Embed Size (px)
Citation preview

PHARMACOIUNETICS AND DRUG DISPOSITION
Immediate- versus controlled-release disopyrarnide: Importance of saturable 1 1
binding Objective: To examine the effects of saturable plasma binding on the pharmacokinetics of immediate- release (IR) and controlled-release (CR) disopyramide. Backflround: Saturable binding causes a lack of correspondence between the pharmacokinetics of total and unbound plasma disopyramide. Levels of total drug may therefore be insensitive to important dif- ferences between formulations. Methods: Patients receiving long-term disopyramide underwent serial blood sampling during withdrawal of equivalent doses of IR and CR disopyramide, and during accumulation of IR disopyramide. Plasma disopyramide was measured by enzyme-multiplied immunoassay technique, protein binding by ultrafil- tration, and a,-acid glycoprotein by radial immunodiffusion. Pharmacologic effect was assessed by use of high-speed ECGs. Values for plasma area under the concentration-time curve and elimination half-life were determined from the log-plasma concentration data; rate of plasma drug accumulation was deter- mined by nonlinear modeling. Results: Saturable plasma binding was evident in all patients. Comparison of total to unbound drug showed that peak-to-trough ratios during steady state were smaller (1.45 versus 2.39; p < 0.001), elim- ination half-life was longer (12.1 versus 4.5 hours; p < 0.001), and the time to achieve 50% of steady- state levels during drug accumulation was shorter (8.1 versus 4.3 hours; p < 0.05). Comparison of IR and CR disopyramide showed that unbound drug levels for CR disopyramide revealed lower peak plasma concentrations (0.75 versus 0.96 pglml) and peak-to-trough ratios (1.83 versus 2.31; p < 0.001). Trough plasma concentrations were similar. Fluctuations in ECG intervals during usual dosing were observed only with IR disopyramide. Conclusimts: Because of saturable plasma binding, total plasma concentrations underestimate fluctuations in unbound disopyramide during usual dosing and are insensitive to significant differences between IR and CR formulations. CR disopyramide provides less interdose variation in free drug levels and more constant pharmacologic effects. (CLIN P-COL THER 1993;54:16-22.)
Richard F. Davies, MD, Lyle A. Siddoway, MD, Leslie Shaw, PhD, J. Toby Barbey, MD, Dan M. Roden, MD, and Raymond L. Woosley, MD Ottawa, Ontario, Canada, and Nashville, Tenn.
From the University of Ottawa Heart Institute, Ottawa, and the Vanderbilt University School of Medicine, Nashville.
Supported by grants from the U.S. Public Health Service, Bethesda, Md. (GM 31304 and General Clinical Research Center RR 0095). Dr. Davies was supported by a fellowship from the Heart and Stroke Foundation of Canada (Ottawa, Ontario).
Received for publication Nov. 25, 1992; accepted Feb. 24, 1993. Reprint requests: Richard F. Davies, MD, University of Ottawa
Heart Institute, 1053 Carling Ave., Ottawa, Ontario KIY 4E9, Canada.
The free fraction of disopyramide is highly depen- dent on its concentration in the patient's blood,'-5 caus- ing a lack of correspondence between the pharmacoki- netics of total and unbound d r ~ g . ' . ~ - ~ (but not a11I2) studies have shown pharmacologic effect to
Copyright 0 1993 by Mosby-Year Book, Inc 0009-92361931$1.00 + 0.10 1311146730

CLINICAL PHARMACOLOGY & THERAPEUTICS VOLUME 54. NUMBER I Davies et al. 17
correspond better to unbound plasma disopyramide con- centrations. Previous studies comparing immediate- release (IR) and controlled-release (CR) disopyramide in norn~al volunteers found peak and trough plasma con- centration of total drug to be similar and plasma clear- ance to be ~ o m ~ a r a b l e . ' ~ - ' ~ However, although satura- ble binding has been shown to influence assessment of the area under the plasma concentration-time curve (AUC) for different formulations of IR di~opyramide, '~ a comparison of IR disopyramide and CR disopyramide formulations by use of measures of unbound drug has not been done previously. We hypothesized that satura- ble plasma binding may make measures of total plasma disopyramide insensitive to potentially important differ- ences in unbound drug.
METHODS This study was approved by the institutional review
board of Vanderbilt University Medical Center, with all subjects providing written informed consent. Twelve patients receiving long-term IR disopyramide for the treatment of symptomatic supraventricular or ventricular arrhythmias participated in the study. Pa- tients were excluded if they had significant renal im- pairment (serum creatinine > 1.5 times laboratory nor- mal), hepatic impairment (serum transaminases > 1.5 times normal), congestive heart failure, second or third degree atrioventricular block, or pulmonary or other systemic disease. Patients were also excluded if they had suffered acute myocardial infarction or coro- nary insufficiency within the previous 6 months.
Elimination pharmacokinetics of ZR disopyramide. Patients were admitted to the Vanderbilt Clinical Re- search Center on the day before testing. A detailed medication history was obtained, and compliance with an every-6-hour dosing schedule was confirmed. On the day of testing, fasting patients were given their usual doses of IR disopyramide at 8 AM with 250 ml water. Patients remained fasting until at least 2 hours after disopyramide administration. Serial blood sam- ples and simultaneous high-speed ECGs were obtained immediately before this dose was given and at 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, and 24 hours afterward during drug withdrawal.
Elimination pharmacokinetics of CR disopyramide. Patients were crossed over to an equivalent dosage of CR disopyramide (Norpace CR, G.D. Searle and Com- pany), discharged home, and asked to return for re- evaluation after at least 2 weeks. The procedure was the same as that used to evaluate IR disopyramide. Subjects were admitted to the Vanderbilt Clinical Re- search Center on the day before testing, and the every-
Table I. Patient characteristics
Age (yr) Gender (MIF) Weight (kg) Diagnosis (No. patients)
Coronary artery disease* Cardiomyopathy Mitral valve prolapse
AAG (mgldl) Mean 2 SD Range
Disopyramide dosage 600 mglday 300 mglday
AAG, a ,-Acid glycoprotein. *Patients within 6 months of myocardial or unstable angina excluded
12-hour dosing schedule was confirmed. On the morn- ing of testing, the usual dosage of CR disopyramide was administered at 8 AM with 250 ml water. Serial blood samples and high-speed ECGs were obtained at equivalent times during 24 hours of drug withdrawal.
Effect of disopyramide on ECG intervals. The pharmacologic effects of disopyramide on ECG inter- vals were measured by use of orthogonal lead (X, Y, Z) high-resolution ECGs recorded at 25, 50, and 100 mrnlsec at the time of each blood sample. This was done on the last seven patients taking part in the IR disopyramide versus CR disopyramide comparison. The PR, QRS, RR, and QT intervals for three simul- taneous leads from each tracing were averaged over 5 to 15 cycles by use of a digitizing pad. The QT inter- val was corrected for heart rate by use of the ~ a z e t t l * formula. ECGs were coded by number and analyzed by an observer who was unaware of the drug treat- ment or time of day associated with the tracing.
Pharmacokinetics of ZR disopyramide during drug accumulation. Five of the patients agreed to discon- tinue CR disopyramide for at least 2 weeks and return for an evaluation of disopyramide pharmacokinetics during drug accumulation. To accomplish this, pa- tients were admitted to the Vanderbilt Clinical Re- search Center, and IR disopyramide therapy was initi- ated in the previous total daily dosage. The drug was administered every 6 hours (k 15 minutes) with 250 ml water. Dosing and meal schedules were adjusted so that patients were fasting for the 2 hours before and after each dose. A trough blood sample was drawn immediately before each scheduled dose.
Determination of plasma drug concentrations and protein binding. Blood samples were drawn with a syringe and transferred directly into polytetrafluoro- ethylene-lined screw-capped tubes that contained hep-
18 Davies et al. CLINICAL PHARMACOLOGY & THERAPEmICS
JULY 1993
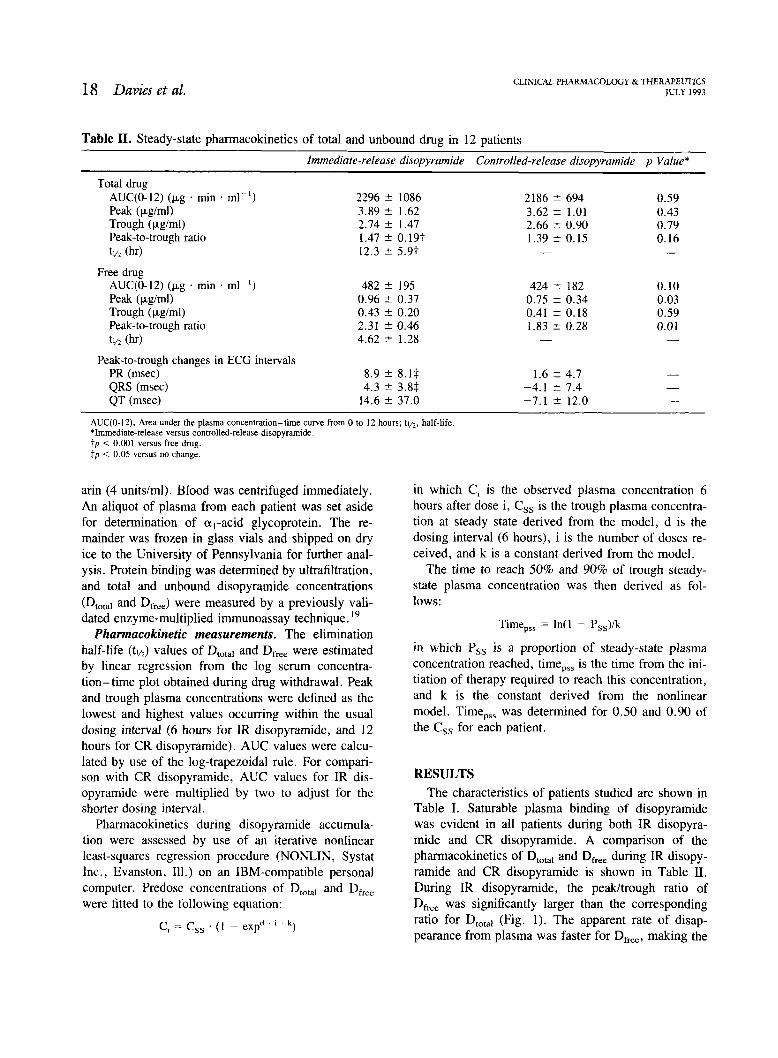
Table 11. Steady-state pharmacokinetics of total and unbound drug in 12 patients
Immediate-release disopyramide Controlled-release disopyramide p Value*
Total drug AUC(0-12) (kg . rnin . ml-I) Peak (pgtrnl) Trough (pglrnl) Peak-to-trough ratio tv2 (hr)
Free drug AUC(0-12) (kg . min . rnl-I) Peak (pgtml) Trough (pglrnl) Peak-to-trough ratio tl/2 (hr)
Peak-to-trough changes in ECCi intervals PR (msec) QRS (msec) QT (rnsec)
AUC(0-12). Area under the plasma concentration-time curve from 0 to 12 hours; tllz, half-life. *Immediate-release versus controlled-release disopyramide. t p < 0.001 versus free drug. fp < 0.05 versus no change
arin (4 unitsiml). Blood was centrifuged immediately. An aliquot of plasma from each patient was set aside for determination of a,-acid glycoprotein. The re- mainder was frozen in glass vials and shipped on dry ice to the University of Pennsylvania for further anal- ysis. Protein binding was determined by ultrafiltration, and total and unbound disopyramide concentrations (DtOta1 and Dfree) were measured by a previously vali- dated enzyme-multiplied immunoassay technique.19
Pharmacokinetic measurements. The elimination half-life (tl/,) values of Dtotal and Dfre, were estimated by linear regression from the log serum concentra- tion-time plot obtained during drug withdrawal. Peak and trough plasma concentrations were defined as the lowest and highest values occurring within the usual dosing interval (6 hours for IR disopyramide, and 12 hours for CR disopyramide). AUC values were calcu- lated by use of the log-trapezoidal rule. For compari- son with CR disopyramide, AUC values for IR dis- opyramide were multiplied by two to adjust for the shorter dosing interval.
Pharmacokinetics during disopyramide accumula- tion were assessed by use of an iterative nonlinear least-squares regression procedure (NONLIN, Systat Inc., Evanston, Ill.) on an IBM-compatible personal computer. Predose concentrations of D,,,, and D,, were fitted to the following equation:
C, = C,, . ( I - expd ' ' ' 1
in which Ci is the observed plasma concentration 6 hours after dose i, Css is the trough plasma concentra- tion at steady state derived from the model, d is the dosing interval (6 hours), i is the number of doses re- ceived, and k is a constant derived from the model.
The time to reach 50% and 90% of trough steady- state plasma concentration was then derived as fol- lows:
Time,,, = In(1 - Pss)/k
in which P,, is a proportion of steady-state plasma concentration reached, time,,, is the time from the ini- tiation of therapy required to reach this concentration, and k is the constant derived from the nonlinear model. Time,,, was determined for 0.50 and 0.90 of the C,, for each patient.
RESULTS The characteristics of patients studied are shown in
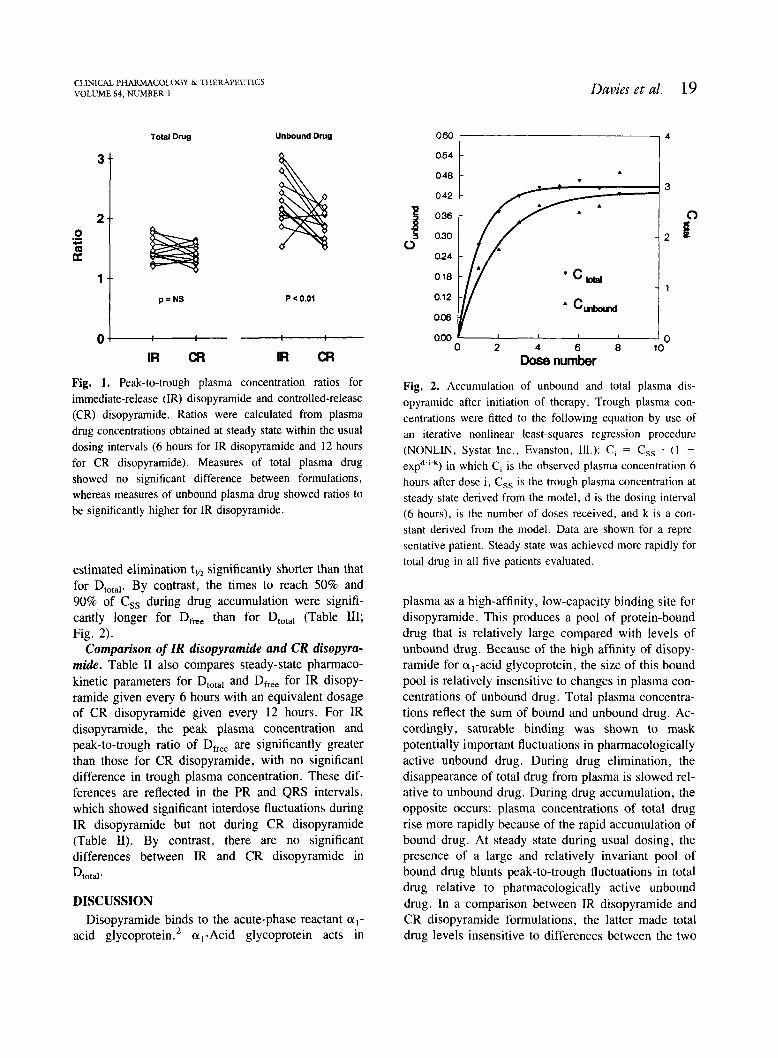
Table I. Saturable plasma binding of disopyramide was evident in all patients during both IR disopyra- mide and CR disopyramide. A comparison of the pharmacokinetics of Dtotal and Dfree during IR disopy- ramide and CR disopyramide is shown in Table 11. During IR disopyramide, the peakltrough ratio of Dfree was significantly larger than the corresponding ratio for DtOta, (Fig. 1). The apparent rate of disap- pearance from plasma was faster for D,,, making the
CLlNICAL PHARMACOLOGY & THERAPEUTICS VOLUME 54, NUMBER 1 Davies e t al. 19
Total Drug Unbound Drug
Fig. 1. Peak-to-trough plasma concentration ratios for immediate-release (IR) disopyramide and controlled-release (CR) disopyramide. Ratios were calculated from plasma drug concentrations obtained at steady state within the usual dosing intervals (6 hours for IR disopyramide and 12 hours for CR disopyramide). Measures of total plasma drug showed no significant difference between formulations, whereas measures of unbound plasma drug showed ratios to be significantly higher for IR disopyramide.
estimated elimination ty, significantly shorter than that for D,,,,,. By contrast, the times to reach 50% and 90% of Css during drug accumulation were signifi- cantly longer for D,, than for DtOta1 (Table 111; Fig. 2).
Comparison of ZR disopyramide and CR disopyra- mide. Table I1 also compares steady-state pharmaco- kinetic parameters for Dm,,, and D,,, for IR disopy- ramide given every 6 hours with an equivalent dosage of CR disopyramide given every 12 hours. For IR disopyramide, the peak plasma concentration and peak-to-trough ratio of D,, are significantly greater than those for CR disopyramide, with no significant difference in trough plasma concentration. These dif- ferences are reflected in the PR and QRS intervals, which showed significant interdose fluctuations during IR disopyramide but not during CR disopyramide (Table 11). By contrast, there are no significant differences between IR and CR disopyramide in Dtotal.
DISCUSSION Disopyramide binds to the acute-phase reactant a,-
acid glycoprotein.2 @,-Acid glycoprotein acts in
Fig. 2. Accumulation of unbound and total plasma dis- opyramide after initiation of therapy. Trough plasma con- centrations were fitted to the following equation by use of an iterative nonlinear least-squares regression procedure (NONLIN, Systat Inc.. Evanston, Ill.): C, = C,, . ( 1 -
e ~ ~ * " ' ~ ) in which Ci is the observed plasma concentration 6 hours after dose i, C,, is the trough plasma concentration at steady state derived from the model, d is the dosing interval (6 hours), is the number of doses received, and k is a con- stant derived from the model. Data are shown for a repre- sentative patient. Steady state was achieved more rapidly for total drug in all five patients evaluated.
0.48 - A .
-
-
I
plasma as a high-affinity, low-capacity binding site for disopyramide. This produces a pool of protein-bound drug that is relatively large compared with levels of unbound drug. Because of the high affinity of disopy- ramide for a,-acid glycoprotein, the size of this bound pool is relatively insensitive to changes in plasma con- centrations of unbound drug. Total plasma concentra- tions reflect the sum of bound and unbound drug. Ac- cordingly, saturable binding was shown to mask potentially important fluctuations in pharmacologically active unbound drug. During drug elimination, the disappearance of total drug from plasma is slowed rel- ative to unbound drug. During drug accumulation, the opposite occurs: plasma concentrations of total drug rise more rapidly because of the rapid accumulation of bound drug. At steady state during usual dosing, the presence of a large and relatively invariant pool of bound drug blunts peak-to-trough fluctuations in total drug relative to pharmacologically active unbound drug. In a comparison between IR disopyramide and CR disopyramide formulations, the latter made total drug levels insensitive to differences between the two
3
2
1
4 0
0 2 6 8 10
Dose number
20 Davies et al. CLINICAL PHARMACOLOGY & THERAPEUTICS
JULY 1993
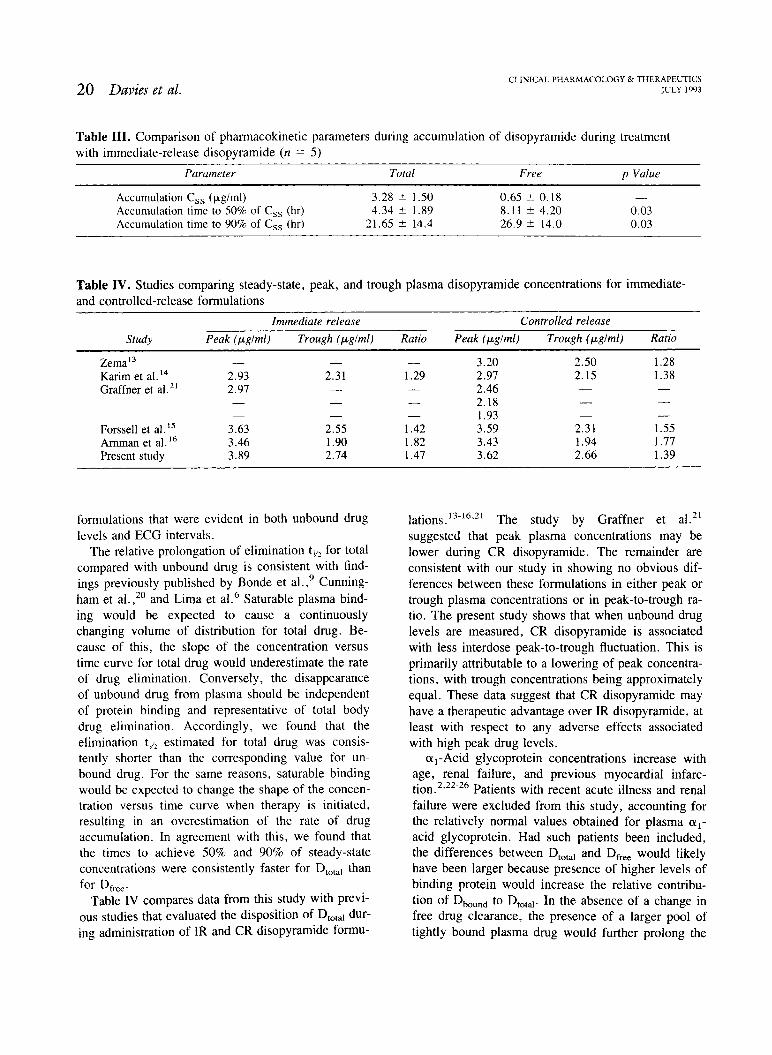
Table 111. Comparison of pharrnacokinetic parameters during accumulation of disopyramide during treatment with immediate-release disopyramide (n = 5)
Parurneter Total Free p Value
Accumulation C,, (yglml) 3.28 * 1.50 0.65 2 0.18 -
Accumulation time to 50% of C,, (hr) 4.34 +- 1.89 8.1 1 * 4.20 0.03 Accumulation time to 90% of C,, (hr) 21.65 14.4 26.9 * 14.0 0.03
Table IV. Studies comparing steady-state, peak, and trough plasma disopyramide concentrations for immediate- and controlled-release formulations
-- -
Immediate release Controlled release
Study Peak (pglrnl) Trough (pglml) Ratio Peak (pglrnl) Trough (pglrnl) Ratio
Zema13 - Karim et a1. l4 2.93 Graffner et a1." 2.97
Forssell et al.I5 3.63 Amman et a1. l 6 3.46 Present study 3.89
formulations that were evident in both unbound drug levels and ECG intervals.
The relative prolongation of elimination tlI2 for total compared with unbound drug is consistent with find- ings previously published by Bonde et al.,9 Cunning- ham et a ~ . , ~ ' and Lima et a1.6 Saturable plasma bind- ing would be expected to cause a continuously changing volume of distribution for total drug. Be- cause of this, the slope of the concentration versus time curve for total drug would underestimate the rate of drug elimination. Conversely, the disappearance of unbound drug from plasma should be independent of protein binding and representative of total body drug elimination. Accordingly, we found that the elimination tlI2 estimated for total drug was consis- tently shorter than the corresponding value for un- bound drug. For the same reasons, saturable binding would be expected to change the shape of the concen- tration versus time curve when therapy is initiated, resulting in an overestimation of the rate of drug accumulation. In agreement with this, we found that the times to achieve 50% and 90% of steady-state concentrations were consistently faster for DtOtal than for D,,.
Table IV compares data from this study with previ- ous studies that evaluated the disposition of DtOta1 dur- ing administration of IR and CR disopyramide formu-
l a t i o n ~ . ' ~ - ' ~ . ~ ~ The study by Graffner et a1.21 suggested that peak plasma concentrations may be lower during CR disopyramide. The remainder are consistent with our study in showing no obvious dif- ferences between these formulations in either peak or trough plasma concentrations or in peak-to-trough ra- tio. The present study shows that when unbound drug levels are measured, CR disopyramide is associated with less interdose peak-to-trough fluctuation. This is primarily attributable to a lowering of peak concentra- tions, with trough concentrations being approximately equal. These data suggest that CR disopyramide may have a therapeutic advantage over IR disopyramide, at least with respect to any adverse effects associated with high peak drug levels.
a,-Acid glycoprotein concentrations increase with age, renal failure, and previous myocardial infarc- tion.2,22-26 Patients with recent acute illness and renal failure were excluded from this study, accounting for the relatively normal values obtained for plasma a , - acid glycoprotein. Had such patients been included, the differences between Dtotal and D,,, would likely have been larger because presence of higher levels of binding protein would increase the relative contribu- tion of D,,,,, to Dtotal. In the absence of a change in free drug clearance, the presence of a larger pool of tightly bound plasma drug would further prolong the

CLINICAL PHARMACOLOGY & THERAPEUTICS VOLUME 54, NUMBER 1 Davies et al. 2 1
estimated tl/, and reduce the interdose peak-to-trough fluctuations in total drug. Previous studies have shown differences in drug pharmacokinetics in patients com- pared with normal v o ~ u n t e e r s . ~ ~ Consideration of pro- tein binding will be of particular importance in assess- ing other drugs which, like disopyramide, bind to acute-phase reactants in a concentration dependent manner. Other drugs that show concentration-depen- dent protein binding include sulfamethazine and its N-acetylated metabo~i te , '~ valproic acid and its ana- logs,29330 pethidine and n ~ r p e t h i d i n e , ~ ~ yohimbine,32 diazepam and ACC-9653 (a phenytoin ~ r o - d r u g ) , ~ ~ cocaine,34 tianeptine and other basic amphoteric
a j m a ~ i n e , ~ ~ and c i c ~ e t a n i n e . ~ ~ Although the potential influence of saturable binding is predictable from the published literature, its effect on pharmaco- kinetic comparisons have not received wide attention. For drugs with this property, pharmacokinetic and pharmacodynamic studies should include measures of unbound drug concentration.
We acknowledge Kimberly Brennan for editorial assis- tance, Patti Hofstetter and Tina Thomas for data collection, and Kathy Foris for assistance with data analysis.
References Giacomini KM, Swezey SE, Turner-Tamiyasu K, B1- aschke TF. The effect of saturable binding to plasma proteins on the pharmacokinetic properties of disopyra- mide. J Pharmacokinet Biopharm 1982;lO: 1- 14. David BM, Madsen BW, Ilett KF. Plasma binding of disopyramide. Br J Clin Pharmacol 1980;9:614-18. Giacomini KM, Blaschke TF. Effect of concentration- dependent binding to plasma proteins on the pharmaco- kinetics and pharmacodynamics of disopyramide. Clin Pharmacokinet 1984;9:42-8. Meffin PJ, Robert EW, Winkle RA, Harapat S, Peters FA, Hamson DC. Role of concentration dependent plasma protein binding in disopyramide disposition. J Pharmacokinet Biopharm 1979;7:29-46. Hinderling P, Bres J, Garrett E. Protein binding and erythrocyte partitioning of disopyramide and its mon- odealkylated metabolite. J Pharm Sci 1974;63:1684-90. Lima JJ, Boudoulas H, Blanford M. Concentration-de- pendence of disopyramide binding to plasma protein and its influence on kinetics and dynamics. J Pharmacol Exp Ther 1981;219:741-7. Bryson SM, Lawrence JR, Steele WH, Campbell BC, Elliott HL, Sumner DJ. The influence of protein bind- ing on disopyramide clearance. Eur J Clin Pharmacol 1982;23:453-6. Braun J, Sorgel F, Gluth WP, Oie S. Does alpha 1-acid glycoprotein reduce the unbound metabolic clearance of
disopyramide in patients with renal impairment? Eur J Clin Pharmacol 1988;35:313-7.
9. Bonde J, Jensen NM, Pedersen LE, et al. Disposition kinetics of disopyramide in human healthy volunteers described by an open three compartment model. Phar- macol Toxicol 1989;64:4 12-6.
10. Lima JJ, Wenzke SC, Boudoulas H, Schaal SF. Antiar- rhythmic activity and unbound concentrations of dis- opyramide enantiomers in patients. Ther Drug Monit 1990;12:23-8.
11. Thibonnier M, Holford NHG, Upton RA, Blume CD, Williams RL. Pharmacokinetic-pharmacodynamic anal- ysis of unbound disopyramide directly measured in se- rial plasma samples in man. J Pharmacokinetic Biop- harm 1984; 12559-73.
12. Holt GW, Noms RLG, Ravenscroft PJ, Bett JHN, Dry- burgh LG, Boyle CM. Effect of disopyramide on left ventricular performance: the relationship of free and to- tal concentrations of the drug and its mono-N-dealky- lated metabolite to noninvasive indices of function. J Cardiovasc Pharmacol 1983;5:5 1-4.
13. Zema MJ. Serum drug concentrations and adverse ef- fects in cardiac patients after administration of a new controlled-release disopyramide preparation. Ther Drug Monit 1984;6: 192-8.
14. Karim A, Schubert EN, Bums TS. Disopyramide plasma concentrations following single and multiple doses of the immediate- and controlled-release cap- sules. Angiology 1983;34:375-92.
15. Forssell G, Graffner C, Nordlander R, Nyquist 0 . Comparative bioavailability of disopyramide after mul- tiple dosing with standard capsules and controlled-re- lease tablets. Eur J Clin Pharmacol 1980; l7:209- 13.
16. Amman K, Graffner C, Rikner L, Ryden L, Voog L. Plasma concentration of disopyramide given as capsules and controlled release tablets. Eur J Clin Pharmacol 1983;24: 199-203.
17. Upton RA, Williams RL. The impact of neglecting nonlinear plasma-protein binding on disopyramide bio- availability studies. J Pharmacokinet Biopharm 1986; 14:365-79.
18. Bazett HC. An analysis of the time relations of electro- cardiograms. Heart 1920;7:353-70.
19. Shaw LM, Altman R, Thompson BC, Fields L. Factors affecting the biding of disopyramide to serum proteins. Clin Chem l985;3 1:616-9.
20. Cunningham JL, Shen DD, Shudo I, Azamoff DL. The effect of non-linear disposition kinetics on the systemic availability of disopyramide. Br J Clin Pharmacol 1978;5:343-6.
21. Graffner C, Lagerstrom PO, Lundborg P, Ronn 0. Rel- ative absorption of disopyramide following acute ad- ministration of standard capsules and controlled release tablets, determined by liquid chromatography. Int J Clin Pharmacol Ther Toxicol 198 1 ; 19:414-20.
22. David BM, Ilett KF, Whitford EG, Stenhouse NS. Pro-

22 Davies et al. CLINICAL PHARMACOLOGY & THERAPEUTICS
JULY 1993
longed variability in plasma protein binding of disopy- ramide after acute myocardial infarction. Br J Clin Pharmacol 1983; 15:435-41.
23. Caplin JL, Johnston A, Hamer J, Camm AJ. The acute changes in serum binding of disopyramide and flecain- ide after myocardial infarction. Eur J Clin Pharmacol 1985;28:253-5.
24. Holt DW, Hayler AM, Healey GF. Effect of age and plasma concentrations of albumin and acid glycoprotein on protein binding of disopyramide. Br J Clin Pharma- col 1983;16:344-5.
25. Piafsky KM, Borga 0 . Plasma protein binding of basic drugs. CLIN PHARMACOL THER 1977;22:545-9.
26. Piafsky KM, Borga 0 , Odar-Cederlof I, Johansson C, Sjoqvist F. Increased plasma protein binding of pro- pranolol and chlorpromazine mediated by disease-in- duced elevations of plasma acid glycoprotein. N Engl J Med 1978;299: 1435-9.
27. Woosley RL, Echt DS, Roden DM. Effects of conges- tive heart failure on the pharmacokinetics and pharma- codynamics of antiarrhythmic agents. Am J Cardiol 1986;57:25B-33B.
28. Tsang YC, Thiessen JJ. Competitive binding of sulfa- methazine and its N-acetylated metabolite. Biopharm Drug Dispos 1989;10:465-79.
29. Liu MJ, Scott KR, Pollack GM. Pharmacokinetics and pharmacodynamics of valproate analogues in rats. I. Spiro[4.6]undecane-2-carboxylic acid. Epilepsia 1990; 3 11465-73.
30. Semmes RL, Shen DD. Comparative pharmacodynam- ics and brain distribution of E-delta 2-valproate and val- proate in rats. Epilepsia 1991;32:232-41.
31. Wong YC, Chan K, Lau OW, Aun C, Houghton IT. Protein binding characterization of pethidine and nor- pethidine and lack of interethnic variability. Methods Find Exp Clin Pharmacol 1991 ;13:273-9.
32. Hubbard JW, Pfister SL, Biediger AM, Herzig TC, Keeton TK. The pharmacokinetic properties of yohim- bine in the conscious rat. Naunyn Schmiedebergs Arch Pharmacol 1988;337:583-7.
33. Hussey EK, Dukes GE, Messenheimer JA, et al. Eval- uation of the pharmacokinetic interaction between diaz- epam and ACC-9653 (a phenytoin prodrug) in healthy male volunteers. Pharm Res l990;7: 1 172-6.
34. Edwards DJ, Bowles SK. Protein binding of cocaine in human serum. Pharm Res 1988;5:440-2.
35. Zini R, Morin D, Salvadori C, Tillement JP. Tianeptine binding to human plasma proteins and plasma from pa- tients with hepatic cirrhosis or renal failure. Br J Clin Pharmacol l990;29:9- 18.
36. Hashimoto Y, Yasuhara M, Kamiya A, Okumura K, Hori R. Pharmacokinetics and dromotropic activity of ajmaline in rats with hyperthyroidism. Br J Pharmacol 1989;96: 163-9.
37. Zini R, Morin D, Jouenne P, Tillement JP. Cicletanine binding to human plasma proteins and erythrocytes, a particular HSA-drug interaction. Life Sci 1988;43: 2103-15.





























