Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1985 by The American Society of Biological Chemists, Inc.
Vol. 260, No. 18, Issue of August 25, pp. 10299-10307,1985 Printed in U.S.A.
Identification of Functional Murine Adenosine Deaminase cDNA Clones by Complementation in Escherichia coli*
(Received for publication, February 25, 1985)
Cho-Yau Yeung$, Diane E. IngoliaS, David B. Roth$, Charles Shoemaker$, Muayyad R. Al-Ubaidi$, Jong-Young Yen$, Chester Ching$, Chefin Bobonis$, Randal J. KaufmanQ, and Rodney E. KellernsSll From SThe Verna and Marrs M c h a n Department of Biochemistry, Baylor College of Medicine, Houston, Texas 77030 and the §Genetics Institute, Boston, Massachusetts 021 15
Total poly(A+) RNA derived from a mouse cell line with amplified adenosine deaminase genes was used as template to synthesize double-stranded cDNA. The cDNAs were inserted into the PstI site of the 8-lacta- mase gene in plasmid pBR322 following G-C tailing. After transformation into adenosine deaminase-defi- cient Escherichia coli hosts, recombinant plasmids con- taining functional murine adenosine deaminase cDNAs were identified by selecting for functional complemen- tation. Analysis of plasmids containing functional adenosine deaminase cDNA sequences strongly sug- gested that adenosine deaminase expression resulted mainly from &lactamase/adenosine deaminase fusion proteins even when the adenosine deaminase codons were out-of-frame with respect to the 8-lactamase gene codons upstream. The nucleotide sequence of a 1.65- kilobase pair cDNA insert in one of the functional recombinant clones was determined and found to con- tain a 1056-nucleotide open reading frame. When this 1056-nucleotide open reading frame was inserted into a mammalian expression vector and introduced into monkey kidney cells, a high level of authentic mouse adenosine deaminase was produced. Nucleic acid blot analysis using a full-length adenosine deaminase cDNA clone as probe revealed that the mouse adenosine de- aminase structural gene was at least 2 1 kilobase pairs in size and encoded three polyadenylated mRNAs. Analysis of the cDNA library from which the func- tional clones were isolated suggested that this approach of cloning functional mammalian adenosine deaminase cDNA clones by genetic complementation of enzyme- deficient bacteria could be accomplished even if the abundance of the adenosine deaminase mRNA se- quences were as low as -0.001%.
Several human genetic immune disorders are associated with a deficiency in any one of three catabolic enzymes which convert adenosine monophosphate to hypoxanthine (for re- view, see Ref. 1). Congenital adenosine deaminase (EC 3.5.4.4) deficiency, in particular, is closely correlated with a geneti-
* This work was supported by National Institutes of Health Grant AI20402, National Science Foundation Grant PCM8104533, Grant Q-893 from the Robert A. Welch Foundation and Grant 6-393 from the March of Dimes (to R. E. K.), and Grant RR05425 from the National Institutes of Health (to C.-Y. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
ll Recipient of Research Career Development Award CA00828 from the National Institutes of Health.
cally recessive and usually fatal form of severe combined immunodeficiency disease which is characterized by the ab- sence of functional T and B cells in affected individuals (2- 5). Pharmacological studies involving adenosine deaminase inhibitors (6-9) have provided additional support for the notion that adenosine deaminase is indispensable for the development and proper functioning of the mammalian im- mune system. This essential relationship between adenosine deaminase expression and immunodevelopment is reflected in the tissue distribution of the enzyme. Although adenosine deaminase is found in all tissues, adenosine deaminase levels in different tissues vary significantly and span 3 orders of magnitude (10-14). The highest adenosine deaminase activi- ties are found in cells of the T-lymphocyte lineage, where adenosine deaminase levels are subject to developmental reg- ulation (15-17) and reach peak values at the cortical thymo- cyte stage. Developmental regulation of adenosine deaminase also occurs in mammalian gastrointestinal tissues where aden- osine deaminase levels increase over 20-fold at the time of weaning (18). These complex and ill-understood functional and regulatory features of the adenosine deaminase gene have provided much of the current impetus for studies on the biochemistry and structure of both the gene and the enzyme.
A prerequisite to studies on adenosine deaminase gene structure and regulation is the isolation of full-length func- tional adenosine deaminase cDNA clones. Such clones will not only provide probes for the isolation and characterization of genomic adenosine deaminase sequences, but would also provide the starting material for constructing functional aden- osine deaminase minigenes for use in both gene replacement therapy and as an amplifiable genetic marker (see “Discus- sion’’). Although we and others (19-24) have reported the isolation of adenosine deaminase cDNA sequences, presum- ably full-length adenosine deaminase cDNA clones (as deter- mined by size) may not be functional due to the presence of unspliced introns (24) and both substitution and inversion cloning artifacts.’ To date, two human adenosine deaminase cDNA clones have been found to be functional (25, 26). We report here the direct isolation of five independent functional murine adenosine deaminase cDNA clones via genetic com- plementation of adenosine deaminase-deficient bacteria. Nu- cleotide sequence analysis of the five functional clones sug- gested that the majority of the adenosine deaminase activity was derived from P-lactamaseladenosine deaminase fusion proteins even when the inserted cDNAs were not in the same reading frame as that of the p-lactamase gene into which the cDNAs had been inserted. One of these clones contained an open reading frame of 1056 nucleotides, coding for a deduced
C.-Y. Yeung et al., manuscript in preparation.
10299

10300 Functional Murine Adenosine Deaminase cDNA Clones
protein of 352 amino acids. An adenosine deaminase minigene containing the entire open reading frame was constructed and shown by transient expression in monkey kidney cells to encode authentic mouse adenosine deaminase. Nucleic acid blot analysis using a full-length cDNA clone as probe revealed that the adenosine deaminase structural gene was at least 21 kb2 in size and encoded three polyadenylated mRNAs. Anal- ysis of the cDNA library from which the functional clones were isolated suggested that this approach of cloning func- tional mammalian adenosine deaminase cDNA clones by ge- netic complementation of enzyme-deficient bacteria may be a generally applicable and highly efficient approach for isolating cDNA clones of average-sized mRNA sequences of as low as -0.001% abundance.
EXPERIMENTAL PROCEDURES
Materials Alanosine was obtained from the Drug Synthesis and Chemistry
Branch, and 2'-deoxycoformycin was provided by the Natural Prod- ucts Branch, Division of Cancer Treatment, National Cancer Insti- tute. [3H]dCTP and [3H]dGTP were from Amersham Corp. ["PI dCTP was from ICN. Avian myeloblastosis virus reverse transcriptase and deoxynucleotide terminal transferase were from Life Sciences. The restriction enzymes PstI, EcoRI, BamHI, HindIII, BglI, BglII, PuuI, PuuII, XhoI, and calf liver tRNA were from Boehringer Mann- heim. S1 nuclease was from Miles Laboratories. Ribonuclease A, adenosine, uridine, inosine, adenine, phenazine methosulfate, nitro- blue tetrazolium, xanthine oxidase, nucleoside phosphorylase, DL- histidine, methionine, thiamine, and heparin were from Sigma. Oligo(dT)-cellulose was from P-L Biochemicals. The SP-64, SP-65 plasmid vectors were from Promega Biotech. The restriction enzymes EcoRV, KpnI, NcoI, RsaI, XbaI were from Biolabs. Klenow fragment was from Bethesda Research Laboratories. The Bradford protein assay kit was from Bio-Rad.
Cell Lines and Culture Conditions
B-l cells were derived from LMTK- Cl-1D mouse cells and selected for resistance to 1lAAU + 2'-deoxycoformycin as previously de- scribed (27). The number following the B-l designation indicates the level of resistance to 2'-deoxycoformycin in micromolar concentra- tion. Maintenance and harvest of these mouse cell lines have been previously described (27). COS-1 cells are monkey kidney CV-1- derived cells which express the SV40 large T antigen constitutively (28). The monkey cells were maintained in Dulbecco's modified Eagle medium supplemented with 15% horse serum (Grand Island Biolog- ical Company).
Bacterial Strains and Culture Conditions
Escherichia colistrain MC1061 (araD139, A[ara-leu]7697, AlacX74, galU, galK, hsr-, hsm+, strA [29]) is a K12-derived methylase-con- taining, restriction enzyme-deficient (M+R-) strain. E. coli strain S4200 (metB, strA, purB, Aadd-uid-man) is a K12-derived adenosine deaminase-deficient strain which is a methionine and adenine auxo- tronh. All bacterial strains were normally maintained in L broth (19). S4200 transformants were selected for adenosine deaminase expres- sion in MATHAG plates which contained 50 mg of methionine, 15 mg of adenine, 15 mg of tetracycline, 100 mg of DL-histidine, 30 mg of adenosine, 2 ml of glycerol, and with or without 50 pmol of 2'- deoxycoformycin/liter of salt buffer. The salt buffer contained 13.6 g of KHzP04, 2 g of (NH&SO,, 0.2 g of MgS04. 7Hz0, 0.01 g of CaCl2, 0.0005 g of FeS04. 7H20, and 1 mg of thiamine in 1 liter of distilled H20 (pH 7.2). Prior to plating the S4200 transformants onto MATHAG plates, the cells were washed twice with MATHAG solu- tion and collected by centrifugation using a microfuge.
Construction of cDNA Library and Isolation of Functional Adenosine Deaminase cDNA Clones
Cloning was performed according to the guidelines for recombinant DNA research from the National Institutes of Health. Construction
The abbreviations used are: kb, kilobase pairs; MATHAG, me- thionine, adenine, tetracycline, histidine, adenosine, glycerol.
of a cDNA library using poly(A+) RNA derived from B-1/50 cells was essentially as previously described (19) with the following modifica- tions to optimize synthesis of full-length cDNA (a) 30 min after the initiation of first-strand synthesis, the amount of reverse transcrip- tase used was increased by 50%, and the reaction was allowed to proceed for an additional 15 min; and (b) the second strands were synthesized by incubating the reaction mixture first with 100 units of avian myeloblastosis virus reverse transcriptase for 30 min at 46 "C, followed by adding 10 units of Klenow fragment and incubating at 37 "C for 30 min.
The double-stranded cDNAs were then treated with S1 nuclease, oligo(dC)-tailed and inserted into pBR322 plasmids which had been linearized with PstI and oligo(dG)-tailed (19). The annealed cDNA- containing plasmids were used to transform the methylase-contain- ing, restriction enzyme-deficient (M+R-) E. coli strain MC1061 using previously published procedures (29). The tetR transformants were pooled, resuspended in 25 ml of L broth, and incubated with shaking for 1 h a t 37 "C. Six ml of resuspended transformants were withdrawn, and the cells were collected by centrifugation in a microfuge. The recombinant plasmids were isolated from mini-lysates (30) of the pelleted cells. These methylated recombinant plasmids were then used to transfect the adenosine deaminase-deficient E. coli strain S4200 using the rubidium chloride (29) procedure. S4200 transform- ants were plated onto MATHAG plates or control Tet (15 rg/ml) plates. After 48 h, tentative positive clones were replica-plated onto MATHAG plates in the presence or absence of 50 p~ 2'-deoxycofor- mycin. Colonies that survived MATHAG selection but were killed by MATHAG + 2'-deoxycoformycin were grown overnight in L broth + tet and analyzed for adenosine deaminase expression.
Screening of the Recombinant MC1061 cDNA Library by Colony Hybridization
Approximately 5000 independent colonies of the primary MC1061 recombinant cDNA library were also screened according to previously published procedures using nick-translated 32P-labeled cDNA insert of pADA5-29 as probe (19). The prehybridization, hybridization, and wash conditions were as previously published (19). Positive colonies were identified by autoradiography.
Enzyme Assays Zymogram Analysis-Recombinant S4200 transformants or con-
trol bacterial cells were pelleted after growing overnight in 30 ml of L broth or L broth + tet. The cells were collected by centrifugation at 3000 X g,,, resuspended in 150 ~1 of homogenizing buffer, and sonicated (27). The sonicates were clarified by centrifugation using a microfuge for 5 min at 4 "C. The supernatants were fractionated by starch gel electrophoresis and histochemically stained for adenosine deaminase or purine nucleoside phosphorylase activities (31).
Adenosine Deaminase Spectrophotometric Assays-Alternatively, the soluble proteins of bacterial and cell extracts (27) were analyzed spectrophotometrically (32), and the protein concentration of the samples was determined using the Bradford protein assay (33).
DNA Sequence Analysis of Cloned cDNA Inserts The entire cDNA nucleotide sequence of pADA5-29 was deter-
mined by subcloning the cDNA into M13 vectors and sequencing using the Sanger dideoxy terminator method (34).
The 5' PstI cDNA fragments of all five functional clones were subcloned with intact PstI sites into the PstI site of the cloning casette in vector pSP64. Following either HindIII or BamHI restric- tion digestion, the linearized plasmids were 32P-labeled either on the 5' ends by kinase or on the 3' ends by Klenow fragment, and both strands were sequenced by the method of Maxam and Gilbert (35).
Expression of Mouse Adenosine Deaminase in Monkey Kidney Cells Construction of the Mouse Adenosine Deaminase Mammalian
Expression Vector p9ADA5-29-A cDNA fragment containing the 1056-nucleotide open reading frame in pADA5-29 was excised follow- ing NcoI and EcoRV digestion. The ends were filled in using Klenow fragment and blunt-end ligated into the EcoRI site of vector p91023 (36).
Transfection and Expression of p9ADA5-29 into COS-1 Cells- p9ADA5-29 was transfected into monkey kidney COS-1 cells using the DEAE-dextran procedure (37). Fifty-two hours after transfection, the COS-1 cells were harvested and sonicated. Clarified cell lysates were fractionated by starch gel electrophoresis and analyzed by his-

Functional Murine Adenosine Deaminase cDNA Clones 10301
tochemical staining for adenosine deaminase and purine nucleoside phosphorylase activities.
Nucleic Acid Blot Analysis
Poly(A+) RNA (Northern) Blot Analysis-Poly(A+) RNA derived from B-1/3.2 cells was isolated according to previously published methods (19). The RNA was fractionated in 1.5% agarose gels in the presence of formaldehyde (38), blotted onto nitrocellulose filters, and probed with nick-translated (39) 32P-labeled pADA5-29 plasmid DNA. Prehybridization and hybridization were performed under pub- lished conditions (19). The most stringent wash condition used was 1 h using 0.1% SSC, 0.1% sodium dodecyl sulfate buffers at 68 "C (19).
DNA (Southern) Blot Analysis-Total genomic DNA derived from B-1/50 cells was isolated under previously published conditions (19). The DNA was digested with the various restriction enzymes under conditions recommended by the vendors. The digested DNA was fractionated in 0.8% agarose gels and blotted onto nitrocellulose filters according to the method of Southern (40). The filters were probed with nick-translated 3ZP-labeled plasmid DNA. Prehybridi- zation, hybridization, and wash conditions were identical to those used for RNA blot hybridizations shown above.
RESULTS
Identification of Functional Adenosine Deaminase cDNA Clones by Genetic Complementation-The feasibility of isolat- ing cDNA clones encoding functional mammalian enzymes via complementation in E. coli has previously been demon- strated on two occasions (29, 41). The general strategy in- volves the insertion of synthetic double-stranded cDNAs into the PstI site of the P-lactamase gene located on the bacterial plasmid pBR322 (see Fig. 20) . Following introduction of the recombinant plasmids into E. coli hosts, the inserted cDNA sequence would be transcribed as part of the p-lactamase transcription unit, which would in turn be translated into protein(s). Plasmid clones containing full-length cDNA se- quences translatable into the functional enzymes of interest would be identified by genetic selection in enzyme-deficient bacterial hosts.
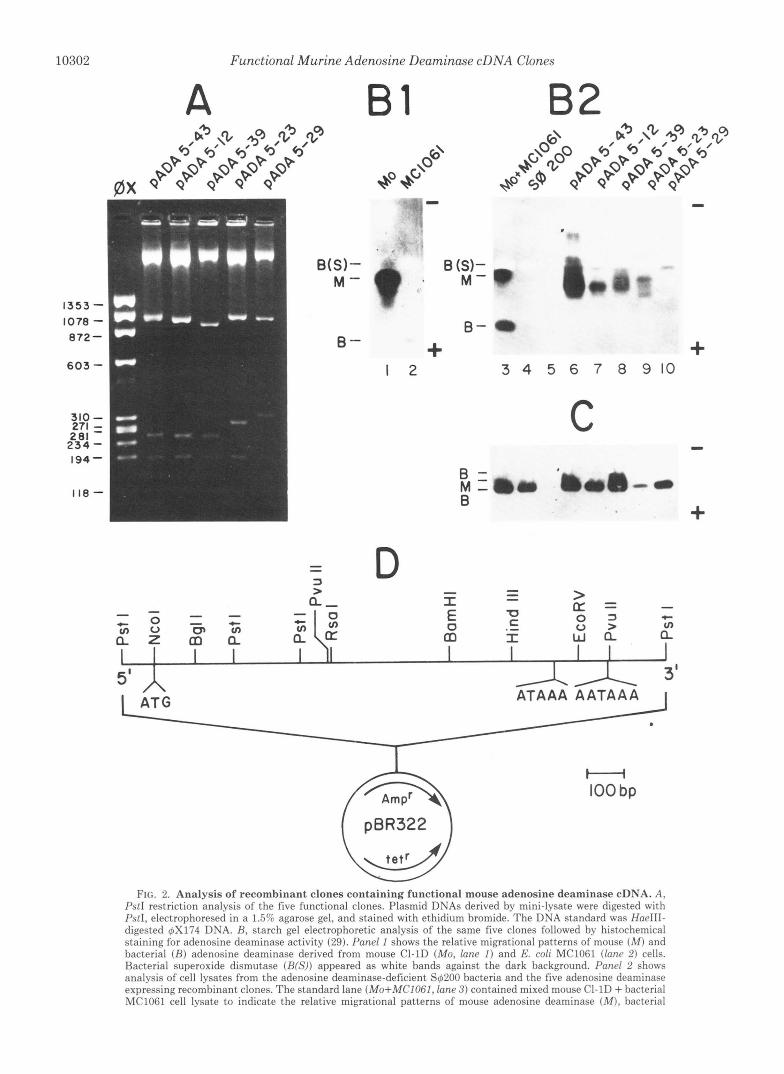
Our cloning of functional murine adenosine deaminase cDNAs was based on the same general strategy with a minor variation involving the use of an adenosine deaminase-specific inhibitor. Double-stranded cDNAs were synthesized essen- tially as previously reported using as template 105 pg of total poly(A+) RNA derived from mouse cells containing amplified adenosine deaminase genes (19). Following the addition of -15 deoxycytidine monophosphate residues at the 3' ends, 0.7% of the cDNAs synthesized were inserted into PstI- digested and dG-tailed pBR322 plasmids. The recombinant plasmids were then methylated following transformation into a methylase-containing, restriction enzyme-deficient (M'R-) E. coli strain, MC1061. Approximately 30,000 independent colonies were obtained. These methylated recombinant plas- mids were pooled and used to transfect the adenosine deami- nase-deficient E. coli strain S4200 (metB, strA, purB, Aadd- uid-man) (42). The transformants were then plated onto MATHAG selection plates (42) which selected specifically for tetR, ADA' transformants (Fig. 1). MATHAG-resistant re- combinant colonies were picked after 42 h and replica-plated onto MATHAG plates in the presence or absence of 50 PM 2'-deoxycoformycin. Colonies found to be resistant to MATHAG selection alone but sensitive to MATHAG + 2'- deoxycoformycin selection were analyzed by adenosine de- aminase and purine nucleoside phosphorylase zymogram analysis (Fig. 2B), and the respective recombinant plasmids were analyzed by PstI restriction analysis (Fig. 2 A ) . The results showed that of 17 clones identified in this way, at least 5 were independent. The restriction maps of all 17 ADA+ clones were identical except for the 5' and 3' ends. A typical
PURINE METABOLISM IN E.COLI MUTANT Sa200
De Novo
x f
+ 11 11
Gua Xan ' Hx Ade '
-X- Pathways blocked by purB mutation
++ Pathway blocked by ADA deletion
Pathway inhibited by histidine FIG. 1. Purine metabolic pathways in the E. coli mutant
S6200. S4200 (met B, strA, pur B, Aadd-uid-man) is a methionine and adenine requiring auxotroph. The pur B mutation affects the adenylosuccinase enzyme which is involved in both the de novo synthesis of 5-aminoimidazole-4-carboxamide ribonucleotide (AZ- CAR) and the conversion of inosine monophosphate (IMP) to aden- osine monophosphate (AMP). The Aadd refers to an adenosine de- aminase (ADA) deletion mutation. By selecting S4200 transformants for growth in MATHAG medium, only cells that had acquired both the tetracyclineR marker gene and a functional adenosine deaminase gene on a plasmid would survive. With histidine blocking the ATP to AICAR pathway, cells that survive must be able to utilize adenosine via adenosine deaminase to supply its guanine nucleotide needs. The adenine and glycerol in the medium supply the adenine nucleotide and carbon needs, respectively, of the resistant cells.
map is shown in Fig. 2 0 . The transformation frequencies are summarized in Table I.
Clone pADA5-29 Contained a 1056-Nucleotide Open Read- ing Frame-Recombinant plasmid pADA5-29 had the longest 5' and 3' PstI restriction fragments, and the cDNA insert was sequenced in its entirety (Fig. 3A). The nucleotide sequence contained an open reading frame comprising 1056 nucleotides which encoded a deduced 39,900-dalton protein of 352 amino acids. The nucleotide and deduced amino acid sequences of this open reading frame showed 80.8 and 83.1% homology, respectively, with two published human adenosine deaminase cDNA sequences (43,44) that were reported to be intron-free. No significant homology to the presumed intron sequence found in another published human clone (24) was located in this open reading frame. The 5' and 3' noncoding regions of pADA5-29 showed 66.7 and 64.9% homology with the corre- sponding nucleotide sequences in the published human clones if the homology was maximized by the introduction of nine gaps of less than 13 nucleotides/gap into the mouse and human sequences.
The ATG codon beginning at nucleotide position +1 (Fig. 3A) was presumed to be the start codon because it is in the preferred sequence context of ANNAUGG (N = nucleotide) which is characteristic of eukaryotic start codons (45). In addition, the ACAC sequence located at -14 to -11 showed reasonable homology with the consensus CACACA sequence found about 10 nucleotides upstream of the AUG initiator codon in yeast mRNAs (46, 47). The consensus polyadenyla- tion signal AATAAA and a partial consensus ATAAA se- quence were found 17 and 179 nucleotides, respectively, up- stream of the poly(A) addition site at +1310.
10302
1353 - I078 - 872 - 603 -
310 - 2 81 271 =
234 - 194-
118 -
A Functional Murine Adenosine Deaminase cDNA Clones
B1 B2
. * I
8- m 9- + +
1 2 3 4 5 6 7 8 9 1 0
C
> a,
p) pBR322 m 100 bp
FIG. 2. Analysis of recombinant clones containing functional mouse adenosine deaminase cDNA. A, PstI restriction analysis of the five functional clones. Plasmid DNAs derived by mini-lysate were digested with M I , electrophoresed in a 1 .55 agarose gel, and stained with ethidium bromide. The DNA standard was HaeIII- digested 6x174 DNA. R, starch gel electrophoretic analysis of the same five clones followed by histochemical staining for adenosine deaminase activity (29). Panel I shows the relative migrational patterns of mouse ( M ) and bacterial ( R ) adenosine deaminase derived from mouse CI-1D (Mo, lane I ) and E. coli MC1061 (Inncl 2 ) cells. Bacterial superoxide dismutase (RW)) appeared as white bands against the dark background. Pand 2 shows analysis of cell lysates from the adenosine deaminase-deficient S6200 bacteria and the five adenosine deaminase expressing recombinant clones. The standard lane (Mo+MCIOGI, lane 3) contained mixed mouse CI-1D + bacterial MClOFl cell lysate to indicate the relative migrational patterns of mouse adenosine deaminase ( M ) , bacterial

Functional Murine Adenosine Deaminase cDNA Clones 10303
TABLE I Selection of functional adenosine deaminase cDNA clones by
expression in adenosine deaminase-deficient E. coli
Selection step Number of
colonies
1st transformation (into MC1061 M+R- strain) 30,000 2nd transformation (into S1#~200) 7,000 MATHAG-resistant transformants 64 Total adenosine deaminase+ transformants 17 Independent adenosine deaminase+ transformants 5
Expression of Mouse Adenosine Deaminase cDNA in Mon- key Kidney COS-1 Cells-To demonstrate unambiguously that the 1056-nucleotide open reading frame in pADA5-29 encoded the entire mouse adenosine deaminase protein, the Ncoll EcoRV cDNA fragment of pADA5-29 (see restriction map in Fig. 2 0 ) was placed into a mammalian expression vector as shown in Fig. 4A. This vector was used to transfect monkey kidney COS-1 cells. Zymogram analysis of lysates from the transfected cells (shown in Fig. 4, B and C) demonstrated that authentic mouse adenosine deaminase was produced at a very high level (XOO-fold above background) in the trans- fected cells. This result proved that the open reading frame of pADA5-29 encoded the entire mouse adenosine deaminase coding sequence and that the ATG codon at position +1 was indeed the start codon for mouse adenosine deaminase mRNA translation (45).
The Mouse Adenosine Deaminase Gene Is at Least 21 Kb in Size and Encodes Three Polyadenylated mRNAs-The full- length plasmid pADA5-29 was used as a hybridization probe to identify adenosine deaminase mRNAs following electro- phoretic fractionation on agarose gels and blot transfer to nitrocellulose filters. Northern analysis (Fig. 5A) showed that three mRNAs of 5.2, 1.7, and 1.5 kb in length hybridized to the full-length clone.
Genomic DNA (Southern) blot analysis was performed to assess the size of the adenosine deaminase structural gene. The results of these experiments show that the full-length cDNA probe hybridizes to a number of EcoRI, BamHT, PstI, and HindIII genomic fragments (Fig. 5C). The orientation of these fragments within the adenosine deaminase structural gene was determined by hybridization with the three PstI fragments of the cDNA. The results (Fig. 5B) indicated, for example, that of the three EcoRI fragments (11, 8.5, and 1.5 kb) identified by the full-length cDNA (Fig. 5C), only the largest hybridized to the 3’ PstI cDNA fragment (Fig. 5B). When EcoRI-digested genomic DNA was probed with the 187-base pair internal PstI fragment (see restriction map in Fig. 2 0 ) of pADA5-29, both the 11- and 8.5-kb fragments showed hybridization (data not shown). This established the order of the EcoRI restriction fragments containing the aden- osine deaminase gene from 5’ to 3’ to be 1.5, 8.5, and 11 kb. Southern analysis of double-digested (EcoRI + BamHI, EcoRI + HindIII, BamHI + HindIII) genomic sequences probed with the 3’ PstI fragment of pADA5-29 generated multiple bands, the sums of which were approximately 10 kb (data not shown). This suggested that the genomic sequence containing the 3‘
two-thirds of the mRNA was at least 10 kb long. Taken as a whole, our results suggested that the minimum size of the adenosine deaminase gene was -21 kb (1.5 + 8.5 + 11 kb). This observation was in good agreement with Southern anal- ysis of HindIII- and BamHI-digested genomic sequences (Fig. 5C), in which the sums of the hybridizing bands were >25 kb. The sum of PstI-digested adenosine deaminase gene frag- ments shown in Fig. 5C was less than 21 kb and probably reflected the presence of multiple PstI restriction sites within introns. The failure of our exon-containing probes to detect these intron fragments would then lead to an underestimation of the gene size based on PstI restriction analysis.
5‘ Sequence Analysis of the Functional Adenosine Deami- nase cDNA Clones-The underlying assumption in our ap- proach to identifying functional adenosine deaminase cDNA clones was that the inserted cDNA sequences would be tran- scribed as part of the P-lactamase transcription unit. The fusion gene transcript could be translated in one of the following ways to yield an enzymatically active adenosine deaminase protein: 1) by initiating translation at the P-lac- tamase start codon to generate a peptide consisting of the amino-terminal segment of the P-lactamase fused to a func- tional (and hence “in-frame” translated) adenosine deaminase peptide, or 2) by initiating translation at the adenosine de- aminase start codon to generate authentic mouse adenosine deaminase. In the latter case, 5’ upstream sequences (includ- ing the poly(dG)tail) could serve as ribosomal binding se- quences as suggested by Chang et al. (48). Starch gel electro- phoretic analysis of five independent functional clones re- vealed that the pattern of adenosine deaminase expression was complex and different for each clone. With the exception of one clone (pADA5-29), authentic mouse adenosine deami- nase accounted for very little, if any, of the adenosine deam- inase produced.
In order to gain insight into the mechanisms responsible for the cDNA-encoded adenosine deaminase expression, the 5’ nucleotide sequences of the various functional clones were determined. The results (Fig. 3B) showed that pADA5-29 was the only clone that retained the authentic translation initia- tion codon ATG. This feature was consistent with the fact that the majority of the adenosine deaminase activity pro- duced by this clone appeared to co-migrate with authentic mouse adenosine deaminase (Fig. 2B). Clone pADA5-23 was the only one among the five clones that contained an adeno- sine deaminase coding sequence that was in-frame with the upstream p-lactamase coding sequence (Fig. 3B). Because of a cloning artifact, the sequence between -39 and +4 of pADA5-23 represented an inverted sequence compared to authentic adenosine deaminase mRNA.l This inversion re- sulted in the removal of the first four nucleotides of the adenosine deaminase-encoding open reading frame, including the ATG start codon. Thus, it was not surprising that no authentic mouse adenosine deaminase was produced by this clone (Fig. 2B). Clones pADA5-12, pADA5-39, and pADA5- 43 were also missing the ATG start codon of the mouse adenosine deaminase sequence (Fig. 3B) and also produced little if any authentic mouse adenosine deaminase (Fig. 2B).
adenosine deaminase (B), and bacterial superoxide dismutase (B(S)). The pADA5-23 lysate was diluted 20-fold relative to the other samples prior to loading. None of the clones showed bacterial adenosine deaminase expression. C , purine nucleoside phosphorylase stain of an identical slice of gel as the one shown in B, Panel 2. This served to provide an indication of how much lysate protein was loaded per lane. D, restriction map of functional mouse adenosine deaminase clones showing the restriction sites of PstI, NcoI, PuuII, BamHI, BglII, RsaI, HindIII, and EcoRV. The enzymes X b d , EcoRI, AuaI, HpaI, KpnI, PuuI, XhoI, and BglII did not cut within the insert. bp, base pair.

10304 Functional Murine Adenosine Deaminase cDNA Clones
A pADA5-29 -7 1 282 635 989 1342
-70 CCACCCTCCC ACCGAGCCM CGCACACCCA GAGAGCTTCG CCCCACAGM CCGGGMCAC GCTCCCMCC -1
1 An: CCC CAC ACA CCC GCA TTC M C M A CCC A M C I A GAG T I A CAC CTC CAC CTG GAT CCA CCC ATC M C CCA CM ACC ATC TTA TAC TTT 90 Met Ala Gln Thr Pro Ala Phe Asn Lys Pro Lys Val Clu Leu His Val His Leu Asp Gly Ala Ile Lys Pro Clu Thr Ile Leu Tyr Phe
91 CCC M C M C AGA CCC ATC CCC CTC CCC CCA GAT ACA G I G GAG GAG CTC CGC M C ATT ATC GGC ATG GAC MC CCC CTC TCC CTC CCA CCC 180 Cly Lys Lys Arg Cly Ile Ala Leu Pro Ala Asp Thr Val Clu Glu Leu A r g Asn Ile Ile Cly k c Asp Lys Pro Leu Ser Leu Pro Gly
181 TTC CTC CCC MC TTT GAC TAC TAC ATG CCT CTG ATT CCG GGC TGC ACA GAG CCC ATC M C ACC ATC CCC TAC CAC "I CTG GAG ATG M G 270 Phe Leu Ala Lys Phe Asp Tyr Tyr Met Pro Val Ile Ala Cly Cy. Arg Glu Ala Ile Lys Arg Ilc Ala Tyr Clu Phe Val Clu Met Lys
271 CCA MG GAG GCC CTC CTC TAT CTC CM GTG CCC TAT AGC CCA CAC CTC CTC CCC M T TCC M C GTC GAC CCA ATC CCC TCC M C CAG ACT 380 Ala Lys Clu Cly Val Val Tyr Val Clu Val Arg Tyr Ser Pro His Leu Leu Ala Asn Ser Lys Val Asp Pro k t Pro Trp A m Gln Thr
361 C M GGG CAC CTC ACC CCT GAT GAC CTT CTC GAT CTT GTG M C CAC CCC CTC CAC CAC CGA CAG CM CCA TTI CCC ATC M C CTC CCC TCC 450 Clu Cly Asp Val Thr Pro Asp Asp Val Val Asp Leu Val Asn Gln Cly Leu Cln Clu Cly Clu Cln Ala Phe Cly Ile Lys Val Arg Ser
451 ATT CTC TCC TCC ATG CCC CAC CAC CCC ACC TCC TCC CTT GAG GTC T I C GAG CTG TCT MC M C TAC M T CAC M C ACC GTC CTC GCT ATC 540 11e Leu Cy8 Cy. Met Arg His Gln Pro Ser Trp Ser Leu Glu Val Leu Glu Leu Cys Lys Lys Tyr Asn Gln Lye Thr Val Val Ala k t
541 CAC TTC GCT GCG GAT GAG ACC ATT CM CCA ACT AGC CTC TTC CCA GGC CAC GTC CM CCC TAT GAG CCC CCA CTA MG M T CCC A T T CAT 630 Asp Leu Ala Cly Asp Clu Thr Ile Glu Gly Ser Ser Leu Phe Pro Gly His Val Clu Ala Tyr Clu Cly Ala Val Lys A m Gly I le His
831 CGG ACC CTC CAC CCT GGC GAG GTC CCC TCT CCT GAG CTT CTC CGT GAG CCT GTC GAC ATC CTC M C ACA GAG ACC CTG GGA CAT CGT TAT 720 Arg Thr Val His Ala Cly Clu Val Cly Ser Pro Clu Val Val Arg Clu Ala Val Asp Ile Leu Lys Thr Clu Arg Val Cly His Cly Tyr
721 CAC ACC ATC GAC GAT CM CCT CTC TAC M C ACA CTA CTC AM G M M C ATG CAC TTI GAG GTC TCC CCC TCC TCC ACC TAC CTC ACA CCC 810 Hi. T k Ile Glu Aap Glu Ala Leu Tyr Asn Arg Leu Leu Lys Glu A m k t His Phe Glu Val Cy. Pro Trp Ser Ser Tyr Leu Thr Cly
811 CCC TCC CAT CCC AM ACC ACC CAT CCC CTT CTT CCC TTC M C M T GAT MC CCC M C TAC TCA CTC M C ACA CAC CAC CCC CTC ATC TTC 900 Ala Trp Asp Pro Lys Thr Thr His Ala Val Val Arg Phe Lys Asn Asp Lys Ala Asn Tyr Ser Leu &n 'Ihr Asp Asp Pro Leu Ile Phe
901 MC TCC ACC CTA GAC ACT GAC TAC CAC ATC ACC M G AM CAC ATG CCC TTC ACT GAG GAG CAG TTC MG CGA CTC M C ATC M C CCA CCC 890 Lys Ser Thr Leu Asp Thr Asp Tyr Cln Met Thr Lye Lya Aep k t Cly Phe Thr Clu Clu Clu Phe Lys Arg Leu A m Ile A m Ala Ala
S@l M G TCA ACC TTC CTC CCA GAG CM GAG M C M C CM CTT CTC C M CGG CTC TAC M A CM TAC CM CCACCACACA CTGACGdCC Lys Ser Ser Phe ~ e u pro Glu Glu Clu Lys ~ y s Clu tu Leu clu Arg Leu Tyr Arg clu Qr Gln 1080
1079

Functional Murine Adenosine Deaminase cDNA Clones
A VA SV40 Sss ADA DHFR 9/40 pBR322
AdMLP
p9ADA5-29 W H “ .5/ c I,
1 2 3 4 5 6 7
PNP
MY - FIG. 4. Expression of mouse adenosine deaminase in mon-
key kidney COS-1 cells following transfection of the expres- sion vector p9ADA5-29. A , p9ADA5-29 contains (from left to right) the adenovirus-VA gene ( V A ) ; the SV40 origin of replication including the 72-base pair enhancer; the adenovirus major later promoter (AdMLP) including the adenovirus tripartite leader and a 5’ splice site; a 3’ splice acceptor site (3’s~); the NcoI/EcoRV cDNA fragment of pADA5-29 which contains the 1056-nucleotide open reading frame (ADA); a dihydrofolate reductase cDNA insert (DHFR) remaining from a previous construct; the SV40 early polyadenylation site (SV40), and the pBR322 sequences needed for propagation in E. coli. COS-1 cells were transfected with p9ADA5-29 and analyzed by starch gel electrophoresis and histochemical staining for adenosine deaminase activity ( B ) and purine nucleoside phosphorylase activity (C). Mouse-specific enzymes are designated Ms and monkey-specific enzymes are designated My. The samples analyzed were: lane 1 , mouse C1-1D cells; lunes 4 and 5, mock-transfected COS-1 cells; lunes 6 and 7, COS-1 cells transfected with p9ADA5-29; and lanes 2 and 3, 100- fold dilutions of the samples in lanes 6 and 7. The transfected cells produced authentic mouse adenosine deaminase at a level that was at least 100-fold that of C1-1D cells, although only approximately 5% of the COS-1 cells were estimated to be transfected.
However, despite the fact that the cDNA coding sequences were out-of-frame with respect to the P-lactamase sequence upstream, all three clones yielded functional adenosine de- aminase peptides. These functional adenosine deaminase pep- tides were unlikely to have been generated by initiation of in frame translation within the cDNA inserts because the first permissable in frame bacterial start codon (an ATT in this case) (45) does not occur until the 49th codon at position +145 (see Fig. 3A). We believe that initiation at this site would be unlikely to yield a functional adenosine deaminase peptide for the following reasons. 1) The first 29 amino acids at the amino terminus showed excellent homology (93.1%) between mouse and human adenosine deaminase compared to the overall amino acid homology of 83.1% for the entire coding region, suggesting that the first 29 amino acids on mouse adenosine deaminase may be essential for enzymatic activity. 2) Although we have identified several clones in the correct orientation but lacking from 50 to 120 nucleotides on the 5’ end of the coding region, none appeared to produce functional adenosine deaminase and the recombinant bacteria were sensitive to MATHAG selection medium. Based on Weiss’ (49) observation that homopolynucleotide sequences are particularly prone to translation frame-shifting in bacte-
A B 81-3 2 ECORl BAMHI PSTl
r“
I
I - 5 2
1yi -17 -1 5
9 7 6.6
4.3
2 2 2 1
0 53
10305
C ECORl BAMHI PSTl HINDIII
- 9 7
- 6 6
2 2
2 1
-0.53
FIG. 5. Blot analysis of poly(A+) RNA and restriction en- zyme-digested genomic DNA derived from mouse cells which overproduce adenosine deaminase. A, Poly(A+) RNA derived from B-1/3.2 cells (27) was fractionated in a 1.5% agarose gel in the presence of formaldehyde (46), blotted onto nitrocellulose filter, and probed with nick-translated 32P-labeled pADA5-29. Three RNA spe- cies of 1.5,1.7, and 5.2 kb in size showed hybridization under stringent hybridization and wash conditions. B, genomic DNA derived from mouse B-1/50 cells was fractionated in a 0.8% agarose gel after digestion with EcoRI, BumHI, or PstI restriction enzymes, respec- tively, blotted onto a nitrocellulose filter, and probed with nick- translated 32P-labeled SpADA5-29/3. SpADA5-29/3 consists of the 3’ 1.1-kb PstI restriction fragment (see restriction map in Fig. 2 0 ) of pADA5-29 inserted into the PstI site of pSP64. One EcoRI restriction fragment (11 kb), two BumHI restriction fragments (8.5 and 4.6 kb), and two PstI fragments (4.6 and 0.53 kb) showed hybridization with the probe under the same stringent hybridization and wash conditions as in A . C, B-1/50-derived genomic DNA was digested with EcoRI, BumHI, PstI, and HindIII restriction enzymes, respectively, fraction- ated in 0.8% agarose, blotted onto a nitrocellulose filter, and probed with nick-translated 32P-labeled pADA5-29. Under the same hybrid- ization and wash conditions as that used in A , three EcoRI restriction fragments (11, 8.5, and 1.5 kb), five BumHI restriction fragments (16.5, 8.5, 5.3, 4.6, and 1 kb), six PstI restriction fragments (4.6, 3.2, 1.4, 1.3, 0.52, and 0.47 kb), and five HindIII restriction fragments (10.5.8.5, 2.5, 1.8, and 1.14 kb) hybridized with the probe.
ria, it is likely that the 5’ poly(dG) “tail” of the cDNA inserts in these three clones provided frame-sliding during transla- tion to allow occasional in-frame translation of the adenosine deaminase coding sequence. Active adenosine deaminase could thus be generated in these clones despite their lack of an authentic ATG start codon in the cDNA and the out-of- frame location of the adenosine deaminase coding region with respect to the upstream p-lactamase sequence. For clone pADA5-23, the in-frame location of the adenosine deaminase sequence with respect to the p-lactamase sequence maximizes the correct translation of the adenosine deaminase mRNA sequence to yield functional P-lactamaseladenosine deami- nase fusion proteins and that may be the sole reason for the 4-fold higher adenosine deaminase activity found in that clone (Table 11). Based on these considerations, we believe that the insertion of the poly(dG) tract 5’ of the cDNA sequence may represent a very advantageous feature of this approach for isolating functional cDNAs by genetic selection in bacteria.
General Applicability of the Bacterial Complementation Cloning Approach-In an attempt to assess the general appli- cability of this approach for isolating functional cDNA clones, we have screened 5,000 of the initial 30,000 independent colonies in the library using the entire cDNA insert of pADA5-

10306 Functional Murine Adenosine Deaminase cDNA Clones
TABLE I1 Comparison of adenosine deaminase expression in the five functional
recombinant clones C1-1D is a mouse LMTK--derived cell line which is used as a
positive control in the assays. Cell strain Specific activity
mmol/min/gprotein relative to C1-ID Cl-1D 0.025 1.0 pADA5-12 0.027 1.1 pADA5-23 0.112 4.5 pADA5-29 0.028 1.1 pADA5-39 0.027 1.1 pADA5-43 0.035 1.4
29 as probe to determine the frequency of both adenosine deaminase clones and possible full-length adenosine deami- nase clones. Seventy-seven transformants cross-hybridized with the cDNA insert. Seventeen (or 22%) of these 77 clones containing adenosine deaminase cDNA sequences were esti- mated to be authentic full-length adenosine deaminase clones based on PstI restriction analysis of the recombinant plas- mids. Eight of the full-length clones were in the same orien- tation as the P-lactamase gene, and nine of them were in the opposite orientation. These observations suggested that the adenosine deaminase mRNA abundance in our starting poly(A+) RNA population was -1.5%. Our cDNA synthesis method yielded 22% full-length clones based on a 1.7-kb mRNA.
DISCUSSION
Based on the work reported here, we believe that the bacterial complementation approach to identifying functional mammalian cDNA clones is feasible for average-sized mRNA of fairly low abundance, provided that an appropriate bacte- rial mutant, a workable selection protocol, and a suitable inhibitor is available. Our cDNA library screening results suggested that the adenosine deaminase mRNA abundance in our starting poly(A+) RNA population was -1.5%. From 7000 recombinant clones analyzed, at least five independent func- tional adenosine deaminase cDNA clones were isolated. Thus, if one started with a 3 x lo6 colony library (which can be easily achieved by starting with 100 pg of poly(A+) RNA), it should be feasible to isolate one functional clone of an average length (-2 kb) mRNA provided that its abundance was higher than 1.5% X 7000/(3 X lo6) X 1/5 or -0.001%. Given the number of different bacterial mutants and complementary selection systems that have been published, this approach to cloning functional mammalian cDNAs-encoding enzymes should have very general applications.
We have noticed that the selection protocol used to isolate adenosine deaminase expressing SqY200 transformants was not very stringent. The inhibition by histidine of the pathway converting ATP to 5-Aminoimidazole-4-carboxamide ribo- nucleotide (see Fig. 1) is lost at a frequency of > 5 x presumably due to feedback-resistant mutants. This, coupled with the observation that numerous satellite colonies which did not express adenosine deaminase could survive the MATHAG selection by cross-feeding from adjacent adenosine deaminase producing colonies, presented a serious problem in terms of the number of clones that could be conveniently handled by this complementation approach. However, by using the MATHAG selection system in conjunction with an inhibitor specific to the target enzyme, we were able to rapidly screen and distinguish adenosine deaminase-producing colo- nies from background mutant transformants which were adenosine deaminase deficient. Thus, even for point mutation
bacterial mutants, which may revert in the W 5 to lo" range, the complementation approach should still be feasible if an inhibitor specific for the target enzyme is available.
By using this approach, we were able to isolate a full-length functional mouse adenosine deaminase cDNA clone. This functional clone was placed in a mammalian expression vector and shown to contain the entire coding sequence with the appropriate start and stop codons. By using this full-length clone as probe, we found the mouse adenosine deaminase gene to be 221 kb in size and to encode at least three polyadeny- lated mRNAs of 1.5, 1.7, and 5.2 kb in size. Of the three adenosine deaminase mRNAs detected, the 1.7-kb mRNA was clearly the most abundant message and may be represented in close to its entirety by the 1.65-kb cDNA insert sequence in clone pADA5-29. The 1.5-kb mRNA may have been gen- erated by utilization of the ATAAA partial consensus penta- nucleotide (Fig. 4A) as a polyadenylation signal. The 162-base pair distance between that sequence and the consensus AA- TAAA sequence downstream was consistent with the size difference between the 1.5 and 1.7-kb mRNAs. Further blot analysis using the sequence between these two consensus signals as probe should clearly resolve that issue. The com- position of the 5.2-kb mRNA remains, at present, unresolved.
Sequence analysis of five independent functional clones revealed that adenosine deaminase expression in these recom- binant bacteria was generated mainly through the production of enzymatically active fi-lactarnase/adenosine deaminase fu- sion proteins. In-frame fusion of p-lactamase and adenosine deaminase sequences appeared to enhance adenosine deami- nase expression 4-fold. However, even when the adenosine deaminase coding sequence was out-of-frame with the /3- lactamase sequence, functional fusion proteins were still pro- duced in sufficient quantity to enable the transformants to grow under selection for adenosine deaminase expression.
The production of a eukaryotic adenosine deaminase expression vector as a dominant selectable and amplifiable marker gene has important implications. Our previous work (19) has demonstrated the feasibility of selecting for cells with a 3000-fold amplification in both the adenosine deaminase gene and protein. We have since been able to isolate cells with an 11,000-fold increase in adenosine deaminase activity relative to parental cells? This enormous amplification of both the gene and protein apparently had no detrimental effect on the cells and is achieved through the use of the very specific adenosine deaminase selection protocol, llAAU, in conjunction with a potent adenosine deaminase inhibitor, 2'- deoxycoformycin. By covalently linking an adenosine deami- nase expression vector to any gene of choice, that gene could be co-amplified with the adenosine deaminase expression vector using the 1lAAU + 2'-deoxycoformycin selection scheme we developed (27). This approach should allow the production of large quantities of specific nucleic acids and proteins in cultured mammalian cells and may be of impor- tance for both research and commercial purposes.
Acknowledgments-Alanosine was kindly provided by the Drug Synthesis Branch, Division of Cancer Treatment, National Cancer Institute. 2'-Deoxycoformycin was generously provided by the Nat- ural Products Branch, Developmental Therapeutics Program, Divi- sion of Cancer Treatment, National Cancer Institute. We are grateful to Dr. David W. Martin, Jr. for valuable discussions and for the E. coli strain MC1061. We are grateful to Dr. Barbara Bachmann for E. coli strain S4200. We also thank Drs. John H. Wilson, Vincent Kid4 Vicky Blakesley, and Amy G. Hook for valuable discussions and Meredith Riddell for assistance in preparing the manuscript.
D. E, Ingolia et al., manuscript in preparation.

Functional Murine Adenosine Deaminase cDNA Clones 10307
Addendum-We have now inserted the NcoIIEcoRV fragment of Kessel, A., De Waard, A., and Van der Eb, A. J. (1983) Gene pADA5-29 into the bacterial expression vector ptac-12 (50), trans- (Amst.) 2 5 , 231-240 fected this bacterial expression vector into E. coli, and showed that 23. Wiginton, D. A,, Adrian, G. S., Friedman, R. L., Suttle, D. P., the unfused murine adenosine deaminase protein synthesized in E. and Hutton, J. J. (1983) Proc. Natl. Acad. Sci. U. S. A. 80, coli appears as a single band and migrates slightly more cathodally 7481-7485 compared to authentic mouse adenosine deaminase following starch 24. Wiginton, D. A,, Adrian, G. S., and Hutton, J . J . (1984) Nucleic gel electrophoresis. This unambiguously established that the adeno- Acids Res. 12, 2439-2446 sine deaminase synthesized in our five adenosine deaminase-produc- 25. Valerio, D., McIvor, R. S., Williams, S. R., Duyvesteyn, M. G. C., ing recombinant clones (see Fig. 2B) were mainly in the form of van Ormondt, H., van der Eb, A. J., and Martin, D. W., Jr. fusion proteins. (1984) Gene (Amst.) 3 1 , 147-153
26. Friedman, R. L. (1985) Proc. Natl. Acad. Sci. U. S. A. 8 2 , 703-
27. Yeung, C. Y., Ingolia, D. E., Bobonis, C., Dunbar, B. S., Riser, M. E., Siciliano, M. J., and Kellems, R. E. (1983) J. Biol. Chem.
REFERENCES 707
1. Boss, G. R., and Seegmiller, J . E. (1982) Annu. Reu. Genet. 16, 297-328
2. Giblett, E. R., Anderson, J. E., Cohen, F., Pollara, B., and 28, ~ l ~ ~ ~ ~ ~ , G. (1981) cell 23 , 175-182 Meuwissen, H. J. (1972) Lancet 2, 1067-1069
3. Parkman, R., Gelfand, E. W., Rosen, F. S., Sanderson, A., and Hirschhorn, R. N. (1975) N. Engl. J . Med. 292 , 714-719
4. Pickering, R. J., Pollara, B., and Meuwissen, H. J. (1974) Clin. Immunol. Immunopathol. 3,301-302
5. Hirschhorn, R., Vawter, G. F., Kirkpatrick, J . A., and Rosen, F. S. (1979) Clin. Immunol. Immurwpathol. 14 , 107-120
6. Smyth, J. F., Young, R. C., and Young, D. M. (1978) Cancer Chemother. Pharmacol. 1,49-51
7. Smyth, J. F.y Paine, R. M.y Jackman, A. L.* Harrap, K. R.* 34. Sanger, F., Nickler, S., and Coulson, A. R. (1977) Proc. Natl. Chassin, M. M., Adamson, R. H., and Johns, D. G. (1980) Cancer Chemother. Pharmacol. 5,93-101
Acad. Sci. U. S. A. 7 4 , 5463-5467
8. Grever, M. R., Siaw, M. F. E., Jacob, D. F., Neidhart, J. A., Miser, 35. Maxam, A., and Gilbert, W. (1980) Methods Enzymol. 6 5 , 499-
560 J. M. s., Hutton% J. J., and Balcerzak, s' p. (lg81) 36. Wong, G. G., Witek, J. F., Temple, P. A., Wilkens, K., Leary, A.
C., Luxenburg, D., Jones, S. S., Brown, E. L., Kay, R. M., Ou, E. C., Shoemaker, C., Golde, D. C., Kaufman, R. J., Hewick, R.
10. BradY, T. G., and O'Donovan, c . (1965) COW. BkXhem. PhYSiol. 37. Kaufman, R. J. (1985) Pro,-. Natl. Acad. Sei. u. S. A. 8 2 , 689- M., Wang, E., and Clark, S . C. (1985) Science 228,810-815
l l . Edwards, y. H.* Hopkinson, D. A.* and H. (lg71) Ann. 38. Rave, N., Crkvenjakov, R., and Boedtker, H. (1979) Nucleic Acids
12. Van der WeYden, w . B.3 a n d K e h ' , w . N. (lg76) J. B i d Chem. 39. Maniatis, T., Jeffrey, A., and Kleid, D. G. (1975) Proc, Natl.
258,8338-8345
29. Goddard, J. M., Caput, D., Williams, S. R., and Martin, D. W., Jr . (1983) Proc. Natl. Acad. Sci. U. S. A. 80,4281-4285
30. Birnboim, H. C., and Doly, J. (1979) Nucleic Acids Res. 7 , 1513- 1525
31. Yeung, C. Y., Riser, M. E., Kellems, R. E., and Siciliano, M. J . (1983) J. Biol. Chem. 2 5 8 , 8330-8337
32. Agarwal, R. P., and Parks, R. E. (1978) Methods Enzymol. 51 , 502-507
33. Bradford, M. M. (1976) Anal. Biochem. 72, 248-254
Blood 57,406-417 9. Polmar, S. H., Wetzler, E. M., Stern, R. C., and Hirschhorn, R.
(1975) Lancet 2, 743-746
14, 101-120 693
Hum. Genet. 35 , 207-214
25 1,5448-5456 13. Adams, A., and Harkness, R. A. (1976) C h . Ezp. Immunol. 2 6 , 40. Southern, E. (1975) J , ~ ~ 1 . ~ i ~ l . 98 , 503-517
647-649 14. Seegmiller, J. E., Thompson, L., Bluestein, H., Willis, R., Mat-
41. Chang, A. C. Y., Nunberg, J. H., Kaufman, R. J., Erlich, H. A., Schimke, R. T., and Cohen, S. N. (1978) Nature 275,617-624
sumoto, s., and Carson, D. (1980) in Biological Basis O f Inmu- 42. Jochimsen, B. P. N., and Vestergaard, T. (1975) Mol. Gen. Genet. nodeficiency (Gelfand, E. D., and Dosch, H. M., eds) pp. 251- 268, Raven Press, New York
15. Barton, R., Martiniuk, F., Hirshhorn, R., and Goldschneider, J. 43. Adrian, G. S., Wiginton, D. A., and Hutton, J. J. (1984) Mol. Cell.
(1980) Cell. Immunol. 4 9 , 208-214 16. Sidi, Y., Umiel, T., Trainin, N., Pinkhas, J., and Sperling, 0.
44. Daddona, P. E., Shewach, D. S., Kelley, W. N., Argos, P., Mark-
(1982) Thymus 4 , 147-154 ham, A., and Orkin, S. H. (1984) J . Biol. Chem. 2 5 9 , 12101- 12106
17. Chechik, B., Schrader, W. P., and Daddona, P. E. (1980) J. Natl. 45. Kozak, M. (1983) Microbial. Re", 4 7 , 1-45 Cancer fnst. 6 4 , 1077-1083 46. Dobson, M. J., Tuite, M., Roberts, N., Kingsman, A,, Kingsman,
18. Lee, P. C. (1973) Deu. Biol. 3 1 , 227-233 S., Perkins, R., Conroy, S., Dunbar, B., and Fothergill, L. A. 19. Yeung, C. Y., Frayne, E. G., Al-Ubaidi, M. R., Hook, A. G., (1982) Nucleic Acids Res. 10, 2625-2637
Ingolia, D. E., Bobonis, c . , Wright, D., and Kellems, R. E. 47. Zalkin, H., and Yanofsky, C. (1982) J. Bioi. Chem. 2 5 7 , 1491- (1983) J. Biol. Chem. 2 5 8 , 15179-15185 1500
20. Hunt, s. w., 111, and Hoffee, p. A. (1983) J. Bd. Chem. 258 , 48. Chang, A. C. Y., Erlich, H. A., Gunsalus, R. P., Nunberg, J. H.,
21. Orkin, S. H., Daddona, P. E., Shewach, D. S., Markham, A. F., Natl. Acad. Sci. U. S. A. 77 , 1442-1446
Res. 6,3559-3567
Acad. Sci. U. S.A . 72 , 1184-1188
143,85-91
Bioi. 4,1712-1717
13185-13192 Kaufman, R. J., Schimke, R. T., and Cohen, S. N. (1980) Proc.
Bruns, G. A,, Goff, S. C., and Kelley, W. N. (1983) J. Biol. 49. Weiss, R. B. (1984) Proc. Natl. Acad. Sei. u. S. A. 8 1 , 5797-5801 Chem. 2 5 8 , 12753-12756 50. Amann, E., Brosins, J., and Ptashne, M. (1983) Gene (Amst.)
22. Valerio, D., Duyvesteyn, M. G. C., Meera Khan, P., Geurts van 864,167-178