Embed Size (px)
Citation preview

1
Assessing the Activity of Cediranib, a VEGFR-2/-3 tyrosine kinase inhibitor, against
VEGFR-1 and members of the structurally related PDGFR-family
Sandra R. Brave1, Kirsty Ratcliffe1, Zena Wilson1, Neil H. James1, Sue Ashton1, Anna
Wainwright1, Jane Kendrew1, Philippa Dudley1, Nicola Broadbent1, Graham Sproat1, Sian
Taylor1, Claire Barnes1, Jeffrey C. Silva2, Charles L. Farnsworth2, Laurent Hennequin3,
Donald J. Ogilvie1,5, Juliane M. Jürgensmeier1, Masabumi Shibuya4, Stephen R. Wedge1,and
Simon T. Barry1
Authors Affiliations: 1Cancer Bioscience, AstraZeneca, Alderley Park, Macclesfield, Cheshire,
SK10 4TG, United Kingdom; 2 Cell Signaling Technology, 3 Trask Lane, Danvers, MA 01923,
United Sates of America; 3AstraZeneca Pharma, Centre de Recherches, Z.I. La Pompelle,
Chemin de Vrilly, Reims, France and 4Department of Molecular Oncology, Tokyo Medical and
Dental University, Tokyo 113-8519, Japan. 5Current address: Paterson Institute for Cancer
Research Wilmslow Rd., Manchester, M20 4BX, United Kingdom
Running Title: Assessment of cediranib selectivity
Key Words: Cediranib, AZD2171, VEGF, VEGF receptor-1, c-Kit, PDGFR,
angiogenesis, tyrosine kinase,
Abbreviations
AML, acute myeloid leukemia
ATP, adenosine triphosphate
BrdU, bromodeoxuridine
BSA, bovine serum albumin
CST, Cell Signaling Technology
DMEM, Dulbecco's modified Eagle's medium
DMSO, dimethyl sulphoxide
ELISA, enzyme-linked immunosorbent assay
FGFR, fibroblast growth factor receptor
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

2
GIST, gastrointestinal stromal tumor
HCl, hydrochloric acid
IC50, half maximal inhibitory concentration
LTQ, linear trap quadrupole
MAPK, mitogen-activated protein kinase
MSD, Meso Scale Discovery
NaF, sodium fluoride
PDGFR, platelet-derived growth factor receptor
PlGF, placental growth factor
SCF, stem cell factor
SCLC, small cell lung cancer
SDS, sodium dodecylsulfate
SEM, standard error of the mean
VEGFR, vascular endothelial growth factor receptor
VSMC, vascular smooth muscle cells
XTT, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium
hydroxide
Financial Support:
Sandra R. Brave, Kirsty Ratcliffe, Zena Wilson, Neil H. James, Sue Ashton, Anna Wainwright,
Jane Kendrew, Philippa Dudley, Nicola Broadbent, Graham Sproat, Sian Taylor, Claire
Barnes, Laurent Hennequin, Donald J. Ogilvie, Juliane M. Jürgensmeier, Stephen R. Wedge,
and Simon T. Barry are current or former employees and shareholders of AstraZeneca.
Jeffrey C. Silva, Charles L. Farnsworth, are employees of Cell Signaling Technology.
The analysis was performed by Cell Signaling Technology funded by AstraZeneca.
Corresponding Authors:
Simon Barry Cancer Bioscience, AstraZeneca, Alderley Park, Macclesfield, Cheshire, SK10
4TG, United Kingdom (+44 1625 513350) e-mail: [email protected]
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

3
Steve Wedge Cancer Bioscience, AstraZeneca, Alderley Park, Macclesfield, Cheshire, SK10
4TG, United Kingdom (+441625 523236) e-mail: [email protected]
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

4
Abstract
Cediranib is a potent inhibitor of the vascular endothelial growth factor receptor (VEGFR)-2
and -3 tyrosine kinases. This study assessed the activity of cediranib against the VEGFR-1
tyrosine kinase and the platelet-derived growth factor receptor (PDGFR)-associated kinases
c-Kit, PDGFR-α and -β. Cediranib inhibited VEGF-A-stimulated VEGFR-1 activation in
AG1-G1-Flt1 cells (IC50, 1.2nM). VEGF-A induced greatest phosphorylation of VEGFR-1 at
tyrosine residues Y1048 and Y1053; this was reversed by cediranib. Potency against
VEGFR-1 was comparable to that previously observed versus VEGFR-2 and R-3. Cediranib
also demonstrated significant activity against wild-type c-Kit in cellular phosphorylation assays
(IC50, 1−3nM) and in a stem cell factor-induced proliferation assay (IC50, 13nM). Furthermore,
phosphorylation of wild-type c-Kit in NCI-H526 tumor xenografts was reduced markedly
following oral administration of cediranib (≥1.5 mg/kg/day) to tumor-bearing nude mice. The
activity of cediranib against PDGFR-β and -α was studied in tumor cell lines, vascular smooth
muscle cells (VSMC) and a fibroblast line using PDGF-AA and PDGF-BB ligands. Both
receptor phosphorylation (IC50, 12–32nM) and PDGF-BB-stimulated cellular proliferation (IC50,
32nM in human VSMC; 64nM in osteosarcoma cells) were inhibited. In vivo, ligand induced
PDGFR-β phosphorylation in murine lung tissue was inhibited by 55% following treatment
with cediranib at 6 mg/kg, but not ≤3 mg/kg. In contrast, in C6 rat glial tumor xenografts in
mice, ligand induced phosphorylation of both PDGFR-α and -β was reduced by 46−61% by
cediranib 0.75 mg/kg. Additional selectivity was demonstrated versus Flt-3, CSF1R, EGFR,
FGFR1 and FGFR4. Collectively, these data indicate that cediranib is a potent pan-VEGFR
kinase inhibitor with similar activity against c-Kit, but is significantly less potent versus
PDGFR-α and –β.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

5
Introduction
Inhibition of vascular endothelial growth factor-A (VEGF) mediated signalling has been found
to give therapeutic benefit in oncology, as monotherapy in the treatment of renal cell cancer
(1, 2), and when combined with certain cytotoxic therapies in other disease settings (3).
Approaches used to inhibit VEGF receptor (VEGFR) signalling include small molecule
inhibitors that bind into the adenosine triphosphate (ATP)-binding pocket within the kinase
domain of VEGFR-2, to prevent ATP catalysis and propagation of receptor signalling. This
receptor predominantly transduces the angiogenic and permeability activity of VEGF-A. Two
other VEGF receptors are also described with differential binding of VEGF family ligands.
Each small molecule differs in its potency against the VEGF receptors, selectivity versus
other kinases, physicochemical properties and pharmacokinetic profile (4, 5). Some VEGFR
tyrosine kinase inhibitors have additional activity against one or more members of the platelet-
derived growth factor receptor (PDGFR) family of kinases (class III), which comprises
PDGFR-α, PDGFR-β, c-Kit, colony-stimulating factor receptor-1 and Flt-3; these receptors
having some structural homology to the VEGFR family members in that each harbors a
kinase-insert sequence in its intracellular domain (6). However, VEGFR kinase inhibitors have
also been described with activity against tyrosine kinases outside of the PDGFR class, and in
some case against serine/threonine kinases. Inhibiting multiple kinases simultaneously may
provide additional therapeutic opportunities in defined disease settings, but may impact
adversely on tolerability, particularly if chronic administration or usage in combination with
concurrent cytotoxic treatment, is required.
Cediranib has been previously shown to be a potent inhibitor of VEGFR-2 and -3 signalling in
cellular assays and to inhibit the growth of both angiogenic blood vessels and lymph-
angiogenic vessels in vivo (7−9). In recombinant kinase assays, cediranib also inhibits
VEGFR-1 kinase activity (IC50 of 5 nM) within a similar concentration range to VEGFR-2 and -
3 (IC50 values of <0.1 nM and ≤3nM respectively) (7). However, a more quantitative
assessment of inhibitor activity against endogenous VEGFR-1 kinase has proven technically
challenging in endothelial cells because of the low intrinsic activity of this receptor. Cediranib
demonstrates selectivity versus other kinases (7). Cediranib has similar potency against c-Kit
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

6
when compared to VEGFR-2 in phosphorylation assays, but less potency against PDGFR-α
and -β, particularly in a PDGF-AA/PDGFR-α driven tumor cell proliferation assay. However,
the activity of cediranib against a c-Kit driven phenotypic endpoint, or PDGFR-α and -β
signalling in normal cell types (which may also influence therapeutic response), or a
comparative inhibition of these targets in vivo, was not previously examined.
This study primarily aimed to further describe the pharmacology of cediranib by (i)
determining activity against VEGFR-1 in cells, (ii) using a wider complement of cell lines and
assays to examine activity against c-Kit and PDGFR-α/-β signalling in vitro, and (iii) to
examine pharmacodynamic inhibition of c-Kit, PDGFR-α and PDGFR-β in vivo over a dose-
range where cediranib has previously shown activity in tumor models. The data confirm that
cediranib is primarily a pan-VEGFR inhibitor that can inhibit wild-type c-Kit. The data also
suggest that cediranib may have some partial pharmacodynamic activity against PDGFR-α
and -β receptor activation in tumors, although this inhibition may be of limited functional
relevance, but does inhibit FGFR1 and FGFR4.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

7
Materials and Methods
Reagents
Cediranib [(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)
quinazoline (AZD2171) (Fig.1) was synthesized according to the processes described in
WO00/47212, in particular those described in Example 240 of WO/47212. The free base of
cediranib was used in these preclinical studies, with a molecular weight of 450.51. For all in
vitro assays, cediranib was prepared initially as a 10 mM stock solution in dimethyl sulphoxide
(DMSO) and diluted in the relevant assay media, such that the final concentration of DMSO
did not exceed 0.01%, with the exception of studies examining direct effects on tumor cells
where 1% DMSO was required to examine higher concentrations of cediranib. All in vivo
studies were conducted by once-daily oral gavage. For studies in mice, cediranib was
suspended in 1% (weight/volume) aqueous polysorbate 80 (polyoxyethylene (20) sorbitan
mono-oleate in deionized water) and dosed at 0.1 ml/10 g of body weight.
Cell culture
NCI-H526 (a human small cell lung cancer [SCLC] line), U118MG (a human glioblastoma
line), MG63 (a human osteosarcoma line) and C6 (a rat glial line) cells were purchased from
the American Type Culture Collection (Manassas, Virginia, USA), no further authentication
was performed on these lines. The M07e (a human acute myelogenous leukemia [AML] line)
cells were purchased from the German Collection of Microorganisms and Cell Cultures
(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (German Collection of
Microorganisms and Cell Cultures); Braunschweig, Germany), no further authentication was
performed. NIH 3T3 (mouse fibroblast) cells were obtained from A. Wong, Jefferson Cancer
Institute, no further authentication was performed. Human aortic and coronary vascular
smooth muscle cells (VSMC) were purchased from PromoCell GmbH (Heidelberg, Germany).
All cell lines were routinely passaged less than 10 times with the exception of the primary
vascular cells which were passaged no more than 4 times. NCI-H526, U118MG, MG63, C6,
M07e, NIH 3T3, human aortic and coronary VSMC were maintained in culture as per
providers recommendation. M07e cells were maintained in culture in the presence of
interleukin-3 (5 ng/ml) and granulocyte macrophage colony stimulating factor (5 ng/ml).
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

8
Inhibition of growth factor stimulated receptor phosphorylation in vitro
The ability of cediranib to inhibit receptor phosphorylation in cells was determined using
Western blotting. Cells were serum starved overnight in the presence (M07e and NCI-H526)
or absence (aortic and coronary VSMC) of 0.1% bovine serum albumin (BSA) or in the
presence of 1% charcoal stripped serum (MG63, U118MG, C6, NIH 3T3 cells). Cells were
then incubated with cediranib for 60−120 min and stimulated with the relevant ligand: stem
cell factor (SCF, 50 ng/ml), PDGF-AA or PDGF-BB (50 ng/ml) for 5−10 min. SCF was
obtained from R&D Systems Inc. (Abingdon, Oxfordshire, UK) and PDGF-AA and PDGF-BB
from Sigma-Aldrich (Poole, Dorset, UK). Cell lysates of NCI-H526, M07e, aortic and coronary
VSMC were made in lysis buffer I (50 mM Tris-HCl pH 7.6; 137 mM sodium chloride; 10%
glycerol; 0.1% Igepal; 0.1% sodium dodecylsulfate [SDS]; 50 mM sodium fluoride; 1mM
sodium orthovanadate and cocktail protease inhibitor tablets [Roche Diagnostics Ltd., Lewes,
UK)). Cell lysates of MG63, U118MG, C6 and NIH 3T3 cells were made in lysis buffer 2 (10%
glycerol, 2% SDS, 50 mM Tris-HCL, 200mM 2-mercaptoethanol).
The protein concentration in the lysates was determined using a bicinchoninic acid (BCA)
assay kit (Pierce, IL, USA) and Western blotting was performed on whole cell lysates (50–75
μg of protein loaded per lane) using standard SDS-PAGE methods with detection by
enhanced chemiluminescence. Total and phosphorylated proteins were measured using
antibodies to c-Kit (CST 3391; Cell Signaling Technology, Inc. [CST], Beverley,
Massachusetts, USA), and phosphorylated c-Kit (SC5535; Santa Cruz Biotechnology Inc,
Santa Cruz, California, USA); PDGFR-α (AF-307, R&D Systems Inc.), PDGFR-α (CST 3164),
and phosphorylated PDGFR-α (SC12910, Santa Cruz); PDGFR-β (AF-385, R&D Systems
Inc.), PDGFR-β (1469-1, Epitomics, Suffolk, UK), phosphorylated PDGFR-β (CST 3161);
mitogen-activated protein kinase (MAPK) (CST 9102) and phosphorylated MAPK (CST 9101).
Phosphorylation was quantitated using the ChemiGenius imaging system for
Chemiluminescence (Syngene, USA) with the exception of the human coronary VSMC which
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

9
were quantified by enzyme-linked immunosorbent assay (ELISA) (Goat Anti-PDGFR Alpha
R&D systems AF-307-NA capture, R&D Systems Inc. Duoset detection).
AG1-G1-Flt-1 cells were established with the permission of the Ethics Committee for scientific
research at the Institute of Medical Science, University of Tokyo, Tokyo, Japan. Briefly, a
human adult benign angioma was excised surgically and plated with F12-nutrient mixture
(Ham) medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS)
(JRH Biosciences, Leneza, Kansas) and 40 μg/ml kanamycin (Wako, Osaka, Japan). A
pEF1α-SV40 large T antigen plasmid was introduced into the cells using dimethyl sulphoxide
(DMSO) and polybrene (SIGMA-Aldrich, St Louis, Missouri). An SV40 large T-positive clone
AG1-G1 cell was isolated, and then pBCMGS-Neo-Flt-1 carrying the full length of Flt-1 cDNA
(10), or the empty vector pBCMGS-Neo plasmid was transfected into AG1-G1 cell by
Effectene Transfection Reagent (QIAGEN, Valencia, CA). Clone selection and culture were
performed with HAMS F-12 medium containing 10% FBS, 40 μg/ml kanamycin and 400 μg/ml
geneticin G418 (Wako). G418 was decreased to 200 μg/ml in regular culture. To examine
inhibition of VEGFR-1 phosphorylation, cells were placed in serum-free media overnight and
then incubated with cediranib for 90 min and stimulated with VEGF 50 ng/ml (R & D Systems
Inc., Abingdon, UK) for the last 5 min of incubation. Cell lysates were made in lysis buffer 1
and phosphorylated VEGFR-1 evaluated using Meso Scale methodology (Meso Scale
Discovery [MSD], Maryland, USA). The phosphorylated VEGFR-1 was analyzed by MSD
ELISA. Total VEGFR-1 antibody (MAB321, R & D Systems Inc., Abingdon, UK) was spotted
onto high-bind MSD plates and incubated for 2 h at room temperature after which time plates
were blocked and then washed. Cell lysates were added and incubated overnight at 4°C.
Plates were washed, the sulphotagged-pY20 detection antibody added and left to incubate for
1 h at room temperature. Following washing, read buffer was added and plates were read
immediately on the SECTOR™ Imager 6000 (MSD, Maryland, USA). To visualize
phosphorylated VEGFR-1 by Western blot, VEGFR1 was immune-precipitated with an
antibody to total VEGFR-1 (SC316, Santa Cruz), and immunoblotted with the anti-
phosphotyrosine antibody PY20 (SC508, Santa Cruz). Levels of total VEGFR-1 were
confirmed by immunoblotting with SC316.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

10
Sample preparation and mass spectrometry for identification of phosphotyrosine
modification of VEGFR-1
Analysis of VEGFR-1 phosphorylated epitope changes was performed using Cell Signaling
Technology’s proprietary PhosphoScan™ methodology. AG1-G1-Flt-1 cells were placed in
serum-free media overnight and stimulated with VEGF (50 ng/ml) or placenta growth factor
(PlGF) (100 ng/ml, R&D Systems) for 5 min, the former also with and without cediranib (100
nM; 85 min pre-incubation and 5 min co-incubation with ligand). Protein extracts from
AG1-G1-Flt1 cells were prepared by suspending cells in Urea Lysis Buffer (20 mM HEPES
[pH 8.0], 9.0 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-
glycerol phosphate). Lysates generated from approximately 2 x 108 cells were prepared for
each sample condition (control, VEGF-treated, PlGF-treated and VEGF- + cediranib-treated.
The resulting protein extracts (40 mg total protein each) were then reduced with dithiothreitol
(4.5 mM), carboxamidomethylated using iodoacetamide (10.0 mM), and subsequently
digested with trypsin (1:100 weight, trypsin to total protein). Peptides were separated from
non-peptide material by solid-phase extraction with Sep-Pak C18 cartridges. Lyophilized
peptides were re-dissolved, and phosphorylated peptides were isolated using a slurry of
immobilized phosphorylated tyrosine antibody (CST #9411) conjugated to protein-G agarose
beads (Roche, Indianapolis, Indiana, USA). Peptides were eluted from antibody-resin into a
total volume of 100 µL in 0.15% trifluoroacetic acid. Eluted peptides were concentrated with
PerfectPure C18 tips immediately prior to liquid chromatography−mass spectrometry (MS)
analysis. Peptides were loaded directly onto a 10 cm x 75 µm PicoFrit capillary column
packed with Magic C18 AQ reversed-phase resin. The column was developed with a 45 min
linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. Tandem mass
spectra were collected with an linear trap quadrupole (LTQ)-Orbitrap hybrid mass
spectrometer, using a top-ten method, a dynamic exclusion repeat count of 1 and a repeat
duration of 30 s. MS spectra were collected in the Orbitrap component of the mass
spectrometer, and MS/MS spectra were collected in the LTQ. MS/MS spectra were evaluated
using TurboSequest in the Proteomics Browser package (v. 27, rev. 13) The ratios of the
integrated peak height intensities for phosphopeptide quantification were obtained using the
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

11
XCalibur software 2.0.7 (ThermoFinnigan, San Jose, CA). A reduction in peak intensity in
VEGF-treated cells compared to control, or PlGF-treated cells compared with control or VEGF
plus cediranib-treated cells compared to control was expressed as a negative fold change
value. All integrated peak intensity calculations were manually reviewed to ensure proper
integration of consistently shaped, co-eluting peaks. Changes in phosphorylated peptide
levels were measured by taking the ratio of raw intensities between control and treated cells,
with the untreated sample as the reference (denominator) in each case. Raw intensity ratios
were normalized using a median adjustment technique whereby the log2 ratios comprising
each binary comparison (inhibitor treated versus control) was independently and globally
adjusted such that the normalized median log2 ratio is zero. The normalized log2 ratios were
then converted to their corresponding normalized fold-changes.
Inhibition of growth factor mediated cellular proliferation
NCI-H526 cells were used to determine the effect of cediranib on SCF-stimulated
proliferation. Cells were seeded at a density of 1x105 per ml in 96-well microtiter plates in
phenol-red free low-serum containing media (0.2% FBS) overnight. The following day cells
were pre-treated with cediranib (0.1–100 nM) for 30 minutes before stimulation with 50 ng/ml
SCF and then incubation for 72 h at 37°C. Cell proliferation was determined using an XTT
endpoint (Roche Diagnostics Ltd., Lewes, UK). All assays were performed in triplicate and the
mean ± standard error of the mean (SEM) was calculated from six independent experiments.
Human aortic VSMC were used to determine the effect of cediranib on PDGF-BB-stimulated
proliferation. Cells were seeded at 10,000 cells/well in black-walled 96-well plates in smooth
muscle cell growth medium 2 (Promocell GmbH Heidelberg, Germany) and incubated
overnight at 37°C. The following day the medium was replaced with Dulbecco's modified
Eagle's medium (DMEM; Gibco, Abingdon, UK) containing 0.1% FBS, PDGF-BB (50 ng/ml)
and cediranib. After 24-h incubation a bromodeoxuridine (BrdU) reagent (Amersham, UK)
was added and cells incubated for a further 24 h at 37oC. Cells were fixed in formalin for 15
min and proliferation assessed by staining for BrdU using the Cell Proliferation Fluorescence
kit (Amersham, UK). Cells were imaged on the ArrayScan (Cellomics, USA). All assays were
performed in triplicate and mean ± SEM was calculated from three independent experiments.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

12
MG63 cells were used to determine the effect of cediranib on PDGF-AA- and PDGF-BB-
stimulated proliferation. Cells were seeded at 1500 cells/well in 96-well plates in phenol red
DMEM containing 1% charcoal stripped serum for 24 h at 37°C. The following day the
medium was replaced with DMEM (Sigma-Aldrich) containing PDGF-AA or PDGF-BB (50
ng/ml) and cediranib for a further 72 h. Cell proliferation was determined as described above.
All assays were performed in triplicate and a mean ± SEM calculated from three independent
experiments.
Inhibition of receptor phosphorylation in vivo
The activity of cediranib was evaluated in a NCI-H526 human SCLC tumor xenograft model.
Tumors were implanted subcutaneously in the hind flank of female nude (nu/nu genotype)
mice of at least 8 weeks of age. When tumors reached a volume 0.36 ± 0.02 cm3, mice were
randomized (8−10 per group) and dosed with cediranib (0.75–6 mg/kg/d) or vehicle
administered once daily by oral gavage. Tumor volumes were assessed by bilateral Vernier
caliper measurement at least twice weekly and calculated using the formula (length x width) x
√ (length x width) x (π/6), where length was taken to be the longest diameter across the tumor
and width the corresponding perpendicular. To remove any size dependency prior to
statistical evaluation (the variance in mean tumor volume data increases proportionally with
volume and is therefore disproportionate between groups), data was log-transformed prior to
statistical evaluation using a one-tailed two-sample t test. NCI-H526 xenograft tumors were
evaluated for c-Kit receptor phosphorylation ex-vivo using immunoprecipitation, following
acute (2 days; eight animals per group) or chronic (17 days; three animals per group)
treatment with cediranib. Tumors were homogenized in lysis buffer I and following a protein
assay, 5 mg of protein from each sample was immunoprecipated overnight at 4°C with
anti-c-Kit conjugated agarose beads. The immune complexes were washed and proteins
eluted by boiling in SDS sample buffer. Standard SDS-Page methods were performed to
enable detection of total and phosphorylated c-Kit using antibodies as previously described.
Protein phosphorylation was quantitated using the ChemiGenius as described above.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

13
The activity of cediranib was also evaluated in a C6 rat glial tumor xenograft model in mice.
Cells were cultured in 199 media (Life Technologies, Abingdon, UK) supplemented with 10%
FCS and 2 mM glutamine and maintained in 7.5% CO2. For all tumor studies, C6 glial cells
were resuspended in sterile phosphate-buffered saline and inoculated subcutaneously
(1 x 105 in 100 μl) in the hind flank of male athymic mice. Tumor volumes were assessed as
described above. Approximately 21 days post implant when the C6 glial tumor volume was
between 0.5−0.8 cm3, mice were randomized into groups of 10 prior to treatment. Mice
received a single bolus oral dose of either cediranib or vehicle control. Animals were
sacrificed 4 h post dose. Five minutes prior to sacrifice, VEGF-A (20 μg per mouse:
AstraZeneca) and PDGF-BB (75 μg per mouse: Biosource, Paisley UK) were co-administered
intravenously (0.3 ml/mouse in PBS) to all animals. Terminal blood samples were taken from
the vena cava into lithium heparin tubes to collect plasma samples for pharmacokinetic
analysis. Tumors and lungs were excised and halved, each tissue half weighed and snap
frozen immediately in liquid nitrogen. Tissue samples were stored at –80°C until processed for
Western blot analysis for total and phosphorylated VEGFR-2 and PDGFR-β. Lungs were
homogenized in lysis buffer I and tumors were homogenized in lysis buffer III (60mM Tris,
150mM sodium chloride, 1mM ethylenediaminetetraacetic acid, 1% Igepal, 0.25% sodium
deoxycholate, 40% glycerol, 8% SDS, 200mM Tris-HCl, Phosphatase Inhibitor Cocktail 1 and
2 [Sigma-Aldrich]).
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

14
Results
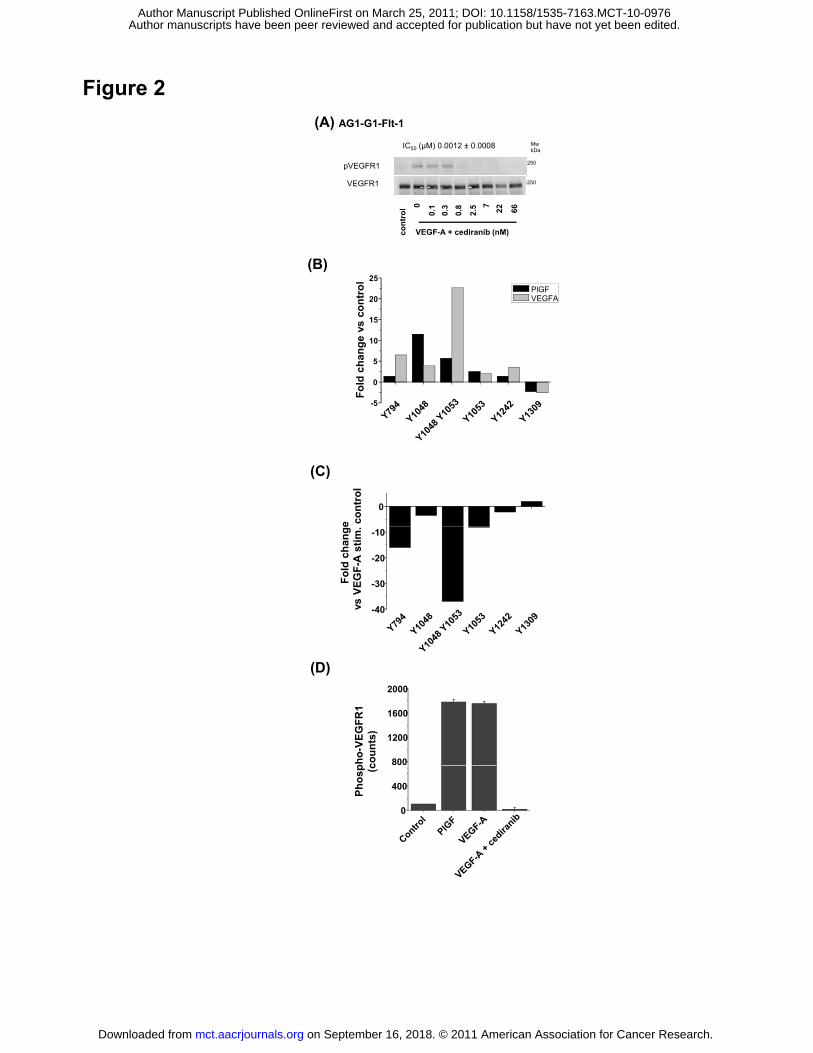
Cediranib is a potent inhibitor of VEGFR-1
To determine the potency of cediranib against VEGFR-1 in cells, a cell line stably transfected
with full length VEGFR-1 (AG1-AG-Flt-1) was used. Cediranib inhibited VEGF-A driven
VEGFR-1 phosphorylation with an IC50 value of 1.2 nM (Fig. 2A). This is comparable with the
cellular potency versus VEGFR-2 (0.5 nM) (7) and VEGFR−3 (<1 nM) (8), and consistent with
the primary pharmacology of the compound being that of a potent pan-inhibitor of VEGFR-1,
-2 and -3 tyrosine kinase activity.
To identify the tyrosine phosphorylation sites on VEGFR-1 modulated by ligand-induced
autophosphorylation and inhibition by cediranib, PhosphoscanTM was performed on VEGFR-1
isolated from the AG1-G1 cells treated with VEGF-A, and VEGF-A in the presence of 100 nM
cediranib. Phosphorylated receptor was enriched via a total phospho-tyrosine
immune-precipitation. The residues phosphorylated on VEGFR-1 in each treated lysate were
examined by specifically identifying phosphorylated peptides corresponding to VEGFR-1 (Fig.
2A, B; Supplementary Table 1). Upon stimulation with VEGF-A or PlGF, significant induction
of phosphorylation of peptides incorporating tyrosine residues Y1053, Y1048/Y1053, Y1048
was observed. Modest induction of phosphorylation was also detected at residues 794, 1242
but the magnitude of change was lower (Fig. 2A). The pattern of ligand induced
phosphorylation by both VEGF-A and PlGF was similar although the magnitude of induction
was higher with VEGF-A than with PlGF. Serine phosphorylated peptides were also detected,
although the significance of these modifications is unclear (Supplementary Table 1). This
shows that under these conditions the phosphorylation status of VEGFR-1 is dynamically
regulated on a restricted number of residues upon engaging VEGF-A or PlGF, with Y1048
and Y1053 showing the greatest fold changes.
To determine which residues were dynamically regulated by cediranib we compared protein
extracts from cells stimulated with VEGF-A with those from cells stimulated with VEGF in the
presence of 100 nM cediranib (Fig. 2C). There was a marked reduction in the relative
abundance of peptides corresponding to Y794, Y1053, Y1053/48, and Y1048 in cediranib-
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

15
treated samples, with a 37-fold reduction in the presence of the peptide corresponding to
pY1053/48 in the cediranib-treated samples (Fig. 2C). The total tyrosine phosphorylation
status of VEGFR-1 in the lysates used for this specific analysis was also assessed by ELISA.
VEGFR-1 from each lysate was captured and the level of tyrosine phosphorylation detected
using an anti-phosphotyrosine antibody (Fig. 2D). Both VEGF-A and PlGF induced significant
phosphorylation of VEGFR-1 in the lysates. Cediranib inhibited the VEGF-A induced
phosphorylation of VEGFR-1.
Cediranib inhibits c-Kit phosphorylation and SCF-induced proliferation
Cediranib inhibits c-Kit with a similar potency to that with which it inhibits the tyrosine kinase
activity of the VEGF receptors (7). The activity of cediranib against c-Kit was tested in two cell
lines, M07e and NCI-H526. SCF-stimulated c-Kit phosphorylation was inhibited with IC50
values of 3 and 1 nM respectively (Fig. 3 A and B). MAPK as a downstream signalling marker
was also inhibited with an IC50 value similar to that for inhibition of receptor phosphorylation
(Fig. 3A and B). The relationship between inhibition of acute ligand-induced phosphorylation
and SCF/c-Kit-dependent proliferation was determined using NCI-H526 cells. Cediranib
inhibited SCF-stimulated proliferation of NCI-H526 cells after 72 h, with an IC50 value of 0.013
nM (Fig. 3C), and full inhibition being achieved at concentrations of between 20 nM and 50
nM. From these experiments, it appears that about 10-fold higher concentrations were
required to inhibit functional consequences of c-Kit signalling (i.e. c-Kit-dependent cellular
proliferation) than for inhibition of receptor phosphorylation.
Mutations in c-Kit are associated with certain tumors such as gastrointestinal stromal tumors
(GIST) and AML where they drive tumor growth. The activity of cediranib against a range of
common c-Kit mutations was also determined using a panel of cell lines that either expressed
mutated c-Kit endogenously, or were transiently transfected with mutated receptors. The c-Kit
mutations assessed were V560G, V559D, W557R, Del 557-558, V654A, T670I, D816V,
D816Y and N822K. To assess the potential of the compound to inhibit phosphorylation of
these receptors, cells were incubated in the presence and absence of 20 nM of cediranib.
Cediranib inhibited phosphorylation of c-Kit mutants V560G, V559D, W557R and Del
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

16
557-558, V654A and N822K markedly, but it did not inhibit constitutive phosphorylation of
c-Kit mutants T670I D816V and D816Y (Supplementary Table 2).
Inhibition of c-Kit phosphorylation by cediranib in vivo
Inhibition of c-Kit phosphorylation was examined in vivo in established NCI-H526 tumor
xenografts, following chronic once-daily dosing of cediranib (17 doses of either 6, 3, 1.5 or
0.75 mg/kg) to tumor-bearing mice (Fig. 3D). The dose-range examined has been previously
determined to result in dose-dependent inhibition of a wide range of human tumor xenograft
models that do not express or have a dependency upon c-Kit. C-Kit was immunoprecipated
and the samples analyzed for phosphorylated and total receptors. In NCI-H526 tumors,
cediranib reduced phosphorylation of the receptor by greater than 80% at doses as low as
0.75 mg/kg. Although NCI-H526 tumor growth has been suggested to be dependent on c-Kit
(11), despite the tumors expressing constitutively phosphorylated c-Kit, no enhanced effects
on growth or survival of the tumor cells was observed in these experiments (Supplementary
Fig. 1) compared to other xenografts not expressing c-Kit.
Cediranib inhibits PDGFR-mediated autophosphorylation and PDGF-driven
proliferation at higher concentrations
In recombinant kinase assays, cediranib has been previously shown to demonstrate lower
potency, 10- and 36-fold for inhibition of PDGFR-α and -β than for VEGF receptors or 2.5-
and 19-fold for c-Kit (7). The activity against PDGFR-α and -β signalling was further explored
using a range of cell types including other tumor cells, VSMC, and fibroblasts (Fig. 4 A-F).
PDGF-AA and PDGF-BB ligands were used in stimulation assays, the former inducing
homodimerization of PDGFR-α and the latter homodimerization of PDGFR-β and
heterodimerization of PDGFR-α and -β. U118MG human glioma cells express both human
PDGFR-α and -β. Cediranib inhibited PDGF-AA-induced phosphorylation of PDGFR-α and
PDGF-BB-induced phosphorylation of PDGFR-β with mean IC50 values of 20 nM and 32 nM,
respectively. In C6 rat glioma cells, a similar IC50 value of 24 nM was observed versus PDGF-
AA stimulation of PDGFR-α (Fig. 4C). In NIH3T3 cells (mouse fibroblast line) cediranib was
slightly more potent, inhibiting PDGF-BB mediated phosphorylation of PDGFR-β with an IC50
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

17
value of 12 nM (Fig. 4D). Comparable activity was found in smooth muscle cell types. In
cultured human coronary VSMC the primary PDGFR receptor is PDGFR-α (data not shown).
Cediranib inhibited PDGF-AA stimulated receptor phosphorylation with an IC50 value of 15 nM
(Fig. 4E). In contrast, in human aortic VSMC the primary PDGFR receptor is PDGFR-β (data
not shown). In these cells cediranib inhibits PDGF-BB-induced phosphorylation of PDGFR-β
with an IC50 value of 23 nM (Fig. 4F). To determine how effectively cediranib inhibits the
functional consequences of PDGFR activation, its potency was assessed in both PDGF-AA
and PDGF-BB driven proliferation assays. In human aortic VSMC cediranib inhibited PDGF-
BB-stimulated proliferation after 48 h with an IC50 value of 36 nM (Fig. 4G) similar to the
potency versus PDGFR-β phosphorylation in the same cells. In MG63 cells, cediranib
inhibited PDGF-BB stimulated proliferation with an IC50 value of 63.5 nM (Fig. 4H), similar to
the previously reported IC50 value of 40 nM versus PDGF-AA-induced proliferation in the
same cell line (7).
Cediranib gives differential inhibition of PDGFR signalling in C6 tumors and murine
lung tissue in ligand-induced acute pharmacodynamic assays
We have previously demonstrated time and dose dependent inhibition of VEGFR-2 in murine
lung tissue using a ligand induced pharmacodynamic assay (12). This approach was taken
because the inter-animal variability in pVEGFR-2 levels was found to be high, making
accurate assessment of inhibitor dose responses extremely difficult. Addition of exogenous
ligand to stimulate receptor phosphorylation results overcame this issue. Here we used a
similar approach to assess the inhibition of PDGFR activation relative to VEGFR-2 to gain
greater insight in to the effects of cediranib on these receptors in vivo. The relative potency of
cediranib versus VEGFR-2 and PDGFR-β was compared directly in vivo in the same animal.
To normalize levels of phosphorylated VEGFR-2 and stimulate PDGFR-α and -β
phosphorylation, animals were injected with both VEGF-A and PDGF-BB immediately prior to
sacrifice. Lungs from animals bearing C6 tumors receiving cediranib 6, 3, 1.5 or 0.75 mg/kg
for 4 h were assessed for levels of phosphorylated VEGFR-2 (Fig. 5A) and phosphorylated
PDGFR-β (Fig. 5B) 4 hours after dosing. This timepoint was chosen as we established the
maximal exposure of cediranib occurs between 2-3 hours in mice (data not shown).
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

18
Consistent with previous data (12), VEGFR-2 phosphorylation in lung was significantly
reduced over a range of doses from 6 mg/kg down to 0.75 mg/kg (Fig. 5A). In contrast,
phosphorylation of PDGFR-β was only significantly inhibited in lung at 6 mg/kg with no
significant inhibition achieved at lower doses (Fig. 5B).
To further explore the relative activity against activity PDGFR-α and PDGFR-β expressed in
tumour cells we examined C6 tumour xenografts. C6 cells express both PDGFR-α and
PDGFR-β. Whilst PDGFR-α is constitutively phosphorylated in C6 cells in vivo, the injection of
PDGF-BB results in the additional phosphorylation of PDGFR-β, enabling inhibition of both
receptors to be studied in the same tumor. In separate studies (acute and chronic) we have
assessed the activity of cediranib against PDGFR-α in C6 tumour xenografts (at 6 hours) and
established that cediranib gives up to 60% inhibition of PDGFR-α phosphorylation. Again due
to variability in phosphorylation between tumours these data were variable (data not shown).
Adopting an acute ligand stimulation approach allowed us to further examine inhibition of both
PDGFRs in tumor and lung within the individual animals. In contrast to the modulation of
receptor phosphorylation in lung tissue, phosphorylation of both PDGFR-α (Fig. 5C) and -β
(Fig. 5D) in C6 tumors, in the same animals, was partially inhibited by all doses of cediranib
examined.
Selectivity against other tyrosine kinase receptors
Some VEGFR tyrosine kinase inhibitors also have activity against other kinases. Selectivity of
cediranib against mutFlt-3 and CSF-1R members of the PDGFR family have been shown
previously (Wedge et al, 2005). In addition to determining the relative activity against
VEGFR-1, c-Kit and PDGFRs, we also tested its activity against wild-type Flt-3 and fibroblast
growth factor receptor FGFR1 and FGFR4 (Supplementary Table 3). The activity against Flt-
3 was determined using OCI-AML-5 cells stimulated with Flt-3 ligand (Flt-3L). The activity
against FGFR1 and FGFR4 was determined by transiently overexpressing the receptor in
Cos-1 cells which resulted in constitutive phosphorylation of the receptor. Cediranib was
inactive against wild-type Flt-3 (IC50 value >1 μM) and had marginal activity versus FGFR-1
and -4 (IC50 values of 0.35 and 2.17 μM respectively). These data indicate that cediranib
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

19
has is significantly less active against these receptors than against the VEGF receptors, or c-
Kit.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

20
Discussion
We have previously shown that cediranib is a potent inhibitor of VEGFR-2, and -3, and that it
reduces growth of a wide range of tumor models by targeting tumor vasculature (7, 8, 12).
Cediranib has selectivity for VEGFR-2 against a wide range of kinases, including the PDGFR-
family members CSF-1R and Flt-3 in cellular phosphorylation assays (420- and >20,000-fold
selectivity, respectively, versus VEGFR-2) (7). Here we explore the pharmacology of
cediranib in more depth, examining its activity against VEGFR-1 in cells and the PDGFR-
family members c-Kit, PDGFR-α and PDGFR-β in vitro and in vivo within a dose-range of
0.75–6 mg/kg which has been routinely examined within preclinical tumor xenograft
experiments.
Establishing the potency of small molecule inhibitors against VEGFR-1 signalling in ligand-
induced endothelial cell assays has proven challenging due to the low intrinsic kinase activity
associated with this receptor. Despite this low kinase activity there is some evidence to
implicate VEGFR-1 signalling in pathological angiogenesis (13−15), and in the recruitment of
macrophages and myeloid prescursor cell recruitment to tumors (16−18); a process which
has been linked with resistance to VEGF signalling inhibitors (19−21). The role of the
VEGFR1 kinase domain in recruitment of BM derived cells into tumours has been confirmed
using VEGFR1 TK-/- transgenic mice (22). Consequently, concurrent inhibition of VEGFR-1
and -2 signalling, may afford added therapeutic benefit. To examine inhibition of VEGFR-1,
we utilized a cell line derived from a human benign angioma into which the full-length receptor
was over-expressed by stable-transfection. This cell line does not express VEGFR-2 and
therefore enables a more accurate assessment of activity against VEGFR-1, by avoiding any
confounds that could result from VEGFR heterodimerization. The inhibition of VEGF-induced
VEGFR-1 phosphorylation by cediranib, as determined by Western blotting, was evident at a
potency that is comparable to that determined against VEGFR-2 and -3 activation in cellular
assays, thereby confirming that cediranib is a pan-VEGFR kinase inhibitor. Whilst it is clear
that VEGFR-1 does induce specific signalling (10, 23, 24), there is limited information on the
residues involved. We therefore also used a MS-based method to examine the residues
activated on VEGFR-1 by VEGF or PlGF in AG1-G1-Flt-1 cells, and inhibition of the VEGF-
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

21
induced response by cediranib. VEGF-A and PlGF were found to induce a broadly similar
pattern of change in VEGFR-1 increasing the phosphorylation of tyrosine residues Y794,
Y1048, Y1053 and Y1242. The greatest fold-change evident was at Y1048 and Y1053 which
are within the tyrosine kinase domain of the receptor. Cediranib treatment abolished all
VEGF-stimulated phosphorylation on the four residues described, but had greatest effect
against Y1048/1053, the signal from this peptide sequence being reduced by nearly 37 fold
when compared to the untreated control, suggesting that phosphorylation at this site was the
most labile. These data contrast with previous studies that have described a number of
VEGFR-1 residues in the C-terminal tail as being modulated by VEGF-A treatment, in
particular Y1213 (25−27), but also Y1327 and Y1333 (27), and Y1309 in response to PlGF
stimulation (24). Although peptides indicating phosphorylation at Y1213 were detected in our
study, these were not modulated by ligand activation. These differences may be attributable
to the cell line examined or technical approach used. We were unable to develop a
pharmacodynamic assay to measure inhibition of VEGFR-1 activity in vivo, due to both the
low level of receptor phosphorylation and the inability to identify selective phosphorylation-
specific antibodies to VEGFR-1. However, given the similar potency of cediranib against each
VEGF receptor in cells, it would be reasonable to assume that it has the ability to inhibit
VEGFR-1 driven signalling responses in vivo.
In addition to having activity against the VEGF receptors, cediranib also inhibits the kinase
activity of c-Kit. Inhibition of wild-type c-Kit signalling in M07e and NCI-H526 cells prevented
downstream MAPK phosphorylation. Cediranib inhibited the SCF-induced proliferation of NCI-
H526 cells and reduced an associated increase in AKT phosphorylation (data not shown).
However, a decrease in potency (of approximately 10-fold) was observed for inhibition of
SCF-induced proliferation, suggesting that up to 90% of SCF signalling through c-Kit and
MAPK needs to be suppressed to deliver a cytostatic effect in NCI-H526 cells. In vivo,
inhibition (~85%) of the constitutive phosphorylation of c-Kit in established NCI-H526
xenograft tumors was observed after 17 days of chronically dosing cediranib at 0.75−6 mg/kg.
This suggests that cediranib may elicit a pharmacodynamic affect great enough to influence
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

22
the phenotypic consequences of c-Kit signalling in vivo, although an enhanced anti-tumor
effect was not observed in xenografts derived from this particular tumor line.
Mutation or aberrant activation of c-Kit and its ligand SCF are associated with the progression
of numerous solid and hematological malignancies, including GIST (28), SCLC (29), and AML
(30). Approximately 95% of GIST cases are c-Kit positive, with 60−70% positive for the c-Kit
exon 11 mutations (V560G, V559D, W557R Del557-558) against which the c-Kit/PDGFR/Abl
kinase inhibitor imatinib shows activity (31). More recently secondary mutations of c-Kit have
been identified that confer acquired resistance to imatininb (32, 33). Cediranib was found to
inhibit phosphorylation of all of the imatinib sensitive c-Kit mutant forms found in GIST, and in
addition inhibited two of the secondary point mutations which confer acquired resistance to
imatinib (V654A, N822K) (34−37). However, cediranib was not active against the T670I gate
keeper mutation in c-Kit (38) or the D816V/D816Y c-Kit mutations (35−37).
Previous data generated in cellular phosphorylation assays showed that cediranib was 10-to
16-fold less active against PDGFR-α and -β, than VEGFR, and this margin increased to 100-
fold when a comparative assessment of ligand-induced proliferation was performed. The in
vitro data generated with cediranib in this study against receptor phosphorylation in multiple
cell types is consistent with our previous data, with IC50 values in the range of 15–32 nM
being slightly higher than existing data in MG63 cells (cediranib IC50 of 5 and 8 nM against
PDGFR-α and -β respectively). A previously observed drop-off in potency of approximately 7-
fold between phosphorylation and proliferation endpoints for PDGF-AA/PDGFR-α signalling in
MG63 cells was also apparent in the present studies when these cells were stimulated with
PDGF-BB. The activity of cediranib against PDGF-BB-induced PDGFR-β phosphorylation and
cellular proliferation was more comparable in the primary VSMCs, suggesting that PDGFR-
mediated signalling responses may be cell-type dependent. In vivo, cediranib inhibited
PDGFR-β signalling in C6 rat tumor xenografts across the dose range examined, in contrast
to an effect only being evident at a dose of 6 mg/kg in normal lung tissue. This apparent
discrepancy is unlikely to be due to species-specificity differences given the high degree of
receptor homology between mouse and rat. Whilst a distribution effect cannot be ruled out,
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

23
the tissue concentrations of cediranib in C6 tumors did not exceed those in normal lung tissue
(data not shown). Neither is the inhibition of PDGFR phosphorylation in C6 tumors due to a
bystander effect that is secondary to the anti-vascular effects of cediranib, since compound
treatment does not affect the phosphorylation of other receptor tyrosine kinases, such as
EGFR in Lovo human colorectal tumor xenografts, at doses that significantly inhibit tumor
growth (data not shown). An alternative explanation for the divergent effect observed in lung
and C6 tumors could relate to the differential regulation or function of PDGFR-β in these
tissue compartments, the receptor driving significant cellular proliferation in C6 tumors but not
in normal lung tissue.
Although cediranib inhibited PDGFR-α and -β phosphorylation in C6 tumors, this model did
not appear to have increased sensitivity to the antitumor effects of the compound. A dose of 3
mg/kg cediranib, which inhibited PDGFR-α and -β phosphorylation by 73 and 76%,
respectively, 4 h after an acute dose, inhibited tumor growth by 52% (± 4% SEM) after 10 to
14 days of continuous once-daily dosing (data not shown): an effect not dissimilar to that
observed in non-PDGFR-dependent tumor models as a consequence of inhibiting VEGF
signalling (5). This finding re-enforces the fact that very significant inhibition of PDGFR
signalling may be required to prevent phenotypic signalling responses. The activity of
cediranib against PDGFR-α and -β would therefore not be expected to contribute significantly
to an effect on tumor growth or survival, unless a tumor has a particularly high dependency on
signalling from these receptors.
This work highlights the significant challenge to accurately describe the relative activity of an
ATP-competitive inhibitor potent against more than one kinase. This requires consideration of
activity at the recombinant kinase level, within multiple cellular phosphorylation and
proliferation assays, and then in vivo potency against the pertinent kinase in target tissues.
These data are all required to fully interpret observations made in pre-clinical models. The in
vivo PD data that show across the dose range cediranib is primarily a VEGF signalling
inhibitor with activity against c-Kit. That a significant drop off in potency is observed between
ligand-induced receptor phosphorylation and cellular proliferation for c-Kit, PDGFR-α and -β,
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

24
but not for VEGFR-2 in endothelial cell assays (7), combined with the relative order of
potency against these targets within a number of in vitro assays suggest cediranib is primarily
a VEGFR inhibitor.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

25
Acknowledgements:
We would like to acknowledge the support of the staff in CDMG for xenograft studies, Susan
Lovick for statistical analysis, Jonathon Orme for FGFR-1 and -4 assays and analyses,
Jonathan Dry for mutation analysis, James Sherwood and John Smith for c-Kit sequencing
and mutation analyses.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

26
References
1. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al.
Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma:
a randomised, double-blind phase III trial. Lancet 2007;370:2103-11.
2. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, et
al. Overall survival and updated results for sunitinib compared with interferon alfa in
patients with metastatic renal cell carcinoma. J Clin Oncol 2009; 27:3584-90.
3. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al.
Bevacizumab plus irinotecan, fluorouracil and leucovorin for metastatic colorectal
cancer. N Engl J Med 2004; 350:2335-42.
4. Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR
signaling Nat Rev Clin Oncol 2009;6:569-79.
5. Wedge SR and Jürgensmeier JM. VEGF receptor tyrosine kinase inhibitors for the
treatment of cancer, In: Tumour angiogenesis - basic mechanisms and cancer
therapy, Springer-Verlag Heidelberg (Eds. Marme, D. and Fusenig, N., ISBN 978-3-
540-33176-6), 2008: Chapter 23: p.395-424.
6. McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell
A , et al. Crystal structure of the kinase domain of human vascular endothelial growth
factor receptor 2: a key enzyme in angiogenesis. Structure 1999;7:319-30.
7. Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al.
AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor
receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res
2005;65:4389-400.
8. Heckman CA, Holopainen T, Wirzenius M, Keskitalo S, Jeltsch M, Ylä-Herttuala S, et
al. The tyrosine kinase inhibitor cediranib blocks ligand-induced vascular endothelial
growth factor receptor-3 activity and lymphangiogenesis. Cancer Res 2008;68:4754-
62.
9. Padera TP, Kuo AH, Hoshida T, Liao S, Lobo J, Kozak KR, et al. Differential
response of primary tumor versus lymphatic metastasis to VEGFR-2 and VEGFR-3
kinase inhibitors cediranib and vandetanib. Mol Cancer Ther 2008;7:2272-79.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

27
10. Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M. A unique
signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial
growth factor VEGF. Oncogene 1995;10:135-47.
11. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT
and platelet-derived growth factor receptor beta in preclinical models of human small
cell lung cancer. Mol Cancer Ther 2003;2:471-78.
12. Smith NR, James NH, Oakley I, Wainwright A, Copley C, Kendrew J, et al. Acute
pharmacodynamic and antivascular effects of the vascular endothelial growth factor
signaling inhibitor AZD2171 in Calu-6 human lung tumor xenografts. Mol Cancer Ther
2007;6:2198-208.
13. Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M et al.
Synergism between vascular endothelial growth factor and placental growth factor
contributes to angiogenesis and plasma extravasation in pathological conditions. Nat
Med. 2001; 7:575-83.
14. Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-
PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy
vessels. Cell 2007;131:463-75.
15. Wu Y, Zhong Z, Huber J, Bassi R, Finnerty B, Corcoran E, et al. Anti-vascular
endothelial growth factor receptor-1 antagonist antibody as a therapeutic agent for
cancer. Clin Cancer Res 2006;12:6573-84.
16. Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, et al.
Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor
angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med 2002;8:831-40.
17. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al.
VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic
niche. Nature 2005;438:820-7
18. Murakami M, Zheng Y, Hirashima M, Suda T, Morita Y, Ooehara J, et al. VEGFR1
tyrosine kinase signaling promotes lymphangiogenesis as well as angiogenesis
indirectly via macrophage recruitment. Arterioscler Thromb Vasc Biol 2008;28:658-
64.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

28
19. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor
refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat
Biotechnol 2007:25:911-920.
20. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-
dependent tumour angiogenesis. Nature 2007;450:825-31.
21. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid
cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy
in mouse models. Proc Natl Acad Sci U S A 2009;106:6742-7.
22. Muramatsu M, Yamamoto S, Osawa T, Shibuya M. Vascular endothelial growth factor
receptor-1 signaling promotes mobilization of macrophage lineage cells from bone
marrow and stimulates solid tumor growth. Cancer Res. 2010 70, 8211-21.
23. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different
signal transduction properties of KDR and Flt1, two receptors for vascular endothelial
growth factor. J Biol Chem 1994;269:26988-95.
24. Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, et al. Role
of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1
and Flk1 Nat Med 2003;9:936-43.
25. Igarashi K, Isohara T, Kato T, Shigeta K, Yamano T, Uno I. Tyrosine 1213 of Flt-1 is a
major binding site of Nck and SHP-2. Biochem Biophys Res Commun 1998;246:95-9
26. Yu Y, Hulmes JD, Herley MT, Whitney RG, Crabb JW, Sato JD. Direct identification
of a major autophosphorylation site on vascular endothelial growth factor receptor Flt-
1 that mediates phosphatidylinositol 3'-kinase binding. Biochem J 2001;358:465-72.
27. Ito N, Wernstedt C, Engström U, Claesson-Welsh L. Identification of vascular
endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of
SH2 domain-containing molecules. J Biol Chem 1998;273:23410-8.
28. Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a
ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61:8118-21.
29. Camps C, Sirera R, Bremnes RM, Garde J, Safont MJ, Blasco A, et al. Analysis of
c-Kit expression in small cell lung cancer: prevalence and prognostic implications.
Lung Cancer 2006;52:343-7.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

29
30. Advani AS. C-Kit as a target in the treatment of acute myelogenous leukemia. Curr
Hematol Rep 2005;4:51-8.
31. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H,et al.
Kinase mutations and imatinib response in patients with metastatic gastrointestinal
stromal tumor. J Clin Oncol 2003;21:4342-9.
32. Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke
T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal
stromal tumors under treatment with imatinib mesylate. Clin Cancer Res
2006;12:1743-9.
33. Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, et al. Clonal
evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal
tumors. Clin Cancer Res 2007;13:5398-405.
34. Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is
more sensitive to Imatinib (STI571) compared with wild-type c-Kit whereas the kinase
domain mutant D816VKit is resistant. Mol Cancer Ther 2002;1:1115-24.
35. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired
resistance to imatinib in gastrointestinal stromal tumor occurs through secondary
gene mutation. Clin Cancer Res 2005;11:4182-90.
36. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis, and
treatment of gastrointestinal stromal tumors. J Gastroenterol 2005;40:775-80.
37. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al.
Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin
Oncol 2006;24:4764-74.
38. Tamborini E, Bonadiman L, Negri T, Greco A, Staurengo S, Bidoli P, et al. Detection
of over-expressed and phosphorylated wild-type kit receptor in surgical specimens of
small cell lung cancer. Clin Cancer Res. 2004; 10:8214-19.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

30
Figure Legends
Figure 1. The structure of cediranib
Figure 2. Cediranib inhibits activation of VEGFR-1 and modulates phosphorylation of specific
sites on VEGFR-1. The activity of cediranib against VEGFR-1 was investigated using AG1-
G1-Flt-1 cells. (A) Serum-starved cells were stimulated with 50 ng/ml VEGF-A for 5 min in the
absence or presence of a range of concentrations of cediranib. Phosphorylation of VEGFR-1
was determined in by Western blotting of VEGFR-1 immunoprecipitated with a total VEGFR-1
antibody and then blotted with a phosphotyrosine antibody. Equal loading was determined by
Western blotting using a total VEGFR-1 antibody. The IC50 was determined using MSD
ELISA; the mean IC50 ± SEM observed over a number of experiments is shown. (B) To
determine the residues on VEGFR-1 that show dynamic changes in phosphorylation upon
activation of VEGFR-1, AG1-G1-Flt1 cells were stimulated with 50 ng/ml VEGF-A and 100
ng/ml PlGF. Lysates were immunoprecipitated with an anti-phosphotyrosine antibody and the
associated proteins analyzed by MS. Fold change in phosphorylation of individual sites
versus unstimulated control samples is represented. (C) To determine the residues on
VEGFR-1 that are phosphorylated upon activation of VEGFR-1 AG1-G1-Flt1 cells were
stimulated with 50 ng/ml VEGF-A in the presence of 100 nM cediranib. Fold change in
phosphorylation versus VEGF-A treated sample on individual sites is represented. (D)
Changes in phosphorylation within the lysates subjected to MS analysis were confirmed using
MSD ELISA. Changes are consistent with those seen in multiple previous experiments.
Figure 3. Cediranib inhibits SCF driven c-Kit phosphorylation and proliferation in tumor cells.
To assess phosphorylated c-Kit inhibition NCI-H526 (A) and M07e (B) cells, serum-starved
cells were incubated in the presence or absence of cediranib as indicated for 90−120 min,
then stimulated with 50 ng/ml of SCF for 5−10 min. Representative Western blots
demonstrating the inhibition of phosphorylated c-Kit and phosphorylated MAPK in (A) NCI-
H526 cells and (B) M07e cells are shown. All data are representative of at least three similar
experiments. The mean IC50 ± SEM observed over a number of experiments is shown. (C) To
determine the ability of cediranib to inhibit SCF-stimulated proliferation NCIH526 cells were
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

31
stimulated with 50 ng/ml cediranib in the presence and absence of cediranib for 72 h and
proliferation assessed by BrdU incorporation. The mean IC50 ± SEM observed over a number
of experiments is shown. (D) To demonstrate cediranib inhibits phosphorylation of c-Kit in
chronically dosed NCI-H526 tumor xenografts, mice bearing NCI-H526 tumors were treated
once-daily orally with 6, 3, 1.5 and 0.75 mg/kg/day of cediranib and the levels of
phosphorylated c-Kit were determined by ex vivo analysis. c-Kit was immunoprecipitated from
tumor lysates using a total c-Kit antibody. c-Kit and phosphorylated c-Kit were detected by
Western blotting and chemiluminescence, a representative Western blot is shown.
Phosphorylated c-Kit was quantified and the mean levels in each group ± SEM are shown.
Figure 4. Cediranib inhibits PDGF-ligand stimulated phosphorylation of PDGFR-α and -β and
inhibits ligand dependent proliferation. To determine the activity of cediranib against PDGFRs
a range of cell lines U118MG (A, B), C6 (C), NIH 3T3 (D), human coronary VSMC (E) and
human aortic VSMC (F) were stimulated with PDGF-AA (50 ng/ml) or PDGF-BB (50 ng/ml) for
5−10 min and then lysed. Inhibition of phosphorylated PDGFR-β in (D) human aortic VSMC
and inhibition of phosphorylated PDGFR-α in (E) human coronary VMSC and C6 cells.
Representative Western blots showing phosphorylated PDGFR-α and -β, as well as levels of
total receptors are shown as indicated. The mean IC50 ± SEM observed over a number of
experiments is shown. To determine the effect of cediranib on PDGF-stimulated proliferation
human aortic VSMC (G) and MG63 (H) cells were stimulated with 50 ng/ml PDGF-BB for 72 h
in the presence or absence of cediranib. The mean IC50 ± SEM observed over a number of
experiments is shown.
Figure 5. Cediranib inhibits phosphorylation of PDGFR-β in lung and tumor tissue and
phosphorylation of VEGFR-2 in lung in established human C6 tumor xenograft model
following a single dose. Acute changes in phosphorylation of VEGFR-2 and PDGFR-α and -β
were determined following a single dose of cediranib at the doses 6, 3, 1.5 and 0.75 mg/kg.
Phosphorylation of PDGFR-α and -β and VEGFR-2 was stimulated by injection of PDGF-BB
and VEGF-A 5 min prior to sacrifice of the animals. (A) Levels of phosphorylated VEGFR-2 in
lung lysates were quantitated and the mean levels ± SEM are represented. A representative
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

32
Western blot showing phosphorylated VEGFR-2 and total VEGFR-2 is inset. (B) Levels of
phosphorylated PDGFR-β in lung were quantitated and the mean levels ± SEM are
represented. A representative Western blot showing phosphorylated PDGFR-β and total
PDGFR-β is inset. (B) Levels of phosphorylated PDGFR-α (C) and phosphorylated PDGFR-β
(D) in tumor tissue lysates were quantitated and the mean levels ± SEM are represented.
Representative Western blots showing phosphorylated PDGFR-α and -β and total
phosphorylated PDGFR-α and -β are inset.
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976
Figure 1
O
N
N
O
O
F
N
N
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976
(A) AG1-G1-Flt-1
pVEGFR1
MwkDa
-250
IC50 (µM) 0.0012 ± 0.0008
Figure 2
VEGFR1
pVEGFR1
cont
rol 0
0.1
0.3
0.8
2.5 7 22 66
-250
VEGF-A + cediranib (nM)
(B)25
PlGFol
-5
0
5
10
15
20PlGFVEGFA
Fold
cha
nge
vs c
ontr
o
(C)
0
e cont
rol
-40
-30
-20
-10
Fold
cha
nge
vs V
EGF-
A s
tim.
(D)
800
1200
1600
2000
pho-
VEG
FR1
coun
ts)
0
400Phos
p (c
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976
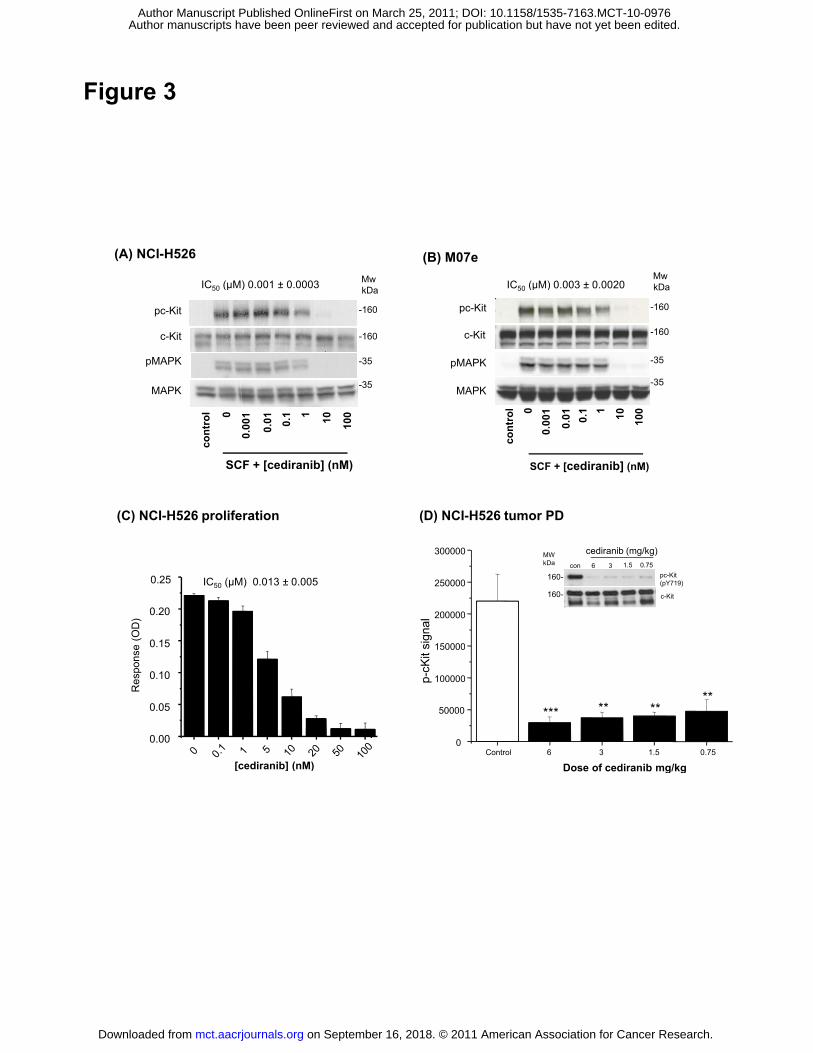
Figure 3
(A) NCI-H526
MwkDIC50 (µM) 0 001 ± 0 0003
(B) M07e
IC50 (µM) 0 003 ± 0 0020MwkDa
-160
-160
-35
-35
pc-Kit
pMAPK
MAPK
c-Kit
kDaIC50 (µM) 0.001 ± 0.0003 IC50 (µM) 0.003 ± 0.0020
-160
-160
-35
-35
pc-Kit
pMAPK
MAPK
c-Kit
kDa
SCF + [cediranib] (nM)
cont
rol 0
0.00
1
0.01 0.
1 1 10 100
SCF + [cediranib] (nM)
cont
rol 0
0.00
1
0.01 0.
1 1 10 100
(C) NCI-H526 proliferation (D) NCI-H526 tumor PD
0 15
0.20
0.25
e (O
D)
IC50 (µM) 0.013 ± 0.005
150000
200000
250000
300000
sign
al
con 6 3 1.5 0.75
cediranib (mg/kg)MWkDa
pc-Kit(pY719)
c-Kit
160-
160-
0.00
0.05
0.10
0.15
Res
pons
e
[cediranib] (nM)Control 6 3 1.5 0.75
0
50000
100000
150000
p-cK
it s
Dose of cediranib mg/kg
*** ** ****
[cediranib] (nM) Dose of cediranib mg/kg
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

Figure 4
MwkDa
co
ntr
ol 0
0.1
50
.46
1.4 4.1 12
37
10
03
00
-250
-250PDGFRα
pPDGFRα
IC50 (µM) 0.020 ± 0.008
(A) U118MG
MwkDa
on
tro
l 00
.15
0.4
61
.44
.1 12
37
10
0
30
0
-250
-250PDGFRβ
pPDGFRβ
IC50 (µM) 0.032 ± 0.010
(B) U118MG
PDGF-AA + cediranib (nM)
c
MwkDa
co
ntr
ol 0
0.1
50
.46
1.4 4.1 12 37
10
03
00
-250
-250PDGFRα
pPDGFRαIC50 (µM) 0.024 ± 0.004
(C) C6MwkDa
co
ntr
ol 0
0.1
50
.46
1.4 4.1 12 37
10
03
00
-250
-250PDGFRβ
pPDGFRβIC50 (µM) 0.012 ± 0.0027
(D) NIH 3T3
PDGF-BB + cediranib (nM)
co
PDGF-AA + cediranib (nM)
c
PDGF-BB + cediranib (nM)
c
MwkDa
-250pPDGFRα
(E) Human coronary VSMC
IC50 (µM) 0.015 ± 0.003MwkDa
-250pPDGFRβ
IC50 (µM) 0.023 ± 0.007
(F) Human aortic VSMC
PDGF-AA + cediranib (nM)
con
tro
l 0
0.3 0.8
2.5 7 22
66
20
0
-250PDGFRα
PDGF-BB + cediranib (nM)
con
tro
l 0
-250PDGFRβ
0.3 0.8 2.5 7 22
66
20
0
(G) Human aortic VSMC (PDGF-BB) (H) MG63 (PDGF-BB)
5
10
15
20
Res
pons
e (B
rDu
Uni
ts)
IC50 (µM) 0.032 ± 0.004 IC50 (µM) 0.0635 ± 0.0009
2
4
6
8
10
12
Res
pons
e (O
D x
100)
0
[cediranib] (nM)
0
2
[cediranib] nM
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976
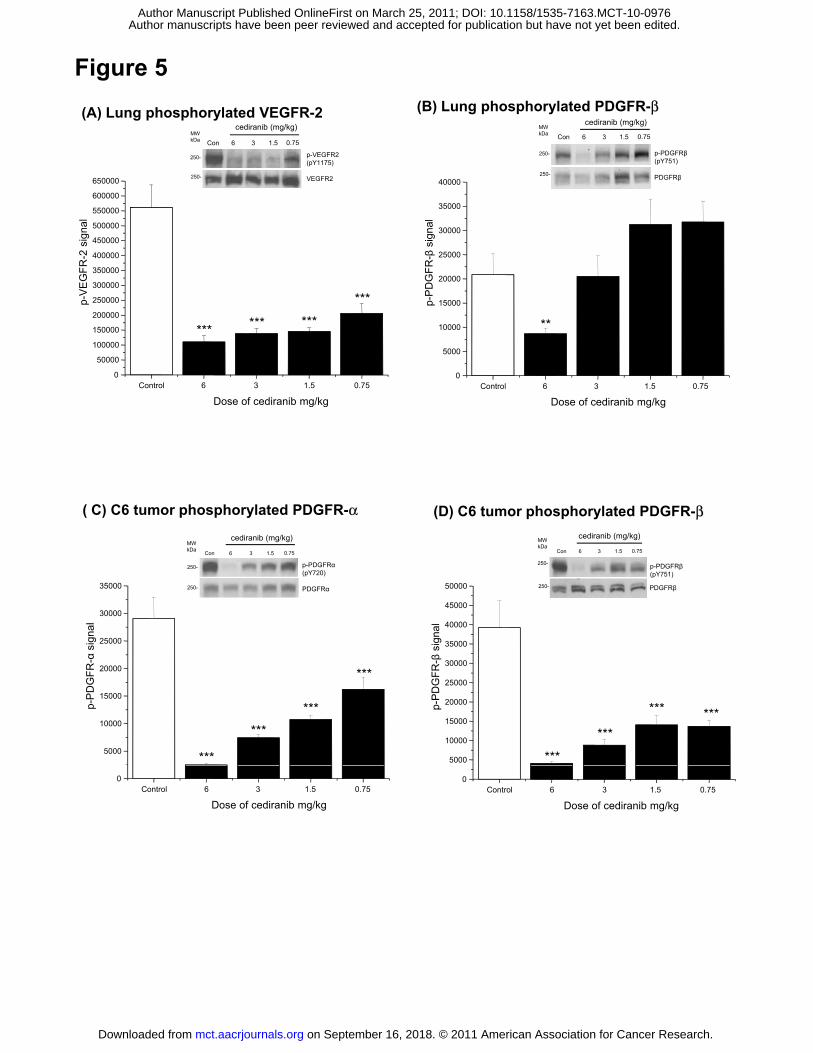
(A) Lung phosphorylated VEGFR-2
p-VEGFR2 (pY1175)
250-
MWkDa Con 6 3 1.5 0.75
cediranib (mg/kg)
(B) Lung phosphorylated PDGFR-β
250-
MWkDa Con 6 3 1.5 0.75
cediranib (mg/kg)
p-PDGFRβ(pY751)
Figure 5
300000
350000
400000
450000
500000
550000
600000
650000
EG
FR-2
sig
nal
VEGFR2250-
20000
25000
30000
35000
40000
DG
FR-β
sign
al
PDGFRβ250-
Control 6 3 1.5 0.750
50000
100000
150000
200000
250000
300000
p-V
E
Dose of cediranib mg/kg
*** *** ***
***
Control 6 3 1.5 0.750
5000
10000
15000p-P
D
Dose of cediranib mg/kg
**
g g
( C) C6 tumor phosphorylated PDGFR-α
g g
(D) C6 tumor phosphorylated PDGFR-β
25000
30000
35000
sign
al
p-PDGFRα (pY720)
PDGFRα
250-
250-
MWkDa
Con 6 3 1.5 0.75
cediranib (mg/kg)
35000
40000
45000
50000
igna
l
PDGFRβ
250-
250-
MWkDa
Con 6 3 1.5 0.75
cediranib (mg/kg)
p-PDGFRβ (pY751)
5000
10000
15000
20000
p-P
DG
FR-α
s
***
***
***
***
5000
10000
15000
20000
25000
30000
35000
p-P
DG
FR-β
s
***
***
*** ***
Control 6 3 1.5 0.750
Dose of cediranib mg/kgControl 6 3 1.5 0.75
0
Dose of cediranib mg/kg
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976

Published OnlineFirst March 25, 2011.Mol Cancer Ther Sandra R Brave, Kirsty Ratcliffe, Zena Wilson, et al. structurally related PDGFR-familykinase inhibitor, against VEGFR-1 and members of the Assessing the Activity of Cediranib, a VEGFR-2/-3 tyrosine
Updated version
10.1158/1535-7163.MCT-10-0976doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2011/03/24/1535-7163.MCT-10-0976.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/early/2011/03/24/1535-7163.MCT-10-0976To request permission to re-use all or part of this article, use this link
on September 16, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 25, 2011; DOI: 10.1158/1535-7163.MCT-10-0976
















![Role of targeted therapy in metastatic colorectal cancer · the VEGF family bind to three variants of receptors, VEGFR-1 (FLT-1), VEGFR-2 (FLK-1/KDR), and VEGFR-3 (FLT-4)[24,25]](https://img.pdfslide.us/doc/110x75/605c4af6a50bd930d55d2836/role-of-targeted-therapy-in-metastatic-colorectal-cancer-the-vegf-family-bind-to.jpg)












