Embed Size (px)
Citation preview

Atmos. Chem. Phys., 8, 1353–1366, 2008www.atmos-chem-phys.net/8/1353/2008/© Author(s) 2008. This work is distributed underthe Creative Commons Attribution 3.0 License.
AtmosphericChemistry
and Physics
Hydrogen isotope fractionation in the photolysis of formaldehyde
T. S. Rhee1, C. A. M. Brenninkmeijer 2, and T. Rockmann3
1Korea Polar Research Institute, Incheon, Korea2Atmospheric Chemistry Division, Max Planck Institute for Chemistry, Mainz, Germany3Institute for Marine and Atmospheric Research Utrecht, Utrecht University, Utrecht, The Netherlands
Received: 2 August 2007 – Published in Atmos. Chem. Phys. Discuss.: 29 August 2007Revised: 31 January 2008 – Accepted: 1 February 2008 – Published: 7 March 2008
Abstract. Experiments investigating the isotopic fractiona-tion in the formation of H2 by the photolysis of CH2O un-der tropospheric conditions are reported and discussed. Thedeuterium (D) depletion in the H2 produced is 500(±20)‰with respect to the parent CH2O. We also observed that com-plete photolysis of CH2O under atmospheric conditions pro-duces H2 that has virtually the same isotope ratio as that ofthe parent CH2O. These findings imply that there must be avery strong concomitant isotopic enrichment in the radicalchannel (CH2O+hν→CHO+H) as compared to the molecu-lar channel (CH2O+hν→H2+CO) of the photolysis of CH2Oin order to balance the relatively small isotopic fractionationin the competing reaction of CH2O with OH. Using a 1-boxphotochemistry model we calculated the isotopic fractiona-tion factor for the radical channel to be 0.22(±0.08), whichis equivalent to a 780(±80)‰ enrichment in D of the remain-ing CH2O. When CH2O is in photochemical steady state, theisotope ratio of the H2 produced is determined not only by theisotopic fractionation occurring during the photolytical pro-duction of H2 (αm) but also by overall fractionation for theremoval processes of CH2O (αf ), and is represented by theratio of αm/αf . Applying the isotopic fractionation factorsrelevant to CH2O photolysis obtained in the present study tothe troposphere, the ratio ofαm/αf varies from∼0.8 to∼1.2depending on the fraction of CH2O that reacts with OH andthat produces H2. This range ofαm/αf can render the H2produced from the photochemical oxidation of CH4 to be en-riched in D (with respect to the original CH4) by the factorof 1.2–1.3 as anticipated in the literature.
Correspondence to:T. S. Rhee([email protected])
1 Introduction
Formaldehyde (CH2O) is a key carbonyl compound in the at-mosphere. Its abundance varies over a wide range from sub-ppb levels to∼100 ppb depending largely on local sources(Warneck, 1999). Its turnover is large in the atmosphere andit is a source of molecular hydrogen (H2), carbon monox-ide (CO), and of the hydroperoxyl radical (HO2), yet lim-ited measurements are available in various atmospheric re-gions. Recent satellite observations of CH2O make it possi-ble to investigate its distribution on regional and global scales(e.g., Martin et al., 2004; Wittrock et al., 2006). While directemissions from fossil fuel combustion, biomass burning, andalso automotive exhaust contribute significantly to the bur-den of atmospheric CH2O (Carlier et al., 1986; Garcia et al.,2005), in situ production of CH2O by photochemical oxida-tion of volatile organic compounds appears to be the domi-nant source on a global scale (Carlier et al., 1986; Warneck,1999). In remote oceanic areas (Wagner et al., 2002; Welleret al., 2000), in the free troposphere (Frost et al., 2002), andin the stratosphere, only the photochemical oxidation of CH4serves as the major source. Apart from the importance ofthe rather simple CH2O molecule in the Earth’s atmosphereand far beyond, it is also subject to fundamental research re-garding for instance the exact processes during its photolysis(e.g., Moore and Weisshaar, 1983; Townsend et al., 2004;Troe, 2007).
CH2O is broken down by photolysis (R1 and R2) and byphotochemical oxidation (R3) in the troposphere (Calvert,1980):
CH2O + hν → CHO+ H (R1)
CH2O + hν → CO+ H2 (R2)
CH2O + OH → CHO+ H2O (R3)
Published by Copernicus Publications on behalf of the European Geosciences Union.

1354 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
Reaction (R1) produces the HO2 radical by the rapid re-action of hydrogen (H) and formyl (CHO) radicals with at-mospheric oxygen (O2), which can lead to the formation ofthe hydroxyl radical (OH) via the reaction with NO or O3in the atmosphere. This is an important propagation of theradical chain. Only reaction (R2) yields H2. All photochem-ical reactions of CH2O do produce CO, while solely reaction(R2) forms H2, which is the topic of our research. In fact,this photochemically produced H2 constitutes∼50 to∼60%of the total source of tropospheric H2 (Novelli et al., 1999;Rhee et al., 2006b).
In the stratosphere, H2 originates both from this in situphotolysis process (R2), albeit under photochemically verydifferent conditions, and from tropospheric import. Recentlyit has been established that stratospheric H2 is enriched indeuterium (D) along with the decrease of CH4 mixing ratioswhilst the H2 mixing ratios remain almost constant (Rahn etal., 2003; Rhee et al., 2006a; Rockmann et al., 2003). It ap-pears that the D enrichment of H2 is much stronger than theconcomitant enrichment for CH4 acompanying its destruc-tion by OH, O(1D), and Cl radicals. This means that theD enrichment of H2 occurs not only by the fractionation inthe reaction of H2 with oxidizing radicals (OH, Cl, O(1D))but is also due to the chain reactions leading from CH4 toH2 (Rhee et al., 2006a). Gerst and Quay (2001) discussedpotential reactions that may lead to the D enrichment alongthe photochemical chain reactions of CH4. However, the de-tailed mechanism by which the D content of H2 is accumu-lated has not yet been elucidated due to the lack of measure-ments for isotopic fractionation factors at each reaction stepand branching, all of which are fundamentally difficult to de-termine.
To address this question, as a first step we have investi-gated the isotopic fractionation occurring during the photol-ysis of CH2O by which H2 is produced for the conditionsat Earth’s surface. In spite of its crucial role in the isotopebudget of H2, as well as CO, in the atmosphere, the isotopicfractionation occurring during photolysis of CH2O has beenrarely investigated in the past (Crounse et al., 2003; Feilberget al., 2005; Feilberg et al., 2007b). Since CH2O is a rela-tively “long-lived” intermediate in the photochemical chainreactions between CH4 and H2, the results will provide es-sential insight into understanding the accumulation of D inH2 produced.
2 Experiments
Formaldehyde (CH2O) was prepared by purifying para-formaldehyde (Merck) in a vacuum system followingthe method of Spence and Wild (1935). Solid para-formaldehyde was heated at∼420 K under vacuum. For pu-rification the evaporating CH2O and impurities were forcedthrough a set of glass U-tubes which were partly immersed inan ethanol sludge (∼160 K) made with liquid nitrogen. Pu-
rified formaldehyde was then collected in a U-tube dippedin liquid nitrogen (77 K). A given amount of pure CH2O(∼3 mbar) was released to a 3-L glass bulb and several 0.1-Lglass flasks simultaneously, all of which were connected tothe same manifold. The pure CH2O in the 0.1-L glass flaskswere used to determine the D/H ratio of the CH2O (see be-low). Afterwards pressure inside the manifold was read bya capacitance manometer (MKS10, Baratron). CH2O-freesynthetic air was then introduced into the 3-L glass bulb toreach about ambient pressure and the final pressure was readby another capacitance manometer (MKS1000, Baratron) todetermine the CH2O mixing ratio. Since these pressure read-ings are essential for determining the CH2O mixing ratio inthe reactors used for the photolysis experiments, the capaci-tance manometers were calibrated accurately by an absolutemanometer (Digiquartz 740, Paroscientific) whenever neces-sary. The CH2O-air mixture was used as a stock for a seriesof CH2O photolysis experiments. The CH2O mixing ratiosin the stock air were usually around 0.3%.
Aliquots of the CH2O stock air were transferred to quartzor glass flasks, diluted to the target mixing ratio with CH2O-free synthetic air, and photolyzed for a few hours to∼17days (Table 1). The CH2O mixing ratios in the reactors wereless than∼2 ppm except in the experiments running for fewhours, for which∼50 ppm of CH2O was used. After photol-ysis we measured the H2 mixing ratio and D/H ratio. TheδDvalues and mixing ratios of the H2 produced were determinedby a recently developed technique involving continuous-flowisotope ratio mass spectrometry (Rhee et al., 2004).
In order to test stability of CH2O in the reactor, we hadonce monitored the pressure inside the 3-L glass bulb for 2days after injecting pure CH2O at ∼3 mbar. No change inpressure inside was found, indicating no absorption or lossof CH2O by polymerization or heterogeneous reactions. Thesame results even at higher pressure of pure CH2O air havebeen reported (e.g.,Horowitz and Calvert, 1978).
All glass used was Duran glass (Schott), thoroughly evac-uated and heated prior to use. Glass bulbs were kept in thedark by wrapping them with aluminum foil or with blackcloth to avoid any photochemical reactions prior to com-mencing CH2O photolysis experiments. CH2O photolysisexperiments in sunlight were carried out on the roof of a3-story building of the Max Planck Institute for Chemistry,Mainz (50◦ N, 8.16◦ E), in August and September of 2003and in March, May and June of 2004 (Table 1). We alsoconducted CH2O photolysis experiments using a xenon (Xe)short arc lamp (XBO 75W/2). A characteristic intensityspectrum of the light sources and the transmission of the re-actor materials are shown in Fig. 1 together with photolyticproperties of CH2O.
The D/H ratio of the original CH2O in the stock air was de-termined by analyzing the isotopic composition of the pureCH2O in the 0.1-L glass flasks, which originated from thesame source of CH2O as that in the stock air (see above).The pure CH2O sample was photolyzed using a mercury
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/
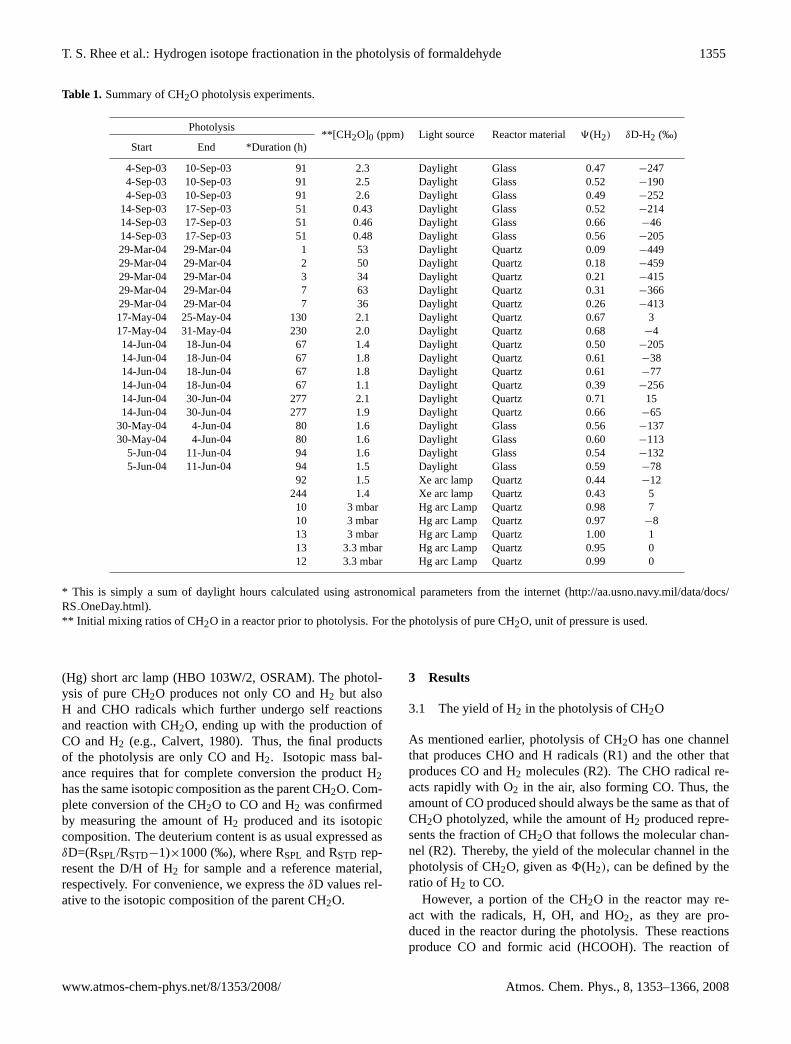
T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1355
Table 1. Summary of CH2O photolysis experiments.
Photolysis**[CH 2O]0 (ppm) Light source Reactor material9(H2) δD-H2 (‰)
Start End *Duration (h)
4-Sep-03 10-Sep-03 91 2.3 Daylight Glass 0.47 −2474-Sep-03 10-Sep-03 91 2.5 Daylight Glass 0.52 −1904-Sep-03 10-Sep-03 91 2.6 Daylight Glass 0.49 −252
14-Sep-03 17-Sep-03 51 0.43 Daylight Glass 0.52 −21414-Sep-03 17-Sep-03 51 0.46 Daylight Glass 0.66 −4614-Sep-03 17-Sep-03 51 0.48 Daylight Glass 0.56 −20529-Mar-04 29-Mar-04 1 53 Daylight Quartz 0.09 −44929-Mar-04 29-Mar-04 2 50 Daylight Quartz 0.18 −45929-Mar-04 29-Mar-04 3 34 Daylight Quartz 0.21 −41529-Mar-04 29-Mar-04 7 63 Daylight Quartz 0.31 −36629-Mar-04 29-Mar-04 7 36 Daylight Quartz 0.26 −41317-May-04 25-May-04 130 2.1 Daylight Quartz 0.67 317-May-04 31-May-04 230 2.0 Daylight Quartz 0.68 −414-Jun-04 18-Jun-04 67 1.4 Daylight Quartz 0.50 −20514-Jun-04 18-Jun-04 67 1.8 Daylight Quartz 0.61 −3814-Jun-04 18-Jun-04 67 1.8 Daylight Quartz 0.61 −7714-Jun-04 18-Jun-04 67 1.1 Daylight Quartz 0.39 −25614-Jun-04 30-Jun-04 277 2.1 Daylight Quartz 0.71 1514-Jun-04 30-Jun-04 277 1.9 Daylight Quartz 0.66 −65
30-May-04 4-Jun-04 80 1.6 Daylight Glass 0.56 −13730-May-04 4-Jun-04 80 1.6 Daylight Glass 0.60 −113
5-Jun-04 11-Jun-04 94 1.6 Daylight Glass 0.54 −1325-Jun-04 11-Jun-04 94 1.5 Daylight Glass 0.59 −78
92 1.5 Xe arc lamp Quartz 0.44 −12244 1.4 Xe arc lamp Quartz 0.43 510 3 mbar Hg arc Lamp Quartz 0.98 710 3 mbar Hg arc Lamp Quartz 0.97 −813 3 mbar Hg arc Lamp Quartz 1.00 113 3.3 mbar Hg arc Lamp Quartz 0.95 012 3.3 mbar Hg arc Lamp Quartz 0.99 0
* This is simply a sum of daylight hours calculated using astronomical parameters from the internet (http://aa.usno.navy.mil/data/docs/RS OneDay.html).** Initial mixing ratios of CH2O in a reactor prior to photolysis. For the photolysis of pure CH2O, unit of pressure is used.
(Hg) short arc lamp (HBO 103W/2, OSRAM). The photol-ysis of pure CH2O produces not only CO and H2 but alsoH and CHO radicals which further undergo self reactionsand reaction with CH2O, ending up with the production ofCO and H2 (e.g., Calvert, 1980). Thus, the final productsof the photolysis are only CO and H2. Isotopic mass bal-ance requires that for complete conversion the product H2has the same isotopic composition as the parent CH2O. Com-plete conversion of the CH2O to CO and H2 was confirmedby measuring the amount of H2 produced and its isotopiccomposition. The deuterium content is as usual expressed asδD=(RSPL/RSTD−1)×1000 (‰), where RSPL and RSTD rep-resent the D/H of H2 for sample and a reference material,respectively. For convenience, we express theδD values rel-ative to the isotopic composition of the parent CH2O.
3 Results
3.1 The yield of H2 in the photolysis of CH2O
As mentioned earlier, photolysis of CH2O has one channelthat produces CHO and H radicals (R1) and the other thatproduces CO and H2 molecules (R2). The CHO radical re-acts rapidly with O2 in the air, also forming CO. Thus, theamount of CO produced should always be the same as that ofCH2O photolyzed, while the amount of H2 produced repre-sents the fraction of CH2O that follows the molecular chan-nel (R2). Thereby, the yield of the molecular channel in thephotolysis of CH2O, given as8(H2), can be defined by theratio of H2 to CO.
However, a portion of the CH2O in the reactor may re-act with the radicals, H, OH, and HO2, as they are pro-duced in the reactor during the photolysis. These reactionsproduce CO and formic acid (HCOOH). The reaction of
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008
1356 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
220 240 260 280 300 320 340 3600
2
4
6
8
10
12
14
Wavelength (nm)
Abso
rptio
n cr
oss
sect
ion
(x10
20 c
m2 m
olec
ule-1
)
0.0
0.2
0.4
0.6
0.8
1.0Li
ght I
nten
sity
(arb
itrar
y un
its)
0
20
40
60
80
100
Tran
smis
sion
(%)
0.0
0.2
0.4
0.6
0.8
1.0
Hg lamp
Sun
Xe lamp
Duran glassQuartz
Φr
Quan
turm
Yie
ld
Φm
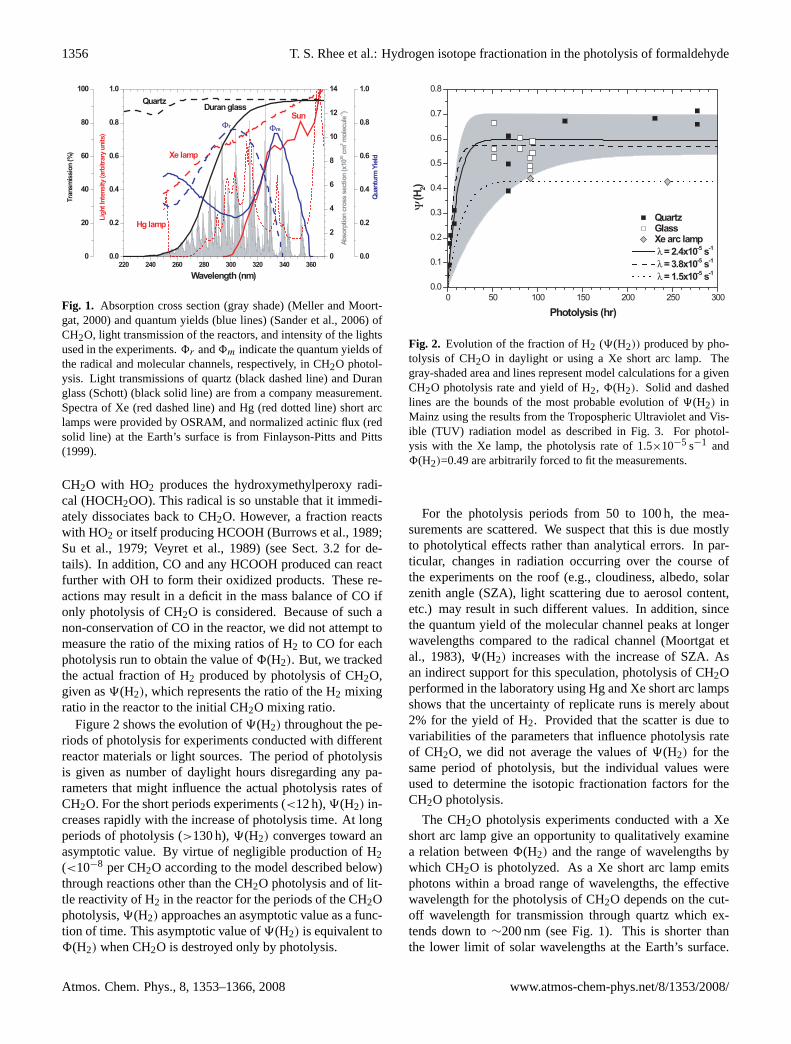
Fig. 1. Absorption cross section (gray shade) (Meller and Moort-gat, 2000) and quantum yields (blue lines) (Sander et al., 2006) ofCH2O, light transmission of the reactors, and intensity of the lightsused in the experiments.8r and8m indicate the quantum yields ofthe radical and molecular channels, respectively, in CH2O photol-ysis. Light transmissions of quartz (black dashed line) and Duranglass (Schott) (black solid line) are from a company measurement.Spectra of Xe (red dashed line) and Hg (red dotted line) short arclamps were provided by OSRAM, and normalized actinic flux (redsolid line) at the Earth’s surface is from Finlayson-Pitts and Pitts(1999).
CH2O with HO2 produces the hydroxymethylperoxy radi-cal (HOCH2OO). This radical is so unstable that it immedi-ately dissociates back to CH2O. However, a fraction reactswith HO2 or itself producing HCOOH (Burrows et al., 1989;Su et al., 1979; Veyret et al., 1989) (see Sect. 3.2 for de-tails). In addition, CO and any HCOOH produced can reactfurther with OH to form their oxidized products. These re-actions may result in a deficit in the mass balance of CO ifonly photolysis of CH2O is considered. Because of such anon-conservation of CO in the reactor, we did not attempt tomeasure the ratio of the mixing ratios of H2 to CO for eachphotolysis run to obtain the value of8(H2). But, we trackedthe actual fraction of H2 produced by photolysis of CH2O,given as9(H2), which represents the ratio of the H2 mixingratio in the reactor to the initial CH2O mixing ratio.
Figure 2 shows the evolution of9(H2) throughout the pe-riods of photolysis for experiments conducted with differentreactor materials or light sources. The period of photolysisis given as number of daylight hours disregarding any pa-rameters that might influence the actual photolysis rates ofCH2O. For the short periods experiments (<12 h),9(H2) in-creases rapidly with the increase of photolysis time. At longperiods of photolysis (>130 h),9(H2) converges toward anasymptotic value. By virtue of negligible production of H2(<10−8 per CH2O according to the model described below)through reactions other than the CH2O photolysis and of lit-tle reactivity of H2 in the reactor for the periods of the CH2Ophotolysis,9(H2) approaches an asymptotic value as a func-tion of time. This asymptotic value of9(H2) is equivalent to8(H2) when CH2O is destroyed only by photolysis.
0 50 100 150 200 250 3000.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Ψ(H
2)
Photolysis (hr)
QuartzGlassXe arc lamp λ = 2.4x10-5 s-1
λ = 3.8x10-5 s-1
λ = 1.5x10-5 s-1
Fig. 2. Evolution of the fraction of H2 (9(H2)) produced by pho-tolysis of CH2O in daylight or using a Xe short arc lamp. Thegray-shaded area and lines represent model calculations for a givenCH2O photolysis rate and yield of H2, 8(H2). Solid and dashedlines are the bounds of the most probable evolution of9(H2) inMainz using the results from the Tropospheric Ultraviolet and Vis-ible (TUV) radiation model as described in Fig. 3. For photol-ysis with the Xe lamp, the photolysis rate of 1.5×10−5 s−1 and8(H2)=0.49 are arbitrarily forced to fit the measurements.
For the photolysis periods from 50 to 100 h, the mea-surements are scattered. We suspect that this is due mostlyto photolytical effects rather than analytical errors. In par-ticular, changes in radiation occurring over the course ofthe experiments on the roof (e.g., cloudiness, albedo, solarzenith angle (SZA), light scattering due to aerosol content,etc.) may result in such different values. In addition, sincethe quantum yield of the molecular channel peaks at longerwavelengths compared to the radical channel (Moortgat etal., 1983),9(H2) increases with the increase of SZA. Asan indirect support for this speculation, photolysis of CH2Operformed in the laboratory using Hg and Xe short arc lampsshows that the uncertainty of replicate runs is merely about2% for the yield of H2. Provided that the scatter is due tovariabilities of the parameters that influence photolysis rateof CH2O, we did not average the values of9(H2) for thesame period of photolysis, but the individual values wereused to determine the isotopic fractionation factors for theCH2O photolysis.
The CH2O photolysis experiments conducted with a Xeshort arc lamp give an opportunity to qualitatively examinea relation between8(H2) and the range of wavelengths bywhich CH2O is photolyzed. As a Xe short arc lamp emitsphotons within a broad range of wavelengths, the effectivewavelength for the photolysis of CH2O depends on the cut-off wavelength for transmission through quartz which ex-tends down to∼200 nm (see Fig. 1). This is shorter thanthe lower limit of solar wavelengths at the Earth’s surface.
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/
T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1357
Jan Apr Jul Oct Jan
30
40
50
60
70(a)
47.8
27.1
SZ
A (°)
0 10 20 30 40 50 60 70 80 90
0.60
0.65
0.70
0.75 (b)
47.827.1
Φ (H
2)
SZA (°)
8
6
4
2
0
J CH
2O (x
105 s
-1)
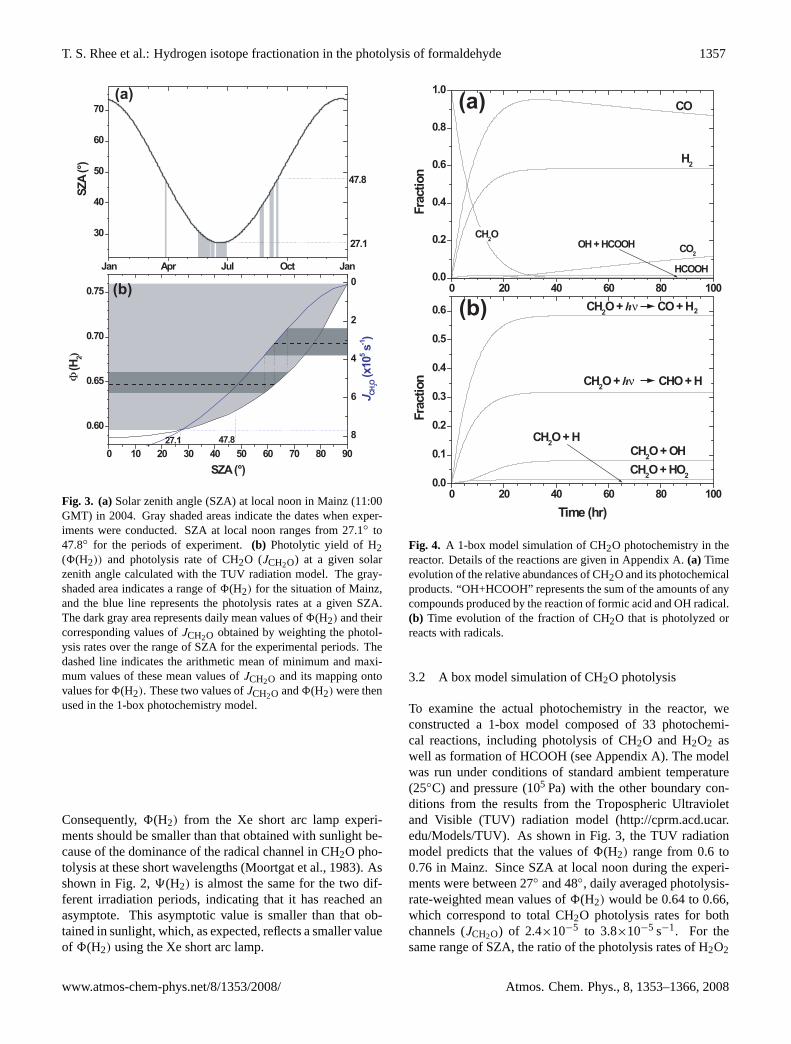
Fig. 3. (a)Solar zenith angle (SZA) at local noon in Mainz (11:00GMT) in 2004. Gray shaded areas indicate the dates when exper-iments were conducted. SZA at local noon ranges from 27.1◦ to47.8◦ for the periods of experiment.(b) Photolytic yield of H2(8(H2)) and photolysis rate of CH2O (JCH2O) at a given solarzenith angle calculated with the TUV radiation model. The gray-shaded area indicates a range of8(H2) for the situation of Mainz,and the blue line represents the photolysis rates at a given SZA.The dark gray area represents daily mean values of8(H2) and theircorresponding values ofJCH2O obtained by weighting the photol-ysis rates over the range of SZA for the experimental periods. Thedashed line indicates the arithmetic mean of minimum and maxi-mum values of these mean values ofJCH2O and its mapping ontovalues for8(H2). These two values ofJCH2O and8(H2) were thenused in the 1-box photochemistry model.
Consequently,8(H2) from the Xe short arc lamp experi-ments should be smaller than that obtained with sunlight be-cause of the dominance of the radical channel in CH2O pho-tolysis at these short wavelengths (Moortgat et al., 1983). Asshown in Fig. 2,9(H2) is almost the same for the two dif-ferent irradiation periods, indicating that it has reached anasymptote. This asymptotic value is smaller than that ob-tained in sunlight, which, as expected, reflects a smaller valueof 8(H2) using the Xe short arc lamp.
0 20 40 60 80 1000.0
0.1
0.2
0.3
0.4
0.5
0.6
0 20 40 60 80 1000.0
0.2
0.4
0.6
0.8
1.0 (a)
OH + HCOOHCH2O
HCOOH
CO2
H2
CO
Frac
tion
(b)
CH2O + H
CH2O + HO2
CH2O + OH
CH2O + hν CHO + H
CH2O + hν CO + H2
Time (hr)
Frac
tion
Fig. 4. A 1-box model simulation of CH2O photochemistry in thereactor. Details of the reactions are given in Appendix A.(a) Timeevolution of the relative abundances of CH2O and its photochemicalproducts. “OH+HCOOH” represents the sum of the amounts of anycompounds produced by the reaction of formic acid and OH radical.(b) Time evolution of the fraction of CH2O that is photolyzed orreacts with radicals.
3.2 A box model simulation of CH2O photolysis
To examine the actual photochemistry in the reactor, weconstructed a 1-box model composed of 33 photochemi-cal reactions, including photolysis of CH2O and H2O2 aswell as formation of HCOOH (see Appendix A). The modelwas run under conditions of standard ambient temperature(25◦C) and pressure (105 Pa) with the other boundary con-ditions from the results from the Tropospheric Ultravioletand Visible (TUV) radiation model (http://cprm.acd.ucar.edu/Models/TUV). As shown in Fig. 3, the TUV radiationmodel predicts that the values of8(H2) range from 0.6 to0.76 in Mainz. Since SZA at local noon during the experi-ments were between 27◦ and 48◦, daily averaged photolysis-rate-weighted mean values of8(H2) would be 0.64 to 0.66,which correspond to total CH2O photolysis rates for bothchannels (JCH2O) of 2.4×10−5 to 3.8×10−5 s−1. For thesame range of SZA, the ratio of the photolysis rates of H2O2
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008
1358 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
-500
-400
-300
-200
-100
0
100 αm = 0.55, = 0.91
J J + (HO2 + CH2O) J + (OH + CH2O) J + (OH + CH2O), αOH = 0.78 J + K, αr = 0.22, αk = 0.78
δD
-H2 (
‰)
Ψ(H2)
αr
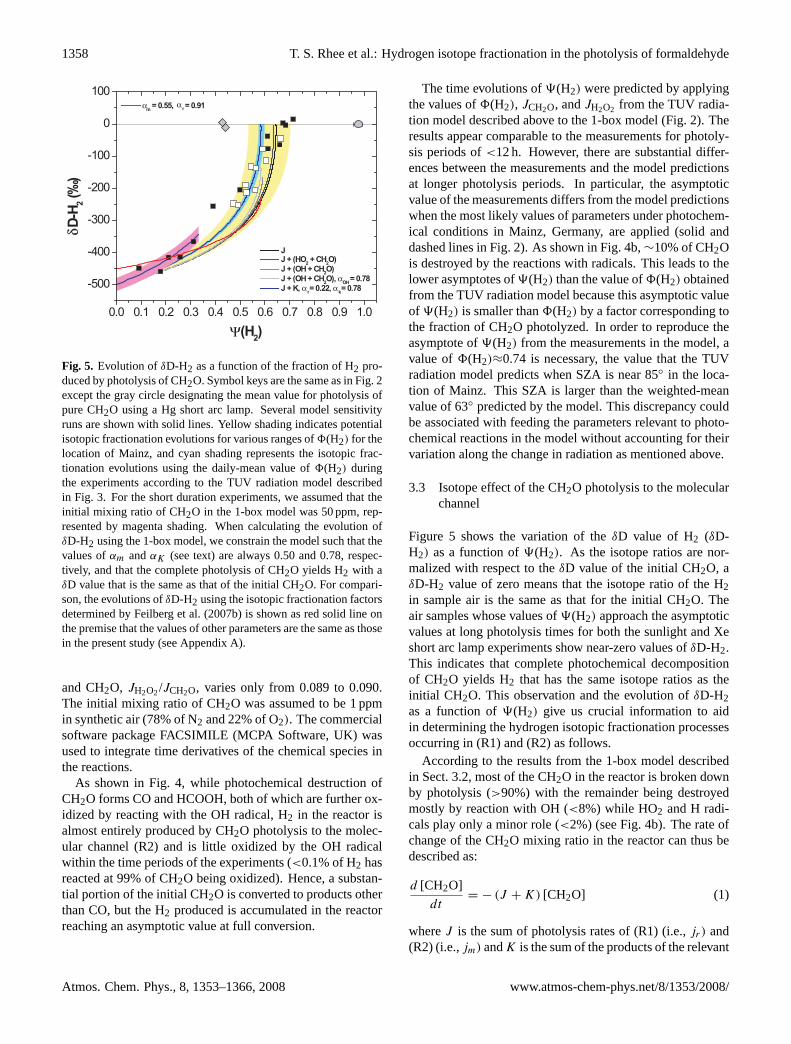
Fig. 5. Evolution ofδD-H2 as a function of the fraction of H2 pro-duced by photolysis of CH2O. Symbol keys are the same as in Fig. 2except the gray circle designating the mean value for photolysis ofpure CH2O using a Hg short arc lamp. Several model sensitivityruns are shown with solid lines. Yellow shading indicates potentialisotopic fractionation evolutions for various ranges of8(H2) for thelocation of Mainz, and cyan shading represents the isotopic frac-tionation evolutions using the daily-mean value of8(H2) duringthe experiments according to the TUV radiation model describedin Fig. 3. For the short duration experiments, we assumed that theinitial mixing ratio of CH2O in the 1-box model was 50 ppm, rep-resented by magenta shading. When calculating the evolution ofδD-H2 using the 1-box model, we constrain the model such that thevalues ofαm andαK (see text) are always 0.50 and 0.78, respec-tively, and that the complete photolysis of CH2O yields H2 with aδD value that is the same as that of the initial CH2O. For compari-son, the evolutions ofδD-H2 using the isotopic fractionation factorsdetermined by Feilberg et al. (2007b) is shown as red solid line onthe premise that the values of other parameters are the same as thosein the present study (see Appendix A).
and CH2O, JH2O2/JCH2O, varies only from 0.089 to 0.090.The initial mixing ratio of CH2O was assumed to be 1 ppmin synthetic air (78% of N2 and 22% of O2). The commercialsoftware package FACSIMILE (MCPA Software, UK) wasused to integrate time derivatives of the chemical species inthe reactions.
As shown in Fig. 4, while photochemical destruction ofCH2O forms CO and HCOOH, both of which are further ox-idized by reacting with the OH radical, H2 in the reactor isalmost entirely produced by CH2O photolysis to the molec-ular channel (R2) and is little oxidized by the OH radicalwithin the time periods of the experiments (<0.1% of H2 hasreacted at 99% of CH2O being oxidized). Hence, a substan-tial portion of the initial CH2O is converted to products otherthan CO, but the H2 produced is accumulated in the reactorreaching an asymptotic value at full conversion.
The time evolutions of9(H2) were predicted by applyingthe values of8(H2), JCH2O, andJH2O2 from the TUV radia-tion model described above to the 1-box model (Fig. 2). Theresults appear comparable to the measurements for photoly-sis periods of<12 h. However, there are substantial differ-ences between the measurements and the model predictionsat longer photolysis periods. In particular, the asymptoticvalue of the measurements differs from the model predictionswhen the most likely values of parameters under photochem-ical conditions in Mainz, Germany, are applied (solid anddashed lines in Fig. 2). As shown in Fig. 4b,∼10% of CH2Ois destroyed by the reactions with radicals. This leads to thelower asymptotes of9(H2) than the value of8(H2) obtainedfrom the TUV radiation model because this asymptotic valueof 9(H2) is smaller than8(H2) by a factor corresponding tothe fraction of CH2O photolyzed. In order to reproduce theasymptote of9(H2) from the measurements in the model, avalue of8(H2)≈0.74 is necessary, the value that the TUVradiation model predicts when SZA is near 85◦ in the loca-tion of Mainz. This SZA is larger than the weighted-meanvalue of 63◦ predicted by the model. This discrepancy couldbe associated with feeding the parameters relevant to photo-chemical reactions in the model without accounting for theirvariation along the change in radiation as mentioned above.
3.3 Isotope effect of the CH2O photolysis to the molecularchannel
Figure 5 shows the variation of theδD value of H2 (δD-H2) as a function of9(H2). As the isotope ratios are nor-malized with respect to theδD value of the initial CH2O, aδD-H2 value of zero means that the isotope ratio of the H2in sample air is the same as that for the initial CH2O. Theair samples whose values of9(H2) approach the asymptoticvalues at long photolysis times for both the sunlight and Xeshort arc lamp experiments show near-zero values ofδD-H2.This indicates that complete photochemical decompositionof CH2O yields H2 that has the same isotope ratios as theinitial CH2O. This observation and the evolution ofδD-H2as a function of9(H2) give us crucial information to aidin determining the hydrogen isotopic fractionation processesoccurring in (R1) and (R2) as follows.
According to the results from the 1-box model describedin Sect. 3.2, most of the CH2O in the reactor is broken downby photolysis (>90%) with the remainder being destroyedmostly by reaction with OH (<8%) while HO2 and H radi-cals play only a minor role (<2%) (see Fig. 4b). The rate ofchange of the CH2O mixing ratio in the reactor can thus bedescribed as:
d [CH2O]
dt= − (J + K) [CH2O] (1)
whereJ is the sum of photolysis rates of (R1) (i.e.,jr) and(R2) (i.e.,jm) andK is the sum of the products of the relevant
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/

T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1359
photochemical reaction rate coefficients (ki) and radical con-centrations (Xi) as follows.
J = jm + jr (2)
K =
∑i
ki [Xi ] (3)
In the same way, for the next abundant isotopologue,CHDO, one obtains:
d [CHDO]
dt= −
(J ′
+ K ′)
[CHDO] (4)
whereJ ′ andK ′ indicate the sums of the photolysis rates andthe photochemical reaction rates for CHDO, respectively.
In terms of non-equilibrium kinetics, the isotopic fraction-ation factor is represented as the kinetic isotope effect (orsimply isotope effect), which is expressed by the ratio of re-action rates for the different isotopologues, one of which hasa rare isotope substituted for the common one (Melander andSaunders, 1980). We define here the isotopic fractionationfactor as the ratio of photochemical reaction rates or photol-ysis rates of an isotopologue which has a single deuteriumto that for the most abundant isotopologue. For instance, theisotopic fractionation factor for the molecular channel,αm is:
αm =j ′m
jm
(5)
Hence,J ′ andK ′ in Eq. (4) have the following relationshipwith the corresponding rates for CH2O by means of isotopicfractionation factor,αi .
J ′= j ′
r + j ′m
= αrjr + αmjm (6)
K ′=
∑i
k′
i [Xi ]
=
∑i
αkiki [Xi ]
= αKK (7)
By definition, the isotopic fractionation factor for CH2O,αf , is
αf =J ′
+ K ′
J + K
= αr ×jr
J×
J
J + K+ αm ×
jm
J×
J
J + K+ αK ×
K
J + K(8a)
In Eq. (8a), the ratio ofjm to J represents the yield of H2from photolysis of CH2O (8(H2)), and the ratioJ /(J+K)
is the fraction of CH2O that is photolyzed. Designating thelatter as0, αf can be rewritten as
αf = αr (1 − 8) 0 + αm80 + αK (1 − 0) (8b)
Or simply,
αf = αhν0 + αK (1 − 0) (8c)
where αhν represents the isotopic fractionation factor forphotolysis of CH2O. Since the amount of radicals producedalong the experiments is not constant,0 is not a constant butvaries as a function of time. In addition, strictly speaking,8(H2) varied during the sunlight experiments as did SZA(Fig. 3b). Accordinglyαf is changing along with the CH2Ophotolysis and photochemical reactions. Nevertheless, as-suming thatαf is constant gives a convenient way to deter-mine the isotopic fractionation factor for the production ofH2, αm.
Integrating Eqs. (1) and (4) and then dividing [CHDO]by [CH2O] leads to the well-known Rayleigh equation(Rayleigh, 1902):
RQ
Ro
= f αf −1 (9)
whereRo is the isotope ratio of the initial CH2O,RQ is thatfor the remaining CH2O along the course of experiment, andf the fraction of the remaining CH2O. Thus, the isotope ratioof the products (Rp) as a function of CH2O photochemicaldestruction can be obtained by mass balance:
Rp
Ro
=1 − f αf
1 − f(10)
ActuallyRp is the sum of the isotope ratios of the productsformed by CH2O photolysis and its photochemical reactionswith radicals. The isotope ratio of the H2, Rm, which is pro-duced from CH2O photolysis to the molecular channel, canbe derived from the following derivatives:
d [H2]
dt= jm [CH2O] (11)
and
d [HD]
dt= j ′
m [CHDO] (12)
Solving Eqs. (11) and (12) with inserting the solutions ofEqs. (1) and (4), respectively, and the definition ofαm inEq. (5),Rm has the following relation withRo:
Rm
Ro
=αm
αf
×1 − f αf
1 − f(13)
By dividing (13) by (10), the ratio of the isotope ratios of H2(Rm) and all products from CH2O photochemistry (Rp) isthe same as the ratios of their isotopic fractionation factors:
Rm
Rp
=αm
αf
(14)
Similar experessions can be derived for the radical channelof CH2O photolysis (15) and for the photochemical reactions(16):
Rr
Rp
=αr
αf
(15)
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008
1360 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
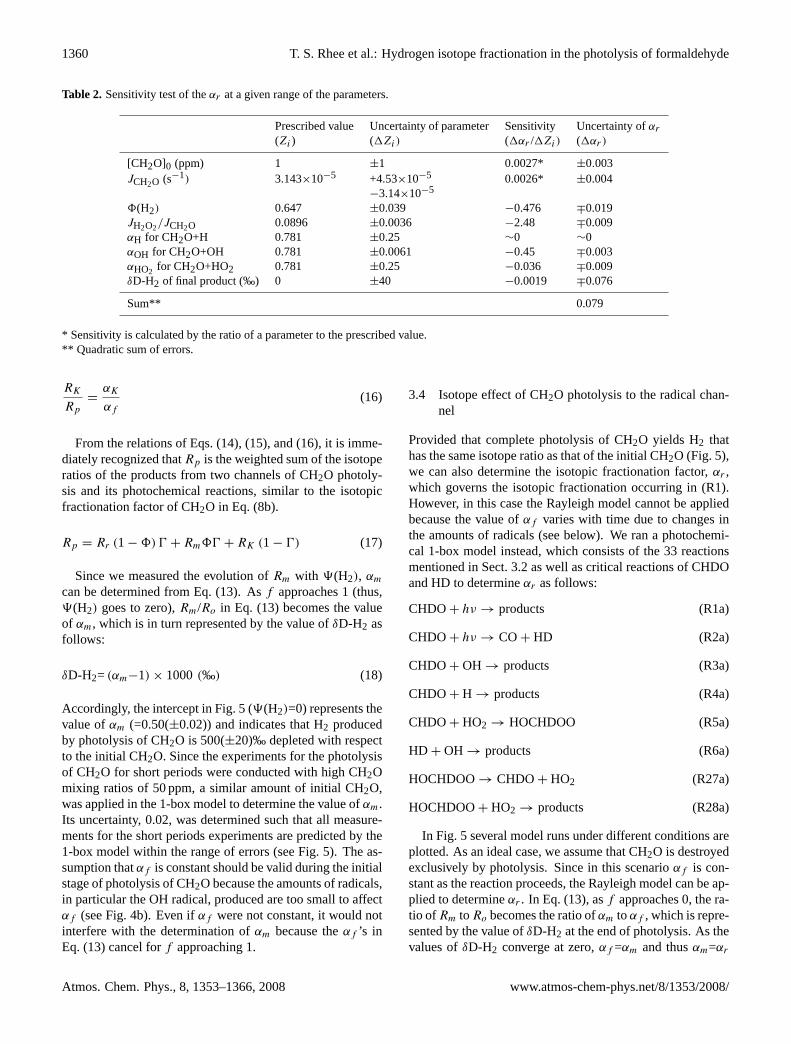
Table 2. Sensitivity test of theαr at a given range of the parameters.
Prescribed value Uncertainty of parameter Sensitivity Uncertainty ofαr
(Zi ) (1Zi) (1αr /1Zi) (1αr )
[CH2O]0 (ppm) 1 ±1 0.0027* ±0.003JCH2O (s−1) 3.143×10−5 +4.53×10−5
−3.14×10−50.0026* ±0.004
8(H2) 0.647 ±0.039 −0.476 ∓0.019JH2O2/JCH2O 0.0896 ±0.0036 −2.48 ∓0.009αH for CH2O+H 0.781 ±0.25 ∼0 ∼0αOH for CH2O+OH 0.781 ±0.0061 −0.45 ∓0.003αHO2 for CH2O+HO2 0.781 ±0.25 −0.036 ∓0.009δD-H2 of final product (‰) 0 ±40 −0.0019 ∓0.076
Sum** 0.079
* Sensitivity is calculated by the ratio of a parameter to the prescribed value.** Quadratic sum of errors.
RK
Rp
=αK
αf
(16)
From the relations of Eqs. (14), (15), and (16), it is imme-diately recognized thatRp is the weighted sum of the isotoperatios of the products from two channels of CH2O photoly-sis and its photochemical reactions, similar to the isotopicfractionation factor of CH2O in Eq. (8b).
Rp = Rr (1 − 8) 0 + Rm80 + RK (1 − 0) (17)
Since we measured the evolution ofRm with 9(H2), αm
can be determined from Eq. (13). Asf approaches 1 (thus,9(H2) goes to zero),Rm/Ro in Eq. (13) becomes the valueof αm, which is in turn represented by the value ofδD-H2 asfollows:
δD-H2= (αm−1) × 1000(‰) (18)
Accordingly, the intercept in Fig. 5 (9(H2)=0) represents thevalue ofαm (=0.50(±0.02)) and indicates that H2 producedby photolysis of CH2O is 500(±20)‰ depleted with respectto the initial CH2O. Since the experiments for the photolysisof CH2O for short periods were conducted with high CH2Omixing ratios of 50 ppm, a similar amount of initial CH2O,was applied in the 1-box model to determine the value ofαm.Its uncertainty, 0.02, was determined such that all measure-ments for the short periods experiments are predicted by the1-box model within the range of errors (see Fig. 5). The as-sumption thatαf is constant should be valid during the initialstage of photolysis of CH2O because the amounts of radicals,in particular the OH radical, produced are too small to affectαf (see Fig. 4b). Even ifαf were not constant, it would notinterfere with the determination ofαm because theαf ’s inEq. (13) cancel forf approaching 1.
3.4 Isotope effect of CH2O photolysis to the radical chan-nel
Provided that complete photolysis of CH2O yields H2 thathas the same isotope ratio as that of the initial CH2O (Fig. 5),we can also determine the isotopic fractionation factor,αr ,which governs the isotopic fractionation occurring in (R1).However, in this case the Rayleigh model cannot be appliedbecause the value ofαf varies with time due to changes inthe amounts of radicals (see below). We ran a photochemi-cal 1-box model instead, which consists of the 33 reactionsmentioned in Sect. 3.2 as well as critical reactions of CHDOand HD to determineαr as follows:
CHDO+ hν → products (R1a)
CHDO+ hν → CO+ HD (R2a)
CHDO+ OH → products (R3a)
CHDO+ H → products (R4a)
CHDO+ HO2 → HOCHDOO (R5a)
HD + OH → products (R6a)
HOCHDOO→ CHDO+ HO2 (R27a)
HOCHDOO+ HO2 → products (R28a)
In Fig. 5 several model runs under different conditions areplotted. As an ideal case, we assume that CH2O is destroyedexclusively by photolysis. Since in this scenarioαf is con-stant as the reaction proceeds, the Rayleigh model can be ap-plied to determineαr . In Eq. (13), asf approaches 0, the ra-tio of Rm toRo becomes the ratio ofαm toαf , which is repre-sented by the value ofδD-H2 at the end of photolysis. As thevalues ofδD-H2 converge at zero,αf =αm and thusαm=αr
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/

T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1361
Table 3. Comparison of the isotope effects determined from CH2O photolysis experiments.
Source Minor αm αr *αOH 8(H2) ** 0 αhν αf αm/αf
isotopologue
This study [CHDO] 0.50(±0.02) 0.22 (±0.08) 0.781(±0.006) 0.65 (±0.04) 0.69 (±0.28) 0.40 (±0.03) 0.51 (±0.11) 0.97 (±0.21)Feilberg et al. (2007b) [CHDO] 0.55(±0.02) 0.91(±0.05) 0.781(±0.006) ***0.77(±0.06) 0.69 (±0.28) 0.63(±0.01) 0.68 (±0.04) 0.81 (±0.06)Crounse et al. (2003) [CHDO] 0.8Feilberg et al. (2007a) [CD2O] 0.333(±0.056)
* Kinetic isotope effect for CH2O + OH from Feilberg et al. (2004).** The value is calculated for the Mainz conditions for the periods of experiments.*** The value was calculated by the relationαhν=αm×8(H2)+αr×(1−8(H2)).
according to the relation in Eq. (8b) since0=1. This scenariois, however, unlikely considering the substantial productionof radicals via the radical channel (R1), which may in turnreact with CH2O in the reactor as described above. Introduc-tion of the reactions of H and/or HO2 with both CH2O andCHDO with and without kinetic isotope effect do not signif-icantly change the evolution ofδD-H2 compared to the idealscenario that only accounts for CH2O photolysis. However,it is apparent that the reaction of OH and CH2O is critical fordetermination ofαr , as theδD-H2 value for the final prod-uct of H2 reaches only∼−170‰. Taking the kinetic isotopeeffect for the reaction of CH2O with OH radicals into ac-count increases theδD-H2 value for the final product a littleto ∼−130‰. Applying the kinetic isotope effect for the re-action of HD with OH does not improve the model to sim-ulate the measurements because of too slow reaction rate ofH2+OH. However, decreasing the value ofαr from 0.50 to0.22 (thus larger isotope effect) makes it possible to reachthe δD-H2 value of the final H2 to zero and significantlyimproves the predicted evolution ofδD-H2 compared to themeasurements. Therefore, provided that the TUV radiationmodel and the reaction rates applied in the 1-box model arecorrect, our best estimate ofαr is 0.22 and the total isotopicfractionation factor of CH2O due to photolysis (αhν) resultsin 0.40 for8(H2)=0.647, the yield of H2 which is the bestestimate from the TUV radiation model for the average con-ditions of Mainz at the times of the experiments (see Fig. 3).
As the value ofαr in the present study is not determineddirectly by measurement, but is based on model calculations,we conducted sensitivity runs to estimate the uncertainty ofαr by varying the values of the various parameters used inthe 1-box model. These parameters are the mixing ratio ofCH2O in the reactor,8(H2), photolysis rates of CH2O andH2O2, kinetic isotope effects for the reaction of CHDO withthe radicals, and the uncertainty ofδD-H2 for the final prod-uct (Table 2). Among themαr is the most sensitive to theratio of the photolysis rate of H2O2 to that for CH2O be-cause large production of OH by photolysis of H2O2 leadsto the increase of the fraction of CH2O that reacts with OHin the reactor, which in turn lowers the value ofαr to com-pensate for it (see Eq. 8b). The same effect can be caused bythe variation ofαOH for CH2O+OH and by8(H2). Sensitiv-ity runs for the potential error in theδD-H2 value of the final
product shows the largest impact toαr among the parametersbecause of its large potential error of 40‰, which includesthe uncertainty of theδD value of the original CH2O (=4‰).Overall most of the uncertainty forαr originates from theuncertainties in8(H2) and theδD-H2 of the final products.The quadratic sum of the errors incurred by these parametersamounts to 0.08.
4 Discussions
4.1 Comparison with previous research
To our knowledge three experiments have been done insunlight to determine the isotopic fractionation factor forformaldehyde photolysis (Table 3): One experiment investi-gated the isotopic fractionation of CH2O itself by measuringtime evolution of the amount of isotopologues, CH2O andCD2O using an optical method (Feilberg et al., 2007a; Feil-berg et al., 2005), another experiment examined the sameisotopic fractionation but for CH2O and CHDO using thesame technique and the D/H ratio of H2 produced by massspectrometry (Feilberg et al., 2007b), and the other mea-sured the D/H ratio of H2 produced from the photolysis ofCH2O which is reported in a conference proceeding abstract(Crounse et al., 2003). In the latter study a similar procedureas in the present study was apparently applied. However, thelack of details of the experiment, in particular the fractionof H2 (9(H2)) and theδD value of the original CH2O usedfor the photolysis experiments, both of which are critical todetermineαm, makes it difficult to inferαm from this sin-gle value ofδD. The authors reported that the photolysis ofCH2O produces isotopically light H2, theδD value of whichis ∼−200‰. If the authors meant the value to be the de-gree of enrichment of the H2 produced,αm is ∼0.8, whichis far larger (so less isotopically fractionated) than what weobtained in this study.
In the case of Feilberg et al. (2005)’s experiments, the ra-tio of photolysis rate of the two isotopologues,JCD2O/JCH2O,was determined as 0.333(±0.056) (Feilberg et al., 2007a)using an optical technique. This value is smaller thanthe value forJCHDO/JCH2O (=αhν) of 0.40(±0.03) deter-mined in the present study as expected from the assumption
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008

1362 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
that double-deuterated formaldehyde is more stable than thesingle-deuterated one due to zero point energy difference.
Recent work reported by the same group (Feilberg et al.,2007b) has a particular interest as the goal of the experimentis the same as the present study, but approaches it in a dif-ferent way. In this experiment, the authors determined thevalues ofαm andαhν as 0.55(±0.02) and 0.63(±0.01), re-spectively. The value ofαm is similar to, while that forαhν
is far larger than, the values determined in the present study.Actually the large discrepancy ofαhν points to a much largerdifference in the value ofαr between Feilberg et al. (2007b)and the present study: 0.91(±0.05) versus 0.22(±0.08). Un-like the previous work (Feilberg et al., 2005), Feilberg etal. (2007b) took into account the CH2O production in thechamber of the facility in determination ofαhν in additionto leakage of the experimental chamber. Notwithstanding,there is still such a large discrepancy in the isotopic frac-tionation factors of CH2O between the two studies. Besidesthe discrepancy in the magnitude ofαr , an interesting resultof Feilberg et al. (2007b) is that the degree of the isotopicfractionation in CH2O photolysis to the molecular channelis larger than that for the radical channel, being opposite tothe results from the present study and from early results byMcQuigg and Calvert (1969).
It is useful to recall the different experimental conditionsin both studies. Feilberg et al. (2007b) performed an iso-topic tracer study using similar amounts of CH2O and CHDOin the EUPHORE reactor in Valencia, Spain, which allowedthem to inferαhν directly by a spectroscopic method.αm wasthen inferred from the isotope-ratio-mass-spectrometric mea-surements of HD and modeling of the H2 yield using a givenquantum yield for CH2O photolysis. The direct determina-tion of αhν using spectroscopic measurement, however, hadto be corrected to account for the losses of CH2O and CHDOby the reaction with OH radical and large leakage of air inthe chamber as well as production of CH2O from the wall.In addition, their values ofαr andαm depend on which valueof the quantum yield for CH2O photolysis are applied. In ourstudy, performed at the level of natural deuterium abundance,αm is the “directly” inferred quantity, andαhν follows fromthe experimental results that the isotopic compositions of theinitial CH2O and of the H2 that are formed from completephotolysis are virtually the same, but it also requires a cor-rection for reaction with radicals. At present we are not ableto identify the reason of the large discrepancy in the isotopicfractionation factors of CH2O between the two studies. Moreexperiments can resolve this issue.
4.2 Atmospheric implication
The determination ofαm and αr may provide an impor-tant insight to comprehend what causes the enrichment in Dthroughout the photochemical oxidation pathway from CH4to H2. The overall composite of isotopic fractionation factors
from CH4 to H2, αCH4-H2, may be defined as:
αCH4-H2 =R0
H2
RCH4
(19)
whereR0H2
represents the isotope ratio of H2 produced byphotochemical oxidation of CH4 andRCH4 is that for CH4.Strictly speaking,αCH4-H2 differs from the general definitionof isotopic fractionation factor in that it is a function of notonly thermodynamic conditions but also environmental pa-rameters such as radiation, radical species and their concen-trations in the atmosphere. Nonetheless, given a system withthese parameters,αCH4-H2 can be considered as an isotopicfractionation factor. Rhee et al. (2006a) estimated the valueof αCH4-H2 to be 1.3 in the troposphere, meaning that the H2produced from CH4 oxidation is enriched in D by 1.3 timesas much as the initial CH4. Gerst and Quay (2001) and Priceet al. (2007) also expected D in the H2 from photochemicaloxidation of CH4 to be enriched by a factor of 1.2–1.3.
As Gerst and Quay (2001) described in detail,αCH4-H2 isthe product of several factors that are associated with pho-tochemical chain reactions from CH4 to H2. These factorsinclude: (1) isotopic fractionation occurring during the reac-tion of CH4 with OH (αCH4), the rate-determining step of thephotochemical chain reactions of CH4, as well as the sub-sequent isotopic fractionation processes occurring along theway to CH2O (α6), (2) the branching ratios in the reactionsof deuterated species, e.g., CH3D, CH2DOOH, and CH2DO,(3) the factor of 2 brought up by the reduction of the numberof hydrogen atoms from CH4 to CH2O, and finally (4) iso-topic fractionation occurring during the photolytical produc-tion of H2 from CH2O. Assuming that CH2O is in a photo-chemical steady state, as it has a far shorter chemical lifetimethan CH4 and H2, point (4) is represented by the ratio of theisotopic fractionation factor of the H2 produced (αm) to thatfor CH2O (αf ), which determines the degree of D enrich-ment of H2 (Rhee et al., 2006a). Note thatαf differs fromαhν by the effect of isotopic fractionation arising from reac-tion with OH radical (αOH) in the troposphere. Combiningall these factors yields:
αCH4-H2 = 2 × αCH4 × βCH4 × α6 × βp ×αm
αf
(20)
whereβCH4 is the branching ratio for the deuterated prod-uct, CH2D, in the reaction of CH3D and OH, andβp is acombined branching ratio for other short-lived intermediates,CH2DOOH, and CH2DO.
Regarding the right-hand side of Eq. (20), the value ofαCH4 is 0.78(±0.07) at 298 K (Gierczak et al., 1997) anddecreases with the decrease of temperature, that forβCH4
is at most unity but most likely is less than unity as Gerstand Quay (2001) speculated, and the same is expected forβp. In the subsequent reactions, there is no compelling ratio-nale that the more deuterated isotopologues react faster thanthe lighter ones considering the theoretical view of lower
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/
T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1363
zero point energy for the isotopically heavier isotopologues.Thus, the value ofα6 may not be larger than unity. Thelast two parameters in Eq. (20),αf and αm, are what weare concerned with here: sinceαf is a combined isotopicfractionation factor due to photolysis and photochemical re-actions of CH2O by the fraction of the reaction routes asshown in Eqs. (8), the value is the weighted mean of theisotopic fractionation factors involved in the reactions. Aslisted in Table 3 under the radiation conditions of Mainz, thebest values ofαm andαr were estimated as 0.50(±0.02) and0.22(±0.08), respectively, from the present study. Feilberget al (2004) determined the value ofαOH as 0.781(±0.006).The optimal values of8(H2) and0 in Mainz were calcu-lated as 0.647(±0.039) and 0.69(±0.28), respectively, forthe periods of experiments using the TUV radiation modelat a weighted mean SZA of 62.7◦ (see Fig. 3). In order todetermine0, we calculated OH radical concentrations andtheir uncertainties from the relationship between the photol-ysis rate of O3 (J (O1D)) and OH concentration by Rohrerand Berresheim (2006) (i.e., [OH]=2.4×J (O1D)+0.13 andσ=0.07×106+0.33×[OH]). By inserting these values intoEq. (8b) the resulting value forαf is 0.51(±0.11). Most ofits uncertainty is carried over from the uncertainty of OH.The resulting ratio ofαm/αf (=0.97(±0.21)) is slightly lowerthan unity, but because of its large uncertainty, arising fromthe uncertainty of the OH concentration, it is not possible topredict with certainty whether the CH2O photolysis leads toa depletion or enrichment of D in the H2 produced with re-spect to the parent CH2O. When using the values of isotopicfractionation factors determined by Feilberg et al. (2007b),the CH2O photolysis leads to the depletion of D in the H2,however, even taking into account the uncertainty ofαm/αf
(see Table 3).We extend the calculation of the ratio ofαm/αf to a range
of values of8(H2) and0, assuming that the values ofαm,αr , andαOH determined from the present study and Feilberget al. (2004) are applicable to the entire troposphere. The po-tential ranges of8(H2) for the troposphere were estimatedusing the TUV radiation model with varying SZA at the alti-tudes of the US standard atmosphere. In order to estimate0
for the troposphere, it is necessary to know the reaction rateof CH2O+OH at a given time and place. The reaction rate co-efficient varies by∼15% in the troposphere due to change intemperature, while the OH concentration varies in the orderof magnitude with its peak occurring at local noon. The peakvalues are well above 107 molecules cm−3 (e.g., Berresheimet al., 2003), leading to0 ∼0.45. Thus, the range of0 islikely to be between 0.4 and 1 in the troposphere. As shownin Fig. 6, the ratios ofαm/αf vary from∼0.8 to∼1.2, whichsuggests that, depending on the values of0 and8(H2) inthe troposphere, the H2 produced from the CH2O photolysiscould be either enriched or depleted in D. For instance, at theEarth’s surface the values ofαm/αf along the track of the sunare likely to be lower than unity, thus yielding the depletedH2 in D with respect to the parent CH2O.
0.55 0.60 0.65 0.70 0.75 0.800.4
0.5
0.6
0.7
0.8
0.9
1.0
0.55 0.60 0.65 0.70 0.75 0.800.4
0.5
0.6
0.7
0.8
0.9
1.0
1.2
1.1
1
0.9
0.8
Φ(H2)
Γ
0 10203040
50
60
8070
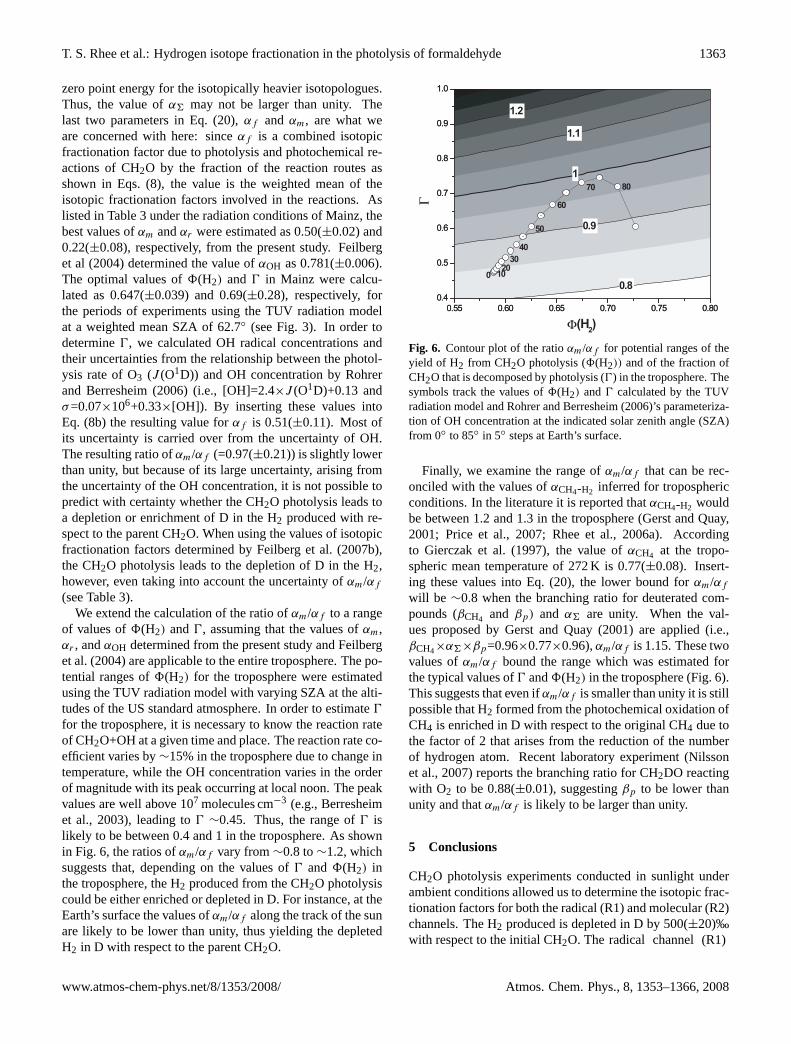
Fig. 6. Contour plot of the ratioαm/αf for potential ranges of theyield of H2 from CH2O photolysis (8(H2)) and of the fraction ofCH2O that is decomposed by photolysis (0) in the troposphere. Thesymbols track the values of8(H2) and0 calculated by the TUVradiation model and Rohrer and Berresheim (2006)’s parameteriza-tion of OH concentration at the indicated solar zenith angle (SZA)from 0◦ to 85◦ in 5◦ steps at Earth’s surface.
Finally, we examine the range ofαm/αf that can be rec-onciled with the values ofαCH4-H2 inferred for troposphericconditions. In the literature it is reported thatαCH4-H2 wouldbe between 1.2 and 1.3 in the troposphere (Gerst and Quay,2001; Price et al., 2007; Rhee et al., 2006a). Accordingto Gierczak et al. (1997), the value ofαCH4 at the tropo-spheric mean temperature of 272 K is 0.77(±0.08). Insert-ing these values into Eq. (20), the lower bound forαm/αf
will be ∼0.8 when the branching ratio for deuterated com-pounds (βCH4 and βp) and α6 are unity. When the val-ues proposed by Gerst and Quay (2001) are applied (i.e.,βCH4×α6×βp=0.96×0.77×0.96),αm/αf is 1.15. These twovalues ofαm/αf bound the range which was estimated forthe typical values of0 and8(H2) in the troposphere (Fig. 6).This suggests that even ifαm/αf is smaller than unity it is stillpossible that H2 formed from the photochemical oxidation ofCH4 is enriched in D with respect to the original CH4 due tothe factor of 2 that arises from the reduction of the numberof hydrogen atom. Recent laboratory experiment (Nilssonet al., 2007) reports the branching ratio for CH2DO reactingwith O2 to be 0.88(±0.01), suggestingβp to be lower thanunity and thatαm/αf is likely to be larger than unity.
5 Conclusions
CH2O photolysis experiments conducted in sunlight underambient conditions allowed us to determine the isotopic frac-tionation factors for both the radical (R1) and molecular (R2)channels. The H2 produced is depleted in D by 500(±20)‰with respect to the initial CH2O. The radical channel (R1)
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008

1364 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
appears to have a much stronger isotopic fractionation thanthe molecular channel (R2), resulting in D enrichment ofthe remaining CH2O by 780(±80)‰. This isotope effect issignificantly larger than the result obtained from the exper-iments in the EUPHORE reaction chamber by Feilberg etal. (2007b), a difference we do not understand at present.
Applying the isotopic fractionation factors obtained fromthe present study to the conditions of Mainz, CH2O photoly-sis may produce the H2 that is slightly depleted in D. How-ever, the large uncertainty in the combined isotope effects ofthe photochemical reactions of CH2O, which primarily orig-inates from the uncertainty of OH concentration, makes itimpossible to precisely define the role of CH2O photolysisin the D enrichment of H2. In the troposphere, CH2O pho-tolysis may produce the H2 either enriched or depleted in Dwith respect to the parent CH2O depending on the fractionof CH2O that reacts with OH or that is photolyzed to H2.Nonetheless, our estimated range ofαm/αf (∼0.8 to∼1.2)in the troposphere can be reconciled with the production ofH2 enriched in D with respect to the original CH4 by the fac-tor reported in the literature.
Appendix A
1-box photochemistry model
The 1-box model is composed of 33 reactions (Table A1)running at 25◦C and 105 Pa of air which is composed of 78%of N2 and 22% of O2. Unless otherwise mentioned, the yieldof H2 in the photolysis of CH2O and the ratio ofJH2O2/JCH2Oare assumed to be 0.647 and 0.0896, respectively, followingthe result from the TUV radiation model in Mainz.
Acknowledgements.We gratefully acknowledge support of this re-search from the Max Planck Society. We thank J. Crowley foradvice concerning photochemistry and his suggestion to conduct asimple experiment and K. Boering for useful comments to the ear-lier version. T. S. R. thanks Korea Meteorological AdministrationResearch and Development Program under Grant CATER 2007-4405 and Korea Polar Research Program under Grant PE08030 forresearch support.
Edited by: J. N. Crowley
Table A1. Photochemical reactions in the model.
No.* Reaction Rate coefficient** Notes
(R1) CH2O + hν → CHO + H 1.109E-5 1(R2) CH2O + hν → CO + H2 2.033E-5 1(R3) CH2O + OH→ CHO + H2O 8.6E-12×exp(166/RT) 2(R3’) CH2O + OH→ HCOOH + H 2.01E-13 9(R4) CH2O + H → CHO + H2 2.14E-12×exp(−9063/RT)×(T/298)1.62 8(R5) CH2O + HO2 → HOCH2OO 6.71E-15×exp(4989/RT) 3(R6) H2 + OH → H + H2O 5.5E-12×exp(−16629/RT) 3(R7) H2O2 + hν → 2OH 2.816E-6 1(R8) O2 + CHO→ CO + HO2 3.5E-12×exp(1164/RT) 3(R9) CHO + CHO→ CH2O + CO 5.0E-11 4(R9’) CHO + CHO→ (CHO)2 5.0E-11 5(R10) CHO + H→ CO + H2 1.13E-10 6(R11) CHO + OH→ CO + H2O 1.69E-10 4(R12) CHO + HO2 → product 5.0E-11 4(R13) H2O + CHO→ CH2O + OH 8.54E-13×exp(−108920/RT) 7(R14) H2O2 + CHO→ CH2O + HO2 1.69E-13×exp(−29018/RT) 7(R15) O2 + H → HO2 M×5.71E-32×(T/298)−1.6 3(R16) H + H→ H2 M×8.85E-33×(T/298)−0.6 4(R17) OH + H→ H2O M×4.38E-30×(T/298)−2.0 4(R18) (CHO)2 + OH → product 1.1E-11 2(R19) HCOOH + OH→ product 4.0E-13 3(R20) CO + OH→ CO2 + H 1.5E-13×(1+0.6×P/1013.25) 3(R21) CO + HO2 → CO2 + OH 5.96E-11×exp(−95616/RT)×(T/298)0.5 10(R22) OH + OH→ H2O2 M×6.20E-31×(T/298)−1 3(R23) HO2 + H → product 8.10E-11 3(R24) HO2 + OH → H2O + O2 4.8E-11×exp(2079/RT) 3(R25) HO2 + HO2 → H2O2 + O2 M×1.7E-33×exp(8314/RT) 3(R26) H2O2 + OH → HO2 + H2O 2.91E-12×exp(−1330/RT) 3(R27) HOCH2OO→ HO2 + CH2O 2.4E12×exp(−58201/RT) 2(R28) HOCH2OO + HO2 → HCOOH + H2O + O2 5.6E-15×exp(19123/RT) 2(R29) 2HOCH2OO→ 2HOCH2O + O2 5.5E-12 11(R29’) 2HOCH2OO→ HCOOH + CH2(OH)2 + O2 5.71E-14×exp(6236/RT) 11(R30) O2 + HOCH2O → HCOOH + HO2 3.5E-14 12
Notes: 1. TUV radiation model; 2. Atkinson et al. (1997); 3. DeMore et al. (1997); 4. Baulch et al. (1992); 5. Stoeckel et al. (1985); 6. Ziemer et al. (1998); 7. Tang and Hampson(1986); 8. Baulch et al. (1994); 9. Yetter et al. (1989); 10. Volman (1996); 11. Atkinson et al. (1992); 12. Veyret et al. (1982)* Prime (’) designates the second reaction.** R andT in rate constant designate gas constant and absolute temperature, respectively.M indicates air concentration in termolecular reaction. The units of the rate coefficientsfor first-, second-, and third-order reactions are s−1, cm3 molecule−1 s−1, and cm6 molecule−2 s−1, respectively.
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/

T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde 1365
References
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson Jr., R. F., Kerr, J.A., Rossi, M. J., and Troe, J.: Evaluated kinetic, photochemicaland heterogeneous data for atmospheric chemistry: supplementV, IUPAC subcommittee on gas kinetic data evaluation for at-mospheric chemistry, J. Phys. Chem. Ref. Data, 26, 521–1011,1997.
Atkinson, R., Baulch, D. L., Cox, R. A., Hampsonm Jr., R. F., Kerr,J. A., and Troe, J.: Evaluated kinetic and photochemical datafor atmospheric chemistry: Supplement IV. IUPAC subcommit-tee on gas kinetic data evaluation for atmospheric chemistry, J.Phys. Chem. Ref. Data, 21(6), 1125–1568, 1992.
Baulch, D. L., Cobos, C. J., Cox, R. A., Esser, C., Frank, P., Just, T.,Kerr, J. A., Pilling, M. J., Troe, J., Walker, R. W., and Warnatz, J.:Evaluated kinetic data for combustion modelling, J. Phys. Chem.Ref. Data, 21(3), 411–734, 1992.
Baulch, D. L., Cobos, C. J., Cox, R. A., Frank, P., Hayman, G., Just,T., Kerr, J. A., Murrells, T., Pilling, M. J., Troe, J., Walker, R.W., and Warnatz, J.: Evaluated kinetic data for combustion mod-elling. Supplement I., J. Phys. Chem. Ref. Data, 23, 847–1033,1994.
Berresheim, H., Plass-Dulmer, C., Elste, T., Mihalopoulos, N., andRohrer, F.: OH in the coastal boundary layer of Crete duringMINOS: Measurements and relationship with ozone photolysis,Atmos. Chem. Phys., 3, 639–649, 2003,http://www.atmos-chem-phys.net/3/639/2003/.
Burrows, J. P., Moortgat, G. K., Tyndall, G. S., Cox, R. A., Jenkin,M. E., Hayman, G. D., and Veyret, B.: Kinetics and mechanismof the photooxidation of formaldehyde. 2. Molecular modulationstudies, J. Phys. Chem., 93(6), 2375–2382, 1989.
Calvert, J. G.: The homogeneous chemistry of formaldehyde gen-eration and destruction within the atmosphere, in: Proceedingsof the NATO advanced study institute on Atmospheric Ozone:Its variation and human influences, edited by: Aikin, A. C. andNicholet, M., 153–190, 1980.
Carlier, P., Hannachi, H., and Mouvier, G.: The Chemistry ofCarbonyl-Compounds in the Atmosphere – a Review, Atmos.Environ., 20(11), 2079–2099, 1986.
Crounse, J. D., Rahn, T., Wennberg, P. O., and Eiler, J.: Isotopiccomposition of molecular hydrogen formed from the photolysisof formaldehyde in sunlight, EOS Transactions, 84(36), F178,2003.
DeMore, W. B., Sander, S. P., Golden, D. M., Hampson, R. F.,Kurylo, M. J., Howard, C. J., Ravishankara, A. R., Kolb, C. E.,and Molina, M. J.: Chemical kinetics and photochemical datafor use in stratospheric modeling, Jet Propulsion Laboratory, JPLPublication 94-4, Pasadena, California, 269 pp., 1997.
Feilberg, K. L., D’Anna, B., Johnson, M. S., and Nielsen, C.: Ad-ditions and Corrections: Relative tropospheric photolysis ratesof HCHO, H13CHO, HCH18O, and DCDO measured at the Eu-ropean Photoreactor facility, J. Phys. Chem. A, 111(5), p. 992,2007a.
Feilberg, K. L., D’Anna, B., Johnson, M. S., and Nielsen, C.J.: Relative tropospheric photolysis rates of HCHO, H13CHO,HCH18O, and DCDO measured at the European Photoreactorfacility, J. Phys. Chem., 109(37), 8314–8319, 2005.
Feilberg, K. L., Johnson, M. S., Bacak, A., Rockmann, T., andNielsen, C. J.: Relative tropospheric photolysis rates of HCHOand HCDO measured at the European Phtotoreactor facility, J.
Phys. Chem., 111(37), 9034–9046, 2007b.Feilberg, K. L., Johnson, M. S., and Nielsen, C. J.: Relative re-
action rates of HCHO, HCDO, DCDO, H13CHO, and HCH18Owith OH, Cl, Br, and NO3 radicals, J. Phys. Chem., 108(36),7393–7398, 2004.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chemistry of the upper andlower atmoshere, Academic Press, San Diego, 1999.
Frost, G. J., Fried, A., Lee, Y.-N., Wert, B., Henry, B., Drummond,J. R., Evans, M. J., Fehsenfeld, F. C., Goldan, P. D., Holloway,J. S., Hubler, G., Jakoubek, R., Jobson, B. T., Knapp, K., Kuster,W. C., Roberts, J., Rudolph, J., Ryerson, T. B., Stohl, A., Stroud,C., Sueper, D. T., Trainer, M., and Williams, J.: Comparisons ofbox model calculations and measurements of formaldehyde fromthe 1997 North Atlantic Regional Experiment, J. Geophys. Res.,107(D8), doi:10.1029/2001JD000896, 2002.
Garcia, A. R., Volkamer, R., Molina, L. T., Molina, M. J., Samuel-son, J., Mellqvist, J., Galle, B., Herndon, S. C., and Kolb, C. E.:Separation of emitted and photochemical formaldehyde in Mex-ico City using a statistical analysis and a new pair of gas-phasetracers, Atmos. Chem. Phys., 6, 4545–4557, 2006,http://www.atmos-chem-phys.net/6/4545/2006/.
Gerst, S. and Quay, P.: Deuterium component of the global molec-ular hydrogen cycle, J. Geophys. Res., 106(D5), 5021–5031,2001.
Gierczak, T., Talukdar, R. K., Herndon, S. C., Vaghjiani, G. L., andRavishankara, A. R.: Rate coefficients for the reactions of hy-droxyl radicals with methane and deuterated methanes, J. Phys.Chem., 101(17), 3125–3134, 1997.
Horowitz, A. and Calvert, J. C.: The quantum efficiency of the pri-mary processes in formaldehyde photolysis at 3130A and 25◦C,Int. J. Chem. Kinet., 10, 713–732, 1978.
Martin, R. V., Parrish, D. D., Ryerson, T. B., Nicks Jr., D. K.,Chance, K., Kurosu, T. P., Jacob, D. J., Sturges, E. D., Fried, A.,and Wert, B. P.: Evaluation of GOME satellite measurements oftropospheric NO2 and HCHO using regional data from aircraftcampaigns in the southeastern United States, J. Geophys. Res.,109, D24307, doi:10.1029/2004JD004869, 2004.
McQuigg, RD., and Calvert, JG.: The photodecomposition ofCH2O, CD2O, CHDO, and CH2O-CD2O mixtures at xenonflash lamp intensities, J. Am. Chem. Soc., 91(7), 1590–1599,1969.
Melander, L. and Saunders Jr., W. H.: Reaction rates of isotopicmolecules, John Wiley & Sons, New York, 391 pp., 1980.
Meller, R. and Moortgat, G. K.: Temperature dependence of the ab-sorption cross sections of formaldehyde between 223 and 323 Kin the wavelength range 225–375 nm, J. Geophys. Res., 105(D6),7089–7101, 2000.
Moore, C. B. and Weisshaar, J. C.: Formaldehyde photochemistry,Annu. Rev. Phys. Chem., 34, 525–555, 1983.
Moortgat, G. K., Seiler, W., and Warneck, P.: Photodissociation ofHCHO in air : CO and H2 quantum yields at 220 and 300K, J.Chem. Phys., 78(3), 1185–1190, 1983.
Nilsson, E. J. K., Johnson, M. S., Taketani, F., Matsumi, Y., Hurley,M. D., and Wallington, T. J.: Atmospheric deuterium fraction-ation: HCHO and HCDO yields in the CH2DO + O2 reaction,Atmos. Chem. Phys., 7, 5873–5881, 2007,http://www.atmos-chem-phys.net/7/5873/2007/.
Novelli, P. C., Lang, P. M., Masarie, K. A., Hurst, D. F., Myers,R., and Elkins, J. W.: Molecular hydrogen in the troposphere:
www.atmos-chem-phys.net/8/1353/2008/ Atmos. Chem. Phys., 8, 1353–1366, 2008

1366 T. S. Rhee et al.: Hydrogen isotope fractionation in the photolysis of formaldehyde
Global distribution and budget, J. Geophys. Res., 104(D23),30 427–30 444, 1999.
Price, H., Jaegle, L., Rice, A., Quay, P., Novelli, P. C., andGammon, R.: Global budget of molecular hydrogen and itsdeuterium content: Constraints from ground station, cruise,and aircraft observations, J. Geophys. Res., 112, D22108,doi:10.1029/2006JD008152, 2007.
Rahn, T., Eiler, J. M., Boering, K. A., Wennberg, P. O., McCarthy,M. C., Tyler, S., Schauffler, S., Donnelly, S., and Atlas, E.: Ex-treme deuterium enrichment in stratospheric hydrogen and theglobal atmospheric budget of H2, Nature, 424, 918–921, 2003.
Rayleigh, L.: On the distillation of binary mixtures, Philos. Mag.,4(23), 521–537, 1902.
Rhee, T. S., Brenninkmeijer, C. A. M., Braß, M., and Bruhl, C.: Theisotopic composition of H2 from CH4 oxidation in the strato-sphere and the troposphere, J. Geophys. Res., 111, D23303,doi:10.1029/2005JD6760, 2006a.
Rhee, T. S., Brenninkmeijer, C. A. M., and Rockmann, T.: Theoverwhelming role of soils in the global atmospheric hydrogencycle, Atmos. Chem. Phys., 6, 1611–1625, 2006b.
Rhee, T. S., Mak, J., Rockmann, T., and Brenninkmeijer, C. A. M.:Continuous-flow isotope analysis of the deuterium/hydrogen ra-tio in atmospheric hydrogen, Rapid Commun. Mass Spectrom.,18(3), 299–306, 2004.
Rockmann, T., Rhee, T. S., and Engel, A.: Heavy hydrogen in thestratosphere, Atmos. Chem. Phys., 3, 2015–2023, 2003,http://www.atmos-chem-phys.net/3/2015/2003/.
Rohrer, F. and Berresheim, H.: Strong correlation between levelsof tropospheric hydroxyl radicals and solar ultraviolet radiation,Nature, 442, 184–187, 2006.
Sander, S. P., Friedl, R. R., Ravishankara, A. R., Golden, D. M.,Kolb, C. E., Kurylo, M. J., Molina, M. J., Moortgat, G. K.,Keller-Rudek, H., Finlayson-Pitts, B. J., Wine, P. H., Huie, R. E.,and Orkin, V. L.: Chemical Kinetics and Photochemical Data forUse in Atmospheric Studies: Evaluation number 15, Jep Propul-sion Laboratory, JPL Publication 06-2, Pasadena, CA, 2006.
Spence, R. and Wild, W.: The preparation of liquid monomericformaldehyde, J. Chem. Soc., 338–340, 1935.
Stoeckel, F., Schuh, M. D., Goldstein, N., and Atkinson, G. H.:Time-resolved intracavity laser spectroscopy: 266 nm photodis-sociation of acetaldehyde vapor to form HCO, Chem. Phys., 95,135–144, 1985.
Su, F., Calvert, J. G., and Shaw, J. H.: Mechanism of the photoox-idation of gaseous formaldehyde, J. Phys. Chem., 83(25), 3185–3191, 1979.
Tang, W. and Hampson, R. F.: Chemical kinetic data base for com-
bustion chemistry. Part I. Methane and related compounds, J.Phys. Chem. Ref. Data, 15, 1087–1279, 1986.
Townsend, D., Lahankar, S. A., Lee, S. K., Chambreau, S. D., Suits,A. G., Zhang, X., Rheinecker, J., Harding, L. B., and Bowman,J. M.: The roaming atom: Straying from the reaction path informaldehyde decomposition, Science, 306(5699), 1158–1161,2004.
Troe, J.: Analysis of quantum yields for the photolysis of formalde-hyde at lambda>310 nm, J. Phys. Chem. A, 111(19), 3868–3874, 2007.
Veyret, B., Lesclaux, R., Rayez, M. T., Rayez, J. C., Cox, R. A.,and Moortgat, G. K.: Kinetics and mechanism of the photooxida-tion of formaldehyde. 1. Flash photolysis study, J. Phys. Chem.,93(6), 2368–2374, 1989.
Veyret, B., Rayez, J. C., and Lesclaux, R.: Mechanism of thephotooxidation of formaldehyde studied by flash photolysis ofCH2O-O2-NO mixtures, J. Phys. Chem., 86, 3424–3430, 1982.
Volman, D. H.: Photochemistry of the gaseous hydrogen peroxide-carbon monoxide system IV. Survey of the rate constant and reac-tion profile for the HO2+CO→CO2+OH reaction, J. Photochem.Photobiol., A, 100(1), 1–3, 1996.
Wagner, V., von Glasow, R., Fischer, H., and Crutzen, P. J.:Are CH2O measurements in the marine boundary layer suit-able for testing the current understanding of CH4 photooxi-dation? : A model study, J. Geophys. Res., 107(D3), 4029,doi:10.1029/2001JD000722, 2002.
Warneck, P.: Chemistry of the natural atmosphere, Academic Press,San Diego, 927 pp., 1999.
Weller, R., Schrems, O., Boddenberg, A., Gab, S., and Gautrois,M.: Meridional distribution of hydroperoxides and formalde-hyde in the marine boundary layer of the Atlantic (48◦ N–35◦ S)measured during the Albatross campaign, J. Geophys. Res.,105(D11), 14 401–14 412, 2000.
Wittrock, F., Richter, A., Oetjen, H., Burrows, J. P., Kanakidou,M., Myriokefalitakis, S., Volkamer, R., Beirle, S., Platt, U., andWagner, T.: Simultaneous global observations of glyoxal andformaldehyde from space, Geophys. Res. Lett., 33(16), L16804,doi:10.1029/2006GL026310, 2006.
Yetter, R. A., Rabitz, H., Dryer, F. L., Maki, R. G., and Klemm, R.B.: Evaluation of the rate constant for the reaction OH+H2CO:Application of modeling and sensitivity analysis techniques fordetermination of the product branching ratio, J. Chem. Phys.,91(7), 4088–4097, 1989.
Ziemer, H., Dobe, S., Wagner, H.G., Olzmann, M., Viskolcz, B.,and Temp, F.: Kinetics of the reactions of HCO with H and Datoms, Ber. Bunsen-Ges. Phys. Chem., 102, 897–905, 1998.
Atmos. Chem. Phys., 8, 1353–1366, 2008 www.atmos-chem-phys.net/8/1353/2008/

![Impacts of aerosols and clouds on photolysis frequencies and ... of aerosols and cloud… · [2] Photolysis reactions play a very important role in atmospheric chemistry. Ozone photolysis](https://img.pdfslide.us/doc/110x75/5f07e35b7e708231d41f41d6/impacts-of-aerosols-and-clouds-on-photolysis-frequencies-and-of-aerosols-and.jpg)



























