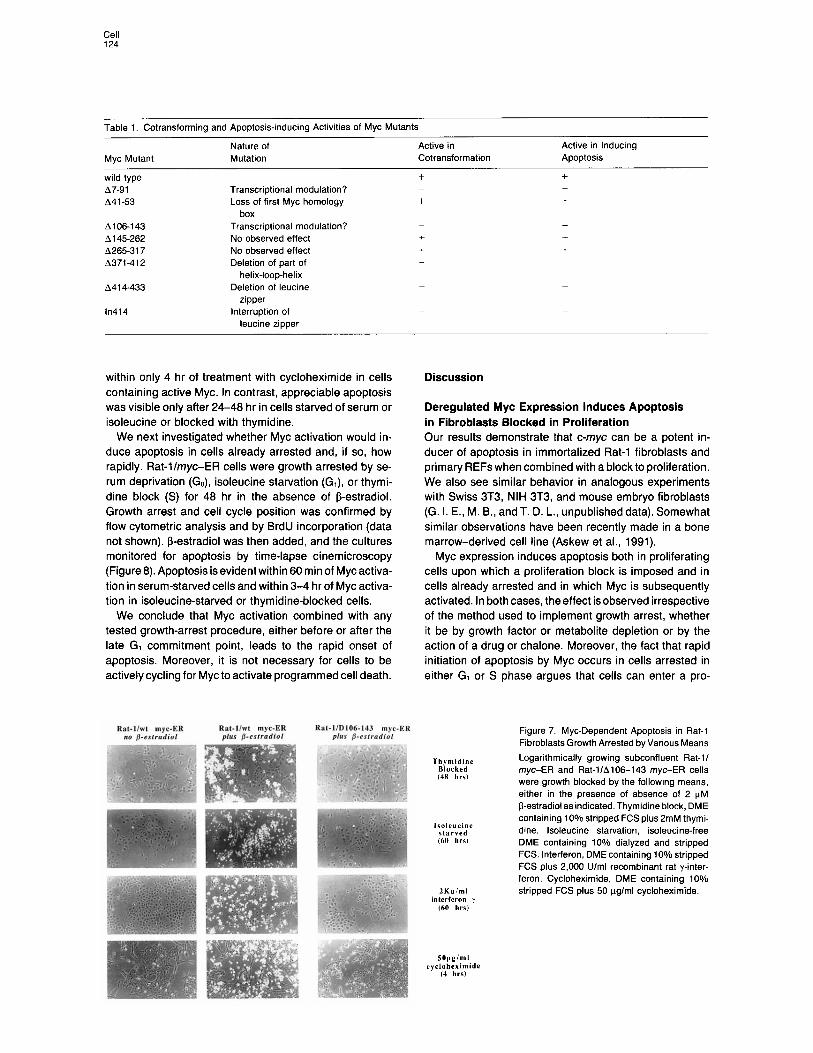
when expressed constitutively in serum-deprived Rat-l cells. The results demonstrate a complete concordance between those regions required for apoptosis and those necessary for cotransformation, autosuppression, and in- hibition of differentiation (Table 1). Thus, the ability of Myc to induce apoptosis is mediated by similar domains of the protein to those involved in other known functions attrib- uted to the Myc protein.
Constitutive Myc Expression Induces Apoptosis in Rat-l Cells Arrested by Various Means at Various Points in the Cell Cycle Fibroblast proliferation can be temporarily blocked in a number of mechanistically different ways whilst main- taining viability (reviewed in Pardee, 1989). These include
Go arrest by serum deprivation, S phase block by thymi- dine excess, late G, block by isoleucine deprivation, inter- feron arrest in G,, and transient treatment with cyclohexi- mide (Zetterberg and Larsson, 1985). We examined whether any of these procedures induced apoptosis in Rat-l cells in a Myc-dependent fashion. Rat-llmyc-ER fibroblasts were maintained in asynchronous subcon- fluent logarithmic cultures for several days and then sub- jected to various types of proliferation block either in the absence or presence of f3-estradiol. Cultures were then examined for apoptosis at various appropriate time points (Figure 7). Application of any of these growth-blocking re- gimes activated apoptosis in a Myc-dependent manner, although the onset of apoptosis varied depending upon the specific treatment. Significant apoptosis was visible
Cell 124
Table 1. Cotransforming and Apoptosis-inducing Activities of Myc Mutants
Myc Mutant Nature of Active in Mutation Cotransformation
Active in Inducing Apoptosis
wild type A7-91 A41 -53
A106-143 A145-262 A26531 7 A371-412
A41 4-433
in414
Transcriptional modulation? Loss of first Myc homology
box Transcriptional modulation? No observed effect No observed effect Deletion of part of
helix-loop-helix Deletion of leucine
zipper interruption of
leucine zipper
+ + - -
+ +
- -
+ + + + - -
- -
- -
within only 4 hr of treatment with cycloheximide in cells containing active Myc. In contrast, appreciable apoptosis was visible only after 24-48 hr in cells starved of serum or isoleucine or blocked with thymidine.
We next investigated whether Myc activation would in- duce apoptosis in cells already arrested and, if so, how rapidly. Rat-l/myc-ER cells were growth arrested by se- rum deprivation (Go), isoleucine starvation (G,), or thymi- dine block (S) for 48 hr in the absence of f3-estradiol. Growth arrest and cell cycle position was confirmed by flow cytometric analysis and by BrdU incorporation (data not shown). 8-estradiol was then added, and the cultures monitored for apoptosis by time-lapse cinemicroscopy (Figure 8). Apoptosis is evident within 60 min of Myc activa- tion in serum-starved cells and within 3-4 hr of Myc activa- tion in isoleucine-starved or thymidine-blocked cells.
We conclude that Myc activation combined with any tested growth-arrest procedure, either before or after the late G, commitment point, leads to the rapid onset of apoptosis. Moreover, it is not necessary for cells to be actively cycling for Myc to activate programmed cell death.
Rat-IhI myc-ER no fl-rsrradiol
Rat-l/W myc-ER plus fl-cslmdiol
Rat-l/D106-143 myc-ER plus 0.eslradiol
Discussion
Deregulated Myc Expression Induces Apoptosis in Fibroblasts Blocked in Proliferation Our results demonstrate that c-myc can be a potent in- ducer of apoptosis in immortalized Rat-l fibroblasts and primary REFs when combined with a block to proliferation. We also see similar behavior in analogous experiments with Swiss 3T3, NIH 3T3, and mouse embryo fibroblasts (G. I. E., M. B., and T. D. L., unpublished data). Somewhat similar observations have been recently made in a bone marrow-derived cell line (Askew et al., 1991).
Myc expression induces apoptosis both in proliferating cells upon which a proliferation block is imposed and in cells already arrested and in which Myc is subsequently activated. In both cases, the effect isobserved irrespective of the method used to implement growth arrest, whether it be by growth factor or metabolite depletion or by the action of a drug or chalone. Moreover, the fact that rapid initiation of apoptosis by Myc occurs in cells arrested in either G, or S phase argues that cells can enter a pro-
Figure 7. Myc-Dependent Apoptosis in Rat-l Fibroblasts Growth Arrested by Various Means
‘Thymidine Blocked (48 hrs)
Logarithmically growing subconfluent Rat-l/ myc-ER and Rat-l/A106-143 myc-ER cells were growth blocked by the following means, either in the presence of absence of 2 uM 6-estradiol as indicated. Thymidine block, DME
lsoleucine starved (60 hrs,
2Kulml interferon y
I60 hrs,
containing 10% stripped FCS plus 2mM thymi- dine. lsoleucine starvation, isoleucine-free DME containing 10% dialyzed and stripped FCS. Interferon, DME containing 10% stripped FCS plus 2,000 U/ml recombinant rat y-inter- feron. Cycloheximide, DME containing 10% stripped FCS plus 50 uglml cycloheximide.
SOpglml rycloheximide
(4 hrs)
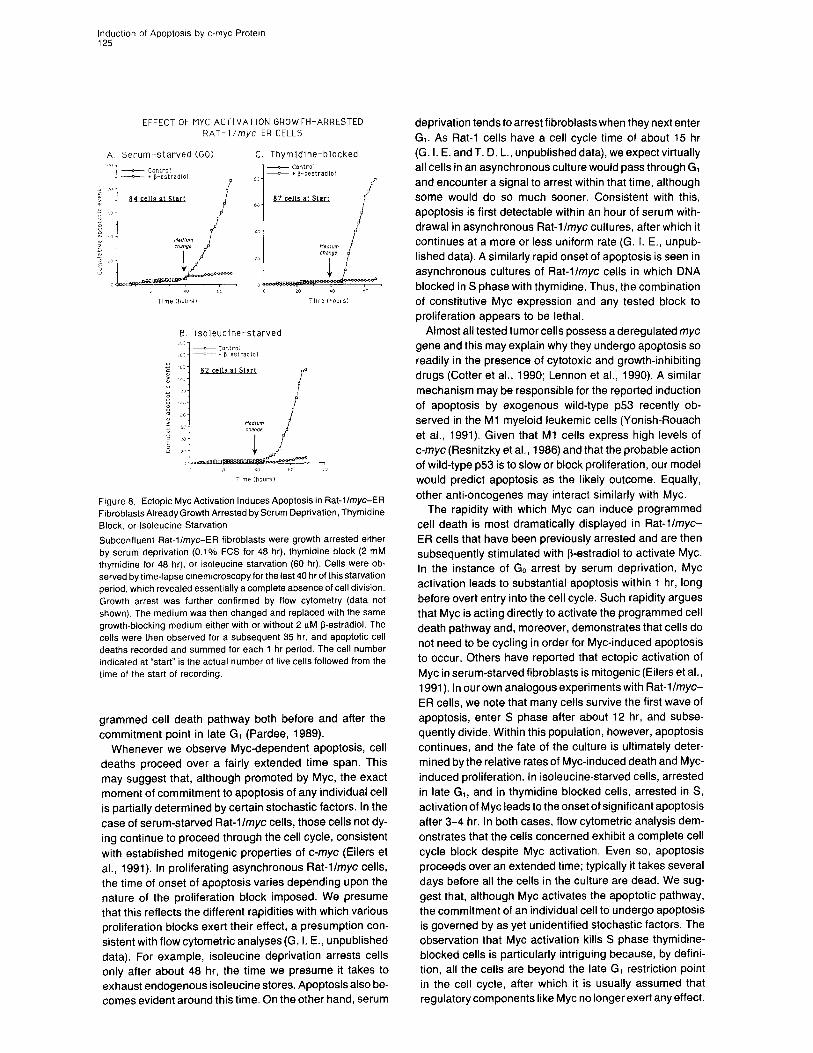
Inductron of Apoptosrs by c-myc Protein 125
EFFECT OF MYC ACTIVATION GROWTH-ARRESTED RAT- I /myc-ER CELLS
A. Serum ~starved (GO) C. Thymidtne-blocked
B. Isoleucine-starved
Figure 8. Ectopic Myc Activation Induces Apoptosis in Rat-llmyc-ER Fibroblasts Already Growth Arrested by Serum Deprivation, Thymidine Block, or lsoleucine Starvation
Subconfluent Rat-l/myc-ER fibroblasts were growth arrested either by serum deprivation (0.1% FCS for 48 hr), thymidine block (2 mM thymidine for 48 hr). or isoleucine starvation (60 hr). Cells were ob- served by time-lapse cmemicroscopy for the last 40 hr of this starvation period, which revealed essentially a complete absence of cell division. Growth arrest was further confirmed by flow cytometry (data not shown). The medium was then changed and replaced with the same growth-blocking medium either with or without 2 pM !3-estradiol. The cells were then observed for a subsequent 35 hr, and apoptotic ceil deaths recorded and summed for each 1 hr period. The cell number indicated at “start” is the actual number of live cells followed from the time of the start of recording.
grammed cell death pathway both before and after the commitment point in late G, (Pardee, 1989).
Whenever we observe Myc-dependent apoptosis, cell deaths proceed over a fairly extended time span. This may suggest that, although promoted by Myc, the exact moment of commitment to apoptosis of any individual cell is partially determined by certain stochastic factors. In the case of serum-starved Rat-llmyc cells, those cells not dy- ing continue to proceed through the cell cycle, consistent with established mitogenic properties of c-myc (Eilers et al., 1991). In proliferating asynchronous flat-llmyc cells, the time of onset of apoptosis varies depending upon the nature of the proliferation block imposed. We presume that this reflects the different rapidities with which various proliferation blocks exert their effect, a presumption con- sistent with flow cytometric analyses (G. I. E., unpublished data). For example, isoleucine deprivation arrests cells only after about 48 hr, the time we presume it takes to exhaust endogenous isoleucine stores. Apoptosis also be- comes evident around this time. On the other hand, serum
deprivation tends to arrest fibroblasts when they next enter GI. As Rat-l cells have a cell cycle time of about 15 hr (G. I. E. and T. D. L., unpublished data), we expect virtually all cells in an asynchronous culture would pass through G, and encounter a signal to arrest within that time, although some would do so much sooner. Consistent with this, apoptosis is first detectable within an hour of serum with- drawal in asynchronous Rat-llmyc cultures, after which it continues at a more or less uniform rate (G. I. E., unpub- lished data). A similarly rapid onset of apoptosis is seen in asynchronous cultures of Rat-llmyc cells in which DNA blocked in S phase with thymidine. Thus, the combination of constitutive Myc expression and any tested block to proliferation appears to be lethal.
Almost all tested tumor cells possess a deregulated myc gene and this may explain why they undergo apoptosis so readily in the presence of cytotoxic and growth-inhibiting drugs (Cotter et al., 1990; Lennon et al., 1990). A similar mechanism may be responsible for the reported induction of apoptosis by exogenous wild-type p53 recently ob- served in the Ml myeloid leukemic cells (Yonish-Rouach et al., 1991). Given that Ml cells express high levels of c-myc (Resnitzky et al., 1986) and that the probable action of wild-type ~53 is to slow or block proliferation, our model would predict apoptosis as the likely outcome. Equally, other anti-oncogenes may interact similarly with Myc.
The rapidity with which Myc can induce programmed cell death is most dramatically displayed in Rat-lImyc- ER cells that have been previously arrested and are then subsequently stimulated with 6-estradiol to activate Myc. In the instance of Go arrest by serum deprivation, Myc activation leads to substantial apoptosis within 1 hr, long before overt entry into the cell cycle. Such rapidity argues that Myc is acting directly to activate the programmed cell death pathway and, moreover, demonstrates that cells do not need to be cycling in order for Myc-induced apoptosis to occur. Others have reported that ectopic activation of Myc in serum-starved fibroblasts is mitogenic (Eilers et al., I 991). In our own analogous experiments with Rat-llmyc- ER cells, we note that many cells survive the first wave of apoptosis, enter S phase after about 12 hr, and subse- quently divide. Within this population, however, apoptosis continues, and the fate of the culture is ultimately deter- mined by the relative rates of Myc-induced death and Myc- induced proliferation. In isoleucine-starved cells, arrested in late G,, and in thymidine blocked cells, arrested in S, activation of Myc leads to the onset of significant apoptosis after 3-4 hr. In both cases, flow cytometric analysis dem- onstrates that the cells concerned exhibit a complete cell cycle block despite Myc activation. Even so, apoptosis proceeds over an extended time; typically it takes several days before all the cells in the culture are dead. We sug- gest that, although Myc activates the apoptotic pathway, the commitment of an individual cell to undergo apoptosis is governed by as yet unidentified stochastic factors. The observation that Myc activation kills S phase thymidine- blocked cells is particularly intriguing because, by defini- tion, all the cells are beyond the late G, restriction point in the cell cycle, after which it is usually assumed that regulatory components like Myc no longer exert any effect.

Cell 126
Myc and the Mechanism of Apoptosis The complete dependence of the apoptosis we describe upon the presence of active Myc is shown by the absolute requirement for exogenous P-estradiol in Rat-llmyc-ER cells. Moreover, both the rate of apoptosis and its sensitiv- ity to induction by serum deprivation depend on the amount of Myc protein present in a cell. In addition, dele- tion mapping shows that the same regions of Myc are involved in inducing apoptosis as are involved in cotrans- formation, autoregulation, and inhibition of differentiation (Freytag et al., 1990; Penn et al., 1990a; Stone et al., 1987). This includes regions mediating heterodimerization with Max and sequence-specific DNA binding (Blackwell et al., 1990; Blackwood and Eisenman, 1991) and those thought to be involved in transcriptional modulation (Kato et al., 1990). Given the above, together with the rapidity with which Myc induces programmed cell death, we deem it likely that Myc is directly involved in initiating apoptosis, presumably by the regulation of specific “apoptotic” genes. This hypothesis can be tested now that probes for some apoptosis-associated genes are available (Owens et al., 1991).
Our results further raise the intriguing possibility that c-myc may comprise a general component of the apoptotic pathway. Such an idea is consistent with results from a number of studies of apoptosis in which c-myc expression has been examined. For example, in a study of experimen- tally induced rodent tumors, Wyllie et al. (1987) reported an elevated level of apoptosis specifically associated with c-myc expression. In two well characterized instances of normal tissue involution through apoptosis, estrogen abla- tion of MCF-7 breast carcinoma xenografts and androgen ablation of rat prostate, c-myc is the only major oncogene whose expression is consistenUy raised during the period of cell death (Buttyan et al., 1988; Kyprianou et al., 1991; Quarmby et al., 1987). Finally, apoptosis of embryonic thymocytes during thymic censorship is initiated by activa- tion of the T cell receptor (Smith et al., 1989) and such activation is known to cause substantial induction c-myc in immature CD4*8+ thymocytes (Riegel et al., 1990). Not- withstanding such circumstantial evidence, the notion that myc genes comprise general components of the pro- grammed cell death pathway, in addition to regulating pro- liferation, is radical and will require further substantiation.
Myc, Proliferation, and Apoptosis What might be the evolutionary sense in having the c-myc oncogene drive both proliferation and programmed cell death, two apparently contradictory processes? Antisense oligonucleotide experiments confirm that Myc is essential for cell proliferation (Heikkila et al., 1987; Loke et al., 1988; Prochownik et al., 1988), whereas it has been shown that induction of Myc activity alone is sufficient to drive quies- cent cells into the cell cycle and maintain them there (Eilers et al., 1991). Myc is therefore both necessary and sufficient for cell proliferation. Clearly, this is a potentially very dan- gerous state of affairs because any mutation that deregu- lated c-myc expression would in principle be oncogenic. If, however, in addition to regulating genes mediating pro-
liferation, Myc also activates genes mediating apoptosis, then all cells expressing Myc would necessarily be primed for programmed cell death. Successful proliferation would then presumably occur only if apoptosis were actively in- hibited, perhaps by activation of complementary signal transduction pathways.
Fibroblasts in the adult are mesenchymal cells in which proliferation is confined to maintenance and repair. Prolif- eration and quiescence are regulated by mitogen availabil- ity and topoinhibition and are accompanied and perhaps mediated by appropriately regulated c-myc expression. In contrast, when many other cell types are deprived of necessary cytokines, they undergo apoptosis (Araki et al., 1990; Williams et al., 1990) and only appear able to enter a quiescent state upon differentiation. We suggest that the reason such cells die is that they are not configured to down regulate c-myc in response to cytokine removal and consequently undergo apoptosis. In this model, the role of differentiation-inducing factors is to turn off c-myc expres- sion, so allowing both differentiation and stable growth arrest. Such a hypothesis is eminently testable in a wide range of experimental systems.
Myc and Carcinogenesis If c-myc is a potent potential inducer of apoptosis, why is its deregulation and overexpression so pervasive, and thus so strongly positively selected, during carcinogene- sis? We propose that the proliferative advantage afforded by c-myc activation is essential for carcinogenesis but, because the proliferative and apoptotic functions of Myc are tightly coupled, it is impossible to select for one without also selecting for the other. According to this argument, deregulation of c-myc is an essential step in carcinogene- sis, but it is lethal should the cell concerned ever be unable to proliferate due to lack of nutrients, space, or necessary cytokines. If this hypothesis is correct, one can envisage three obvious mechanisms that might allow a cell with deregulated c-myc to survive. The first involves acquisition of a second mutation that renders the affected cell inde- pendent of previously obligate cytokines. We suggest that activation of an appropriate cooperating oncogene might exemplify this. The second involves acquisition of a lesion in the programmed cell death pathway. Indeed, an exam- ple of such a mechanism is already known. The activated bcl-2 proto-oncogene cooperates with c-myc and is known to block programmed cell death in lymphoid tissues (Hock- enbery et al., 1990). Third, a cell with deregulated myc might survive if subjected to continuous mitogenic stimula- tion. This might occur in chronic degenerative, hyperplas- tic, and inflammatory situations, all of which are known to predispose to neoplasia.
Conclusion: a Model for Myc Function in the Determination of Cell Fate On the basis of ourdataand the arguments detailed above, we venture to propose the following model. Myc drives two coupled functions: proliferation and programmed cell death. Successful proliferation in normal cells requires the active suppression of programmed cell death, thereby pro-

InductIon of Apoptosts by c-myc Protein 127
viding an inbuilt failsafe to guard against uncontrolled pro- liferation and so allowing the same basic machinery to regulate the two necessarily linked processes of prolifera- tion and programmed cell death. Given that c-myc expres- sion is necessary for proliferation and that down regulation of myc appears necessary for growth arrest and differenti- ation, the c-myc proto-oncogene would appear to be an important central regulator determining the various fates of a cell: proliferation, arrest, differentiation, and death.
Experimental Procedures
Cell Culture and Cell Lines The preparation of recombinant retroviruses directing constitutive ex- pression of c-myc, various c-myc mutants, and myc-ER chimeras has already been described, as has the isolation of appropriately infected Rat-l cells (Eilers et al., 1989. 1991; Penn, et al., 1990a, 1990b). Cells were assayed for constitutive expression of both c-myc mRNA by RNAase protection and Northern blot analysis (Penn, et al., 1990a, 1990b) and for c-myc protein by ELISA and semiquantitaive immuno- fluorescent confocal microscopy (Moore et al., 1987; Waters et al., 1991). All cells were maintained in Dulbecco’s modified E4 medium (DME) supplemented with 10% FCS and 1 mglml Geneticin. Cells were passaged by standard trypsinization and seeded directly onto tissue culture plastic. Ecotropic viruses directing expression of chime- ras between Myc and truncated ER were a kind gift from Dr. Martin Eilers and Professor J. Michael Bishop (University of California, San Francisco). Rat-l cells were infected with viruses encoding Myc-ER chimeras, and Rat-l lines expressing wild-type Myc-ER and A106- 143 Myc-ER were isolated as described for Rat-llmyc lines (Penn, et al., 1990a, 1990b). Myc-ER and A106-143 Myc-ER clones were maintained in phenol red-free DME supplemented with 10% char- coal-dextran stripped FCS and 1 mglml Geneticin. Myc was function- ally activated by the addition of b-estradiol to the medium at a final concentration of 2 uM.
Biochemical and Analytical Techniques To examine nucleosome laddering, equal numbers of cells were estab- lished in 25 cm3tissue culture flasks. Medium was then changed in each flask, as indicated, and at various time points dying cells were harvested by virtue of their reduced adherence. As a consequence, very few cells were obtained from nondying cultures, with conse- quently little DNA. The experiment was thus normalized according to starting cell number rather than to amount of DNA extracted. We chose this method of normalization because the proportion of laddered chro- matin at any one time is quite small relative to intact chromatin present in those nonapoptotic cells in the culture, and this large excess of intact DNA obscures any ladders present. The assay therefore shows when chromatin laddering occurs, but the results are in no way quantitative. Instead, quantitation of apoptosis was carried out by time-lapse cinemi- croscopy (see below). DNA extraction and fractionation on 1.5% agar- ose gels were performed as described (Smith et al., 1989).
Standard electron microscopic procedures were used. Time-lapse cinemicroscopy was conducted using a Olympus in-
verted phase contrast microscope, and images were collected on 16 mm monochrome tine film with a tine camera regulated by an external timer. Cell division events were scored at the time at which septa formed between two daughter cells. Apoptotic cell death events were scored midway between the last appearance of normality and the point at whtch the cell became fully detached and rounded. This corresponds to about t+ 15 min in Figure 3A.
Acknowledgments
We are Indebted to Derek Davies for his invaluable and expert help in flow cytometric analysis. We are grateful to all our many scientific colleagues for their advice and help, and in particular to Drs. Richard Treisman, Mike Owen, Mair Churchill, Mary Collins, Ron Laskey. Paul Nurse, and Kathy Weston. Dedicated to Tam and Theo.
The costs of publication of this article were defrayed in part by
the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 16 USC Section 1734 solely to indicate this fact.
Received October 22, 1991; revised January 14, 1992
References
Araki, S., Shimada, Y., Kaji, K., and Hayashi, H. (1990). Apoptosis of vascular endothelial cells by fibroblast growth factor deprivation. Biochem. Biophys. Res. Commun. 768, 1194-1200.
Askew, D., Ashmun, R., Simmons, B., and Cleveland, J. (1991). Consti- tutivec-mycexpression in IL-9dependent myeloid cell linesuppresses cycle arrest and accelerates apoptosis. Oncogene 6, 1915-1922.
Blackwell, T. K., Kretzner. L., Blackwood, E. M., Eisenman, R. N., and Weintraub, H. (1990). Sequence-specific DNA binding by the c-rnyc protein. Science 250, 1149-I 151.
Blackwood, E. M., and Eisenman, R. N. (1991). Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211-1217.
Bursch, W., Kleine, L., and Tenniswood, M. (1990). The biochemistry of cell death by apoptosis. Biochem. Cell Biol. 68, 1071-1074.
Buttyan, R., Zakeri, Z., Lockshin, R.. and Wolgemuth, D. (1988). Cas- cade induction of c-fos, c-rnyc, and heat shock 70K transcripts during regression of the rat ventral prostate gland. Mol. Endocrinol. 2, 650- 657.
Cotter, T. G., Lennon, S. V., Glynn, J. G., and Martin, S. J. (1990). Cell death via apoptosis and its relationship to growth, development and differentiation of both tumour and normal cells. Anticancer Res. 70, 1153-1159. Dean, M., Levine, R. A., Ran, W.. Kindy, M. S.. Sonenshein, G. E.. and Campisi, J. (1986). Regulation of c-myc transcription and mRNA abundance by serum growth factors and cell contact. J. Biol. Chem. 267, 9161-9166.
Eilers. M., Picard, D., Yamamoto, K. R., and Bishop, M. J. (1989). Chimaeras of Myc oncoprotein and steroid receptors cause hormone- dependent transformation of cells. Nature 340, 66-68.
Eilers, M., Schirm, S., and Bishop, J. M. (1991). The Myc protein acti- vates transcription of the alpha-prothymosin gene. EMBO J. 70, 133- 141.
Field, J. K.. and Spandidos, D. A. (1990). The role of ras and myc oncogenes in human solid tumours and their relevance in diagnosis and prognosis (review). Anticancer Res. 70, l-22.
Freytag, S. 0. (1988). Enforced expression of the c-myc oncogene inhibits cell differentiation by precluding entry into a distinct prediffer- entiation state in GO/Gl. Mol. Cell. Biol. 8, 1614-1624.
Freytag, S. O., Dang, C. V., and Lee, W. M. F. (1990). Definition of the activities and properties of c-myc required to inhibit cell differentiation. Cell Growth Diff. 7, 339-343.
Heikkila, R., Schwab, G.. Wickstrom, E., Loke, S. L.. Pluznik, D. H.. Watt, R., and Neckers, L. M. (1987). A c-myc antisense oligodeoxy- nucleotide inhibits entry into S phase but not progress from GO to Gl. Nature 328, 445-449.
Hockenbery, D., Nunez, G., Milliman, C., Schreiber, R. D., and Kors- meyer, S. J. (1990). BcCP is an inner mitochondrial membrane protein that olocks programmed cell death. Nature 348, 334-336.
Kate, G. J., Barrett, J., Villa, G. M., and Dang, C. V. (1990). An amino- terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 70, 5914-5920.
Kyprianou, N., English, H. F.. Davidson, N. E., and Isaacs, J. T. (1991). Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 57, 162-166.
Landschulz, W. Ii., Johnson, P. F., and McKnight. S. L. (1988). The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240, 1759-l 764.
Lennon, S. V., Martin, S. J., and Cotter, T. G. (1990). Induction of apoptosis (programmed cell death) in tumour cell lines by widely di- verging stimuli. Biochem. Sot. Trans. 78, 343-345.

Cell 128
Loke, S. L., Stein, C., Zhang, X., Avigan, M., Cohen, J., and Neckers, L. M. (1988). Delivery of c-mycantisensephosphorothioateoligodeoxy- nucleotides to hematopoietic cells in culture by liposome fusion: spe- cific reduction in c-myc protein expression correlates with inhibition of cell growth and DNA synthesis. Curr. Top. Microbial. Immunol. 741, 262-289.
and Oren, M. (1991). Wild-type p53 induces apoptosis of myeloid leu- kaemic cells that is inhibited by interleukin-6. Nature 352, 345-347.
Zetterberg, A., and Larsson, 0. (1985). Kinetic analysis of regulatory events in GI leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 82, 5365-5369.
Moore, J. P., Hancock, D. C., Littlewood, T. D., and Evan, G. I. (1987). A sensitive and quantitative enzyme-linked immunosorbence assay for the c-myc and N-myc oncoproteins. Oncogene Res. 2, 65-80.
Murre, C., McCaw, P. S., and Baltimore, D. (1989). A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughter- less, &fyoD, and myc proteins. Cell 56, 777-783.
Ormerod, M. G. (1990). Analysisof DNA. In Flow Cytometry, A Practical Approach, M. G. Ormerod, ed. (Oxford: IRL Press), pp. 69-87.
Owens, G., Hahn, W., and Cohen, J. (1991). Identification of mRNAs associated with programmed cell death in immature thymocytes. Mol. Cell. Biol. 17, 4177-4188.
Note Added in Proof
Please address correspondence to Dr. G. I. Evan.
Pardee, A. B. (1989). GI events and regulation of cell proliferation. Science 246, 603-608.
Penn, L., Brooks, M., Laufer, E., Littlewood, T., Morgenstern, J., Evan, G., Lee, W., and Land, H. (1990a). Domains of human c-myc protein required for autosuppression and cooperation with ras oncogenes are overlapping. Mol. Cell. Biol. 70, 4961-4966.
Penn, L. J. Z., Brooks, M. W., Laufer, E. M., and Land, H. (1990b). Negative autoregulation of c-myc transcription. EMBO J. 9, 1113- 1121.
Persson, H., Gray, H. E., Godeau, F., Braunhut, S., and Bellve, A. Ft. (1986). Multiple growth-associated nuclear proteins immunoprecipi- tated by antisera raised against human c-myc peptide antigens, Mol. Cell Biol. 6, 942-949.
Prendergast, G., and Ziff, E. (1991). Methylation-sensitive sequence- specific DNA binding by the c-myc basic region. Science 251, 186- 189.
Prochownik, E. V., Kukowska, J., and Rodgers, C. (1988). c-myc anti- sense transcripts accelerate differentiation and inhibit GI progression in murine erythroleukemia cells. Mol. Cell. Biol. 8, 3883-3695.
Ouarmby, V., Beekman, W., Wilson, E., and French, F. (1987). Andro- gen regulation of c-myc messenger ribonucleic acid levels in rat ventral prostate. Mol. Endocrinol. 7, 865-874.
Resnitzky, D., Yarden, A., Zipori, D., and Kimchi, A. (1986). Autocrine p-related interferon controls c-myc suppression and growth arrest dur- ing hematopoietic cell differentiation. Cell 46, 31-40.
Riegel, J. S., Richie, E. R., and Allison, J. P. (1990). Nuclear events afler activation of CD4+8+ thymocytes. J. Immunol. 744, 361 l-3618.
Smith, C. A., Williams, G. T., Kingston, R., Jenkinson, E. J., andowen, J. J. T. (1989). Antibodies to CDS/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature 337, 181- 184.
Spencer, C. A., and Groudine, M. (1991). Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 56, l-48.
Stone, J., de Lange, T., Ramsay, G., Jakobvits, E., Bishop, J. M., Varmus, H., and Lee, W. (1987). Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol. Cell. Biol. 7, 1697-1709.
Waters, C., Littlewood, T., Hancock, D., Moore, J., and Evan, G. (1991). c-myc protein expression in untransformed fibroblasts. Oncogene 6, 101-109.
Williams, G. T., Smith, C. A., Spooncer, E., Dexter, T. M., and Taylor, D. R. (1990). Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature 343, 76-79.
Wyllie, A. H. (1987). Apoptosis: cell death under homeostatic control. Arch. Toxicol. Suppl. 7 1, 3-10.
Wyllie, A. H., Rose, K. A., Morris, R. G., Steel, C. M., Foster, E., and Spandidos, D. A. (1987). Rodent fibroblast tumours expressing human myc and raas genes: growth, metastasis and endogenous oncogene expression. Br. J. Cancer 56, 251-259.
Yonish-Rouach, E., Resnitzky. D., Lotem, J., Sachs, L.. Kimchi, A.,
![MYC 2012-2013 Application Packet - Wichita, Kansas€¦ · Web viewIn the subject line, please type, “[First Name] [Last Name] – MYC Application.” Example: John Doe – MYC](https://img.pdfslide.us/doc/110x75/5f09a1057e708231d427bfd9/myc-2012-2013-application-packet-wichita-kansas-web-view-in-the-subject-line.jpg)