Embed Size (px)
Citation preview

Loss of the Aryl Hydrocarbon Receptor InducesHypoxemia, Endothelin-1, and Systemic
Hypertension at Modest AltitudeAmie K. Lund, Larry N. Agbor, Nan Zhang, Amy Baker, Huawei Zhao,
Gregory D. Fink, Nancy L. Kanagy, Mary K. Walker
Abstract—The aryl hydrocarbon receptor (AHR) is a basic helix-loop-helix Per-Arnt-Sim transcription factor that mediatesinduction of metabolic enzymes and toxicity of certain environmental pollutants. Although AHR knockout (KO) micedevelop cardiac hypertrophy, conflicting reports associate this pathology with hypotension or endothelin (ET)-1–dependent hypertension. Because hypertension occurred at modest altitude, we tested the hypothesis that loss of AHRincreases the sensitivity to hypoxia-induced ET-1, contributing to systemic hypertension. We found that AHR KO micewere hypertensive at modest altitude (1632 m) but hypotensive at low altitude (225 m). When AHR KO mice residingat 1632 m were exposed to the partial pressure of inspired oxygen (PIO2) at sea level for 11 days, blood pressure declinedto levels measured at 225 m. Although plasma ET-1 in AHR KO mice was significantly elevated at 1632 m anddecreased at 225 m and sea level PIO2, pulmonary prepro-ET-1 mRNA was significantly reduced at 1632 m anddecreased further at 225 m and sea level PIO2. Blood gas analysis revealed that AHR KO mice were hypoxemic,hypercapnic, and acidotic at 1632 m, values that were attenuated and normalized after 24 hours and 11 days under sealevel PIO2, respectively. Lastly, AHR inactivation in endothelial cells by small interfering RNA significantly reducedbasal prepro-ET-1 mRNA but did not alter hypoxia-induced expression. Our studies establish the AHR KO mouse asa model in which modest decreases in PIO2 lead to hypoxemia, increased plasma ET-1, and systemic hypertensionwithout increased pulmonary prepro-ET-1 mRNA expression. (Hypertension. 2008;51:803-809.)
Key Words: blood pressure � hypertension � endothelin � oxygen � gene regulation
The aryl hydrocarbon receptor (AHR) is a ligand-activatedtranscription factor belonging to the basic helix-loop-
helix Per-Arnt-Sim family of DNA binding proteins, whichalso includes hypoxia-inducible factors.1 Although the AHRis known to mediate induction of drug-metabolizing enzymesand toxicity after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin, recent evidence has revealed a physiological role forAHR in cardiovascular homeostasis. AHR knockout (KO)mice develop cardiac hypertrophy,2,3 which is mediated, inpart, by elevated plasma angiotensin II and endothelin-1(ET-1).4,5 There are conflicting reports, however, of whetherthe cardiac hypertrophy is associated with systemic hyper-tension. Lund et al4,5 reported that cardiac hypertrophy inAHR KO mice is preceded by hypertension, whereasVasquez et al6 and Ichihara et al7 reported that AHR KO miceare hypotensive and normotensive, respectively. The expla-nation for the disparate blood pressure values among thesestudies is unclear.
AHR and hypoxia-inducible factor-1� share a commondimerization partner, AHR nuclear translocator (hypoxia-
inducible factor-1�), as well as other transactivators, andstudies have shown that these 2 signal transduction path-ways can exhibit reciprocal inhibitory cross-talk.8,9 Themechanism by which this functional interference occurs isnot clear, nor has the physiological relevance of theseinteractions been defined. If AHR functions physiological-ly to attenuate hypoxia-induced responses, then AHR KOmice might exhibit an increased sensitivity to hypoxia-mediated gene induction and changes in physiology. Evi-dence supporting this idea was published recently showingthat AHR KO mice are more responsive to the induction ofvascular endothelial growth factor and neovasculogenesisafter hindlimb ischemia.7 Because hypoxia is a potentstimulus of the vasoconstricting peptide, ET-1,10,11 andbecause ET-1– dependent hypertension was reported inAHR KO mice residing at a modest altitude (Albuquerque,NM, 1632 m), we reasoned that the differences in bloodpressure reported in AHR KO mice may result fromdifferences in how the mice respond to changes in thepartial pressure of inspired oxygen (PIO2). Thus, we tested
Received August 26, 2007; first decision September 18, 2007; revision accepted December 21, 2007.From the College of Pharmacy (A.K.L., L.N.A., N.Z., A.B., M.K.W.) and Department of Cell Biology and Physiology, School of Medicine (N.L.K.,
M.K.W.), University of New Mexico Health Sciences Center, Albuquerque; and the Department of Pharmacology and Toxicology (H.Z., G.D.F.), Collegeof Human Medicine, Michigan State University, East Lansing.
Correspondence to Mary K. Walker, College of Pharmacy, MSC09 5360, 2703 Frontier NE, University of New Mexico, Albuquerque, NM 87131.E-mail [email protected]
© 2008 American Heart Association, Inc.
Hypertension is available at http://hypertension.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.107.100586
803
Hypoxemia and Hypertension
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from
by guest on M
ay 13, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from
by guest on M
ay 13, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from
by guest on M
ay 13, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from
by guest on M
ay 13, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

the hypothesis that loss of AHR enhances hypoxia-inducedET-1 expression and increases blood pressure in animalsexposed to modest hypoxia.
MethodsAn expanded Methods section is available in the online DataSupplement at http://hyper.ahajournals.org.
AnimalsAHR KO mice were obtained from Dr Frank Gonzalez (NationalCancer Institute, Bethesda, Md) and backcrossed 11 generations toC57Bl/6N, whereas AHR WT mice were purchased from Harlan at6 to 8 weeks of age. All of the study protocols were reviewed andapproved by the University of New Mexico Institutional AnimalCare and Use Committee and were in accordance with the NationalInstitutes of Health Guide for the Care and Use of Laboratory Animals.
Blood Pressure AnalysisArterial blood pressure was measured using radiotelemetry (DataSciences International). At the University of New Mexico, bloodpressure was continuously recorded for 6 days at environmental PIO2
(122 mm Hg) and continued to be recorded when mice were exposedto simulated sea level PIO2 (150 mm Hg) for 11 days.
Analysis of Blood Gases, Hemoglobin,and HematocritArterial pO2, pCO2, oxygen saturation, pH, and total CO2 weremeasured with an i-STAT (Abbott Point of Care Inc) from a carotidblood sample taken 2 to 3 days after catheter implantation. Bloodgases were analyzed in mice residing at 1632 m and after 24 hoursor 11 days of exposure to simulated sea level PIO2. Venoushematocrit and hemoglobin were measured with the i-STAT frommice residing at 1632 m and after 11 days of exposure to simulatedsea level PIO2.
Analysis of ET-1 ExpressionET-1 in plasma and cell culture media was assayed by radioimmu-noassay (Amersham Pharmacia) and ELISA (R&D Systems), re-spectively. Pulmonary prepro-ET-1 mRNA was analyzed from totalRNA using quantitative PCR with SYBR green detection, an Icycler(Bio-Rad), and phosphoglycerate kinase mRNA as an internalnormalization control.
Cell Culture StudiesHuman umbilical vein endothelial cells and microvascular endothe-lial cells isolated from lung were purchased from Lonza. Cells weretransfected with control or AHR small interfering RNA (siRNA;Dharmacon) using Lipofectamine 2000 (Invitrogen) alone or incombination with 2.5% or 1.0% O2.
Statistical AnalysisAll of the values were expressed as means�SEMs, and P�0.05 wasconsidered significant.
ResultsAHR KO Mice Are Hypotensive at Low Altitudeand Hypertensive at Modest AltitudeTo investigate whether blood pressure of AHR KO mice wasaffected by altitude, blood pressure of AHR wild-type (WT)and KO male mice was measured by radiotelemetry atMichigan State University (225 m) and the University of NewMexico (1632 m). All of the mice were born and raised atthe University of New Mexico, and those studied at lowaltitude were shipped to Michigan State University at 3months of age. Mean arterial pressure (MAP) of AHR WT
mice did not differ with altitude. However, compared withage-matched AHR WT controls, MAP of AHR KO micewas significantly higher at a modest altitude of 1632 m butwas significantly lower at 225 m (Figure 1). In addition,changes in MAP with altitude were paralleled by similarchanges in diastolic and systolic blood pressure, but therewere no differences in heart rate between genotypes orlocations (data not shown).
Plasma ET-1 in AHR KO Mice Is Increased atLow Altitude and Is Increased Further atModest AltitudeBecause previous research demonstrated that hypertension inAHR KO mice at modest altitude is associated with elevatedET-14 and mediated by the endothelin A receptor,5 wemeasured plasma ET-1 from AHR WT and KO mice at 225and 1632 m. Plasma ET-1 was significantly increased in AHRKO mice at both altitudes, compared with AHR WT mice;however, the levels of ET-1 were significantly higher in AHRKO mice at 1632 m compared with AHR KO mice at 225 m(Figure 2A).
Pulmonary Prepro-ET-1 mRNA in AHR KO MiceIs Decreased at Modest Altitude and Is DecreasedFurther at Low AltitudeTo determine whether the increased circulating ET-1 wasassociated with increased prepro-ET-1 mRNA expression, wemeasured pulmonary prepro-ET-1 mRNA from AHR WTand KO mice at 225 and 1632 m. In contrast to plasma ET-1levels, prepro-ET-1 mRNA expression was significantly re-duced in AHR KO mice at modest and low altitude comparedwith AHR WT mice. However, as was observed for plasmaET-1, the levels of prepro-ET-1 mRNA ET-1 were signifi-cantly higher in AHR KO mice at 1632 m compared withAHR KO mice at 225 m (Figure 2B).
MAP and ET-1 Expression in AHR KO MiceVary With PIO2To establish whether differences in PIO2 contributed to thechanges in MAP and ET-1 levels in AHR KO mice atdifferent altitudes, baseline MAP in 4-month–old AHR WTand KO mice was measured by radiotelemetry for 6 days at
Figure 1. MAP in 4-month-old AHR WT and KO mice measuredby radiotelemetry when residing at Michigan State University(225 m; n�8 per genotype) or the University of New Mexico(1632 m; n�7 to 12 per genotype). Two-way ANOVA demon-strated significant differences based on location (P�0.009) andlocation-genotype interaction (P�0.001).
804 Hypertension March 2008
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from
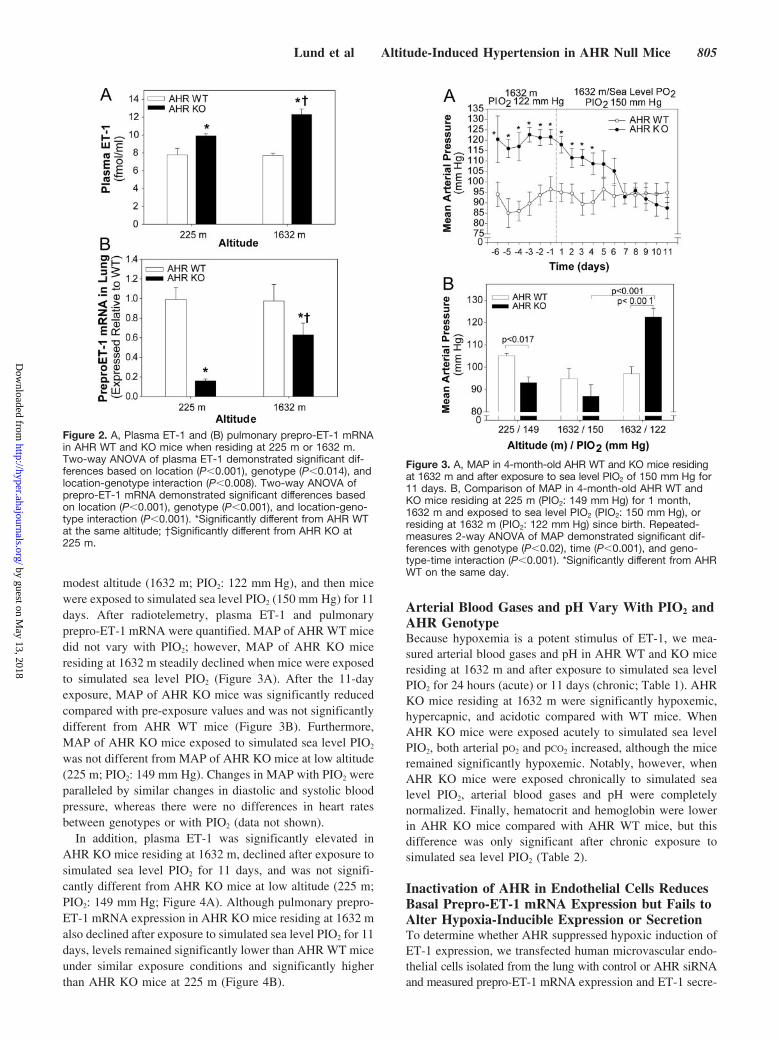
modest altitude (1632 m; PIO2: 122 mm Hg), and then micewere exposed to simulated sea level PIO2 (150 mm Hg) for 11days. After radiotelemetry, plasma ET-1 and pulmonaryprepro-ET-1 mRNA were quantified. MAP of AHR WT micedid not vary with PIO2; however, MAP of AHR KO miceresiding at 1632 m steadily declined when mice were exposedto simulated sea level PIO2 (Figure 3A). After the 11-dayexposure, MAP of AHR KO mice was significantly reducedcompared with pre-exposure values and was not significantlydifferent from AHR WT mice (Figure 3B). Furthermore,MAP of AHR KO mice exposed to simulated sea level PIO2
was not different from MAP of AHR KO mice at low altitude(225 m; PIO2: 149 mm Hg). Changes in MAP with PIO2 wereparalleled by similar changes in diastolic and systolic bloodpressure, whereas there were no differences in heart ratesbetween genotypes or with PIO2 (data not shown).
In addition, plasma ET-1 was significantly elevated inAHR KO mice residing at 1632 m, declined after exposure tosimulated sea level PIO2 for 11 days, and was not signifi-cantly different from AHR KO mice at low altitude (225 m;PIO2: 149 mm Hg; Figure 4A). Although pulmonary prepro-ET-1 mRNA expression in AHR KO mice residing at 1632 malso declined after exposure to simulated sea level PIO2 for 11days, levels remained significantly lower than AHR WT miceunder similar exposure conditions and significantly higherthan AHR KO mice at 225 m (Figure 4B).
Arterial Blood Gases and pH Vary With PIO2 andAHR GenotypeBecause hypoxemia is a potent stimulus of ET-1, we mea-sured arterial blood gases and pH in AHR WT and KO miceresiding at 1632 m and after exposure to simulated sea levelPIO2 for 24 hours (acute) or 11 days (chronic; Table 1). AHRKO mice residing at 1632 m were significantly hypoxemic,hypercapnic, and acidotic compared with WT mice. WhenAHR KO mice were exposed acutely to simulated sea levelPIO2, both arterial pO2 and pCO2 increased, although the miceremained significantly hypoxemic. Notably, however, whenAHR KO mice were exposed chronically to simulated sealevel PIO2, arterial blood gases and pH were completelynormalized. Finally, hematocrit and hemoglobin were lowerin AHR KO mice compared with AHR WT mice, but thisdifference was only significant after chronic exposure tosimulated sea level PIO2 (Table 2).
Inactivation of AHR in Endothelial Cells ReducesBasal Prepro-ET-1 mRNA Expression but Fails toAlter Hypoxia-Inducible Expression or SecretionTo determine whether AHR suppressed hypoxic induction ofET-1 expression, we transfected human microvascular endo-thelial cells isolated from the lung with control or AHR siRNAand measured prepro-ET-1 mRNA expression and ET-1 secre-
Figure 2. A, Plasma ET-1 and (B) pulmonary prepro-ET-1 mRNAin AHR WT and KO mice when residing at 225 m or 1632 m.Two-way ANOVA of plasma ET-1 demonstrated significant dif-ferences based on location (P�0.001), genotype (P�0.014), andlocation-genotype interaction (P�0.008). Two-way ANOVA ofprepro-ET-1 mRNA demonstrated significant differences basedon location (P�0.001), genotype (P�0.001), and location-geno-type interaction (P�0.001). *Significantly different from AHR WTat the same altitude; †Significantly different from AHR KO at225 m.
Figure 3. A, MAP in 4-month-old AHR WT and KO mice residingat 1632 m and after exposure to sea level PIO2 of 150 mm Hg for11 days. B, Comparison of MAP in 4-month-old AHR WT andKO mice residing at 225 m (PIO2: 149 mm Hg) for 1 month,1632 m and exposed to sea level PIO2 (PIO2: 150 mm Hg), orresiding at 1632 m (PIO2: 122 mm Hg) since birth. Repeated-measures 2-way ANOVA of MAP demonstrated significant dif-ferences with genotype (P�0.02), time (P�0.001), and geno-type-time interaction (P�0.001). *Significantly different from AHRWT on the same day.
Lund et al Altitude-Induced Hypertension in AHR Null Mice 805
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

tion after exposure of cells to 21.0,% 2.5%, or 1.0% O2.Successful AHR inactivation was confirmed using methodsreported elsewhere.12 Inactivation of AHR in human micro-vascular endothelial cells isolated from lung significantlyreduced prepro-ET-1 mRNA expression under normoxicconditions (Figure 5A) but failed to alter hypoxia-inducedexpression (Figure 5B) and had no effect on hypoxia-inducedET-1 secretion (data not shown). Similar results were ob-tained in studies conducted with human umbilical veinendothelial cells (data not shown).
DiscussionThese results establish the AHR KO mouse as a geneticmodel where systemic blood pressure varies with a smallchange in inspired pO2, and systemic hypertension develops ata modest altitude. This response is unique, because bothhumans and animal models typically develop pulmonaryhypertension, right ventricular hypertrophy, and polycythe-mia with increases in altitude and decreases in PIO2. Hypox-ia-induced pulmonary hypertension is associated with in-creased circulating and pulmonary ET-1 expression,13–15 anddisease pathogenesis is ET-1-dependent.16 In AHR KO mice,hypoxia induces circulating ET-1 and pulmonary prepro-ET-1mRNA, although the pulmonary mRNA expression remainssignificantly lower than AHR WT mice, and AHR KO micedevelop systemic hypertension in the absence of symptoms ofpulmonary hypertension. AHR KO mice do not exhibitpolycythemia and, in fact, show a slight decrease in hemat-ocrit. In addition, AHR KO mice do not exhibit an increase inright ventricle weight (Table S1), characteristic of increasedpulmonary arterial pressure, but rather exhibit an increase inleft ventricle weight, characteristic of increased systemicarterial pressure. Although most studies report pulmonaryhypertension after chronic exposure to severe hypoxia (PIO2
of 72 mm Hg), �3 genetic models develop pulmonaryhypertension after chronic exposure to modest hypoxia (PIO2:122 mm Hg).17–19 Thus, it is possible that AHR KO micedevelop hypoxia-induced systemic hypertension, rather thanpulmonary hypertension, because of the failure to inducepulmonary prepro-ET-1 mRNA expression above basal levelsobserved in WT mice.
There is a limited number of reports of systemic hyperten-sion after chronic exposure to severe hypoxia (PIO2 of 72 to85 mm Hg).20–23 These increases in systemic blood pressureare associated with increases in ET-1 and pulmonary arterialpressure, and neither a nonselective endothelin A/B receptorantagonist nor a selective endothelin A receptor antagonistreduces the systemic hypertension. Thus, the mechanismunderlying the development of systemic hypertension inAHR KO mice at modest altitude appears to differ from thatassociated with other models of altitude-induced systemichypertension. Nonetheless, the PIO2-dependent changes inblood pressure in the AHR KO mice provide a potentialexplanation for different blood pressure values reported by
Figure 4. A, Plasma ET-1 and (B) pulmonary prepro-ET-1 mRNAin AHR WT and KO mice residing at 225 m (PIO2: 149 mm Hg),1632 m and exposed to sea level PIO2 (PIO2: 150 mm Hg), or1632 m (PIO2: 122 mm Hg). Two-way ANOVA of plasma ET-1demonstrated significant differences based on PIO2 (P�0.005),genotype (P�0.001), and PIO2-genotype interaction (P�0.02).Two-way ANOVA of prepro-ET-1 demonstrated significant dif-ferences based on PIO2 (P�0.002), genotype (P�0.001), andPIO2-genotype interaction (P�0.001). *Significantly different fromAHR WT at the same altitude; †Significantly different from AHRKO at 225 m; ‡Significantly different from AHR KO at 1632 mexposed to sea level PIO2.
Table 1. Arterial Blood Gases, pH, and Total CO2 of AHR WT and KO Mice Residing at 1632 m and After Acute (24-Hour) or Chronic(11-Day) Exposure to Simulated Sea Level PIO2
Arterial Blood Parameter
1632 m Acute Simulated Sea Level PIO2 Chronic Simulated Sea Level PIO2
AHR WT (N�6) AHR KO (N�5) AHR WT (N�6) AHR KO (N�5) AHR WT (N�6) AHR KO (N�5)
PO2, mm Hg 71�3.1 54�4.1* 86�4.2 65�4.6†‡ 78.0�2.1 74.8�2.0‡
PCO2, mm Hg 27.3�0.8 31.3�1.7* 32.2�2.2 34.9�1.7‡ 35.8�2.6* 36.2�1.4
O2 saturation, % 94.0�1.1 81.6�3.2* 96.2�0.6 88.4�2.8 94.2�0.6 94.8�0.9‡
pH 7.41�0.03 7.26�0.03* 7.37�0.03 7.29�0.06 7.34�0.04 7.41�0.03‡
TCO2, mmol/L 18.2�0.8 15.2�1.8* 19.5�0.7 18.6�2.6 24.4�1.2 22.6�1.1
TCO2 indicates total CO2.*P�0.05 vs AHR WT at 1632 m.†P�0.05 vs AHR WT at acute simulated sea level PIO2.‡P�0.05 vs AHR KO at 1632 m.
806 Hypertension March 2008
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

others studying this KO model.6,7 Furthermore, our studies, aswell as those of others, provide evidence that the AHRsignaling pathway may play a role in the pathogenesis ofsystemic hypertension. Both the AHR and AHR-regulatedgene cytochrome P4501A1 map to quantitative trait lociassociated with hypertension,24 –26 and human hypertensionis associated with polymorphisms in both AHR and cyto-chrome P4501A1,27 as well as chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin.28 Thus, the mechanism bywhich AHR signaling regulates blood pressure and impactsthe etiology and pathogenesis of hypertension requires furtherinvestigation.
The elevated levels of plasma ET-1 in AHR KO mice atlow altitude suggest that there is increased production and/ordecreased clearance of ET-1 under normoxic conditions. Ifproduction of ET-1 is increased, the source remains to beidentified; however, our data would suggest that the lung isnot the source, because pulmonary prepreo-ET-1 mRNA issignificantly reduced at low altitude, and ET-1 has beenshown to be regulated primarily at the transcriptional level.29
Alternatively, if clearance is reduced, one possible explana-tion is that it results from a significantly smaller liver (TableS1). Adult AHR KO mice exhibit a liver that is 25% to 30%smaller than WT mice as a result of a persistent ductusvenosus, which shunts blood away from the liver.30 Althoughthe lung is the primary site for ET-1 clearance, studies haveshown that the liver is an important site for ET-1 clearance,and circulating ET-1 increases significantly when hepaticclearance is impaired.31 Thus, it is possible that alterations inET-1 and blood pressure regulation may result from devel-opmental defects resulting from the genetic deletion of Ahr.
The elevated levels of plasma ET-1 in AHR KO mice atmodest altitude are consistent with the degree of hypoxemiaexhibited by these mice. Hypoxemia is a potent inducer of
ET-1 in humans and experimental animals,14,32–35 and arterialpO2 �65 mm Hg is associated with significant increases inplasma ET-1 in humans.33 Furthermore, the degree of hypox-emia correlates with induction of pulmonary prepro-ET-1mRNA expression in the AHR KO mice, although the levelsremain significantly less than WT mice. Thus, the hypoxemiaexhibited by AHR KO mice at modest altitude could beresponsible for the increase observed in plasma ET-1 abovethe already elevated basal levels.
Our data do not support the hypothesis that AHR KO miceare generally more sensitive to hypoxic induction of geneexpression. Although AHR KO mice were significantlyhypoxemic, hematocrit was not increased as might be ex-pected if the mice were more sensitive to hypoxic inductionof erythropoietin. Furthermore, AHR deficiency in pulmo-nary endothelial cells does not alter the sensitivity to hypoxicinduction of prepro-ET-1 mRNA. These observations are incontrast to the enhanced response of AHR KO mice toischemia-induced expression of vascular endothelial growthfactor and neovasculogenesis,7 suggesting that the sensitivityof AHR KO mice to hypoxic induction of gene expressionmay be tissue and/or gene dependent.
The presence of mild hypoxemia and hypercapnia in AHRKO mice at modest altitude was unexpected, and the mech-anism underlying this observation is not known. Decreases inarterial pO2 are detected rapidly by the carotid body, whichstimulates a persistent increase in ventilation, increasingarterial pO2 and decreasing arterial pCO2.36 Based on thearterial blood gas data, this ventilatory response appears tofunction normally in AHR WT mice at modest altitude butmay not in AHR KO mice. Alternatively, the hypoxemia inAHR KO mice could result from alterations in lung morphol-ogy that reduce diffusion capacity or from a ventilation-
Figure 5. Prepro-ET-1 mRNA in humanmicrovascular endothelial cells isolatedfrom lung (HMVEC-L) exposed to controlor AHR siRNA and incubated at (A) 21.0%or (B) 21.0%, 2.5%, or 1.0% O2 for 18hours. Two-way ANOVA demonstratedsignificant differences based on O2
(P�0.001) and siRNA (P�0.02) but no evi-dence of O2-siRNA interaction (P�0.58).*Significantly different from control siRNA;†Significantly different from 21.0% O2.
Table 2. Hematocrit and Hemoglobin of AHR WT and KO Mice Residing at 1632 m and AfterChronic (11-Day) Exposure to Simulated Sea Level PIO2
Venous Blood Parameter
1632 m Chronic Simulated Sea Level PIO2
AHR WT (N�9) AHR KO (N�8) AHR WT (N�9) AHR KO (N�8)
Hematocrit 0.49�0.01 0.45�0.01 0.49�0.02 0.44�0.02*
Hemoglobin, g/dL 16.5�0.4 15.4�0.4 16.6�0.5 14.8�0.6*
*P�0.005 vs AHR WT at chronic simulated sea level PIO2.
Lund et al Altitude-Induced Hypertension in AHR Null Mice 807
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

perfusion mismatch. Future studies will need to investigatethe contributions of these factors to the hypoxemia.
Finally, the reason for the acidosis observed in AHR KOmice at modest altitude also remains to be determined butprobably is not a result of hypercapnia. Although arterial pHdecreases as arterial pCO2 increases, the degree of acidosis inAHR KO mice is more severe than would be predicted by themild degree of hypercapnia. Given that chronic exposure tosimulated sea level PIO2 normalizes the acidosis, it is possiblethat hypoxemia-induced lactic acidosis may be the underlyingcause, although this needs to be investigated further.
In conclusion, our studies establish the AHR KO mouseas a genetic model in which modest decreases in PIO2 leadto significant hypoxemia, increased ET-1, and systemichypertension in the absence of symptoms of pulmonaryhypertension.
PerspectivesAlthough studies show that people living at high altitudestend to have lower systemic blood pressure, these differencesare only observed after years of residence.37 In contrast, shortvisits to modest or high altitudes induce significant increasesin systemic blood pressure,38 and preexisting genetic factorsmay predispose low altitude residents to altitude-inducedhypertension.39 Our data suggest that the Ahr may represent acandidate gene for altitude-induced hypertension. Furtherstudy of AHR KO mice may reveal novel pathways in theregulation of systemic blood pressure by hypoxia and identifyphysiological mechanisms that contribute to altitude-inducedsystemic hypertension.
AcknowledgmentsWe thank Drs Scott Burchiel, Benjimen R. Walker, Tom Resta,Laura Gonzalez-Bosc, and Matthew Campen for their technicalassistance and critical discussions.
Sources of FundingThis work was supported by National Institutes of Health grantsP30ES12072 to the University of New Mexico and HL078914 toM.K.W., American Heart Association Grant-in-Aid 0550028Z toM.K.W. and US Environmental Protection Agency Science to AchieveResults Predoctoral Fellowship U91621501 to A.K.L.
DisclosuresNone.
References1. Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of
environmental and developmental signals. Annu Rev PharmacolToxicol. 2000;40:519–661.
2. Thackaberry EA, Gabaldon D, Walker MK, Smith SM. Aryl hydro-carbon receptor null mice exhibit cardiac hypertrophy and increasedcardiac hypoxia-inducible factor 1-� in the absence of cardiac hyp-oxia. Cardiovasc Tox. 2002;4:263–273.
3. Fernandez-Salguero P, Ward JM, Sundberg JP, Gonzalez FJ. Lesions ofaryl-hydrocarbon receptor-deficient mice. Vet Pathol. 1997;34:605–614.
4. Lund AK, Goens MB, Kanagy NL, Walker MK. Cardiac hypertrophy inaryl hydrocarbon receptor (AhR) null mice is correlated with elevatedangiotensin II, endothelin-1 and mean arterial blood pressure. ToxicolAppl Pharmacol. 2003;193:177–187.
5. Lund AK, Goens MB, Nunez B, Walker MK. Characterizing the role ofendothelin-1 in the progression of cardiac hypertrophy in aryl hydro-carbon receptor (AHR) null mice. Toxicol Appl Pharmacol. 2006;212:127–135.
6. Vasquez A, Atallah-Yunes N, Smith FC, You X, Chase SE, SilverstoneAE, Vikstrom KL. A role for the aryl hydrocarbon receptor in cardiacphysiology and function as demonstrated by AhR knockout mice.Cardiovasc Toxicol. 2003;3:153–163.
7. Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, GonzalezFJ, Murohara T. A role for the aryl hydrocarbon receptor in regulation ofischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1297–1304.
8. Chan WK, Yao G, Gu YZ, Bradfield CA. Cross-talk between the arylhydrocarbon receptor and hypoxia inducible factor signaling pathways.Demonstration of competition and compensation. J Biol Chem. 1999;274:12115–12123.
9. Pollenz RS, Davarinos NA, Shearer TP. Analysis of aryl hydrocarbonreceptor-mediated signaling during physiological hypoxia reveals lack ofcompetition for the aryl hydrocarbon nuclear translocator transcriptionfactor. Mol Pharmacol. 1999;56:1127–1137.
10. Minchenko A, Caro J. Regulation of endothelin-1 gene expression inhuman microvascular endothelial cells by hypoxia and cobalt: role ofhypoxia responsive element. Mol Cellular Biochem. 2000;208:53–62.
11. Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecularregulation of the endothelin-1 gene by hypoxia. Contributions of hypox-ia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP.J Biol Chem. 2001;276:12645–12653.
12. Zhang N, Walker MK. Crosstalk between the aryl hydrocarbon receptorand hypoxia on the constitutive expression of cytochrome P4501A1mRNA. Cardiovasc Toxicol. 2007;7:282–290.
13. Goerre S, Wenk M, Bartsch P, Luscher TF, Niroomand F, Hohenhaus E,Oelz O, Reinhart WH. Endothelin-1 in pulmonary hypertension asso-ciated with high-altitude exposure. Circulation. 1995;91:359–364.
14. Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS.Enhanced endothelin-1 and endothelin receptor gene expression inchronic hypoxia. J Appl Physiol. 1994;77:1451–1459.
15. Elton TS, Oparil S, Taylor GR, Hicks PH, Yang RH, Jin H, Chen YF.Normobaric hypoxia stimulates endothelin-1 gene expression in the rat.Am J Physiol. 1992;263:R1260–R1264.
16. Eddahibi S, Raffestin B, Clozel M, Levame M, Adnot S. Protection frompulmonary hypertension with an orally active endothelin receptor antag-onist in hypoxic rats. Am J Physiol. 1995;268:H828–H835.
17. Fagan KA, Fouty BW, Tyler RC, Morris KG Jr, Hepler LK, Sato K,LeCras TD, Abman SH, Weinberger HD, Huang PL, McMurtry IF,Rodman DM. The pulmonary circulation of homozygous or heterozygouseNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest. 1999;103:291–299.
18. Nagaoka T, Muramatsu M, Sato K, McMurtry I, Oka M, Fukuchi Y. Mildhypoxia causes severe pulmonary hypertension in fawn-hooded but not inTester Moriyama rats. Respir Physiol. 2001;127:53–60.
19. Ivy DD, Yanagisawa M, Gariepy CE, Gebb SA, Colvin KL, McMurtryIF. Exaggerated hypoxic pulmonary hypertension in endothelin Breceptor-deficient rats. Am J Physiol Lung Cell Mol Physiol. 2002;282:L703–L712.
20. Schweda F, Blumberg FC, Schweda A, Kammerl M, Holmer SR, RieggerGAJ, Pfeifer M, Kramer BK. Effects of chronic hypoxia on renal reningene expression in rats. Nephrol Dial Transplant. 2000;15:11–15.
21. Ni Z, Bemanian S, Kivlighn SD, Vaziri ND. Role of endothelin and nitricoxide imbalance in the pathogenesis of hypoxia-induced arterial hyper-tension. Kidney Int. 1998;54:188–192.
22. Vaziri ND, Wang ZQ. Sustained systemic arterial hypertension inducedby extended hypobaric hypoxia. Kidney Int. 1996;49:1457–1463.
23. Modesti PA, Vanni S, Morabito M, Modesti A, Marchetta M, Gamberi T,Sofi F, Savia G, Mancia G, Gensini GF, Parati G. Role of endothelin-1 inexposure to high altitude: Acute Mountain Sickness and Endothelin-1(ACME-1) study. Circulation. 2006;114:1410–1416.
24. Garrett MR, Saad Y, Dene H, Rapp JP. Blood pressure QTL that differ-entiate Dahl salt-sensitive and spontaneously hypertensive rats. PhysiolGenomics. 2000;3:33–38.
25. Stoll M, Kwitek-Black AE, Cowley AW Jr, Harris EL, Harrap SB,Krieger JE, Printz MP, Provoost AP, Sassard J, Jacob HJ. New targetregions for human hypertension via comparative genomics. Genome Res.2000;10:473–482.
26. Ramos A, Moisan MP, Chaouloff F, Mormede C, Mormede P. Identifi-cation of female-specific QTLs affecting an emotionality-related behaviorin rats. Mol Psychiatry. 1999;4:453–462.
27. Gambier N, Marteau JB, Batt AM, Marie B, Thompson A, Siest G,Foernzler D, Visvikis-Siest S. Interaction between CYP1A1 T3801C and
808 Hypertension March 2008
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

AHR G1661A polymorphisms according to smoking status on bloodpressure in the Stanislas cohort. J Hypertens. 2006;24:2199–2205.
28. Kang HK, Dalager NA, Needham LL, Patterson DG Jr, Lees PS, Yates K,Matanoski GM. Health status of Army Chemical Corps Vietnam veteranswho sprayed defoliant in Vietnam. Am J Ind Med. 2006;49:875–884.
29. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, MitsuiY, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptideproduced by vascular endothelial cells. Nature. 1988;332:411–415.
30. Lahvis GP, Lindell SL, Thomas RS, McCuskey RS, Murphy C, Glover E,Bentz M, Southard J, Bradfield CA. Portosystemic shunting and per-sistent fetal vascular structures in aryl hydrocarbon receptor-deficientmice. Proc Natl Acad Sci U S A. 2000;97:10442–10447.
31. Tran Duc AT, Schwab AJ, Simard A, Villeneuve L, Dupuis J. Reductionin hepatic endothelin-1 clearance in cirrhosis. Clin Sci (Lond). 2003;105:227–234.
32. Aguirre JI, Morrell NW, Long L, Clift P, Upton PD, Polak JM, WilkinsMR. Vascular remodeling and ET-1 expression in rat strains with dif-ferent responses to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol.2000;278:L981–L987.
33. Faller M, Kessler R, Sapin R, Chaouat A, Ehrhart M, Ducolone A,Weitzenblum E. Regulation of endothelin-1 at rest and during a short
steady-state exercise in 21 COPD patients. Pulm Pharmacol Ther. 1998;11:151–157.
34. Cargill RI, Kiely DG, Clark RA, Lipworth BJ. Hypoxaemia and releaseof endothelin-1. Thorax. 1995;50:1308–1310.
35. Trakada G, Nikolaou E, Pouli A, Tsiamita M, Spiropoulos K.Endothelin-1 levels in interstitial lung disease patients during sleep. SleepBreath. 2003;7:111–118.
36. Prabhakar NR, Jacono FJ. Cellular and molecular mechanisms associatedwith carotid body adaptations to chronic hypoxia. High Alt Med Biol.2005;6:112–120.
37. Marticorena E, Ruiz L, Severino J, Galvez J, Penaloza D. Systemic bloodpressure in white men born at sea level: changes after long residence athigh altitudes. Am J Cardiol. 1969;23:364–368.
38. Palatini P, Businaro R, Berton G, Mormino P, Rossi GP, Racioppa A,Pessina AC, Dal PC. Effects of low altitude exposure on 24-hour bloodpressure and adrenergic activity. Am J Cardiol. 1989;64:1379–1382.
39. Kumar R, Qadar Pasha MA, Khan AP, Gupta V, Grover SK, Norboo T,Srivastava KK, Selvamurthy W, Brahamchari SK. Association of high-altitude systemic hypertension with the deletion allele-of the angioten-sin-converting enzyme (ACE) gene. Int J Biometeorol. 2003;48:10–14.
Lund et al Altitude-Induced Hypertension in AHR Null Mice 809
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

Nancy L. Kanagy and Mary K. WalkerAmie K. Lund, Larry N. Agbor, Nan Zhang, Amy Baker, Huawei Zhao, Gregory D. Fink,
Hypertension at Modest AltitudeLoss of the Aryl Hydrocarbon Receptor Induces Hypoxemia, Endothelin-1, and Systemic
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2008 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.107.1005862008;51:803-809; originally published online January 22, 2008;Hypertension.
http://hyper.ahajournals.org/content/51/3/803World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2008/01/22/HYPERTENSIONAHA.107.100586.DC1Data Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 13, 2018
http://hyper.ahajournals.org/D
ownloaded from

1
Loss of the Aryl Hydrocarbon Receptor Induces Hypoxemia, Endothelin-1, and
Systemic Hypertension at Modest Altitude
Amie K. Lund, Larry N. Agbor, Nan Zhang, Amy Baker, Huawei Zhao, Gregory D. Fink, Nancy
L. Kanagy and Mary K. Walker
From the College of Pharmacy (A.K.L., L.N.A., N.Z., A.B., M.K.W.) and Department of Cell
Biology and Physiology, School of Medicine (N.L.K., M.K.W.), University of New Mexico
Health Sciences Center, Albuquerque, NM; and Department of Pharmacology and Toxicology
(H.Z., G.D.F.), College of Human Medicine, Michigan State University, East Lansing, MI
Running title: Altitude-induced Hypertension in AHR Null Mice
On-line supplement
Address correspondence to: Mary K. Walker College of Pharmacy MSC09 5360 2703 Frontier NE University of New Mexico Albuquerque, NM 87131 T: 505-272-0580 F: 505-272-0704 Email: [email protected]

2
Online Data Supplement
Expanded Materials and Methods
Chemicals. Lipofectamine 2000 and Opti-MEM I reduced-serum media were purchased from
Invitrogen (Carlsbad, CA).
Animals. The aryl hydrocarbon receptor knockout (AHR KO) mice were obtained from Dr.
Frank Gonzalez (National Cancer Institute) and backcrossed 11 generations to C57Bl/6N
(Harlan). Mice were fed ad libitum and maintained on a 12/12 light/dark cycle. C57Bl/6N AHR
wildtype (WT) mice were purchased from Harlan at 6-8 wks of age. At the time of euthanasia,
mice were deeply anesthetized with ketamine/xylazine (80 mg/kg and 4 mg/kg, respectively) and
blood was collected into a heparinized syringe by cardiac puncture. Lungs were removed and
snap frozen. Blood was centrifuged for 10 min at 950 x g to separate plasma, which was stored
at -20° until analyzed.
Blood Pressure Analysis. To implant the radiotelemetry transmitters (PA-C20), mice were
injected with buphrenorphine (0.1 mg/kg, subq) 30 min prior to ketamine (91 mg/kg, ip) and
acepromazine (0.9 mg/kg, ip) to induce anesthesia. The left carotid artery was exposed, and after
separating jugular vein and nerves, the artery was securely ligated with sutures and catheter
inserted to 1.0 cm from the carotid artery bifurcation. A tunnel was created under the skin and
body of the transmitter was placed subcutaneously in the right abdominal area. The mouse was
kept warm and observed until completely conscious. Beginning seven days after surgery,

3
baseline blood pressure values, including mean, systolic, and diastolic; and heart rate were
recorded for 10 s every 15 min for 24 hr/d for a total of 6 d (Data Sciences International,
Minneapolis, MN). At UNM, after recording baseline blood pressure values the mice were
placed into a Plexiglass chamber where the PIO2 was maintained at 150 mm Hg for 11 d by
infusion of oxygen and blood pressure values continued to be recorded.
Arterial Blood Gas Analysis. Microrenathane (Braintree Scientific, Braintree, MA) was
stretched to < 400 µm using a heat gun and sesame oil, and a suture anchor point was added
using silastic adhesive. Mice were injected with buphrenorphine (0.1 mg/kg, subq) and
anesthetized with isoflurane. The catheter was filled with a heparin/glycerol (50 U/ml), inserted
into the left carotid artery, and tunneled subcutaneously where it exited loosely between the
scapula. Catheters were flushed twice per day with heparinized saline (50 U/ml). In the first
group of mice residing at 1632 m, an arterial blood sample was drawn from conscious animals
two days post surgery and PO2, PCO2, oxygen saturation, pH, and total CO2 were measured
using an i-STAT 1 analyzer (Abbott Point of Care Inc., East Windsor, NJ). These same mice
were then placed into a Plexiglass chamber where PIO2 was maintained at simulate sea level
(150 mm Hg) and after 24 hr a second arterial blood sample was drawn without removing the
animal from the chamber, and blood gases analyzed. In the second group of mice residing at
1632 m, mice were placed into the oxygen chamber and exposed to simulated sea level PIO2 for
11 d. On day 9 the mice were removed from the chamber to implant an arterial catheter (~45
min) and then returned to the chamber for an additional two days. After a total of 11 d at
simulated sea level PIO2, an arterial blood sample was drawn and analyzed. In a third group of
mice, a blood sample was collected from the mandibular vein and then the mice were placed into

4
the oxygen chamber and exposed to simulated sea level PIO2 for 11 d when a second blood
sample was collected. These samples were analyzed for hematocrit and hemoglobin with the i-
STAT analyzer.
Analysis of ET-1 Expression. Plasma ET-1 was concentrated using C2 Amprep minicolumns
(Amersham Pharmacia, Piscataway, NJ) and assayed by RIA (Amersham Pharmacia), while ET-
1 from cell culture media was analyzed by ELISA (R&D Systems, Minneapolis, MN). For
preproET-1 mRNA analysis, total RNA was isolated from the lung (Trizol, Sigma-Aldrich, St.
Louis, MO) and endothelial cells (RNeasy®, QIAGEN, Valencia, CA), cDNA was synthesized
(iScript Select, Bio-Rad, Hercules, CA) with random primers at 42°C for 30 min, and
quantitative real time PCR analysis conducted with SYBR green PCR Master mix (Bio-Rad), a
Bio-Rad Icycler optical system, and phosphoglycerate kinase (PGK1) mRNA as the internal
normalization control for lung tissue and 18s rRNA for the cultured endothelial cells.
Primers were designed using Beacon Designer software and PCR reaction efficiency was
determined (efficiency = eln10/-s -1, s=standard curve slope) and optimized >99%. Amplification
of a single, appropriately sized band will be confirmed by gel electrophoresis and melt-curve
analyses. Amplification of target and PGK cDNA were run side-by-side in triplicate. Threshold
crossing value (CT) was calculated using well factor background subtraction, and gene
expression normalized to PGK1 or 18s for each sample. The mouse PCR primer sequences
were: ET-1 sense: 5’ AAGACCATCTGTGTGGCTTCTAC 3’; ET-1 antisense: 5’
CAGCCTTTCTTGGTGTTTGGAT 3’; PGK1 sense: 5’ CAAGCTACTGTGGCCTCTGGT 3’;
and PGK1 antisense: 5’ CGGCATATTTCTTGCTGCTCTC 3’,while the human PCR primer

5
sequences were: ET-1 sense: 5’ CATTGGTGACAGACCTTCGGG 3’; ET-1 antisense: 5’
GATGCTCCTGCTCTGATCCCA 3’; 18s sense: 5’ CGGAGGTTCGAAGACGATCAGATA
3’; 18s antisense: 5’ TTGGTTTCCCGGAAGCTGCC 3’.
Cell Culture and siRNA Transfection. HUVEC and HMVEC-L were purchased from Lonza
(Walkersville, MD) and cultured in microvascular endothelial cell media-2 (EGM-2MV, Lonza),
which contained 5% fetal bovine serum, 0.1% antibiotics (GA-1000) and other supplements, at
37°C in 21% O2 and 5% CO2. For transfections, cells were plated at 2 x 105 per well (~ 80%
confluence) in EGM-2MV without antibiotics and the next day Lipofectamine 2000, mixed with
Opti-MEM I reduced-serum media and either control siRNA or AHR ON-TARGETplus
SMARTpool siRNA (Dharmacon, Chicago, IL), was added to cells for 48 hrs. When the cells
were exposed to siRNA and hypoxia, hypoxia exposures were performed during the last 18 hrs
of the 48-hr siRNA treatment period. For hypoxia, cells were exposed to 2.5 or 1% O2 using
nitrogen infusion in a Napco 7001H incubator (Fisher Scientific, Pittsburgh, PA).
Statistical Analysis. Altitude- and genotype-related changes on MAP and ET-1 were analyzed
by two-way analysis of variance (ANOVA) with post hoc Holm-Sidak comparisons. Inspired
PIO2-, time-, and genotype-related changes on MAP were analyzed by repeated measures, two-
way ANOVA with post hoc Holm-Sidak comparisons. Arterial blood gases were compared
between genotypes by an unpaired Student’s t-test and within a genotype by a paired and
unpaired Student’s t-test when appropriate. Oxygen- and siRNA-related changes on AHR and
preproET-1 mRNA expression, and ET-1 secretion in endothelial cells were analyzed by two-
way ANOVA with post hoc Holm-Sidak comparisons. P<0.05 was considered statistically

6
significant in all cases.

7
Table S1. Body, heart, and liver weights (g) of AHR WT and KO mice at 4 mo of age.
Genotype N BW HW HW/BW % LV+S LV+S/BW % RV RV/BW % RV/LV+S Liver
WT 8 27.8±0.7 0.117±0.003 0.42±0.02 0.088±0.002 0.32±0.01 0.029±0.001 0.10±0.01 0.33±0.02 0.052±0.0007
KO 6 30.1±1.1 0.143±0.006* 0.48±0.02 0.112±0.004* 0.37±0.02* 0.031±0.003 0.10±0.01 0.28±0.02 0.036±0.0003*
Abbreviations: BW, body weight; HW, heart weight; LV+S, left ventricle plus septum; RV, right ventricle
*P<0.05 compared to AHR WT





























