Embed Size (px)
Citation preview

DOI: 10.1126/science.1243640, 428 (2014);343 Science
et al.Kathryn M. MonroeAbortively Infected with HIVIFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells
This copy is for your personal, non-commercial use only.
clicking here.colleagues, clients, or customers by , you can order high-quality copies for yourIf you wish to distribute this article to others
here.following the guidelines
can be obtained byPermission to republish or repurpose articles or portions of articles
): February 2, 2014 www.sciencemag.org (this information is current as of
The following resources related to this article are available online at
http://www.sciencemag.org/content/343/6169/428.full.htmlversion of this article at:
including high-resolution figures, can be found in the onlineUpdated information and services,
http://www.sciencemag.org/content/suppl/2013/12/18/science.1243640.DC1.html can be found at: Supporting Online Material
http://www.sciencemag.org/content/343/6169/428.full.html#relatedfound at:
can berelated to this article A list of selected additional articles on the Science Web sites
http://www.sciencemag.org/content/343/6169/428.full.html#ref-list-1, 9 of which can be accessed free:cites 34 articlesThis article
http://www.sciencemag.org/content/343/6169/428.full.html#related-urls1 articles hosted by HighWire Press; see:cited by This article has been
http://www.sciencemag.org/cgi/collection/immunologyImmunology
subject collections:This article appears in the following
registered trademark of AAAS. is aScience2014 by the American Association for the Advancement of Science; all rights reserved. The title
CopyrightAmerican Association for the Advancement of Science, 1200 New York Avenue NW, Washington, DC 20005. (print ISSN 0036-8075; online ISSN 1095-9203) is published weekly, except the last week in December, by theScience
on
Feb
ruar
y 2,
201
4w
ww
.sci
ence
mag
.org
Dow
nloa
ded
from
o
n F
ebru
ary
2, 2
014
ww
w.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
on
Feb
ruar
y 2,
201
4w
ww
.sci
ence
mag
.org
Dow
nloa
ded
from
o
n F
ebru
ary
2, 2
014
ww
w.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
on
Feb
ruar
y 2,
201
4w
ww
.sci
ence
mag
.org
Dow
nloa
ded
from
o
n F
ebru
ary
2, 2
014
ww
w.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
IFI16 DNA Sensor Is Required forDeath of Lymphoid CD4 T CellsAbortively Infected with HIVKathryn M. Monroe,1* Zhiyuan Yang,1* Jeffrey R. Johnson,1,2 Xin Geng,1 Gilad Doitsh,1Nevan J. Krogan,1,2,3 Warner C. Greene1,2,4†
The progressive depletion of quiescent “bystander” CD4 T cells, which are nonpermissive to HIVinfection, is a principal driver of the acquired immunodeficiency syndrome (AIDS). These cellsundergo abortive infection characterized by the cytosolic accumulation of incomplete HIV reversetranscripts. These viral DNAs are sensed by an unidentified host sensor that triggers an innateimmune response, leading to caspase-1 activation and pyroptosis. Using unbiased proteomicand targeted biochemical approaches, as well as two independent methods of lentiviral shorthairpin RNA–mediated gene knockdown in primary CD4 T cells, we identify interferon-g–inducibleprotein 16 (IFI16) as a host DNA sensor required for CD4 T cell death due to abortiveHIV infection. These findings provide insights into a key host pathway that plays a centralrole in CD4 T cell depletion during disease progression to AIDS.
HIV-AIDS is a devastating global epidemicwith over 70 million infections and35 million deaths (according to theWorld
Health Organization). AIDS is primarily causedby loss of the quiescent “bystander” CD4 T cellsthat populate lymphoid organs. These cells arenot permissive for viral replication, which re-sults in abortive infection and the accumulationof incomplete DNA transcripts (1). These cyto-solic viral DNAs trigger an innate immune re-sponse that activates cell death. Here, we soughtto identify the host DNA sensor that initiates celldeath in abortively infected CD4 T cells.
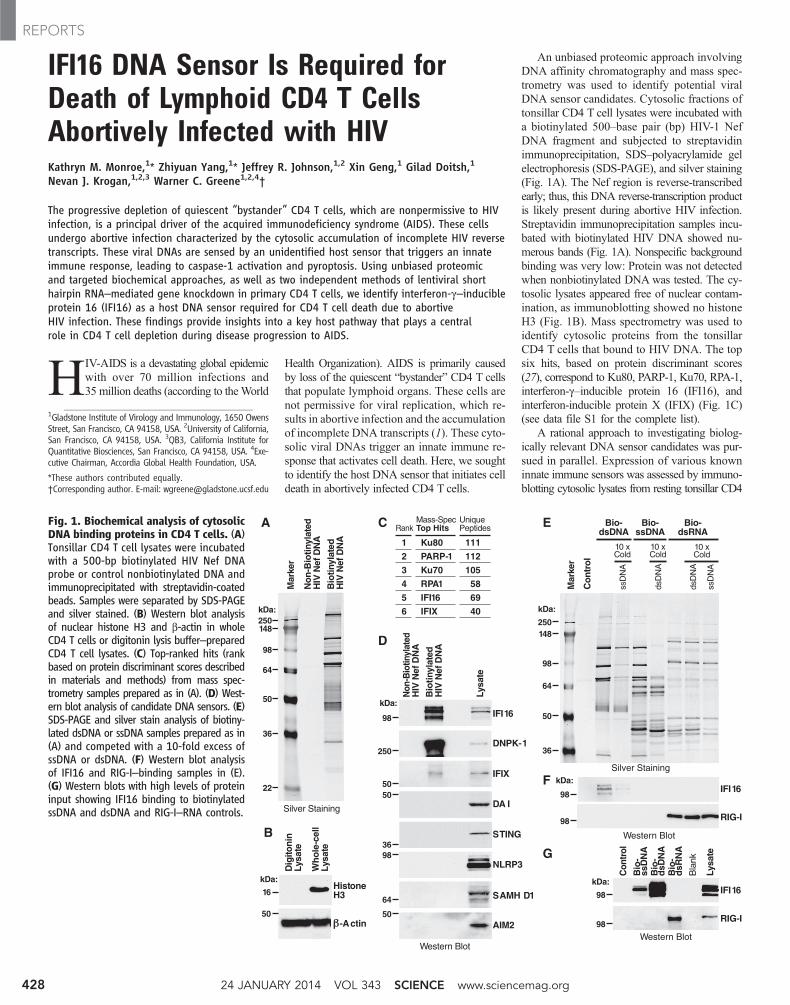
An unbiased proteomic approach involvingDNA affinity chromatography and mass spec-trometry was used to identify potential viralDNA sensor candidates. Cytosolic fractions oftonsillar CD4 T cell lysates were incubated witha biotinylated 500–base pair (bp) HIV-1 NefDNA fragment and subjected to streptavidinimmunoprecipitation, SDS–polyacrylamide gelelectrophoresis (SDS-PAGE), and silver staining(Fig. 1A). The Nef region is reverse-transcribedearly; thus, this DNA reverse-transcription productis likely present during abortive HIV infection.Streptavidin immunoprecipitation samples incu-bated with biotinylated HIV DNA showed nu-merous bands (Fig. 1A). Nonspecific backgroundbinding was very low: Protein was not detectedwhen nonbiotinylated DNA was tested. The cy-tosolic lysates appeared free of nuclear contam-ination, as immunoblotting showed no histoneH3 (Fig. 1B). Mass spectrometry was used toidentify cytosolic proteins from the tonsillarCD4 T cells that bound to HIV DNA. The topsix hits, based on protein discriminant scores(27), correspond to Ku80, PARP-1, Ku70, RPA-1,interferon-g–inducible protein 16 (IFI16), andinterferon-inducible protein X (IFIX) (Fig. 1C)(see data file S1 for the complete list).
A rational approach to investigating biolog-ically relevant DNA sensor candidates was pur-sued in parallel. Expression of various knowninnate immune sensors was assessed by immuno-blotting cytosolic lysates from resting tonsillar CD4
1Gladstone Institute of Virology and Immunology, 1650 OwensStreet, San Francisco, CA 94158, USA. 2University of California,San Francisco, CA 94158, USA. 3QB3, California Institute forQuantitative Biosciences, San Francisco, CA 94158, USA. 4Exe-cutive Chairman, Accordia Global Health Foundation, USA.
*These authors contributed equally.†Corresponding author. E-mail: [email protected]
Fig. 1. Biochemical analysis of cytosolicDNA binding proteins in CD4 T cells. (A)Tonsillar CD4 T cell lysates were incubatedwith a 500-bp biotinylated HIV Nef DNAprobe or control nonbiotinylated DNA andimmunoprecipitated with streptavidin-coatedbeads. Samples were separated by SDS-PAGEand silver stained. (B) Western blot analysisof nuclear histone H3 and b-actin in wholeCD4 T cells or digitonin lysis buffer–preparedCD4 T cell lysates. (C) Top-ranked hits (rankbased on protein discriminant scores describedin materials and methods) from mass spec-trometry samples prepared as in (A). (D) West-ern blot analysis of candidate DNA sensors. (E)SDS-PAGE and silver stain analysis of biotiny-lated dsDNA or ssDNA samples prepared as in(A) and competed with a 10-fold excess ofssDNA or dsDNA. (F) Western blot analysisof IFI16 and RIG-I–binding samples in (E).(G) Western blots with high levels of proteininput showing IFI16 binding to biotinylatedssDNA and dsDNA and RIG-I–RNA controls.
Ku80
PARP-1
Ku70
RPA1
IFI16
IFIX
111
112
105
58
69
40
Mass-SpecTop Hits
UniquePeptides
IFI16
DNPK-1
IFIX
DA I
NLRP3
AIM2
STING
SAMH D1
A
D
B
Non
-Bio
tinyl
ated
HIV
Nef
DN
A
Bio
tinyl
ated
HIV
Nef
DN
A
No
n-B
iotin
ylat
edH
IV N
ef D
NA
Bio
tinyl
ated
HIV
Nef
DN
A
Lysa
te
Mar
ker
HistoneH3
-Actin
Dig
iton
inLy
sate
Wh
ole
-cel
lLy
sate
250kDa:
148
64
50
98
36
22
250
kDa:
148
64
50
98
36
1
2
3
4
5
6
Rank
Silver Staining
Western Blot
Silver Staining
Western Blot
Bio-dsDNA
Mar
ker
Co
ntr
ol
Con
trol
Bio
-ss
DN
AB
io-
dsD
NA
Bio
-d
sRN
A
Bla
nk
Lysa
te
10 xCold
dsD
NA
ssD
NA
ssD
NA
10 xCold
Bio-ssDNA
dsD
NA
10 xCold
Bio-dsRNA
IFI16F
G
E
98
kDa:
98 RIG-I
Western Blot
IFI1698
kDa:
98RIG-I
16
kDa:
50
98
250
5050
3698
64
50
kDa:
C
24 JANUARY 2014 VOL 343 SCIENCE www.sciencemag.org428
REPORTS
T cells, which confirmed the presence of IFI16(2,3);AIM2(4–7);DAI(8);STING(9–11);DNPK-1(12); NLRP3 (13–15); and IFIX (PYHIN-1)(16) (Fig. 1D). Cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS)(17, 18) was detected neither at the protein levelin tonsillar CD4 T cells (fig. S1D) nor in the af-finity chromatography–mass spectrometry experi-ments (data file S1).Wewere intriguedwith IFI16because it was identified in both approaches andshown to form an inflammasome (3, 16). Of theknown inflammasome DNA sensors, IFI16, butnot AIM2, bound HIV-1 DNA (Fig. 1D). BecauseAIM2 binds DNA in a non–sequence-specificmanner, we had expected that AIM2 would be atop candidate, but it was not identified by massspectrometry (data file S1). IFI16 mRNA levelsare five times those of AIM2 mRNA in restingtonsillar CD4 T cells (fig. S1A). Of note, all threeIFI16 isoforms were detected in the cytosol andnucleus of primary tonsillar CD4 Tcells (fig. S1B).
Reverse transcription of the HIV RNA ge-nome initially generates single-stranded DNA(ssDNA) and then double-stranded DNA (dsDNA);
either might be sensed during abortive infection.A biotinylated dsDNA probe was incubated withcytosolic extracts from tonsillar CD4 T cells with10-fold excess of unlabeled ssDNA as a com-petitor (Fig. 1E). IFI16 effectively bound dsDNA(Fig. 1F) as described (2, 19) and was in com-petition with “cold” ssDNA. Biotinylated ssDNAwas subjected to binding and competition withcold dsDNA, but IFI16 was not initially detectedby immunoblotting. However, further analysiswith higher protein input confirmed that IFI16binds to ssDNA, albeit more weakly than todsDNA (Fig. 1G). Retinoic acid–inducible geneI (RIG-I) selectively bound dsRNA as a control(Fig. 1, F and G).
Standard methods, including liposome-mediateddelivery of small interfering RNAs (siRNAs) orinfection with vesicular stomatitis virus glycopro-tein (VSVG)–pseudotyped lentiviruses encodingshort hairpin RNAs (shRNAs) are ineffective fortargeted gene knockdown in resting CD4 T cells(20, 21). siRNA nucleofection is highly variable,often toxic, and associated with extensive cell deathin tonsillar cultures. To overcome these challenges
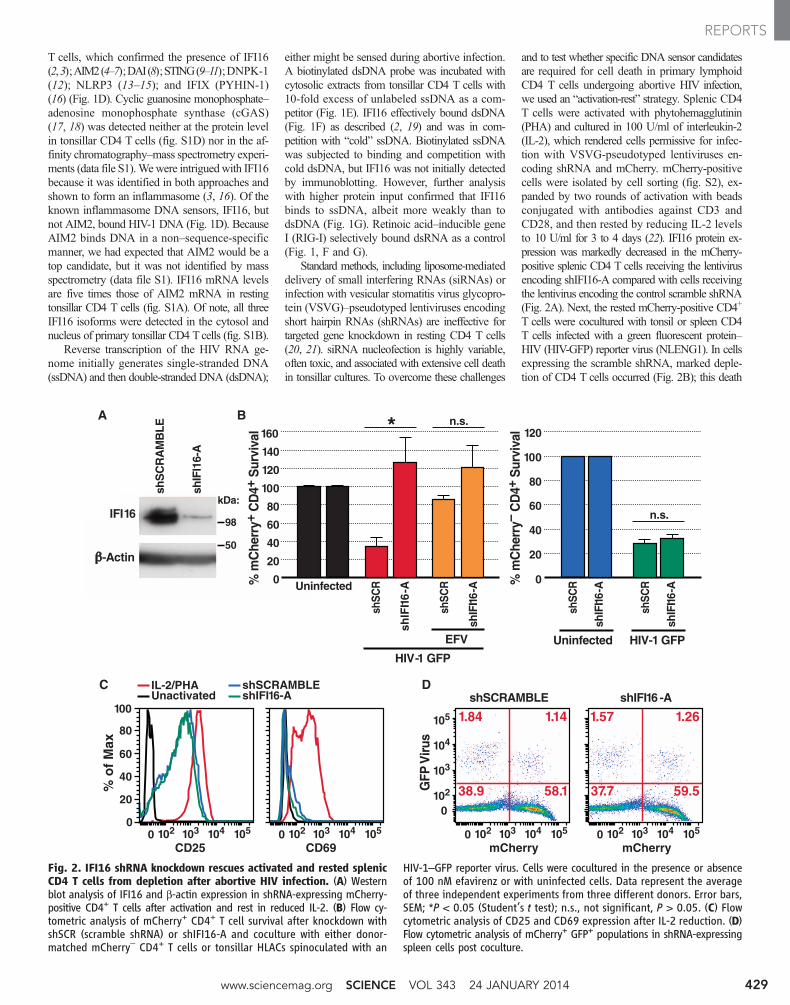
and to test whether specific DNA sensor candidatesare required for cell death in primary lymphoidCD4 T cells undergoing abortive HIV infection,we used an “activation-rest” strategy. Splenic CD4T cells were activated with phytohemagglutinin(PHA) and cultured in 100 U/ml of interleukin-2(IL-2), which rendered cells permissive for infec-tion with VSVG-pseudotyped lentiviruses en-coding shRNA and mCherry. mCherry-positivecells were isolated by cell sorting (fig. S2), ex-panded by two rounds of activation with beadsconjugated with antibodies against CD3 andCD28, and then rested by reducing IL-2 levelsto 10 U/ml for 3 to 4 days (22). IFI16 protein ex-pression was markedly decreased in the mCherry-positive splenic CD4 T cells receiving the lentivirusencoding shIFI16-A compared with cells receivingthe lentivirus encoding the control scramble shRNA(Fig. 2A). Next, the rested mCherry-positive CD4+
T cells were cocultured with tonsil or spleen CD4T cells infected with a green fluorescent protein–HIV (HIV-GFP) reporter virus (NLENG1). In cellsexpressing the scramble shRNA, marked deple-tion of CD4 T cells occurred (Fig. 2B); this death
BA
C D
% m
Che
rry+
CD
4+ S
urvi
val
0
20
40
60
80
100
120
140
160
Uninfected
shSC
R
shSC
R
shIF
I16
-A
shSC
R
shIF
I16-A
shSC
R
shIF
I16-A
shIF
I16-A
HIV-1 GFP
shSCRAMBLE shIFI16 -A
% m
Che
rry–
CD
4+ S
urvi
val
0
20
40
60
80
*100
120
CD69 mCherry mCherry
1.84 1.57
38.9 37.7
1.14 1.26
58.1 59.5
102 1030 104 105102 1030 104 105 102 1030
GFP
Viru
s
104 105
102
103
104
105
0
CD25
40
60
80
100
0
20
IL-2/PHAUnactivated shIFI16-A
shSCRAMBLE
102 1030 104 105
% o
f M
ax
IFI16
-Actin
shS
CR
AM
BLE
shIF
I16-
A
EFV Uninfected HIV-1 GFP
n.s.
n.s.98
kDa:
50
Fig. 2. IFI16 shRNA knockdown rescues activated and rested splenicCD4 T cells from depletion after abortive HIV infection. (A) Westernblot analysis of IFI16 and b-actin expression in shRNA-expressing mCherry-positive CD4+ T cells after activation and rest in reduced IL-2. (B) Flow cy-tometric analysis of mCherry+ CD4+ T cell survival after knockdown withshSCR (scramble shRNA) or shIFI16-A and coculture with either donor-matched mCherry– CD4+ T cells or tonsillar HLACs spinoculated with an
HIV-1–GFP reporter virus. Cells were cocultured in the presence or absenceof 100 nM efavirenz or with uninfected cells. Data represent the averageof three independent experiments from three different donors. Error bars,SEM; *P < 0.05 (Student’s t test); n.s., not significant, P > 0.05. (C) Flowcytometric analysis of CD25 and CD69 expression after IL-2 reduction. (D)Flow cytometric analysis of mCherry+ GFP+ populations in shRNA-expressingspleen cells post coculture.
www.sciencemag.org SCIENCE VOL 343 24 JANUARY 2014 429
REPORTS
was rescued by adding a nonnucleoside reverse-transcriptase inhibitor, efavirenz (EFV), whichimplicates abortive HIV infection as previouslydescribed (1). In sharp contrast, introduction ofshIFI16-A resulted in survival of the mCherry-positive CD4+ T cells. In the same experiments,mCherry-negative CD4+ T cells were markedly de-pleted, which suggested that they had returnedto a sufficient state of rest to undergo abortiveinfection.
To exclude more formally the possibility that the“activated and rested” CD4 T cells were dying as aresult of productive infection, we assessed theactivation status of these cells. Flow cytometricanalysis revealed that CD4 T cells cultured in re-duced concentrations of IL-2 had lower levels ofCD25 and CD69 than cells activated with 100 U/mlIL-2 and 10 mg/ml PHA (Fig. 2C). However, CD25levels were higher than found in unactivated cells,which indicated that these cells had not fullyreturned to a resting state. This finding likely relates
in part to the up-regulation of CD25 expressionby IL-2 (23). To directly test how permissive thesecells were to productive HIV infection, we utilizedan HIV-1–GFP reporter virus. In cells express-ing shScramble or shIFI16-A, only ~1 to 2% ofthe mCherry-positive cells and ~1 to 2% ofmCherry-negative cells were productively infected,as indicated by GFP expression (Fig. 2D). Thus,the 60 to 70% depletion of CD4 T cells observedwas not due to high levels of productive viralinfection.
To confirm IFI16 as an HIV-1 DNA sensorand to test a broader array of potential candidates,we used a second, more rapid shRNA knock-down strategy. Virus-like particles (VLPs) werepackaged with the simian immunodeficiency virus(SIV) accessory protein Vpx that degrades theSAMHD1 restriction factor and renders cells sus-ceptible to lentiviral infection (24, 25). This meth-od was adapted for use in resting CD4 T cellson the basis of prior success in cells of myeloid
origin (26, 27). Twenty-four hours after VLP-Vpx spinoculation, complete tonsillar HLACswere spinoculated with shRNA-mCherry lenti-viral vectors pseudotyped with HIV glycoprotein160 (gp160) envelope protein (Env) (fig. S3) (28).Cells were cocultured 3 days later with humanembryonic kidney (HEK) 293T cells producingor not producing HIV-1 virions. CD4 T cell deathwas assessed 1 or 2 days later in mCherry-positiveCD4+ T cells expressing the shRNA and mCherry-negative CD4+ T cells lacking the shRNA. Inparallel, EFV was added to select wells.
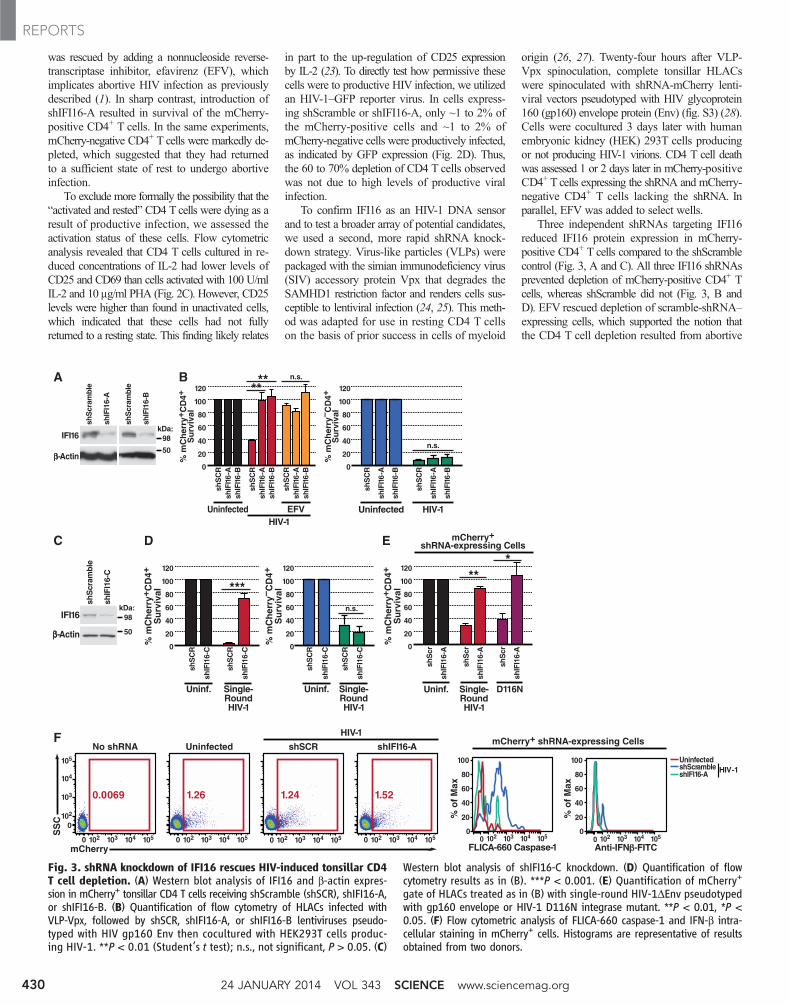
Three independent shRNAs targeting IFI16reduced IFI16 protein expression in mCherry-positive CD4+ T cells compared to the shScramblecontrol (Fig. 3, A and C). All three IFI16 shRNAsprevented depletion of mCherry-positive CD4+ Tcells, whereas shScramble did not (Fig. 3, B andD). EFV rescued depletion of scramble-shRNA–expressing cells, which supported the notion thatthe CD4 T cell depletion resulted from abortive
0
20
40
60
80
100
120
0
20
40
60
80
100
120BA
C
% m
Ch
erry
+ CD
4+S
urv
ival
% m
Ch
erry
– CD
4+S
urv
ival
shS
CR
Uninfected
shIF
I16-
Bsh
IFI1
6-A
shS
CR
shIF
I16-
Bsh
IFI1
6-A
shS
CR
shIF
I16-
B
shIF
I16-
A
shS
CR
shIF
I16-
B
shIF
I16-
A
shS
CR
shIF
I16-
Bsh
IFI1
6-A
0
0
20
40
60
80
100
120
F
% m
Ch
erry
+ CD
4+S
urv
ival
mCherry+shRNA-expressing Cells
shS
cr
shIF
I16-
A
shS
cr
shIF
I16-
A
shS
cr
shIF
I16-
A
Uninf.
HIV-1UninfectedHIV-1
EFV
D116NSingle-RoundHIV-1
0
20
40
60
80
100
120
0
20
40
60
80
100
120
D E
% m
Ch
erry
+ CD
4+S
urv
ival
% m
Ch
erry
– CD
4+S
urv
ival
shS
CR
shIF
I16-
C
shS
CR
shIF
I16-
C
Uninf. Single-RoundHIV-1
shS
CR
shIF
I16-
C
shS
CR
shIF
I16-
C
Uninf. Single-RoundHIV-1
IFI16
-Actin
shS
cram
ble
shIF
I16-
A
IFI16
-Actin
shS
cram
ble
shIF
I16-
C
shS
cram
ble
shIF
I16-
B
mCherry+ shRNA-expressing Cells
mCherry
SS
C % o
f M
ax
% o
f M
ax
1051041031020 1051041031020
No shRNA shSCR
Anti-IFN -FITC
102
103
104
105
0
1051041031020
Uninfected
1051041031020
shIFI16-A
HIV-1
20
40
60
80
Uninfected100
0
20
40
60
80
100
FLICA-660 Caspase-110510410310201051041031020
n.s.
n.s.**
n.s.
***
**
***
shScrambleshIFI16-A HIV-1
98kDa:
50
98kDa:
50
1.521.241.260.0069
Fig. 3. shRNA knockdown of IFI16 rescues HIV-induced tonsillar CD4T cell depletion. (A) Western blot analysis of IFI16 and b-actin expres-sion in mCherry+ tonsillar CD4 T cells receiving shScramble (shSCR), shIFI16-A,or shIFI16-B. (B) Quantification of flow cytometry of HLACs infected withVLP-Vpx, followed by shSCR, shIFI16-A, or shIFI16-B lentiviruses pseudo-typed with HIV gp160 Env then cocultured with HEK293T cells produc-ing HIV-1. **P < 0.01 (Student’s t test); n.s., not significant, P > 0.05. (C)
Western blot analysis of shIFI16-C knockdown. (D) Quantification of flowcytometry results as in (B). ***P < 0.001. (E) Quantification of mCherry+
gate of HLACs treated as in (B) with single-round HIV-1DEnv pseudotypedwith gp160 envelope or HIV-1 D116N integrase mutant. **P < 0.01, *P <0.05. (F) Flow cytometric analysis of FLICA-660 caspase-1 and IFN-b intra-cellular staining in mCherry+ cells. Histograms are representative of resultsobtained from two donors.
24 JANUARY 2014 VOL 343 SCIENCE www.sciencemag.org430
REPORTS
infection (Fig. 3B). Moreover, mCherry-negativeCD4+Tcellswere depleted regardless of the shRNA,which demonstrated that experimental conditionswere sufficient for abortive infection in all infectedsamples (Fig. 3, B and D). Thus, using an inde-pendent method for shRNA knockdown, we con-firmed that IFI16 is required for lymphoid CD4 Tcell depletion by HIVafter abortive HIV infection.
To confirm that shScramble mCherry-positiveCD4 T cells die via abortive infection, which re-quires reverse transcription but not integration(1), cells were cocultured with HEK293T cellsto produce single-round HIV-1 (DEnv with gp160coexpressed) or HIV containing a disabling in-tegrase mutation, D116N, in which aspartic acidat codon 116 is replaced by asparagine (Fig. 3E).These replication-defective, nonspreading virusesinduced depletion of mCherry-positive CD4 Tcells expressing shScramble. In contrast, intro-duction of shIFI16-A rescued cells from HIV-1–mediated depletion. Thus, neither productive
infection nor HIV integration is required for celldeath. Knockdown of IFI16 decreased caspase-1activation in the mCherry-positive cells, whereasinterferon-b (IFN-b) was induced in HIV-infectedcells with the shScramble control but not in cellsexpressing shIFI16-A (Fig. 3F). These findingssuggest that IFI16 is required to sense incom-plete DNA reverse transcripts that accumulatein abortively infected cells; their accumulationleads to caspase-1 activation, which results in thesubsequent death of these cells via pyroptosis(29). IFI16 sensing also leads to IFN-b induction.
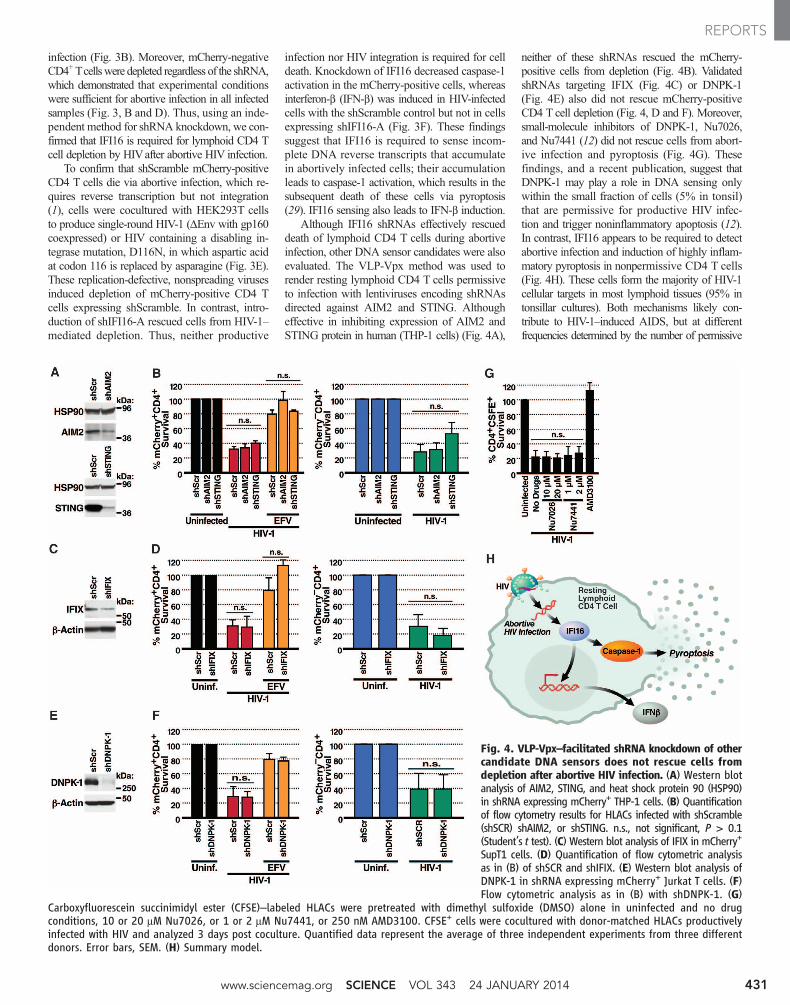
Although IFI16 shRNAs effectively rescueddeath of lymphoid CD4 T cells during abortiveinfection, other DNA sensor candidates were alsoevaluated. The VLP-Vpx method was used torender resting lymphoid CD4 T cells permissiveto infection with lentiviruses encoding shRNAsdirected against AIM2 and STING. Althougheffective in inhibiting expression of AIM2 andSTING protein in human (THP-1 cells) (Fig. 4A),
neither of these shRNAs rescued the mCherry-positive cells from depletion (Fig. 4B). ValidatedshRNAs targeting IFIX (Fig. 4C) or DNPK-1(Fig. 4E) also did not rescue mCherry-positiveCD4 T cell depletion (Fig. 4, D and F). Moreover,small-molecule inhibitors of DNPK-1, Nu7026,and Nu7441 (12) did not rescue cells from abort-ive infection and pyroptosis (Fig. 4G). Thesefindings, and a recent publication, suggest thatDNPK-1 may play a role in DNA sensing onlywithin the small fraction of cells (5% in tonsil)that are permissive for productive HIV infec-tion and trigger noninflammatory apoptosis (12).In contrast, IFI16 appears to be required to detectabortive infection and induction of highly inflam-matory pyroptosis in nonpermissive CD4 T cells(Fig. 4H). These cells form the majority of HIV-1cellular targets in most lymphoid tissues (95% intonsillar cultures). Both mechanisms likely con-tribute to HIV-1–induced AIDS, but at differentfrequencies determined by the number of permissive
Fig. 4. VLP-Vpx–facilitated shRNA knockdown of othercandidate DNA sensors does not rescue cells fromdepletion after abortive HIV infection. (A) Western blotanalysis of AIM2, STING, and heat shock protein 90 (HSP90)in shRNA expressing mCherry+ THP-1 cells. (B) Quantificationof flow cytometry results for HLACs infected with shScramble(shSCR) shAIM2, or shSTING. n.s., not significant, P > 0.1(Student’s t test). (C) Western blot analysis of IFIX in mCherry+
SupT1 cells. (D) Quantification of flow cytometric analysisas in (B) of shSCR and shIFIX. (E) Western blot analysis ofDNPK-1 in shRNA expressing mCherry+ Jurkat T cells. (F)Flow cytometric analysis as in (B) with shDNPK-1. (G)
Carboxyfluorescein succinimidyl ester (CFSE)–labeled HLACs were pretreated with dimethyl sulfoxide (DMSO) alone in uninfected and no drugconditions, 10 or 20 mM Nu7026, or 1 or 2 mM Nu7441, or 250 nM AMD3100. CFSE+ cells were cocultured with donor-matched HLACs productivelyinfected with HIV and analyzed 3 days post coculture. Quantified data represent the average of three independent experiments from three differentdonors. Error bars, SEM. (H) Summary model.
www.sciencemag.org SCIENCE VOL 343 24 JANUARY 2014 431
REPORTS

versus nonpermissive cellular targets residing withinvarious lymphoid tissues.
IFI16 evolved as an antiviral DNA sensor(2, 3). In addition, IFI16 exerts novel antiviral ac-tivity, including restriction of herpesvirus repli-cation by inhibiting viral gene expression (30).That IFI16 is targeted for degradation by herpes-viruses (31) further highlights an evolutionarypressure to counteract its activity. Our studies re-veal that IFI16 initiates an innate immune responsethat, rather than protecting the host, drives thedebilitating CD4 T cell depletion that underliesprogression to AIDS in untreated HIV-infectedindividuals. The cycle of abortive infection, in-flammatory death, and recruitment of new cellslikely explains how this innate host response isundermined and, in fact, centrally contributes toHIV pathogenesis. Our findings now identify IFI16as a criticalDNAsensor required for cell death duringabortiveHIV-1 infection. Therapies directed againstthis host death pathwaymight preserve CD4 Tcellsand reduce chronic inflammation––two signaturepathologies in HIV infection.
References and Notes1. G. Doitsh et al., Cell 143, 789–801 (2010).2. L. Unterholzner et al., Nat. Immunol. 11, 997–1004 (2010).3. N. Kerur et al., Cell Host Microbe 9, 363–375 (2011).4. T. L. Roberts et al., Science 323, 1057–1060 (2009).5. T. Bürckstümmer et al., Nat. Immunol. 10, 266–272 (2009).
6. T. Fernandes-Alnemri, J. W. Yu, P. Datta, J. Wu,E. S. Alnemri, Nature 458, 509–513 (2009).
7. V. Hornung et al., Nature 458, 514–518 (2009).8. A. Takaoka et al., Nature 448, 501–505 (2007).9. D. L. Burdette et al., Nature 478, 515–518 (2011).10. H. Ishikawa, Z. Ma, G. N. Barber,Nature 461, 788–792 (2009).11. H. Ishikawa, G. N. Barber, Nature 455, 674–678 (2008).12. A. Cooper et al., Nature 498, 376–379 (2013).13. S. Mariathasan et al., Nature 440, 228–232 (2006).14. F. S. Sutterwala et al., Immunity 24, 317–327 (2006).15. T. D. Kanneganti et al., Nature 440, 233–236 (2006).16. R. L. Brunette et al., J. Exp. Med. 209, 1969–1983 (2012).17. L. Sun, J. Wu, F. Du, X. Chen, Z. J. Chen, Science 339,
786–791 (2013).18. D. Gao et al., Science 341, 903–906 (2013).19. T. Jin et al., Immunity 36, 561–571 (2012).20. L. M. Agosto et al., J. Virol. 83, 8153–8162 (2009).21. D. Yu, W. Wang, A. Yoder, M. Spear, Y. Wu, PLOS Pathog.
5, e1000633 (2009).22. A. Bosque, V. Planelles, Blood 113, 58–65 (2009).23. J. M. Depper et al., Proc. Natl. Acad. Sci. U.S.A. 82,
4230–4234 (1985).24. B. Descours et al., Retrovirology 9, 87 (2012).25. H. M. Baldauf et al., Nat. Med. 18, 1682–1689 (2012).26. G. Berger et al., Nat. Protoc. 6, 806–816 (2011).27. G. S. Bobadilla, N. Sunseri, N. R. Landau, Gene Ther. 20,
514–520 (2013).28. Materials and methods are available as supplementary
materials in Science Online.29. G. Doitsh et al., Nature 10.1038/nature12940 (2013).30. G. R. Gariano et al., PLOS Pathog. 8, e1002498 (2012).31. M. H. Orzalli, N. A. DeLuca, D. M. Knipe, Proc. Natl.
Acad. Sci. U.S.A. 109, E3008–E3017 (2012).
Acknowledgments: We are grateful for technical assistancefrom the Gladstone Flow Core, including M. Cavrois, M. Gesner,
and J. Tawney. We thank M. Spindler for the generous gift of thepSicoR-mCherry vector, and D. N. Levy for the NLENG1 and D116Nplasmids. Efavirenz and azidothymidine were obtained from theAIDS Reagent Program, Division of AIDS, National Institute ofAllergy and Infectious Diseases, NIH. We thank L. Chavez,I. Munoz-Arias, and N. Byers for technical advice; T. Johnson fortechnical assistance; G. Howard and A. L. Lucido for editorialassistance; J.C.W. Carroll for graphic arts; and R. Givens andS. Cammack for administrative assistance. The data presentedin this manuscript are tabulated in the main paper and thesupplementary materials. A patent application has been filed(U.S. provisional serial number 61/511,023) regarding theidentification of abortive HIV infection as a driver of CD4 T celldepletion involving the activation by IFI16 of caspase-1 ininflammasomes leading to pyroptosis, an intensely inflammatoryform of programmed cell death. This work was made possible withsupport from the University of California San Francisco–GladstoneCenter for AIDS Research (CFAR), an NIH-funded program (P30AI027763), with additional funding from the Gladstone Institutes.This work was also supported by NIH R21 AI102782, U19 AI096113(Martin Delaney CARE Collaboratory), and 1DP1036502 to W.C.G.and NIH P50 GM082250, P01 AI090935, and P50 GM081879to N.J.K. N.J.K. is a Keck Young Investigator. K.M.M. is anA. P. Giannini Fellow supported by the A. P. Giannini Foundation.
Supplementary Materialswww.sciencemag.org/content/343/6169/428/suppl/DC1Materials and MethodsFigs. S1 to S3Table S1Data File S1References (32–34)
23 July 2013; accepted 4 December 2013Published online 19 December 2013;10.1126/science.1243640
Adaptation of Innate LymphoidCells to a Micronutrient DeficiencyPromotes Type 2 Barrier ImmunityS. P. Spencer,1,2,3* C. Wilhelm,1,2* Q. Yang,3 J. A. Hall,1,2† N. Bouladoux,1,2 A. Boyd,4T. B. Nutman,4 J. F. Urban Jr.,5 J. Wang,6 T. R. Ramalingam,1,7 A. Bhandoola,3T. A. Wynn,1,7 Y. Belkaid1,2‡
How the immune system adapts to malnutrition to sustain immunity at barrier surfaces, suchas the intestine, remains unclear. Vitamin A deficiency is one of the most common micronutrientdeficiencies and is associated with profound defects in adaptive immunity. Here, we found thattype 3 innate lymphoid cells (ILC3s) are severely diminished in vitamin A–deficient settings,which results in compromised immunity to acute bacterial infection. However, vitamin Adeprivation paradoxically resulted in dramatic expansion of interleukin-13 (IL-13)–producingILC2s and resistance to nematode infection in mice, which revealed that ILCs are primary sensorsof dietary stress. Further, these data indicate that, during malnutrition, a switch to innatetype 2 immunity may represent a powerful adaptation of the immune system to promote hostsurvival in the face of ongoing barrier challenges.
Maintenance of barrier defense is an ab-solute requirement for mammalian hosthealth and survival. Intestinal immu-
nity has evolved in the context of constitutiveexposure to microbial pressures. In this regard,the gastrointestinal (GI) tract is home to an es-timated 100 trillion commensals, and more than2 billion humans are chronically infected with par-asitic worms (1, 2). Over the course of evolution,maintenance of barrier immunity had to adaptto unstable nutritional availability in the face of
these ongoing challenges. Although dietary lig-ands can be sensed by immune cells (3–6), it isnot clear how the immune system integrates die-tary cues in order to tune immune responses onthe basis of the nutritional state of the host.
Malnutrition remains the primary cause ofimmunosuppression worldwide (7). Nonethe-less, despite profoundly impaired adaptive im-munity associated with malnutrition, most humanscan survive for extended periods of severe die-tary restriction. We postulated that compensatory
mechanisms might be in place to sustain de-fined branches of immunity and, in particular,responses associated with the protection of bar-rier tissues. Vitamin A deficiency is one of themost common nutrient deficiencies, affecting anestimated 250 million children in regions of theworld where chronic worm infections are prev-alent (8). This essential micronutrient supportsadaptive immunity through its metabolite, retinoicacid (RA), which is highly enriched in the gas-trointestinal tract (9, 10). Here, we examine thepossibility that innate lymphoid cells (ILCs)—potent mediators of barrier maintenance, tissuerepair, and host defense (11, 12)—may be primary
1Immunity at Barrier Sites Initiative, Laboratory of ParasiticDiseases, National Institute of Allergy and Infectious Disease,NIH, Bethesda 20892, USA. 2Mucosal Immunology Section,Laboratory of Parasitic Diseases, National Institute of Allergyand Infectious Disease, NIH, Bethesda, MD 20892, USA. 3De-partment of Pathology and Laboratory Medicine, Institutefor Immunology, Perelman School of Medicine, University ofPennsylvania, Philadelphia, PA 19104, USA. 4Helminth Im-munology Section, Laboratory of Parasitic Diseases, NationalInstitute of Allergy and Infectious Disease, NIH, Bethesda, MD20892, USA. 5U.S. Department of Agriculture, Beltsville HumanNutrition Research Center, Diet, Genomics, and ImmunologyLaboratory, Beltsville, MD 20705, USA. 6Program in MetabolicBiology, Nutritional Science and Toxicology, University of Cal-ifornia, Berkeley, CA 94720, USA. 7Immunopathogenesis Section,Laboratory of Parasitic Diseases, National Institute of Allergy andInfectious Disease, NIH, Bethesda, MD 20892, USA.
*These authors contributed equally to this work.†Present address: Molecular Pathogenesis Program, The KimmelCenter for Biology and Medicine of the Skirball Institute, NewYork University School of Medicine, New York, NY 10016, USA.‡Corresponding author. E-mail: [email protected]
24 JANUARY 2014 VOL 343 SCIENCE www.sciencemag.org432
REPORTS


























