Embed Size (px)
Citation preview

DMD #33159
1
In Vitro Hepatic Metabolism of Cediranib, a Potent Vascular Endothelial
Growth Factor Tyrosine Kinase Inhibitor: Interspecies Comparison and Human
Enzymology
Timothy Schulz-Utermoehl1, Michael Spear1, Christopher R.J. Pollard, Christine Pattison,
Helen Rollison, Sunil Sarda, Michelle Ward, Nick Bushby, Angela Jordan & Mike Harrison
Clinical Pharmacology & DMPK Department, Alderley Park, AstraZeneca UK Ltd,
Macclesfield, Cheshire SK10 4TG, United Kingdom.
DMD Fast Forward. Published on July 15, 2010 as doi:10.1124/dmd.110.033159
Copyright 2010 by the American Society for Pharmacology and Experimental Therapeutics.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
2
Running title: In Vitro Hepatic Metabolism of Cediranib
Corresponding Author:
Dr Timothy Schulz-Utermoehl, Clinical Pharmacology and DMPK Department, Alderley
Park, AstraZeneca UK Ltd, Macclesfield, Cheshire SK10 4TG, United Kingdom.
Telephone: +44-1625-233632
Fax: +44-1625-230614
Email: [email protected]
Number of Text Pages : 35
Number of Tables : 3
Number of Figures : 6
Number of References : 29
Number of Words
Abstract : 248
Introduction : 232
Discussion : 1065
Non-standard abbreviations: 1-ABT, 1-aminobenzotriazole; CRC, colorectal cancer; HLM,
human liver microsomes; HPLC-MS, high performance liquid chromatography-mass
spectrometry; KIT, stem cell factor receptor; TKI, tyrosine kinase inhibitor; UGT; uridine
glucuronosyl transferase; UDPGA, uridine diphosphate glucuronic acid; VEGF, vascular
endothelial growth factor
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
3
Abstract
The in vitro metabolism of cediranib, a vascular endothelial growth factor (VEGF) tyrosine
kinase inhibitor (TKI) of all three VEGF receptors in late stage development for the treatment
of colorectal cancer and recurrent glioblastoma was investigated in hepatic proteins from
pre-clinical species and humans using radiolabelled material. In human hepatocyte cultures,
oxidative and conjugative metabolic pathways were identified, with pyrrolidine
N+-glucuronidation being the major route. The primary oxidative pathways were di-and
tri-oxidations and pyrrolidine N-oxidation. All metabolites with the exception of the
N+-glucuronide metabolite were observed in rat and cynomolgus monkey hepatocyte
preparations. Additional metabolism studies in liver microsomes from these or other
preclinical species (CD-1 mouse, Han Wistar rat, Dunkin Hartley guinea-pig, Göttingen
mini-pig, New Zealand White rabbit, Beagle dog, cynomolgus and Rhesus monkey) indicated
that the N+-glucuronide metabolite was not formed in these additional species. Incubations
with recombinant flavin-containing monooxygenase (FMO) and uridine
glucuronosyltransferase (UGT) enzymes and inhibition studies using the non-selective
cytochrome P450 (CYP450) chemical inhibitor 1-aminobenzotriazole (1-ABT) in human
hepatocytes indicated that FMO1 and FMO3 contributed to cediranib N-oxidation, whilst
UGT1A4 had a major role in cediranib N+-glucuronidation. CYP450 enzymes contributed
only a minor role to the metabolism of cediranib. In conclusion, species differences in the
formation of the N+-glucuronide metabolite of cediranib were observed. All other
metabolites of cediranib found in humans were also detected in rat and cynomolgus monkey.
Non-CYP450 enzymes are predominantly involved in the metabolism of cediranib and this
suggests that clinical drug interactions involving other co-administered drugs are unlikely.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
4
Introduction
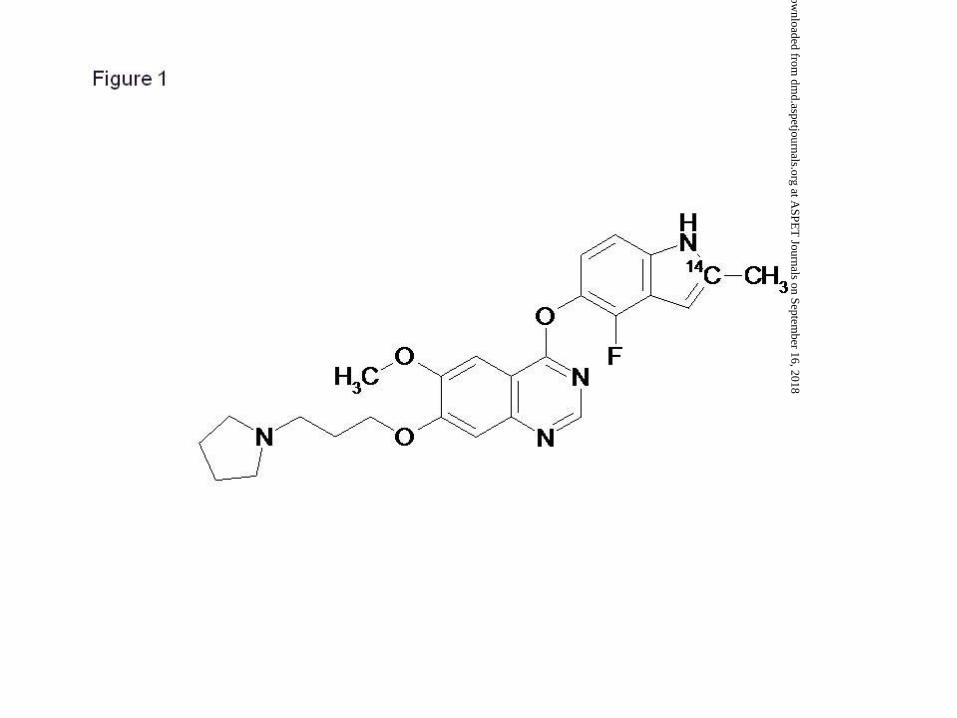
Cediranib (4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-
pyrrolidinyl)propoxy]quinazoline; RECENTIN™; AZD2171)) (Fig. 1) is a highly potent
vascular endothelial growth factor (VEGF) inhibitor, with activity against all three VEGF
receptor tyrosine kinases, and stem cell factor receptor (KIT). It is currently in Phase III
clinical trials for the treatment of colorectal cancer (CRC) and recurrent glioblastoma. VEGF
is involved in the regulation of key processes throughout the angiogenic cascade, and over-
production of VEGF in tumours facilitates tumour progression by stimulating angiogenesis
and increasing vascular permeability. Inhibition of VEGF receptor tyrosine kinases stabilizes
the progression of tumours by disrupting tumour-induced angiogenesis (Kim et al., 1993).
Pre-clinical studies have shown that cediranib prevents VEGF-induced angiogenesis in vivo
and inhibits the growth of established human tumour xenografts in athmic mice in a
dose-dependent manner (Wedge et al., 2005). In the clinic, cediranib has exhibited activity in
patients with advanced CRC and recurrent glioblastoma (Batchelor et al., 2007; Drevs et al.,
2007; Chen et al., 2009).
To gain an understanding of the species differences in the disposition of cediranib and
pre-clinical coverage of human metabolites, the hepatic metabolism of [14C]-cediranib was
investigated in liver preparations from preclinical species and human. The in vitro metabolic
profiles and identity of cediranib metabolites were assessed and a proposed metabolic
pathway suggested. The identity of the hepatic human Phase I and II enzymes involved in
the oxidation and conjugation of cediranib were confirmed and are reported here.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
5
Materials and Methods
Chemicals & Materials. Dexamethasone, insulin, penicillin-streptomycin, Williams’ E
media, β-NADPH, alamethicin, D-(+)-glucuronic acid, acetaminophen, 1-aminobenzotriazole
(1-ABT), DMSO-d6, [14C]-7-ethoxycoumarin (specific activity, 56 mCi/mmol) and
[14C]-acetaminophen (specific activity, 5.4 mCi/mmol) were purchased from Sigma-Aldrich
(Poole, UK). 10 % foetal bovine serum, gentamycin, Leibovitz L15 media and HEPES buffer
were purchased from Invitrogen (Paisley, UK). Matrigel was obtained from BD Biosciences
(San Jose, CA). Ultima Flo-M scintillation cocktail was purchased from Packard Instruments,
(Pangbourne, UK). [14C]-cediranib maleate (4-[(4-fluoro-[2-14C]-2-methyl-1H-indol-5-
yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline maleate) (batch 1R4, specific
activity of 26.9 mCi/mmol; batch 1R5, specific activity of 32.9 mCi/mmol; batch SEL/1713
(1R6), specific activity of 59 mCi/mmol; batch 016, specific activity of 56 mCi/mmol) was
synthesised by the Isotope Chemistry Group at AstraZeneca UK Ltd (batches 1R4, 1R5 and
016; Macclesfield, UK) or by Selcia Limited (batch SEL/1713 (1R6); Ongar, UK). The
radiochemical purity of 1R4, 1R5, 1R6 and 016 was >98, >98, 99 and >98%, respectively.
An authentic metabolite standard, cediranib pyrrolidine N-oxide, was synthesised by
AstraZeneca UK Ltd (Macclesfield, UK). The N+-glucuronide of cediranib was
biosynthesised by Novacta Biosystems (Welwyn Garden City, UK). Fisher Scientific
supplied acetonitrile and methanol. All other chemicals or solvents were purchased from
commercial suppliers and were of analytical grade or the best equivalent.
Hepatocytes. Viable fresh hepatocytes were isolated from male Han-Wistar rats
(AstraZeneca Alderley Park; Macclesfield, UK) using the standard collagenase perfusion
technique. Freshly isolated human hepatocytes from 2 male (donor 1 - unknown race aged
39; donor 3 - Asian, aged 38) and 3 female donors (donor 2 - unknown race, aged 68; donor 4
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
6
- Caucasian, aged 60 years and donor 5 - unknown race, aged 45 years) were purchased from
the United Kingdom Human Tissue Bank (UKHTB; Leicester, UK). All human liver-derived
samples were obtained with permission from the relevant local ethical committees. Freshly
isolated cynomolgus monkey hepatocytes were purchased from Covance (Harrogate, UK).
Cell yield and viability were assessed using Trypan Blue.
Liver Microsomes. Pooled human liver microsomes (28 male donors and 5 female donors;
lot 63103) were obtained from BD Biosciences (San Jose, CA). Pooled liver microsomes
from cynomolgus monkey (8 male donors; lot 3), Dunkin-Hartley guinea-pig (8 male donors;
lot 12635), Göttingen mini-pig (3 male donors; lot 71911), Rhesus monkey (4 male donors;
lot 1), New Zealand White rabbit (3 male donors; lot 98358), CD-1 mouse (150 male donors;
lot 80662), beagle dog (20 male donors; lot 11969) and Han-Wistar rat (30 male donors;
lot 37975) were purchased from BD Biosciences (San Jose, CA). These samples were stored
at -80°C in a controlled tissue collection until use.
Recombinant Enzymes. Samples of microsomal fractions prepared from insect cells
transfected with plasmid vectors expressing human UGT1A1 (lot 13), UGT1A3 (lot 13),
UGT1A4 (lots 9, 10, 11, 34582 and 59840), UGT1A6 (lot 10), UGT1A7 (lot 41931),
UGT1A8 (lot 36867), UGT1A9 (lot 8), UGT1A10 (lot 51067), UGT2B4 (lot 4), UGT2B7
(lot 55297), UGT2B15 (lot 8) and UGT2B17 (lot 4) were purchased from BD Biosciences
(San Jose, CA). Microsomes prepared from insect cells that expressed human FMO1
(lot 52151), FMO3 (lot 64186) and FMO5 (lot 56381) and microsomes from cells that
contained the empty transfection vector were also obtained from BD Biosciences.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
7
Characterisation of [14C]-cediranib. Stock solutions of [14C]-cediranib were diluted in
either 0.2% (v/v) formic acid in water: 0.2% formic acid in acetonitrile 95:5 (v/v) or
water: acetonitrile 80:20 (v/v) to concentrations in the range of 74 to 100 µM (dependent on
specific activity of the batch used for each experiment), before the radiochemical purity was
assessed by HPLC with radiochemical detection (HPLC-RAD).
Hepatocyte Suspension Incubations. Human hepatocytes (donors 1, 2 and 3; approximately
1 X 106 cells/mL) were pre-incubated at 37°C in Leibovitz L15 media in the absence and
presence of 1-aminobenzotriazole (1-ABT; 1 mM final) for 15 min prior to incubation with
[14C]-cediranib. [14C]-cediranib (prepared as a 3 mM solution in methanol) was diluted 1 in
300 with the microsomal suspension to achieve a final concentration of 10 µM (arbitrary
concentration). Incubations with [14C]-cediranib were maintained at 37°C for up to 4 hours
in a shaking water bath and were terminated with an equal volume of ethanol. Appropriate
control incubations were also performed. The positive control compound, [14C]-7-
ethoxycoumarin (20 µM), was incubated in parallel to [14C]-cediranib incubations. All
samples were vortex mixed, centrifuged at 14171 g for up to 10 min and the resulting
supernatants stored at –20°C until analysis by HPLC with radiometric (RAD) and mass
spectrometry detection (MSn). Assays were performed in duplicate.
Hepatocyte Culture Incubations. Incubations of hepatocytes (donors 4 and 5) with
[14C]-cediranib were based on the method described previously (LeCluyse et al., 2000) with
minor modifications. Hepatocytes (132,000 viable cells/cm2) were plated onto 12 well plates
(growth areas of 3.8 cm2 per well) pre-coated with collagen I (BD Biosciences, San Jose, CA)
and sandwiched with a top layer of Matrigel (250 µg/mL). Following a 12 to 24 hour
stabilisation period, hepatocytes were incubated with [14C]-cediranib (10 µM final (arbitrary
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
8
concentration); initially dissolved in methanol as a 3 mM stock solution) in Williams’ E
media containing dexamethasone (30 nM), gentamycin (50 µg/mL), HEPES buffer (10 mM),
insulin (170 nM) and penicillin-streptomycin (102 U/mL; 100 µg/mL). The total incubation
volume was 1 mL. Hepatocytes were incubated for up to 72 hours (24 hours for donor 4) at
37°C in an atmosphere of 5 % CO2 in air. Reactions were terminated with one half volume
of ethanol. Appropriate control incubations were also performed. The positive control
compound, [14C]-7-ethoxycoumarin (20 µM), was incubated in parallel to [14C]-cediranib
incubations. Samples were vortexed and centrifuged 14171 g for 10 min to remove
precipitated protein/cell pellets, and the resulting supernatants were stored at –20°C until
analysis by HPLC with radiometric (RAD) and mass spectrometry detection (MSn). Assays
were performed in duplicate.
Hepatic Subcellular Fractions Incubations. [14C]-cediranib (10 µM final (arbitrary
concentration); initially dissolved in methanol as a 2 mM stock solution) was incubated with
human liver microsomes, cytosol and S9. The incubations were carried out at a protein
concentration of 1 mg/mL in phosphate buffer (100 mM; pH 7.4), with 1 mM NADPH or
NAD. Incubations were maintained at 37°C for up to 120 min in a shaking water bath and
terminated with an equal volume of ethanol. All samples were centrifuged and stored in the
dark at -20°C until analysis by HPLC with radiometric detection.
Phase II Hepatic Microsomal and Recombinant UGT Enzyme Incubations. Incubations
(1 mL) of liver microsomes from human, cynomolgus monkey, Dunkin-Hartley guinea-pig,
Göttingen mini-pig, rhesus monkey, New Zealand White rabbit, CD-1 mouse, beagle dog and
Han-Wistar rat (all at 1 mg protein/mL) with [14C]-cediranib (10 µM final (arbitrary
concentration); initially dissolved in methanol as a 3 mM stock solution) were conducted in
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
9
phosphate buffer (100 mM; pH 7.4) containing 3.3 mM magnesium chloride (MgCl2) and
50 µg alamethicin/mg protein. The solvent attributable to the alamethicin was evaporated to
dryness under a stream of nitrogen at 35°C in the incubation tubes prior to the addition of the
remaining pre-incubation components. After a 5 min pre-incubation period, the reactions
were initiated by the addition of UDPGA (4 mM final). Following a 60-min incubation at
37°C, aliquots were removed into an equal volume of ethanol. Appropriate control
incubations were also performed. The positive control compound, [14C]-acetaminophen
(10 µM), was incubated in parallel to [14C]-cediranib incubations. Samples were centrifuged
(14171 g) for 10 min to remove precipitated protein and the resulting supernatant stored at
-80°C until subjected to analysis by HPLC-RAD and HPLC-MSn. Assays were performed in
duplicate.
For incubations with individual recombinant UGT enzymes, the incubations mixtures
contained the same components, except that a final protein concentration of
0.25 mg UGT protein/mL was used. In addition, following a 60-min incubation at 37°C,
aliquots were removed into an equal volume of 0.05% formic acid in ethanol.
Recombinant FMO Enzyme Incubations. Microsomes prepared from insect cell lines that
heterologously expressed human FMO1, FMO3 and FMO5 (all at 0.2 mg/mL) were
incubated with [14C]-cediranib (5 µM final (arbitrary concentration); initially dissolved in
methanol as a 1 mM stock solution) in phosphate buffer (100 mM; pH 7.4), with 1 mM
NADPH. Incubations were maintained at 37°C for 120 min and terminated with an equal
volume of ethanol. Additionally, control incubations were performed with microsomes from
cells that contained the empty transfection vector. All samples were centrifuged and stored in
the dark at -20°C until analysis by HPLC with radiometric detection.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
10
Enzyme Kinetics of [14C]-cediranib N-oxidation in recombinant FMO1 and FMO3
Enzymes. The N-oxidation of [14C]-cediranib was conducted under linear conditions with
respect to incubation time and protein concentration. A range of concentrations (0.2 to
50 µM) of [14C]-cediranib was incubated with either 0.02 mg/mL FMO1 or 0.2 mg/mL
FMO3 protein in phosphate buffer (100 mM; pH 7.4) for 10 (FMO1) or 20 min (FMO3) at
37°C in the presence of NADPH (1 mM). Incubations were terminated by the addition of an
equal volume of ethanol followed by centrifugation at 14171 g for 10 min. The resultant
supernatants were stored at –20°C until analysis by HPLC with radiochemical detection.
Metabolite Profiling by HPLC-RAD. [14C]-cediranib and its metabolites were separated on
an ACE AQ column (4.6 X 150 mm, 5 µm; Advanced Chromatography Technologies (ACT),
Aberdeen, UK) using Perkin Elmer 200 Series pumps (Beaconsfield, UK) at a flow rate of 1
mL/min. The aqueous mobile phase (solvent A) consisted of 0.2% (v/v) formic acid in
deionised water while the organic mobile phase (solvent B) consisted of 0.2% (v/v) formic
acid in acetonitrile. The initial mobile phase consisted of 5% solvent B, which was increased
to 20% over a period of 15 min, then further increased to 25% in 20 min, maintained for
25 min, and finally to 50% over 5 min. This value was held for 10 min before returning to
5% solvent B for column equilibration for 10 min before the next injection. Radiochemical
detection was performed using a Packard Radiomatic Flo-One Beta 500 TR series detector
(Packard Instruments, Pangbourne, UK) with a 500 µL liquid flow cell and Ultima Flo-M
scintillation cocktail (Packard Instruments, Pangbourne, UK) running at 3 mL/min. The data
were collected and analysed using either FLO-ONE version 3.65 (Packard Instruments,
Pangbourne, UK) or LAURA version 3.4.1.10e (Lablogic Systems Ltd, Sheffield, UK).
Recovery of sample radioactivity following centrifugation of the quenched incubates was
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
11
determined prior to injection onto the HPLC system. To ensure the system suitability, the
recovery of radioactivity from the column was evaluated by comparing the radioactive
content (determined by liquid scintillation counting) of the post-column eluent to the pre-
column eluent. No loss of radioactivity on the column was found for any of the incubated
samples.
For the UGT and FMO phenotyping experiments and Phase II microsomal incubate samples,
a shorter HPLC gradient was used. [14C]-cediranib and its metabolites were separated on a
Polaris C18-A column (4.6 X 150 mm, 5 µm, Varian, Yarnton, UK) using either Jasco
PU-1580 Intelligent (Jasco (UK), Great Dunmow, UK) or Agilent 1200 Series (Agilent
Technologies UK Ltd, Stockport, UK) HPLC pumps. The initial mobile phase consisted of
5% solvent B, which was maintained for 2 min, increased to 25% over a period of 4.5 min,
further increased to 29% in 15.5 min, to 33% in 4 min, and finally to 95% in 1.5 min. This
value was held for 4.5 min before returning to 5% solvent B for column equilibration for
4.9 min before the next injection.
[14C]-acetaminophen and its metabolites were separated on an Eclipse XDB C18 column
(4.6 X 150 mm, 5 µm; Agilent Technologies UK Ltd, Stockport, UK) using Agilent 1200
Series HPLC pumps (Stockport, UK) with a flow rate of 1 mL/min. The aqueous mobile
phase (solvent A) consisted of ammonium acetate in deionised water (10 mM; pH 5.5) while
the organic mobile phase (solvent B) consisted of methanol. The initial mobile phase
consisted of 5% solvent B, which was maintained for 15 min and increased to 95% over a
period of 1 min. This value was returned to 5% solvent B within a 1 min for column
equilibration for 3 min before the next injection.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
12
[14C]-7-ethoxycoumarin and its metabolites were separated on a Zorbax SB-C8 column
(4.6 X 150 mm, 5 µm; Agilent Technologies UK Ltd, Stockport, UK) using Jasco PU-2080
Intelligent HPLC pumps (Jasco (UK), Great Dunmow, UK) with a flow rate of 1 mL/min.
The aqueous mobile phase (solvent A) consisted of ammonium acetate in deionised water
(10 mM; pH 5.5) while the organic mobile phase (solvent B) consisted of methanol. The
initial mobile phase consisted of 10% solvent B, which was increased to 17% over a period of
7 min, further increased to 29% in 2min, to 55% in 10 min and finally to 70% in 6 min. This
value was returned to 10% solvent B within a 5 min for column equilibration for 5 min before
the next injection.
Metabolite identification by HPLC-MSn. [14C]-Cediranib and its metabolites were
separated on a Polaris C18 column (4.6x150 mm, 5 μm, Varian, Yarnton, UK) using Agilent
HP1200SL binary pumps (Agilent Technologies UK Ltd, Stockport, UK) with a flow rate of
1 mL/min. The aqueous mobile phase (solvent A) consisted of 0.2% formic acid in deionised
water while the organic mobile phase (solvent B) consisted of 0.2% formic acid in
acetonitrile. Mass spectrometry data were collected either using Xcalibur version 2.0
(Thermo Fisher Scientific, Waltham, MA) on a Thermo Finnigan LTQ ion trap instrument
(Thermo Fisher Scientific, Waltham, MA) fitted with an electrospray ionisation (ESI) source
operating in positive mode under optimised conditions (voltage 3.5 kV; sheath gas nitrogen,
collision energy 20 eV, capillary temperature 350 °C), or using the MassLynx v4.0 software
(Waters) on a Waters Micromass Quattro MicroTM API instrument (Waters, Elstree, UK)
fitted with an electrospray ionisation (ESI) source operating in positive ion mode under
optimised conditions (cone voltage 20 V; collision energy 20 eV).
Metabolite Identification/Characterisation by 1H NMR spectroscopy. 1H NMR spectra
were acquired on a Bruker DRX500 NMR spectrometer operating at 500 MHz resonance
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
13
frequency). 1H NMR data were collected and analysed using Topspin software (version 2.0).
Cediranib pyrrolidine N-oxide was reconstituted in DMSO-d6.
Data Analysis. The assessment of the metabolism of [14C]-cediranib was made by both
qualitative and quantitative analysis of the chromatographic patterns. Radiolabelled
components were assumed to have the same specific activity as parent molecule.
MSn data were obtained for the metabolites identified in the analysis by HPLC-MSn with
radiometric detection. Components were identified as being derived from [14C]-cediranib if
they demonstrated elements of the characteristic isotopic and fragmentation patterns observed
with the parent molecule.
Enzyme kinetic parameters (Km and Vmax) were determined by non-linear regression using
Grafit version 5.0.10 (Erithacus Software Limited; Horley, UK).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
14
Results
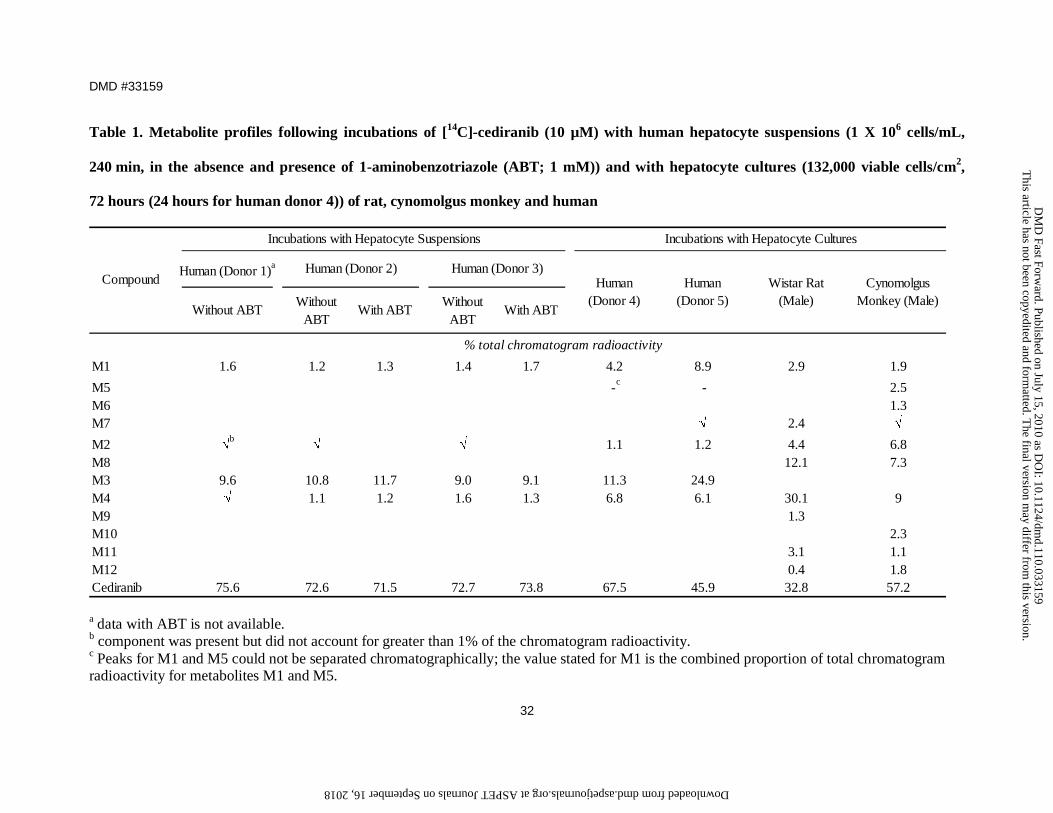
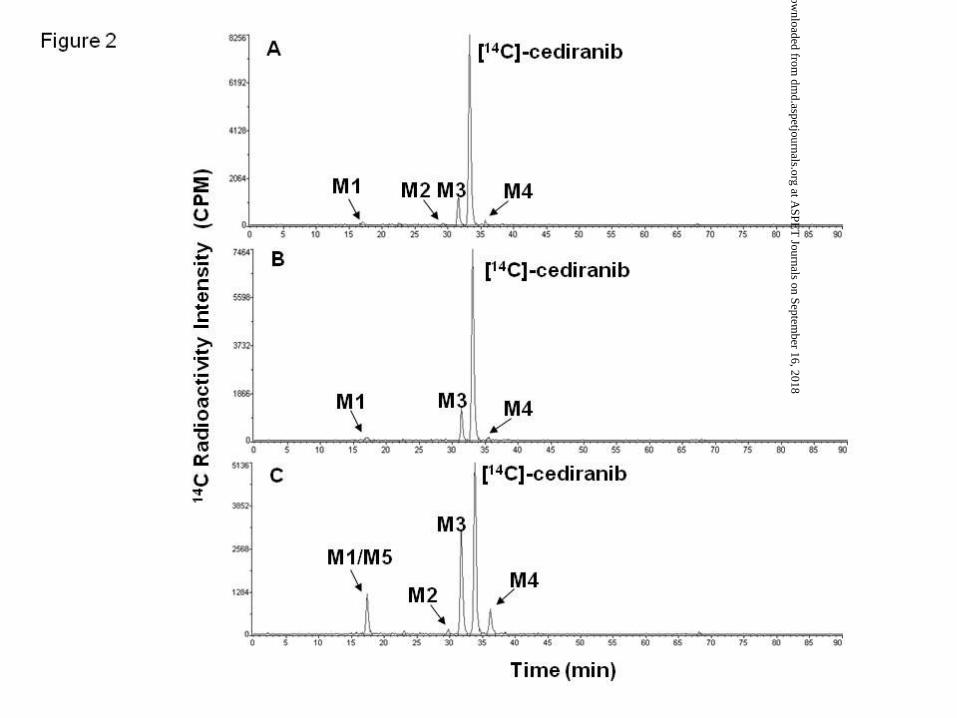
Metabolism profile of [14C]-cediranib in hepatocytes. The metabolic profiles of
[14C]-cediranib in human hepatocyte suspension and culture are highlighted in Figures 2 and
3, respectively. In human hepatocyte suspension incubations, only one major metabolite,
M3, was detected, which accounted for up to 10.8% of total chromatogram radioactivity
(Figure 2A; Table 1). Minor components included metabolites M1, M2 and M4. The
formation of M3 as well as M1 and M4 were not inhibited in the presence of the
non-selective CYP450 chemical inhibitor, 1-ABT (Figure 2B).
Consistent with the findings in human hepatocyte suspensions, [14C]-cediranib was
metabolised to these 4 components as well as to two further minor metabolites M5 and M7,
in human hepatocyte cultures (Figure 2C; Table 1). With the exception of metabolite M3, all
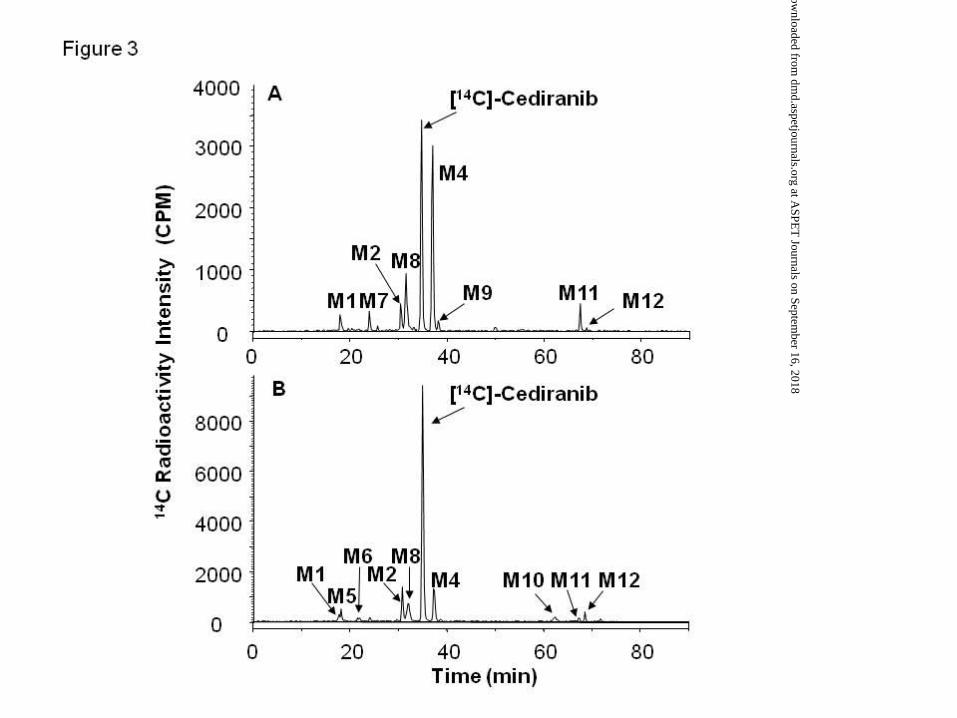
other metabolites detected in human hepatocytes were also observed in either rat and/or
cynomolgus monkey hepatocyte cultures (Figure 3). Unlike human, M4 was the major
metabolite in both rat and cynomolgus hepatocyte incubations and accounted for 30 and 9%,
respectively, of total chromatogram radioactivity. Metabolites M8, M10 and M11 were
detected in rat and cynomolgus monkey hepatocyte samples but not in human hepatocytes,
whilst metabolites M6 and M10 were detected only in cynomolgus monkey hepatocytes
(Table 1). The mass spectral data for cediranib and its metabolites are summarised in
Table 2.
The recovery of sample radioactivity from the HPLC column for human hepatocyte
suspension (donor 3) and culture (donor 5) incubations was 102 and 99.3%, respectively. For
rat and cynomolgus monkey hepatocyte cultures, these values were 98.5 and 94.3%,
respectively.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
15
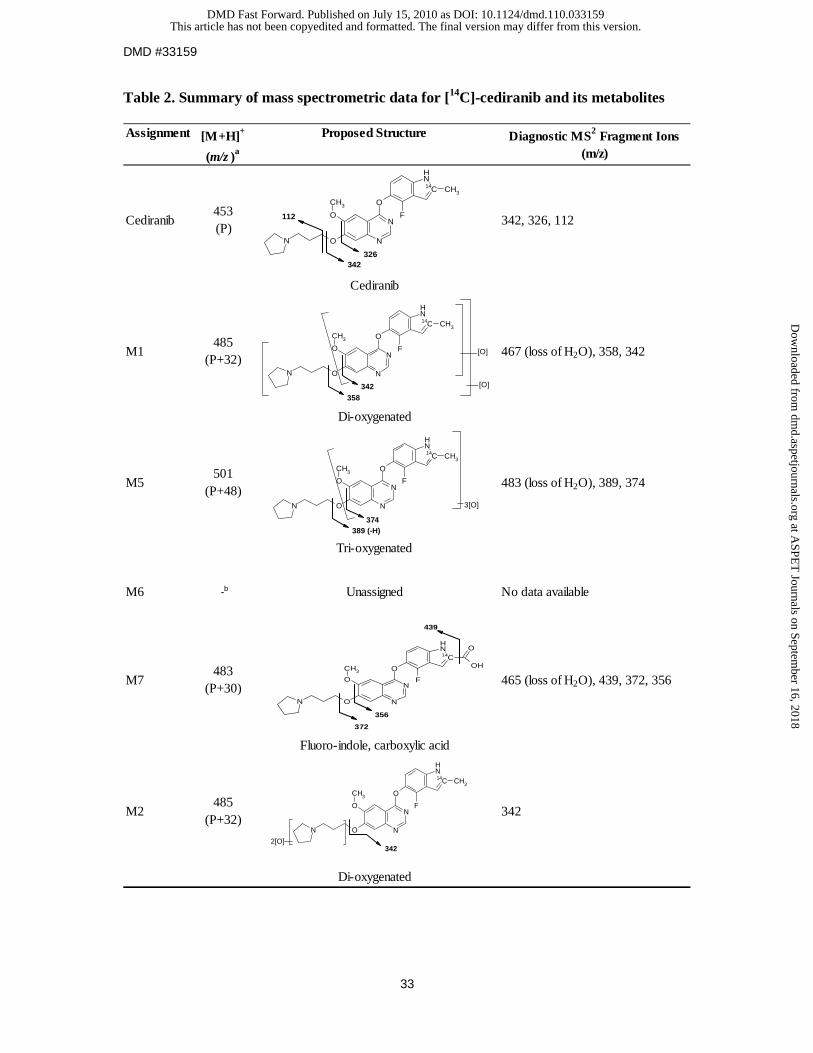
Metabolite identification in hepatocytes. Based on mass spectrometric data, metabolite M3
showed a [M+H]+ ion of m/z 629 corresponding to a gain of 176 atomic mass units (amu)
from [14C]-cediranib (m/z 453) (Table 2), consistent with the addition of a glucuronic acid
moiety to the molecule. The exact site of conjugation in metabolite M3 was confirmed by
LC-MSn and [1H]-NMR, which indicated conjugation on the pyrrolidine nitrogen (Lenz et al.,
(2010)). The proton NMR chemical shifts and coupling constants of a bio-synthesised
standard of metabolite M3 are included in Table 3.
For metabolite M4, an [M+H]+ ion at m/z 469 was characteristic of mono-oxygenation. With
a major fragment at m/z 128, corresponding to a gain of 16 amu on the propyl pyrrolidinyl
moiety (m/z 112), this suggested that oxidation occurred on this group (Table 2). The
synthetic standard for cediranib pyrrolidine N-oxide showed a similar retention time and mass
fragmentation profile, thus confirming that the structural identity of metabolite M4 and
cediranib N-oxide were identical. The proton NMR chemical shifts and coupling constants of
the synthetic standard for cediranib pyrrolidine N-oxide (M4) are included in Table 3.
The mass spectrum of metabolite M7 showed an [M+H]+ ion at m/z 483, 30 amu higher than
that of protonated [14C]-cediranib. With two major fragments at m/z 356 and 372,
corresponding to increments of +30 amu on the fluoro-indole quinazoline moiety (m/z 326
and 342 respectively), this suggested that oxidations had occurred on this group, resulting in
the formation of the carboxylic acid M7 (Table 2).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
16
Metabolites M1, M2 and M5 had mass spectral profile characteristics of di- and
tri-oxygenations, although precise positional assignment of these oxidations was not possible
based on the MSn fragmentation data.
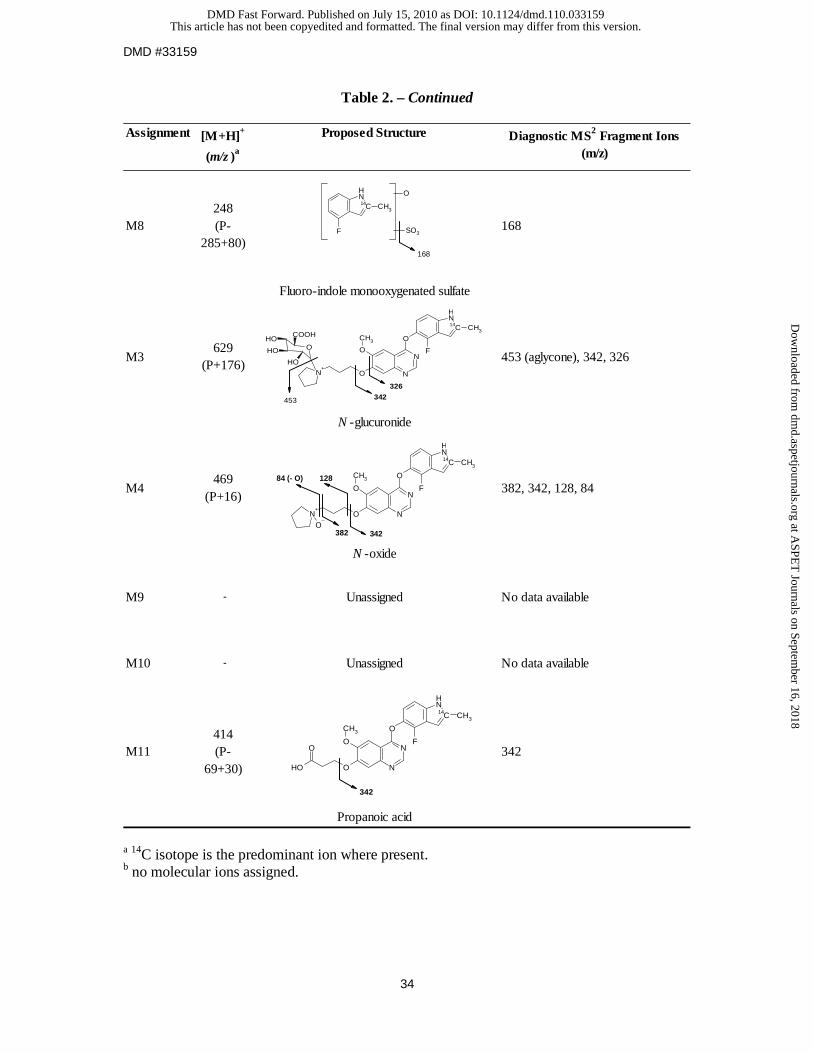
The mass spectrum of metabolite M8 showed an [M+H]+ ion at m/z 248, 205 amu less than
that of protonated [14C]-cediranib. The MS/MS of protonated M8 produced one major
fragment at m/z 168, corresponding to the loss of a sulfate moiety. The results suggest that
metabolite M8 is a fluoro-indole monooxygenated sulfate.
Metabolite M11 displayed an [M+H]+ ion of m/z 414 corresponding to a loss of 39 amu from
[14C]-cediranib (m/z 453) (Table 2), and a predominant MS/MS fragment at m/z 342. This is
consistent with the loss of the pyrrolidine ring and oxidation to form the propanoic acid.
No molecular ion assignments were possible for metabolites M6, M9, M10 and M12.
Oxidative metabolic profiles of [14C]-cediranib in human hepatic microsomal fractions.
Minor metabolism (<12% depletion of [14C]-cediranib total chromatogram radioactivity) was
observed following incubation of [14C]-cediranib with human liver microsomes and S9 in the
presence of NADPH (data not shown). Up to five components were observed, however the
structure of only one could be elucidated. This identified component in S9 accounted for
6.7 % of the total radiochromatogram radioactivity and the mass fragmentation profile was
consistent with that for metabolite M4.
The oxidative metabolites M1, M2 and M5 that were detected in human hepatocytes were not
formed in the presence of NADPH in either human liver microsomes or S9. No metabolism
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
17
was observed in the human cytosolic incubate samples. Similarly, no metabolites were
detected in the incubation of [14C]-cediranib with human liver microsomes or S9 in the
presence of the cofactor, NAD.
Involvement of FMO enzymes in the metabolism of [14C]-cediranib. A role for
flavin-containing monooxygenase (FMO) enzymes in the metabolism of [14C]-cediranib was
anticipated based on the role of these enzymes in N-oxidation reactions (Cashman, 1995) and
on the inability of the non-selective CYP inhibitor, 1-ABT, to inhibit the formation of the
N-oxide metabolite of [14C]-cediranib (metabolite M4) in human hepatocytes (Figure 2).
Incubation of [14C]-cediranib (5 μM) with microsomes prepared from cell lines expressing a
single FMO isoform indicated that in the presence of FMO1, extensive metabolism of
[14C]-cediranib was observed, with only metabolite M4 (accounting for 85.0% of the total
chromatogram radioactivity), being generated (data not shown). Similarly for FMO3, only
metabolite M4 was detected, which accounted for 60.6% of the total chromatogram
radioactivity. FMO5 and insect control enzymes displayed no catalytic activity for the
formation of metabolite M4.
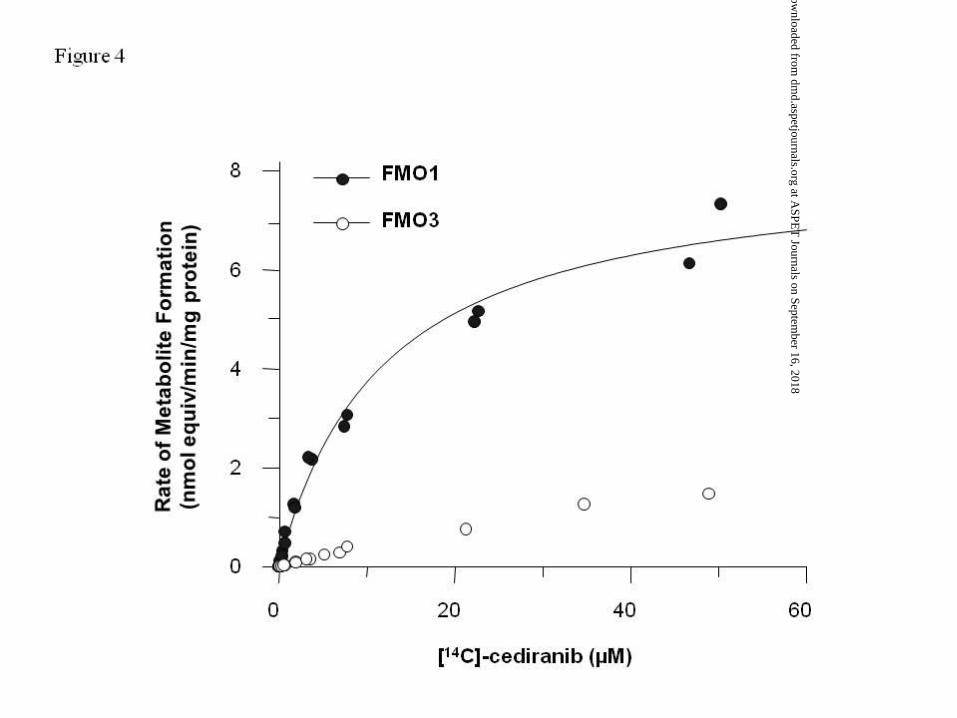
Enzyme kinetic experiments for [14C]-cediranib N-oxide (metabolite M4) in recombinant
FMO1 showed that the rate of formation conformed to Michaelis-Menten kinetics
characterised by values for apparent Km of 11.9 µM and Vmax of 8.2 nmol equiv/min/mg
protein (Figure 4). The kinetic parameters for metabolite M4 in recombinant FMO3 could
not be calculated, as the rate of formation did not reach Vmax over the concentration range
tested (Figure 4). Solubility limitations prevented the use of higher [14C]-cediranib
concentrations.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
18
Phase II metabolic profiles of [14C]-cediranib in liver microsomes. Human liver
microsomal incubations containing [14C]-cediranib and UDPGA resulted in the formation of
a single metabolite (M3; 15.2% of total chromatogram radioactivity), as observed by HPLC
with radiochemical detection (results not shown). A small component representing
approximately 1% of the total chromatogram radioactivity with a retention time of 27.5 min
was also detected in the HLM sample, however as this component was also present in the
control incubates, it was considered not of metabolic origin. No metabolites were detected
following incubation of [14C]-cediranib in microsomes from cynomolgus monkey,
Dunkin-Hartley guinea-pig, Göttingen mini-pig, rhesus monkey, New Zealand White rabbit,
CD-1 mouse, beagle dog or Han Wister rat in the presence of UDPGA (data not shown).
Involvement of UGT enzymes in the metabolism of [14C]-cediranib. The identification of
the UGT enzymes responsible for the N+-glucuronidation of [14C]-cediranib was performed
using recombinant UGT enzymes. UGT enzyme identification studies in human liver
microsomes were not performed due to the limited availability of a complete set of UGT
selective chemical inhibitors (Miners et al., 2004; Uchaipichat et al., 2006).
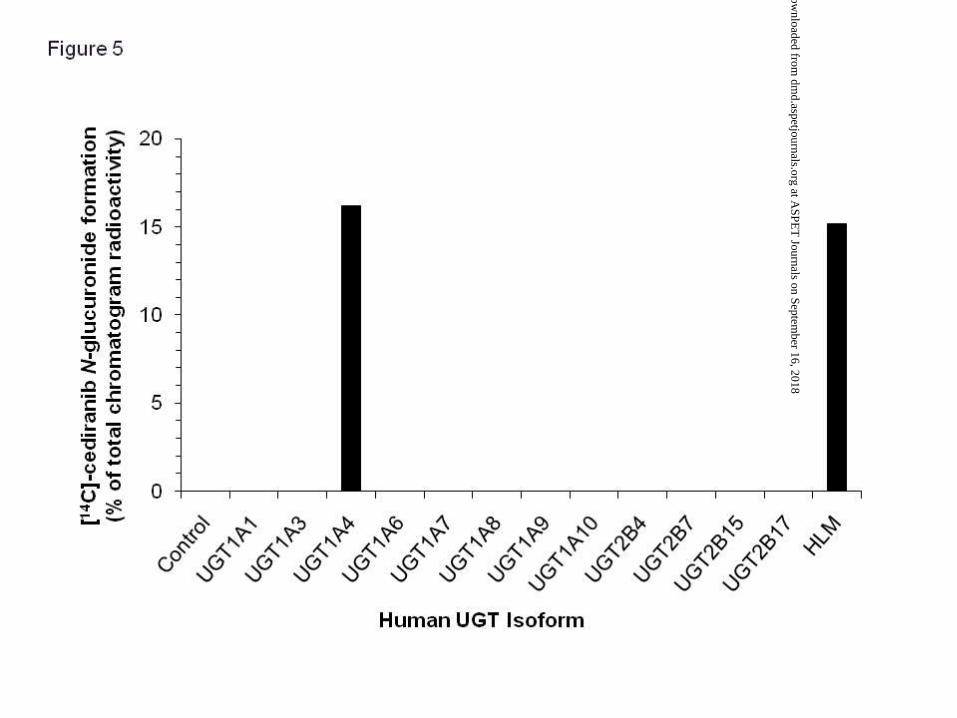
Incubation of [14C]-cediranib (10 μM) with microsomes prepared from cell lines expressing a
single UGT isoform indicated that the metabolism of [14C]-cediranib was limited to UGT1A4
(Figure 5), with a reduction observed in the amount of chromatogram radioactivity
represented by [14C]-cediranib and the formation of one major component (M3; 16.2 % of
total chromatogram radioactivity). Incubations with UGTs 1A1, 1A3, 1A6, 1A7, 1A8, 1A9,
1A10, 2B4, 2B7, 2B15 and 2B17 showed no evidence of metabolism of [14C]-cediranib
(Figure 5).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
19
Discussion
The present study was undertaken to elucidate the biotransformation pathways and identify
the enzymes involved in the in vitro metabolism of cediranib. It has been demonstrated that
the in vitro metabolism of cediranib proceeds predominantly via non-CYP450 enzymes, with
the UGT and FMO enzymes playing a major role.
In vitro metabolism studies have shown that cediranib was metabolised to at least 11
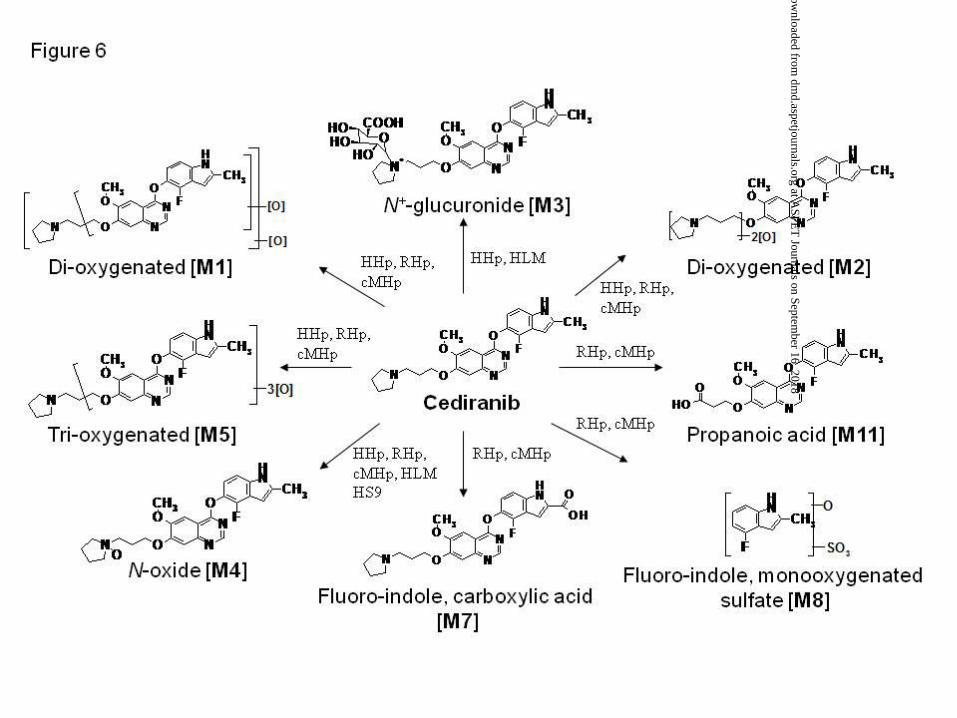
components in hepatic proteins from human, rat and cynomolgus monkey. A summary of the
proposed metabolic pathway of [14C]-cediranib is highlighted in Figure 6.
Incubations with hepatic in vitro systems have highlighted that marked species differences in
the N+-glucuronidation of cediranib was observed. The formation of the pyrrolidine
N+-glucuronide metabolite (M3) was only produced in human and was not detected in the
other species tested. This finding was supported by previous studies where quaternary
N-glucuronidation of cyclic tertiary amines were shown to exhibit distinct species
differences, with a general trend that only human and higher primates have the capability of
catalysing such reactions (Chaudhuri et al., 1976; Fisher et al., 1980; Delbressine et al., 1992;
Chiu & Huskey, 1998). These studies have shown that several cyclic tertiary amine-
containing drugs, the anti-histamines cyproheptadine and tripelennamine, and the
anti-depressant mianserin, also undergo human-specific N+-glucuronidation. Of all the
species studied, the N+-glucuronide metabolites of these drugs were only detected in the urine
of higher primates and/or human. For cediranib, the N+-glucuronide metabolite (M3) has
been identified in the plasma and faeces of humans following a single oral dose of 45 mg
[14C]-cediranib; however, it has not been detected in vivo in plasma, urine or faeces following
a single oral dose of 0.5 mg/kg [14C]-cediranib to beagle dogs, in vivo in urine or faeces
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
20
following a single oral dose of 1.0 mg/kg [14C]-cediranib to cynomolgus monkeys or in vivo
in urine, plasma or bile following a single oral dose of 30 mg/kg [14C]-cediranib to Sprague
Dawley rats (unpublished data). Additional data to be published at a later date will support
the conclusion that M3 is a major metabolite in human and should be considered in this
context.
The enzyme catalysing the N+-glucuronidation of cediranib was shown using recombinant
proteins to be human UGT1A4. This observation is consistent with previous studies
confirming that N+-glucuronidation of cyclic tertiary amines usually proceeds via UGT1A4
(Green et al., 1995; Green et al., 1998; Green & Tephly, 1996; 1998; Kubota et al., 2007).
Information on the interspecies variation in the expression of this enzyme is limited, although
it is well established that in the rat, a pseudo-gene is present in the UGT1 gene complex that
encodes for an incomplete UGT1A4 protein, leaving the rat without a functional UGT1A4
enzyme (Green & Tephly 1998). This could potentially provide an explanation for the lack
of N+-glucuronidation observed in this species. With the exception of rabbit, little data is
available on UGT1A4 expression levels in cynomolgus monkey and the other laboratory
species and thus the inability of the other species to catalyse the formation of a quaternary
N+-glucuronide of cediranib remains unexplained (Green & Tephly 1998).
Functional polymorphisms of hepatic UGT1A4 that are predicted to alter the hepatic
metabolism and detoxification of selective xenobiotics and steroids have been identified in
German Caucasian and Japanese populations (Ehmer et al., 2004; Mori et al., 2005; Saeki et
al., 2005). The frequencies of two genetic variations, P24T (UGT1A4.2) and L48V
(UGT1A4.3) were up to 8 and 17%, respectively (Benoit-Biancamano et al., 2009). Whether
these UGT1A4 polymorphisms could potentially influence the inter-individual variability in
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
21
cediranib disposition in vivo and impact on an individual’s response to cediranib treatment
remains to be assessed, given that functional investigations of UGT1A4 polymorphisms to
date have been limited to in vitro systems. In addition, it has also been suggested that the
effects of these functional polymorphisms may be dependent on the xenobiotic administered
(Mori et al., 2005).
In this study, the human FMO enzymes were shown to be primarily involved in the
N-oxidation of cediranib to metabolite M4. Amongst the FMO enzymes tested, only human
FMO1 and FMO3 were able to catalyse this reaction in vitro, with FMO1 being most
efficient in catalysing this reaction. Although FMO1 is the most prominent isoform in adult
kidney, its expression is very low in adult liver, with FMO3 being the major isoform in this
tissue (Cashman & Zhang, 2006). Based on the kinetic parameters and absolute levels of
each enzyme, it is conceivable that in vivo, both enzymes may be involved to a similar extent
in the N-oxidation of cediranib. A role for the CYP450 enzymes in the formation of this
metabolite was excluded, due to a lack of significant inhibition of cediranib N-oxide
formation by 1-ABT.
The human enzyme(s) responsible for the metabolism of cediranib to metabolite M2 were
CYP450 enzymes. As for metabolite M1, the enzymes involved in its formation are
unknown and have yet to be determined. However, it is clear that CYP450 enzymes do not
play a role, given the lack of inhibition of metabolite formation in the presence of 1-ABT.
Based on the structure of cediranib, a possible role for aldehyde oxidase may exist (Beedham,
2001). Whilst no metabolism of cediranib was observed following incubation with human
cytosolic fractions, it should be noted that this may be due to the instability of aldehyde
oxidase upon preparation and storage of the human hepatic tissue (Lang & Kalgutkar, 2003).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
22
For the formation of metabolite M11, which was only formed in rat and cynomolgus monkey,
a potential mechanism would involve the hydroxylation of the methylene group adjacent to
the pyrrolidine nitrogen, followed by the intermediate formation of the aldehyde through
N-dealkylation, which would then be rapidly oxidised to the carboxylic acid M11.
In conclusion, the present study has shown that the major biotransformation pathways of
cediranib in humans involve UGT1A4-mediated N+-glucuronidation and
non-CYP450-mediated oxidations, with the N-oxidation of cediranib catalysed by the FMO
enzymes. With the exception of the N+-glucuronide, all other metabolites of cediranib found
in humans were also detected in rat and cynomolgus monkey. As the metabolism of
cediranib proceeds via conjugative and non-CYP450 oxidative pathways, it is predicted that
cediranib may be less prone to potential drug-drug interactions from other co-administered
xenobiotics. The FMO enzymes are not readily inhibited or induced by xenobiotics
(Cashman & Zhang, 2006, Mitchell, 2008) and the magnitude of UGT-based drug
interactions is significantly less when compared to CYP450-mediated interactions (Williams
et al., 2004; Kiang et al., 2005).
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
23
Acknowledgements
We thank Drs Ian Wilson and Eva Lenz (Clinical Pharmacology & DMPK department) for
critically reviewing this manuscript. We also thank Ian McFarlane (Pharmaceutical
Development department), Eva Lenz and Selcia Limited (Ongar, UK) for the preparation of
the authentic cediranib N-oxide metabolite standard, for providing support in the preparation
of the NMR data, and for the preparation of radiolabelled cediranib (batch 1R6), respectively.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
24
References
Batchelor TT, Sorensen AG, di Tomaso E, Zhang Q-T, Duda DG, Cohen KS, Kozak KR,
Cahill DP, Chen P-J, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Dappatz J, Louis DN,
Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY and Jain RK (2007) CEDIRANIB, a pan-
VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema
in glioblastoma patients. Cancer Cell, 11: 83-95.
Beedham C (2001) Molybdenum hydroxylases. Enzyme systems that metabolise drugs and
other xenobiotics. Edited by Costas Ioannides John Wiley & Sons Ltd
Benoit-Biancamano MO, Adam JP, Bernard O, Court MH, Leblanc MH, Caron P and
Guillemette (2009) A pharmacogenetics study of the human glucuronosyltransferase
UGT1A4. Pharmacogenet Genom, 19(12): 945-954.
Cashman JR and Zhang J (2006) Human flavin-containing monooxygenases. Annu Rev
Pharmacol Toxicol, 46: 65-100.
Chaudhuri NK, Servano OA, Manniullo MJ, Luders RC, Chao DK and Bartlett MF (1976)
Metabolism of tripelennamine in man. Drug Metab Dispos, 4: 372-378.
Chen E, Jonker D, Gauthier I, MacLean M, Wells J, Powers J and Seymour L (2009) Phase I
study of cediranib in combination with oxaliplatin and infusional 5-fluorouracil in patients
with advanced colorectal cancer. Clin Cancer Res, 15(4): 1481-1486.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
25
Chiu SL & Huskey SW (1998) Species differences in N-glucuronidation. Drug Metab
Dispos, 26(9): 838-847.
Delbressine PC, Moonen MEG, Kaspersen FM, Jacobs PL and Wagenaars GL (1992)
Biotransformation of mianserin in laboratory animals and man. Xenobiotica, 22: 227-336.
Drevs J, Siegert P, Medinger M, Mross K, Zirrgiebel U, Harder J, Blum H, Robertson J,
Jurgensmeier JM, Puchalski TA, Young H, Saunders O and Unger C (2007) Phase I Clinical
Study of cediranib, an oral vascular endothelial growth factor signalling inhibitor, in patients
with advanced solid tumours. J Clin Oncol, 25(20): 3045-3054.
Ehmer U, Vogel A, Shuette JK, Krone B, Manns MP and Strassburg CP (2004) Variation of
hepatic glucuronidation: Novel functional polymorphisms of the
UDP-glucuronosyltransferase UGT1A4. Hepatology, 39(4): 970-977.
Fisher LJ, Thies RL, Charkowski D and Donham KJ (1980) Formation and urinary excretion
of cryptoheptadine glucuronide in monkeys, chimpanzees and humans. Drug Metab Dispos,
8: 422-424.
Green MD, Bishop WP and Tephly (1995) Expressed human UGT1.4 protein catalyzes the
formation of quaternary ammonium-linked glucuronides. Drug Metab Dispos, 23(3):
299-302.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
26
Green MD and Tephly TR (1998) Glucuronidation of amine and hydroxylated xenobiotics
and Endobiotics catalyzed by expressed human UGT1.4 protein. Drug Metab Dispos, 24:
356-363.
Green MD and Tephly TR (1998) Glucuronidation of amine substrates by purified and
expressed UDP-glucuronosyltransferase proteins. Drug Metab Dispos, 26: 860-867.
Hawes EM (1998) N+-glucuronidation, a common pathway in human metabolism of drugs
with a tertiary amine group. Drug Metab Dispos, 26(9): 830-837.
Kiang TKL, Ensom MHH and Chang TKH (2005) UDP-glucuronosyltransferases and
clinical drug-drug interactions. Pharmacol Therapeutics, 106: 97-132.
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS and Ferrara N (1993) Inhibition
of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vi-
vo. Nature 362, 841–844.
Kubota T, Lewis BC, Elliot DJ, Mackenzie PI and Miners JO (2007) Critical roles of residues
36 and 40 in the phenol and tertiary amine aglycone substrate Selectivities of UDP-
glucuronosyltransferases 1A3 and 1A4. Mol Pharmacol, 72(4): 1054-1062.
LeCluyse E, Madan A, Hamilton G, Carroll K, DeHaan R and Parkinson A (2000)
Expression and regulation of cytochrome P450 enzymes in primary cultures of human
hepatocytes. J Biochem Mol Toxicol, 14(4): 177-188.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
27
Land D and Kalgutkar AS (2003) Non-P450 mediated oxidative metabolism of xenobiotics.
In Drug metabolizing enzymes: cytochrome P450 and other enzymes in drug discovery and
development by JS Lee, RS Obach and MB Fisher. FontisMedia SA and Marcel Dekker Inc.
Lenz A, Spear, M, Drake C, Ward M, Schulz-Utermoehl T and Harrison M (2010)
Characterisation and identification of the N+-glucuronide metabolite of cediranib. J
Pharmaceut Biomed, doi:10.1016/j.jpba.2010.03.023.
Luffer-Atlas D (2008) Unique/major human metabolites:why, how, and when to test for
safety in animals. Drug Metab Rev, 40: 447-463.
Miners JO, Smith PA, Sorich MJ, McKinnon RA & Mackenzie PI (2004) Predicting drug
glucuronidation parameters: application of in vitro and in silico modelling approaches. Annu
Rev Pharmacol Toxicol, 44: 1-25.
Mitchell SC (2008) Flavin Mono-oxygenase (FMO) – the “other” oxidase. Current Drug
Metab, 9: 280-284.
Mori A, Maruo Y, Iwai M, Sato H and Takeuchi Y (2005) UDP-glucuronosyltransferase 1A4
polymorphisms in a Japanese population and kinetics of clozapine glucuronidation. Drug
Metab Dispos, 33:672-675.
Saeki M, Saito Y, Jinno H, Sai K, Hachisuka A, Kaniwa N, Ozawa S, Kawamoto M,
Kamatani N, Shirao K, Minami H, Ohtsu A, Yoshida T, Saijo N, Komamura K, Kotake T,
Morshita H, Kamakura S, Kitakaze M, Tomoike H and Sawada J (2005) Genetic Variations
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
28
and Haplotypes of UGT1A4 in a Japanese Population. Drug Metab Pharmacokinet, 20(2):
144-151.
Uchaipichat V, Mackenzie PI, Elliot DJ & Miners JO (2006) Selectivity of substrate
(trifluoperazine) and inhibitors (amitriptyline, adrosterone, canrenoic acid, hecogenin,
phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human UDP-
glucuronosyltransferase. Drug Metab Dispos, 34:449-456.
Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James
NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S,
Richmond GH, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S,
Jürgensmeier JM and Ogilvie DJ (2005) Cediranib: a highly potent, orally bioavailable,
vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of
cancer. Cancer Res, 65(10): 4389-4400.
Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR and
Ball SE (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: a
pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug
Metab Dispos, 32: 1201-1208.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
29
Footnotes
Parts of this work were previously presented at the following conference: 16th North
American Regional Meeting, International Society for the Study of Xenobiotics (ISSX),
Baltimore, MD
Reprint requests to:
Dr Timothy Schulz-Utermoehl, Clinical Pharmacology and DMPK Department, Alderley
Park, AstraZeneca UK Ltd, Macclesfield, Cheshire SK10 4TG, United Kingdom.
Email: [email protected]
1Joint First Authors
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
30
Legends for figures
Figure 1. The chemical structure of [14C]-cediranib.
Figure 2. Representative HPLC-RAD metabolite profiles following incubation of
[14C]-cediranib (10 μM) with human hepatocyte suspensions (donor 2; 1 X 106 cells/mL, at
37°C for 240 min) in the absence (A) and presence (B) of 1-aminobenzotriazole (1-ABT;
1 mM) and with human hepatocyte cultures (C) (donor 5, 132,000 viable cells/cm2, at 37°C
for 72 hours; 5% CO2). The proportion of radioactivity attributed to each metabolite is
shown in Tables 1 and 2.
Figure 3. Representative HPLC-RAD metabolite profiles following incubation (at 37°C for
72 hours; 5% CO2) of [14C]-cediranib (10 μM) with rat (A) and cynomolgus monkey (B)
hepatocyte cultures (132,000 viable cells/cm2). The proportion of radioactivity attributed to
each metabolite is shown in Table 1.
Figure 4. Formation kinetics of the N-oxide metabolite (M4) over the [14C]-cediranib
concentration range of 0.2 to 50 µM in recombinant FMO1 (solid circle) and FMO3 (open
circle). Vmax and Km values of 8.2 nmol equiv/min/mg protein and 11.9 µM for recombinant
FMO1 were obtained. The corresponding values for recombinant FMO3 could not be
calculated.
Figure 5. Formation of [14C]-cediranib N-glucuronide (M3) by recombinant human UGT
enzymes and human liver microsomes. Incubations were carried out at 37°C with 10 µM
[14C]-cediranib for 60 min using 0.25 (recombinant UGT enzymes) and 1 mg/mL (human
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
31
liver microsomes) protein concentration. Each column represents the mean of duplicate
determinations.
Figure 6. Proposed metabolic pathway of [14C]-cediranib in hepatocyte suspensions, cultures
and hepatic subcellular fractions from human and pre-clinical species. HHp, human
hepatocytes; RHp, rat hepatocytes; cMHp, cynomolgus monkey hepatocytes; HLM, human
liver microsomes; HS9, human liver S9.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
DMD #33159
32
Table 1. Metabolite profiles following incubations of [14C]-cediranib (10 µM) with human hepatocyte suspensions (1 X 106 cells/mL,
240 min, in the absence and presence of 1-aminobenzotriazole (ABT; 1 mM)) and with hepatocyte cultures (132,000 viable cells/cm2,
72 hours (24 hours for human donor 4)) of rat, cynomolgus monkey and human
Human (Donor 1)a
Without ABTWithout
ABTWith ABT
Without ABT
With ABT
M1 1.6 1.2 1.3 1.4 1.7 4.2 8.9 2.9 1.9
M5 -c - 2.5M6 1.3M7 √ 2.4 √
M2 √b
√ √ 1.1 1.2 4.4 6.8M8 12.1 7.3M3 9.6 10.8 11.7 9.0 9.1 11.3 24.9M4 √ 1.1 1.2 1.6 1.3 6.8 6.1 30.1 9M9 1.3M10 2.3M11 3.1 1.1M12 0.4 1.8Cediranib 75.6 72.6 71.5 72.7 73.8 67.5 45.9 32.8 57.2
Incubations with Hepatocyte Suspensions Incubations with Hepatocyte Cultures
Human (Donor 4)
% total chromatogram radioactivity
Compound Human (Donor 5)
Wistar Rat (Male)
Cynomolgus Monkey (Male)
Human (Donor 2) Human (Donor 3)
a data with ABT is not available. b component was present but did not account for greater than 1% of the chromatogram radioactivity. c Peaks for M1 and M5 could not be separated chromatographically; the value stated for M1 is the combined proportion of total chromatogram radioactivity for metabolites M1 and M5.
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
DM
D Fast Forw
ard. Published on July 15, 2010 as DO
I: 10.1124/dmd.110.033159
at ASPET Journals on September 16, 2018 dmd.aspetjournals.org Downloaded from
DMD #33159
33
Table 2. Summary of mass spectrometric data for [14C]-cediranib and its metabolites
Assignment [M+H]+
(m/z )a
Proposed Structure Diagnostic MS2 Fragment Ions (m/z)
Cediranib453 (P)
342, 326, 112
Cediranib
M1485
(P+32)467 (loss of H2O), 358, 342
Di-oxygenated
M5501
(P+48)483 (loss of H2O), 389, 374
Tri-oxygenated
M6 -b Unassigned No data available
M7483
(P+30)465 (loss of H2O), 439, 372, 356
Fluoro-indole, carboxylic acid
M2485
(P+32)342
Di-oxygenated
C14NH
N N
NO
O
O
CH3
F
CH3
342326
112
C14NH
N N
NO
O
O
CH3
F
CH3
2[O]342
C14NH
N N
NO
O
O
CH3
F
CH3
3[O]
389 (-H)374
N O
O
CH3
N
N
O
NH
C14
F
O
OH
372
356
439
N O
O
CH3
N
N
O
NH
C14 CH3
F
[O]
[O]
358342
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
DMD #33159
34
Table 2. – Continued
Assignment [M+H]+
(m/z )a
Proposed Structure Diagnostic MS2 Fragment Ions (m/z)
M8248 (P-
285+80)168
Fluoro-indole monooxygenated sulfate
M3629
(P+176)453 (aglycone), 342, 326
N -glucuronide
M4469
(P+16)382, 342, 128, 84
N -oxide
M9 - Unassigned No data available
M10 - Unassigned No data available
M11414 (P-
69+30)342
Propanoic acid
C14NH
N+
N
NO
O
O
CH3
F
CH3
O342
84 (- O) 128
382
C14NH
N
NO
O
O
CH3
F
CH3
O
OH
342
N+
O
O
CH3
N
N
O
NH
C14 CH3
FOOH
OH
OH
COOH
342
326
453
NH
C14 CH3
F
O
SO3
168
a 14C isotope is the predominant ion where present. b no molecular ions assigned.
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from

DMD #33159
35
Table 3. 1H NMR characteristics of cediranib and its metabolites M3 and M4
Cediranib M4 M3
(multiplicity) 1-2 NR NR NR
3 7.43 (s) 7.42 (s) 7.44 (s)4 7.65 (s) 7.60 (s) 7.67 (s)
5-7 NR NR NR8 8.53 (s) 8.53 (s) 8.53 (s)
9-15 NR NR NR16 7.17 (d) 7.2 (d) 7.18 (d)17 6.99 (dd) 7.01 (dd) 7.00 (dd)
18-19 NR NR NR20 6.25 (s) 6.26 (s) 6.25 (s)21 NR NR NR22 11.35 11.36 11.3523 2.42 (s) 2.43 (s) 2.42 (s)24 NR NR NR25 4.33 (t) 4.39 (t) 4.28 (t)26 2.24 (m) 2.52 (m) 2.41 (m)27 3.35 (t) 3.94 3.60, 3.7028 NR NR NR29 3.37 (m, br) 3.67 (m, br) 3.83 (m, br)30 1.97 (m, br) 2.09 (m, br) 2.08 (m, br)31 1.97 (m, br) 2.23 (m, br) 2.21 (m, br)32 3.37 (m, br) 3.93 (m, br) 3.92 (m, br)33 4.01 (s) 3.97 (s) 4.0134 NR NR 4.6835 NR NR 3.6736 NR NR 3.3237 NR NR 3.2038 NR NR 3.45
MetabolitePosition
1H (ppm)
NH
N+
N
NO
O
O
FOOH
OH
OH
COOH
1
23
45
67
8
9
10
13
14
17
1618
22
21 23
201915
24
11
33
12
25
2
27
28
29
30
31 32
M3
35
36
3738
34
NH
N N
NO
O
O
F1
23
45
67
8
9
10
13
14
17
1618
22
21 23
201915
24
11
33
12
25
2
27
28
29
30
31 32
CediranibNH
N+
N
NO
O
O
F
O
1
23
45
67
8
9
10
13
14
17
1618
22
21 23
201915
24
11
33
12
25
2
27
28
29
30
31 32
M4
Notations: br, broad peak; d, doublet; dd, doublet of a doublet; m, multiplet; NR, no result; s, singlet
This article has not been copyedited and form
atted. The final version m
ay differ from this version.
DM
D Fast Forw
ard. Published on July 15, 2010 as DO
I: 10.1124/dmd.110.033159
at ASPET Journals on September 16, 2018 dmd.aspetjournals.org Downloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from
This article has not been copyedited and formatted. The final version may differ from this version.DMD Fast Forward. Published on July 15, 2010 as DOI: 10.1124/dmd.110.033159
at ASPE
T Journals on Septem
ber 16, 2018dm
d.aspetjournals.orgD
ownloaded from





























