Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 91, pp. 12882-12886, December 1994Biochemistry
Transcription of histone H4, H3, and Hi cell cycle genes: Promoterfactor HiNF-D contains CDC2, cyclin A, and an RB-related protein
(cell proliferation/gene expression/cyclin-dependent kinase/tumor suppressor/protein phosphorylation)
ANDRE J. VAN WIJNEN*, FARAH AzIz*, XAVIER GRANiAt, ANTONIO DE LUCAt, RAJESH K. DESAI*,KAREN JAARSVELD*, THOMAS J. LAST*, KENNETH SOPRANO§, ANTONIO GIORDANOt, JANE B. LIAN*,JANET L. STEIN*, AND GARY S. STEIN**Department of Cell Biology, University of Massachusetts Medical School and Cancer Center, 55 Lake Avenue North, Worcester, MA 01655; tFels Institutefor Cancer Research and Molecular Biology, Temple University School of Medicine, 3307 North Broad Street, Philadelphia, PA 19140; §Department ofMicrobiology and Immunology, Temple University, 3400 North Broad Street, Philadelphia, PA 19140; and tInstitute for Cancer Research and MolecularMedicine, Jefferson Cancer Institute, Thomas Jefferson University, Philadelphia, PA 19107
Communicated by Sheldon Penman, August 25, 1994
ABSTRACT Cell cycle-controlled human histone genes arecoordinately expressed during S phase, and transcriptionalregulation involves a series of trans-acting factors (HiNFs). Theproliferation-specific factor HiNF-D interacts with multiplerecognition motifs in histone H4, H3, and Hi promoters. Usinggel shift immunoassays, we show that CDC2, cyclin A, and anRB-related protein are ubiquitous subunits of HiNF-D bindingactivity isolated from several cell types. HiNF-D levels in vivoare sensitive to okadaic acid and staurosporine, indicating thatHiNF-D activity and/or assembly is influenced by phosphor-ylation status. Thus, HiNF-D appears to be a multicomponentphosphoprotein that participates in coordinate control of mul-tiple histone H4, H3, and Hi genes during the cell cycle. Thepresence of cell cycle mediators in the HiNF-D complex suggestslinkage between transcriptional control of histones, enzymesinvolved in DNA synthesis, and the onset of DNA replicationduring the G1/S phase transition.
The onset of proliferation from quiescence occurs via asequential series of biochemical regulatory events that cul-minate in cell cycle progression into S phase (1). Transitionfrom G1 to S phase involves the transcriptional and posttran-scriptional regulation of genes encoding enzymes used innucleotide metabolism and DNA synthesis (2), as well ashistone genes encoding proteins that are absolutely requiredfor the packaging of newly replicated DNA (3, 4). Control ofG1 phase progression into S phase is determined in part bytumor-suppressor proteins, including p53 and members oftheRB family (5-10). These important regulatory factors governboth directly and indirectly the transcription of cell cyclestage-specific genes by an intricate molecular interplay withmultiple distinct cyclins (11-14), CDC2-related kinases (15-20), E2F-related DNA-binding activities (21-23), and inhib-itory factors. The activities of cyclin-dependent kinases arerequired for cell cycle regulatory events during the G1/S andG2/M transitions.
Cell cycle-controlled human histone H4, H3, H2B, H2A,and Hi genes represent a series of five multigene familieswith each comprising 10-20 distinct gene copies (3). Histonegene regulation is mediated by distinct arrays of promoterelements that determine maximal levels of transcriptionduring early S phase (24, 25). The specific complement ofpromoter factors, as well as the arrangement of the cognateelements, is different for each gene, which suggests a degreeof heterogeneity in transcriptional control of individual his-tone genes. However, recently we have established recog-nition sites for a shared, proliferation-specific promoter fac-
tor, HiNF-D, interacting with multiple histone (H4, H3, andH1) genes (26-28). Regulation of the interaction of HiNF-Dwith these genes correlates with modulations in histone geneexpression (29-34).
Determination of the molecular composition of HiNF-D isnecessary to establish the role of this factor in the coordina-tion of histone gene transcription and to functionally linkcontrol of histone gene expression with the molecular cir-cuitry involved in cell cycle progression. We have used anextensive set of immunological reagents to assess a relation-ship between HiNF-D and cell cycle related proteins, and weshow data suggesting HiNF-D is a multisubunit phosphopro-tein containing CDC2, cyclin A, and an RB-related protein.
MATERIALS AND METHODS
Immunological Reagents. Preparation of specific anti-C-terminal peptide antibodies to CDC2 (G6) (15) and CDK2(35), as well as monoclonal anti-cyclin A (C160) (11) andmonoclonal antibodies to RB (XZ77, XZ104, and XZ133)(36), has been described. Polyclonal antibodies directedagainst cyclins A, B, Dl, D2, and D3 were produced againstglutathione S-transferase fusion proteins. Polyclonal anti-cyclin E, anti-Schizosaccharomycespombe CDC2 (G8), anti-PSTAIRE, -ERK III, and -CDC2/CT, and anti-p107 andmonoclonal antibodies to p107 (SD2, SD6, and SD9) werekindly provided.
Gel Shift Assays. Nuclear extracts containing HiNF-Dbinding activity, 32P-labeled probes, and competitor oligonu-cleotides (Table 1) were prepared as described (26-28).Binding reaction mixtures for HiNF-D were electrophoresedin 4% (80:1 acrylamide/N,N'-methylenebisacrylamideweight ratio) polyacrylamide gels in 0.5x TBE buffer (37).Gel supershift assays were carried out by preincubation ofnuclear proteins (10 ,ul; 2-5 jig) with antibody (1 ,ul) for 15 minon ice, followed by addition of 10 Al ofDNA mixture (27) andincubation for 15 min at room temperature prior to electro-phoresis. Binding-site selection by immunoprecipitation wasperformed with nuclear proteins after preclearance withrabbit preimmune serum and protein A-Sepharose. Pre-cleared samples were incubated for 15 min with antiserum (inthe same protein-to-antibody ratio as in supershift assays)and a mixture of 32P-labeled wild-type (nt -97 to -38 pluslinker at nt -38; 72 bp) and mutant (SUB-11; nt -93 to -53plus linkers, 49 bp) HiNF-D binding sites. After immunopre-cipitation of protein/DNA complexes, the pellet was boiledin SDS sample buffer and retention of wild-type and mutantDNA fragments was monitored by electrophoresis in nonde-naturing 12% (30:1) polyacrylamide gels.
12882
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 91 (1994) 12883
Table 1. OligonucleotidesBinding*
Name Sequence (5' to 3') D M PTM-3 CGCTTTC GGTTTTCAAT CTGGTCCGAT ACTCTTGTAT ATCA + + +SUB-11 CGCTTTC GGTTcTCAgT tcGGTCCGcc ACTCTTGTAT ATCA - + +INS-10 CGCTTTCaGGTTTTCAAT CTGGTCCGATaACTCTTGTAT ATCA + - +NH-6 CGCTTTCaGGTTcTCAgT tcGGTCCGccaACTgTcGTAT Aaag - - +MC-7 CGCTgTC taTgTTacAT aTGGTCCGAT ACTCTTGTAT ATCA ± - -GT-9 CGCTTTC GGTTTTCAAT Cgtcgaatgc ACTCTTGTAT ATCA + + -ST-8 CGCTTTC GGTTTTCAAT CTGGTCCGAT ctgtcacgog cTCA ± + +Spl CCCGGCC CCGCCCATCC CCGGCCCCGC CCATCCG - - -
The wild-type TM-3 oligonucleotide spans nt -93 to -53, encompassing site II of the H4 gene. Theremaining oligonucleotides (except nonspecific competitor Spl) contain specific mutations within thissequence (lowercase).*Binding characteristics of HiNF-D, -M, and -P with each oligonucleotide: +, wild-type; binding ±,decreased binding; -, no binding.
RESULTS
Histone gene transcription factor HiNF-D binds in conjunc-tion with HiNF-M and HiNF-P at the multipartite cell cycledomain of the H4 promoter (site II) (Fig. 1) (38-40). HiNF-Pis a cell growth-regulated factor, similar to the H4 subtype-specific factor H4-TF2 (27, 34, 41). HiNF-M interacts with a
key regulatory motif in the distal part of site II (26, 27, 40) andis a growth-responsive IRF-related 48-kDa protein that re-cently has been purified to homogeneity (P. S. Vaughan,J.L.S., and G.S.S.). HiNF-D is a ubiquitous, proliferation-specific factor (26-29) and mediates a protein/DNA complexof very low mobility in gel shift assays (Fig. 2).
Cyclin-Dependent Kinase CDC2 Is a Subunit ofHiNF-D. Weanalyzed immunoreactivity of the HiNF-D complex in gelshift assays with several antibodies directed against thecyclin-dependent kinases CDC2 and CDK2. HiNF-D activitywas consistently immunoreactive with a CDC2-specific pep-tide antiserum (designated G6), as reflected by a selectiveantibody-dependent supershift in HiNF-D mobility (Fig. 2A).No effects on HiNF-D mobility were observed with CDK2antiserum. Extending the duration of electrophoresis in-creased resolution between the HiNF-D and HiNF-D/antibody complexes and revealed a substantial decrease inmobility of the HiNF-D complex in the presence of theCDC2/G6 antiserum (Fig. 2B). Heat-inactivated antiserumhad no effect on HiNF-D mobility (Fig. 2C), which rules outionic effects on the migration of the HiNF-D complex.
HiFiN iF-P
- H4-Site H4-Site 1)-971 -38
F-DH3
3-ite 1H3H3 iteite~~~~~~~~~~~~~-200-------- 0
Hil -SF rHN F-B
Hi
Site Ii-213 -78
FIG. 1. Protein/DNA interactions in H4, H3, and Hi genepromoters. The probe for the human H4 gene (thick line) bindsfactors HiNF-D, -M, and -P and spans the in vivo protein/DNAinteraction domain H4 site II (rectangle) that is essential for cell cycleregulation (24). Probes for the H3 and Hi promoters span analogousHiNF-D binding domains (hatching) which are composed of corebinding elements and auxiliary sequences that enhance efficiency ofbinding (28). Additional transcription factors are indicated withcircles.
Incubation of HiNF-D with antibody in the presence of theimmunogenic peptide abolished the supershift of HiNF-D(Fig. 2C), indicating that this peptide competed with theHiNF-D epitope for the CDC2 antibody. Thus, CDC2 is anintrinsic subunit ofthe HiNF-D complex which interacts withthe H4 gene cell cycle domain.
Mutation of six specific nucleotides (mutant SUB-il) in H4site II abolished formation of both the HiNF-D complex andthe corresponding HiNF-D/antibody complex (data notshown). Also, mutant SUB-il failed to compete for HiNF-D-binding to site II (e.g., Fig. 2A). To assess whetherCDC2-containing complexes can discriminate between theintact site II cell cycle domain and the mutant site II se-quences, we designed a variant binding-site selection proce-dure. A mixture of radiolabeled wild-type and mutantHiNF-D binding-site oligonucleotides were incubated in thepresence of several antisera (Fig. 2D). After immunoabsorp-tion of ternary DNA/protein/antibody complexes with pro-tein A-Sepharose beads, we observed preferential retentionof the wild-type HiNF-D binding site in the precipitate onlywhen the anti-CDC2 G6 antiserum) was present. We con-clude that CDC2 complexes can selectively recognize the siteII cell cycle domain, and that at least six nucleotides knownto influence HiNF-D binding are required for this CDC2/H4gene interaction.
Cyclin A and an RB-Related Protein Are Additional Com-ponents of the HiNF-D Complex. The presence of the cyclin-dependent kinase CDC2 in the HiNF-D complex could pre-dict a cyclin component. Indeed, highly concentrated cyclinA monoclonal antibody (C160) selectively blocked formationof the HiNF-D complex, but inhibition was not observedwhen the antibody was heat inactivated (Fig. 2C). Otherinvestigators have also shown that the cyclin A antibodyC160 disrupts cyclin A-containing protein/DNA complexes(42). Immunoreactivity was not observed with several anti-sera directed against cyclins B, Dl, D2, D3, and E (Fig. 2Cand data not shown).We also explored the presence of RB-related proteins in
the HiNF-D complex. Similar to results obtained for cyclinA, we observed inhibition of HiNF-D binding activity with amonoclonal antibody, XZ104 (36), against the RB family ofproteins (Fig. 2E). Weak inhibition was also observed for RBmonoclonal antibodies XZ77 and XZ133, as well as a p107polyclonal antiserum, although a control hybridoma super-natant or a panel of monoclonal antibodies directed againstthe p107 protein did not disrupt the HiNF-D complex (Fig. 2Eand data not shown). Thus, cyclin A and an RB-relatedprotein appear to be closely associated with HiNF-D bindingactivity.HiNF-D-Related Complexes with H3 and Hi Promoters Also
Contain CDC2, Cyclin A and an RB-Related Protein. Cross-competition assays have revealed that low mobility com-
Biochemistry: van Wijnen et al.

12884 Biochemistry: van Wijnen et al.
A DNA competitors+
.+ +
-i2+ 2+E+or- o----
C) >0 :
antibodies
co0_X C\ C\J
>-0 00C) Y>5 Q a
...... ..
....
1 2 3 4 5 6 7 8
Dantibodies
c'N W
-0
0coz
CooC) rCo EGXKREB/X2
C) F-v LOa
C\!
..
~~~vpw.mMUT
1 2 3 4 5
B DNA comretitors antibodies+
C 0(5
m+ X,-J CaY
.. .... ..~~~~~1 2 345 67 8
Z104 CONTROLr
uCoOC Loo oo -0- N. . PI
C antibodies
C)< Co C\ +\-jz ow
.............::-
........ . ..
+AT-1W
w_
--D1 2 3 4 5
F antibodies
-: 0 0 jZj m o DNA competitors0000 m -0
c+ I C S N'
DW .O *11 ".W *w 32p=H3
*D 0 D
1 2 3 4 5 6 7 8 9 10
32P=H1
1 2 3 4 5 6 7 8 9 10
FIG. 2. HiNF-D contains CDC2, cyclin A, and an RB-related protein. (A) Gel shift assay with the H4 probe and HeLa protein (5 ,ug).Competition analysis (Left) was performed with a 100-fold molar excess of the following oligonucleotides: lanes 2-5, TM-3, GT-9, INS-10, andSUB-11 (see Table 1), respectively; lane 1, no competitor DNA (control, CTRL). These DNA fragments contain specific point mutations whichabolish binding ofHiNF-D, -M, or -P as indicated by minus signs above the lanes (27). (Right) Immunoreactivity ofHiNF-D with CDC2 antiserum(G6), but not a CDK2 antiserum (1 ,ul of each), is shown in the right portion (lanes 6-8). Complexes mediated by HiNF-D and -M, as well asthe supershifted HiNF-D complex (D+a) are indicated by arrowheads. No effects on mobility of HiNF-D were observed with polyclonalantibodies directed against the entire CDC2 protein (G8) or a C-terminal region of CDC2 (CDC2/CT) (data not shown). (B) Binding reactionswith antibodies (lanes 6-8) and competition analysis using oligonucleotides TM-3, SUB-11, INS-10, and NH-6 (lanes 2-5, respectively) wereset up as in A, but electrophoresis time was extended. (C) Gel shift assay as in panel A, using antibodies against cyclin A (C160) (aCLN A),cyclin B (aCLN B), and CDC2 (G6) (aCDC2). Antibodies were directly incubated with nuclear protein (Upper; -AT) or subjected to prior heatinactivation (Lower, +AT). Absence of a HiNF-D supershift was observed upon coincubation of the CDC (G6) antibody in the presence of theimmunogenic peptide (aCDC2+PEP) (Upper, lane 5) or upon heat inactivation of the CDC2 (G6) antibody (Lower, lane 4). HiNF-D bindingwas inhibited with cyclin A (C160) antibody (Upper, lane 2) but not when the antibody was heat inactivated (Lower, lane 2). Antibodies directedagainst cyclins B, Di, D2, D3, and E were not reactive (lanes 3 and data not shown). (D) Binding-site selection assay monitoring retention ofa mixture of wild-type (WT, 72 bp) and mutant (MUT, 49 bp) DNA fragments upon immunoprecipitation ofbinding reaction mixtures. The MUTfragment was initially present in 2-fold molar excess over the WT fragment (lane 1, input DNA mixture). Selective retention ofWT was observedin the presence of the supershifting CDC2 (G6) antibody (lane 2), but not with control antibodies (lanes 3-5, respectively, aCDC2/G8,aRB/XZ104, and aCLN E). (E) Gel shift assay as in A, but monitoring the inhibitory effects of increasing amounts (0-1.0 gl) of RB (XZ104)antibody (lanes 1-5) and the absence of this effect with a control hybridoma supematant (lanes 6-10). Weak immunoreactivity was observedwith RB antibodies XZ77 and XZ133, as well as with a p107 polyclonal antibody (data not shown). (F) Gel shift assay with the H3 (Upper) andHi (Lower) probes and a HiNF-D fraction from HeLa cells (27). Sequence specificity of HiNF-D binding with each gene is illustrated bycompetition analysis [lanes 9-11, respectively, no competitor (C), specific (S) TM-3 fragment, and nonspecific (N) Spl oligonucleotide]. Thesame antibodies reactive with HiNF-D were used as in C and E. The HiNF-D complexes (D) and the corresponding supershifts with CDC2 (G6)antibody (a+D) are indicated by arrowheads.
plexes forming on H3 and Hi promoter fragments in gel shiftassays are mediated by the same DNA-binding activityintrinsic to the H4 gene/HiNF-D complex (28). To investi-gate whether additional components are shared, we analyzedimmunoreactivity ofthe H3 and Hi promoter complexes withthe CDC2 (G6), cyclin A (C160), and RB (XZ104) antibodies.The results indicate that HiNF-D complexes observed withall three promoters react specifically with the same antibod-ies (Fig. 2F and data not shown). Thus, the principal HiNF-Dcomplex for each of the three histone genes contains at leastthree distinct shared non-DNA-binding subunits.
Recognition of H4 Site II by a 76 kDa Factor. To assess themolecular weights of proteins directly binding to the HiNF-Drecognition sequence, we performed UV crosslinking with aprobe spanning H4 site II (Fig. 3A). To enhance the speci-ficity of the assay(we designed a probe that contains a majorcluster of 32P-labeled dG nucleotides immediately adjacent tocrosslinkable BrdU residues. This cluster of radioactive and
crosslinking labels is located exactly in the region of site IIrequired for sequence-specific binding of HiNF-D based onmethylation-interference and mutational analysis (26, 27).The results show reproducible crosslinking ofa 76-kDa factor(p76) (Fig. 3A). Neither molecular -size nor competitionspecificity of this factor match that of HiNF-M (48 kDa) orHiNF-P/H4-TF2 (65 kDa). The competition characteristicsof p76 with- several site II wild-type and mutant binding-siteoligonucleotides are consistent with this factor recognizing(and crosslinking to) sequences required for the HiNF-D/siteII interaction. Thus, this factor may represent an intrinsicDNA-binding component of HiNF-D.
Association of HiNF-D Components into Protein/ProteinComplexes. The identification of three non-DNA-bindingcomponents (CDC2/p34, cyclin A/p60, and an RB-relatedprotein) and the putative p76 DNA-binding subunit, as wellas maintenance of HiNF-D binding activity during severalchromatography procedures (27), suggests that HiNF-D
Proc. Natl. Acad. Sci. USA 91 (1994)
Proc. Natl. Acad. Sci. USA 91 (1994) 12885
AA DNA competitorsn CO o) CD 'N~CDo HcoI)(r~Zc)
kDa-97
p76_o - -66
-45
1 2 3 4 5 6 7
UV-crosslinking probe:
5-- GATCCGC TTTCGGTTTTCAATC TGGTCCGAT ---3r-CBAGGCGAAAGCCAAAAGBBAGACCAGGCBA ---
O** * * *00 * ** 0 --,* * s es primer
key recognition basepairs
B
15Gradient fraction
C
c,ji
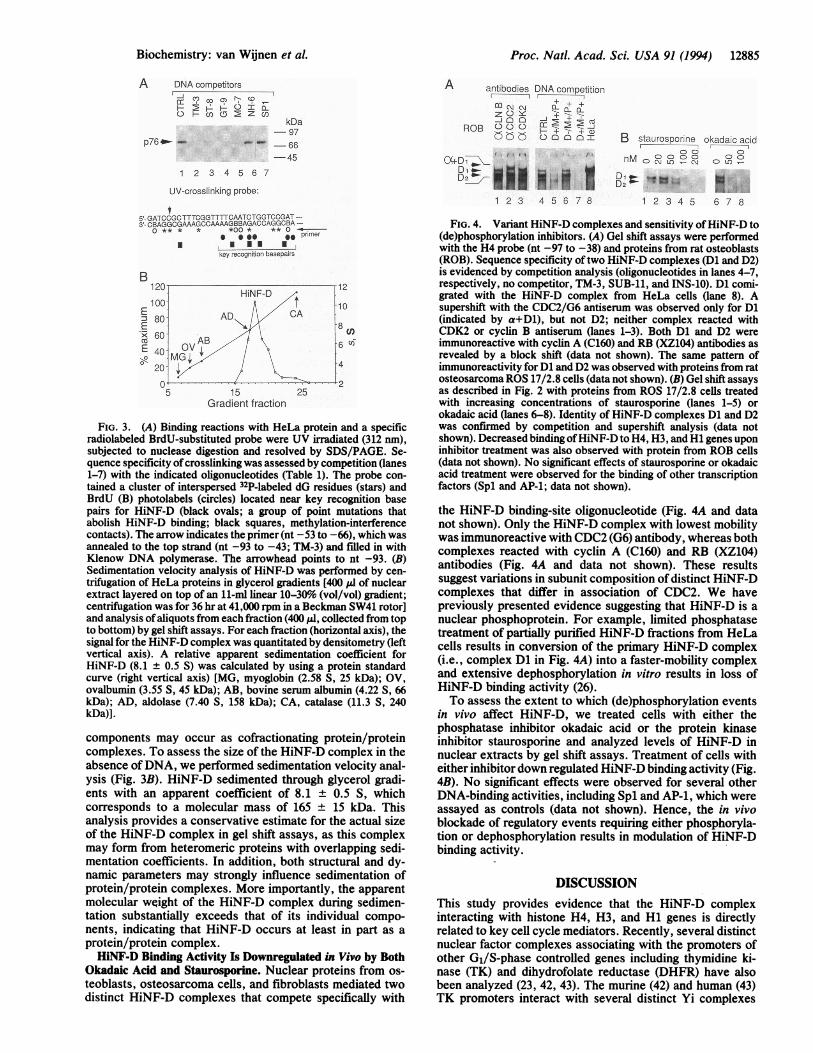
FIG. 3. (A) Binding reactions with HeLa protein and a specificradiolabeled BrdU-substituted probe were UV irradiated (312 nm),subjected to nuclease digestion and resolved by SDS/PAGE. Se-quence specificity ofcrosslinking was assessed by competition (lanes1-7) with the indicated oligonucleotides (Table 1). The probe con-tained a cluster of interspersed 32P-labeled dG residues (stars) andBrdU (B) photolabels (circles) located near key recognition basepairs for HiNF-D (black ovals; a group of point mutations thatabolish HiNF-D binding; black squares, methylation-interferencecontacts). The arrow indicates the primer (nt -53 to -66), which wasannealed to the top strand (nt -93 to -43; TM-3) and filled in withKlenow DNA polymerase. The arrowhead points to nt -93. (B)Sedimentation velocity analysis of HiNF-D was performed by cen-trifugation of HeLa proteins in glycerol gradients [400 PI of nuclearextract layered on top of an 11-ml linear 10-30% (vol/vol) gradient;centrifugation was for 36 hr at 41,000 rpm in a Beckman SW41 rotor]and analysis ofaliquots from each fraction (400 ,ud, collected from topto bottom) by gel shift assays. For each fraction (horizontal axis), thesignal for the HiNF-D complex was quantitated by densitometry (leftvertical axis). A relative apparent sedimentation coefficient forHiNF-D (8.1 ± 0.5 S) was calculated by using a protein standardcurve (right vertical axis) [MG, myoglobin (2.58 S, 25 kDa); OV,ovalbumin (3.55 S, 45 kDa); AB, bovine serum albumin (4.22 S, 66kDa); AD, aldolase (7.40 S, 158 kDa); CA, catalase (11.3 S, 240kDa)].
components may occur as cofractionating protein/proteincomplexes. To assess the size of the HiNF-D complex in theabsence ofDNA, we performed sedimentation velocity anal-ysis (Fig. 3B). HiNF-D sedimented through glycerol gradi-ents with an apparent coefficient of 8.1 ± 0.5 S, whichcorresponds to a molecular mass of 165 ± 15 kDa. Thisanalysis provides a conservative estimate for the actual sizeof the HiNF-D complex in gel shift assays, as this complexmay form from heteromeric proteins with overlapping sedi-mentation coefficients. In addition, both structural and dy-namic parameters may strongly influence sedimentation ofprotein/protein complexes. More importantly, the apparentmolecular weight of the HiNF-D complex during sedimen-tation substantially exceeds that of its individual compo-nents, indicating that HiNF-D occurs at least in part as aprotein/protein complex.HiNF-D Binding Activity Is Downregulated in Vivo by Both
Okadaic Acid and Staurosporine. Nuclear proteins from os-teoblasts, osteosarcoma cells, and fibroblasts mediated twodistinct HiNF-D complexes that compete specifically with
antibodies DNA competitionI I
z C) ve CLCmmcn d-iROB 0 c)0D I0 , :
5.5C cD oI
0(+D1 lID1E O..X..
i
12 3 4 5 6 7 8
B staurosporine okadaic acid
CD 0 C "!
1 2 3 4 5 6 7 8
FIG. 4. Variant HiNF-D complexes and sensitivity ofHiNF-D to(de)phosphorylation inhibitors. (A) Gel shift assays were performedwith the H4 probe (nt -97 to -38) and proteins from rat osteoblasts(ROB). Sequence specificity oftwo HiNF-D complexes (Di and D2)is evidenced by competition analysis (oligonucleotides in lanes 4-7,respectively, no competitor, TM-3, SUB-11, and INS-10). Di comi-grated with the HiNF-D complex from HeLa cells (lane 8). Asupershift with the CDC2/G6 antiserum was observed only for Di(indicated by a+D1), but not D2; neither complex reacted withCDK2 or cyclin B antiserum (lanes 1-3). Both Di and D2 wereimmunoreactive with cyclin A (C160) and RB (XZ104) antibodies asrevealed by a block shift (data not shown). The same pattern ofimmunoreactivity for Di and D2 was observed with proteins from ratosteosarcoma ROS 17/2.8 cells (data not shown). (B) Gel shift assaysas described in Fig. 2 with proteins from ROS 17/2.8 cells treatedwith increasing concentrations of staurosporine (lanes 1-5) orokadaic acid (lanes 6-8). Identity of HiNF-D complexes Di and D2was confirmed by competition and supershift analysis (data notshown). Decreased binding ofHiNF-D to H4, H3, and Hi genes uponinhibitor treatment was also observed with protein from ROB cells(data not shown). No significant effects of staurosporine or okadaicacid treatment were observed for the binding of other transcriptionfactors (Spl and AP-1; data not shown).
the HiNF-D binding-site oligonucleotide (Fig. 4A and datanot shown). Only the HiNF-D complex with lowest mobilitywas immunoreactive with CDC2 (G6) antibody, whereas bothcomplexes reacted with cyclin A (C160) and RB (XZ104)antibodies (Fig. 4A and data not shown). These resultssuggest variations in subunit composition of distinct HiNF-Dcomplexes that differ in association of CDC2. We havepreviously presented evidence suggesting that HiNF-D is anuclear phosphoprotein. For example, limited phosphatasetreatment of partially purified HiNF-D fractions from HeLacells results in conversion of the primary HiNF-D complex(i.e., complex Di in Fig. 4A) into a faster-mobility complexand extensive dephosphorylation in vitro results in loss ofHiNF-D binding activity (26).To assess the extent to which (de)phosphorylation events
in vivo affect HiNF-D, we treated cells with either thephosphatase inhibitor okadaic acid or the protein kinaseinhibitor staurosporine and analyzed levels of HiNF-D innuclear extracts by gel shift assays. Treatment of cells witheither inhibitor down regulated HiNF-D binding activity (Fig.4B). No significant effects were observed for several otherDNA-binding activities, including Spl and AP-1, which wereassayed as controls (data not shown). Hence, the in vivoblockade of regulatory events requiring either phosphoryla-tion or dephosphorylation results in modulation of HiNF-Dbinding activity.
DISCUSSIONThis study provides evidence that the HiNF-D complexinteracting with histone H4, H3, and Hi genes is directlyrelated to key cell cycle mediators. Recently, several distinctnuclear factor complexes associating with the promoters ofother Gu/S-phase controlled genes including thymidine ki-nase (TK) and dihydrofolate reductase (DHFR) have alsobeen analyzed (23, 42, 43). The murine (42) and human (43)TK promoters interact with several distinct Yi complexes
Biochemistry: van Wijnen et al.

12886 Biochemistry: van Wijnen et al.
that are immunoreactive with cyclin A, RB, and/or CDC2antibodies depending on the particular complex analyzed.HiNF-D migrates substantially slower than Yi factors in gelshift assays, and the main form ofHiNF-D contains cyclin A,CDC2, and an RB-related protein within the same protein/DNA complex. We have also observed HiNF-D variants thatare not associated with CDC2. The similarities in composi-tion of Yi complexes binding to the TK gene and HiNF-Dcomplexes interacting with distinct histone genes may reflectsimilarities in regulation of these two classes of genes.Factor HiNF-D is an unusual sequence-specific DNA-
binding activity that interacts in overlapping arrangementswith the cell cycle regulatory promoter sequences of histoneH4, H3, and Hi genes (26-28). HiNF-D binding activity isregulated in conjunction with modulation of histone geneexpression under many different biological circumstances(29-34). Here, we find that the phosphatase-inhibitor okadaicacid and the protein kinase inhibitor staurosporine eachselectively inhibit HiNF-D binding activity. These resultssupport the concept that specific phosphorylation-relatedevents determine HiNF-D binding activity. As staurosporineis a potent inhibitor ofCDC2 kinase (44), the possibility arisesthat inhibition of the enzymatic activity of CDC2 (perhaps ina cyclin A-dependent manner) may negatively affect HiNF-Dbinding activity.
In conclusion, our results corroborate the concept thatHiNF-D is an important multicomponent phosphoprotein(containing CDC2, cyclin A, an RB-like protein, and aputative p76 DNA-binding subunit) that participates in co-ordinate control of multiple H4, H3, and Hi histone genesduring the cell cycle. Furthermore, the presence of cellcycle-related components in HiNF-D/histone gene com-plexes suggests a direct link between transcriptional regula-tion of histone proteins, enzymes involved in nucleotidemetabolism and DNA synthesis, and the start of DNAreplication during the G1/S phase transition.
We thank Mark Birnbaum, Laura McCabe, Rauf Shakoori, RosaMastrototaro, Jack Green, Elizabeth Buffone, Christine Dunshee,Shirwin Pockwinse, and Elizabeth Bronstein for conceptual, tech-nical, and/or editorial support. We thank A. Koff, Y. Xiong, S.Pelech, M. Ewen, and N. Dyson for supplying antibodies. Thesestudies were supported by grants from the National Institutes ofHealth (GM32010, AR39588, AR42262). A.G. received support fromthe W. W. Smith Charitable trust, the Milheim Foundation, and theNational Institutes of Health (CA60999). X.G. was supported by aLady Tata Memorial Award.
1. Pardee, A. (1989) Science 246, 603-608.2. Hofbauer, R. & Denhardt, D. T. (1991) Crit. Rev. Eukaryotic
Gene Expression 1, 247-300.3. Stein, G. S., Stein, J. L. & Marzluff, W. F., eds. (1984) His-
tone Genes (Wiley, New York).4. Osley, M. A. (1991) Annu. Rev. Biochem. 60, 827-861.5. Momand, J., Zambetti, G. P., Olson, D. C., George, D. &
Levine, A. J. (1992) Cell 69, 1237-1245.6. Zhu, L., van den Heuvel, S., Helin, K., Fattaey, A., Ewen, M.,
Livingston, D., Dyson, N. & Harlow, E. (1993) Genes Dev. 7,1111-1125.
7. Mayol, X., Grana, X., Baldi, A., Sang, N., Hu, Q. & Giordano,A. (1993) Oncogene 8, 2561-2566.
8. Ewen, M., Xing, Y. & Livingston, D. (1991) Cell 66, 1155-1164.9. Hannon, D. J., Demetrick, D. & Beach, D. (1993) Genes Dev.
7, 2378-2391.10. Li, Y., Graham, C., Lacy, S., Duncan, A. M. V. & Whyte, P.
(1993) Genes Dev. 7, 2366-2377.11. Giordano, A., Whyte, P., Harlow, E., Franza, B. R., Jr.,
Beach, D. & Draetta, G. (1989) Cell 58, 981-990.12. Koff, A., Cross, F., Fisher, A., Schumacher, J., Leguellec, K.,
Philippe, M. & Roberts, J. M. (1991) Cell 66, 1217-1228.
13. Giordano, A., McCall, C., Whyte, P. & Franza, B. R. (1991)Oncogene 6, 481-485.
14. Matsushime, H., Ewen, M. E., Strom, D. K., Kato, J. Y.,Hanks, S. K., Roussel, M. F. & Sherr, C. J. (1992) Cell 71,323-334.
15. Draetta, G. & Beach, D. (1988) Cell 54, 17-26.16. D'Urso, G., Marraccino, R. L., Marshak, D. R. & Roberts,
J. M. (1990) Science 250, 786-791.17. Elledge, S. J., Richman, R., Hall, F. L., Williams, R. T.,
Lodgson, N. & Harper, J. W. (1992) Proc. Natl. Acad. Sci.USA 89, 2907-2911.
18. Meyerson, M., Endres, G. H., Wu, C. L., Su, L. K., Gorka,C., Nelson, C., Harlow, E. & Tsai, L. H. (1992) EMBO J. 70,337-350.
19. Grania, X., deLuca, A., Sang, N., Fu, Y., Claudio, P. P.,Rosenblatt, J., Morgan, D. 0. & Giordano, A. (1994) Proc.Natl. Acad. Sci. USA 91, 3834-3838.
20. Grafia, X., Claudio, P. P., deLuca, A., Sang, N. & Giordano,A. (1994) Oncogene 9, 2097-2103.
21. Kaelin, W. G., Jr., Krek, W., Sellers, W. R., DeCaprio, J. A.,Ajchenbaum, F., Fuchs, C. S., Li, Y., Farnham, P. J., Blanar,M. A., Livingston, D. M. & Flemington, E. K. (1992) Cell 70,351-364.
22. Lees, J. A., Saito, M., Vidal, M., Valentine, M., Look, T.,Harlow, E., Dyson, N. & Helin, K. (1993) Mol. Cell. Biol. 13,7813-7825.
23. Girling, R., Partridge, J. F., Bandara, L. S., Burden, N., Totty,N. F., Hsuan, J. J. & La Thangue, N. B. (1993) Nature (Lon-don) 362, 83-87.
24. Stein, G. S., Stein, J. L., van Wijnen, A. J. & Lian, J. B. (1994)J. Cell. Biochem. 54, 393-404.
25. Heintz, N. (1991) Biochim. Biophys. Acta 1088, 327-339.26. van Wijnen, A. J., Ramsey-Ewing, A., Bortell, R., Owen,
T. A., Lian, J. B., Stein, J. L. & Stein, G. S. (1991) J. Cell.Biochem. 46, 174-189.
27. van Wijnen, A. J., van den Ent, F. M. I., Lian, J. B., Stein,J. L. & Stein, G. S. (1992) Mol. Cell. Biol. 12, 3273-3287.
28. van den Ent, F. M. I., van Wijnen, A. J., Lian, J. B., Stein,J. L. & Stein, G. S. (1994) J. Cell. Physiol. 159, 515-530.
29. van Wijnen, A. J., Wright, K. L., Lian, J. B., Stein, J. L. &Stein, G. S. (1989) J. Biol. Chem. 264, 15034-15042.
30. Holthuis, J., Owen, T. A., van Wijnen, A. J., Wright, K. L.,Ramsey-Ewing, A., Kennedy, M. B., Carter, R., Cosenza, S.,Soprano, K. J., Lian, J. B., Stein, J. L. & Stein, G. S. (1990)Science 247, 1454-1457.
31. van Wijnen, A. J., Lian, J. B., Stein, J. L. & Stein, G. S. (1991)J. Cell. Physiol. 47, 337-351.
32. van Wijnen, A. J., Choi, T. K., Owen, T. A., Wright, K. L.,Lian, J. B., Jaenisch, R., Stein, J. L. & Stein, G. S. (1991)Proc. Natl. Acad. Sci. USA 88, 2573-2577.
33. Wright, K. L., Dell'Orco, R. T., van Wijnen, A. J., Stein, J. L.& Stein, G. S. (1992) Biochemistry 31, 2812-2818.
34. van den Ent, F. M. I., van Wijnen, A. J., Last, T. J., Bortell,R., Stein, J. L., Lian, J. B. & Stein, G. S. (1993) Cancer Res.53, 2399-2409.
35. Rosenblatt, J., Gu, Y. & Morgan, D. 0. (1992) Proc. Natl.Acad. Sci. USA 89, 2824-2828.
36. Hu, Q., Bautista, C., Edwards, G., Defeo-Jones, D., Jones, R.& Harlow, E. (1991) Mol. Cell. Biol. 11, 5792-5799.
37. Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D.,Seidman, J. G., Smith, J. A. & Struhl, K., eds. (1987) CurrentProtocols in Molecular Biology (Wiley, New York).
38. Pauli, U., Chrysogelos, S., Stein, G., Stein, J. & Nick, H.(1987) Science 236, 1308-1311.
39. Kroeger, P. E., van Wijnen, A. J., Pauli, U., Wright, K. L.,Stein, G. S. & Stein, J. L. (1995) J. Cell. Biochem., in press.
40. Ramsey-Ewing, A. L., van Wijnen, A. J., Stein, G. S. & Stein,J. L. (1994) Proc. Natl. Acad. Sci. USA 91, 4475-4479.
41. Dailey, L., Roberts, S. B. & Heintz, N. (1988) Genes Dev. 7,4582-4584.
42. Li, L.-J., Naeve, G. S. & Lee, A. S. (1993) Proc. Natl. Acad.Sci. USA 90, 3554-3558.
43. Dou, Q.-P., Markell, P. & Pardee, A. B. (1992) Proc. Natl.Acad. Sci. USA 89, 3256-3260.
44. Gadbois, D. M., Hamaguchi, J. R., Swank, R. A. & Bradbury,E. M. (1992) Biochem. Biophys. Res. Commun. 184, 80-85.
Proc. Natl. Acad. Sci. USA 91 (1994)





























