Embed Size (px)
Citation preview

Identification of 15 Different Candidate Causal Point Mutations and Three Polymorphisms in 19 Patients With Protein S Deficiency Using a Scanning
Method for the Analysis of the Protein S Active Gene
By S. Gandrille, D. Borgel, V. Eschwege-Gufflet, Mf. Aillaud, M. Dreyfus, C. Matheron, P. Gaussem, J.F. Abgrall, B. Jude, P. Sie, P. Toulon, and M. Aiach
To screen for point mutations causing protein S deficiency, we used a sequence of techniques specifically for the study of the protein S active gene, PSa. This strategy comprises amplification of exons and intron/exon junctions by means of the polymerase chain reaction (PCR) and electrophoresis of the amplified fragments in polyacrylamide gel containing a gradient of denaturing agents (denaturing gradient gel electrophoresis). Only fragments with altered melting be- havior are sequenced after asymmetric PCR. Beside the fre-
ENOMIC ABNORMALITIES responsible for coagula- tion-protein disorders have been the subject of exten-
sive studies during the past few years. Various disease-caus- ing changes, including large deletions and insertions,' recombination with homologous genes,'.' and point muta- t i o n ~ , ~ . ~ have been identified. The latter are the most frequent defects in hemophilia B: mild hemophilia A: antithrombin I11 deficiencies,'j and protein C defi~iencies,~ whereas recom- binations are frequent in severe hemophilia A.'
Hereditary protein S deficiency is an important risk factor for th romb~sis .~*~ It is present in 4% to 8% of patients with unexplained venous thrombosis and is sometimes associated with arterial thrombosis.'o,''
Two homologous genes for protein S PSa and PS@ have been assigned to chromosome 3.'' The PSa gene (or PROS 1) spans over 80 kilobases and comprises 15 ex on^.'^-'^ The PS0 gene (or PROS 2) presents a high degree of homology with the PSa gene: 97% and 95.4% homology in the coding and noncoding parts, respectively. It doubles exons I1 to XV of the PSa gene. The multiple base changes observed in the PSP sequences corresponding to the exons of PSa generate stop codons and frameshifts. Because PSP has no open read-
G
From CJF INSERM 91-01. UFR des Sciences Pharmaceutiques et Biologiques, and French network INSERM on the "Molecular Mechanisms Responsible for Protein C and Protein S Hereditary Dejiciencies. "
Submitted February 23, 1994; accepted September I S , 1994. Supported by Institut National de la Sante' et de la Recherche
Me'dicale (INSERM), and by grants from the Association Frangaise contre les Myopathies (AFM) and Diagnostica Stago.
This work has been presented in preliminary reports to the 65th Scientijic Sessions of the American Heart Association, New Orleans, L A , November 1992 and appeared in abstract form in Circulation 86:1, 1992 (abstr no. 3240); and to the XIVth Congress of the International Society on Thrombosis and Haemostasis, New-York, NY, July 1993, and appeared in abstract form in Thromb Haemost 69:790, 1993 (abstr no. 883).
Address reprint requests to S. Gandrille, PhD, INSERM CJF 91- 01, UFR des Sciences Pharmaceutiques et Biologiques, 4, Avenue de I'Observatoire, F-75270 Paris cedex 0 6 , France.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. section 1734 solely to indicate this fact. 0 1995 by The American Society of Hematology. 0006-4971/95/8501-0027$3.00/0
130
quent polymorphism already described on Pro 626, we de- tected 18 different sequence variations by studying exons 11. IV. V, VIII, X, and XV in 19 of 100 consecutive patients with protein S deficiency. Fifteen were candidate causal mu- tations, 4 of which were associated with a qualitative defi- ciency (type lla or Ilb). The remaining three sequence varia- tions were probably polymorphisms. 0 7995 by The American Society of Hematology.
ing frame, it is probably a pseudogene, and circulating pro- tein S is thus the product of PSa, the active gene.
The 5' part of the PSa gene shows strong homology with the genes of vitamin K-dependent proteins, particularly pro- tein C. In contrast, the 3' part of the gene, encoding the C- terminal part of the protein, is homologous to that of sex hormone-binding globulin (SHBG).
Two large deletions of the PSa gene have been shown to be associated with PS deficiency, one in the middle,''j and the other spanning 5.3 kb and including exon XIII." A few point mutations have recently been described'R-'2; the delete- rious effect of one is contr~versial . '~~'~
To elucidate the molecular basis of protein S deficiency, by analogy with hereditary protein C deficiencies that are frequently caused by point mutations, we sought point muta- tions in the coding sequence of the PSa gene. We describe our screening strategy based on denaturing gradient gel elec- trophoresis (DGGE) of amplified genomic fragments and direct sequencing, and report 15 novel point mutations and three polymorphisms.
MATERIALS AND METHODS
Patients. Plasma and DNA samples were obtained from normal subjects and from patients with a quantitative (type I) or qualitative (types IIa and IIb) protein S deficiency, recruited by a French net- work on the behalf of INSERh4. The normal subjects were white (like the patients), except for three who were black.
Protein S antigen was measured using an immunoenzymatic method before (total antigen) and after (free antigen) treatment with polyethyleneglycol (PEG). Protein S activity was determined using the Staclot kit (Diagnostica Stago, Asnikres, France). Normal ranges in the three assays ranged between 70% and 130%.
The protein S deficiency was diagnosed on the basis of low protein S activity and/or low free or total protein S antigen levels. According to Comp:' type I deficiency is characterized by low total and free protein S antigen levels, whereas type IIa is characterized by low free protein S but normal total protein S antigen levels. In type IIb. only protein S activity is decreased.
Blood samples. Venous blood samples were collected in evacu- ated tubes containing 0.129 mol/L trisodium citrate ( 1 : 10) for protein S assays and the plasma was kept frozen until use.
Venous blood was collected in ethylene diamine tetracetic acid for DNA studies and kept at 4°C. Leukocytes were isolated within 48 hours and stored frozen until DNA extraction as described by Bell et al.24
Materials. Thermus aquaticus (Taq) polymerase (5 UIpL) was from Perkin Elmer Cetus Instruments (Norwalk, CT).
The four deoxynucleotides deoxyguanosine triphosphate (dGTP),
Blood, Vol 85, No 1 (January l), 1995: pp 130-138
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom

MUTATIONS IN THE ACTIVE PROTEIN S GENE 131
S IX X XI1 xlll XIV
- * 0 ~ ~ 8 1 0 1 p6m
" QCp61m p6128
- Qccps14A PS140
*
" GcPSu PS- p611A PS11c GCp611B
" - PSIY GcPSI3B
QCPSIX PS190 - * "
PSIW Qcp61m "
Fig 1. Location of the ampliiiwtion primers on the protein S active gene. Exons are symbolized by boxes. The 5' part of exon 1, which is noncoding, is striped. Primers with a GC-clamp at their 5' end are marked by an open bar. The sequences of the amplification primers are indicated below, from 5' to 3'; the oligonucleotides used as sequencing primers are indicrted by an asterisk P). The sequences belwwn brackets correspond to the GC-clamp sequence. Exon I: PS 1A (5' untrantlated region) AGGCTCGCTGGGTCGCTGGC and PS 18. (NS a) ACGCAG lTCAGTCGACAAT0; Exon II GC PS 2A (IVS a1 [GCCGCCCCGCCCGGCCCGTCCCCCTGGCGCCCGGCACCCGFGATTAATTCATATAAACTG and PS 28. (NS b) TAGATGTGTGAAClTGATAG; Exon 111 PS 3A (NS b) G G T ~ G C T M G A T A T ~ and GC PS 38 (NS c) [CCCCCGCCCOCCCGGAGC- GGCCGGCGCCCG~ATGTCACATGGTAATTAAAG; Exon W: GC PS 4A (IVS c) [GCCCGGGCGCCCTCCCCCGCGCGTCCGGCGlACCTClTGGGACAG- TTCCTA and PS4B* (IVS d) O T T m G l l l T G T l l l l T C A ; Exon V PS 5A* (IVS dl GTACACACAATmArrm and GC PS MI (NS e) [GGGCGCCGG- CCCGGCCCGACCCCCGCGCGTCCGGCGCCCGIGAGAAClllTCAGGAGACCA; Exon VI: PS 6A (IVS e) GCAGlTCATAGTATCGTCGGTTATATT- ACllTAAAAAT and GC PS 0B (NS f ) [CCCCCGCCCGCCCGGAGCGGCCGGCGCCCG1TTATTCTCACATAGTAAATG; Exon Vll: PS 7A* (NS f l AAACAAGATC CAGGAAACAC and GC PS 7 8 (NS g) [CCGCGGCCCGCCGTCCCCGCCCGGCCCTGC~GGCGCCCO~~ATCAGTAA- TGATACCACCA; Exon VIII: PS 8A* (IVS g) GGATATTAAAGllTGTGTGC and GC PS 8 8 (IVS h) [GGGCCGGCCGGGCCGGGCTGGGGGCGCGCA- GGCCGCGGGCITCTGTAlllTCCTGAClTAG; Exon IX GC PS SA (IVS h) [GCCGCCCCGCCCGGCCCGTCCCCCTGGCGCCCGGCACCCGKAAACA- TAAGCAATAACAT and PS SB (IVS i) AlTATCATTGGTAlTGGTTC; Exon X GC PS 1OA (NS i) [GCCCGCCGGCCCGACCCC C G C G C G T c c O G ~ ~ A G l G C l l l C T G T A ~ A C T C and PS lOB* (IVS j) AAAGTGGGAAGATAlTGATG; Exon XI: GC PS 118 (NS k) [GGG- CCGGCCGGGCCGGGCTGGGGGCGCGCAGGCCGCGGGCICTAlTACAGACAAAAGGAAC and PS 11A (IVS j) AAAATGATTACllTACAGAA or PS 11C (exon XI, aa 366 to 372) TAAATAAACCTGGACCm; Exon XII: GC PS 12A (IVS k) [GCCGCCCCGCCCGGCCCOICCCCCTOOCOCCCOOCA- CCCGFCATAATCGAGCCACTGllT and PS 128 (IVS 1) TAGATACTCAATAATGllTC; Exon XWI: PS 13A (IVS 1) TGlTAATAATAATTCCTTCTGA and GC PS 138 (exon XIII, aa 485 to 478) [GGGCCGGCCGGGCCGGGCTGGGGGCGCGCAGGCCGCGCCCGGClAG~GCATAACACCAGTG/ GP PS 13C (exon XIII, aa 466 to 472) [GCCCGGCCCCGCCGCCCCGCCCGGCCCGTCC~GGCGCCCGGCACCCO~ATGTAAATGTGA~GAAT and PS 13D (NS m) GTAAATACTGCTATGTATAC; Exon XIV GC PS 14A (NS m) [GCCGCCCCOCCCGGCCCGTCCCCTGGC~GGCACCCGIAGCAGC- AlTACTClTACTCC and PS 148 (IVS n) ATCGGTTTGATTAAAATATA Exon XV: PS 15A* (NS n) CAAACAAGATGCTAMAGTC and GC PS 15B (3' untrandated region) [GGGCCGGCCGGGCCGGGCTGGGGGCGCGCAGGCCGCGGGCJAAACATAAGTATAATTACAC. The undodined and bold sequonco of PS 6A corresponds to an upstream oligonucleotide sequence added to obtain a better melting curve. The undorllned and bold nucleothie of GC PS SA corrmonds to a smuence variation introduced to create a restriction site within the amplified product of the gene PS&
deoxycytosine triphosphate (dCTP), deoxyadenosine triphosphate (dATP), and deoxythimidine triphosphate ( d m ) were from Phar- macia Fine Chemicals (Uppsala, Sweden). The Sequenase kit was from United States Biochemicals Corporation (Cleveland, OH). The Centricon 100 apparatus was obtained from Amicon (Denver, CO). (Y-'~S(~ATP) and Y-'~P(~ATP) were purchased from Amersham (Buckinghamshire, United Kingdom). The restriction endonucleases were from Biolabs (Ozyme, Montigny-le-Bretonneux, France).
Oligonucleotides were from Genset (Paris, France). Experimental design. Each PSa gene exon and its splice junc-
tions were amplified enzymatically as described by Saiki et al,= using a set of oligonucleotides suited to DGGE analysis and permit- ting specific amplification of the PSa gene.
Computer analysis was performed with the MELT 87 and SQHTX programs written and provided by Drs Lerman and Silverstein.x
The Melt 87 algorithm predicts the melting behavior of a DNA fragment according to its nucleotide sequence and base composition. We used the information provided by this program to select the positions of polymerase chain reaction (PCR) primers that would generate amplified fragments suitable for DGGE. Because the se- quence of interest has to be located within the first melting domain of the fragment, a G + C-rich oligomer was attached to the 5' extremity of one of the amplification primers. This GC-rich sequence artificially creates a high-temperature melting d0main,2~-'~ thereby positioning the sequence of interest in the lower stability domain. The location of the amplification primers and their nucleotide se- quences are given in Fig 1. Because they are composed of two
melting domains, exons XI and XI11 were studied using two sets of primers (see legend of Fig 1 and Table 1).
DGGE could not be applied to the analysis of exon I because of its very high melting temperature. This exon was therefore studied by direct sequencing after asymmetric PCR.
The PSa gene was selectively amplified by applying the principle of the amplification-refractory mutation system": primers were posi- tioned such that the 3' end of at least one of the two primers was complementary to the PSa sequence and not that of PSP.
DNA amplification. Each PCR mixture contained 30 pmol up- stream and downstream primer, 200 pmolR. each M, 1 pg of genomic DNA, 1X PCR buffer, and 2.5 U of Taq polymerase, in a final volume of 100 pL. In addition, 5% DMSO was added to the PCR mixtures in the case of exons 111 to V, VI1 to X, XI1 and XI11 fragment A.
The 1X PCR buffer was 10 mmolR. TRIS-HCI, pH 8.3.50 mmol/ L KCI, 0.01% (wt/vol) gelatin, and 1.5 mmoVL MgC12 for exons I1 to VIII, X, XI fragment B, XIII fragment B, XIV, and XV, 2 mmoY L MgCI2 for exons IX, XI fragment A, and XII, and 2.5 mmoVL MgCI2 for exon XJU fragment A.
The thermal cycles comprised 5 minutes of denaturation at 94"C, followed by 30 (exons m, IV, VI, VIII, IX, X, XII, and XIV), 35 (exons II, V, VII, XI fragments A, exon XIII fragments A and B, and XV), or 40 cycles (exon XI fragment B) of denaturation for 1 minute at 94"C, annealing for 1 minute at a temperature depending on the primer set, and extension for 1 minute at 72°C. The annealing temperatures were 46°C for exons 11, VI, XI fragment A (amplified
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom
132 GANDRILLE ET AL
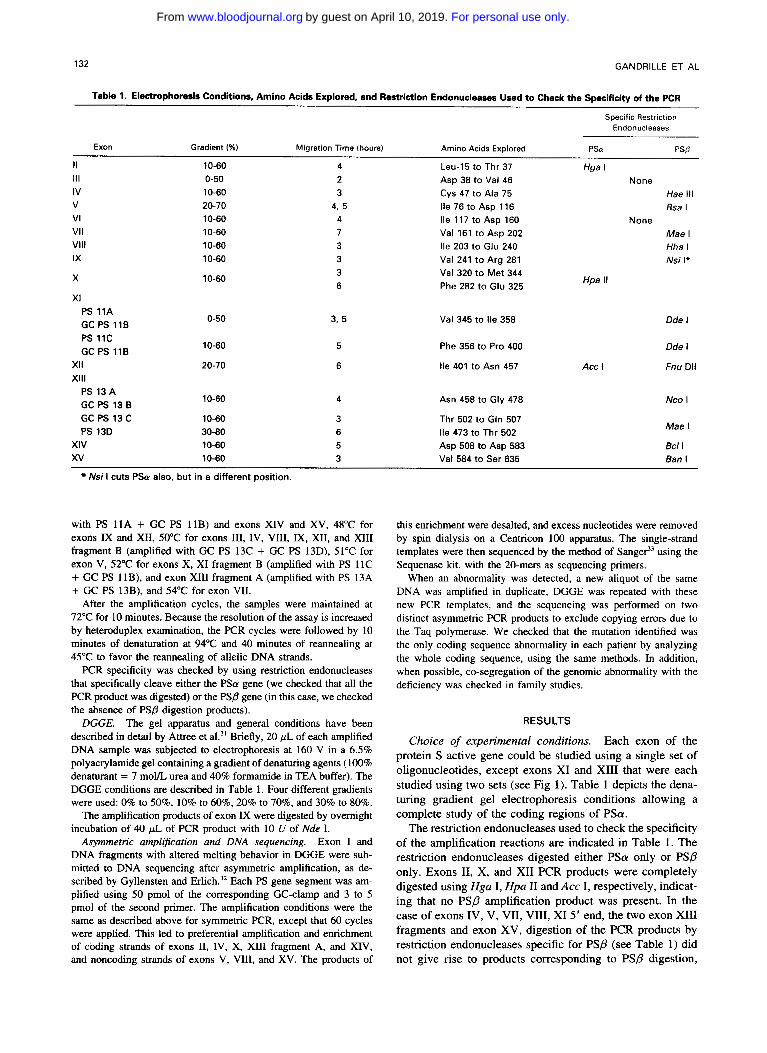
Table 1. Electrophoresis Conditions. Amino Acids Explored, and Restriction Endonucleases Used to Check the Specificity of the PCR
Specific Restrlction Endonucleases
Exon Gradient (%) Migration Time (hours) Amino Acids Explored PSa PSB
II 10-60 4 111 2 IV 3 V
Leu-l5 to Thr 37 Asp 38 to Val 46 Cys 47 to Ala 75
0-50 Hga I
10-60 None
20-70 Hae Ill
4. 5 Ile 76 to Asp 116 Rsa I VI 10-60 4 Ile 117 to Asp 160 VI1 Vlll IX
X 10-60
XI
None 10-60 7 Val 161 to Asp 202 Mae I
Hha I 10-60 3 Ile 203 to Glu 240 10-60 3 Val 241 to Arg 281 Nsi I*
3 Val 320 to Met 344 6 Phe 282 to Glu 325
Hpa II
PS 11A GC PS l l B PS 11c GC PS l l B
XI1 Xlll
PS 13 A GC PS 13 B GC PS 13 C PS 13D
XIV xv
0-50
10-60
20-70
10-60
10-60 30-80 10-60 10-60
3. 5
5
6
Val 345 to Ile 358
Phe 356 to Pro 400
Ile 401 to Asn 457
Asn 458 to Gly 478
Thr 502 to Gln 507 Ile 473 to Thr 502 Asp 508 to Asp 583 Val 584 to Ser 635
Acc l
Dde I
Dde I
Fnu Dl1
NCO I
Mae I
BC/ l Ban l
Nsi I cuts PSa also, but in a different position.
with PS 11A + GC PS 11B) and exons XIV and XV, 48°C for exons IX and XII, 50°C for exons 111, IV, VIII, M, XII, and XI11 fragment B (amplified with GC PS 13C + GC PS 13D), 51°C for exon V, 52°C for exons X, XI fragment B (amplified with PS 11C + GC PS 1 lB), and exon XI11 fragment A (amplified with PS 13A + GC PS 13B), and 54°C for exon VII.
After the amplification cycles, the samples were maintained at 72°C for 10 minutes. Because the resolution of the assay is increased by heteroduplex examination, the PCR cycles were followed by 10 minutes of denaturation at 94°C and 40 minutes of reannealing at 45°C to favor the reannealing of allelic DNA strands.
PCR specificity was checked by using restriction endonucleases that specifically cleave either the PSa gene (we checked that all the PCR product was digested) or the PSP gene (in this case, we checked the absence of PSP digestion products).
DGGE. The gel apparatus and general conditions have been described in detail by Attree et al.” Briefly, 20 pL of each amplified DNA sample was subjected to electrophoresis at 160 V in a 6.5% polyacrylamide gel containing a gradient of denaturing agents (100% denaturant = 7 m o m urea and 40% formamide in TEA buffer). The DGGE conditions are described in Table 1. Four different gradients were used: 0% to 50%. 10% to 60%, 20% to 70%, and 30% to 80%.
The amplification products of exon IX were digested by overnight incubation of 40 pL of PCR product with 10 U of Nde I.
Asymmetric amplification and DNA sequencing. Exon I and DNA fragments with altered melting behavior in DGGE were sub- mitted to DNA sequencing after asymmetric amplification, as de- scribed by Gyllensten and Erli~h.~’ Each PS gene segment was art-
plified using 50 pm01 of the corresponding GC-clamp and 3 to 5 pmol of the second primer. The amplification conditions were the same as described above for symmetric PCR, except that 60 cycles were applied. This led to preferential amplification and enrichment of coding strands of exons 11, IV, X, XI11 fragment A, and XIV, and noncoding strands of exons V, VIII, and XV. The products of
this enrichment were desalted, and excess nucleotides were removed by spin dialysis on a Centricon 100 apparatus. The single-strand templates were then sequenced by the method of Sanger” using the Sequenase kit, with the 20-mers as sequencing primers.
When an abnormality was detected, a new aliquot of the same DNA was amplified in duplicate, DGGE was repeated with these new PCR templates, and the sequencing was performed on two distinct asymmetric PCR products to exclude copying errors due to the Taq polymerase. We checked that the mutation identified was the only coding sequence abnormality in each patient by analyzing the whole coding sequence, using the same methods. In addition, when possible, co-segregation of the genomic abnormality with the deficiency was checked in family studies.
RESULTS
Choice of experimental conditions. Each exon of the protein S active gene could be studied using a single set of oligonucleotides, except exons XI and XI11 that were each studied using two sets (see Fig I) . Table 1 depicts the dena- turing gradient gel electrophoresis conditions allowing a complete study of the coding regions of P S a .
The restriction endonucleases used to check the specificity of the amplification reactions are indicated in Table 1. The restriction endonucleases digested either PSa only or PSP only. Exons 11, X, and XI1 PCR products were completely digested using Hga I, Hpa I1 and Acc I, respectively, indicat- ing that no PS/? amplification product was present. In the case of exons IV, V, VII, VIII, XI 5’ end, the two exon XI11 fragments and exon XV, digestion of the PCR products by restriction endonucleases specific for PS@ (see Table 1) did not give rise to products corresponding to PS/? digestion,
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom
MUTATIONS IN THE ACTIVE PROTEIN S GENE 133
Exon II Exon IV Exon V
Normal subjects
Exon Vlll Exon X Exon XV
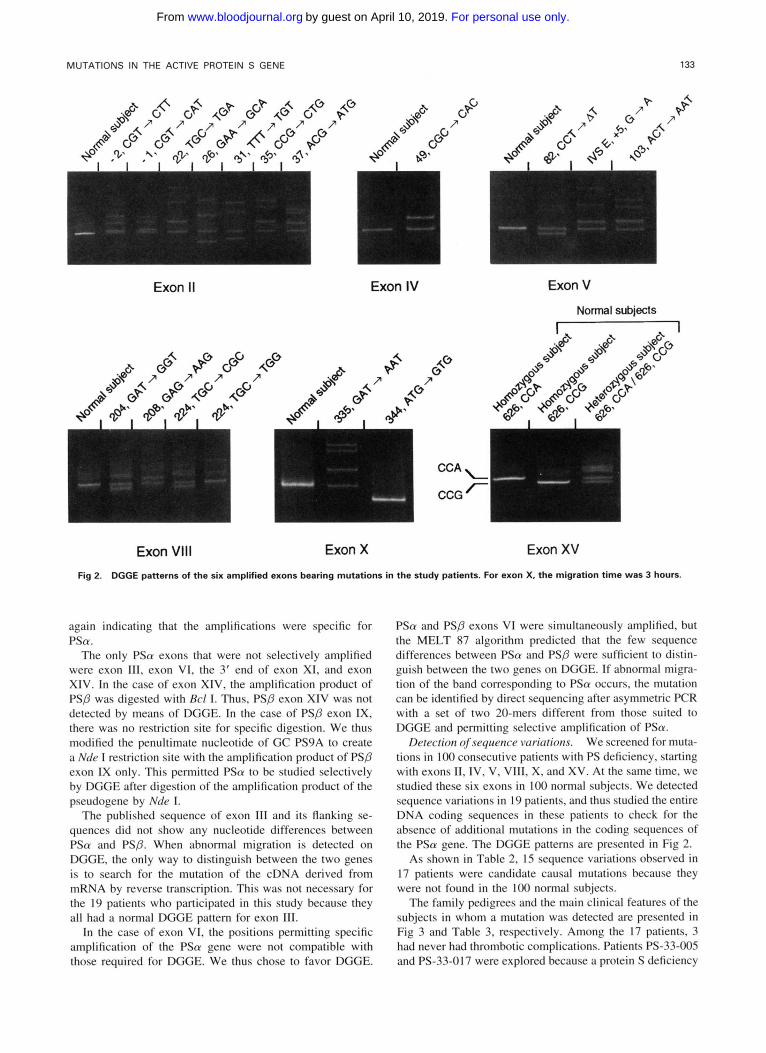
Fig 2. DGGE patterns of the six amplified exons bearing mutations in the study patients. For exon X, the migration time was 3 hours.
again indicating that the amplifications were specific for PSa.
The only PSa exons that were not selectively amplified were exon 111, exon VI, the 3’ end of exon X!, and exon XIV. In the case of exon XIV, the amplification product of PSP was digested with RC/ I . Thus, PS0 exon XIV was not detected by means of DGGE. In the case of PS0 exon !X, there was no restriction site for specific digestion. We thus modified the penultimate nucleotide of GC PS9A to create a Nde ! restriction site with the amplification product of PSP exon IX only. This permitted P S a to be studied selectively by DGGE after digestion of the amplification product of the pseudogene by Nde 1.
The published sequence of exon I11 and its flanking se- quences did not show any nucleotide differences between PSa and PSP. When abnormal migration is detected on DGGE. the only way to distinguish between the two genes is to search for the mutation of the cDNA derived from mRNA by reverse transcription. This was not necessary for the 19 patients who participated in this study because they all had a normal DGGE pattern for exon 111.
In the case of exon VI, the positions permitting specific amplification of the PSa gene were not compatible with those required for DGGE. We thus chose to favor DGGE.
PSa and PS0 exons VI were simultaneously amplified, but the MELT 87 algorithm predicted that the few sequence differences between PSa and PS0 were sufficient to distin- guish between the two genes on DGGE. If abnormal migra- tion of the band corresponding to PSa occurs, the mutation can be identified by direct sequencing after asymmetric PCR with a set of two 20-mers different from those suited to DGGE and permitting selective amplification of PSa.
Detection of sequence variations. We screened for muta- tions in 1 0 0 consecutive patients with PS deficiency, starting with exons 11, IV, V, VII!, X, and XV. At the same time, we studied these six exons in 1 0 0 normal subjects. We detected sequence variations in I9 patients. and thus studied the entire DNA coding sequences in these patients to check for the absence of additional mutations in the coding sequences of the PSa gene. The DGGE patterns are presented in Fig 2.
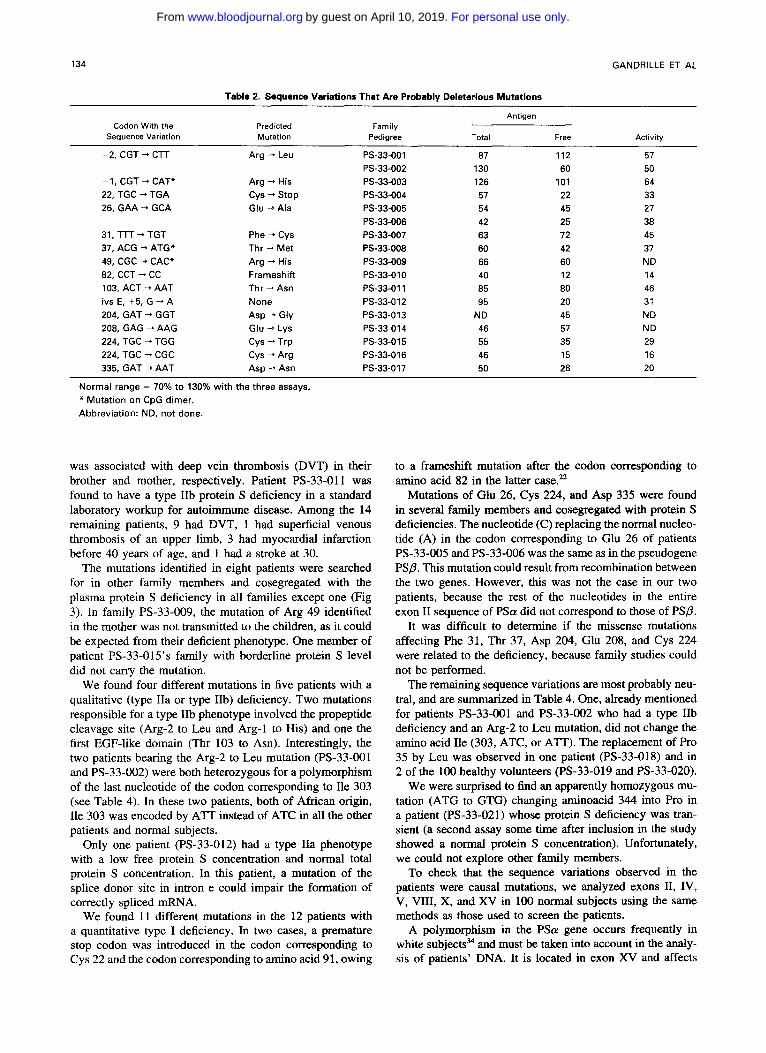
As shown in Table 2, IS sequence variations observed in 17 patients were candidate causal mutations because they were not found in the 1 0 0 normal subjects.
The family pedigrees and the main clinical features of the subjects in whom a mutation was detected are presented in Fig 3 and Table 3, respectively. Among the 17 patients. 3 had never had thrombotic complications. Patients PS-33-005 and PS-33-017 were explored because a protein S deficiency
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom
134 GANDRILLE ET AL
Table 2. Sequence Variations That Are Probably Deleterious Mutations
Antigen Codon With the Predicted
Sequence Variation Mutation Pedigree Total Free Activlty Family
-2, CGT - CTT Arg -+ Leu PS-33-001 87 112 57 PS-33-002 130 60 50
-1, CGT -* CAT* Arg -+ His PS-33-003 126 101 64 22, TGC + TGA cys -+ stop PS-33-004 57 22 33 26, GAA - GCA Glu +Ala PS-33-005 54 45 27
31, TTT -* TGT Phe -+ Cys PS-33-007 63 72 45 37, ACG -, ATG* Thr + Met PS-33-008 60 42 37 49, CGC -+ CAC* Arg - His PS-33-009 66 60 ND 82, CCT + CC Frameshift PS-33-010 40 12 14 103, ACT + AAT Thr + Asn PS-33-01 1 85 80 46 ivs E, +5, G + A None PS-33-012 95 20 31 204, GAT + GGT Asp -+ Gly PS-33-013 ND 45 ND 208, GAG + AAG Glu -+ Lys PS-33.014 46 57 ND 224, TGC - TGG Cys 4 Trp PS-33-015 55 35 29 224, TGC + CGC Cys -+ Arg PS-33-016 46 15 16 335, GAT + AAT Asp -+ Asn PS-33-017 50 28 20
PS-33-006 42 25 38
Normal range = 70% to 130% with the three assays. * Mutation on CpG dimer. Abbreviation: ND, not done.
was associated with deep vein thrombosis (DVT) in their brother and mother, respectively. Patient PS-33-011 was found to have a type 1% protein S deficiency in a standard laboratory workup for autoimmune disease. Among the 14 remaining patients, 9 had DVT, 1 had superficial venous thrombosis of an upper limb, 3 had myocardial infarction before 40 years of age, and 1 had a stroke at 30.
The mutations identified in eight patients were searched for in other family members and cosegregated with the plasma protein S deficiency in all families except one (Fig 3). In family PS-33-009, the mutation of Arg 49 identified in the mother was not transmitted to the children, as it could be expected from their deficient phenotype. One member of patient PS-33-015’s family with borderline protein S level did not carry the mutation.
We found four different mutations in five patients with a qualitative (type IIa or type IIb) deficiency. Two mutations responsible for a type IIb phenotype involved the propeptide cleavage site (Arg-2 to Leu and Arg-l to His) and one the first EGF-like domain (Thr 103 to Am). Interestingly, the two patients bearing the Arg-2 to Leu mutation (PS-33-001 and PS-33-002) were both heterozygous for a polymorphism of the last nucleotide of the codon corresponding to Ile 303 (see Table 4). In these two patients, both of African origin, Ile 303 was encoded by A’IT instead of ATC in all the other patients and normal subjects.
Only one patient (PS-33-012) had a type IIa phenotype with a low free protein S concentration and normal total protein S concentration. In this patient, a mutation of the splice donor site in intron e could impair the formation of correctly spliced mRNA.
We found 11 different mutations in the 12 patients with a quantitative type I deficiency. In two cases, a premature stop codon was introduced in the codon corresponding to Cys 22 and the codon corresponding to amino acid 91, owing
to a frameshift mutation after the codon corresponding to amino acid 82 in the latter case.”
Mutations of Glu 26, Cys 224, and Asp 335 were found in several family members and cosegregated with protein S deficiencies. The nucleotide (C) replacing the normal nucleo- tide (A) in the codon corresponding to Glu 26 of patients PS-33-005 and PS-33-006 was the same as in the pseudogene PSP. This mutation could result from recombination between the two genes. However, this was not the case in our two patients, because the rest of the nucleotides in the entire exon I1 sequence of PSa did not correspond to those of PSP.
It was difficult to determine if the missense mutations affecting Phe 31, Thr 37, Asp 204, Glu 208, and Cys 224 were related to the deficiency, because family studies could not be performed.
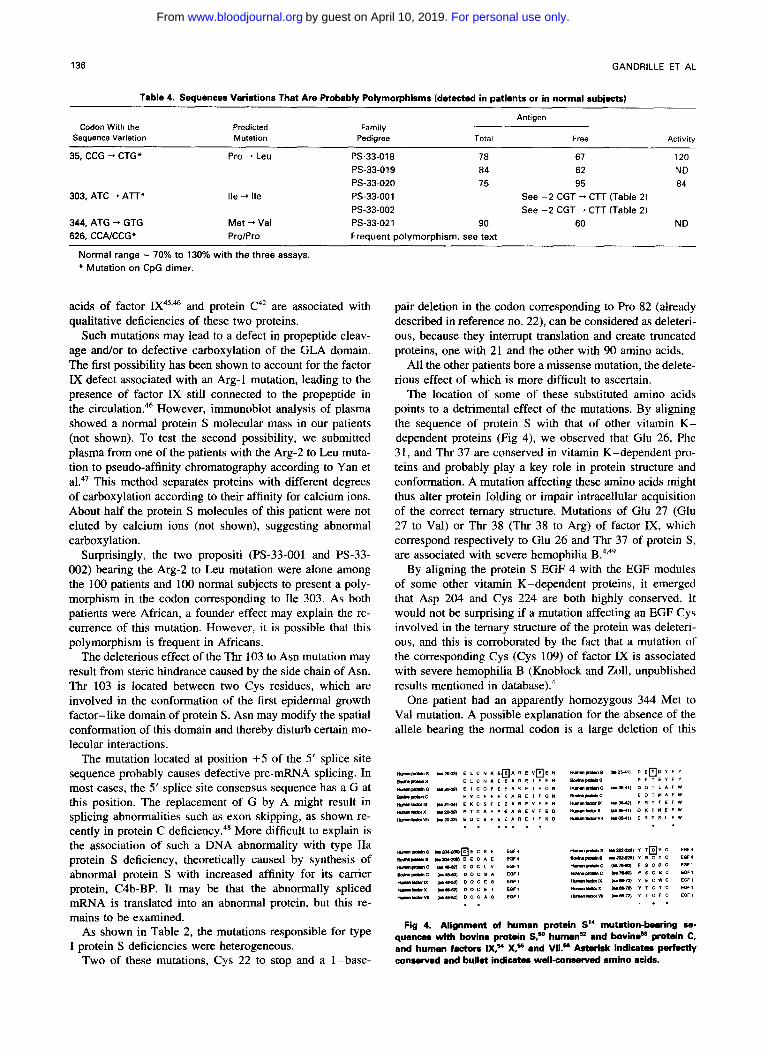
The remaining sequence variations are most probably neu- tral, and are summarized in Table 4. One, already mentioned for patients PS-33-001 and PS-33-002 who had a type IIb deficiency and an Arg-2 to Leu mutation, did not change the amino acid Ile (303, ATC, or A n ) . The replacement of Pro 35 by Leu was observed in one patient (PS-33-018) and in 2 of the 100 healthy volunteers (PS-33-019 and PS-33-020).
We were surprised to find an apparently homozygous mu- tation (ATG to GTG) changing aminoacid 344 into Pro in a patient (PS-33-021) whose protein S deficiency was tran- sient (a second assay some time after inclusion in the study showed a normal protein S concentration). Unfortunately, we could not explore other family members.
To check that the sequence variations observed in the patients were causal mutations, we analyzed exons 11, IV, V, VIII, X, and XV in 100 normal subjects using the same methods as those used to screen the patients.
A polymorphism in the PSa gene occurs frequently in white subjects” and must be taken into account in the andy- sis of patients’ DNA. It is located in exon XV and affects
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom

MUTATIONS IN THE ACTIVE PROTEIN S GENE 135
Fig 3. Family pedigrees of patients with mutations identified in the protein S gene. The number indicates the propositus of each family and corresponds to the number in Tables 2 and 4, ie, patient in whom protein S gene coding regions were entirely studied. In the other family memben, only the fragment corresponding to tha mu- tated fragment in the propositus was screened by DGGE. Asterisks indicate subjects with thrombotic complications. 0 0, male and famale with normal protein S; D @, male and female with low pro- tein S and a protein S gene mutation; m @, unexplored; !Zf @, decaased; B @, male and female with protein S deficiency and in whom the mutation observed in the other family member($) was not found; 0 @, male and female with low protein S, unexplored for protein S gene mutation.
the codon corresponding to Pro 626 (CCA or CCG). Such a single A/G nucleotide transition yields a significant differ- ence in melting behavior. As shown in Fig 2, three different DGGE patterns were obtained for exon XV: two patterns typical of homozygous subjects (CCA, upper band, or CCG, lower band), and a pattern typical of heterozygous subjects, with each allele migrating as a separate homoduplex. In addition, heteroduplexes are formed by reannealing between the CCG- and CCA-containing alleles during PCR.
We established the allele frequency of this polymorphism in the French population by studying 100 normal subjects. CCA was detected in 57% of cases and CCG in 43%. The observed heterozygosity (52%) was very close to the calcu- lated value (49%) and the polymorphism information content (PIC) value was from 0.37.
DISCUSSION
To identify point mutations responsible for protein S defi- ciencies, we adapted denaturing gradient gel electrophoresis to the analysis of the protein S gene. DGGE is a powerful tool to detect gene fragments bearing mutations as small as a single base substitution, and has been successfully applied to many different genes including coagulation factor IX30*35
factor VIII:*36 cystic fibrosis tran~regulator,~'.~~ low-density lipoprotein receptor,4o and protein C4"'' genes.
The main obstacle to studying the protein S active gene (PSa) is the presence of a pseudogene, PSP, which has strong homology with PSa (97% in the coding regions, 95.4% in the intronic sequences) and doubles exons I1 to XV. We overcame this problem by choosing the amplifica- tion primers according to the principle of the amplification- refractory mutation system (ARMS). We were thus able to amplify selectively most PSa exons (exons 11, IV, V, VII, VIII, X, XI 5' part, XII, XIII, XIV, and XV).
We first performed DGGE of the PCR products corre-
sponding to exons XV, to check that we could readily detect the frequent polymorphism described, Pro 626 encoded ei- ther by CCA or by CCG.34 The allele frequencies of the exon XV polymorphism in the French population were somewhat different from those in the Dutch population, as the CCA and CCG alleles were present in, respectively, 57% and 43% of the French subjects tested (v 48% and 52% in Dutch subjects).
We started our study of the PSa gene by screening exons 11, IV, V, VIII, X, and XV of 100 consecutive patients with qualitative (type IIa and IIb) or quantitative (type I) protein S deficiencies. We detected abnormal DGGE patterns of these six exons in 19 patients and identified the sequence variations by direct sequencing of asymmetric PCR products. We checked that the mutations were the only coding se- quence abnormality in these 19 patients by analyzing the all exons and boundary regions. Using the same methods as those used to study the deficient subjects, we also screened 100 normal subjects for the observed mutations.
In one patient (PS-33-018), the detrimental effect of the mutation could be ruled out, as the predicted Pro 35 by Leu substitution was also present in two normal subjects (PS-33-
The mutations we detected in the patients were associated with type I deficiency, except one (IVS E, +5 G to A) associated with a type IIa phenotype and three (Arg-2 to Leu, Arg-l to His and Thr 103 to Asn) associated with a type IIb phenotype.
Four propositi had a type IIb protein S deficiency. Three families bore mutations affecting the codon corresponding to Arg-2 (two families) or Arg-l (one family), amino acids belonging to the cleavage site of the propeptide. The third mutation affected the codon corresponding to Thr 103.
The Arg-2 to Leu and Arg- 1 to His mutations are probably deleterious. Indeed, mutations on the corresponding amino
019 and PS-33-020).
Table 3. Main Clinical Features Observed in 17 Patients With a Candidate Causal Mutation
Family Age Symptomatology (age of Familial History of Pedigree (in years1 the first thrombotic event) Thrombosis
PS-33-001 PS-33-002 PS-33-003 PS-33-004 PS-33-005 PS-33-006 PS-33-007 PS-33-008 PS-33-009 PS-33-010 PS-33-0 11 PS-33-0 12 PS-33-013 PS-33-01 4 PS-33-01 5 PS-33-01 6 PS-33-017
30 14 44 24 33 29 34 45 39 34 24 27 39 30 56 36 42
MI (27) DVT (12) DVT + PE (40) DVT + PE (21) None DVT (20) DVT (22) DVT (25) MI DVT + PE (26) None MI (23) DVT + PE (35) Stroke (25) DVT + PE (42) SVT after perfusion None
Yes No Yes Yes Yes Yes Yes Yes No Yes No No
Yes Yes
(28) Yes Yes
Abbreviations: MI, myocardial infarction; DVT, deep venous throm- bosis; SVT, superficial venous thrombosis; and PE, pulmonary embo- lism.
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom
136 GANDRILLE ET AL
Table 4. Sequences Variations That Are Probably Polymorphisms (detected in patients or in normal subjects)
Codon With the Sequence Variation
35, CCG "* CTG* Pro --t Leu PS-33-018 78 67 120
Antigen Predicted Family Mutation Pedigree Total Free Activity
PS-33-019 84 62 ND PS-33.020 75 95 84
303, ATC --t A n * Ile + Ile PS-33-001 See -2 CGT "* ClT (Table 2)
344, ATG "* GTG Met "* Val PS-33.021 90 60 ND 626, CCAJCCG* Pro/Pro Frequent polymorphism, see text
PS-33-002 See -2 CGT "* CTT (Table 2)
Normal range = 70% to 130% with the three assays. * Mutation on CpG dimer.
acids of factor IX45,46 and protein C4* are associated with qualitative deficiencies of these two proteins.
Such mutations may lead to a defect in propeptide cleav- age and/or to defective carboxylation of the GLA domain. The first possibility has been shown to account for the factor IX defect associated with an Arg-l mutation, leading to the presence of factor IX still connected to the propeptide in the c i rc~la t ion .~~ However, immunoblot analysis of plasma showed a normal protein S molecular mass in our patients (not shown). To test the second possibility, we submitted plasma from one of the patients with the Arg-2 to Leu muta- tion to pseudo-affinity chromatography according to Yan et al.47 This method separates proteins with different degrees of carboxylation according to their affinity for calcium ions. About half the protein S molecules of this patient were not eluted by calcium ions (not shown), suggesting abnormal carboxylation.
Surprisingly, the two propositi (PS-33-001 and PS-33- 002) bearing the Arg-2 to Leu mutation were alone among the 100 patients and 100 normal subjects to present a poly- morphism in the codon corresponding to Ile 303. As both patients were African, a founder effect may explain the re- currence of this mutation. However, it is possible that this polymorphism is frequent in Africans.
The deleterious effect of the Thr 103 to Asn mutation may result from steric hindrance caused by the side chain of Asn. Thr 103 is located between two Cys residues, which are involved in the conformation of the first epidermal growth factor-like domain of protein S. Asn may modify the spatial conformation of this domain and thereby disturb certain mo- lecular interactions.
The mutation located at position +5 of the 5' splice site sequence probably causes defective pre-mRNA splicing. In most cases, the 5' splice site consensus sequence has a G at this position. The replacement of G by A might result in splicing abnormalities such as exon skipping, as shown re- cently in protein C defi~iency.~' More difficult to explain is the association of such a DNA abnormality with type IIa protein S deficiency, theoretically caused by synthesis of abnormal protein S with increased affinity for its carrier protein, C4b-BP. It may be that the abnormally spliced mRNA is translated into an abnormal protein, but this re- mains to be examined.
As shown in Table 2, the mutations responsible for type I protein S deficiencies were heterogeneous.
Two of these mutations, Cys 22 to stop and a l-base-
pair deletion in the codon corresponding to Pro 82 (already described in reference no. 22), can be considered as deleteri- ous, because they interrupt translation and create truncated proteins, one with 21 and the other with 90 amino acids.
All the other patients bore a missense mutation, the delete- rious effect of which is more difficult to ascertain.
The location of some of these substituted amino acids points to a detrimental effect of the mutations. By aligning the sequence of protein S with that of other vitamin K- dependent proteins (Fig 4), we observed that Glu 26, Phe 31, and Thr 37 are conserved in vitamin K-dependent pro- teins and probably play a key role in protein structure and conformation. A mutation affecting these amino acids might thus alter protein folding or impair intracellular acquisition of the correct ternary structure. Mutations of Glu 27 (Glu 27 to Val) or Thr 38 (Thr 38 to Arg) of factor IX, which correspond respectively to Glu 26 and Thr 37 of protein S , are associated with severe hemophilia
By aligning the protein S EGF 4 with the EGF modules of some other vitamin K-dependent proteins, it emerged that Asp 204 and Cys 224 are both highly conserved. It would not be surprising if a mutation affecting an EGF Cys involved in the ternary structure of the protein was deleteri- ous, and this is corroborated by the fact that a mutation of the corresponding Cys (Cys 109) of factor IX is associated with severe hemophilia B (Knoblock and Zoll, unpublished results mentioned in databa~e).~
One patient had an apparently homozygous 344 Met to Val mutation. A possible explanation for the absence of the allele bearing the normal codon is a large deletion of this
Fig 4. Alignment of human protein S" mutation-bearing se- quences with bovine protein S,- human- and bovine" protdn C, and human factors Iny X,% and VII.' Asterisk indicates perfectly Conserved and bullet indicates well-conserved amino acids.
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom

MUTATIONS IN THE ACTIVE PROTEIN S GENE 131
part of the gene, making the patient hemizygous. However, the low protein S level found before inclusion of the patient in this series was not confirmed several months later. In the absence of other available family members it is difficult to clarify this issue.
Finally, the mutation Arg 49 to His detected in patient PS-33-009 did not cosegregate with the protein S deficiency, as the two children of the prepositus were PS-deficient and did not bear the mutation. However, this mutation affects an Arg that is conserved among different mammalian PSs0”’ and could thus be considered as deleterious. It is possible that the children bear another deleterious mutation transmitted by their father. Experiments are underway to find mutations in another part of the PS gene of the two children.
In conclusion, we applied a strategy that permits the screening of large series of patients for mutations of the coding sequences of the PSa gene. Using this tool, we de- tected three novel polymorphisms and identified 15 different candidate mutations in 19 patients with protein S deficien- cies. This shows that point mutations, especially missense mutations, can cause protein S deficiency.
ACKNOWLEDGMENT
We thank Drs Lerman and Silverstein who kindly provided us with the computer programs MELT 87 and SQHTX.
REFERENCES 1. Tuddenham EGD, Cooper DN, Gitschier J, Higuchi M, Hoyer
LW, Yoshioka A, Peake IR, Schwaab R, Olek K, Kazazian HH, Lavergne JM, Giannelli F, Antonarakis SE: Haemophilia A: Data- base of nucleotide substitutions, deletions, insertions and re- arrangements of the factor VIII gene. Nucl Acid Res 19:4821, 1991
2. Olds RJ, Lane DA, Chowdhury V, De Stefan0 V, Leone G, Thein SL: Complete nucleotide sequence of the antithrombin gene: Evidence for homologous recombination causing thrombophilia. Biochemistry 32:4216, 1993
3. Reitsma PH, Poort SR, Bernardi F, Gandrille S, Long GL, Sala N. Cooper DN: Protein C deficiency: A database of mutations. For the protein C and S subcommittee of the scientific and standardiza- tion committee of the international society on thrombosis and haemo- stasis. Thromb Haemost 69:77, 1993
4. Giannelli F, Green PM, High KA, Sommer S, Poon MC, Lud- wig M, Schwaab R, Reitsma PH, Goossens M, Yoshioka A, Brownlee G G : Haemophilia B: Database of point mutations and short additions and deletions-fourth edition, 1993. Nucl Acid Res 21:3075, 1993
5. Higuchi M, Antonarakis SE, Kasch L, Oldenburg J, Econo- mou-Petersen E, Olek K, Arai M, Inaba H, Kazazian HH Jr: Molecu- lar characterization of mild-to-moderate hemophilia A: Detection of the mutation in 25 of 29 patients by denaturing gradient gel electrophoresis. Proc Natl Acad Sci USA 88:8307, 1991
6. Lane DA, Olds RI, Boisclair M, Chowdhury V, Thein SL, Cooper DN, Blajchman M, Perry D, Emmerich J, Aiach M: Anti- thrombin I11 mutation database: First update. For the thrombin and its inhibitors subcommittee of the scientific and standardization com- mittee to the international society on thrombosis and haemostasis. Thromb Haemost 70:361, 1993
7. Lakich D, Kazazian HH, Antonarakis SE, Gitschier J: Inver- sions disrupting the factor VI11 gene are a common cause of severe haemophilia A. Nat Genet 5:236, 1993
8. Comp P, Esmon C: Recurrent venous thromboembolism in patients with a partial deficiency of protein S. N Engl J Med 31 1:1525, 1984
9. Schwartz H, Fischer M, Hopmeier P, Batard MA, Griffin JH: Familial protein S deficiency is associated with recurrent thrombosis. Blood 64:1297, 1984
10. Girolami A, Simioni P, Lazzaro AR, Cordiano I: Severe arte- rial cerebral thrombosis in a patient with protein S deficiency (mod- erately reduced total and markedly reduced free protein S): A family study. Thromb Haemost 61:144, 1989
11. Allaart C, Aronson D, Ruys TH, Rosendaal F, Bockel J, Bertina RM, Briet E: Hereditary protein S deficiency in young adults with arterial occlusive disease. Thromb Haemost 64:206, 1990
12. Watkins PC, Eddy R, Fukushima Y, Byers MG, Cohen EH, Dackowski WR, Wydro RM, Shows EH: The gene for protein S maps near the centromere of human chromosome 3. Blood 71:238, 1988
13. Ploos van Amstel HK, Reitsma PH, van der Logt PE, Bertina RM: Intron-exon organization of the active human protein S gene PSa and its pseudogene PS@ Duplication and silencing during pri- mate evolution. Biochemistry 29:7853, 1990
14. Schmidel DK, Tatro AV, Tomczak JA, Long GL: Organiza- tion of the human protein S genes. Biochemistry 29:7845, 1990
15. Edenbrandt CM, Lundwall A, Wydro R, Stenflo J: Molecular analysis of the gene for vitamin K dependent protein S and its pseudogene. Cloning and partial gene organization. Biochemistry 29:7861, 1990
16. Ploos van Amstel HK, Huisman MV, Reitsma PH, ten Cate JW, Bertina RM: Partial protein S gene deletion in a family with hereditary thrombophilia. Blood 73:479, 1989
17. Schmidel DK, Nelson RM, Broxson EH Jr, Comp PC, Marlar RA, Long GL: A 5.3-kb deletion including exon XI11 of the protein S a gene occurs in two protein S-deficient families. Blood 77:551, 1991
18. Yamazaki T, Sugiura I, Matsushita T, Kojima T, Kagami K, Takamatsu J, Saito H: A phenotypically neutral dimorphism of pro- tein S: The substitution of Lys 155 by Glu in the second EGF domain predicted by an A to G base change in the gene. Thromb Res 70:395, 1993
19. Hayashi T, Nishioka J, Shigekiyo T, Saito S, Suzuki K: Pro- tein S Tokushima: Abnormal molecule with a substitution of Glu for Lys-155 in the second epidermal growth factor-like domain of protein S. Blood 83:683, 1994
20. Reitsma PH, Ploos van Amstel HK, Bertina RM: Three novel mutations in five unrelated subjects with hereditary protein S defi- ciency type I. J Clin Invest 93:486, 1994
21. Gomez E, Ledford MR, Pegelow CH, Reitsma PH, Bertina RM: Homozygous protein S deficiency due to a one base pair dele- tion that leads to a stop codon in exon 111 of the protein S gene. Thromb Haemost 71:723, 1994
22. Borgel D, Gandrille S, Gouault-Heilmann M, Aiach M: First frameshift mutation in the active protein S gene associated with a quantitative hereditary deficiency. Blood Coagul Fibrinolysis 5593, 1994
23. Comp PC: Laboratory evaluation of protein S status. Semin Thromb Hemost. 16:177, 1990
24. Bell G , Karam J, Rutter W: Polymorphic DNA region adja- cent to the 5‘ end of the human insuline gene. Proc Natl Acad Sci USA 78:5759, 1981
25. Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA: Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487, 1988
26. Lerman LS, Silverstein K: Computational simulation of DNA melting and its application to denaturing gradient gel electrophoresis, in Methods in Enzymology. Wu R (ed) New York, NY, Academic Press, 1987, pp 482-501
27. Sheffield VC, Cox DR, Lerman LS, Myers RM: Attachment of a 40-base-pair G + C-rich sequence (GC-clamp) to genomic DNA
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom

138 GANDRILLE ET AL
fragments by the polymerase chain reaction results in improved detection of single-base changes. Proc Natl Acad Sci USA 86:232, 1989
28. Myers RM, Fischer SG, Maniatis T, Lerman LS: Modification of the melting properties of duplex DNA by attachment of a GC- rich DNA sequence as determined by denaturing gradient gel electro- phoresis. Nucl Acid Res 13:311 I , 1985
29. Myers RM, Fischer SG, Lerman LS, Maniatis T: Nearly all single base substitutions in DNA fragments joined to a GC-clamp can be detected by denaturing gradient gel electrophoresis. Nucl Acid Res 13:3131, 1985
30. Newton C, Graham A, Heptinstall L, Powell S, Summers C, Kalshener N. Smith J, Markham A: Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucl Acid Res 17:2503, 1989
3 1. Attree 0, Vidaud D, Vidaud M, Amselem S , Lavergne JM, Goossens M: Mutations in the catalytic domain of human coagula- tion factor IX: Rapid characterization by direct genomic sequencing of DNA fragments displaying an altered melting behavior. Genomics 4:266, 1989
32. Gyllensten UB, Erlich HA: Generation of single-stranded DNA by the polymerase chain reaction and its application to direct sequencing of the HLA-DQA locus. Proc Natl Acad Sci USA 85:7652, 1988
33. Sanger F, Nicklen S , Coulson AR: DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463, 1977
34. Diepstraten CM, Ploos Van Amstel JK, Reitsma PH, Bertina RM: A CCNCCG neutral polymorphism in the codon for Pro 626 of the human protein S gene PS alpha (PROSI). Nucl Acid Res 19:5091, 1991
35. Tartary M, Vidaud D, Pia0 Y, Costa JM, Bahnak BR, Fressi- naud E, Congard B, Laurian Y, Meyer D, Lavergne JM: Detection of a molecular defect in 40 of 44 patients with haemophilia B by PCR and denaturing gradient gel electrophoresis. Br J Haematol 84:662, 1993
36. Kogan S , Gitschier J: Mutations and a polymorphism in the factor VI11 gene discovered by denaturing gradient gel electrophore- sis. Proc Natl Acad Sci USA 87:2092, 1990
37. Ghanem N, Girodon E, Vidaud M, Martin J, Fanen P, Plassa F, Goossens M: A comprehensive scanning method for rapid detec- tion of a-globin gene mutations and polymorphisms. Hum Mut I :229, 1992
38. Ferec C, Audrezet MP, Guillermit H, Moullier P, Quere I , Verlingue C: Detection of over 98% cystic fibrosis mutations in a Celtic population. Nat Genet 1:188, 1992
39. Fanen P, Ghanem N, Vidaud M, Besmond C, Martin J, Costes B, Plassa F, Goossens M: Molecular characterization of cystic fibro- sis: 16 Novel mutations identified by analysis of the whole cystic fibrosis conductance transmembrane regulator (CFTR) coding re- gions and splice site junctions. Genomics 13:770, 1992
40. Top B, Van Der Zee A, Havekes LM, Van’t Hooft FM, Frants RR: Identification of a splice-site mutation in the low density lipo- protein receptor gene by denaturing gradient gel electrophoresis. Hum Genet 91:480, 1994
41, Gandrille S, Vidaud M, Aiach M, Alhenc-Gelas M, Fischer
AM, Gouault-Heilmann M, Toulon P, Fiessinger JN, Goossens M: Two novel mutations responsible for hereditary type I protein C deficiency. Characterization by denaturing gradient gel electrophore- sis. Hum Mut 1:491, 1992
42. Gandrille S , Alhenc-Gelas M, Gaussem P, Aillaud MF, Du- puy E, Juhan-Vague I, Aiach M: Five novel mutations located in exons TI1 and IX of the protein C gene in patients with defective protein C anticoagulant activity. Blood 82:159, 1993
43. Gandrille S , Jude B, Alhenc-Gelas M, Aiach M: Compound heterozygosity in a family with quantitative protein C deficiency illustrating the complexity of underlaying molecular mechanism. Thromb Haemost 70:747, 1993
44. Gandrille S , Goossens M, Aiach M: Scanning method to es- tablish the molecular basis of protein C deficiencies. Hum Mut 4:20, I994
45. Thompson AR, Schoof JF, Weinmann AF, Chen SH: Factor IX mutations: Rapid, direct screening methods for 20 new families with hemophilia B. Thrumb Res 65:289, 1992
46. Diuguid DL, Rabiet MJ, Furie BC, Liebman HA, Furie B: Molecular basis of hemophilia B: A defective enzyme due to an unprocessed propeptide is caused by a point mutation in the factor IX precursor. Proc Natl Acad Sci USA 83:5803, 1986
47. Yan SCB, Razzano P, Chao YB, Walls JD, Berg DT, McClure DB, Grinnell BW: Characterization and novel purification of recom- binant human protein C from three mammalian cell lines. Biotech- nology 8:655, 1990
48. Lind B, van Solinge WW, Schwaxtz M, Thorsen S : Splice site mutation in the human protein C gene associated with venous thrombosis: Demonstration of exon skipping by ectopic transcript analysis. Blood 82:2423, 1993
49. Wang NS, Zhang NS, Thompson AR, Chen SH: Factor IX Chongqing: A new mutation in the calcium-binding domain of factor IX resulting in severe hemophilia B. Thromb Haemost 63:24, 1990
SO. Dahlback B, Lundwall A, Stenflo J: Primary structure of bo- vine vitamin K-dependent protein S. Proc Natl Acad Sci USA 83:4199, 1986
5 1. Chu MD, Sun J, Bird P: Cloning and sequencing of a cDNA encoding the murine vitamin K-dependent protein S. Biochem Bio- phys Acta 1217:325, 1994
52. Foster D, Davie EA: Characterization of a cDNA coding for human protein C. Proc Natl Acad Sci USA 81:4766, 1984
53. Long G, Belegaje RM, MacGillivray RTS: Cloning and se- quencing of liver cDNA coding for bovine protein C. Proc Natl Acad Sci USA 81:5653, 1984
54. Yoshitake S, Schach BC, Foster D, Davie EW, Kurachi K: Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 24:3736, 1985
55. Leytus SP, Foster DC, Kurachi K, Davie EW: Gene for human factor X: A blood coagulation factor whose gene organization is essentially identical with that of factor IX and protein C. Biochemis- try 25:5098, 1986
56. Hagen F, Gray CL, O’Hara P, Grant FJ, Saari GC, Woodbuy RG, Hart CE, Insley M, Kisiel W, Kurachi K, Davie EW: Character- ization of a cDNA coding for human factor VII. Proc Natl Acad Sci USA 83:2412, 1986
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom

1995 85: 130-138
Abgrall, B Jude and P SieS Gandrille, D Borgel, V Eschwege-Gufflet, M Aillaud, M Dreyfus, C Matheron, P Gaussem, JF scanning method for the analysis of the protein S active genethree polymorphisms in 19 patients with protein S deficiency using a Identification of 15 different candidate causal point mutations and
http://www.bloodjournal.org/content/85/1/130.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on April 10, 2019. by guest www.bloodjournal.orgFrom





























