Embed Size (px)
Citation preview

Identification and characterization of novel regulators in
mitochondria related cell death
Ph.D. Thesis
Enikő Hocsák
Program leader: Professor Balázs Sümegi, DSci
University of Pécs, Medical School
Department of Biochemistry and Medical Chemistry
Hungary
2010

Hocsák, E., Identification and characterization of novel regulators
2
1.Contents
1.Contents 2
2. Abbreviations 4
3. Introduction 6
3.1. The role of mitochondria in cell death 6
3.1.1. Mitocondrial Membrane Polarization (∆ψ) 7
3.1.2. The mitochondrial permeability transition (MPT) 7
3.1.3. BH3 domain proteins, the Bcl-2 family 9
3.2. The TIP47 protein 11
3.3. SOUL/HBP2, a novel BH3 domain protein 13
4. Aims of the study 15
5. Materials and methods 16
5.1. Chemicals and antibodies 16
5.2. Preparation of polyclonal antibodies 16
5.3. Construction of expression plasmids, expression and purification 17
5.4. Stable transfection 17
5.5. Cell culture and sample preparation 17
5.6. Western blot analysis 18
5.7. Suppression of TIP47 expression by diced siRNA technique 18
5.8. Cell viability assay 19
5.9. Determination of intracellular reactive oxygen species (ROS) 19
5.10. Detecting mitochondrial membrane potential (∆ψ) 19
5.11. Isolation of mitochondria for the in vitro study 20
5.12. Annexin V-FITC assay 20
5.13. Flow cytometry analysis 21
5.14. Statistical analysis 21
6. Results 22
6.1. TIP47 protects mitochondrial membrane integrity and inhibits cell death 22
6.1.1. Effect of expression of TIP47 on H2O2 or taxol-induced cell death 22
6.1.2. The effect of taxol treatment on the cell viability of HeLa cells
transfected with TIP47 siRNA 23
6.1.3. Induction of apoptosis by H2O2, Taxol and etoposide in NIH3T3 cells 24

Hocsák, E., Identification and characterization of novel regulators
3
6.1.4. Detection of H2O2 induced apoptosis by flow cytometry 26
6.1.5. Localization of TIP47 in oxidative stress 27
6.1.6. Effect of recombinant TIP47 on the mPT and ∆ψ in Vitro 27
6.1.7. The effect of TIP47 on release of mitochondrial intermembrane
space proteins from living cells 30
6.1.8. Effect of TIP47 on the mitochondrial membrane potential in NIH3T3
cells 31
6.1.9. Effect of TIP47 on ROS-production 32
6.1.10. The effect of TIP47 on the pro- and anti-apoptotic signaling
pathways 33
6.2. Cell death in oxidative stress by a novel BH3 domain protein 35
6.2.1. Characterization of SOUL protein 35
6.2.2.Determination of the role of mPT complex in the SOUL-induced
sensitization toward oxidative stress 35
6.2.3. Effect of deletion of the putative BH3 domain on the cell death
facilitating effect of SOUL 37
6.2.4. Effect of SOUL on cell death is inhibited by the overexpression of
anti-apoptotic proteins 38
6.2.5. Effect of SOUL on the hydrogen-peroxide-induced collapse of MMP 42
7. Discussion 44
8. Conclusions 49
9. Acknowledgements 52
10. References 53
11. List of Publications 63

Hocsák, E., Identification and characterization of novel regulators
4
2. Abbreviations
A23187 5-(methylamino)-2-({(2R,3R,6S,8S,9R,11R)-3,9,11-trimethyl-8-[(1S)-1-
methyl-2-oxo-2-(1H-pyrrol-2-yl)ethyl]-1,7-dioxaspiro[5.5]undec2-yl}methyl)-
1,3-benzoxazole-4- carboxylic acid
ADRP adipose differentiation-related protein
AIF apoptosis inducing factor
ANT adenine nucleotide translocase
Bcl-2 B-cell lymphoma
C-400 5-(and-6)-carboxy-2', 7’-dichlorodihydrofluorescein diacetate
CIN cervical intraepithelial neoplasia
CypD cyclophilin D
cyt-c cytochrome c
CsA, cyclosporine A
DIABLO direct IAP–binding protein of low isoelectric point [pI]
DMEM Dulbecco’s modified Eagle’s medium
dsiRNA diced small interfering ribonucleic acid
Endo G endonuclease G
Etoposide 4' - demethyl - epipodophyllotoxin 9 - [4,6-O-(R)-ethylidene-beta-D-
glucopyranoside], 4' - (dihydrogen phosphate)
FCS fetal calf serum
FITC fluorescein isothiocyanate
G418 (2R,3S,4R,5R,6S)-5-amino-6-[(1R,2S,3S,4R,6S)-4,6-diamino-3-
[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-methylaminooxan-2-yl]oxy-2-
hydroxycyclohexyl]oxy-2-(1-hydroxyethyl)oxane-3,4-diol
GFP green fluorescent protein
GST glutathione S-transferase
H2O2 hydrogen-peroxide
HBP22 heme binding protein 1
HIV-1 Human Immunodeficiency Virus
IAP inhibitor of apoptosis binding protein
IPTG isopropyl-β-D-thiogalactopyranoside
JC-1 5,5,6,6-tetracloro-1,1,3,3 -tetraethylbenzimidazolyl-carbocyanine iodide

Hocsák, E., Identification and characterization of novel regulators
5
M6PR mannose 6-phosphate receptors
MMP mitochondrial membrane permeabilization
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
mPT mitochondrial permeability transition
∆ψ mitochondrial membrane potential
NCBI National Center for Biotechnology Information
PAT perilipin, adipophilin, and TIP47
PBS phosphate buffered saline
PCR Polymerase Chain Reaction
pI isoelectric point
PI propidium iodide
PP17b placental tissue protein 17b
PTP permeability transition pore
PTPC permeability transition pore complex
Rh-123 rhodamine-1,2,3
ROS reactive oxygen species
siRNA small interfering RNA
Smac second mitochondrial activator of caspases
SOUL/HBP2 heme binding protein 2
Taxol (2α,4α,5β,7β,10β,13α)-4,10-bis (acetyloxy)-13-{[(2R,3S)-3-(benzoylamino)-2-
hydroxy-3-phenylpropanoyl]oxy} -1,7-dihydroxy-9-oxo-5,20-epoxytax-11-
en-2-yl benzoate
TIP47 tail interacting protein of 47 kDa
t-TIP47 truncated-TIP47
VDAC voltage-dependent anionic channel

Hocsák, E., Identification and characterization of novel regulators
6
3. Introduction
For the past 15 years our laboratory has been involved of identified several genes with
previously unknown functions that affected cell death processes such as tail interacting protein
of 47 kDa (TIP47) or Heme Binding Protein 2 (HBP2/ SOUL). In this study the effect of these
proteins in mitochondrial releated cell death is described.
This work provides compelling evidence of the protective effect of TIP47 which exerts an
antiapoptotic effect on cell survival by inhibiting the activation of proapoptotic proteins, by
assisting the effect of anti-apoptotic proteins and by the direct stabilization of the
mitochondrial membrane potential.
We identified genes, which in contrast, destabilize the mitochondrial membrane system,
such as SOUL, which sensitized fibroblast cells to cell death in the absence of elevated ROS
formation. In the presence of subthreshold calcium, recombinant SOUL provoked
mitochondrial permeability transition (mPT) in vitro, which was inhibited by cyclosporine A
(CsA). In vivo, SOUL overexpression enhanced the dissipation of mitochondrial membrane
potential, as well as SOUL promoted necrotic death in etoposide and A23187 treated cells,
which effect was prevented by CsA.
This Thesis presents my own individual research efforts with my colleague team members.
3.1. The role of mitochondria in cell death
The cell’s decision to die from necrosis or apoptosis is dictated at least in part by the
abundance of intracellular energy stores. Indeed, whereas apoptosis requires a minimal amount
of intracellular ATP, necrosis is generally accompanied by its total depletion [1-2].
The intrinsic cell death pathway involves the initiation of apoptosis as a result of a
disturbance of intracellular homeostasis. In this pathway, mitochondria are critical for the
execution of cell death, and so this pathway has been referred to as the mitochondrial cell
death pathway.

Hocsák, E., Identification and characterization of novel regulators
7
3.1.1. Mitocondrial Membrane Polarization (∆ψ)
Mitochondrial membrane permeabilization (MMP) is frequently the decisive event that
delimits the frontier between survival and death. Local players that determine the propensity to
MMP include the pro- and antiapoptotic members of the B-cell lymphoma (Bcl-2) family,
proteins from the mitochondrial permeability transition pore complex, as well as a plethora of
interacting partners including mitochondrial lipids. The inhibition of MMP constitutes an
important strategy for the pharmaceutical prevention of unwarranted cell death. Conversely,
induction of MMP in tumor cells constitutes the goal of anticancer chemotherapy [3].
MMP is characterized by several hallmarks that include:
1) the release of cytochrome c (cyt-c), second mitochondria-derived activator of caspase/direct
inhibitor of apoptosis binding protein (IAP) with a low isoelectric point (pI) (Smac/DIABLO),
through the mitochondrial outer membrane and the subsequent activation of effector caspases;
2) the release of caspase-independent apoptogenic death effectors, such as apoptosis inducing
factor (AIF) and endonuclease G (EndoG);
3) a bioenergetic catastrophe including the arrest of oxidative phosphorylation, as well as the
accumulation of reactive oxygen species (ROS),
4) an alteration of the ∆ψ of the mitochondrial inner membrane.
According to current knowledge, the principal mechanism leading to inner membrane
permeabilization is the so-called “permeability transition.”
3.1.2. The mitochondrial permeability transition (mPT)
The mitochondrial permeability transition (mPT) refers to a transition in the permeability
of the mitochondrial inner membrane that occurs when mitochondria in vitro are treated with
calcium [Ca2+] or reagents that increase oxidative stress [4-10].

Hocsák, E., Identification and characterization of novel regulators
8
Fig.1. PTPC architecture [11]. Although the exact molecular composition of the permeability transition pore complex (PTPC) has not been clearly established yet, a consensus has started to emerge about the proteins involved in its scaffold structure, which builds up at the contact sites between the mitochondrial outer and inner membranes (OM and IM). In addition to the VDAC and the adenine nucleotide translocase (ANT), which represent the main PTPC components, these include hexokinase (HK, interacting with VDAC from the cytosol), creatine kinase (CK, interacting with PTPC from the intermembrane space, IMS), peripheral-type benzodiazepine receptor (PBR, interacting with PTPC from OM), and cyclophilin D (CypD, interacting with ANT from the mitochondrial matrix). HK and CK seemingly associate with the PTPC scaffold in a mutually exclusive fashion. Both anti- and proapoptotic members of the Bcl-2 family modulate the activity of PTPC, through direct interactions with ANT or VDAC.
The exact molecular nature of the permeability transition pore, the PTP is still a matter of
debate, although an emerging consensus considers a multicomponent protein complex, the
PTPC, and not a single protein, as being responsible for the opening of PTP. PTPC is
assembled at the contact sites of the mitochondrial membranes and that its scaffold structure is
based on the dynamic interaction between VDAC, ANT, and CypD [1, 10, 12]. The voltage-
dependent anionic channel, VDAC protein is a pore in the mitochondrial outer membrane that
allows low molecular weight solutes to pass and gain access to inner membrane transport
systems. The adenine nucleotide translocator, ANT is a structural component of the MPT, and
matrix cyclophilin D (CyP-D) bound to complexes of VDAC and the ANT to form the MPT
[7].
Hocsák, E., Identification and characterization of novel regulators
9
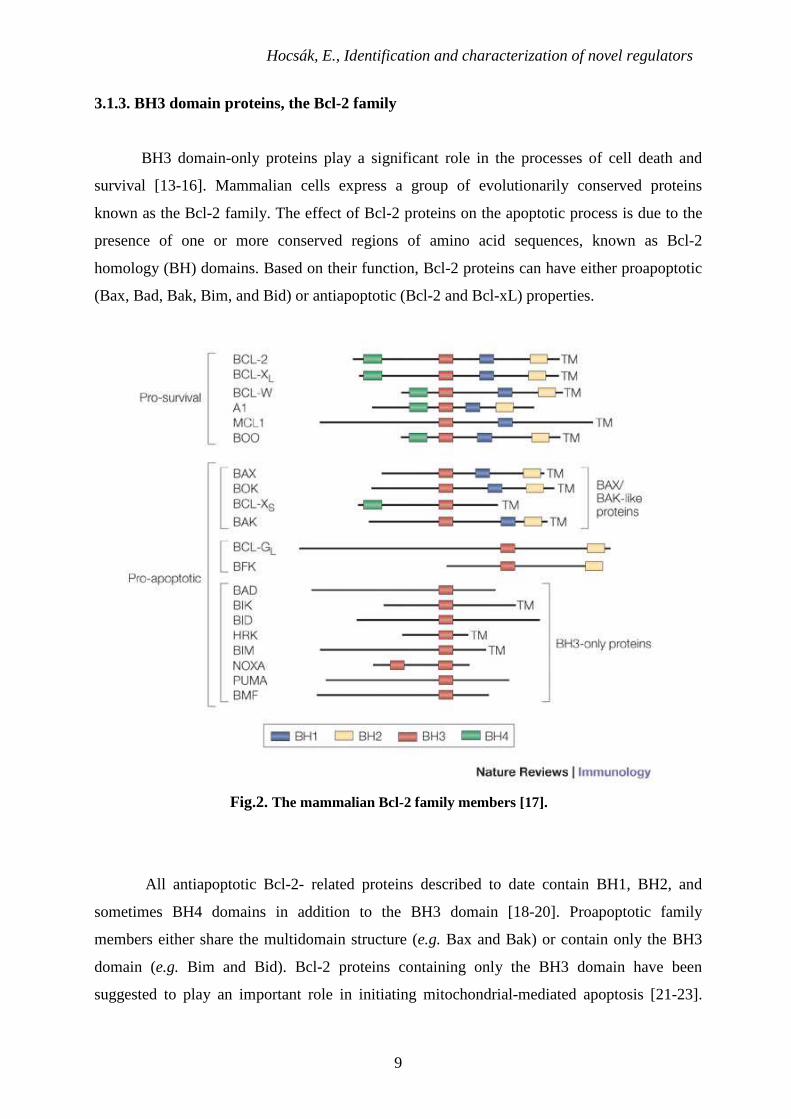
3.1.3. BH3 domain proteins, the Bcl-2 family
BH3 domain-only proteins play a significant role in the processes of cell death and
survival [13-16]. Mammalian cells express a group of evolutionarily conserved proteins
known as the Bcl-2 family. The effect of Bcl-2 proteins on the apoptotic process is due to the
presence of one or more conserved regions of amino acid sequences, known as Bcl-2
homology (BH) domains. Based on their function, Bcl-2 proteins can have either proapoptotic
(Bax, Bad, Bak, Bim, and Bid) or antiapoptotic (Bcl-2 and Bcl-xL) properties.
Fig.2. The mammalian Bcl-2 family members [17].
All antiapoptotic Bcl-2- related proteins described to date contain BH1, BH2, and
sometimes BH4 domains in addition to the BH3 domain [18-20]. Proapoptotic family
members either share the multidomain structure (e.g. Bax and Bak) or contain only the BH3
domain (e.g. Bim and Bid). Bcl-2 proteins containing only the BH3 domain have been
suggested to play an important role in initiating mitochondrial-mediated apoptosis [21-23].

Hocsák, E., Identification and characterization of novel regulators
10
Proapoptotic BH3 domain-containing proteins are generally regarded as latent death factors
that are normally held in check and must be activated to exhibit their death-inducing functions.
Several mechanisms appear to contribute to the activation of the prodeath function of Bcl-2
family proteins. Bax undergoes conformational changes, relocates to mitochondria, may
oligomerize with other Bax molecules in the mitochondrial membrane, and can be cleaved by
calpain to enhance its proapoptotic effects [24-28]. Bid and Bim share a common mode of
action via BH3-domain-mediated binding to Bax-type proteins at the outer mitochondrial
membrane [26]. This physical interaction is believed to trigger a conformational change of the
multidomain proapoptotic members, resulting in their intramembranous oligomerization and
permeabilization of the outer mitochondrial membrane [27]. In addition, interactions between
proapoptotic Bcl-2 family members and lipid bilayers have an important contribution to this
process. Specific lipids can promote the membrane association of activated forms of Bax and
Bid and can induce mitochondrial cyt-c release [29]. Specifically, cardiolipin increases binding
of both cBid and tBid to pure lipid vesicles as well as to the outer mitochondrial membranes
[30], and myristoylation of tBid further enhances its membrane avidity [31]. Furthermore,
other possible mechanisms of action have been proposed for proapoptotic BH3 domain-only
proteins, including (i) binding to and neutralization or reversal of prosurvival Bcl-2-type
family member functions [13] and (ii) modulation of resident mitochondrial channels, such as
VDAC and ANT [32]. It was demonstrated that VDACs are not an absolutely necessary
component of the mPT complex [33], but when VDAC is present, it can play a role in the
regulation of mPT [34-36]. There are data indicating that proapoptotic Bcl-2 homologues can
activate mPT in oxidative stress showing that an oxidant-damaged mitochondrial membrane
system can react differently to BH3 domain proteins [36-39]. In addition, the oligomer Bax
alone can induce mitochondrial permeability transition and complete cyt-c release without
oxidative stress, indicating that under certain conditions, BH3 domain proteins can contribute
to mitochondrial inner membrane permeabilization, mPT, and necrotic death [35-40].

Hocsák, E., Identification and characterization of novel regulators
11
Fig.3. The molecular basis of apoptosis [41].
It was also demonstrated that antiapoptotic Bcl-xL can bind to the VDAC-1 barrel
laterally at strands 17 and 18 and can influence mitochondrial permeabilization [42-44].
Therefore, antiapoptotic Bcl-2 protein can also influence (protect) the inner mitochondrial
system, probably via interaction with VDAC [42-43, 45-46].
3.2. The TIP47 protein
Tail-interacting protein (TIP47) is a member of the perilipin/ adipophilin/TIP47 (PAT)
family, which also includes perilipin, adipose differentiation-related protein (ADRP), S3-12
and OXPAT proteins [47-53]. PAT family surface proteins are components of the plasma
membrane and they influence cellular lipid metabolism and also insulin signaling, by means of
controlling access of lipases to stored lipid esters [52]. PAT proteins are characterized by
primary sequence homology, which is evolutionary conserved across all species [54]. The
distribution of PAT proteins varies among different tissues [48]: while the expression of

Hocsák, E., Identification and characterization of novel regulators
12
perilipin and S3-12 proteins is restricted to adipose and steroidogenic tissues, ADRP and
TIP47 are found ubiquitously [47, 55-57]. The critical role of PAT proteins in the packaging
and storage of neutral lipids and as components of lipid droplets has been established by a
number of studies [49, 52, 57-62].
TIP47 was primarily identified as a possible cargo protein involved in the delivery of
mannose 6-phosphate receptors (M6PR) from endosomes to the trans-Golgi network [63-64].
It was shown to bind to the cytoplasmic domains of cation-independent and cation-dependent
M6PR [65-66] and to be transported to late endosomes by binding to Rab9 GTPase [67-68].
The recognition of a sequence homology between ADRP and TIP47 [57, 69] led to the
discovery that TIP47, like other PAT family members, associates with lipid droplets [57].
Although this finding was initially questioned [58], the role of TIP47 in lipid metabolism was
subsequently confirmed by various sources [49, 54, 61]. Besides its apparent function in lipid
metabolism, TIP47, along with other PAT proteins, influences insulin signaling [52]. The
combined knockout of ADRP and TIP47 was shown to lead to decreased insulin sensitivity
[70-71]. A recent study reported the possible role of TIP47 in Human Immunodeficiency Virus
(HIV-1) infection [72]. TIP47, as a cellular cofactor, was found to play a part in Env
incorporation, allowing the encounter and physical association between HIV-1 Gag and Env
proteins during the viral assembly process [72].
Parallel to studies conducted by other laboratories, our work-group carried out
experiments to elucidate the role of TIP47, which shed new light on the function of this
controversial protein. TIP47 was found to be identical to a soluble, pregnancy-related placental
tissue protein 17b (PP17b) [73]. Of particular significance was the discovery, that, compared
to normal cervical tissue, TIP47 is overexpressed in cervix carcinoma, as is its messenger
RNA in the HeLa cancer cell line [73-75]. Moreover, a correlation could be detected between
the level of TIP47 expression and the different stages of cervical intraepithelial neoplasia
(CIN) [76]. Moderate immunohistochemical staining of the dysplastic cells was apparent in
CIN I-II, whereas strong staining was observed in CIN III and in situ carcinoma [76].
Recently, the role of TIP47 as a potential tumor marker of cervical carcinoma was suggested
[77]. Since TIP47 is secreted into the serum, and its level is elevated in the progression of the
tumor to invasive or metastatic carcinoma and mitigated after treatment, the monitoring of
TIP47 by immunological methods may be beneficial for cervical cancer patients [77].
However, to this date, there is no data available for its possible role in tumor
development, or in regulation of cell death.

Hocsák, E., Identification and characterization of novel regulators
13
3.3. SOUL/HBP2, a novel BH3 domain protein
Role of heme-containing proteins in the regulation of cell death and survival are well
characterized. They can affect formation of reactive oxygen species hence induction of direct
oxidative damages as well as induction of mitochondrial permeability transition (mPT) [78-
79].
Heme-binding protein 1 (HBP22) is an ubiquitously expressed protein having high
affinity for heme and protoporphyrine [79-80]. Human SOUL (SOUL) is a 23 kDa haem-
binding protein that was first identified as the PP23 protein isolated from human full-term
placentas. [79-80]. SOUL is expressed just in some specific tissues, has more than 40%
sequence homology with HBP22, and has much higher binding affinity for porphyrines than
HBP22 [81].
SOUL facilitates mitochondrial permeability transition and cell death without affecting
reactive oxygen production [79]. The data base search indicated that heme-binding protein
2/SOUL has a BH3 domain-like structure. The presence of a BH3 domain in SOUL and the
above data indicate that this protein besides binding heme may have a role in the processes of
cell death and survival. In present we provide evidence for the sensitization effect of SOUL in
hydrogen-peroxide-induced cell death, the facilitation of the release of pro-apoptotic
mitochondrial proteins and the promotion of the collapse of MMP both in living cells and in
vitro in isolated mitochondria. We show that SOUL promote the permeabilization of both
outer and inner mitochondrial membranes in oxidative stress and its effect can be reversed by
deleting the putative BH3 sequence from SOUL as well as by inhibition of mitochondrial
permeability transition either by cypD suppression or overexpression of anti-apoptotic Bcl-2
members.
SOUL can facilitate cell death in Taxol and A23187-treated cells [79]. Taxol is a
mitotic inhibitor used in cancer chemotherapy. Paclitaxel stabilizes microtubules and as a
result, interferes with the normal breakdown of microtubules during cell division [82-88].
A23187 is a mobile ion-carrier that forms stable complexes with divalent cations (ions with a
charge of +2). A23187 is also known as Calcium ionophore. The ionophore is used to increase
intracellular Ca2+ levels in intact cells and to induce apoptosis in some cells [89].
Analyzing the protein sequence data base, we found that SOUL contained a sequence
with a high similarity to BH3 domains, suggesting that this feature may account for its

Hocsák, E., Identification and characterization of novel regulators
14
observed cell death-facilitating effect. Previously, SOUL was reported to be strongly
expressed in retina, the pineal gland, and liver [90-91]. We found that it was expressed in
various tissues to different extents.

Hocsák, E., Identification and characterization of novel regulators
15
4. Aims of the study
The aim of this work was to further clarify the function of TIP47 protein. In addition,
we investigated interactions of SOUL, a novel BH3 domain protein between Bcl-2 family
members in oxidative stress.
The present study is aimed at a more in-depth study of these two regulator proteins,
including:
• To describe the effect of TIP47 on cell death by measuring cell viability of TIP47
overexpressing and control cells treated by different types of cell death inducing agents
• To show the effect of TIP47 suppression by siRNA technique on cell death in
overexpressing and control cells using a Hela cell modell
• To analyse in role of TIP47 of both apoptotic and necrotic cell death
• To find intracellular localization of TIP47 in oxidative stress
• To describe the effect of TIP47 on the mitochondrial permeability transition and on the
stability of the Mitochondrial Membrane Potential (∆ψ)
• To elucidate the effect of TIP47 on ROS-production in TIP47 overexpressing and
control cells
• To find the effect of TIP47 on the pro- and anti-apoptotic signaling pathways
• To show the effect of deletion of the putative BH3 domain on the cell death
• To determine of the role of mPT complex in the SOUL-induced sensitization toward
oxidative stress
• To describe the effect of SOUL on cell death is inhibited by the overexpression of anti-
apoptotic proteins
• To describe the effect of SOUL on the hydrogen-peroxide-induced collapse of MMP

Hocsák, E., Identification and characterization of novel regulators
16
5. Materials and methods
5.1. Chemicals and antibodies
All the chemicals for cell culture, β-D-1-thiogalactopyranoside (IPTG), Glutathione
Sepharose 4B, methylthiazolyldiphenyl-tetrazolium bromide (MTT), propidium iodide (PI)
and rhodamine-1,2,3 (Rh-123), hydrogen-peroxide (H2O2) were purchased from Sigma-
Aldrich Kft (Budapest, Hungary), Taxol (paclitaxel) was obtained from ICN Biomedicals Inc.
(Aurora, OH). Fluorescent dyes 5,5’,6,6’-tetracloro-1,1’,3,3’-tetraethyl-
benzimidazolylcarbocyanine iodide (JC-1) and fluorescein isothiocyanate (FITC)-conjugated
Annexin V were purchased from Molecular probes (Leiden, Netherlands). The antibodies for
Western blots were purchased as follows: anti-Bax, anti-Bcl-2 and anti-Caspase-3 from Cell
Signaling Technology (Beverly, MA); anti rabbit IgG from Sigma-Aldrich Kft (Budapest,
Hungary), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Delta Biolabs
(Gilroy, CA); anti-AIF and anti-endonuclease G polyclonal antibodies from Oncogene (San
Diego, CA). Mammalian vector pcDNA3.1 was obtained from Invitrogen (Carlsbad, CA,
USA), pEGFP-C1 vector was from BD Biosciences (Franklin Lakes, NJ, USA), Bcl-2-
overexpressing clones were ordered from RZPD GmbH (Berlin, Germany) and pEGFP-C1
vector was purchased from BD Biosciences (Franklin Lakes, NJ, USA). ∆BH3-SOUL plasmid
was ordered from MrGene.com (Mr. Gene GmbH, Regensburg, Germany).
5.2. Preparation of polyclonal antibodies
Two rabbits were immunized subcutaneously at multiple sites with 100 pg of
recombinant TIP47/Glutation-S-transferase (GST) or SOUL/GST fusion protein (with GST
located at the N-terminal of the fusion protein) in Freund's complete adjuvant (Sigma). Four
subsequent booster injections at 4-week intervals were given with 50 pg of protein in Freund's
incomplete adjuvant (Sigma). Blood was collected 14 days after boosting, and the antiserums
were stored at −20˚C. The IgG fraction was isolated from the sera by protein G-Sepharose
chromatography and affinity purified using recombinant TIP47 or SOUL protein bound to a
CNBr-activated Sepharose 4B column and affinity chromatography was carried out as
described previously [92].

Hocsák, E., Identification and characterization of novel regulators
17
5.3. Construction of expression plasmids, expression and purification
The whole open reading frame of TIP47, t-TIP47 and SOUL were cloned into pGEX-
4T-1 expression vector (Pharmacia, Uppsala, Sweden). The TIP47-, t-TIP47-, SOUL-pGEX-
4T-1 expression vectors were first transformed into E. coli DH5α then into E. coli BL21
competent host strain. Bacteria were induced with isopropyl-b-D-thiogalactopyranoside, and
the expressed TIP47-GST, t-TIP47-GST or SOUL-GST fusion proteins were subsequently
purified with Glutathione Sepharose 4B column (Pharmacia) in the presence of glutathione.
∆BH3-SOUL was cloned into pcDNA3.1.
5.4. Stable transfection
Full length TIP47 cDNA, t-TIP47 cDNA, SOUL cDNA and ∆BH3-SOUL cDNA were
PCR amplified and the construct was subcloned into a pcDNA3.1 (Invitrogen) mammalian
vector containing a Geneticin (G-418) resistance gene. The vectors containing TIP47, t-TIP47,
SOUL, ∆BH3-SOUL or the empty pcDNA3.1 or pEGFP-C1 (BD Biosciences) vector was
transfected into NIH3T3 cells with Lipofectamin 2000 according to the manufacturer’s
protocol (Invitrogen). Cells were then grown in selective media (10% FCS-DMEM containing
500µg/ml G-418). Cell clones were subsequently screened by Western blot analysis for an
increase in TIP47 or SOUL protein expression relative to that in the pcDNA-transfected cells.
TIP47-, t-TIP47-, SOUL- or ∆BH3-SOUL overexpressing clones were selected for further
experiments.
5.5. Cell culture and sample preparation
NIH3T3 and HeLa cells were purchased from the American Type Culture Collection
(Manassas, VA). All cell lines were maintained in Dulbecco's Modified Eagle Medium
(DMEM) supplemented with 10% FCS, 2 units/ml penicillin-streptomycin mixture (Flow
Laboratories, Rockville, MD) and incubated in 5% CO2 - 95% air at 37°C. Cells were
harvested and low-speed centrifuged, then the pellet was dispersed by vortexing in lysis buffer

Hocsák, E., Identification and characterization of novel regulators
18
(50 mM Tris pH 7.4, 1 mM PMSF) for 10 min at 4ºC. After further cell disruption in a
Teflon/glass homogenizer, the homogenate was pelleted, and the supernatant was measured by
BCA reagent and equalized for 1 mg/ml protein content in Laemmli solution for Western
blotting.
Isolation of subcellular fractions (cytosol, nuclear and mitochondrial fractions) was
carried out by standard lab protocols as described previously [93-94].
5.6. Western blot analysis
100 ng/lane protein of cell culture or mitochondrion samples were subjected to 12 %
(w/v) SDS/PAGE (BioRad, Hercules, Ca, USA). Immunoblots were blocked, and developed
with polyclonal anti-TIP47 or anti-SOUL antibody [77] or polyclonal antibodies against the
cytosolic marker glycerinaldehyde-3-phosphate dehydrogenase (GA3PD), the mitochondrial
marker pyruvate decarboxylase 1α (PDC-1α) and the nuclear marker histon H1 (Histon H1), or
polyclonal antibodies against the signal transduction pathway proteins and horseradish-
peroxidase-labeled secondary IgG as described in [94]. Protein bands were revealed by ECL
chemiluminescence system. All experiments were repeated four times.
5.7. Suppression of TIP47 expression by diced siRNA technique
The whole coding sequence of TIP47 was Polymerase Chain Reaction (PCR)
amplified, double-stranded RNA (dsRNA) was generated by the BLOCK-IT RNAi TOPO
Transcription KIT, the dsRNA was diced by BLOCK-IT Dicer RNAi Transfection KIT and
the result was dsiRNA. The whole procedure was done according to the manufacturer’s
manual (Invitrogen). HeLa cells were transiently transfected with the dsiRNA in OPTI-MEM I
Reduced Serum Medium (Invitrogen) using Lipofectamine 2000 (Invitrogen). The effect of
suppression was tested by Western blot using polyclonal anti-TIP47 primary antibody.

Hocsák, E., Identification and characterization of novel regulators
19
5.8. Cell viability assay
Cells were seeded into 96-well plates at a starting density of 104cells/well and cultured
overnight before H2O2 or Taxol was added to the medium at a concentration and composition
indicated in the figure legends. After the incubation period, the media were removed and
replaced with DMEM containing an appropriate amount of the MTT solution (Chemicon Inc.,
El Segundo, CA) to each well for 4h. The MTT reaction was terminated by adding HCl to the
medium at a final concentration of 10 mM. The amount of water-insoluble blue formasan dye
formed from MTT was proportional to the number of live cells and was determined with an
Anthos Labtech 200 enzyme-linked immunosorbent assay reader at 550 nm wavelength after
dissolving the blue formasan precipitate in 10% SDS. All experiments were run in at least four
parallels and repeated three times.
5.9. Determination of intracellular reactive oxygen species (ROS)
Intracellular ROS were determined using the oxidation-sensitive C-400 fluorescent
dye. Cells were seeded into 96-well plates at a starting density of 104 cell/well and cultured
overnight, and then were subjected to 0, 0.5, 0.8, 1.0 or 1.5 mM H2O2 for 3 h. Cells were
rinsed, labelled with the fluorescent dye for 15 min at 37 ºC, and analyzed using Anthos
Labtech 200 enzyme-linked immunosorbent assay reader at 485 or 578 nm excitation and 555
or 597 nm emission wavelengths for C-400.
5.10. Detecting mitochondrial membrane potential (∆ψ)
The changes in ∆ψ were assayed using JC-1 dye, which is taken up by the
mitochondria. Empty pcDNA-, t-TIP47-, TIP47-, Bcl-2- SOUL- or ∆BH3-SOUL- transfected
NIH3T3 cells were seeded at 1 x 106 cells/well in a 6-well plate containing coverslips and
cultured at least overnight before the experiment. After subjecting the cells to the appropriate
treatment (indicated in the figure legends), the coverslips were rinsed twice in PBS then placed
upside down on the top of a small chamber formed by a microscope slide, filled with PBS
supplemented with 0.5% FCS and containing 5 µg/ml JC-1-dye (Molecular probes). Cells

Hocsák, E., Identification and characterization of novel regulators
20
were imaged with a Zeiss Axiovert 25 fluorescent microscope equipped with a ProgRes C12
Plus CCD camera using a 63 x objective and epifluorescent illumination. For JC-1
fluorescence, the cells were loaded with the dye for 15 min, then the same microscopic field
was imaged first with 546 nm bandpass excitation and 590 nm emission (red), then with green
filters. Under these conditions we did not observe considerable bleed-through between the red
and green images.
5.11. Isolation of mitochondria for the in vitro study
Rats were sacrificed and the mitochondria were isolated from the liver by differential
centrifugation as described by a standard protocol [95]. All isolated mitochondria were
purified by Percoll gradient centrifuging [96], and the mitochondrial protein concentrations
were determined by Biuret method with bovine serum albumin as standard. Mitochondrial
membrane potential was monitored by fluorescence of Rh-123, released from the mitochondria
following the induction of mitochondrial permeability transition (mPT) at room temperature
using a Perkin-Elmer fluorimeter (London, UK) at an excitation wavelength of 495 and an
emission wavelength of 535 nm. Briefly, mitochondria at the concentration of 1 mg protein/ml
were preincubated in the assay buffer (70 mM sucrose, 214 mM mannitol, 20 mM N-2-
hydroxyethyl piperasine-N'-2-ethanesulfonic acid, 5 mM glutamate, 0.5 mM malate, 0.5 mM
phosphate) containing 1 µM Rh-123 and recombinant TIP47 for 60 seconds. Alteration of the
∆ψ was induced by the addition of calcium at the indicated concentration. Changes of
fluorescence intensity were detected for 4 min. The results are demonstrated by representative
original registration curves from five independent experiments, each repeated three times.
5.12. Annexin V-FITC assay
Fluorescent microscopy was performed to visualize the early and late apoptotic cells.
H2O2 treated cells were resuspended in binding buffer and stained with annexin V-FITC/PI for
10-10 min, the cells were then mounted over glass slide and observed under fluorescent
microscope. The same microscopic field was imaged first with 546 nm bandpass excitation
and 590 nm emission (red), then with green filters.

Hocsák, E., Identification and characterization of novel regulators
21
5.13. Flow cytometry analysis
Cell death was induced with 500 µM hydrogen peroxide (Sigma) for 3 hours in sham-
transfected, t-TIP47-, TIP47- or SOUL-overexpressing NIH3T3 cells. SOUL-overexpressing
NIH3T3 cells were either co-overexpressing or not Bcl-2 or Bcl-xL proteins. After treatment,
cells were rinsed and harvested. The annexin V FLUOS staining kit (Roche Applied Science)
was used to stain cells with fluorescent isothiocyanate-conjugated annexin V and PI according
to the manufacturer’s protocol. The cells were analyzed by flow cytometry on a BD
FacsCalibur flow cytometer (BD Biosciences). Quadrant dot plots were created by Cellquest
software (BD Biosciences) to identify living and necrotic cells and cells in the early or late
phase of apoptosis [97]. Cells in each category are expressed as a percentage of the total
number of stained cells counted and are presented as pie charts.
5.14. Statistical analysis
Each experiment was repeated at least three times. Values in the figures, tables and text
are expressed as mean ± SEM of n observations. Statistical analysis was performed by analysis
of variance followed by Student’s t-test. Statistical significance was set at p<0,05.

Hocsák, E., Identification and characterization of novel regulators
22
6. Results
6.1. TIP47 protects mitochondrial membrane integrity and inhibits cell
death
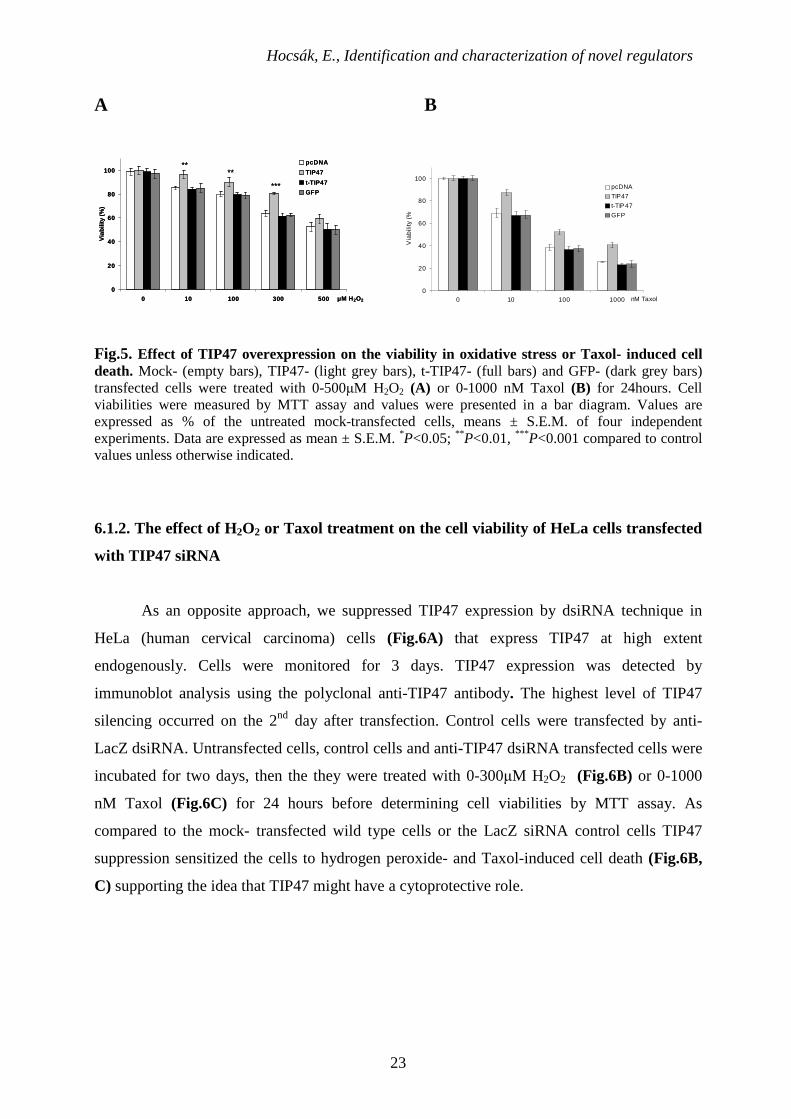
6.1.1. Effect of expression of TIP47 on H2O2- or Taxol-induced cell death
To evaluate possible physiological functions of TIP47, we transfected NIH3T3 cells
that endogenously express TIP47 at low extent with empty pcDNA3.1 vector, pEGFP-C1 and
a construct with pcDNA3.1 containing the full-length TIP47 or t-TIP47 cDNA. Effectiveness
of the transfection was assessed by immunoblotting utilizing a custom- made anti-TIP47
primary antibody [77].
Transfected cells were treated with 0-500 µM H2O2 (Fig.5A) or 0-1000 nM Taxol
(Fig.5B) for 24hours. Cell viabilities were measured by MTT assay and values were presented
in a bar diagram.
TIP47 overexpression significantly increased TIP47 levels (Fig.4) and the resistance of
NIH3T3 cells against hydrogen peroxide induced cell death (Fig.5A) or Taxol induced cell
death (Fig.5B) compared to the mock-, or t-TIP47 - transfected cells and even GFP-
overexpressing cells, which were also used as controls.
- β-actin
- t-TIP47
- TIP47
1: pcDNA2: GFP3: t-TIP474: TIP47
- β-actin
- t-TIP47
- TIP47
1: pcDNA2: GFP3: t-TIP474: TIP47
Fig.4. Level of TIP47 in NIH3T3 cells. Level of TIP47 expression was assessed by immunoblot analysis (A) utilizing custom-made anti-TIP47 primary antibody in mock-transfected (1), GFP-expressing (2), t-TIP47-overexpressing (3) or TIP47-overexpressing (4) NIH3T3 cells.
Hocsák, E., Identification and characterization of novel regulators
23
A B
0
20
40
60
80
100
0 10 100 300 500 µM H2O2
Via
bilit
y (%
)
pcDNA
TIP47
t-TIP47
GFP
****
***
0
20
40
60
80
100
0 10 100 300 500 µM H2O2
Via
bilit
y (%
)
pcDNA
TIP47
t-TIP47
GFP
****
***
0
20
40
60
80
100
0 10 100 1000 nM Taxol
Via
bili
ty (
%)
pcDNA
TIP47
t-TIP47
GFP
Fig.5. Effect of TIP47 overexpression on the viability in oxidative stress or Taxol- induced cell death. Mock- (empty bars), TIP47- (light grey bars), t-TIP47- (full bars) and GFP- (dark grey bars) transfected cells were treated with 0-500µM H2O2 (A) or 0-1000 nM Taxol (B) for 24hours. Cell viabilities were measured by MTT assay and values were presented in a bar diagram. Values are expressed as % of the untreated mock-transfected cells, means ± S.E.M. of four independent experiments. Data are expressed as mean ± S.E.M. *P<0.05; ** P<0.01, *** P<0.001 compared to control values unless otherwise indicated.
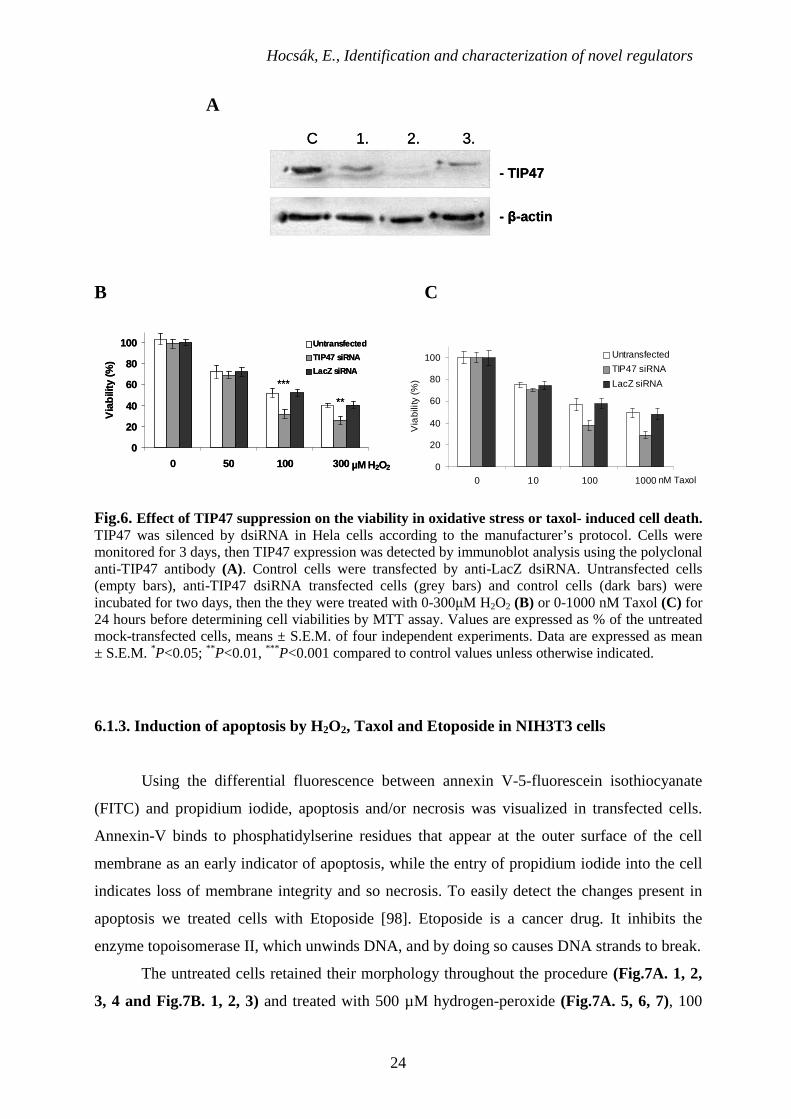
6.1.2. The effect of H2O2 or Taxol treatment on the cell viability of HeLa cells transfected
with TIP47 siRNA
As an opposite approach, we suppressed TIP47 expression by dsiRNA technique in
HeLa (human cervical carcinoma) cells (Fig.6A) that express TIP47 at high extent
endogenously. Cells were monitored for 3 days. TIP47 expression was detected by
immunoblot analysis using the polyclonal anti-TIP47 antibody. The highest level of TIP47
silencing occurred on the 2nd day after transfection. Control cells were transfected by anti-
LacZ dsiRNA. Untransfected cells, control cells and anti-TIP47 dsiRNA transfected cells were
incubated for two days, then the they were treated with 0-300µM H2O2 (Fig.6B) or 0-1000
nM Taxol (Fig.6C) for 24 hours before determining cell viabilities by MTT assay. As
compared to the mock- transfected wild type cells or the LacZ siRNA control cells TIP47
suppression sensitized the cells to hydrogen peroxide- and Taxol-induced cell death (Fig.6B,
C) supporting the idea that TIP47 might have a cytoprotective role.
Hocsák, E., Identification and characterization of novel regulators
24
A
C 1. 2. 3.
- TIP47
- β-actin
C 1. 2. 3.
- TIP47
- β-actin
B C
0
20
40
60
80
100
0 50 100 300 µM H2O2
Via
bilit
y (%
)
Untransfected
TIP47 siRNA
LacZ siRNA
***
**
0
20
40
60
80
100
0 50 100 300 µM H2O2
Via
bilit
y (%
)
Untransfected
TIP47 siRNA
LacZ siRNA
***
**
0
20
40
60
80
100
0 10 100 1000 nM Taxol
Via
bilit
y (%
)
Untransfected
TIP47 siRNA
LacZ siRNA
Fig.6. Effect of TIP47 suppression on the viability in oxidative stress or taxol- induced cell death. TIP47 was silenced by dsiRNA in Hela cells according to the manufacturer’s protocol. Cells were monitored for 3 days, then TIP47 expression was detected by immunoblot analysis using the polyclonal anti-TIP47 antibody (A). Control cells were transfected by anti-LacZ dsiRNA. Untransfected cells (empty bars), anti-TIP47 dsiRNA transfected cells (grey bars) and control cells (dark bars) were incubated for two days, then the they were treated with 0-300µM H2O2 (B) or 0-1000 nM Taxol (C) for 24 hours before determining cell viabilities by MTT assay. Values are expressed as % of the untreated mock-transfected cells, means ± S.E.M. of four independent experiments. Data are expressed as mean ± S.E.M. *P<0.05; ** P<0.01, *** P<0.001 compared to control values unless otherwise indicated.
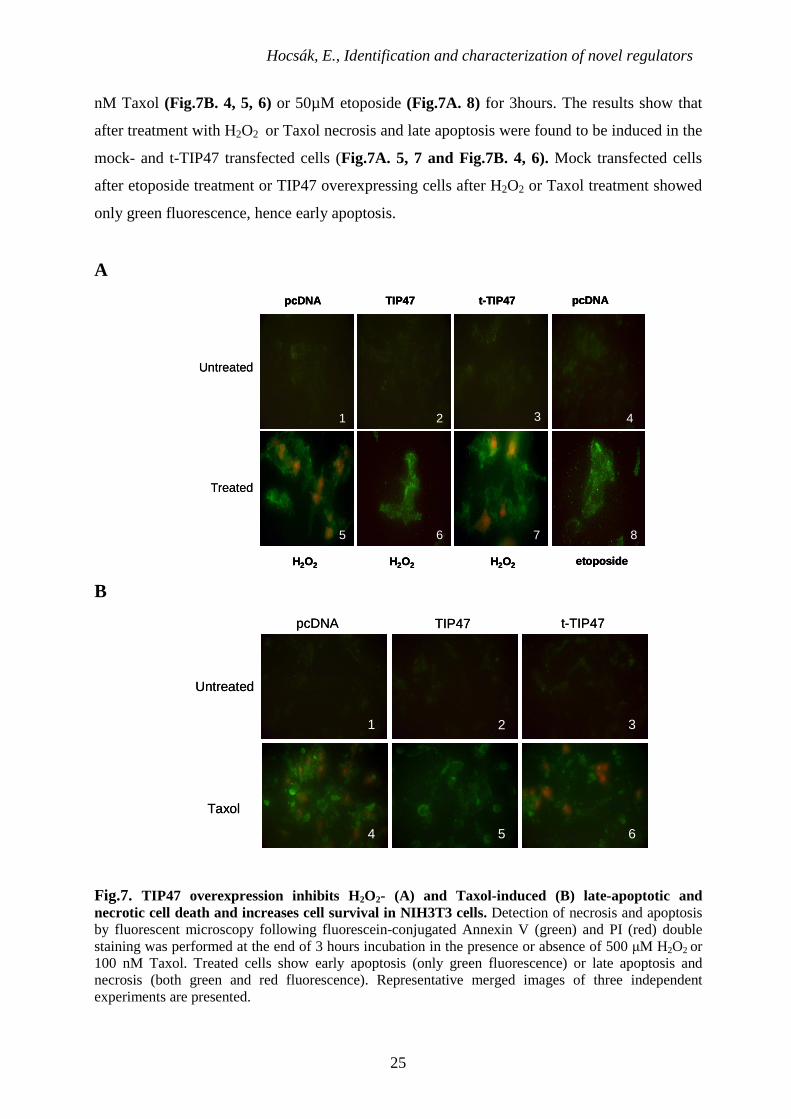
6.1.3. Induction of apoptosis by H2O2, Taxol and Etoposide in NIH3T3 cells
Using the differential fluorescence between annexin V-5-fluorescein isothiocyanate
(FITC) and propidium iodide, apoptosis and/or necrosis was visualized in transfected cells.
Annexin-V binds to phosphatidylserine residues that appear at the outer surface of the cell
membrane as an early indicator of apoptosis, while the entry of propidium iodide into the cell
indicates loss of membrane integrity and so necrosis. To easily detect the changes present in
apoptosis we treated cells with Etoposide [98]. Etoposide is a cancer drug. It inhibits the
enzyme topoisomerase II, which unwinds DNA, and by doing so causes DNA strands to break.
The untreated cells retained their morphology throughout the procedure (Fig.7A. 1, 2,
3, 4 and Fig.7B. 1, 2, 3) and treated with 500 µM hydrogen-peroxide (Fig.7A. 5, 6, 7), 100
Hocsák, E., Identification and characterization of novel regulators
25
nM Taxol (Fig.7B. 4, 5, 6) or 50µM etoposide (Fig.7A. 8) for 3hours. The results show that
after treatment with H2O2 or Taxol necrosis and late apoptosis were found to be induced in the
mock- and t-TIP47 transfected cells (Fig.7A. 5, 7 and Fig.7B. 4, 6). Mock transfected cells
after etoposide treatment or TIP47 overexpressing cells after H2O2 or Taxol treatment showed
only green fluorescence, hence early apoptosis.
A
H2O2
1 2
7
3 4
85 6
etoposide
pcDNA t-TIP47TIP47 pcDNA
H2O2 H2O2
Untreated
Treated
H2O2
1 2
7
3 4
85 6
etoposide
pcDNA t-TIP47TIP47 pcDNA
H2O2 H2O2H2O2
1 2
7
3 4
85 6
etoposide
pcDNA t-TIP47TIP47 pcDNA
H2O2 H2O2
Untreated
Treated
B
Taxol
pcDNA TIP47 t-TIP47
1 2 3
4 5 6
Untreated
Taxol
pcDNA TIP47 t-TIP47
1 2 3
4 5 6
Untreated
Fig.7. TIP47 overexpression inhibits H2O2- (A) and Taxol-induced (B) late-apoptotic and necrotic cell death and increases cell survival in NIH3T3 cells. Detection of necrosis and apoptosis by fluorescent microscopy following fluorescein-conjugated Annexin V (green) and PI (red) double staining was performed at the end of 3 hours incubation in the presence or absence of 500 µM H2O2 or 100 nM Taxol. Treated cells show early apoptosis (only green fluorescence) or late apoptosis and necrosis (both green and red fluorescence). Representative merged images of three independent experiments are presented.
Hocsák, E., Identification and characterization of novel regulators
26
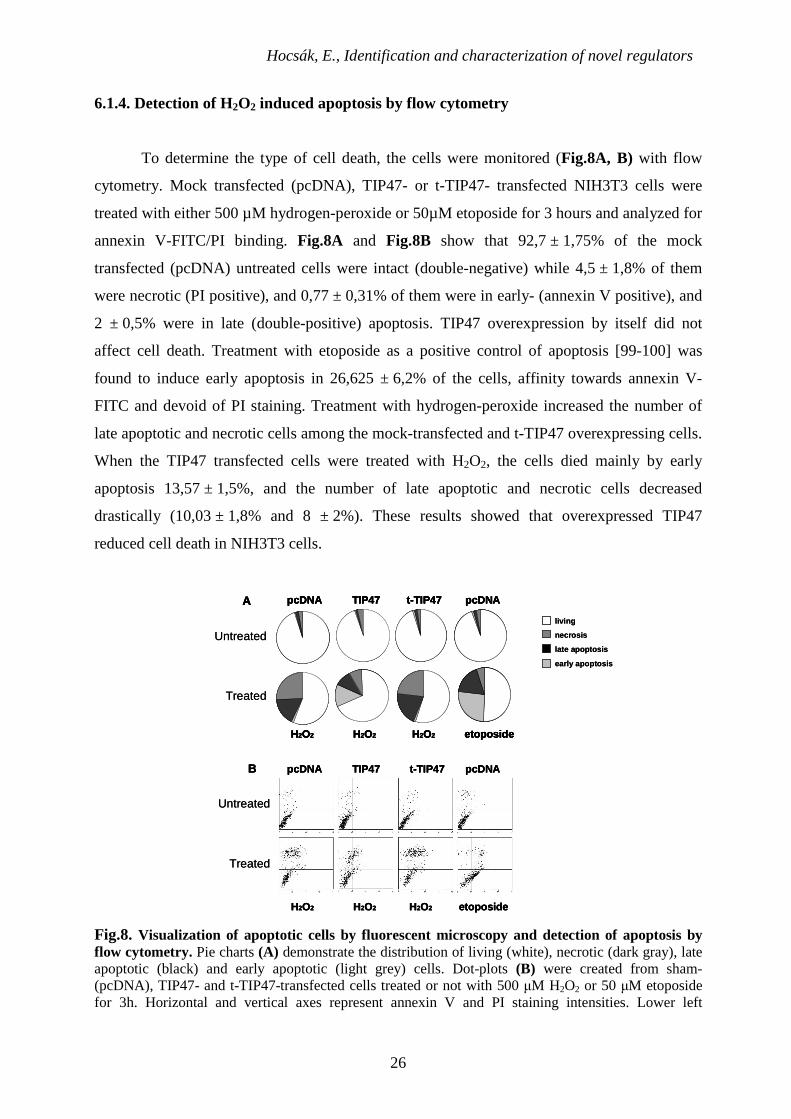
6.1.4. Detection of H2O2 induced apoptosis by flow cytometry
To determine the type of cell death, the cells were monitored (Fig.8A, B) with flow
cytometry. Mock transfected (pcDNA), TIP47- or t-TIP47- transfected NIH3T3 cells were
treated with either 500 µM hydrogen-peroxide or 50µM etoposide for 3 hours and analyzed for
annexin V-FITC/PI binding. Fig.8A and Fig.8B show that 92,7 ± 1,75% of the mock
transfected (pcDNA) untreated cells were intact (double-negative) while 4,5 ± 1,8% of them
were necrotic (PI positive), and 0,77 ± 0,31% of them were in early- (annexin V positive), and
2 ± 0,5% were in late (double-positive) apoptosis. TIP47 overexpression by itself did not
affect cell death. Treatment with etoposide as a positive control of apoptosis [99-100] was
found to induce early apoptosis in 26,625 ± 6,2% of the cells, affinity towards annexin V-
FITC and devoid of PI staining. Treatment with hydrogen-peroxide increased the number of
late apoptotic and necrotic cells among the mock-transfected and t-TIP47 overexpressing cells.
When the TIP47 transfected cells were treated with H2O2, the cells died mainly by early
apoptosis 13,57 ± 1,5%, and the number of late apoptotic and necrotic cells decreased
drastically (10,03 ± 1,8% and 8 ± 2%). These results showed that overexpressed TIP47
reduced cell death in NIH3T3 cells.
B pcDNA TIP47 t-TIP47 pcDNA
H2O2 H2O2 H2O2 etoposide
pcDNA TIP47 t-TIP47 pcDNAA
H2O2 H2O2 H2O2 etoposide
living
necrosis
late apoptosis
early apoptosis
Untreated
Treated
Treated
Untreated
B pcDNA TIP47 t-TIP47 pcDNA
H2O2 H2O2 H2O2 etoposide
B pcDNA TIP47 t-TIP47 pcDNA
H2O2 H2O2 H2O2 etoposide
pcDNA TIP47 t-TIP47 pcDNAA
H2O2 H2O2 H2O2 etoposide
pcDNA TIP47 t-TIP47 pcDNAA
H2O2 H2O2 H2O2 etoposide
pcDNA TIP47 t-TIP47 pcDNAA
H2O2 H2O2 H2O2 etoposide
living
necrosis
late apoptosis
early apoptosis
Untreated
Treated
Treated
Untreated
Fig.8. Visualization of apoptotic cells by fluorescent microscopy and detection of apoptosis by flow cytometry. Pie charts (A) demonstrate the distribution of living (white), necrotic (dark gray), late apoptotic (black) and early apoptotic (light grey) cells. Dot-plots (B) were created from sham- (pcDNA), TIP47- and t-TIP47-transfected cells treated or not with 500 µM H2O2 or 50 µM etoposide for 3h. Horizontal and vertical axes represent annexin V and PI staining intensities. Lower left

Hocsák, E., Identification and characterization of novel regulators
27
quadrant; living cells, lower right quadrant; early apoptotic cells, upper left quadrant; necrotic cells, upper right quadrant; late apoptotic cells. Values are means of 3 independent experiments.
6.1.5. Localization of TIP47 in oxidative stress
To demonstrate the redistribution of TIP47 during the cell death process, we performed
immunoblot experiments on fractionated HeLa cells treated or not with 500 µM H2O2 for 3
hours. Fig.9. shows that in untreated HeLa cells, TIP47 was present in the cytoplasm and not
detected in the mitochondrial or the nuclear fraction. However, upon a short exposure to
oxidative stress, there was a significant increase of TIP47 protein in the mitochondrial
subfraction of the cells, accompanied by a slight decrease in the cytosolic fraction. This data
indicate that in response to oxidative stress, TIP47 can relocate to the mitochondria.
C M N C M N
TIP47
Control H 2O2
47 kDa
GA3PD
PDC1αααα
Histon H1
C M N C M N
TIP47
Control H 2O2
47 kDa
GA3PD
PDC1αααα
Histon H1
Fig.9. Intracellular localization of TIP47. Western blot analysis of cytosolic (C) mitochondrial (M) and nuclear (N) fractions of HeLa cell treated (H2O2) or not (Control) with 500 µM H2O2 for 3 hours. Western blotting was developed as described previously using anti-TIP47 primary antibody, the cytosolic marker glycerinaldehyde-3-phosphate dehydrogenase (GA3PD), the mitochondrial marker pyruvate decarboxylase 1α (PDC-1α) and the nuclear marker histon H1 (Histon H1). Photomicrographs demonstrate representative blots of three independent experiments.
6.1.6. Effect of recombinant TIP47 on the mPT and ∆ψ in Vitro
In order to demonstrate the direct effect of TIP47 on mitochondrial permeability
transition, we monitored ∆ψ and mitochondrial swelling from isolated, Percoll gradient-
purified rat liver mitochondria. Supplemented calcium (150 µM) induced the decrease of the
∆ψ, as detected by the release of the membrane potential-sensitive dye, Rh-123, from isolated
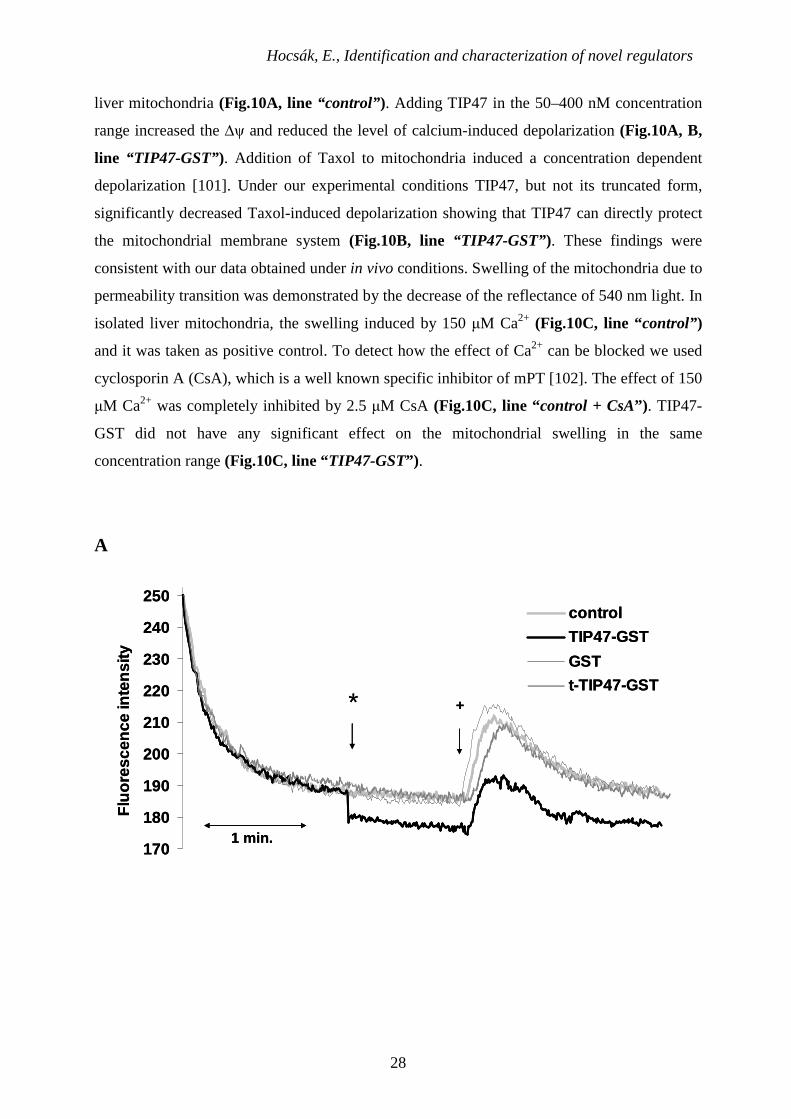
Hocsák, E., Identification and characterization of novel regulators
28
liver mitochondria (Fig.10A, line “control” ). Adding TIP47 in the 50–400 nM concentration
range increased the ∆ψ and reduced the level of calcium-induced depolarization (Fig.10A, B,
line “TIP47-GST”). Addition of Taxol to mitochondria induced a concentration dependent
depolarization [101]. Under our experimental conditions TIP47, but not its truncated form,
significantly decreased Taxol-induced depolarization showing that TIP47 can directly protect
the mitochondrial membrane system (Fig.10B, line “TIP47-GST” ). These findings were
consistent with our data obtained under in vivo conditions. Swelling of the mitochondria due to
permeability transition was demonstrated by the decrease of the reflectance of 540 nm light. In
isolated liver mitochondria, the swelling induced by 150 µM Ca2+ (Fig.10C, line “control” )
and it was taken as positive control. To detect how the effect of Ca2+ can be blocked we used
cyclosporin A (CsA), which is a well known specific inhibitor of mPT [102]. The effect of 150
µM Ca2+ was completely inhibited by 2.5 µM CsA (Fig.10C, line “control + CsA”) . TIP47-
GST did not have any significant effect on the mitochondrial swelling in the same
concentration range (Fig.10C, line “TIP47-GST”) .
A
170
180
190
200
210
220
230
240
250
Flu
ores
cenc
e in
tens
ity
control
TIP47-GST
GST
t-TIP47-GST+*
1 min.170
180
190
200
210
220
230
240
250
Flu
ores
cenc
e in
tens
ity
control
TIP47-GST
GST
t-TIP47-GST+*
1 min.
Hocsák, E., Identification and characterization of novel regulators
29
B
150
160
170
180
190
200
210
220
230
240
250
Flu
ores
cenc
e in
tens
ity
t-TIP47GSTGSTTIP47 GSTTaxolAddition of
proteins
1 min.
0
10
20
30
40
50
0 50 100 200 400 nM
∆ fl
uore
scen
ce in
tens
ity o
f TIP
47-
GS
T h
yper
pola
rizat
ion
0
5
10
15
20
25
TIP47-GST t-TIP47-GST GST
∆ fl
uore
scen
ce in
tens
ity o
f Tax
ol tr
eatm
ent
150
160
170
180
190
200
210
220
230
240
250
Flu
ores
cenc
e in
tens
ity
t-TIP47GSTGSTTIP47 GSTTaxolAddition of
proteins
1 min.150
160
170
180
190
200
210
220
230
240
250
Flu
ores
cenc
e in
tens
ity
t-TIP47GSTGSTTIP47 GSTTaxolAddition of
proteins
1 min.
0
10
20
30
40
50
0 50 100 200 400 nM
∆ fl
uore
scen
ce in
tens
ity o
f TIP
47-
GS
T h
yper
pola
rizat
ion
0
5
10
15
20
25
TIP47-GST t-TIP47-GST GST
∆ fl
uore
scen
ce in
tens
ity o
f Tax
ol tr
eatm
ent
C
1 0 0
1 5 0
2 0 0
2 5 0
3 0 0
3 5 0
4 0 0
4 5 0
5 0 0
Flu
ores
cenc
e in
tens
ity
c o n tro lc o n tro l + C s AT IP 4 7 -G S TG S Tt-T IP 4 7 -G S T
2 min.
* +
1 0 0
1 5 0
2 0 0
2 5 0
3 0 0
3 5 0
4 0 0
4 5 0
5 0 0
Flu
ores
cenc
e in
tens
ity
c o n tro lc o n tro l + C s AT IP 4 7 -G S TG S Tt-T IP 4 7 -G S T
2 min.
* +
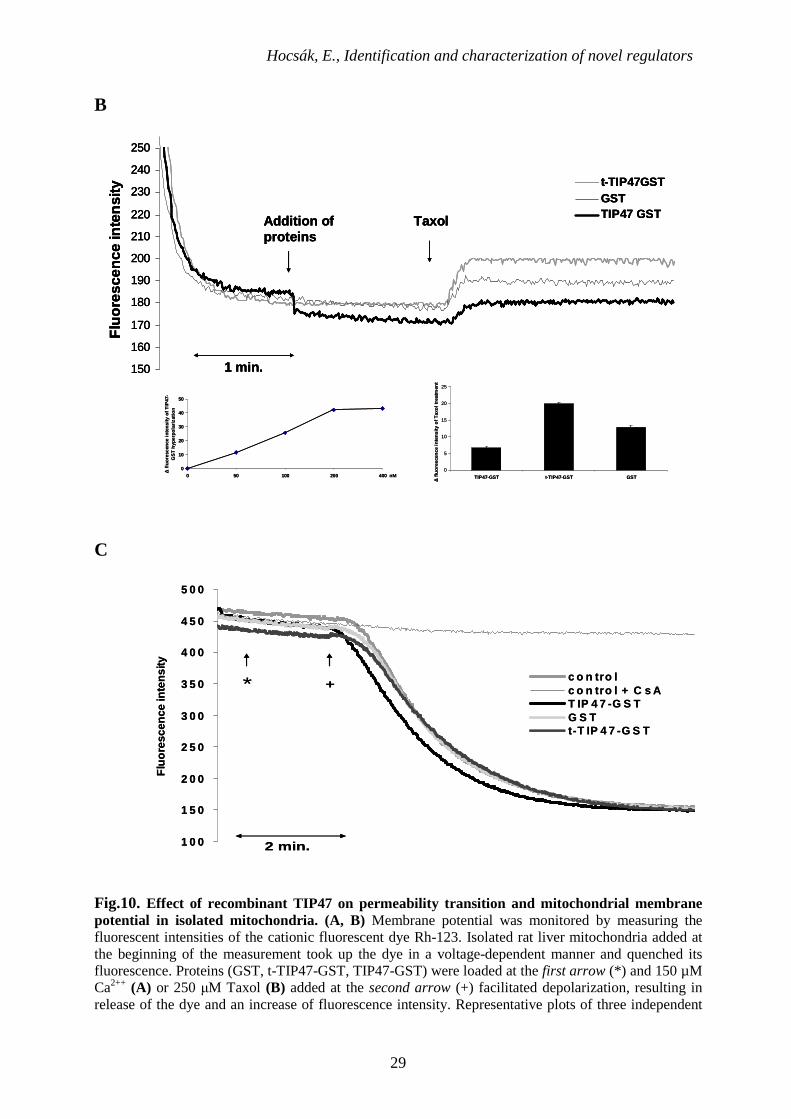
Fig.10. Effect of recombinant TIP47 on permeability transition and mitochondrial membrane potential in isolated mitochondria. (A, B) Membrane potential was monitored by measuring the fluorescent intensities of the cationic fluorescent dye Rh-123. Isolated rat liver mitochondria added at the beginning of the measurement took up the dye in a voltage-dependent manner and quenched its fluorescence. Proteins (GST, t-TIP47-GST, TIP47-GST) were loaded at the first arrow (*) and 150 µM Ca2++ (A) or 250 µM Taxol (B) added at the second arrow (+) facilitated depolarization, resulting in release of the dye and an increase of fluorescence intensity. Representative plots of three independent

Hocsák, E., Identification and characterization of novel regulators
30
experiments are presented. (C), permeability transition demonstrated by monitoring E540 in isolated rat liver mitochondria was induced by adding 150 µM Ca2++ at the second arrow (+). Separately, 200 nM GST, t-TIP47-GST, TIP47-GST or 2.5 µM CsA were loaded at the first arrow (*). Representative plots of three independent experiments are presented.
6.1.7. The effect of TIP47 on release of mitochondrial intermembrane space proteins
from living cells
Following apoptotic stimuli and the collapse of the mitochondrial membrane potential,
mitochondrial proteins are released into the cytosol or the nucleus where they play a part in the
process of apoptosis. AIF and EndoG, which induce apoptosis in a caspase-independent
manner, translocate to the nucleus, whereas cyt-c, a member of caspase-dependent apoptosis,
is released into the cytosol. The exposure of sham-transfected cells to Taxol resulted in the
intracellular translocation of the mitochondrial proteins, AIF, EndoG and cyt-c. However,
TIP47 overexpression conferred resistance to taxol-induced cell death by inhibiting the
translocation of AIF and EndoG (Fig.11). Interestingly, though, TIP47 overexpression had no
effect on cyt-c release following taxol treatment (data not shown).
1 2 3 4AIF
EndoG
Cytosol
Histon H1
PDC 1α
GAPDH
1: pcDNA
2: pcDNA - taxol treated
3: TIP47
4: TIP47 - taxol treated
Nucleus
Mitochondrion 1 2 3 4
1 2 3 4
AIF
AIF
EndoG
EndoG
Fig.11. TIP47 overexpression inhibited the Taxol-induced nuclear translocation of EndoG and AIF in NIH3T3 cells. Immuno-blot analysis of the pro-apoptotic mitochondrial proteins EndoG and AIF in TIP47-transfected NIH3T3 cells was carried out following a 6 h treatment with taxol at the concentration indicated. Cells transfected with pcDNA served as controls. Even loading was checked by Histon H1, PDC-1α and GAPDH immunoreactivity in the appropriate subcellular fractions. Representative blots of three independent experiments are presented.
Hocsák, E., Identification and characterization of novel regulators
31
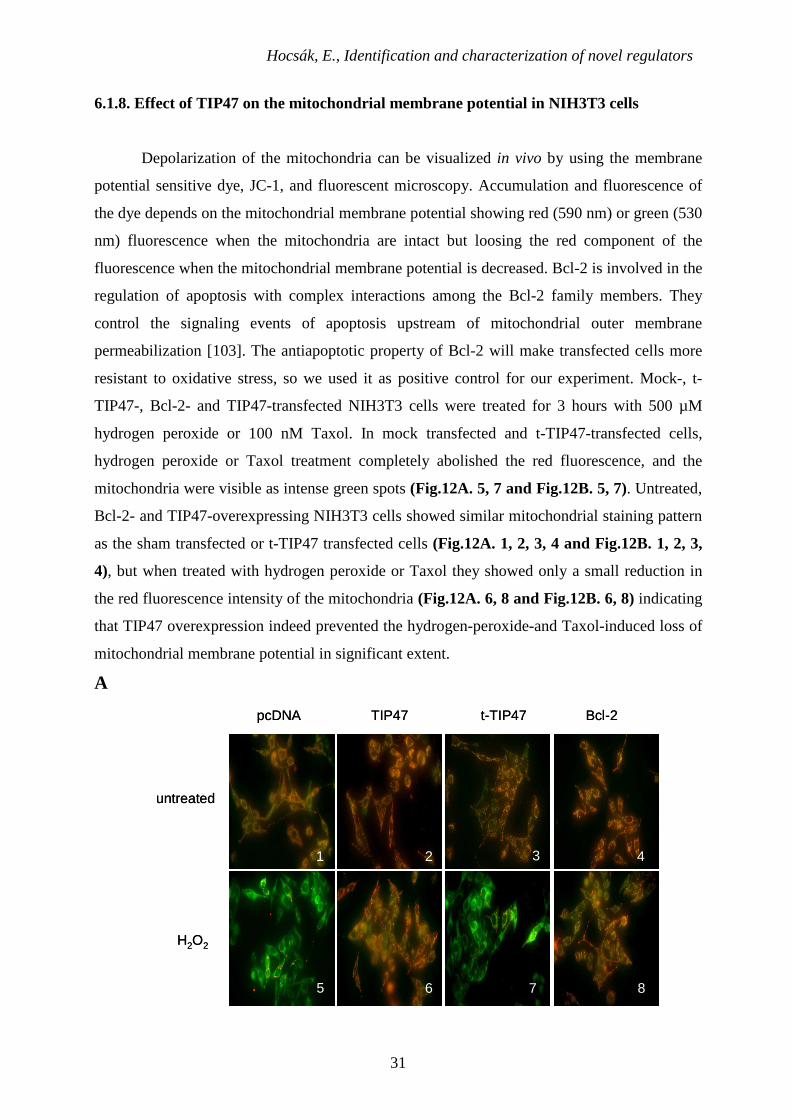
6.1.8. Effect of TIP47 on the mitochondrial membrane potential in NIH3T3 cells
Depolarization of the mitochondria can be visualized in vivo by using the membrane
potential sensitive dye, JC-1, and fluorescent microscopy. Accumulation and fluorescence of
the dye depends on the mitochondrial membrane potential showing red (590 nm) or green (530
nm) fluorescence when the mitochondria are intact but loosing the red component of the
fluorescence when the mitochondrial membrane potential is decreased. Bcl-2 is involved in the
regulation of apoptosis with complex interactions among the Bcl-2 family members. They
control the signaling events of apoptosis upstream of mitochondrial outer membrane
permeabilization [103]. The antiapoptotic property of Bcl-2 will make transfected cells more
resistant to oxidative stress, so we used it as positive control for our experiment. Mock-, t-
TIP47-, Bcl-2- and TIP47-transfected NIH3T3 cells were treated for 3 hours with 500 µM
hydrogen peroxide or 100 nM Taxol. In mock transfected and t-TIP47-transfected cells,
hydrogen peroxide or Taxol treatment completely abolished the red fluorescence, and the
mitochondria were visible as intense green spots (Fig.12A. 5, 7 and Fig.12B. 5, 7). Untreated,
Bcl-2- and TIP47-overexpressing NIH3T3 cells showed similar mitochondrial staining pattern
as the sham transfected or t-TIP47 transfected cells (Fig.12A. 1, 2, 3, 4 and Fig.12B. 1, 2, 3,
4), but when treated with hydrogen peroxide or Taxol they showed only a small reduction in
the red fluorescence intensity of the mitochondria (Fig.12A. 6, 8 and Fig.12B. 6, 8) indicating
that TIP47 overexpression indeed prevented the hydrogen-peroxide-and Taxol-induced loss of
mitochondrial membrane potential in significant extent.
A
H2O2
1 2 3
5 6 7
4
8
pcDNA t-TIP47TIP47 Bcl-2
untreated
H2O2
1 2 3
5 6 7
4
8
1 2 3
5 6 7
4
8
pcDNA t-TIP47TIP47 Bcl-2
untreated
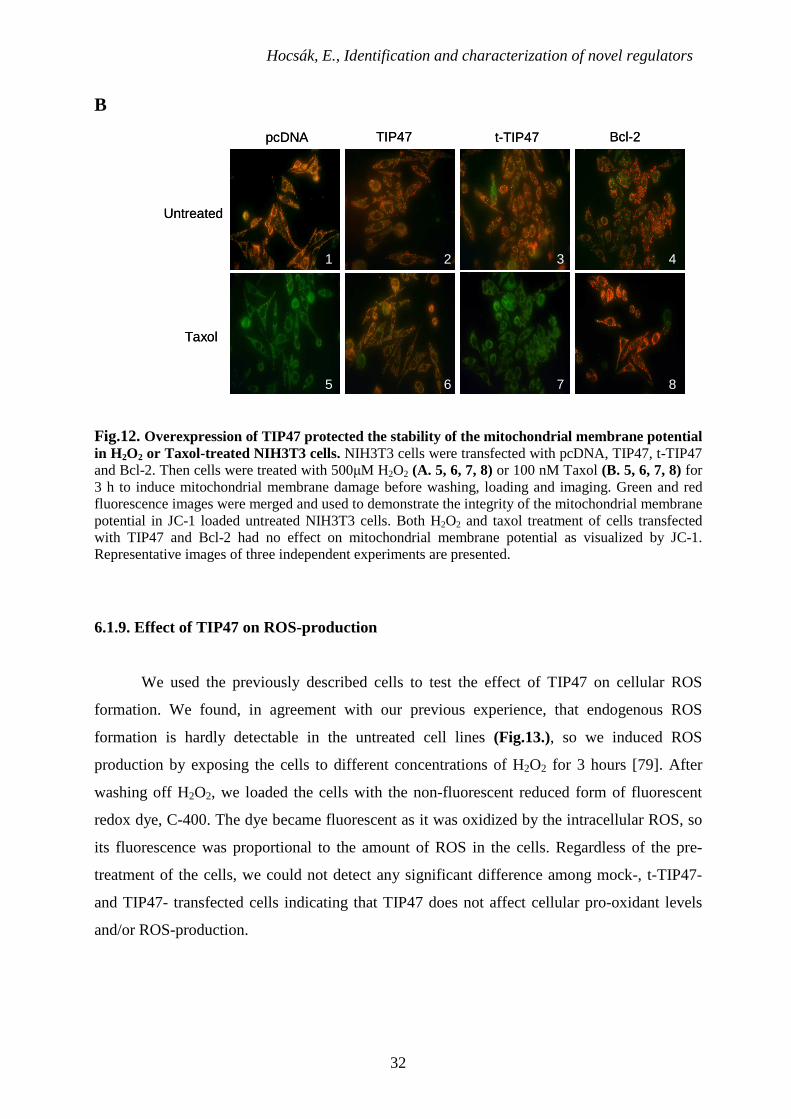
Hocsák, E., Identification and characterization of novel regulators
32
B
1
8765
432
Untreated
Taxol
pcDNA Bcl-2t-TIP47TIP47
1
8765
432
Untreated
Taxol
pcDNA Bcl-2t-TIP47TIP47
Fig.12. Overexpression of TIP47 protected the stability of the mitochondrial membrane potential in H2O2 or Taxol-treated NIH3T3 cells. NIH3T3 cells were transfected with pcDNA, TIP47, t-TIP47 and Bcl-2. Then cells were treated with 500µM H2O2 (A. 5, 6, 7, 8) or 100 nM Taxol (B. 5, 6, 7, 8) for 3 h to induce mitochondrial membrane damage before washing, loading and imaging. Green and red fluorescence images were merged and used to demonstrate the integrity of the mitochondrial membrane potential in JC-1 loaded untreated NIH3T3 cells. Both H2O2 and taxol treatment of cells transfected with TIP47 and Bcl-2 had no effect on mitochondrial membrane potential as visualized by JC-1. Representative images of three independent experiments are presented.
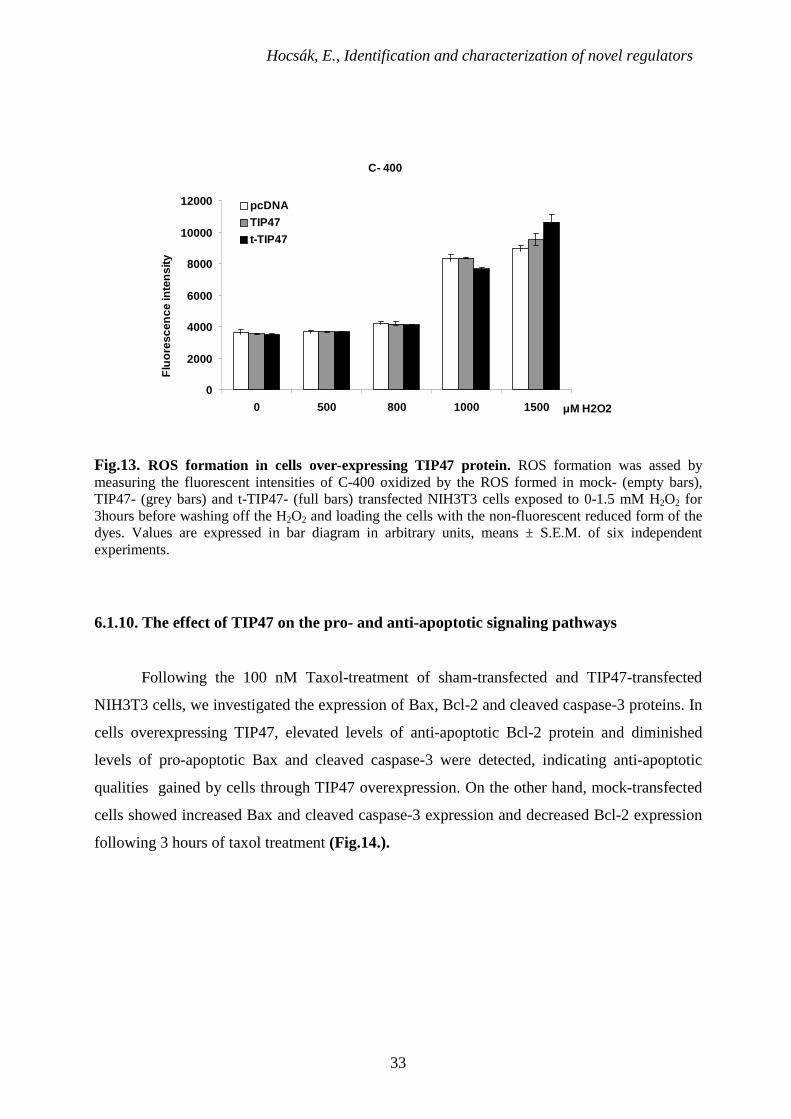
6.1.9. Effect of TIP47 on ROS-production
We used the previously described cells to test the effect of TIP47 on cellular ROS
formation. We found, in agreement with our previous experience, that endogenous ROS
formation is hardly detectable in the untreated cell lines (Fig.13.), so we induced ROS
production by exposing the cells to different concentrations of H2O2 for 3 hours [79]. After
washing off H2O2, we loaded the cells with the non-fluorescent reduced form of fluorescent
redox dye, C-400. The dye became fluorescent as it was oxidized by the intracellular ROS, so
its fluorescence was proportional to the amount of ROS in the cells. Regardless of the pre-
treatment of the cells, we could not detect any significant difference among mock-, t-TIP47-
and TIP47- transfected cells indicating that TIP47 does not affect cellular pro-oxidant levels
and/or ROS-production.
Hocsák, E., Identification and characterization of novel regulators
33
Fig.13. ROS formation in cells over-expressing TIP47 protein. ROS formation was assed by measuring the fluorescent intensities of C-400 oxidized by the ROS formed in mock- (empty bars), TIP47- (grey bars) and t-TIP47- (full bars) transfected NIH3T3 cells exposed to 0-1.5 mM H2O2 for 3hours before washing off the H2O2 and loading the cells with the non-fluorescent reduced form of the dyes. Values are expressed in bar diagram in arbitrary units, means ± S.E.M. of six independent experiments.
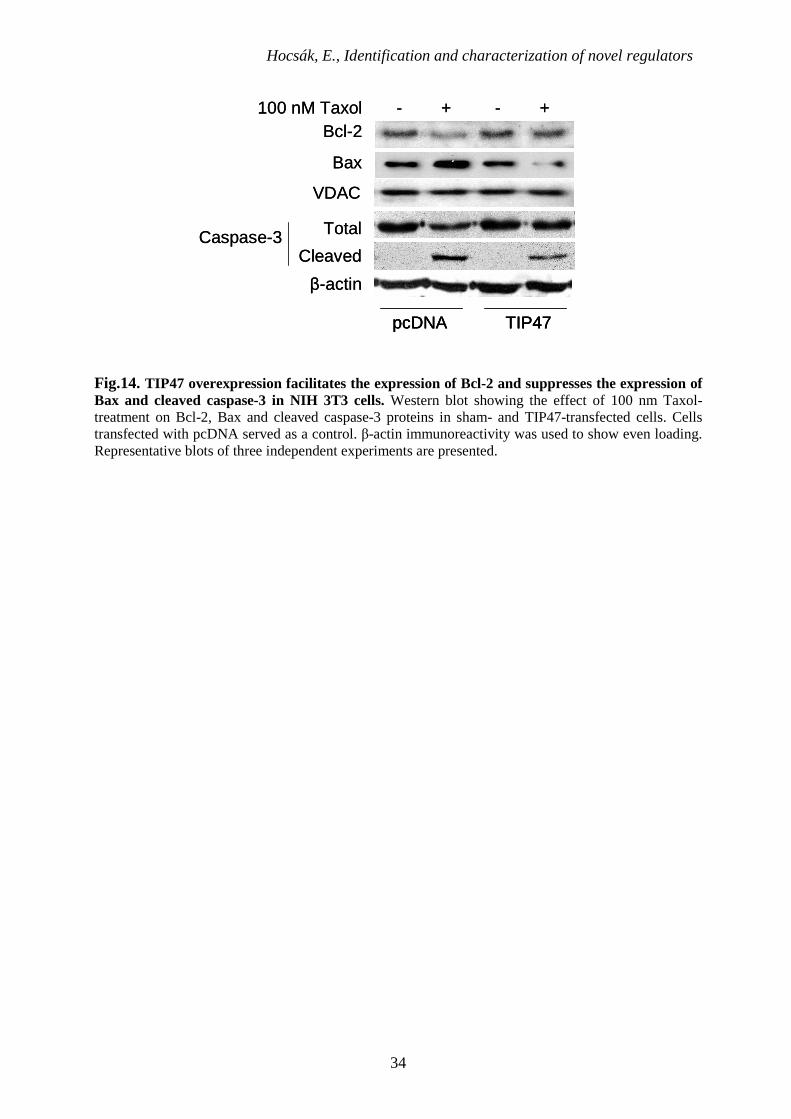
6.1.10. The effect of TIP47 on the pro- and anti-apoptotic signaling pathways
Following the 100 nM Taxol-treatment of sham-transfected and TIP47-transfected
NIH3T3 cells, we investigated the expression of Bax, Bcl-2 and cleaved caspase-3 proteins. In
cells overexpressing TIP47, elevated levels of anti-apoptotic Bcl-2 protein and diminished
levels of pro-apoptotic Bax and cleaved caspase-3 were detected, indicating anti-apoptotic
qualities gained by cells through TIP47 overexpression. On the other hand, mock-transfected
cells showed increased Bax and cleaved caspase-3 expression and decreased Bcl-2 expression
following 3 hours of taxol treatment (Fig.14.).
C- 400
0
2000
4000
6000
8000
10000
12000
0 500 800 1000 1500 µM H2O2
Flu
ores
cenc
e in
tens
ity
pcDNA
TIP47
t-TIP47
Hocsák, E., Identification and characterization of novel regulators
34
Bax
- + - +
β-actin
Bcl-2
Caspase-3
TIP47pcDNA
100 nM Taxol
Total
Cleaved
VDAC
Bax
- + - +
β-actin
Bcl-2
Caspase-3
TIP47pcDNA TIP47pcDNA
100 nM Taxol
Total
Cleaved
VDAC
Fig.14. TIP47 overexpression facilitates the expression of Bcl-2 and suppresses the expression of Bax and cleaved caspase-3 in NIH 3T3 cells. Western blot showing the effect of 100 nm Taxol-treatment on Bcl-2, Bax and cleaved caspase-3 proteins in sham- and TIP47-transfected cells. Cells transfected with pcDNA served as a control. β-actin immunoreactivity was used to show even loading. Representative blots of three independent experiments are presented.

Hocsák, E., Identification and characterization of novel regulators
35
6.2. Cell death in oxidative stress by a novel BH3 domain protein
6.2.1. Characterization of SOUL protein
NCBI Database analysis showed that there is a BH3 domain in SOUL protein, which is
a member of the heme binding protein family. A multiple alignment of the BH3 domains of
Bcl-2 associated proteins show high homology to a sequence found in SOUL (Fig.15).
Fig.15. Comparison of the BH3 domains amongst SOUL and Bcl-2 family members. Identical amino acids are black-shaded, dark-gray-shaded amino acids indicate conserved substitutions and light-gray-shaded amino acids mean semi-conserved substitutions. Sequences of Bcl-xL, Bcl-2, Bcl-W, Bim, Bfk, Bik, Bid, Bax, Bad, Bcl-2-KSHV proteins were accessed from NCBI Protein database.
6.2.2. Determination of the role of mPT complex in the SOUL-induced sensitization
toward oxidative stress
We used siRNA to silence cyclophilin D, a constitutional element of the mitochondrial
transition pore. SOUL-overexpressing and sham-transfected control NIH3T3 cells were
pretreated or not with 30 nM cyclophilin D specific siRNA. Efficacy of the suppression of
cyclophilin D and expression of SOUL was tested by Western blotting (Fig 16A). All cells
were treated with different concentrations of hydrogen peroxide ranging from 0 to 500 µM.
We observed that SOUL-overexpressing cells are sensitized to hydrogen-peroxide while the

Hocsák, E., Identification and characterization of novel regulators
36
cyclophilin D suppressed cells had similar sensitivity toward it than the sham-transfected cells
(Fig. 16B). Similar experiments were performed with ∆BH3-SOUL overexpressing cells,
however, we did not find any significant difference between these cells and mock-transfected
cells (data not shown). That is, blocking mPT by suppressing cyclophilin D inhibited
mitochondrial permeability transition and blocked the cell death sensitizing effect of SOUL in
oxidative stress. To confirm our findings, we double-stained the above mentioned cells with
Annexin V conjugated FITC and propidium iodide, and used flow cytometer to detect cell
death. Mock-transfected and SOUL-overexpressing NIH3T3 cells were co-transfected (cycD
siRNA) or not (Control) with 30nM cyclophilin D siRNA, and were treated or not with 300
µM hydrogen peroxide for 24 hours (Fig. 16C). Among control, untreated cells, 94.92±3.9%
were intact (annexin V and PI negative), 4.74±2.3% were necrotic (annexin V negative, PI
positive) and 2.08±1.2% were apoptotic (annexin V positive and PI positive or negative). An
increase in the number of necrotic cells (6.74±2.7%) was observed in the SOUL-
overexpressing untreated cells. Treatment with H2O2 increased the number of necrotic cells in
both, the control and in the SOUL-overexpressing cells (44.88±4.1% and 70.42±4.8%,
respectively). When cyclophilin D was silenced, control cells showed basically no change in
survival (90.73±4.2% living). However, unlike in control cells, SOUL overexpression failed to
increase (p=0.17, n=3) percentage of necrotic cells as compared to cyclophilin-D-silenced
mock-transfected cells (41.47±4.8% vs. 36.37±3.2%, respectively) upon H2O2 treatment.
These results indicate that SOUL protein facilitates necrotic cell death in oxidative stress, and
this effect can be specifically inhibited by suppressing cyclophilin D indicating that SOUL
seems to exert its effect by interacting with the mitochondrial permeability transition pore.
A

Hocsák, E., Identification and characterization of novel regulators
37
B C
0
25
50
75
100
0 100 300 500 µM H2O2
%
pcDNASOULpcDNA+CycD siRNASOUL+CycD siRNA
Control
CycDsiRNA
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecrosisapop tosis
Control
CycDsiRNA
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecrosisapop tosis
FIG .16. Suppression of cyclophilin D abolished the effect of SOUL on H2O2-treated NIH3T3 cells. Suppression of cyclophilin D (CycD) was achieved by co-transfecting or not (Control) sham-transfected (pcDNA) and SOUL-expressing (SOUL) NIH3T3 cells with 30 nM specific siRNA-expressing vector (CycD siRNA) according to the manufacturer’s recommendation. The cells were treated for 24 hours with hydrogen peroxide at concentrations ranging from 0 to 500 µM. (A) Demonstration of expression of SOUL and cyclophilin D in the cells was performed by Western blotting utilizing anti-SOUL and anti-cyclophilin D as well as anti-actin (loading control) primary antibodies. Photomicrographs demonstrate representative blots of three independent experiments. (B) Comparison of the effect of H2O2 on survival of NIH3T3 cells transfected with pcDNA3.1 (black bars), SOUL (light gray bars), pcDNA3.1+CycD siRNA (white bars) or SOUL + CycD siRNA (dark grey bars). Survival was measured by the MTT method and was expressed as % of the survival of untreated double sham-transfected cells. Values are means ± S.E.M. of three independent experiments. Significant differences are indicated above the bar; *P<0.01; **P<0.001. (C) Detection of necrosis and apoptosis was by flow cytometry following Fluorescein-conjugated Annexin V and PI double-staining performed at the end of a 24-hour incubation in the presence or absence of 300 µM H2O2. Pie charts demonstrate the distribution of living (white), necrotic (gray) and apoptotic (black) cells. Values are means of 3 independent experiments.
6.2.3. Effect of deletion of the putative BH3 domain on the cell death facilitating effect of
SOUL
Although not exclusively, BH3 domain is required in most of the cases for the function
of the BH3-only proteins. In order to determine the role of the putative BH3 domain we found
in SOUL by multiple sequence alignment, we deleted 9 amino acids (LREDGKVFD) between
position 162 and 170 as it was described previously for Bak [104]. Mock-transfected, ∆BH3-
SOUL- and SOUL-expressing cells were treated with different concentrations of hydrogen
peroxide ranging from 0 to 500 µM for 24 h, then viability was measured by the MTT method.
We found comparable expression level for ∆BH3-SOUL and SOUL by Western blotting
utilizing anti-SOUL primary polyclonal antibody (Fig 17A). Fig.17B shows that SOUL-
overexpressing cells are sensitized to hydrogen-peroxide while the cells overexpressing
∆BH3-SOUL had similar sensitivity toward it than the sham-transfected cells. That is, when a
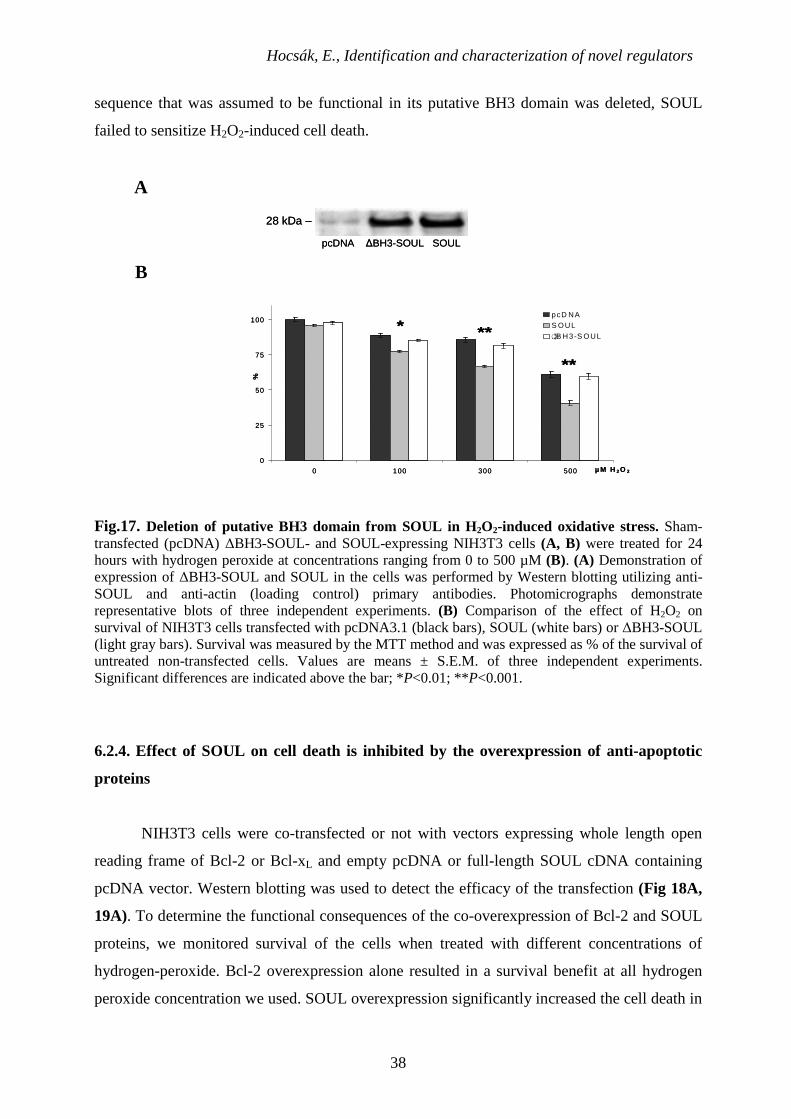
Hocsák, E., Identification and characterization of novel regulators
38
sequence that was assumed to be functional in its putative BH3 domain was deleted, SOUL
failed to sensitize H2O2-induced cell death.
A
28 kDa –
pcDNA ∆BH3-SOUL SOUL
28 kDa –
pcDNA ∆BH3-SOUL SOUL
B
0
25
50
75
100
0 100 300 500 µM H 2 O 2
%
pcD NAS OUL
..B H3-S OUL**
**
*�
0
25
50
75
100
0 100 300 500 µM H 2 O 2
%
pcD NAS OUL
..B H3-S OUL**
**
*�
Fig.17. Deletion of putative BH3 domain from SOUL in H2O2-induced oxidative stress. Sham-transfected (pcDNA) ∆BH3-SOUL- and SOUL-expressing NIH3T3 cells (A, B) were treated for 24 hours with hydrogen peroxide at concentrations ranging from 0 to 500 µM (B). (A) Demonstration of expression of ∆BH3-SOUL and SOUL in the cells was performed by Western blotting utilizing anti-SOUL and anti-actin (loading control) primary antibodies. Photomicrographs demonstrate representative blots of three independent experiments. (B) Comparison of the effect of H2O2 on survival of NIH3T3 cells transfected with pcDNA3.1 (black bars), SOUL (white bars) or ∆BH3-SOUL (light gray bars). Survival was measured by the MTT method and was expressed as % of the survival of untreated non-transfected cells. Values are means ± S.E.M. of three independent experiments. Significant differences are indicated above the bar; *P<0.01; **P<0.001.
6.2.4. Effect of SOUL on cell death is inhibited by the overexpression of anti-apoptotic
proteins
NIH3T3 cells were co-transfected or not with vectors expressing whole length open
reading frame of Bcl-2 or Bcl-xL and empty pcDNA or full-length SOUL cDNA containing
pcDNA vector. Western blotting was used to detect the efficacy of the transfection (Fig 18A,
19A). To determine the functional consequences of the co-overexpression of Bcl-2 and SOUL
proteins, we monitored survival of the cells when treated with different concentrations of
hydrogen-peroxide. Bcl-2 overexpression alone resulted in a survival benefit at all hydrogen
peroxide concentration we used. SOUL overexpression significantly increased the cell death in

Hocsák, E., Identification and characterization of novel regulators
39
the 0 – 500 µM hydrogen peroxide concentration range (p<0.01) demonstrating that SOUL
facilitated oxidative stress induced cell death. But, when cells were co-overexpressing the anti-
apoptotic Bcl-2 protein and SOUL, the effect of SOUL protein was counteracted, and survival
of these cells were similar to mock-transfected cells (Fig 18B). Similar data were observed
when Bcl-xL protein was used instead of Bcl-2 in an identical experimental setup (Fig 19). In
the presence of overexpressed Bcl-xL, SOUL could not decrease the percentage of surviving
cells compared to controls (Fig. 19B). Similar experiments were performed with ∆BH3-SOUL
overexpressing cells, however, we did not find any significant difference between these cells
and mock-transfected cells (data not shown). Data from flow cytometry (Fig. 18C, 19C)
following Annexin V-FITC and PI double staining supported our findings, and showed that the
cell death facilitated by SOUL was mainly necrotic. When cells were co-overexpressing Bcl-2
or Bcl-xL, overexpressed SOUL could not facilitate necrotic cell death induced by H2O2
(70.42±4.8% vs. 28.02±1.8% or 36.69±2.5% respectively) while mock-transfected cells
showed virtually no change in survival (96.11±4.7% and 95.28±3.8% living respectively).
These results indicate that SOUL promoted necrotic cell death induced by oxidative stress, and
this effect was counteracted by the presence of anti-apoptotic Bcl-2 homologues.
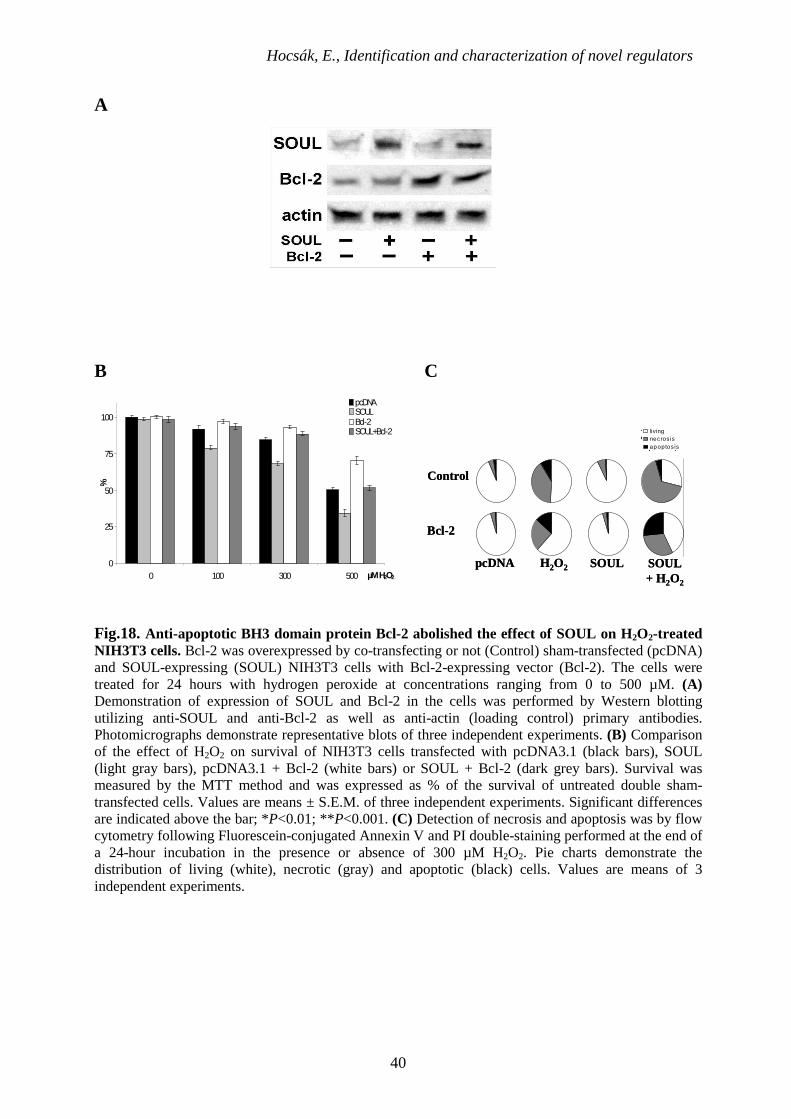
Hocsák, E., Identification and characterization of novel regulators
40
A
B C
0
25
50
75
100
0 100 300 500 µM H2O2
%
pcDNASOULBcl-2SOUL+Bcl-2
Control
Bcl-2
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecrosisapoptosis
Control
Bcl-2
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecrosisapoptosis
Fig.18. Anti-apoptotic BH3 domain protein Bcl-2 abolished the effect of SOUL on H2O2-treated NIH3T3 cells. Bcl-2 was overexpressed by co-transfecting or not (Control) sham-transfected (pcDNA) and SOUL-expressing (SOUL) NIH3T3 cells with Bcl-2-expressing vector (Bcl-2). The cells were treated for 24 hours with hydrogen peroxide at concentrations ranging from 0 to 500 µM. (A) Demonstration of expression of SOUL and Bcl-2 in the cells was performed by Western blotting utilizing anti-SOUL and anti-Bcl-2 as well as anti-actin (loading control) primary antibodies. Photomicrographs demonstrate representative blots of three independent experiments. (B) Comparison of the effect of H2O2 on survival of NIH3T3 cells transfected with pcDNA3.1 (black bars), SOUL (light gray bars), pcDNA3.1 + Bcl-2 (white bars) or SOUL + Bcl-2 (dark grey bars). Survival was measured by the MTT method and was expressed as % of the survival of untreated double sham-transfected cells. Values are means ± S.E.M. of three independent experiments. Significant differences are indicated above the bar; *P<0.01; **P<0.001. (C) Detection of necrosis and apoptosis was by flow cytometry following Fluorescein-conjugated Annexin V and PI double-staining performed at the end of a 24-hour incubation in the presence or absence of 300 µM H2O2. Pie charts demonstrate the distribution of living (white), necrotic (gray) and apoptotic (black) cells. Values are means of 3 independent experiments.
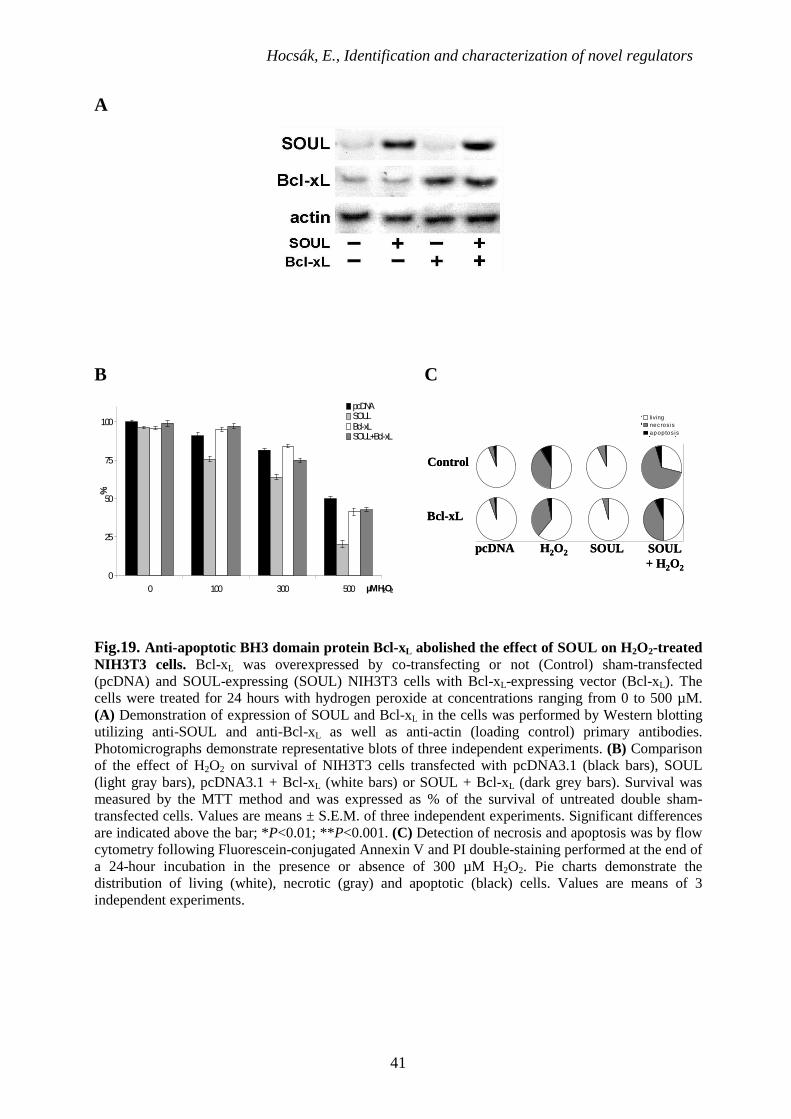
Hocsák, E., Identification and characterization of novel regulators
41
A
B C
0
25
50
75
100
0 100 300 500 µM H2O2
%
pcDNASOULBcl-xLSOUL+Bcl-xL
Control
Bcl-xL
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecros isapoptos is
Control
Bcl-xL
pcDNA H2O2 SOUL + H2O2
SOUL
livingnecros isapoptos is
Fig.19. Anti-apoptotic BH3 domain protein Bcl-xL abolished the effect of SOUL on H2O2-treated NIH3T3 cells. Bcl-xL was overexpressed by co-transfecting or not (Control) sham-transfected (pcDNA) and SOUL-expressing (SOUL) NIH3T3 cells with Bcl-xL-expressing vector (Bcl-xL). The cells were treated for 24 hours with hydrogen peroxide at concentrations ranging from 0 to 500 µM. (A) Demonstration of expression of SOUL and Bcl-xL in the cells was performed by Western blotting utilizing anti-SOUL and anti-Bcl-xL as well as anti-actin (loading control) primary antibodies. Photomicrographs demonstrate representative blots of three independent experiments. (B) Comparison of the effect of H2O2 on survival of NIH3T3 cells transfected with pcDNA3.1 (black bars), SOUL (light gray bars), pcDNA3.1 + Bcl-xL (white bars) or SOUL + Bcl-xL (dark grey bars). Survival was measured by the MTT method and was expressed as % of the survival of untreated double sham-transfected cells. Values are means ± S.E.M. of three independent experiments. Significant differences are indicated above the bar; *P<0.01; **P<0.001. (C) Detection of necrosis and apoptosis was by flow cytometry following Fluorescein-conjugated Annexin V and PI double-staining performed at the end of a 24-hour incubation in the presence or absence of 300 µM H2O2. Pie charts demonstrate the distribution of living (white), necrotic (gray) and apoptotic (black) cells. Values are means of 3 independent experiments.
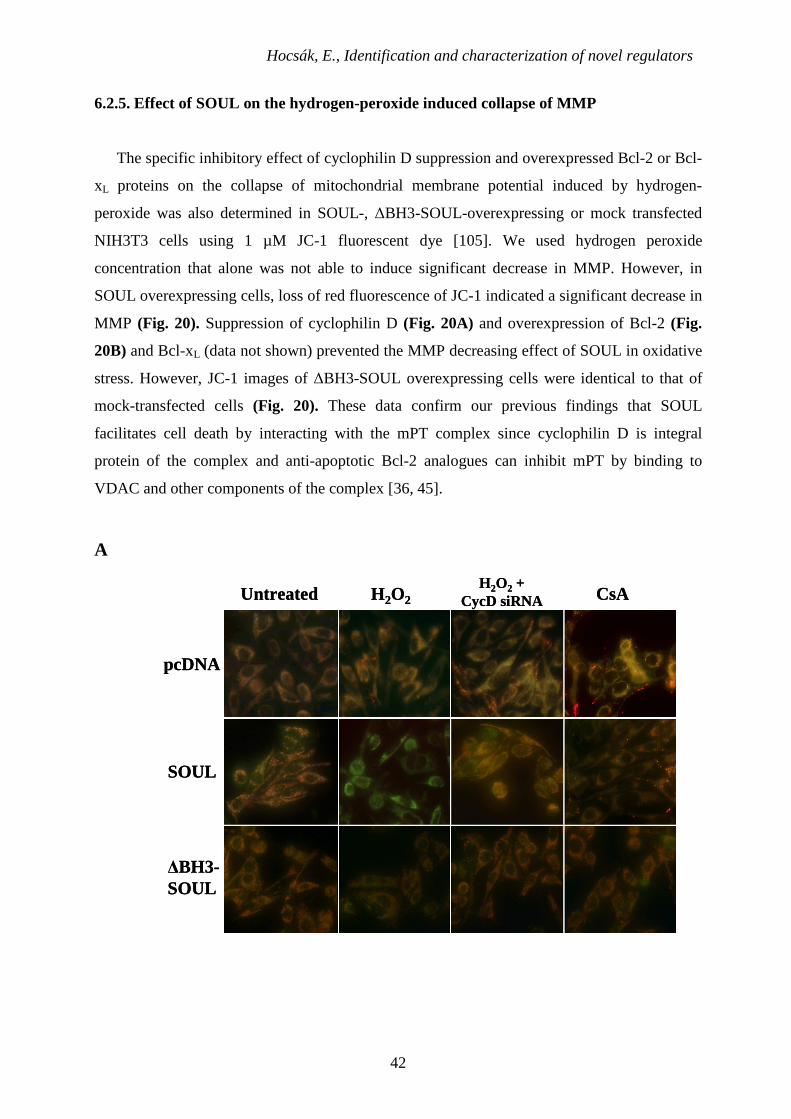
Hocsák, E., Identification and characterization of novel regulators
42
6.2.5. Effect of SOUL on the hydrogen-peroxide induced collapse of MMP
The specific inhibitory effect of cyclophilin D suppression and overexpressed Bcl-2 or Bcl-
xL proteins on the collapse of mitochondrial membrane potential induced by hydrogen-
peroxide was also determined in SOUL-, ∆BH3-SOUL-overexpressing or mock transfected
NIH3T3 cells using 1 µM JC-1 fluorescent dye [105]. We used hydrogen peroxide
concentration that alone was not able to induce significant decrease in MMP. However, in
SOUL overexpressing cells, loss of red fluorescence of JC-1 indicated a significant decrease in
MMP (Fig. 20). Suppression of cyclophilin D (Fig. 20A) and overexpression of Bcl-2 (Fig.
20B) and Bcl-xL (data not shown) prevented the MMP decreasing effect of SOUL in oxidative
stress. However, JC-1 images of ∆BH3-SOUL overexpressing cells were identical to that of
mock-transfected cells (Fig. 20). These data confirm our previous findings that SOUL
facilitates cell death by interacting with the mPT complex since cyclophilin D is integral
protein of the complex and anti-apoptotic Bcl-2 analogues can inhibit mPT by binding to
VDAC and other components of the complex [36, 45].
A
pcDNA
SOUL
H2O2H2O2 +
CycD siRNA CsAUntreated
∆BH3-SOUL
pcDNA
SOUL
H2O2H2O2 +
CycD siRNA CsAUntreated
∆BH3-SOUL
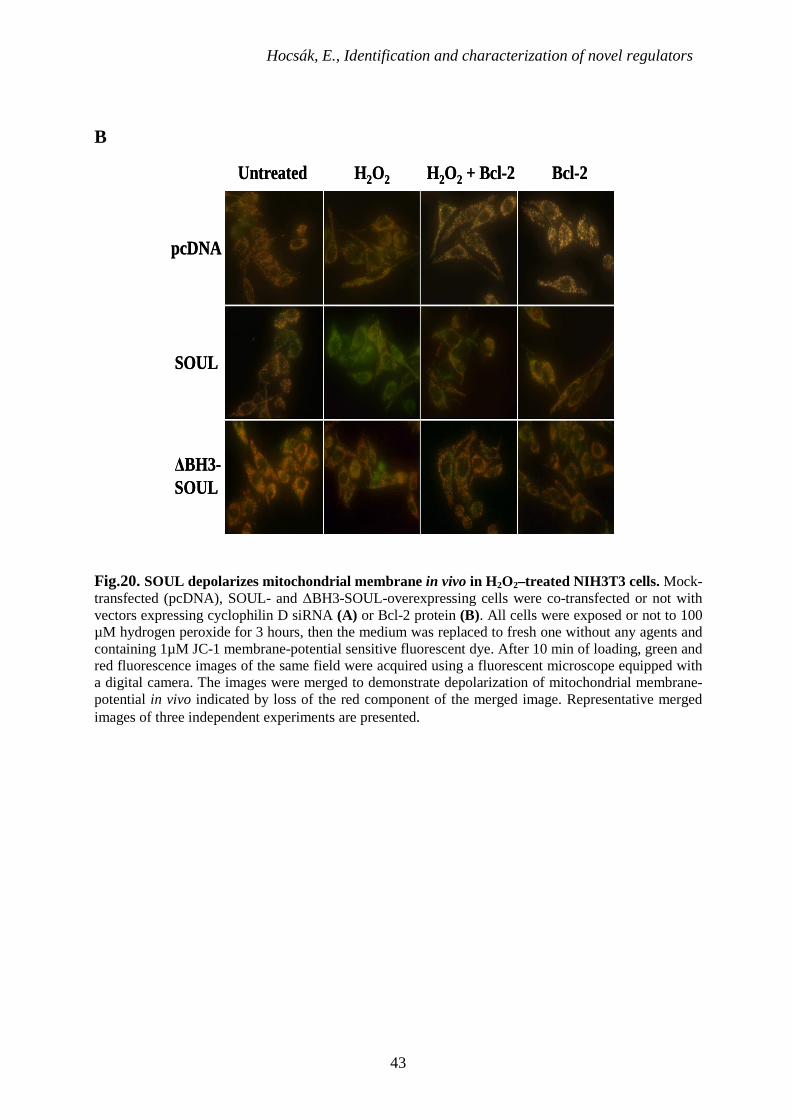
Hocsák, E., Identification and characterization of novel regulators
43
B
H2O2 H2O2 + Bcl-2 Bcl-2
pcDNA
SOUL
Untreated
∆BH3-SOUL
H2O2 H2O2 + Bcl-2 Bcl-2
pcDNA
SOUL
Untreated
∆BH3-SOUL
Fig.20. SOUL depolarizes mitochondrial membrane in vivo in H2O2–treated NIH3T3 cells. Mock-transfected (pcDNA), SOUL- and ∆BH3-SOUL-overexpressing cells were co-transfected or not with vectors expressing cyclophilin D siRNA (A) or Bcl-2 protein (B). All cells were exposed or not to 100 µM hydrogen peroxide for 3 hours, then the medium was replaced to fresh one without any agents and containing 1µM JC-1 membrane-potential sensitive fluorescent dye. After 10 min of loading, green and red fluorescence images of the same field were acquired using a fluorescent microscope equipped with a digital camera. The images were merged to demonstrate depolarization of mitochondrial membrane-potential in vivo indicated by loss of the red component of the merged image. Representative merged images of three independent experiments are presented.

Hocsák, E., Identification and characterization of novel regulators
44
7. Discussion
Mitochondria play a key role in the process of both apoptotic and necrotic cell death
[106]. We induced cell death either with etoposide that promotes apoptosis [98], we also used
hydrogen-peroxide or Taxol to induce cell deth with different mechanisms. Taxol, a potent
chemotherapeutic agent and currently applied in the treatment of ovarian, lung and breast
cancer [107], is known to bring on cell death by altering microtubule assembly and arresting
the cell cyle in G2/M phase [88].
Despite the controversy surrounding the main function of TIP47, the
oncodevelopmental significance of this protein has been established TIP47 is strongly
involved in cancer progression [74-77, 108-109]. We hypothesized that its lipid-trafficking-
related function might not play a role in tumor development, so we investigated its possible
effect in the regulation of cell death. We overexpressed TIP47 in NIH3T3 cell line that
produces this protein in a very low extent endogenously and used its spliced variant, the
truncated-TIP47 (t-TIP47 or PP17a) for internal control. t-TIP47 was expressed in placenta
[73] and it lacks 183 amino acids from the N-terminus of TIP47. We found that TIP47 or t-
TIP47 overexpression had no effect on the survival of NIH3T3 cells, but when cells were
exposed to oxidative stress TIP47 overexpression resulted in increased cell viability while t-
TIP47 had no protective function at all (Fig.5A, B). Interestingly, Orlicky et al. reported that
the N-terminal region of TIP47 is responsible for its lipid related function [60].
To prove its specific effect we investigated the endogenous rather than overexpressed
TIP47 on the cell death process in HeLa cell line that expresses TIP47 in a high extent
(Fig6A). Suppression of TIP47 by dsiRNA technique reached it maximum on the second day
after transfection, but we also found, that complete TIP47 depletion could not be reached
[110]. Nevertheless, suppression of TIP47 made cells less viable to oxidative stress (Fig6B, C)
indicating its specific effect on cell death.
To understand the cellular mechanism by which TIP47 protects cells from oxidative
stress we investigated the cell death in detail. We found that in TIP47-overexpressing cells
treated with H2O2 or Taxol less necrotic cell death occur; they rather showed the signs of early
apoptosis similarly to etoposide treated cells while the overexpression of t-TIP47 had no effect
whatsoever (Fig.7A, B, Fig.8).

Hocsák, E., Identification and characterization of novel regulators
45
AIF and EndoG can be activated by various stress stimuli, including chemotherapeutic
agents [111-112]. When activated and released from the mitochondria, they are translocated to
the nucleus, where they cause the degradation of DNA [113]. Taxol treatment activated AIF
and Endo G and induced their translocation in mock-transfected fibroblast cells. It is of interest
that, in TIP47-transfected cells treatment with taxol did not alter the intracellular localization
of these proteins (Fig.11).
The type of cell death occurring is related to the strength of the insult. During
mitochondrial permeability transition mitochondrial intermembrane proteins could be released
leading to cytochrome c-triggered apoptosis or in case of the permeability transition pore-
dependent failure of ATP generation to necrosis [106]. Since we found that TIP47 can occur in
the mitochondrial subcellular fraction of H2O2 treated cells (Fig.9) we were concerned about
its effect on mitochondria. In vitro we observed that TIP47 did not induce significant swelling
of mitochondria, but it hyperpolarized the mitochondrial membrane while t-TIP47 evoked no
such effect (Fig.10). By using a mitochondrial membrane potential-sensitive fluorescent dye,
JC-1 in NIH3T3 cells we confirmed that TIP47 could stabilize mitochondrial membrane
potential (Fig.12). Several proteins exist that stabilize the mitochondrial membrane.
Endothelin-1 has been reported to be a potent survival factor against myocardial cell death
[114]. Mpv17l overexpression can also stabilize the mitochondrial membrane, but it also forms
complex with HtrA2 that is capable of sensing and regulating ROS net balance in response to
internal and external stress [115]. Since the increase of ROS generation can result in the
collapse of the inner mitochondrial membrane potential and subsequent membrane
permeabilization with the release of cytochrome c into the cytosol, and so the triggering
cellular apoptosis cascades, we determined the effect of TIP47 on intracellular ROS formation.
Surprisingly, we could not observe any significant difference between controls or t-TIP47
transfected or TIP47-overexpressing cells indicating that TIP47 did not contribute to ROS-
formation (Fig.13.)
According to our result TIP47 can stabilize the mitochondrial membrane and influence
cell death without affecting intracellular ROS-formation. This function of TIP47 might be
related to its N-terminal sequence. Overexpression of TIP47 could be a serious advantage for
tumor cells to grow. As we have earlier shown, there is a correlation between the invasiveness
of a cervical tumor and the amount of TIP47 in the cells and the sera [77].

Hocsák, E., Identification and characterization of novel regulators
46
Taxol has been shown to regulate the expression of apoptosis-related proteins,
including Bcl-2, Bax, Bcl-x, p21-waf and tumor necrosis factor alpha (TNF-alpha) [83, 116],
and so, to induce apoptosis [117]. Furthermore, a recent study reported that taxol induces
caspase-independent, AIF-dependent apoptosis in SKOV3 cells [107]. To investigate the effect
of TIP47 on the major apoptotic pathways, TIP47-transfected and mock-transfected NIH3T3
cells were subjected to Taxol treatment, and the levels of Bax, Bcl-2, total caspase-3 and
cleaved caspase-3 were determined by Western blot. The anti-apoptotic effect of Bcl-2,
exerted in part by the inhibition of translocation of Bax to the mitochondria, is well
documented, while Bax and caspase-3 are key participants of the caspase-dependent apoptotic
pathway [118]. Cells transfected with TIP47 acquired an anti-apoptotic quality, since Taxol
exposure in these cells led to increased levels of Bcl-2 and reduced levels of Bax and cleaved
caspase-3, as opposed to the control cells (Fig.14).
In our second study, based on multiple-sequence alignment that indicated that SOUL
has a BH3 domain-like structure, we assumed that the cell deathpromoting effect of SOUL
might be related to this structure (Fig.15). To prove this concept, we disrupted the putative
BH3 domain of SOUL by deleting 9 amino acids from the functional core similarly as it was
done for disrupting BH3 domain function in Bak [104, 119]. We found that the resulting
protein failed to facilitate H2O2-induced cell death. Since cyclophilin D is an essential integral
protein of the PTPC [106, 119], it seems likely that SOUL exerts its effect by interacting with
the complex itself. When cell death was induced under the conditions of cyclophilin D
suppression (Fig.16) or Bcl-2 as well as Bcl-xL overexpression (Figs.18 and 19), BH3-SOUL-
expressing cells did not differ significantly from the sham-transfected ones. Furthermore, it did
not promote H2O2-induced mitochondrial membrane depolarization, as was revealed by JC-1
staining (Fig.20). Because excision of this short sequence resulted in loss of all cell death-
related properties of SOUL, and this sequence showed homology to the BH3 domains of
established Bcl-2 family proteins, SOUL could represent a novel BH3 domain-only protein.
It was reported that BH3 domain proteins, such as the proapoptotic Bcl-2 family
member tBid, by forming a complex with another family member, Bax, can induce cyclophilin
D-independent outer mitochondrial membrane permeabilization and release of proapoptotic
mitochondrial proteins [37]. Oligomer Bax can evoke even mitochondrial permeability
transition and mitochondrial swelling [40, 120]. Despite the accompanying release of

Hocsák, E., Identification and characterization of novel regulators
47
proapoptotic mitochondrial proteins, mPT induced by oligomer Bax can lead to cyclophilin D-
sensitive necrotic cell death [40, 120]. Besides tBid and Bax, Bak was reported to bind to
VDAC and facilitate necrotic and apoptotic cell death [119-124]. It is very likely that SOUL as
a BH3 domain-only protein exerts its effect in a similar manner. This is specifically true under
the conditions of oxidative stress where mPT is induced, and both apoptotic and necrotic cell
death pathways can be activated [106].
Antiapoptotic Bcl-2 homologues can bind to VDAC and inhibitmPTand cell death [36,
45, 119, 125]. We determined whether these Bcl-2 homologues can reverse the action of
SOUL. Our data clearly showed that both Bcl-2 and Bcl-xL could prevent SOUL-promoted
cell death (Figs.18 and 19). Therefore, antiapoptotic Bcl-2 proteins counteract the effect of
both SOUL and the proapoptotic Bcl-2 proteins in basically the same manner. Similarly to
oligomer Bax [40], SOUL promoted the permeabilization of not only the outer but also the
inner mitochondrial membrane, and as a consequence, SOUL facilitated predominantly
necrotic cell death.
Previously, it was reported that the recombinant SOUL protein oligomerizes and forms
hexamers in aqueous solution [81]. It is also known that Bax and Bak can oligomerize [126],
and their oligomer forms are very effective in inducing cell death [40, 120]. Therefore, it is
likely that oligomerization plays a role in the effect of SOUL.
The best studied BH3 domain-only proteins, Bim and Bid, permeabilize the outer
mitochondrial membrane and release proapoptotic mitochondrial proteins without affecting the
integrity of the inner membrane; therefore, they induce cyclophilin D-insensitive apoptotic
death [37, 102, 127]. In contrast, SOUL could not induce cell death by itself like Bim and Bid,
but when cell death was induced by various stimuli, such as Taxol, A23187 [79] or H2O2,
SOUL enhanced it and directed it mainly toward necrosis. The effects on the cell death process
of oligomer Bax and SOUL are very similar. Both are capable of permeabilizing the inner
together with the outer mitochondrial membrane, inducing the release of proapoptotic
mitochondrial proteins and promoting cell death. However, Bax can induce both necrotic and
apoptotic cell death by itself [37, 40, 120], whereas SOUL can only facilitate cell death
induced by other stimuli and direct it toward a necrotic mechanism, presumably by amplifying
mitochondrial impairment.

Hocsák, E., Identification and characterization of novel regulators
48
All together, we provided evidence that TIP47 and SOUL can affect the cell death
process in a different way.
We proved, that SOUL senzitizes cells to necrotic cell death, not only permeabilized
the outer mitochondrial membrane like proapoptotic Bcl-2 homologues but also facilitated an
inner membrane permeabilization in a cyclophilin D-dependent manner and the collapse of
mitochondrial membrane potential. Most likely, this difference is responsible for the observed
properties of SOUL, namely that it cannot induce cell death alone like other proapoptotic BH3
domain-only proteins but facilitates predominantly necrotic cell death. Thus, SOUL plays a
much more complex role in cell physiology than its mere possible involvement in binding and
transporting heme intermediates.
This study provides convincing evidence of the protective effect of TIP47 which exerts
an anti-apoptotic effect on cell survival by inhibiting the activation of proapoptotic proteins,
by assisting the effect of anti-apoptotic proteins and by the direct stabilization of the
mitochondrial membrane potential. Our findings are of particular importance, since they
propose an explanation for the observed cytostatic resistance against taxol-induced cell death
in cancer cells overexpressing TIP47, and thus, lend further insight into the mechanisms used
by cancer cells to evade chemotherapy.
Using our new results we can better understand how and why would elevated TIP47
levels result in the progression of cervical tumors, but still further studies are required to
exploit this property of the protein in the fight against cancer.

Hocsák, E., Identification and characterization of novel regulators
49
8. Conclusions
1. Effect of TIP47 on cell death
We examined effect of TIP47 in different conditions by treating cells with cell death
inducing agents and detecting the living using MTT assay. Hydrogen-peroxide causes
oxidative stress and Taxol affects the microtubule system. In the presence of excess TIP47
cells seemed to have increased resistance against the above mentioned drugs in our
experiment. This effect was statistically significant and was detected with all kinds of drugs
used.
2. Effect of TIP47 suppression by siRNA technique on cell death
Gene silencing is generally used to describe the "switching off" of a gene by a
mechanism other than genetic modification. This process caused by the lack of this specific
protein. When TIP47 was silenced using specific dsiRNAs in cells endogenously expressing it
significant sensitization was observed against the cell death inducing agents as expected.
3. Role of TIP47 in cell death
We used flow cytometry and fluorescent microscopy in this experiment. Cells were
treated with different agent and were stained with propidium iodide and FITC-conjugated
Annexin V to distinguish between the two types of cell death. When we treated the TIP47
transfected cells, the cells died mainly by early apoptosis, and the number of late apoptotic and
necrotic cells decreased drastically. Taxol treatment activated AIF and Endo G and induced
their translocation in mock-transfected fibroblast cells. It is of interest that, in TIP47-
transfected cells treatment with Taxol did not alter the intracellular localization of these
proteins.
4. Intracellular localization of TIP47 in oxidative stress
We detected TIP47’s subcellular localization with Western blot after fractionating cells
endogenously producing high amount of TIP47. According to our results TIP47 was found in

Hocsák, E., Identification and characterization of novel regulators
50
the cytoplasm, but after hydrogen peroxide treatment it was also present in the mitochondrial
fraction in a smaller quantity.
5. Effect of TIP47 on the mitochondrial permeability transition and on the
Mitochondrial Membrane Potential (∆ψ)
We provide that TIP47 can bind to mitochondria and protect mitochondrial membrane
integrity, as well as prevent oxidative stress induced cell death. First we tracked the change of
the mitochondrial membrane potential in TIP47-overexpressing NIH3T3 cells using
fluorescent microscopy. In the TIP47 transfected cells the mitochondrial membrane potential
remained stable after treatment. This result reised our interest and we isolated mitochondria
from rat liver to detect direct effect of recombinant TIP47 in vitro.Recombinant TIP47 not
induced mitochondrial permeability transition when we added subthreshold concentrations of
calcium. These data supports our view that TIP47 protects cell by stabilizing mitochondrial
membrane.
6. Effect of TIP47 on ROS-production
We could not observe any significant difference between controls or TIP47-
overexpressing cells indicating that TIP47 did not contribute to ROS-formation.
7. Effect of TIP47 on the pro- and anti-apoptotic signaling pathways
We investigated the effect of TIP47 on cell viability and on the pro- and anti-apoptotic
signaling pathways in cells, following treatment with the chemotherapeutic agent, Taxol. We
found, that TIP47 over-expressed cells increased levels of Bcl-2 and reduced levels of Bax and
cleaved caspase-3, as opposed to the control cells.
8. Role of mPT complex in the SOUL-induced sensitization toward oxidative stress
We observed that SOUL-overexpressing cells are sensitized to hydrogen-peroxide
while the cyclophilin D suppressed cells had similar sensitivity toward it than the sham-
transfected cells. We performed similar experiments with ∆BH3-SOUL overexpressing cells.

Hocsák, E., Identification and characterization of novel regulators
51
Blocking mPT by suppressing cyclophilin D inhibited mitochondrial permeability transition
and blocked the cell death sensitizing effect of SOUL in oxidative stress.
9. Effect of deletion of the putative BH3 domain on the cell death
We showed that SOUL promotes the permeabilization of both outer and inner
mitochondrial membranes in oxidative stress and that its effect can be reversed by deleting the
putative BH3 sequence from SOUL. When we deleted a 9 amino acid (LREDGKVFD)
sequence that was assumed to be functional in its putative BH3 domain, SOUL failed to
sensitize H2O2 induced cell death.
10. Effect of SOUL on cell death is inhibited by the overexpression of anti-apoptotic
proteins
When cells were co-overexpressing Bcl-2 or Bcl-xL, overexpressed SOUL could not
facilitate necrotic cell death induced by hydrogen-peroxide. Data from flow cytometry showed
that the cell death facilitated by SOUL was mainly necrotic. SOUL promoted necrotic cell
death induced by oxidative stress, and this effect was counteracted by the presence of anti-
apoptotic Bcl-2 homologues.
11. Effect of SOUL on the H2O2-induced collapse of MMP
SOUL facilitated cell death by the promotion of the collapse of mitochondrial
membrane potential (MMP) in living cells. This protein senzitized the cells to cell death by
interacting with the mPT complex since cyclophilin D is integral protein of the complex and
anti-apoptotic Bcl-2 analogues can inhibit mPT by binding to VDAC and other components of
the complex.

Hocsák, E., Identification and characterization of novel regulators
52
9. Acknowledgements
These studies were carried out at the Department of Biochemistry and Medical Chemistry,
Medical School of the University of Pécs between 2006-2010.
I want to express my thanks to my teacher and project leader, Professor Dr. Balázs Sümegi
who managed my studies and gave support and useful advises during my work.
I would like to thank Prof. Dr. Ferenc Gallyas for his tutorial work.
I am grateful to my closest colleagues who assisted me in my experimental studies and
gave a hand with a part of the experiments: Dr. András Szigeti, Dr. Szabolcs Bellyei, Dr.
Boglárka Rácz and Alíz Szabó. I wish to thank all participants of the experiments for their
work, all the doctors, PhD students and assistants of the Institute Biochemistry and Medical
Chemistry participating in the project for their theoretical and technical help.
At last but not least, this work would have never been realized without the support and
help of my family.
This work was supported by AOKKA-34039-1/2009 and 34039-23/2009 as well as
Hungarian Research Grants OTKA 68469, K-73738 and Science, Please! Research Team on
Innovation (SROP-4.2.2/08/1/2008-0011).

Hocsák, E., Identification and characterization of novel regulators
53
10. References
1. Kroemer G, Galluzzi L, and Brenner C., Mitochondrial membrane permeabilization in cell death. Physiol Rev, 2007. 87(1): p. 99-163.
2. Nicotera P, Leist M, and Ferrando-May E., Intracellular ATP, a switch in the decision
between apoptosis and necrosis. Toxicol Lett, 1998. 102-103: p. 139-42. 3. Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G.,
Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis, 2007. 12(5): p. 803-13.
4. Brustovetsky N and Klingenberg M., Mitochondrial ADP/ATP carrier can be
reversibly converted into a large channel by Ca2+. Biochemistry, 1996. 35(26): p. 8483-8.
5. Crompton M., The mitochondrial permeability transition pore and its role in cell
death. Biochem J, 1999. 341 ( Pt 2): p. 233-49. 6. Crompton M, Virji S, Doyle V, Johnson N, Ward JM., The mitochondrial permeability
transition pore. Biochem Soc Symp, 1999. 66: p. 167-79. 7. Crompton M, Virji S, and Ward JM., Cyclophilin-D binds strongly to complexes of the
voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem, 1998. 258(2): p. 729-35.
8. Haworth RA and Hunter DR., The Ca2+-induced membrane transition in
mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys, 1979. 195(2): p. 460-7.
9. Woodfield K, Rück A, Brdiczka D, Halestrap AP., Direct demonstration of a specific
interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J, 1998. 336 ( Pt 2): p. 287-90.
10. Zoratti M and Szabo I., The mitochondrial permeability transition. Biochim Biophys
Acta, 1995. 1241(2): p. 139-76. 11. Abou-Sleiman PM, Muqit MM, and Wood NW., Expanding insights of mitochondrial
dysfunction in Parkinson's disease. Nat Rev Neurosci, 2006. 7(3): p. 207-19. 12. Zoratti M and Szabo I., Electrophysiology of the inner mitochondrial membrane. J
Bioenerg Biomembr, 1994. 26(5): p. 543-53. 13. Bouillet P and Strasser A., BH3-only proteins - evolutionarily conserved proapoptotic
Bcl-2 family members essential for initiating programmed cell death. J Cell Sci, 2002. 115(Pt 8): p. 1567-74.

Hocsák, E., Identification and characterization of novel regulators
54
14. Shimizu S and Tsujimoto Y., Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A, 2000. 97(2): p. 577-82.
15. Hossini AM and Eberle J., Apoptosis induction by Bcl-2 proteins independent of the
BH3 domain. Biochem Pharmacol, 2008. 76(11): p. 1612-9. 16. Youle RJ and Strasser A., The BCL-2 protein family: opposing activities that mediate
cell death. Nat Rev Mol Cell Biol, 2008. 9(1): p. 47-59. 17. Strasser A., The role of BH3-only proteins in the immune system. Nat Rev Immunol,
2005. 5(3): p. 189-200. 18. Adams J.M and Cory S., The Bcl-2 protein family: arbiters of cell survival. Science,
1998. 281(5381): p. 1322-6. 19. Hsu SY and Hsueh AJ., Tissue-specific Bcl-2 protein partners in apoptosis: An ovarian
paradigm. Physiol Rev, 2000. 80(2): p. 593-614. 20. Martinou JC and Green DR., Breaking the mitochondrial barrier. Nat Rev Mol Cell
Biol, 2001. 2(1): p. 63-7. 21. Gross A, McDonnell JM, and Korsmeyer SJ., BCL-2 family members and the
mitochondria in apoptosis. Genes Dev, 1999. 13(15): p. 1899-911. 22. Kelekar A and Thompson CB., Bcl-2-family proteins: the role of the BH3 domain in
apoptosis. Trends Cell Biol, 1998. 8(8): p. 324-30. 23. Reed JC., Bcl-2 family proteins. Oncogene, 1998. 17(25): p. 3225-36. 24. Altznauer F, Conus S, Cavalli A, Folkers G, Simon HU., Calpain-1 regulates Bax and
subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J Biol Chem, 2004. 279(7): p. 5947-57.
25. Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews
DW., Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J, 2005. 24(12): p. 2096-103.
26. Epand RF., Martinou JC, Montessuit S, Epand RM., Transbilayer lipid diffusion
promoted by Bax: implications for apoptosis. Biochemistry, 2003. 42(49): p. 14576-82. 27. Soan L and Fiskum G., Inhibition of mitochondrial neural cell death pathways by
protein transduction of Bcl-2 family proteins. J Bioenerg Biomembr, 2005. 37(3): p. 179-90.
28. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ., Bad, a
heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell, 1995. 80(2): p. 285-91.

Hocsák, E., Identification and characterization of novel regulators
55
29. Liu J, Weiss A, Durrant D, Chi NW, Lee RM., The cardiolipin-binding domain of Bid affects mitochondrial respiration and enhances cytochrome c release. Apoptosis, 2004. 9(5): p. 533-41.
30. Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A,
Basañez G., Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem, 2004. 279(29): p. 30081-91.
31. de Jonge HR, Hogema B, and Tilly BC., Protein N-myristoylation: critical role in
apoptosis and salt tolerance. Sci STKE, 2000. 2000(63): p. pe1. 32. Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM.,
Bid, but not Bax, regulates VDAC channels. J Biol Chem, 2004. 279(14): p. 13575-83. 33. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. , Voltage-dependent
anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol, 2007. 9(5): p. 550-5.
34. Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C., Glycogen synthase kinase 3
inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res, 2008. 103(9): p. 983-91.
35. Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C.,
Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene, 2008. 27(3): p. 285-99.
36. Tsujimoto Y, Nakagawa T, and Shimizu S., Mitochondrial membrane permeability
transition and cell death. Biochim Biophys Acta, 2006. 1757(9-10): p. 1297-300. 37. Brustovetsky N, Dubinsky JM, Antonsson B, Jemmerson R., Two pathways for tBID-
induced cytochrome c release from rat brain mitochondria: BAK- versus BAX-dependence. J Neurochem, 2003. 84(1): p. 196-207.
38. Wang X, Ryter SW, Dai C, Tang ZL, Watkins SC, Yin XM, Song R, Choi AM.,
Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J Biol Chem, 2003. 278(31): p. 29184-91.
39. Wu S, Xing D, Gao X, Chen WR., High fluence low-power laser irradiation induces
mitochondrial permeability transition mediated by reactive oxygen species. J Cell Physiol, 2009. 218(3): p. 603-11.
40. Li T, Brustovetsky T, Antonsson B, Brustovetsky N., Oligomeric BAX induces
mitochondrial permeability transition and complete cytochrome c release without oxidative stress. Biochim Biophys Acta, 2008. 1777(11): p. 1409-21.
41. Tilly JL., Commuting the death sentence: how oocytes strive to survive. Nat Rev Mol
Cell Biol, 2001. 2(11): p. 838-48.

Hocsák, E., Identification and characterization of novel regulators
56
42. Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G., Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science, 2008. 321(5893): p. 1206-10.
43. Malia TJ and Wagner G., NMR structural investigation of the mitochondrial outer
membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry, 2007. 46(2): p. 514-25.
44. Tafani M, Minchenko DA, Serroni A, Farber JL., Induction of the mitochondrial
permeability transition mediates the killing of HeLa cells by staurosporine. Cancer Res, 2001. 61(6): p. 2459-66.
45. Armstrong JS, The role of the mitochondrial permeability transition in cell death.
Mitochondrion, 2006. 6(5): p. 225-34. 46. Tsujimoto Y and Shimizu S., Role of the mitochondrial membrane permeability
transition in cell death. Apoptosis, 2007. 12(5): p. 835-40. 47. Blanchette-Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM,
Theodorakis JL, Greenberg AS, Londos C., Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res, 1995. 36(6): p. 1211-26.
48. Sztalryd C, Bell M, Lu X, Mertz P, Hickenbottom S, Chang BH, Chan L, Kimmel AR,
Londos C., Functional compensation for adipose differentiation-related protein (ADFP) by Tip47 in an ADFP null embryonic cell line. J Biol Chem, 2006. 281(45): p. 34341-8.
49. Wolins NE, Quaynor BK, Skinner JR, Schoenfish MJ, Tzekov A, Bickel PE., S3-12,
Adipophilin, and TIP47 package lipid in adipocytes. J Biol Chem, 2005. 280(19): p. 19146-55.
50. Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V,
Yao-Borengasser A, Rasouli N, Kern PA, Finck BN, Bickel PE., OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes, 2006. 55(12): p. 3418-28.
51. Wolins NE, Skinner JR, Schoenfish MJ, Tzekov A, Bensch KG, Bickel PE., Adipocyte
protein S3-12 coats nascent lipid droplets. J Biol Chem, 2003. 278(39): p. 37713-21. 52. Bickel PE, Tansey JT, and Welte MA., PAT proteins, an ancient family of lipid droplet
proteins that regulate cellular lipid stores. Biochim Biophys Acta, 2009. 1791(6): p. 419-40.
53. Lu X, Gruia-Gray J, Copeland NG, Gilbert DJ, Jenkins NA, Londos C, Kimmel AR.,
The murine perilipin gene: the lipid droplet-associated perilipins derive from tissue-specific, mRNA splice variants and define a gene family of ancient origin. Mamm Genome, 2001. 12(9): p. 741-9.
54. Miura S, Gan JW, Brzostowski J, Parisi MJ, Schultz CJ, Londos C, Oliver B, Kimmel
AR., Functional conservation for lipid storage droplet association among Perilipin,

Hocsák, E., Identification and characterization of novel regulators
57
ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J Biol Chem, 2002. 277(35): p. 32253-7.
55. Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C.,
Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res, 1997. 38(11): p. 2249-63.
56. Servetnick DA, Brasaemle DL, Gruia-Gray J, Kimmel AR, Wolff J, Londos C.,
Perilipins are associated with cholesteryl ester droplets in steroidogenic adrenal cortical and Leydig cells. J Biol Chem, 1995. 270(28): p. 16970-3.
57. Wolins NE, Rubin B, and Brasaemle DL., TIP47 associates with lipid droplets. J Biol
Chem, 2001. 276(7): p. 5101-8. 58. Barbero P, Buell E, Zulley S, Pfeffer SR., TIP47 is not a component of lipid droplets. J
Biol Chem, 2001. 276(26): p. 24348-51. 59. Muthusamy K, Halbert G, and Roberts F., Immunohistochemical staining for
adipophilin, perilipin and TIP47. J Clin Pathol, 2006. 59(11): p. 1166-70. 60. Orlicky DJ, Degala G, Greenwood C, Bales ES, Russell TD, McManaman JL.,
Multiple functions encoded by the N-terminal PAT domain of adipophilin. J Cell Sci, 2008. 121(Pt 17): p. 2921-9.
61. Robenek H, Lorkowski S, Schnoor M, Troyer D., Spatial integration of TIP47 and
adipophilin in macrophage lipid bodies. J Biol Chem, 2005. 280(7): p. 5789-94. 62. Robenek H, Robenek MJ, Buers I, Lorkowski S, Hofnagel O, Troyer D, Severs NJ.,
Lipid droplets gain PAT family proteins by interaction with specialized plasma membrane domains. J Biol Chem, 2005. 280(28): p. 26330-8.
63. Diaz E and Pfeffer SR., TIP47: a cargo selection device for mannose 6-phosphate
receptor trafficking. Cell, 1998. 93(3): p. 433-43. 64. Sincock PM, Ganley IG, Krise JP, Diederichs S, Sivars U, O'Connor B, Ding L, Pfeffer
SR., Self-assembly is important for TIP47 function in mannose 6-phosphate receptor transport. Traffic, 2003. 4(1): p. 18-25.
65. Krise JP, Sincock PM, Orsel JG, Pfeffer SR., Quantitative analysis of TIP47-receptor
cytoplasmic domain interactions: implications for endosome-to-trans Golgi network trafficking. J Biol Chem, 2000. 275(33): p. 25188-93.
66. Orsel JG, Sincock PM, Krise JP, Pfeffer SR., Recognition of the 300-kDa mannose 6-
phosphate receptor cytoplasmic domain by 47-kDa tail-interacting protein. Proc Natl Acad Sci U S A, 2000. 97(16): p. 9047-51.
67. Aivazian D, Serrano RL, and Pfeffer S., TIP47 is a key effector for Rab9 localization. J
Cell Biol, 2006. 173(6): p. 917-26.

Hocsák, E., Identification and characterization of novel regulators
58
68. Carroll KS, Hanna J, Simon I, Krise J, Barbero P, Pfeffer SR., Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science, 2001. 292(5520): p. 1373-6.
69. Heid HW, Moll R, Schwetlick I, Rackwitz HR, Keenan TW., Adipophilin is a specific
marker of lipid accumulation in diverse cell types and diseases. Cell Tissue Res, 1998. 294(2): p. 309-21.
70. Bell M, Wang H, Chen H, McLenithan JC, Gong DW, Yang RZ, Yu D, Fried SK,
Quon MJ, Londos C, Sztalryd C., Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes, 2008. 57(8): p. 2037-45.
71. Ohsaki Y, Maeda T, Maeda M, Tauchi-Sato K, Fujimoto T., Recruitment of TIP47 to
lipid droplets is controlled by the putative hydrophobic cleft. Biochem Biophys Res Commun, 2006. 347(1): p. 279-87.
72. Lopez-Vergès S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C., Tail-
interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci U S A, 2006. 103(40): p. 14947-52.
73. Than NG, Sumegi B, Than GN, Kispal G, Bohn H., Cloning and sequence analysis of
cDNAs encoding human placental tissue protein 17 (PP17) variants. Eur J Biochem, 1998. 258(2): p. 752-7.
74. Than NG, Sumegi B, Than GN, Kispal G, Bohn H., Cloning and sequencing of human
oncodevelopmental soluble placental tissue protein 17 (PP17): homology with adipophilin and the mouse adipose differentiation-related protein. Tumour Biol, 1999. 20(4): p. 184-92.
75. Than NG, Sumegi B, Than GN, Kispal G, Bohn H., Is placental tissue protein
17b/TIP47 a new factor in cervical cancer genesis? Anticancer Res, 1999. 19(6B): p. 5255-8.
76. Than GN, Turóczy T, Sümegi B, Than NG, Bellyei S, Bohn H, Szekeres G.,
Overexpression of placental tissue protein 17b/TIP47 in cervical dysplasias and cervical carcinoma. Anticancer Res, 2001. 21(1B): p. 639-42.
77. Szigeti A, Minik O, Hocsak E, Pozsgai E, Boronkai A, Farkas R, Balint A, Bodis J,
Sumegi B, Bellyei S., Preliminary study of TIP47 as a possible new biomarker of cervical dysplasia and invasive carcinoma. Anticancer Res, 2009. 29(2): p. 717-24.
78. Hermes-Lima M., How do Ca2+ and 5-aminolevulinic acid-derived oxyradicals
promote injury to isolated mitochondria? Free Radic Biol Med, 1995. 19(3): p. 381-90. 79. Szigeti A, Bellyei S, Gasz B, Boronkai A, Hocsak E, Minik O, Bognar Z, Varbiro G,
Sumegi B, Gallyas F Jr., Induction of necrotic cell death and mitochondrial permeabilization by heme binding protein 2/SOUL. FEBS Lett, 2006. 580(27): p. 6447-54.

Hocsák, E., Identification and characterization of novel regulators
59
80. Jacob Blackmon B, Dailey TA, Lianchun X, Dailey HA., Characterization of a human and mouse tetrapyrrole-binding protein. Arch Biochem Biophys, 2002. 407(2): p. 196-201.
81. Sato E, Sagami I, Uchida T, Sato A, Kitagawa T, Igarashi J, Shimizu T., SOUL in
mouse eyes is a new hexameric heme-binding protein with characteristic optical absorption, resonance Raman spectral, and heme-binding properties. Biochemistry, 2004. 43(44): p. 14189-98.
82. Blagosklonny MV, Schulte T, Nguyen P, Trepel J, Neckers LM., Taxol-induced
apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer Res, 1996. 56(8): p. 1851-4.
83. Haldar S, Chintapalli J, and Croce CM., Taxol induces bcl-2 phosphorylation and
death of prostate cancer cells. Cancer Res, 1996. 56(6): p. 1253-5. 84. Horwitz SB., Mechanism of action of taxol. Trends Pharmacol Sci, 1992. 13(4): p. 134-
6. 85. Ibrado AM, Huang Y, Fang G, Bhalla K., Bcl-xL overexpression inhibits taxol-induced
Yama protease activity and apoptosis. Cell Growth Differ, 1996. 7(8): p. 1087-94. 86. Rowinsky EK and Donehower RC., Paclitaxel (taxol). N Engl J Med, 1995. 332(15): p.
1004-14. 87. Rowinsky EK, Onetto N, Canetta RM, Arbuck SG., Taxol: the first of the taxanes, an
important new class of antitumor agents. Semin Oncol, 1992. 19(6): p. 646-62. 88. Schiff PB, Fant J, and Horwitz SB., Promotion of microtubule assembly in vitro by
taxol. Nature, 1979. 277(5698): p. 665-7. 89. Abbott BJ, Fukuda DS, Dorman DE, Occolowitz JL, Debono M, Farhner L., Microbial
transformation of A23187, a divalent cation ionophore antibiotic. Antimicrob Agents Chemother, 1979. 16(6): p. 808-12.
90. Taketani S, Adachi Y, Kohno H, Ikehara S, Tokunaga R, Ishii T., Molecular
characterization of a newly identified heme-binding protein induced during differentiation of urine erythroleukemia cells. J Biol Chem, 1998. 273(47): p. 31388-94.
91. Zylka MJ and Reppert SM., Discovery of a putative heme-binding protein family
(SOUL/HBP) by two-tissue suppression subtractive hybridization and database searches. Brain Res Mol Brain Res, 1999. 74(1-2): p. 175-81.
92. Abe Y, Kimura K, Horiuchi A, Miyake M, Kimura S., Improvement of ELISA
sensitivity by allogeneric adsorption of polyclonal antibodies--a technical note for non-experts. Clin Chim Acta, 1994. 224(1): p. 103-5.
93. Bellyei S, Szigeti A, Boronkai A, Pozsgai E, Gomori E, Melegh B, Janaky T, Bognar
Z, Hocsak E, Sumegi B, Gallyas F Jr., Inhibition of cell death by a novel 16.2 kD heat

Hocsák, E., Identification and characterization of novel regulators
60
shock protein predominantly via Hsp90 mediated lipid rafts stabilization and Akt activation pathway. Apoptosis, 2007. 12(1): p. 97-112.
94. Tapodi A, Debreceni B, Hanto K, Bognar Z, Wittmann I, Gallyas F Jr, Varbiro G,
Sumegi B., Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J Biol Chem, 2005. 280(42): p. 35767-75.
95. Hogeboom GH and Schneider WC., Cytochemical studies of mammalian tissues. III.
Isocitric dehydrogenase and triphosphopyridine nucleotide-cytochrome c reductase of mouse liver. J Biol Chem, 1950. 186(2): p. 417-27.
96. Sims NR., Rapid isolation of metabolically active mitochondria from rat brain and
subregions using Percoll density gradient centrifugation. J Neurochem, 1990. 55(2): p. 698-707.
97. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C., A novel assay for
apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods, 1995. 184(1): p. 39-51.
98. Sentürker S, Tschirret-Guth R, Morrow J, Levine R, Shacter E., Induction of apoptosis
by chemotherapeutic drugs without generation of reactive oxygen species. Arch Biochem Biophys, 2002. 397(2): p. 262-72.
99. Wozniak AJ and Ross WE., DNA damage as a basis for 4'-
demethylepipodophyllotoxin-9-(4,6-O-ethylidene-beta-D-glucopyranoside) (etoposide) cytotoxicity. Cancer Res, 1983. 43(1): p. 120-4.
100. Roos WP and Kaina B., DNA damage-induced cell death by apoptosis. Trends Mol
Med, 2006. 12(9): p. 440-50. 101. Varbiro G, Veres B, Gallyas F Jr, Sumegi B., Direct effect of Taxol on free radical
formation and mitochondrial permeability transition. Free Radic Biol Med, 2001. 31(4): p. 548-58.
102. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill
EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD., Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature, 2005. 434(7033): p. 658-62.
103. Wong WW and Puthalakath H., Bcl-2 family proteins: the sentinels of the
mitochondrial apoptosis pathway. IUBMB Life, 2008. 60(6): p. 390-7. 104. Simonian PL, Grillot DA, and Nunez G., Bak can accelerate chemotherapy-induced
cell death independently of its heterodimerization with Bcl-XL and Bcl-2. Oncogene, 1997. 15(15): p. 1871-5.

Hocsák, E., Identification and characterization of novel regulators
61
105. Reers M, Smith TW, and Chen LB., J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry, 1991. 30(18): p. 4480-6.
106. Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA,
Cascio WE, Bradham CA, Brenner DA, Herman B., The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta, 1998. 1366(1-2): p. 177-96.
107. Ahn HJ, Kim YS, Kim JU, Han SM, Shin JW, Yang HO., Mechanism of taxol-induced
apoptosis in human SKOV3 ovarian carcinoma cells. J Cell Biochem, 2004. 91(5): p. 1043-52.
108. Pauloin A, Ollivier-Bousquet M, and Chanat E., [The double-play of PP17/TIP47].
Med Sci (Paris), 2004. 20(11): p. 1020-5. 109. Than NG, Sumegi B, Bellyei S, Berki T, Szekeres G, Janaky T, Szigeti A, Bohn H,
Than GN., Lipid droplet and milk lipid globule membrane associated placental protein 17b (PP17b) is involved in apoptotic and differentiation processes of human epithelial cervical carcinoma cells. Eur J Biochem, 2003. 270(6): p. 1176-88.
110. Ganley IG, Carroll K, Bittova L, Pfeffer S., Rab9 GTPase regulates late endosome size
and requires effector interaction for its stability. Mol Biol Cell, 2004. 15(12): p. 5420-30.
111. Chen Q, Crosby M, and Almasan A., Redox Regulation of Apoptosis before and after
Cytochrome C Release. Korean J Biol Sci, 2003. 7(1): p. 1-9. 112. Lorenzo HK, Susin SA, Penninger J, Kroemer G., Apoptosis inducing factor (AIF): a
phylogenetically old, caspase-independent effector of cell death. Cell Death Differ, 1999. 6(6): p. 516-24.
113. Singh M and Singh N., Molecular mechanism of curcumin induced cytotoxicity in
human cervical carcinoma cells. Mol Cell Biochem, 2009. 325(1-2): p. 107-19. 114. Iwai-Kanai E, Hasegawa K, Adachi S, Fujita M, Akao M, Kawamura T, Kita T.,
Effects of endothelin-1 on mitochondrial function during the protection against myocardial cell apoptosis. Biochem Biophys Res Commun, 2003. 305(4): p. 898-903.
115. Krick S, Shi S, Ju W, Faul C, Tsai SY, Mundel P, Böttinger EP., Mpv17l protects
against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc Natl Acad Sci U S A, 2008. 105(37): p. 14106-11.
116. Tudor G, Aguilera A, Halverson DO, Laing ND, Sausville EA., Susceptibility to drug-
induced apoptosis correlates with differential modulation of Bad, Bcl-2 and Bcl-xL protein levels. Cell Death Differ, 2000. 7(6): p. 574-86.
117. Fan W., Possible mechanisms of paclitaxel-induced apoptosis. Biochem Pharmacol,
1999. 57(11): p. 1215-21.

Hocsák, E., Identification and characterization of novel regulators
62
118. Gupta S., Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review). Int J Oncol, 2003. 22(1): p. 15-20.
119. Szigeti A, Hocsak E, Rapolti E, Racz B, Boronkai A, Pozsgai E, Debreceni B, Bognar
Z, Bellyei S, Sumegi B, Gallyas F Jr., Facilitation of mitochondrial outer and inner membrane permeabilization and cell death in oxidative stress by a novel Bcl-2 homology 3 domain protein. J Biol Chem, 2010. 285(3): p. 2140-51.
120. Carvalho AC, Sharpe J, Rosenstock TR, Teles AF, Youle RJ, Smaili SS., Bax affects
intracellular Ca2+ stores and induces Ca2+ wave propagation. Cell Death Differ, 2004. 11(12): p. 1265-76.
121. Banerjee J and Ghosh S., Bax increases the pore size of rat brain mitochondrial
voltage-dependent anion channel in the presence of tBid. Biochem Biophys Res Commun, 2004. 323(1): p. 310-4.
122. Lemasters JJ and Holmuhamedov E., Voltage-dependent anion channel (VDAC) as
mitochondrial governator--thinking outside the box. Biochim Biophys Acta, 2006. 1762(2): p. 181-90.
123. Shoshan-Barmatz V, Keinan N, and Zaid H., Uncovering the role of VDAC in the
regulation of cell life and death. J Bioenerg Biomembr, 2008. 40(3): p. 183-91. 124. Sugiyama T, Shimizu S, Matsuoka Y, Yoneda Y, Tsujimoto Y., Activation of
mitochondrial voltage-dependent anion channel by apro-apoptotic BH3-only protein Bim. Oncogene, 2002. 21(32): p. 4944-56.
125. Kowaltowski AJ, Vercesi AE, and Fiskum G., Bcl-2 prevents mitochondrial
permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ, 2000. 7(10): p. 903-10.
126. Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH., Pro-apoptotic
cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ, 2000. 7(12): p. 1166-73.
127. Smaili SS, Hsu YT, Sanders KM, Russell JT, Youle RJ., Bax translocation to
mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ, 2001. 8(9): p. 909-20.

Hocsák, E., Identification and characterization of novel regulators
63
11. List of Publications
This work is based on the following articles:
1. Eniko Hocsak, Boglarka Racz, Aliz Szabo, Laszlo Mester, Edit Rapolti, Szaniszlo Javor,
Szabolcs Bellyei, Ferenc Gallyas Jr., Balazs Sumegi, Andras Szigeti. TIP47 protects
mitochondrial membrane integrity and inhibits oxidative-stress-induced cell death. FEBS
Lett. 2010 Jul 2;584(13):2953-60.
IF: 3,541 (2009)
Cit.: 0
2. Eniko Hocsak, Boglarka Racz, Aliz Szabo, Eva Pozsgai, Andras Szigeti, Edit Szigeti,
Ferenc Gallyas Jr., Balazs Sumegi, Szaniszlo Javor, Szabolcs Bellyei. TIP47 confers
resistance to taxol- induced cell death by preventing the nuclear translocation of AIF and
Endonuclease G. Eur JCB. in press.
IF: 3,314 (2009)
Cit.: 0
3. Andras Szigeti, Eniko Hocsak, Edit Rapolti, Boglarka Racz, Arpad Boronkai, Eva
Pozsgai, Balazs Debreceni, Zita Bognar, Szabolcs Bellyei, Balazs Sumegi, and Ferenc
Gallyas, Jr. Facilitation of Mitochondrial Outer and Inner Membrane Permeabilization and
Cell Death in Oxidative Stress by a Novel Bcl-2 Homology 3 Domain Protein. J Biol
Chem. 2010 Jan 15;285(3):2140-51.
IF: 5,328 (2009)
Cit.: 0

Hocsák, E., Identification and characterization of novel regulators
64
Further publications:
1. Mester L, Szabo A, Atlasz T, Szabadfi K, Reglodi D, Kiss P, Racz B, Tamas A, Gallyas F
Jr, Sumegi B, Hocsak E, Gabriel R, Kovacs K. Protection against chronic hypoperfusion-
induced retinal neurodegeneration by PARP inhibition via activation of PI-3-kinase Akt
pathway and suppression of JNK and p38 MAP kinases. Neurotox Res. 2009 Jul; 16(1):68-
76.
IF: 2,43
Cit.: 1
2. Szigeti A, Minik O, Hocsak E, Pozsgai E, Boronkai A, Farkas R, Balint A, Bodis J,
Sumegi B, Bellyei S.: Preliminary study of TIP47 as a possible new biomarker of cervical
dysplasia and invasive carcinoma. Anticancer Res. 2009; 29: 717-724.
IF: 1,428
Cit.: 1
3. Bellyei S, Szigeti A, Pozsgai E, Boronkai A, Gomori E, Hocsak E, Farkas R, Sumegi B,
Gallyas F Jr.: Preventing apoptotic cell death by a novel small heat shock protein. Eur J
Cell Biol. 2007 Mar; 86(3):161-71.
IF: 3,224
Cit.: 10
4. Bellyei S, Szigeti A, Boronkai A, Pozsgai E, Gomori E, Melegh B, Janaky T, Bognar Z,
Hocsak E, Sumegi B, Gallyas F Jr.: Inhibition of cell death by a novel 16.2 kD heat shock
protein predominantly via Hsp90 mediated lipid rafts stabilization and Akt activation
pathway. Apoptosis. 2007 Jan; 12(1):97-112.
IF: 3,043
Cit.: 7
5. Szigeti A, Bellyei S, Gasz B, Boronkai A, Hocsak E, Minik O, Bognar Z, Varbiro G,
Sumegi B, Gallyas F Jr.: Induction of necrotic cell death and mitochondrial
permeabilization by heme binding protein 2/SOUL. FEBS Lett. 2006 Nov 27;
580(27):6447-54.
IF: 3,372
Cit.: 3

Hocsák, E., Identification and characterization of novel regulators
65
6. Bányai K, Forgách P, Erdélyi K, Martella V, Bogdán A, Hocsák E, Havasi V, Melegh B,
Szucs G.: Identification of the novel lapine rotavirus genotype P[22] from an outbreak of
enteritis in a Hungarian rabbitry. Virus Res. 2005 Nov; 113(2):73-80.
IF: 2,562
Cit.: 7
Total IF: 28,242
Total citations: 29
h-index: 3

Hocsák, E., Identification and characterization of novel regulators
66
Abstracts
1. Hocsák Enikő, Rácz Boglárka, Szabó Alíz, Bellyei Szabolcs, Szigeti Edit, Mester László,
ifj. Gallyas Ferenc, Sümegi Balázs, Szigeti András: A TIP47 fehérje a mitokondrium
membrán stabilizálásával részt vesz a mitokondrium mediálta sejthalál szabályozásában,
40. Membrán-Transzport Konferencia, Sümeg, 2010. május 18-21.
2. Hocsák Enikő, Rápolti Edit, Jávor Szaniszló, Rácz Boglárka, Szigeti András, Szabó Alíz,
Sümegi Balázs: A TIP47 fehérje gátolja az oxidatív stressz indukált apoptózist és
nekrózist. 39. Membrán-Transzport Konferencia, Sümeg, 2009. május 19-22.
3. Rápolti Edit, Szigeti András, Boronkai Árpád, Hocsák Enikő, Rácz Boglárka, Szabó Alíz,
Pozsgai Éva, Bognár Zita, Bellyei Szabolcs, Sümegi Balázs, ifj. Gallyas Ferenc: Egy új,
BH3 domén fehérje oxidative stresszben elősegíti a mitokondrium belső és külső
membránjának permeabilizációját. 39. Membrán-Transzport Konferencia, Sümeg, 2009.
május 19-22.
4. Rácz Boglárka, Rápolti Edit, Hocsák Enikő, Gasz Balázs, Gallyas Ferenc Jr., Kiss Péter,
Horváth Gabriella, Tamás Andrea, Lubics Andrea, Tóth Gábor, Sümegi Balázs, Reglődi
Dóra: Effects of pituitary adenylate cyclase activating polypeptide (PACAP) in ischemic
and oxidative stress-induced cardiomyocyte damage. 39. Membrán-Transzport
Konferencia, Sümeg, 2009. május 19-22.
5. Mester László, Szabó Alíz, Atlasz Tamás, Szabadfi Krisztina, Reglődi Dóra, Kiss Péter,
Rácz Boglárka, Tamás Andrea, Ifj. Gallyas Ferenc, Sümegi Balázs, Hocsák Enikő,
Gábriel Róbert, Kovács Krisztina: Protection against chronic hypoperfusion-induced
retinal neurodegeneration by PARP inhibition via activation of PI-3-kinase Akt pathway
and suppression of JNK and p38 MAP kinases. 39. Membrán-Transzport Konferencia,
Sümeg, 2009. május 19-22.
6. Szigeti A., Bellyei Sz., Boronkai Á., Pozsgai É., Rápolti E., Hocsák E., Mester L., Sümegi
B.,:A SOUL-fehérje mitokondriális permeabilitás-tranzíción keresztül nekrotikus sejthalált
indukál. 38. Membrán-Transzport Konferencia, Sümeg, 2008. május 20-23.

Hocsák, E., Identification and characterization of novel regulators
67
7. Németh Viktória, Montskó Gegő, Solti Izabella, Vető Sára, Tucsek Zsuzsanna, Hocsák
Enikő, Márk László: Komplex biológiai minták GSH tartalmának meghatározása
ioncsapdás tömegspektrometria segítségével. A Magyar Biokémiai Egyesület 2006. évi
Vándorgyűlése, Pécs, 2006. augusztus 30- szeptember2.
8. Bognár Eszter, Solti Izabella, Németh Viktória, Bartha Éva, Tucsek Zsuzsanna, Vető Sára,
Hocsák Enikő, Nagyné Kiss Gyöngyi, Sümegi Balázs, Berente Zoltán: A glükózfelvétel
és a kapcsolódó intracelluláris jelátviteli utak vizsgálata izolált szívmodellen. A Magyar
Biokémiai Egyesület 2006. évi Vándorgyűlése, Pécs, 2006. augusztus 30- szeptember2.
9. Zsuzsanna Tucsek, Balazs Radnai, Zoltan Szabo, Tamas Dolowschiak, Eniko Hocsak,
Aliz Szabo, Jr. Ferenc Gallyas, Tamas Lorand, and Balazs Sumegi: The effect of the IK11,
an E - 4 - arylidene-3-isochromanone on the Hep G2 human hepatocellular carcinoma cell
line. Semmelweis Symposium – Nitric Oxide and Nitrosative stress in the Cardiovascular
System, Budapest, 2006. okt. 29-31.
10. Balazs Radnai, Zsuzsanna Tucsek, BalazsVeres, Katalin Hanto, Peter Jakus, Balazs
Debreceni, Zita Bognar, Eniko Hocsak, Denes Grasz, Ferenc Gallyas Jr. and Balazs
Sumegi: Effect of a PARP inhibitor, HO-3089 on the gene expression profiles of LPS
stimulated RAW 264,7 murine macrophages. Semmelweis Symposium – Nitric Oxide and
Nitrosative stress in the Cardiovascular System, Budapest, 2006. okt. 29-31.
11. Tucsek Zsuzsanna, Radnai Balázs, Szabó Zoltán, Dolowschiák Tamás, Hocsák Enikő,
Szabó Alíz, Solti Izabella, Bognár Eszter, Vető Sára, ifj.Gallyas Ferenc, Loránd Tamás és
Sümegi Balázs: A PJ34 protektív hatása az IK11 indukálta oxidatív stresszben HepG2
sejtvonalon. 36. Membrán-Transzport Konferencia, Sümeg, 2006. május 23-26.
12. Hocsák Enikő, Bellyei Szabolcs, Szigeti András, Boronkai Árpád, Tucsek Zsuzsanna,
Szabó Alíz, Vető Sára, Berki Tímea, Sümegi Balázs: A PP17B fehérje strukturális és
funkcionális vizsgálatai. 36. Membrán-Transzport Konferencia, Sümeg, 2006. május 23-
26.

Hocsák, E., Identification and characterization of novel regulators
68
13. Bellyei Sz., Szigeti A., Boronkai Á., Bognar Z., Hocsák E., Tucsek Zs., Pozsgai É.,
Gallyas F. Jr., Sumegi B.: Egy új 16,2 kDa nagyságú kis molekulasúlyú hő-sokk szerű
fehérje bemutatása. 36. Membrán-Transzport Konferencia, Sümeg, 2006. május 23-26.
14. Boronkai Árpád, Bellyei Szabolcs, Szigeti András, Hocsák Enikő, Tucsek Zsuzsanna,
Sümegi Balázs: A galectin-13 jellemzése, sejthalál indukáló hatása. 36. Membrán-
Transzport Konferencia, Sümeg, 2006. május 23-26.
15. Szigeti András, Bellyei Szabolcs, Boronkai Árpád, Bognár Zita, Gasz Balázs, Szabó
Zoltán, Tucsek Zsuzsanna, Hocsák Enikő, Komlósi Katalin, Várbíró Gábor, Melegh Béla,
Janaky Tamás, Sümegi Balázs, ifj. Gallyas Ferenc: MPTIP1, az első csak BH3 domént
tartalmazó permeability transition-t indukáló fehérje. 36. Membrán-Transzport
Konferencia, Sümeg, 2006. május 23-26.
16. Sara Veto, Izabella Solti, Aliz Szabo, Zsuzsanna Tucsek, Viktoria Nemeth, Eniko Hocsak,
Balazs Veres, Zoltan Berente, Balazs Sumegi: Involvement of Akt/protein kinase B
pathway induction in the protective effect of poly – (ADP-ribose) polymerase 1 inhibition
in endotoxin – induced septic shock. 36. Membrán-Transzport Konferencia, Sümeg, 2006.
május 23-26.
17. Solti Izabella, Bognár Zita, Németh Viktória, Bognár Eszter, Vető Sára, Hocsák Enikő,
Nagyné Kiss Gyöngyi, Szántó Árpád, Várbíró Gábor, Sümegi Balázs: Taxol hatása a
mitokondriumra és a szabadgyök képződésre. 36. Membrán-Transzport Konferencia,
Sümeg, 2006. május 23-26.
18. Aliz Szabo, Zita Bognar, Arpad Szanto, Eniko Hocsak, Katalin Hanto, Edina Pandur,
Judit Nagy, Viktor Poor, Balazs Sumegi: Induction of NFKB dependent COX-2
expresssion in liver cells by amiodarone. 36. Membrán-Transzport Konferencia, Sümeg,
2006. május 23-26.
19. Ferenc Gallyas Jr., Balazs Veres, Balazs Radnai, Eniko Hocsak, Balazs Sumegi:
Significant role of cytoprotective kinase signalling mechanisms in the protective effect of
PARP inhibitors in a murine septic shock model. 30th FEBS Congress - 9th IUBMB
Conference Budapest, Hungary 2nd-7th July, 2005. FEBS J. 2005; 272: 424-425.

Hocsák, E., Identification and characterization of novel regulators
69
20. Anita Palfi, Ambrus Toth, Katalin Hanto, Eniko Hocsak, Robert Halmosi, Eszter
Szabados, Kalman Hideg, Balazs Sumegi, Kalman Toth: Contribution of intracellular
signaling cascades to the cardioprotective effect of poly (ADP-ribose) polymerase
inhibitors in myocardial ischemia-reperfusion. 30th FEBS Congress - 9th IUBMB
Conference Budapest, Hungary 2nd-7th July, 2005. FEBS J. 2005; 272: 311-311.
Presentations
1. Hocsák Enikő, Szabó Alíz, Szigeti András, Ifj. Gallyas Ferenc, Sümegi Balázs: A TIP47
fehérje gátolja az oxidatív stressz indukált sejthalált, Biológus Doktoranduszok
Konferenciája, Pécs, 2009. November 12-13.
2. Ifj. Gallyas Ferenc, Hocsák Enikő, Pozsgai Éva, Szigeti András, Bellyei Szabolcs, Sümegi
Balázs: Mitokondriális membrán-permeabilitást szabályozó új mechanizmusok, 39.
Membrán-Transzport Konferencia, Sümeg, 2009. május 19-22.
3. Hocsák Enikő: A SOUL fehérje mitokondriális permeabilitás tranzíción keresztül
nekrotikus sejthalált indukál. PTE Grastyán Endre VII. Országos Interdiszciplináris
Konferencia, Pécs, 2009. március 23 – 25.
4. Hocsák Enikő: A SOUL fehérje mitokondriális permeabilitás tranzíción keresztül
nekrotikus sejthalált indukál. XIV. Korányi Frigyes Tudományos Fórum, Budapest, 2009.
március 26.
5. Szigeti András, Hocsák Enikő, Sümegi Balázs, ifj. Gallyas Ferenc: SOUL: Egy új BH3-
domaint tartalmazó fehérje, amely regulálja a mitokondriális permeabilitás tranzíciós
pórust. A Magyar Biokémiai Egyesület 2008. évi Vándorgyűlése, Szeged, 2008 aug. 31-
szept. 3.

Hocsák, E., Identification and characterization of novel regulators
70
6. Hocsák Enikő, Mester László, Szabó Alíz, Krisztina Kovács, Szigeti András, Bellyei
Szabolcs, Sümegi Balázs, Gallyas Ferenc Jr.: Mitokondrium mediálta sejthalál indukciója
egy új BH3 domént tartalmazó fehérjével. Magyar Humángenetikai Társaság VII.
Kongresszusa, Pécs, július 11-13.
7. Mester László, Hocsák Enikő, Szabó Alíz, Krisztina Kovács, Bellyei Szabolcs, Szigeti
András, Boronkai Árpád, Pozsgai Éva, Gallyas Ferenc Jr., Sümegi Balázs: Egy új-16.2 kD-
kis hősokk fehérje azonosítása és a jelátviteli folyamatokra gyakorolt szerepének a
vizsgálata. Magyar Humángenetikai Társaság VII. Kongresszusa, Pécs, július 11-13.
8. Hocsák Enikő, Jávor Szaniszló, Bellyei Szabolcs, Szigeti András, Boronkai Árpád,
Sümegi Balázs: A PP17b/TIP47 fehérje strukturális és funkcionális vizsgálatai. PTE
Grastyán Endre VI. Országos Interdiszciplináris Konferencia, Pécs, 2008. március 26 – 28.
9. Radnai Balázs, Tucsek Zsuzsanna, Hocsák Enikő, Vető Sára, Németh Viktória, Bognár
Eszter, Grász Dénes, Berente Zoltán, ifj.Gallyas Ferenc, Sümegi Balázs: A ferulaldehid
hatása az LPS indukálta endotoxikus sokkra egerekben. A Magyar Biokémiai Egyesület
2006. évi Vándorgyűlése, Pécs, 2006. augusztus 30- szeptember2.
10. Balazs Sumegi, Sara Veto, Zsuzsanna Tucsek, Izabella Solti, Eniko Hocsak, Eszter
Bognar, Aliz Szabo, Ferenc Gallyas Jr.: PARP and inflammatory and kinase pathways:
relevance for endotoxic shock. Semmelweis Symposium – Nitric Oxide and Nitrosative
stress in the Cardiovascular System, Budapest, 2006. okt. 29-31.
11. Sümegi Balázs, Kovács Krisztina, Tapodi Antal, Hantó Katalin, Radnai Balázs, ifj. Gallyas
Ferenc, Hocsák Enikő, Hideg Kálmán: Az ADP-riboziláció szerepe a PI3-kináz, Akt és
MAPk jelátviteli útvonalak szabályozásában. XIII. Sejt és Fejlődésbiológiai Napok, Eger,
2005. ápr. 10-12.
12. Bellyei Sz., Szigeti A., Boronkai Á., Bognár Z., Hocsák E., Tucsek Zs., Pozsgai É.,
Gallyas F. Jr., Sümegi B.:Egy új 16.2 kDa nagyságú kis molekulasúlyú hő-sokk szerű
fehérje bemutatása. 36. Membrán-Transzport Konferencia, Sümeg, 2006. május 23-26.

Hocsák, E., Identification and characterization of novel regulators
71
13. Szigeti András, Bellyei Szabolcs, Gasz Balázs, Boronkai Árpád, Hocsák Enikő, Minik
Orsolya, Bognár Zita, Várbíró Gábor, Sümegi Balázs, Ifj. Gallyas Ferenc: Egy, BH3
domén-t tartalmazó, mitokondriális permeability transition-t indukáló fehérje azonosítása.
Magyar Biokémiai Egyesület (MBKE) 2006. évi vándorgyűlése, Pécs, 2006. augusztus 30-
szeptember2
14. Bellyei Sz., Szigeti A., Boronkai Á., Bognár Z., Hocsák E., Pozsgai É., Gallyas F. Jr.,
Sümegi B.:Egy új 16.2 kDa nagyságú kis molekulasúlyú hő-sokk szerű fehérje
funkcionális analízise. Magyar Biokémiai Egyesület (MBKE) 2006. évi vándorgyűlése,
Pécs, 2006. augusztus 30- szeptember2
15. Boronkai Árpád, Bellyei Szabolcs, Szigeti András, Hocsák Enikő, Németh Viktória,
Sümegi Balázs: A galectin-13 indukálta apoptózis molekuláris mechanizmusai. Magyar
Biokémiai Egyesület (MBKE) 2006. évi vándorgyűlése, Pécs, 2006. augusztus 30. -
szeptember 2.





























