Embed Size (px)
Citation preview

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
TOX IC I TY SCREEN ING
1StanfordCardiovascular Institute, StanfordUniversity School ofMedicine, Stanford, CA94305, USA. 2Institute for Stem Cell Biology and Regenerative Medicine, Stanford Uni-versity School of Medicine, Stanford, CA 94305, USA. 3Division of Cardiology, Depart-ment of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA.4Department of Pharmacology and Center for Pharmacogenomics, Northwestern Uni-versity School of Medicine, Chicago, IL 60611, USA. 5Sanford Burnham Prebys MedicalDiscovery Institute, La Jolla, CA 92037, USA. 6Graduate School of Biomedical Sciences,Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA 92037, USA. 7Depart-ment of Mechanical and Aerospace Engineering, University of California, San Diego, LaJolla, CA 92092, USA. 8Division of Oncology, Department of Medicine, Stanford Univer-sity School of Medicine, Stanford, CA 94305, USA. 9Division of Cardiovascular Medicine,Cardio-Oncology Program, Vanderbilt-Ingram Cancer Center, Vanderbilt UniversityMedical Center, Nashville, TN 37212, USA.*These authors contributed equally to this work.†Corresponding author. Email: [email protected]
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
2017 © The Authors,
some rights reserved;
exclusive licensee
American Association
for the Advancement
of Science.
http://stm.sc
Dow
nloaded from
High-throughput screening of tyrosine kinase inhibitorcardiotoxicity with human induced pluripotent stem cellsArun Sharma,1,2,3* Paul W. Burridge,1,2,4* Wesley L. McKeithan,1,5,6 Ricardo Serrano,7
Praveen Shukla,1,2,3 Nazish Sayed,1,2,3 Jared M. Churko,1,2,3 Tomoya Kitani,1,2,3 Haodi Wu,1,2,3
Alexandra Holmström,1,2,3 Elena Matsa,1,2,3 Yuan Zhang,1,2,3 Anusha Kumar,1,2,3 Alice C. Fan,8
Juan C. del Álamo,7 Sean M. Wu,1,2,3 Javid J. Moslehi,9 Mark Mercola,1,3,5 Joseph C. Wu1,2,3†
Tyrosine kinase inhibitors (TKIs), despite their efficacy as anticancer therapeutics, are associated with cardiovascularside effects ranging from induced arrhythmias to heart failure. We used human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs), generated from 11 healthy individuals and 2 patients receiving cancer treat-ment, to screen U.S. Food and Drug Administration–approved TKIs for cardiotoxicities by measuring alterations incardiomyocyte viability, contractility, electrophysiology, calcium handling, and signaling. With these data, we gener-ated a “cardiac safety index” to reflect the cardiotoxicities of existing TKIs. TKIs with low cardiac safety indices exhibitcardiotoxicity in patients. We also derived endothelial cells (hiPSC-ECs) and cardiac fibroblasts (hiPSC-CFs) to ex-amine cell type–specific cardiotoxicities. Using high-throughput screening, we determined that vascular endo-thelial growth factor receptor 2 (VEGFR2)/platelet-derived growth factor receptor (PDGFR)–inhibiting TKIscaused cardiotoxicity in hiPSC-CMs, hiPSC-ECs, and hiPSC-CFs. With phosphoprotein analysis, we determined thatVEGFR2/PDGFR-inhibiting TKIs led to a compensatory increase in cardioprotective insulin and insulin-like growthfactor (IGF) signaling in hiPSC-CMs. Up-regulating cardioprotective signaling with exogenous insulin or IGF1 im-proved hiPSC-CM viability during cotreatment with cardiotoxic VEGFR2/PDGFR-inhibiting TKIs. Thus, hiPSC-CMscan be used to screen for cardiovascular toxicities associated with anticancer TKIs, and the results correlate withclinical phenotypes. This approach provides unexpected insights, as illustrated by our finding that toxicity can bealleviated via cardioprotective insulin/IGF signaling.
ien
by guest on August 10, 2019cem
ag.org/
INTRODUCTIONSmall-molecule tyrosine kinase inhibitors (TKIs) have markedly im-proved life expectancy for cancer patients (1). Since the U.S. Food andDrug Administration (FDA) approval of imatinib for treating chronicmyeloid leukemia, dozens of TKIs have been developed. TKIs inhibitthe phosphorylation activity of hyperactive receptor tyrosine kinases (RTKs)in cancer cells, stymieing enhanced cell survival, proliferation, and migrationphenotypes associated with cancer progression. However, some TKIsare linked to severe cardiotoxicities including heart failure, reduced leftventricular ejection fraction, myocardial infarction, or arrhythmias (2, 3).Given these life-threatening complications, new approaches are neededto assess the cardiotoxicity of anticancer drugs.
Preclinical platforms for evaluating drug cardiotoxicity use animalmodels, which inaccurately predict human cardiac pathophysiology be-cause of interspecies differences in cardiac structure, electrophysiology,and genetics (4). In vitro drug cardiotoxicity assessments also use non-human cells transfected with the human ether-à-go-go–related gene(hERG), which encodes a cardiac potassium channel, to evaluate
drug-induced alterations in cardiac electrophysiology (5). Primary hu-man cardiomyocytes, which are ideal for assessing drug cardiotoxici-ties, are difficult to procure and maintain (6). Because primary humancardiomyocytes are terminally differentiated, it is impossible to obtainsufficient quantities for cardiotoxicity screening. Human induced plu-ripotent stem cells (hiPSCs), however, provide an alternative (7). Humancardiomyocytes can be mass-produced from hiPSCs with chemicallydefined differentiation (8). Patient-specific hiPSC-derived cardiomyocytes(hiPSC-CMs) recapitulate cardiovascular disease phenotypes for dilatedcardiomyopathy, hypertrophic cardiomyopathy, left ventricular noncom-paction, long QT syndrome, viral cardiomyopathy, and others (9–14).
Here, we used patient-specific hiPSC-CMs, hiPSC-derived endothe-lial cells (hiPSC-ECs), and hiPSC-derived cardiac fibroblasts (hiPSC-CFs) from 11 healthy individuals and 2 cancer patients receiving TKIsto evaluate the cardiotoxicities of 21 FDA-approved TKIs. We alsoused cytotoxicity and high-throughput cell contractility assessmentsto establish a TKI “cardiac safety index.”
RESULTSExpression of cardiomyocyte markers and RTKsin hiPSC-CMsEleven hiPSC lines were produced from the somatic tissues of 11healthy individuals by cellular reprogramming with lentivirus or Sendaivirus–based vectors expressing the transcription factors OCT4, SOX2,KLF4, and MYC. These individuals were a diverse group of males andfemales of various ages. Two additional hiPSC lines were created fromtwo individuals receiving TKIs for cancer treatment (fig. S1A). AllhiPSC lines expressed pluripotency markers (fig. S1B). hiPSC-CMswere produced with a chemically defined differentiation protocol(Fig. 1A). The hiPSC-CMs expressed standard cardiomyocyte markers
1 of 13
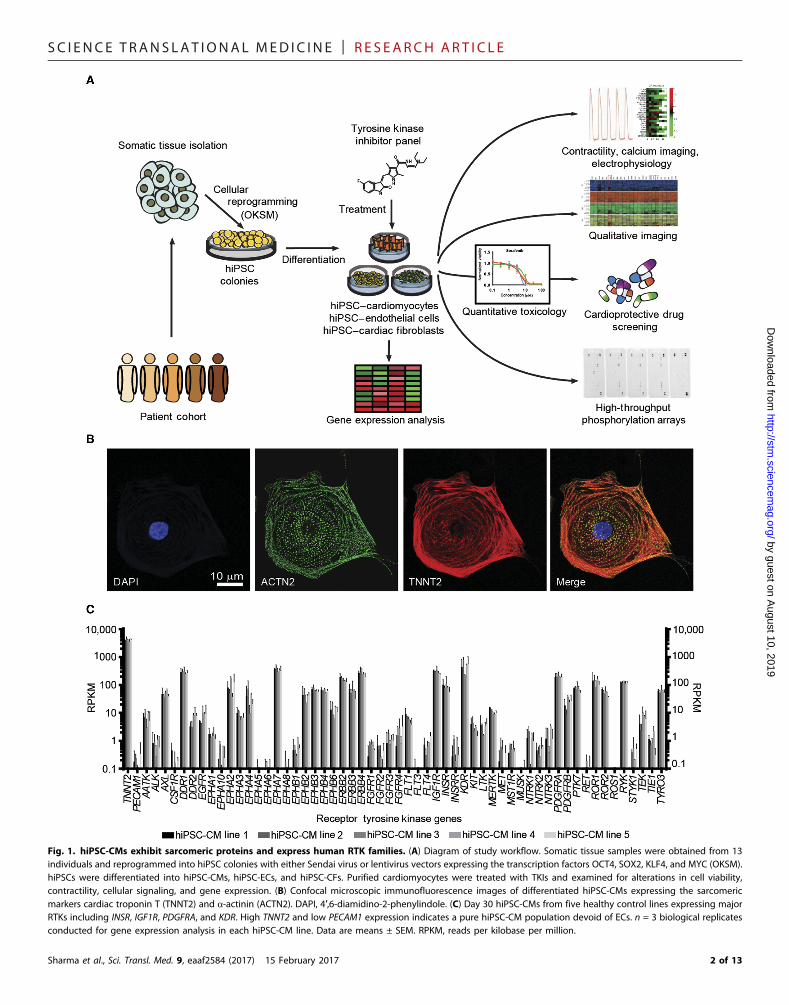
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 1. hiPSC-CMs exhibit sarcomeric proteins and express human RTK families. (A) Diagram of study workflow. Somatic tissue samples were obtained from 13individuals and reprogrammed into hiPSC colonies with either Sendai virus or lentivirus vectors expressing the transcription factors OCT4, SOX2, KLF4, and MYC (OKSM).hiPSCs were differentiated into hiPSC-CMs, hiPSC-ECs, and hiPSC-CFs. Purified cardiomyocytes were treated with TKIs and examined for alterations in cell viability,contractility, cellular signaling, and gene expression. (B) Confocal microscopic immunofluorescence images of differentiated hiPSC-CMs expressing the sarcomericmarkers cardiac troponin T (TNNT2) and a-actinin (ACTN2). DAPI, 4′,6-diamidino-2-phenylindole. (C) Day 30 hiPSC-CMs from five healthy control lines expressing majorRTKs including INSR, IGF1R, PDGFRA, and KDR. High TNNT2 and low PECAM1 expression indicates a pure hiPSC-CM population devoid of ECs. n = 3 biological replicatesconducted for gene expression analysis in each hiPSC-CM line. Data are means ± SEM. RPKM, reads per kilobase per million.
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017 2 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
(Fig. 1B) (8). Cardiomyocytes exhibited spontaneous beating and werepurified for downstream assays (movie S1). Five healthy controlhiPSC-CM lines were chosen for RTK expression analysis; all exhibitednear-identical RTK expression (Fig. 1C). KDR encoding vascular endo-thelial growth factor receptor 2 (VEGFR2), PDGFRA encoding platelet-derived growth factor receptor a (PDGFRa), INSR encoding insulinreceptor, and IGF1R encoding insulin-like growth factor 1 (IGF1) re-ceptor were highly expressed.
High-throughput analysis of TKI-induced cytotoxicity andcontractility in hiPSC-CMsTwenty-one small-molecule TKIs were used for a high-throughputcardiotoxicity screen in hiPSC-CMs (table S1). Many TKIs inhibitmultiple RTK families and induce cardiotoxicities including left ven-tricular dysfunction, myocardial infarction, and arrhythmias. Howev-er, the net benefit with respect to cancer treatment outweighs theserisks, and these drugs are frequently prescribed at major cancer treat-ment centers (table S2). We included the highly cardiotoxic anthra-cycline doxorubicin as a positive control for toxicity. Using thePrestoBlue cell viability assay, we found that the VEGFR2/PDGFR-inhibiting TKIs sorafenib, regorafenib, and ponatinib induced themost cell death in hiPSC-CMs, with median lethal dose (LD50) valuesof 3.4, 7.1, and 4.3 mM, respectively (Fig. 2A). Doxorubicin was ex-tremely cytotoxic to hiPSC-CMs at an LD50 of 0.78 mM. TKIs notstrongly associated with cytotoxicity, such as imatinib or erlotinib,had LD50 values of 78.20 and 87.60 mM, respectively. Sorafenib, regor-afenib, and ponatinib were highly cytotoxic in all 11 healthy controlhiPSC-CM lines, as measured with quantitative and qualitative viabil-ity assays (figs. S2, A to C, and S3). We also performed cytotoxicityassays in hiPSC-CMs and hiPSC-ECs derived from two individualswith kidney cancer (fig. S4). These individuals received two TKIs each:sunitinib as first-line treatment and axitinib as second-line treatment.These patients experienced no significant clinical cardiotoxicity fromeither agent. As expected, we did not observe a significant difference incytotoxicity between TKI-receiving patient hiPSC-CMs and healthycontrol hiPSC-CM lines after subjecting them to sunitinib or axitinib.
To avoid lab-to-lab biases and variations in hiPSC-CM quality, weperformed contractility assessment in CMs derived from commercial-ly available, healthy control hiPSCs. We observed alterations inhiPSC-CM beating rate and other parameters at doses lower thanthe LD50 cytotoxicity values after treatment with multiple TKIs suchas nilotinib and vandetanib, suggesting that irregular beating arisesbefore cardiomyocyte death (Fig. 2B and fig. S5). We also determinedhiPSC-CM contractility parameters in response to increasing TKIconcentrations, effective drug concentrations at which contractility al-terations initially appeared, and TKI concentrations at which hiPSC-CM contraction ceased (Fig. 2C and figs. S6 and S7). To accuratelyassess TKI toxicity, we investigated whether toxic TKI concentrationsobserved in cytotoxicity and contractility assays matched dosesexperienced by patients. We obtained patient Cmax values from FDAliterature, providing an estimate of maximum TKI blood plasma con-centrations in patients (Fig. 2C). By normalizing our in vitro data oncessation of beating, effective concentration, and LD50 cytotoxicityvalues to literature-reported Cmax values, we developed a cardiac safetyindex, a metric that identifies clinically cardiotoxic TKIs (see Materialsand Methods, Fig. 2C, and fig. S7 for details).
Three of seven compounds with cardiac safety indices at or be-low 0.10 (doxorubicin, nilotinib, and vandetanib) were previouslylabeled with FDA black box cardiotoxicity warnings. A safety index
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
value of 0.10 was chosen as our threshold for highly cardiotoxic drugsbecause it marked a separation in the safety index between clinicallycardiotoxic, black boxed drugs (doxorubicin, nilotinib, and vandetanib)and other compounds not commonly associated with cardiotoxicity.Nilotinib and vandetanib, which cause QT interval prolongation andarrhythmias, were selected for further analysis. Three of the TKIs withsafety indices under 0.10 were VEGFR2/PDGFR-inhibiting TKIs (re-gorafenib, sorafenib, and vandetanib). Regorafenib and sorafenib hadsafety indices comparable to that of the cardiotoxic anthracycline dox-orubicin. Thus, VEGFR2/PDGFR-inhibiting TKIs induced cardiotoxi-cities in hiPSC-CMs at clinically relevant concentrations comparableto the doses that patients experience. In patients, VEGFR2/PDGFR-inhibiting TKIs cause various toxicities including hypertension, heartfailure, and QT interval prolongation (15).
Confirmation of toxicity for the known QT interval–prolonging TKIs nilotinib and vandetanib in hiPSC-CMsQT interval prolongation remains a major concern during drug devel-opment (16). Because nilotinib and vandetanib cause dangerous QTinterval prolongation and arrhythmias clinically, we conducted addi-tional contractility, calcium imaging, and electrophysiological analysesin four healthy control hiPSC-CM lines treated with nilotinib or van-detanib (fig. S8). We selected dimethyl sulfoxide (DMSO) and axitinib,which are not associated with contractility abnormalities at clinicallyrelevant doses prescribed to patients (per clinical literature and ourprevious data), as negative controls for toxicity. We observed a pro-longation in cardiomyocyte contraction time after nilotinib or vande-tanib treatment at clinically relevant concentrations as low as 3.7 mM(fig. S8). Neither DMSO nor axitinib elicited contraction irregularitiesat clinically relevant concentrations.
We also conducted calcium imaging of hiPSC-CMs after a 2-hournilotinib or vandetanib treatment (Fig. 3A). At clinically relevant con-centrations (Fig. 2C and table S1), nilotinib and vandetanib prolonged cal-cium transient duration and decreased beat rate in hiPSC-CMs (Fig. 3, Band C). The electrophysiologically “safe” drugs (DMSO control, imatinib,and axitinib) did not significantly alter calcium transient amplitude, tran-sient duration, or beating rate (Fig. 3C); nilotinib and vandetanib alteredcardiomyocyte electrophysiology (Fig. 4). We subjected hiPSC-CMs to anacute TKI treatment up to 10min or to a longer treatment for 2 hours andrecorded cellular electrophysiologywith patch clamping (Fig. 4A).Withacute treatment, TKIs at clinically relevant concentrations did not alteraction potential (AP) duration (Fig. 4B). However, after 2 hours, theQT interval–prolonging TKIs nilotinib and vandetanib significantly pro-longed AP duration and decreased cellular beating rate (Fig. 4, C and D).Drugs not known to alter cardiac electrophysiology, such as DMSO,imatinib, and axitinib, did not alter AP duration. These results demon-strate that detrimental arrhythmogenic effects of known QT interval–prolonging TKIs such as nilotinib and vandetanib can be recapitulatedin hiPSC-CMs, as assessed by contractility assays, calcium imaging, andpatch clamp electrophysiology.
Analysis of TKI cardiotoxicity in hiPSC-ECs and hiPSC-CFsWe next derived endothelial cells (hiPSC-ECs) and cardiac fibroblasts(hiPSC-CFs) from hiPSCs to determine cell type–specific differencesin cardiotoxicity. hiPSC-ECs were produced with a chemically defineddifferentiation protocol using small-molecule Wnt signaling modula-tors, fibroblast growth factor (FGF) and VEGF stimulation, andmagnetic-activated cell sorting of CD31+/CD144+ populations (fig.S9A). These hiPSC-ECs exhibited standard ECmorphologies, markers,
3 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 2. High-throughput analysis of TKI toxicity in purified hiPSC-CMs allows for the development of a TKI cardiac safety index. (A) Dose-response curvesquantifying cytotoxicity after a 72-hour TKI treatment of five healthy control hiPSC-CM lines using a PrestoBlue viability assay. n = 5 biological replicates conducted perline. Data are means ± SEM. (B) Evaluation of hiPSC-CM contractility after a 72-hour TKI treatment with the IC200 Kinetic Imaging Cytometer. Average results fromtriplicate wells shown at each concentration. Red indicates decreased contraction rate, whereas green indicates increased contraction rate. (C) Values gathered fromcytotoxicity and contractility analyses in hiPSC-CMs. Green shading indicates values associated with less cardiotoxicity. Red shading indicates values associated withhigher cardiotoxicity. Cessation of beating is the concentration at which >50% of triplicate wells ceased beating. Effective concentration is the concentration at which asignificant alteration in all listed contractility parameters was detected (see fig. S7 and Materials and Methods for details). Amplitude of effect is the degree to which alllisted contractility parameters were altered at the effective concentration (see Materials and Methods for details). LD50 is the TKI concentration at which a 50% loss inviability is observed from viability assays, averaged across patient hiPSC-CM lines. Patient Cmax represents the maximum TKI blood plasma concentration experiencedby patients reported in FDA literature. The cardiac safety index is a value from 0 to 1 that normalizes contractility and viability parameters to patient Cmax and combinesthese parameters to provide a relative metric for TKI cardiotoxicity. Highlighted drugs (surrounded by a red rectangle) have a safety index at or below 0.10, ourthreshold for highly cardiotoxic compounds. Clinically reported cardiotoxicities are alterations in patient cardiac function (see table S1). QT, QT interval prolongation;Hy, hypertension; LV, left ventricular ejection fraction decrease; HF, heart failure; MI, myocardial infarction; TdP, Torsades de pointes; SCD, sudden cardiac death; Brady,bradycardia; PE, pericardial effusion; Vas, vascular abnormalities; Afib, atrial fibrillation; **cardiovascular toxicity–associated boxed warning; #noncardiovascular toxicity–associated boxed warning.
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017 4 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
and functionality (fig. S9, B to D). As in hiPSC-CMs, sorafenib, regor-afenib, and ponatinib were the most cytotoxic TKIs in hiPSC-ECs (fig.S9E). We also developed a custom hiPSC-CF differentiation protocolusing small-molecule Wnt signaling modulation, FGF2 and VEGFAstimulation, and negative sorting for ECs (fig. S10A). These hiPSC-CFs were negative for cardiomyocyte markers, expressed mesenchy-mal and myofibroblast markers, and were morphologically similarto primary cardiac fibroblasts (fig. S10, B to E). These hiPSC-CFs ex-hibited TKI cytotoxicity profiles similar to those of hiPSC-CMs andhiPSC-ECs, with sorafenib, regorafenib, and ponatinib eliciting thehighest cytotoxicities (fig. S10F). We next treated undifferentiatedhiPSCs with our TKI panel to determine whether noncardiovascularcell types exhibit toxicities similar to hiPSC-CMs, hiPSC-ECs, andhiPSC-CFs (fig. S11). hiPSCs exhibited a unique TKI cytotoxicityprofile, showing higher toxicity from VEGFR2/PDGFR dual inhibitorsthan did cardiovascular derivatives (hiPSC-CMs, hiPSC-ECs, and
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
hiPSC-CFs). For example, axitinib, the least cytotoxic TKI in hiPSC-CMs, was extremely toxic to hiPSCs. Doxorubicin was also substan-tially more toxic in hiPSCs than in cardiovascular cell types. Theseresults suggest that the VEGFR2/PDGFR-inhibiting TKIs sorafenib,regorafenib, and ponatinib exhibit cell type–specific cytotoxicities thatdiffer between cardiovascular and noncardiovascular cell types.
Evaluation of RTK phosphorylation status in hiPSC-CMs afterTKI treatmentTo elucidate TKI-induced signaling alterations, we used an RTK pro-teome profiler to assess RTK phosphorylation after treatment withVEGFR2/PDGFR-inhibiting TKIs (Fig. 5 and fig. S12). Drugs wereadded to hiPSC-CMs at subcytotoxic concentrations. We observed adose-dependent decline in VEGFR2 and PDGFRa phosphorylationafter VEGFR2/PDGFR-inhibiting TKI treatment, with axitinib elicit-ing the strongest dual inhibition, suggesting that these TKIs can inhibitfunctionally relevant signaling pathways in hiPSC-CMs. ErbB2, ErbB4,and epidermal growth factor receptor 2 (EGFR2) phosphorylation re-mained constant over increasing TKI concentrations. Cabozantinib, aknown Axl inhibitor, decreased Axl phosphorylation. Notably, we ob-served increased INSR and IGF1R phosphorylation after treatmentwith ponatinib and axitinib, suggesting a compensatory augmentationin insulin/IGF signaling during VEGFR2/PDGFR inhibition.
Evaluation of insulin- and IGF1-mediated compensatorycardioprotection during TKI treatmentTreatment with insulin or IGF1 can enhance cardiac function duringadverse events (17, 18). Given that insulin/IGF signaling was up-regulated after treatment with VEGFR2/PDGFR-inhibiting TKIs,we hypothesized that this compensatory up-regulation protectshiPSC-CMs from TKI toxicity. To determine whether exogenous in-sulin or IGF1 could enhance cardioprotective signaling in hiPSC-CMs,we used a high-throughput kinase phosphorylation array (Fig. 6A andfig. S13). Both IGF1 and insulin enhanced phosphorylation of theantiapoptotic Akt protein network. Cell survival was enhanced whenhiPSC-CMs exposed to ponatinib were concurrently treated withIGF1 or insulin (Fig. 6B). This observation was confirmed quantita-tively with CellTiter-Glo viability assays (Fig. 6C). Additionally, we ob-served that IGF1 and insulin treatment rescued hiPSC-CMs fromdoxorubicin cytotoxicity (fig. S14). To confirm that the effect of insulin/IGF1 was a result of enhanced cardiomyocyte survival rather thanproliferation, we assessed the cell number at early time points afterTKI treatment. We observed an increase in viability merely 12 hoursafter TKI treatment, confirming that insulin and IGF augment cardi-omyocyte survival (fig. S15). We next evaluated the gene expressionresponse in hiPSC-CMs during treatment with the VEGFR2/PDGFR-inhibiting TKIs sorafenib, regorafenib, and ponatinib and observed anincrease in growth factor receptor gene expression (Fig. 7). NRP2, en-coding for the noncanonical VEGFR neuropilin 2, was up-regulated inour microarray. We subsequently conducted RNA sequencing (RNA-seq) analysis of five healthy control hiPSC-CM lines treated with 1 mMVEGFR2/PDGFR-inhibiting TKI sorafenib for 72 hours and observedincreased expression of KDR, encoding for the VEGFR2 receptor, andVEGFC, encoding for the VEGFC ligand (fig. S16). These gene expres-sion analyses suggest that noncanonical VEGF-binding receptors andVEGF signaling pathway members are up-regulated to compensate forlosing canonical VEGFR signaling after treatment with VEGFR2/PDGFR-inhibiting TKIs. A summary of the compensatory cardiopro-tective signaling model is shown in Fig. 8. Together, our data suggest
Fig. 3. hiPSC-CMs exhibit alterations in intracellular calcium handling after a2-hour treatment with known QT interval–prolonging TKIs. (A) Schematic il-lustrating TKI treatment regimen for hiPSC-CMs before calcium imaging. (B) Rawline scans of individual hiPSC-CM calcium transients after TKI treatment at indi-cated clinically relevant concentrations and calcium dye treatment over multiplebeats. (C) Quantification of hiPSC-CM calcium imaging parameters after a 2-hourTKI treatment. n = 10 cells recorded for each condition. Data are presented asbox-and-whisker plots showing the minimum, first quartile, median, mean, thirdquartile, and maximum of the data set. Student’s t test indicates significance com-pared to control (*P < 0.05 and **P < 0.01).
5 of 13
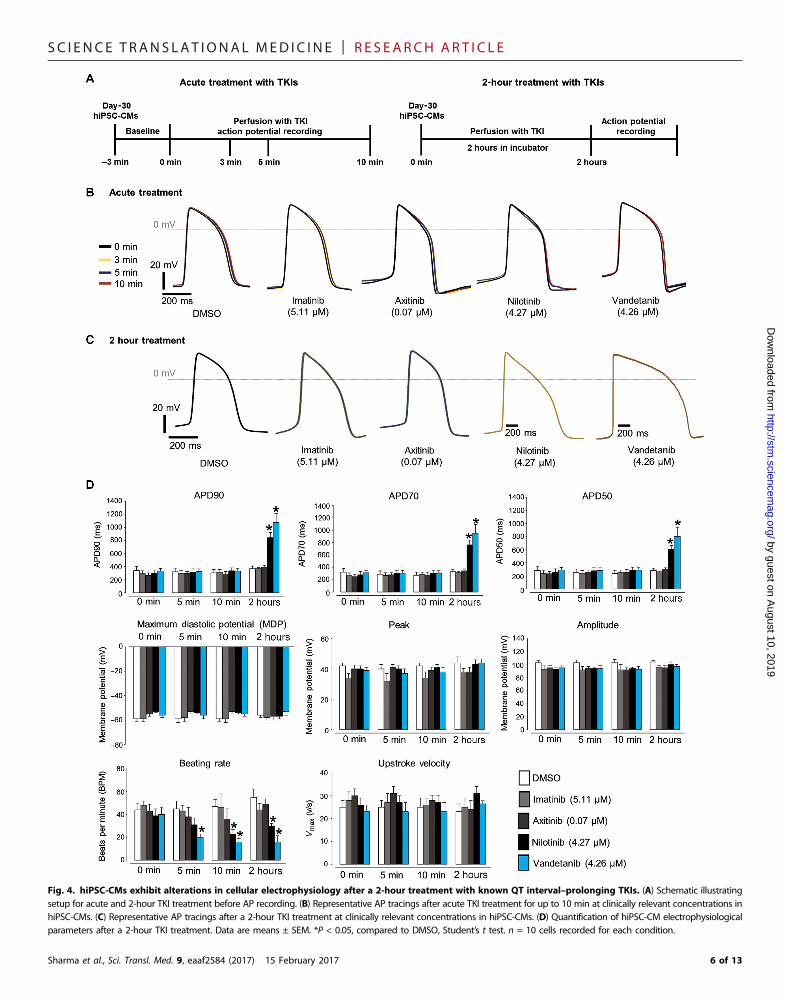
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
Fig. 4. hiPSC-CMs exhibit alterations in cellular electrophysiology after a 2-hour treatment with known QT interval–prolonging TKIs. (A) Schematic illustratingsetup for acute and 2-hour TKI treatment before AP recording. (B) Representative AP tracings after acute TKI treatment for up to 10 min at clinically relevant concentrations inhiPSC-CMs. (C) Representative AP tracings after a 2-hour TKI treatment at clinically relevant concentrations in hiPSC-CMs. (D) Quantification of hiPSC-CM electrophysiologicalparameters after a 2-hour TKI treatment. Data are means ± SEM. *P < 0.05, compared to DMSO, Student’s t test. n = 10 cells recorded for each condition.
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017 6 of 13
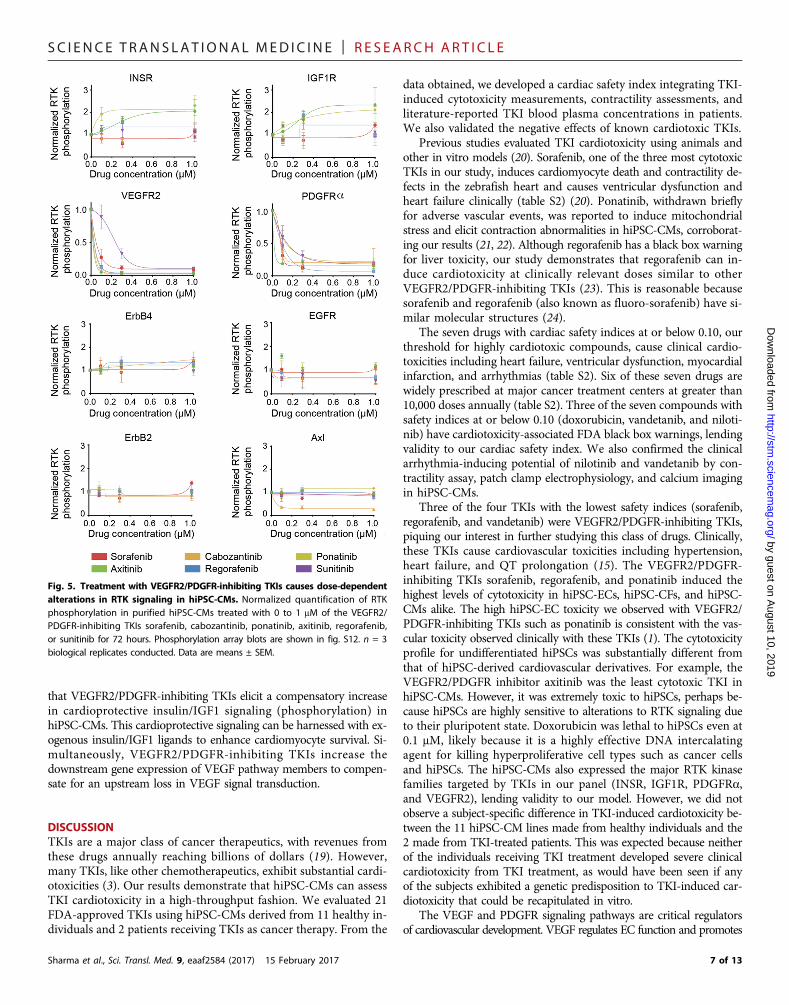
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
that VEGFR2/PDGFR-inhibiting TKIs elicit a compensatory increasein cardioprotective insulin/IGF1 signaling (phosphorylation) inhiPSC-CMs. This cardioprotective signaling can be harnessed with ex-ogenous insulin/IGF1 ligands to enhance cardiomyocyte survival. Si-multaneously, VEGFR2/PDGFR-inhibiting TKIs increase thedownstream gene expression of VEGF pathway members to compen-sate for an upstream loss in VEGF signal transduction.
DISCUSSIONTKIs are a major class of cancer therapeutics, with revenues fromthese drugs annually reaching billions of dollars (19). However,many TKIs, like other chemotherapeutics, exhibit substantial cardi-otoxicities (3). Our results demonstrate that hiPSC-CMs can assessTKI cardiotoxicity in a high-throughput fashion. We evaluated 21FDA-approved TKIs using hiPSC-CMs derived from 11 healthy in-dividuals and 2 patients receiving TKIs as cancer therapy. From the
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
data obtained, we developed a cardiac safety index integrating TKI-induced cytotoxicity measurements, contractility assessments, andliterature-reported TKI blood plasma concentrations in patients.We also validated the negative effects of known cardiotoxic TKIs.
Previous studies evaluated TKI cardiotoxicity using animals andother in vitro models (20). Sorafenib, one of the three most cytotoxicTKIs in our study, induces cardiomyocyte death and contractility de-fects in the zebrafish heart and causes ventricular dysfunction andheart failure clinically (table S2) (20). Ponatinib, withdrawn brieflyfor adverse vascular events, was reported to induce mitochondrialstress and elicit contraction abnormalities in hiPSC-CMs, corroborat-ing our results (21, 22). Although regorafenib has a black box warningfor liver toxicity, our study demonstrates that regorafenib can in-duce cardiotoxicity at clinically relevant doses similar to otherVEGFR2/PDGFR-inhibiting TKIs (23). This is reasonable becausesorafenib and regorafenib (also known as fluoro-sorafenib) have si-milar molecular structures (24).
The seven drugs with cardiac safety indices at or below 0.10, ourthreshold for highly cardiotoxic compounds, cause clinical cardio-toxicities including heart failure, ventricular dysfunction, myocardialinfarction, and arrhythmias (table S2). Six of these seven drugs arewidely prescribed at major cancer treatment centers at greater than10,000 doses annually (table S2). Three of the seven compounds withsafety indices at or below 0.10 (doxorubicin, vandetanib, and niloti-nib) have cardiotoxicity-associated FDA black box warnings, lendingvalidity to our cardiac safety index. We also confirmed the clinicalarrhythmia-inducing potential of nilotinib and vandetanib by con-tractility assay, patch clamp electrophysiology, and calcium imagingin hiPSC-CMs.
Three of the four TKIs with the lowest safety indices (sorafenib,regorafenib, and vandetanib) were VEGFR2/PDGFR-inhibiting TKIs,piquing our interest in further studying this class of drugs. Clinically,these TKIs cause cardiovascular toxicities including hypertension,heart failure, and QT prolongation (15). The VEGFR2/PDGFR-inhibiting TKIs sorafenib, regorafenib, and ponatinib induced thehighest levels of cytotoxicity in hiPSC-ECs, hiPSC-CFs, and hiPSC-CMs alike. The high hiPSC-EC toxicity we observed with VEGFR2/PDGFR-inhibiting TKIs such as ponatinib is consistent with the vas-cular toxicity observed clinically with these TKIs (1). The cytotoxicityprofile for undifferentiated hiPSCs was substantially different fromthat of hiPSC-derived cardiovascular derivatives. For example, theVEGFR2/PDGFR inhibitor axitinib was the least cytotoxic TKI inhiPSC-CMs. However, it was extremely toxic to hiPSCs, perhaps be-cause hiPSCs are highly sensitive to alterations to RTK signaling dueto their pluripotent state. Doxorubicin was lethal to hiPSCs even at0.1 mM, likely because it is a highly effective DNA intercalatingagent for killing hyperproliferative cell types such as cancer cellsand hiPSCs. The hiPSC-CMs also expressed the major RTK kinasefamilies targeted by TKIs in our panel (INSR, IGF1R, PDGFRa,and VEGFR2), lending validity to our model. However, we did notobserve a subject-specific difference in TKI-induced cardiotoxicity be-tween the 11 hiPSC-CM lines made from healthy individuals and the2 made from TKI-treated patients. This was expected because neitherof the individuals receiving TKI treatment developed severe clinicalcardiotoxicity from TKI treatment, as would have been seen if anyof the subjects exhibited a genetic predisposition to TKI-induced car-diotoxicity that could be recapitulated in vitro.
The VEGF and PDGFR signaling pathways are critical regulatorsof cardiovascular development. VEGF regulates EC function and promotes
Fig. 5. Treatment with VEGFR2/PDGFR-inhibiting TKIs causes dose-dependentalterations in RTK signaling in hiPSC-CMs. Normalized quantification of RTKphosphorylation in purified hiPSC-CMs treated with 0 to 1 mM of the VEGFR2/PDGFR-inhibiting TKIs sorafenib, cabozantinib, ponatinib, axitinib, regorafenib,or sunitinib for 72 hours. Phosphorylation array blots are shown in fig. S12. n = 3biological replicates conducted. Data are means ± SEM.
7 of 13
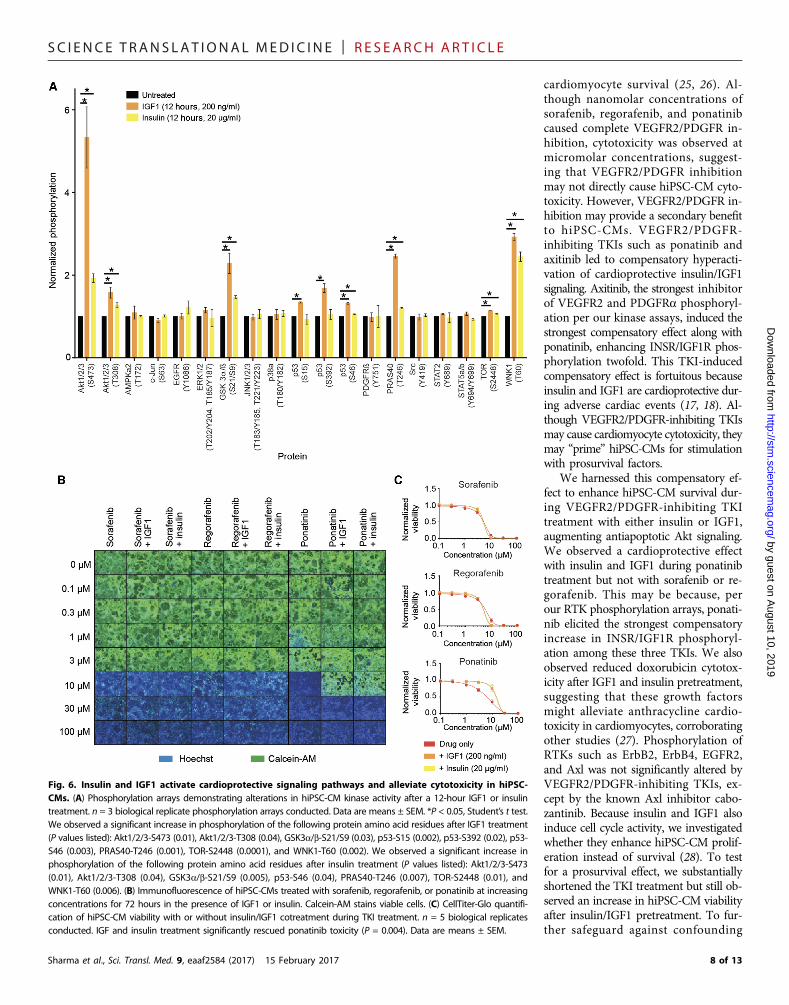
SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
cardiomyocyte survival (25, 26). Al-though nanomolar concentrations ofsorafenib, regorafenib, and ponatinibcaused complete VEGFR2/PDGFR in-hibition, cytotoxicity was observed atmicromolar concentrations, suggest-ing that VEGFR2/PDGFR inhibitionmay not directly cause hiPSC-CM cyto-toxicity. However, VEGFR2/PDGFR in-hibition may provide a secondary benefitto hiPSC-CMs. VEGFR2/PDGFR-inhibiting TKIs such as ponatinib andaxitinib led to compensatory hyperacti-vation of cardioprotective insulin/IGF1signaling. Axitinib, the strongest inhibitorof VEGFR2 and PDGFRa phosphoryl-ation per our kinase assays, induced thestrongest compensatory effect along withponatinib, enhancing INSR/IGF1R phos-phorylation twofold. This TKI-inducedcompensatory effect is fortuitous becauseinsulin and IGF1 are cardioprotective dur-ing adverse cardiac events (17, 18). Al-though VEGFR2/PDGFR-inhibiting TKIsmay cause cardiomyocyte cytotoxicity, theymay “prime” hiPSC-CMs for stimulationwith prosurvival factors.
We harnessed this compensatory ef-fect to enhance hiPSC-CM survival dur-ing VEGFR2/PDGFR-inhibiting TKItreatment with either insulin or IGF1,augmenting antiapoptotic Akt signaling.We observed a cardioprotective effectwith insulin and IGF1 during ponatinibtreatment but not with sorafenib or re-gorafenib. This may be because, perour RTK phosphorylation arrays, ponati-nib elicited the strongest compensatoryincrease in INSR/IGF1R phosphoryl-ation among these three TKIs. We alsoobserved reduced doxorubicin cytotox-icity after IGF1 and insulin pretreatment,suggesting that these growth factorsmight alleviate anthracycline cardio-toxicity in cardiomyocytes, corroboratingother studies (27). Phosphorylation ofRTKs such as ErbB2, ErbB4, EGFR2,and Axl was not significantly altered byVEGFR2/PDGFR-inhibiting TKIs, ex-cept by the known Axl inhibitor cabo-zantinib. Because insulin and IGF1 alsoinduce cell cycle activity, we investigatedwhether they enhance hiPSC-CM prolif-eration instead of survival (28). To testfor a prosurvival effect, we substantiallyshortened the TKI treatment but still ob-served an increase in hiPSC-CM viabilityafter insulin/IGF1 pretreatment. To fur-ther safeguard against confounding
Fig. 6. Insulin and IGF1 activate cardioprotective signaling pathways and alleviate cytotoxicity in hiPSC-CMs. (A) Phosphorylation arrays demonstrating alterations in hiPSC-CM kinase activity after a 12-hour IGF1 or insulintreatment. n = 3 biological replicate phosphorylation arrays conducted. Data are means ± SEM. *P < 0.05, Student’s t test.We observed a significant increase in phosphorylation of the following protein amino acid residues after IGF1 treatment(P values listed): Akt1/2/3-S473 (0.01), Akt1/2/3-T308 (0.04), GSK3a/b-S21/S9 (0.03), p53-S15 (0.002), p53-S392 (0.02), p53-S46 (0.003), PRAS40-T246 (0.001), TOR-S2448 (0.0001), and WNK1-T60 (0.002). We observed a significant increase inphosphorylation of the following protein amino acid residues after insulin treatment (P values listed): Akt1/2/3-S473(0.01), Akt1/2/3-T308 (0.04), GSK3a/b-S21/S9 (0.005), p53-S46 (0.04), PRAS40-T246 (0.007), TOR-S2448 (0.01), andWNK1-T60 (0.006). (B) Immunofluorescence of hiPSC-CMs treated with sorafenib, regorafenib, or ponatinib at increasingconcentrations for 72 hours in the presence of IGF1 or insulin. Calcein-AM stains viable cells. (C) CellTiter-Glo quantifi-cation of hiPSC-CM viability with or without insulin/IGF1 cotreatment during TKI treatment. n = 5 biological replicatesconducted. IGF and insulin treatment significantly rescued ponatinib toxicity (P = 0.004). Data are means ± SEM.
8 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
effects of insulin/IGF1-induced proliferation, we used day 30 postdiffer-entiation hiPSC-CMs, which exhibit lower mitotic activity than do youn-ger cells (28). Finally, we conducted gene expression analysis to elucidategene networks altered by TKI treatment. The VEGFR2/PDGFR-inhibiting TKIs sorafenib, regorafenib, and ponatinib induced a twofoldincrease in the expression of neuropilin 2, a noncanonical VEGFR (29).Thus, hiPSC-CMs may compensate for VEGFR2 signaling loss by up-regulating auxiliary and canonical VEGFR expression, thus facilitat-ing VEGF signaling in the absence of VEGFR2 kinase activity. RNA-seq analysis revealed an up-regulation of KDR (VEGFR2 receptor)and an up-regulation in the expression of VEGF ligands. This suggeststhat in hiPSC-CMs, the upstream blockade of the phosphorylationcascade by VEGFR2/PDGFR-inhibiting TKIs causes a downstream in-crease in the gene expression of VEGF pathway members, potentiallyto compensate for the loss of upstream VEGFR signaling (Fig. 8).
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
In summary, we used hiPSC-CMs, hiPSC-ECs, and hiPSC-CFs toscreen for the cardiotoxicity of 21 FDA-approved TKIs andestablished a cardiac safety index for TKI toxicity. We also validatedthe known cardiotoxicities of FDA black boxed TKIs in our hiPSC-CMplatform. Although TKIs target various RTK families, we observed thatVEGFR2/PDGFR-inhibiting TKIs exhibited high cardiotoxicity onmultiple cardiovascular cell types. These cardiotoxicities could be res-cued by co-opting compensatory prosurvival insulin/IGF1 signalingpathways. There is substantial interest in deriving hiPSCs from a broadercohort of individuals to establish a population-level, prospective, pre-clinical toxicity assessment to better inform drug development. Aspresented here, a combination of electrophysiological, functional,and genetic assays and continued advances in hiPSC biology will en-able the development of efficient high-throughput platforms forpreemptively screening potential chemotherapeutic compounds forcardiotoxicities.
Fig. 7. Treatment with sorafenib, regorafenib, or ponatinib leads to hyper-activation of compensatory signaling through noncanonical VEGFRs. (A) Micro-array heat map illustrating differentially expressed genes after a 72-hour sorafenib,regorafenib, and ponatinib treatment in hiPSC-CMs. Cells were treated with 1 mM TKIto avoid cytotoxicity at higher doses. Red indicates high gene expression, and blueindicates low gene expression. (B) Graph represents fold expression change (com-pared to control) of significantly altered genes after drug treatment. Significantlyaltered genes defined by P < 0.05 compared to untreated control. Multiple P-valuecomparisons made using one-way between-subject analysis of variance (ANOVA).
Fig. 8. Model for the activation of compensatory survival signaling in hiPSC-CMs in response to treatment with VEGFR2/PDGFR-inhibiting TKIs. (A) InhiPSC-CMs, the RTKs VEGFR2, PDGFRa, INSR, and IGF1R are upstream of prosurvi-val signaling pathways. (B) Our results suggest that VEGFR2/PDGFR-inhibiting TKIsup-regulate INSR and IGF1R signaling (phosphorylation) to compensate for theloss of VEGFR/PDGFR signaling (phosphorylation). This compensatory effect canaugment cardiomyocyte survival during TKI treatment via introduction of exoge-nous insulin and IGF1 ligands. We observed an increase in the downstream geneexpression of noncanonical VEGF-binding receptors and VEGFR pathway mem-bers, presumably to compensate for VEGFR2/PDGFR-inhibiting TKI-induced lossin upstream VEGFR signaling (phosphorylation).
9 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
MATERIALS AND METHODSStudy designTo investigate the cardiotoxicity of FDA-approved TKIs in hiPSC-CMs, 11 healthy individuals and 2 individuals receiving TKIs forcancer therapy were recruited and consented. Either skin punch biop-sies or blood draws were performed to obtain primary tissue samplesfor hiPSC production. Reprogramming of either skin fibroblasts orperipheral blood mononuclear cells to hiPSCs was conducted accord-ing to previously published protocols (8, 30). A minimum of n = 3biological replicates were conducted for each experiment, with de-tails in the figure legends.
Chemically defined differentiation of hiPSC-CMsThe hiPSCs were differentiated into hiPSC-CMs using a chemicallydefined protocol and maintained in medium supplemented with hu-man albumin and ascorbic acid (8). This cardiomyocyte maintenancemedium was devoid of growth factors such as insulin and IGF1. Dif-ferentiated cells were glucose-starved and supplemented with 5 mMsodium DL-lactate to metabolically select hiPSC-CMs (31). When re-plating, hiPSC-CMs were dissociated with TrypLE Express (Life Tech-nologies) and reseeded on Matrigel-coated plates.
TKI stocks and cardioprotective growth factor treatmentsTKI stocks (LC Laboratories) were resuspended in 10 mM DMSOand stored at −80°C. Insulin (Life Technologies) was stored at −20°C,and Long-R3 IGF1 (Sigma-Aldrich) was stored at 4°C. For cell viabil-ity rescue experiments, day 30 to 35 postdifferentiation hiPSC-CMswere pretreated with insulin or IGF1 for 12 hours before TKI treat-ment, when insulin or IGF1 was resupplemented.
High-throughput imaging and quantitative viability assaysDay 30 to 35 postdifferentiation hiPSC-CMs were plated on Matrigelat 25,000 cells per well of a 384-well plate (Greiner Bio-One). Cellswere treated with TKIs at 0 to 100 mM for 72 hours unless otherwisespecified. Immunostaining qualitatively assessed cell viability per pre-vious protocols (8). For quantitative viability measurements, cells weretreated with CellTiter-Glo Viability Assay (Promega), CCK8 (Dojin-do), or PrestoBlue reagent (Life Technologies) per manufacturer-recommended procedures. High-throughput imaging and viabilityassays were conducted using a Cytation 5 plate reader/imager (BioTekInstruments). Prism (GraphPad) was used for curve fitting, LD50 cal-culations, and statistical analysis.
High-throughput hiPSC-CM contractility assessmentAfter a 72-hour TKI treatment, hiPSC-CMs were washed with Tyrode’ssolution. Imaging dye was prepared by diluting Hoechst 33258 (H3569,Life Technologies) to 4 mg/ml and wheat germ agglutinin–Alexa Flu-or 488 conjugate (W11261, Life Technologies) to 5 mg/ml in Tyrode’ssolution. Solution was added to hiPSC-CMs and incubated at 37°Cand 5% CO2 for 15 min. After rewashing with Tyrode’s solution,hiPSC-CMs were incubated for 15 min before imaging. The IC200Kinetic Imaging Cytometer (Vala Sciences) recorded a 6.5-s time se-ries of contracting hiPSC-CMs at 100 Hz at 20× magnification perwell of a 384-well plate.
Safety coefficients based on contractility parametersSafety coefficients presented in this paper are computed as follows.Cessation of beating is the concentration at which >50% of tripli-cate wells ceased beating. Effective concentration for the ith metric
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
(ECi) is a concentration that presents a statistically significant con-tractility difference (P < 0.05) from baseline. Amplitude of the effect(AEi) quantifies the magnitude of such departures from baseline as thelog2 of the ratio of the metric value EVi at concentration ECi, hereinafterreferred to as effective value (EVi), to the value of the metric BVi atbaseline concentration. Additionally, i = 1…M, where M is the numberof metrics obtained from the contractility analysis (M = 9 in this paper:Tpeak, Trise, Tfall, total contraction time, Dpeak, Dvalley, Dp2v, contractionrate, and relaxation rate).
AEi ¼ log2EVi
BMi
� �
Average amplitude of effect (AE) is obtained by averaging the dif-ferent AEi’s.
AE ¼ lM∑M
i¼lAEi
Average effective concentration (EC) is a weighted average of thedifferent effective concentrations using each respective amplitude of ef-fect as weight.
EC ¼∑M
i¼lECi⋅AEi
∑M
i¼lAEi
The rationale for this EC expression is that it considers all differentECs and introduces a bias toward the ECs corresponding to metrics thatmost prominently alter normal cell behavior.
Calcium imagingDay 30 to 40 postdifferentiation hiPSC-CMs were reseeded in Matrigel-coated eight-well Lab-Tek II chambers (Nalge Nunc International) andwere treated with TKIs for 2 hours. Cells were treated with 5 mM Fluo-4AM and 0.02% Pluronic F-127 (Molecular Probes) in Tyrode’s solutionfor 15 min at 37°C and washed with Tyrode’s solution afterward. Ca2+
imaging was conducted using a Zeiss LSM 510 Meta confocal micro-scope (Carl Zeiss AG) and analyzed using Zen software. SpontaneousCa2+ transients were obtained at 37°C using a single-cell line scan mode.
ElectrophysiologyWhole-cell APs were recorded with patch clamp technique, as previ-ously described (8). Cultured hiPSC-CMs were dissociated using Try-pLE and plated as single cells on glass coverslips coated with Matrigel.Cells were placed in an RC-26C recording chamber (Warner) andmounted onto an inverted microscope (Nikon). The chamber wascontinuously perfused with warm (35° to 37°C) extracellular solutionof the following composition: 150 mM NaC1, 5.4 mM KCl, 1.8 mMCaCl2, 1.0 mMMgCl2, 1.0 mMNa pyruvate, 15 mMHepes, and 15 mMglucose; pH was adjusted to 7.4 with NaOH. Glass micropipettes (2-to 3-megohm tip resistance) were fabricated from standard wall boro-silicate glass capillary tubes (Sutter BF 100-50-10) and filled with thefollowing intracellular solution: 120 mM KCl, 1.0 mMMgCl2, 10 mMHepes, 10 mM EGTA, and 3 mM Mg-ATP; pH was adjusted to 7.2
10 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
with KOH. Single-beating cardiomyocytes were selected, and APs wererecorded in whole-cell current clampmode using an EPC 10 patch clampamplifier (HEKA). External solution containing 0.1% DMSO (vehicle)was applied to establish the baseline. Then, cells were treated with TKIsolution containing imatinib, axitinib, nilotinib, or vandetanib (LC Lab-oratories). Data were acquired using PATCHMASTER software (HEKA),digitized at 1.0 kHz, and analyzed using FITMASTER (HEKA), Igor Pro(WaveMetrics), and Prism 5 (GraphPad). For recordings on differentiatedventricular-like hiPSC-CMs, the maximum diastolic potential of singlecardiomyocytes varied from −70 to −50 mV, action potential amplitude(APA) was greater than 90 mV, and action potential duration (APD)90/APD50 was less than 1.20. Only cardiomyocytes satisfying the aforemen-tioned criteria were ventricular-like cardiomyocytes and selected for as-sessing the effects of TKIs. Baseline APs were recorded for 3 min beforeapplication of the drug and at 3, 5, and 10 min while keeping a con-tinuous perfusion with the drug. In a separate series of experiments, thedrugs were added for 2 hours at 37°C before patching. Average responsesof n = 10 APs were analyzed per treatment. Significant APD90 prolonga-tion is defined as >10% change in APD90.
Kinase phosphorylation profilingPhosphorylation of human RTKs and other phosphoproteins wasdetermined using Human Phospho-RTK Array or HumanPhospho-Kinase Antibody Array (R&D Systems). Day 30 to 35 post-differentiation hiPSC-CMs were treated with TKIs for 72 hours andlysed. Lysate was incubated overnight on an RTK or phosphokinasepanel and subsequently with an anti–phosphotyrosine–horseradishperoxidase antibody to assess phosphorylation. Blots were developedusing Gel Doc XR (Bio-Rad). Phosphorylation intensity was deter-mined using ImageJ software.
Gene expressionRTK expression in hiPSC-CMs was determined using Ion AmpliSeq(Life Technologies). RNA was extracted using the RNeasy Micro Kit(Qiagen). Complementary DNA libraries were synthesized using theIon AmpliSeq Transcriptome Human Gene Expression Kit. Librarieswere added to Ion PI chips and loaded onto an Ion Chef instrumentfor template preparation. Transcriptome sequencing was conductedon an Ion Proton sequencing system (Life Technologies). For expres-sion analysis of hiPSC-CMs after TKI treatment, a GeneChip Hu-man Gene 1.0 ST DNA microarray was used (Affymetrix).
Statistical analysisData are presented as means ± SEM unless otherwise specified. Com-parisons are conducted via Student’s t test, unless otherwise specified,with significant differences defined by *P < 0.05 or **P < 0.01. Formicroarray experiments, multiple P-value comparisons were madeusing a one-way between-subject ANOVA (P < 0.05) and AffymetrixTranscriptome Analysis Console 2.0 software.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/9/377/eaaf2584/DC1Materials and MethodsFig. S1. hiPSCs exhibit characteristic morphologies and markers of pluripotent stem cells.Fig. S2. Quantitative and qualitative cell viability assays illustrate sorafenib, regorafenib, andponatinib cytotoxicity in hiPSC-CMs.Fig. S3. Quantitative cell viability assays on additional hiPSC-CM lines demonstrate VEGFR2/PDGFR-inhibiting TKI toxicity.Fig. S4. Quantitative cell viability assays in hiPSC-CMs and hiPSC-ECs derived from patientsreceiving TKI treatment.
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
Fig. S5. Commercially available, healthy control hiPSC-CMs exhibit alterations in cellularcontractility after a 72-hour TKI treatment.Fig. S6. Heat maps of high-throughput contractility analysis on commercially available, healthycontrol hiPSC-CMs treated with TKIs.Fig. S7. Extended calculations for TKI safety index after a 72-hour TKI treatment oncommercially available, healthy control hiPSC-CMs.Fig. S8. hiPSC-CMs exhibit alterations in cellular contractility after a 72-hour treatment withknown QT interval–prolonging TKIs.Fig. S9. hiPSC-ECs exhibit EC characteristics and demonstrate cytotoxicity in response to TKI treatment.Fig. S10. hiPSC-CFs exhibit properties of adult cardiac fibroblasts and demonstrate cytotoxicityin response to TKI treatment.Fig. S11. hiPSCs demonstrate a TKI cytotoxicity profile that is unique from those of hiPSC-CMs,hiPSC-ECs, and hiPSC-CFs.Fig. S12. VEGFR2/PDGFR-inhibiting TKI treatment in hiPSC-CMs results in activation ofcompensatory insulin/IGF1 signaling.Fig. S13. IGF1 and insulin treatment activates cardioprotective Akt signaling in hiPSC-CMs.Fig. S14. IGF1 and insulin treatment rescues doxorubicin toxicity in hiPSC-CMs.Fig. S15. IGF1 and insulin treatment rescues ponatinib toxicity at early time points in hiPSC-CMs.Fig. S16. RNA-seq of hiPSC-CMs treated with the VEGFR2/PDGFR-inhibiting TKI sorafenibillustrates compensatory hyperactivation of VEGF signaling.Table S1. Small-molecule TKIs selected for high-throughput cardiotoxicity screen.Table S2. Adverse cardiac events associated with small-molecule TKIs selected for high-throughput cardiotoxicity screen.Movie S1. hiPSC-CMs before purification via glucose deprivation.References (32–50)
REFERENCES AND NOTES1. J. J. Moslehi, M. Deininger, Tyrosine kinase inhibitor–associated cardiovascular toxicity in
chronic myeloid leukemia. J. Clin. Oncol. 33, 4210–4218 (2015).2. E. T. H. Yeh, C. L. Bickford, Cardiovascular complications of cancer therapy: Incidence,
pathogenesis, diagnosis, and management. J. Am. Coll. Cardiol. 53, 2231–2247 (2009).3. T. Force, D. S. Krause, R. A. Van Etten, Molecular mechanisms of cardiotoxicity of tyrosine
kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007).4. H. R. Lu, R. Mariën, A. Saels, F. De Clerck, Species plays an important role in drug-induced
prolongation of action potential duration and early afterdepolarizations in isolatedPurkinje fibers. J. Cardiovasc. Electrophysiol. 12, 93–102 (2001).
5. P. Liang, F. Lan, A. S. Lee, T. Gong, V. Sanchez-Freire, Y. Wang, S. Diecke, K. Sallam,J. W. Knowles, P. J. Wang, P. K. Nguyen, D. M. Bers, R. C. Robbins, J. C. Wu, Drug screeningusing a library of human induced pluripotent stem cell-derived cardiomyocytes revealsdisease-specific patterns of cardiotoxicity. Circulation 127, 1677–1691 (2013).
6. J. S. Mitcheson, J. C. Hancox, A. J. Levi, Cultured adult cardiac myocytes: Futureapplications, culture methods, morphological and electrophysiological properties.Cardiovasc. Res. 39, 280–300 (1998).
7. K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda, S. Yamanaka,Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell131, 861–872 (2007).
8. P. W. Burridge, E. Matsa, P. Shukla, Z. C. Lin, J. M. Churko, A. D. Ebert, F. Lan, S. Diecke,B. Huber, N. M. Mordwinkin, J. R. Plews, O. J. Abilez, B. Cui, J. D. Gold, J. C. Wu, Chemicallydefined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014).
9. A. Sharma, C. Marceau, R. Hamaguchi, P. W. Burridge, K. Rajarajan, J. M. Churko, H. Wu,K. I. Sallam, E. Matsa, A. C. Sturzu, Y. Che, A. Ebert, S. Diecke, P. Liang, K. Red-Horse,J. E. Carette, S. M. Wu, J. C. Wu, Human induced pluripotent stem cell-derivedcardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis andantiviral drug screening platform. Circ. Res. 115, 556–566 (2014).
10. F. Lan, A. S. Lee, P. Liang, V. Sanchez-Freire, P. K. Nguyen, L. Wang, L. Han, M. Yen,Y. Wang, N. Sun, O. J. Abilez, S. Hu, A. D. Ebert, E. G. Navarrete, C. S. Simmons, M. Wheeler,B. Pruitt, R. Lewis, Y. Yamaguchi, E. A. Ashley, D. M. Bers, R. C. Robbins, M. T. Longaker,J. C. Wu, Abnormal calcium handling properties underlie familial hypertrophiccardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell StemCell 12, 101–113 (2013).
11. H. Wu, J. Lee, L. G. Vincent, Q. Wang, M. Gu, F. Lan, J. M. Churko, K. I. Sallam, E. Matsa,A. Sharma, J. D. Gold, A. J. Engler, Y. K. Xiang, D. M. Bers, J. C. Wu, Epigenetic regulation ofphosphodiesterases 2A and 3A underlies compromised b-adrenergic signaling in an iPSCmodel of dilated cardiomyopathy. Cell Stem Cell 17, 89–100 (2015).
12. A. Moretti, M. Bellin, A. Welling, C. B. Jung, J. T. Lam, L. Bott-Flügel, T. Dorn, A. Goedel,C. Höhnke, F. Hofmann, M. Seyfarth, D. Sinnecker, A. Schomig, K.-L. Laugwitz, Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363,1397–1409 (2010).
13. P. W. Burridge, Y. F. Li, E. Matsa, H. Wu, S.-G. Ong, A. Sharma, A. Holmström, A. C. Chang,M. J. Coronado, A. D. Ebert, J. W. Knowles, M. L. Telli, R. M. Witteles, H. M. Blau,
11 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from
D. Bernstein, R. B. Altman, J. C. Wu, Human induced pluripotent stem cell–derivedcardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556 (2016).
14. K. Kodo, S.-G. Ong, F. Jahanbani, V. Termglinchan, K. Hirono, K. InanlooRahatloo,A. D. Ebert, P. Shukla, O. J. Abilez, J. M. Churko, I. Karakikes, G. Jung, F. Ichida, S. M. Wu,M. P. Snyder, D. Bernstein, J. C. Wu, iPSC-derived cardiomyocytes reveal abnormal TGF-bsignalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 18,1031–1042 (2016).
15. W. Li, K. Croce, D. P. Steensma, D. F. McDermott, O. Ben-Yehuda, J. Moslehi, Vascular andmetabolic implications of novel targeted cancer therapies: Focus on kinase inhibitors.J. Am. Coll. Cardiol. 66, 1160–1178 (2015).
16. M. G. Fradley, J. Moslehi, QT prolongation and oncology drug development. Card.Electrophysiol. Clin. 7, 341–355 (2015).
17. A. K. Jonassen, M. N. Sack, O. D. Mjøs, D. M. Yellon, Myocardial protection by insulin atreperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 89, 1191–1198 (2001).
18. M. Buerke, T. Murohara, C. Skurk, C. Nuss, K. Tomaselli, A. M. Lefer, Cardioprotective effectof insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc. Natl.Acad. Sci. U.S.A. 92, 8031–8035 (1995).
19. A. Hill, D. Gotham, J. Fortunak, J. Meldrum, I. Erbacher, M. Martin, H. Shoman, J. Levi,W. G. Powderly, M. Bower, Target prices for mass production of tyrosine kinase inhibitorsfor global cancer treatment. BMJ Open 6, e009586 (2016).
20. H. Cheng, G. Kari, A. P. Dicker, U. Rodeck, W. J. Koch, T. Force, A novel preclinical strategyfor identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ. Res.109, 1401–1409 (2011).
21. V. Prasad, S. Mailankody, The accelerated approval of oncologic drugs: Lessons fromponatinib. JAMA 311, 353–354 (2014).
22. D. R. Talbert, K. R. Doherty, P. B. Trusk, D. M. Moran, S. A. Shell, S. Bacus, A multi-parameterin vitro screen in human stem cell-derived cardiomyocytes identifies ponatinib-inducedstructural and functional cardiac toxicity. Toxicol. Sci. 143, 147–155 (2015).
23. G. D. Demetri, P. Reichardt, Y.-K. Kang, J.-Y. Blay, P. Rutkowski, H. Gelderblom,P. Hohenberger, M. Leahy, M. von Mehren, H. Joensuu, G. Badalamenti, M. Blackstein,A. Le Cesne, P. Schöffski, R. G. Maki, S. Bauer, B. B. Nguyen, J. Xu, T. Nishida, J. Chung,C. Kappeler, I. Kuss, D. Laurent, P. G. Casali; GRID Study Investigators, Efficacy and safetyof regorafenib for advanced gastrointestinal stromal tumours after failure of imatiniband sunitinib (GRID): An international, multicentre, randomised, placebo-controlled,phase 3 trial. Lancet 381, 295–302 (2013).
24. S. M. Wilhelm, J. Dumas, L. Adnane, M. Lynch, C. A. Carter, G. Schutz, K.-H. Thierauch,D. Zopf, Regorafenib (BAY 73–4506): A new oral multikinase inhibitor of angiogenic,stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumoractivity. Int. J. Cancer 129, 245–255 (2011).
25. S. J. Kattman, A. D. Witty, M. Gagliardi, N. C. Dubois, M. Niapour, A. Hotta, J. Ellis, G. Keller,Stage-specific optimization of activin/nodal and BMP signaling promotes cardiacdifferentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240(2011).
26. F. J. Giordano, H. P. Gerber, S. P. Williams, N. VanBruggen, S. Bunting, P. Ruiz-Lozano,Y. Gu, A. K. Nath, Y. Huang, R. Hickey, N. Dalton, K. L. Peterson, J. Ross Jr., K. R. Chien,N. Ferrara, A cardiac myocyte vascular endothelial growth factor paracrine pathway isrequired to maintain cardiac function. Proc. Natl. Acad. Sci. U.S.A. 98, 5780–5785(2001).
27. B. S. Lee, J. Oh, S. K. Kang, S. Park, S. H. Lee, D. Choi, J. H. Chung, Y. W. Chung, S.-M. Kang,Insulin protects cardiac myocytes from doxorubicin toxicity by Sp1-mediatedtransactivation of survivin. PLOS ONE 10, e0135438 (2015).
28. T. C. McDevitt, M. A. Laflamme, C. E. Murry, Proliferation of cardiomyocytes derived fromhuman embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway.J. Mol. Cell. Cardiol. 39, 865–873 (2005).
29. B. Favier, A. Alam, P. Barron, J. Bonnin, P. Laboudie, P. Fons, M. Mandron, J. P. Herault,G. Neufeld, P. Savi, J.-M. Herbert, F. Bono, Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 108, 1243–1250(2006).
30. J. M. Churko, P. W. Burridge, J. C. Wu, Generation of human iPSCs from human peripheralblood mononuclear cells using non-integrative Sendai virus in chemically definedconditions. Methods Mol. Biol. 1036, 81–88 (2013).
31. S. Tohyama, F. Hattori, M. Sano, T. Hishiki, Y. Nagahata, T. Matsuura, H. Hashimoto,T. Suzuki, H. Yamashita, Y. Satoh, T. Egashira, T. Seki, N. Muraoka, H. Yamakawa, Y. Ohgino,T. Tanaka, M. Yoichi, S. Yuasa, M. Murata, M. Suematsu, K. Fukuda, Distinct metabolic flowenables large-scale purification of mouse and human pluripotent stem cell-derivedcardiomyocytes. Cell Stem Cell 12, 127–137 (2013).
32. I. Banerjee, K. Carrion, R. Serrano, J. Dyo, R. Sasik, S. Lund, E. Willems, S. Aceves, R. Meili,M. Mercola, J. Chen, A. Zambon, G. Hardiman, T. A. Doherty, S. Lange, J. C. del Álamo,V. Nigam, Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth,and expression while repressing Tgf-b signaling. J. Mol. Cell. Cardiol. 79, 133–144 (2015).
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
33. R. B. Natale, S. Thongprasert, F. A. Greco, M. Thomas, C.-M. Tsai, P. Sunpaweravong,D. Ferry, C. Mulatero, R. Whorf, J. Thompson, F. Barlesi, P. Langmuir, S. Gogov,J. A. Rowbottom, G. D. Goss, Phase III trial of vandetanib compared with erlotinib inpatients with previously treated advanced non–small-cell lung cancer. J. Clin. Oncol. 29,1059–1066 (2011).
34. E. A. Perez, M. Koehler, J. Byrne, A. J. Preston, E. Rappold, M. S. Ewer, Cardiac safety oflapatinib: Pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin. Proc. 83,679–686 (2008).
35. O. Gunnarsson, N. R. Pfanzelter, R. B. Cohen, S. M. Keefe, Evaluating the safety andefficacy of axitinib in the treatment of advanced renal cell carcinoma. Cancer Manag. Res.7, 65–73 (2015).
36. W. T. A. van der Graaf, J. Y. Blay, S. P. Chawla, D.-W. Kim, B. Bui-Nguyen, P. G. Casali,P. Schöffski, M. Aglietta, A. P. Staddon, Y. Beppu, A. Le Cesne, H. Gelderblom, I. R. Judson,N. Araki, M. Ouali, S. Marreaud, R. Hodge, M. R. Dewji, C. Coens, G. D. Demetri,C. D. Fletcher, A. P. Dei Tos, P. Hohenberger, E. S. Tissue, G. Bone Sarcoma; EORTC SoftTissue and Bone Sarcoma Group and the PALETTE Study Group, Pazopanib for metastaticsoft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3trial. Lancet 379, 1879–1886 (2012).
37. J. E. Cortes, D.-W. Kim, J. Pinilla-Ibarz, P. le Coutre, R. Paquette, C. Chuah, F. E. Nicolini,J. F. Apperley, H. J. Khoury, M. Talpaz, J. DiPersio, D. J. DeAngelo, E. Abruzzese, D. Rea,M. Baccarani, M. C. Müller, C. Gambacorti-Passerini, S. Wong, S. Lustgarten, V. M. Rivera,T. Clackson, C. D. Turner, F. G. Haluska, F. Guilhot, M. W. Deininger, A. Hochhaus,T. Hughes, J. M. Goldman, N. P. Shah, H. Kantarjian; PACE Investigators, A phase 2 trial ofponatinib in Philadelphia chromosome–positive leukemias. N. Engl. J. Med. 369,1783–1796 (2013).
38. A. Grothey, E. Van Cutsem, A. Sobrero, S. Siena, A. Falcone, M. Ychou, Y. Humblet,O. Bouché, L. Mineur, C. Barone, A. Adenis, J. Tabernero, T. Yoshino, H.-J. Lenz,R. M. Goldberg, D. J. Sargent, F. Cihon, L. Cupit, A. Wagner, D. Laurent; CORRECT StudyGroup, Regorafenib monotherapy for previously treated metastatic colorectal cancer(CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial.Lancet 381, 303–312 (2013).
39. M. Schmidinger, C. C. Zielinski, U. M. Vogl, A. Bojic, M. Bojic, C. Schukro, M. Ruhsam,M. Hejna, H. Schmidinger, Cardiac toxicity of sunitinib and sorafenib in patients withmetastatic renal cell carcinoma. J. Clin. Oncol. 26, 5204–5212 (2008).
40. J. Zang, S. Wu, L. Tang, X. Xu, J. Bai, C. Ding, Y. Chang, L. Yue, E. Kang, J. He, Incidence andrisk of QTc interval prolongation among cancer patients treated with vandetanib: Asystematic review and meta-analysis. PLOS ONE 7, e30353 (2012).
41. H. M. Kantarjian, J. E. Cortes, D. W. Kim, H. J. Khoury, T. H. Brummendorf, K. Porkka,G. Martinelli, S. Durrant, E. Leip, V. Kelly, K. Turnbull, N. Besson, C. Gambacorti-Passerini,Bosutinib safety and management of toxicity in leukemia patients with resistance orintolerance to imatinib and other tyrosine kinase inhibitors. Blood 123, 1309–1318 (2014).
42. M. Talpaz, N. P. Shah, H. Kantarjian, N. Donato, J. Nicoll, R. Paquette, J. Cortes, S. O’Brien,C. Nicaise, E. Bleickardt, M. A. Blackwood-Chirchir, V. Iyer, T.-T. Chen, F. Huang,A. P. Decillis, C. L. Sawyers, Dasatinib in imatinib-resistant Philadelphia chromosome–positive leukemias. N. Engl. J. Med. 354, 2531–2541 (2006).
43. R. Kerkela, L. Grazette, R. Yacobi, C. Iliescu, R. Patten, C. Beahm, B. Walters, S. Shevtsov,S. Pesant, F. J. Clubb, A. Rosenzweig, R. N. Salomon, R. A. Van Etten, J. Alroy, J.-B. Durand,T. Force, Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 12,908–916 (2006).
44. T. D. Kim, P. le Coutre, M. Schwarz, P. Grille, M. Levitin, S. Fateh-Moghadam, F. J. Giles,B. Dörken, W. Haverkamp, C. Köhncke, Clinical cardiac safety profile of nilotinib.Haematologica 97, 883–889 (2012).
45. E. Bronte, G. Bronte, G. Novo, F. Bronte, M. G. Bavetta, G. Lo Re, G. Brancatelli, V. Bazan,C. Natoli, S. Novo, A. Russo, What links BRAF to the heart function? New insights from thecardiotoxicity of BRAF inhibitors in cancer treatment. Oncotarget 6, 35589–35601 (2015).
46. G. Kim, A. E. McKee, Y.-M. Ning, M. Hazarika, M. Theoret, J. R. Johnson, Q. C. Xu, S. Tang,R. Sridhara, X. Jiang, K. He, D. Roscoe, W. D. McGuinn, W. S. Helms, A. M. Russell,S. P. Miksinski, J. F. Zirkelbach, J. Earp, Q. Liu, A. Ibrahim, R. Justice, R. Pazdur, FDAapproval summary: Vemurafenib for treatment of unresectable or metastatic melanomawith the BRAFV600E mutation. Clin. Cancer Res. 20, 4994–5000 (2014).
47. J. R. Infante, L. A. Fecher, G. S. Falchook, S. Nallapareddy, M. S. Gordon, C. Becerra,D. J. DeMarini, D. S. Cox, Y. Xu, S. R. Morris, V. G. R. Peddareddigari, N. T. Le, L. Hart,J. C. Bendell, G. Eckhardt, R. Kurzrock, K. Flaherty, H. A. Burris III, W. A. Messersmith, Safety,pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitortrametinib: A phase 1 dose-escalation trial. Lancet Oncol. 13, 773–781 (2012).
48. J. C. Byrd, J. R. Brown, S. O’Brien, J. C. Barrientos, N. E. Kay, N. M. Reddy, S. Coutre,C. S. Tam, S. P. Mulligan, U. Jaeger, S. Devereux, P. M. Barr, R. R. Furman, T. J. Kipps,F. Cymbalista, C. Pocock, P. Thornton, F. Caligaris-Cappio, T. Robak, J. Delgado,S. J. Schuster, M. Montillo, A. Schuh, S. de Vos, D. Gill, A. Bloor, C. Dearden, C. Moreno,J. J. Jones, A. D. Chu, M. Fardis, J. McGreivy, F. Clow, D. F. James, P. Hillmen; RESONATEInvestigators, Ibrutinib versus ofatumumab in previously treated chronic lymphoidleukemia. N. Engl. J. Med. 371, 213–223 (2014).
12 of 13

SC I ENCE TRANS LAT IONAL MED I C I N E | R E S EARCH ART I C L E
Dow
nloa
49. A. Tartarone, G. Gallucci, C. Lazzari, R. Lerose, L. Lombardi, M. Aieta, Crizotinib-inducedcardiotoxicity: The importance of a proactive monitoring and management. Future Oncol.11, 2043–2048 (2015).
50. E. T. Yeh, MD Anderson Practices in Onco-Cardiology (University of Texas MD AndersonCancer Center, 2016).
Acknowledgments: We thank K. Red-Horse for assistance with confocal imaging. We alsothank E. Yeh from the MD Anderson Cancer Center for information regarding TKI usage andcardiotoxicity prevalence. We acknowledge the Stanford High-Throughput Bioscience Center forassistance with high-throughput imaging and plate reader assays. We thank A. Olsonfrom the Stanford Neuroscience Microscopy Service for help with calcium imaging. Funding:We acknowledge support from the American Heart Association Predoctoral Fellowship(13PRE15770000) and NSF Graduate Research Fellowship (DGE-114747) (A.S.); NIH (K99/R00HL121177) and American Heart Association Beginning Grant-in-Aid (14BGIA20480329) (P.W.B.); NIHDirector’s Pioneer Award, American Heart Association Grant-in-Aid, and Endowed Faculty ScholarAward of the Lucile Packard Foundation for Children and Child Health Research Institute at Stanford(S.M.W.); and Burroughs Wellcome Foundation Innovation in Regulatory Science, American HeartAssociation Established Investigator Award, and NIH (R01 HL132875, R01 HL130020, R01 HL128170,R01 HL123968, and R24 HL117756) (J.C.W.). Author contributions: A.S., P.W.B., and J.C.W.designed the study and participated in data analysis and manuscript writing. A.S. and P.W.B.participated in all experimental work. W.L.M. and R.S. conducted high-throughput contractilityassessments. P.S. performed patch clamp electrophysiology. N.S. assisted with deriving hiPSC-CMs and hiPSC-ECs from patients receiving TKI treatment. J.M.C. performed microarray andRNA-seq analysis. T.K. assisted with high-throughput toxicity analysis. H.W. conducted calcium
Sharma et al., Sci. Transl. Med. 9, eaaf2584 (2017) 15 February 2017
imaging. A.H. conducted hiPSC-EC differentiation and characterization. E.M. assisted with geneexpression analysis. Y.Z. performed immunocytochemistry and edited the manuscript. A.K.performed immunocytochemistry and edited the manuscript. A.C.F. assisted with patientrecruitment. J.C.d.A., S.M.W., J.J.M., M.M., and J.C.W. assisted with study design and manuscriptediting. Competing interests: M.M. holds equity in and is on the scientific advisory board forVala Sciences, a company offering high-content screening services, particularly instrumentationused for measuring the electrical and contractile physiology of cardiomyocytes, and is on thescientific advisory board of Stem Cell Theranostics, a company that uses patient-specifichiPSC-CMs for drug discovery. J.C.W. is a cofounder and is on the scientific advisory board ofStem Cell Theranostics. Other authors declare that they have no competing interests. Dataand materials availability: The gene expression data are found at Gene Expression Omnibuswith accession no. GSE8894 and GSE89411.
Submitted 14 January 2016Resubmitted 21 July 2016Accepted 21 November 2016Published 15 February 201710.1126/scitranslmed.aaf2584
Citation: A. Sharma, P. W. Burridge, W. L. McKeithan, R. Serrano, P. Shukla, N. Sayed,J. M. Churko, T. Kitani, H. Wu, A. Holmström, E. Matsa, Y. Zhang, A. Kumar, A. C. Fan,J. C. del Álamo, S. M. Wu, J. J. Moslehi, M. Mercola, J. C. Wu, High-throughput screening oftyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl.Med. 9, eaaf2584 (2017).
ded
13 of 13
by guest on August 10, 2019
http://stm.sciencem
ag.org/ from

induced pluripotent stem cellsHigh-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human
Juan C. del Álamo, Sean M. Wu, Javid J. Moslehi, Mark Mercola and Joseph C. WuChurko, Tomoya Kitani, Haodi Wu, Alexandra Holmström, Elena Matsa, Yuan Zhang, Anusha Kumar, Alice C. Fan, Arun Sharma, Paul W. Burridge, Wesley L. McKeithan, Ricardo Serrano, Praveen Shukla, Nazish Sayed, Jared M.
DOI: 10.1126/scitranslmed.aaf2584, eaaf2584.9Sci Transl Med
anticancer drugs are cardiotoxic.limit the toxicity of this class of drug. This screening method is expected to reveal early on whether potential up-regulating protective insulin/IGF pathways, prompting the authors to devise a combination treatment that mayanalysis even revealed that VEGFR2-inhibiting drugs caused cells to try to compensate for the toxic effects by highly informative, with low values corresponding to those drugs known to cause heart problems in patients. Thetransduction and integrating the results, they calculated a drug-specific ''cardiac safety index.'' This index proved inhibitors altered their physiology. By measuring cell death, contraction, excitability, calcium dynamics, and signalcells from human induced pluripotent stem cells and then examined how a battery of anticancer tyrosine kinase
saving money, time, and perhaps lives. To this end, Sharma and colleagues derived heart−−halt development Discovery early in its life cycle that an anticancer drug causes heart damage (a common side effect) can
Failing fast for tyrosine kinase inhibitors
ARTICLE TOOLS http://stm.sciencemag.org/content/9/377/eaaf2584
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2017/02/13/9.377.eaaf2584.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/11/497/eaav1386.fullhttp://stm.sciencemag.org/content/scitransmed/11/478/eaau8866.fullhttp://stm.sciencemag.org/content/scitransmed/10/435/eaah5457.full
REFERENCES
http://stm.sciencemag.org/content/9/377/eaaf2584#BIBLThis article cites 49 articles, 16 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.Science Translational Medicinetitle licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. TheScience, 1200 New York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
by guest on August 10, 2019
http://stm.sciencem
ag.org/D
ownloaded from





























