Embed Size (px)
Citation preview

14 Regulations for Medical Devices and Application toPlastics Suppliers: History and Overview
Vinny R. Sastri
O U T L I N E
14.1 History and Introduction 337
14.2 United States Regulations 338
14.2.1 FDA Master Files 340
14.3 ISO 13485 (European and Global
Standard) 34014.3.1 European Union Medical Device
Directive 341
14.4 Other Countries 34314.4.1 Japan 343
14.4.2 China 343
14.4.3 Australia 343
14.4.4 India 344
14.4.5 South America 344
14.5 Global Harmonization Task Force
(GHTF) 344
14.6 Applicability of the Regulations to Material
Suppliers 345
14.7 Conclusion 345
References 346
14.1 History and Introduction
Over the past 2000 years, many devices have
been developed and used in the mitigation and
diagnosis of diseases. The materials used in these
devices have ranged from stone, wood, metal, cera-
mics, and most recently plastics. Medical devices
have also evolved in sophistication and complexity
over time. With the formalization of the scientific
method in the seventeenth century such devices
became more prevalent [1]. Many medical devices
were manufactured by doctors or small companies
and sold directly to the public with no government
standards or oversight. With the explosion of medi-
cal technology in the early twentieth century, sev-
eral intermediaries had evolved between the
medical device industry and the public. In 1879, Dr
E.R. Squibb, in an address to the Medical Society
of the State of New York, proposed the enactment
of a national statute to regulate food and drugs [2].
It was not until 27 years later that the Food and
Drug Act of 1906 was introduced into the Congress
and signed into law by President Theodore
Roosevelt [3]. At that time, devices that were harm-
ful to human safety and health proliferated the
market but regulation of medical devices by the
Bureau of Chemistry (the precursor to the Food and
Drug Administration—FDA) was limited to chal-
lenging commercial products only after they had
been released into the market. Devices in the mar-
ketplace that were defective, adulterated, or mis-
branded were seized and the device manufacturers
were prosecuted in a court of law, but only after
the products were sold in the market and caused
harm to the end users. Thus, there was a strong
need for regulating the devices before they entered
the marketplace. An FDA report [4], issued in
September 1970, detailed as many as 10,000 inju-
ries and 731 deaths from ineffective medical
devices. The report recommended the formation of
a regulatory system and body that would enforce
the production and sale of safe and effective
devices to the public. All medical devices already
on the market would be inventoried and classified
into a three-tiered system based on their criticality
of end use. It also detailed requirements for records
and reports, registration and inspection of establish-
ments, and uniform quality assurance programs
called good manufacturing practices (GMP). After
much lobbying by the FDA, Senate bill SR 510,
337Handbook of Polymer Applications in Medicine and Medical Devices. DOI: http://dx.doi.org/10.1016/B978-0-323-22805-3.00014-1
© 2010 Vinny Sastri. Published by Elsevier Inc. All rights reserved. Reproduced from a chapter in: Sastri, Plastics in Medical Devices (2010).

“The Medical Device Amendments of 1973” was
introduced by Senator Edward M. Kennedy and
was passed by the Senate in 1975. House bill HR
11124, introduced by Representative Paul Rogers,
was passed by the House in 1976. These bills even-
tually became the Medical Device Amendments of
1976, and were signed into law by President Nixon.
The Medical Device Amendments of 1976 became
the basis for the medical device regulation in the
United States to control and regulate the production
of finished devices and thus the device manufac-
turers themselves.
The GMP requirements for medical devices came
into effect on December 18, 1978. This regulation
was designed to specify general requirements for all
manufacturers as well as special requirements for
what were termed “critical devices.” Yet, between
1978 and 1990 a number of studies and data from
recalls of medical devices [5,6] indicated that a sig-
nificant number of recalls were due to improper,
faulty, or ineffective designs. On November 28,
1990, Congress passed the Safe Medical Device Act
(SMDA), providing the FDA with the authority to
add pre-production design controls to the GMP regu-
lation. This meant that device manufacturers would
need to have controls over their design and develop-
ment processes including strict controls of the raw
materials and components used to manufacture the
finished device. It also incorporated a provision to
include the oversight of foreign countries selling
products into the United States. Quality System
Regulation 21 CFR Parts 820 [7] was then drafted.
Efforts were made to harmonize this regulation with
both the ISO 9001:1994 entitled “Quality systems:
Model for quality assurance in design, development,
production, installation and servicing” and with ISO
13485:1996 entitled “Quality Systems—Medical
Devices—Particular requirements for the application
of ISO 9001.” This new regulation removed the
term “critical devices” and allowed manufacturers to
tailor their quality systems commensurate with the
risk associated with their device during end use. For
example, an implantable device will need more strin-
gent (design, development, and production) controls
compared to a simple tongue depressor and these
will differ in the level of detail and complexity of
their respective quality system requirements.
The purpose of the regulations is to ensure that
manufacturers of medical devices have the appropri-
ate procedures and processes in place to design,
develop, and produce consistent, safe, and effective
devices for their intended use. The regulations are a
framework for manufacturers and are flexible enough
to allow them to formulate and implement those parts
of the regulation that are applicable to their products
and processes and the risk of their products.
14.2 United States Regulations
The design, development, production, distribu-
tion, and use of medical devices in the United
States of America are regulated by the Federal
Drug and Cosmetics Act in the Code of Federal
Regulations (CFR)—21 CFR Parts 820 [7]. This
regulation is entitled “The Quality System
Regulation.” To sell medical devices in the United
States of America, all (domestic or international)
finished medical device manufacturers must register
with the Federal Drug Administration (FDA), must
be willing to comply with the regulation, and must
be willing to let the FDA inspect their facilities.
The intent of 21 CFR Parts 820 is that “quality
must be designed and built into components
through the application of proper quality systems”
[8]. The regulation requires that medical device
manufacturers establish and implement an appropri-
ate quality system that encompasses the design,
manufacture, packaging, labeling, storage, installa-
tion, and servicing of the finished device intended
for commercial use and distribution in the United
States. Effective quality systems will ensure that
manufacturers are in a “state of control” and pro-
duce consistent, safe, and effective devices for their
intended use. The FDA monitors and inspects the
complaints, data, and records from both end users
and manufacturers to track and determine the safety
and efficacy of a device.
Table 14.1 details the various sections of the reg-
ulation with a brief description of each section.
This regulation pays particular attention to the
design controls that were added to the latest (1997)
version.
Suppliers of raw materials or components do not
have to comply with the regulations, but are subject
to the purchasing controls of the regulations.
Finished device manufacturers must establish
procedures and controls with their suppliers for
essential raw materials that include quality metrics,
material performance and purity specifications,
assurance of supply, and notification of any formu-
lation or process changes.
338 HANDBOOK OF POLYMER APPLICATIONS IN MEDICINE AND MEDICAL DEVICES
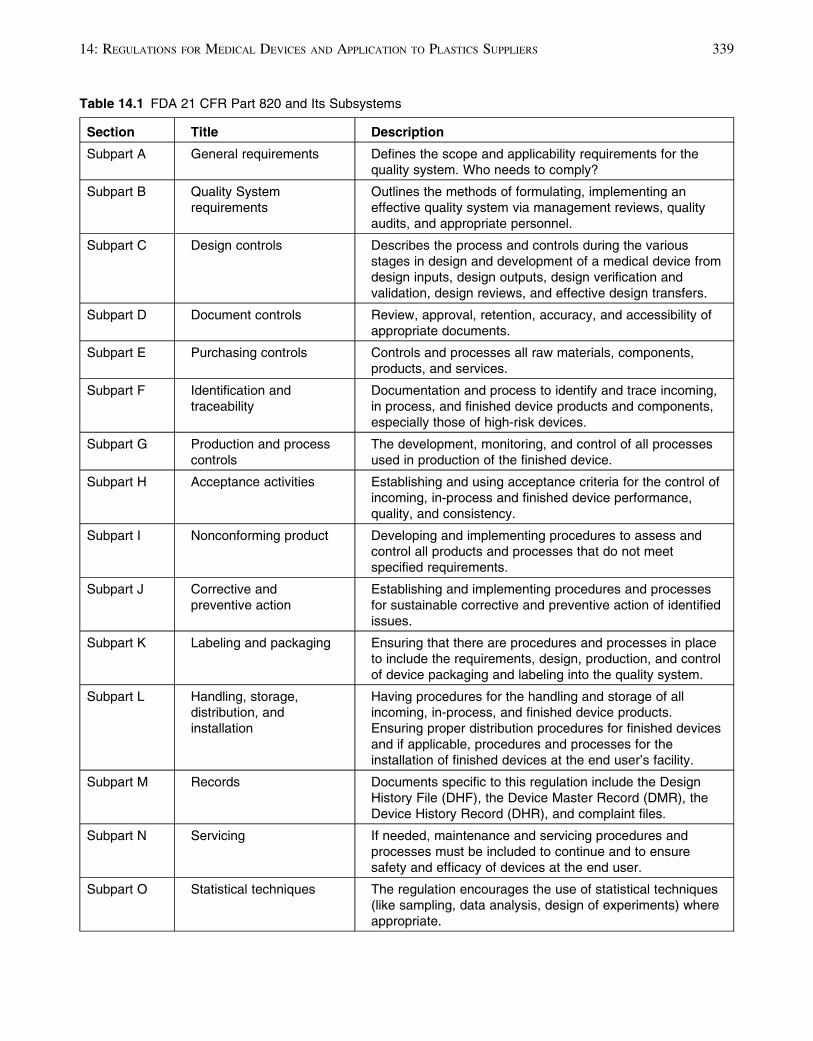
Table 14.1 FDA 21 CFR Part 820 and Its Subsystems
Section Title Description
Subpart A General requirements Defines the scope and applicability requirements for thequality system. Who needs to comply?
Subpart B Quality Systemrequirements
Outlines the methods of formulating, implementing aneffective quality system via management reviews, qualityaudits, and appropriate personnel.
Subpart C Design controls Describes the process and controls during the variousstages in design and development of a medical device fromdesign inputs, design outputs, design verification andvalidation, design reviews, and effective design transfers.
Subpart D Document controls Review, approval, retention, accuracy, and accessibility ofappropriate documents.
Subpart E Purchasing controls Controls and processes all raw materials, components,products, and services.
Subpart F Identification andtraceability
Documentation and process to identify and trace incoming,in process, and finished device products and components,especially those of high-risk devices.
Subpart G Production and processcontrols
The development, monitoring, and control of all processesused in production of the finished device.
Subpart H Acceptance activities Establishing and using acceptance criteria for the control ofincoming, in-process and finished device performance,quality, and consistency.
Subpart I Nonconforming product Developing and implementing procedures to assess andcontrol all products and processes that do not meetspecified requirements.
Subpart J Corrective andpreventive action
Establishing and implementing procedures and processesfor sustainable corrective and preventive action of identifiedissues.
Subpart K Labeling and packaging Ensuring that there are procedures and processes in placeto include the requirements, design, production, and controlof device packaging and labeling into the quality system.
Subpart L Handling, storage,distribution, andinstallation
Having procedures for the handling and storage of allincoming, in-process, and finished device products.Ensuring proper distribution procedures for finished devicesand if applicable, procedures and processes for theinstallation of finished devices at the end user’s facility.
Subpart M Records Documents specific to this regulation include the DesignHistory File (DHF), the Device Master Record (DMR), theDevice History Record (DHR), and complaint files.
Subpart N Servicing If needed, maintenance and servicing procedures andprocesses must be included to continue and to ensuresafety and efficacy of devices at the end user.
Subpart O Statistical techniques The regulation encourages the use of statistical techniques(like sampling, data analysis, design of experiments) whereappropriate.
33914: REGULATIONS FOR MEDICAL DEVICES AND APPLICATION TO PLASTICS SUPPLIERS

14.2.1 FDA Master Files
For the submission of a Premarket Approval
(PMA) [9], a 510(k) [10] for substantially equivalent
devices or an Investigational Device Exemption
(IDE) [11] a finished device manufacturer submits an
application to the FDA containing substantive data of
the finished device including performance, chemical
resistance, biocompatibility, toxicity, and clinical
data. In many cases, the finished device or compo-
nents are made from a supplier’s product or raw
materials. In order that a sound scientific evaluation
may be made of the PMA, 510(k) or the IDE, a
review of data and other information related to the
supplier’s product, facility, or manufacturing proce-
dures is required. While suppliers may be willing to
have the FDA review this information, they may not
want their proprietary information in the hands of
their customers (the finished device manufacturers).
A system for the submission of Master Files was
developed by the FDA to permit the suppliers of the
materials to provide confidential product information
directly to the FDA for its review without disclosing
the confidential information to the customer or manu-
facturer. If the same raw material is used in various
applications, components, or devices, only one Master
File is required.
There are various types of master files depending
upon the intended use.
• Device master files (MAF)—Supporting data
on material used in medical devices (informa-
tion for pre-manufacturing notices, 510(k)s
and Investigational Device Exemptions);
• Drug Master File (DMF)—Supporting data on
material used in drugs; (information for
Investigational New Drug Applications (IND),
New Drug Applications (NDA), and
Abbreviated New Drug Applications (ANDA));
• Biologics Master Files�Supporting data for
material used in applications contacting blood
or blood products (information for notices of
claimed Investigational Exemption for an
Investigational New Drug (IND) for biologics
and biologic licenses);
• Food Master Files (FMF)—Supporting data mate-
rial used in food applications (information for
Food Additive and Color Additive Petitions); and
• Veterinary Medicine Master Files—Supporting
data for materials used in animal drug and
devices (Investigational New Animal
Exemptions (INAD) and New Animal Drug
Applications (NADA)).
The content of a Master File includes the
following:
• Company Name,
• Product Name,
• Manufacturing Address,
• Statement of Commitment,
• Product Formulation,
• Product Specification, and
• Test methods and results (physical, chemical,
biocompatibility, and toxicity).
The information provided in the master file gives
the device manufacturer and the FDA a level of
comfort that the raw material being used in the
device will pass the specific physical, chemical,
biocompatibility, and toxicity tests. The FDA must
be notified of any changes to the formulation and
subsequent properties of the material and the
Master File must be updated. Failure to notify and
comply will render the finished device “adulter-
ated” and may not be subjected for sale or use.
MAFs may be submitted for various types of
operations and products and can be grouped by the
following types:
• facilities and manufacturing procedures and
controls;
• synthesis, formulation, purification, and specifi-
cations for chemicals, materials (e.g., an alloy,
plastic, etc.), or subassemblies for a device;
• packaging materials;
• contract packaging and other manufacturing
(e.g., sterilization);
• nonclinical study data; and
• clinical study data.
14.3 ISO 13485 (European andGlobal Standard)
The international standard for medical devices is
ISO 13485:2003 entitled “Medical devices—Quality
340 HANDBOOK OF POLYMER APPLICATIONS IN MEDICINE AND MEDICAL DEVICES
management systems—Requirements for regula-
tory purposes” [12]. Though geared specifically
toward medical device manufacturers, the ISO
13485 standard is harmonized with ISO 9001:2000
with some differences. ISO 13485:2003 includes
particular requirements for medical devices and
excludes some of the requirements of ISO
9001:2000 that are not appropriate as regulatory
requirements with respect to medical devices.
Thus, organizations which conform to ISO
13485:2003 cannot claim that they conform to
ISO 9001:2000 or vice versa unless their quality
management systems conform to all the require-
ments of ISO 9001:2000. Risk management is a
key part of ISO 13485 [13]. Terms like customer
satisfaction and continuous improvement have
been removed from this document (compared to
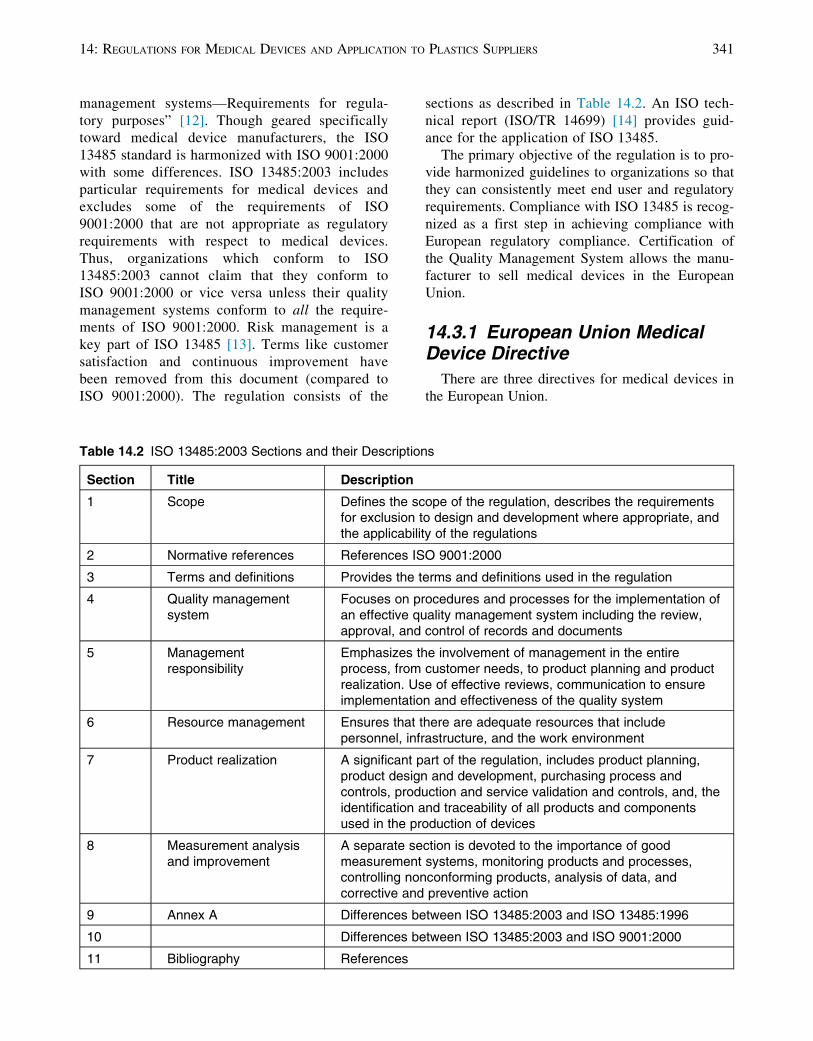
ISO 9001:2000). The regulation consists of the
sections as described in Table 14.2. An ISO tech-
nical report (ISO/TR 14699) [14] provides guid-
ance for the application of ISO 13485.
The primary objective of the regulation is to pro-
vide harmonized guidelines to organizations so that
they can consistently meet end user and regulatory
requirements. Compliance with ISO 13485 is recog-
nized as a first step in achieving compliance with
European regulatory compliance. Certification of
the Quality Management System allows the manu-
facturer to sell medical devices in the European
Union.
14.3.1 European Union MedicalDevice Directive
There are three directives for medical devices in
the European Union.
Table 14.2 ISO 13485:2003 Sections and their Descriptions
Section Title Description
1 Scope Defines the scope of the regulation, describes the requirementsfor exclusion to design and development where appropriate, andthe applicability of the regulations
2 Normative references References ISO 9001:2000
3 Terms and definitions Provides the terms and definitions used in the regulation
4 Quality managementsystem
Focuses on procedures and processes for the implementation ofan effective quality management system including the review,approval, and control of records and documents
5 Managementresponsibility
Emphasizes the involvement of management in the entireprocess, from customer needs, to product planning and productrealization. Use of effective reviews, communication to ensureimplementation and effectiveness of the quality system
6 Resource management Ensures that there are adequate resources that includepersonnel, infrastructure, and the work environment
7 Product realization A significant part of the regulation, includes product planning,product design and development, purchasing process andcontrols, production and service validation and controls, and, theidentification and traceability of all products and componentsused in the production of devices
8 Measurement analysisand improvement
A separate section is devoted to the importance of goodmeasurement systems, monitoring products and processes,controlling nonconforming products, analysis of data, andcorrective and preventive action
9 Annex A Differences between ISO 13485:2003 and ISO 13485:1996
10 Differences between ISO 13485:2003 and ISO 9001:2000
11 Bibliography References
34114: REGULATIONS FOR MEDICAL DEVICES AND APPLICATION TO PLASTICS SUPPLIERS

• The Active Implantable Medical Device
(AIMD) Directive—90/385/EEC;
• The Medical Device Directive (MDD)—93/42/
EEC; and
• The In Vitro Diagnostic Directive (IVD)—98/
79/EC.
After June 14, 1998, medical devices could
not be offered for sale in the European Union
without “CE marking” and a “declaration of con-
formity.” The letters CE stand for “Conformite
Europeene” in French literally meaning “European
Conformity.” For many products CE marking and a
declaration of conformity may only be affixed with
proof of a certified quality system and/or product
testing based on its end use. The quality systems
certification, the CE marking, and the declaration
of conformity are provided by a “Notified Body”
which is an organization appointed by the national
accreditation authorities and which “notifies” the
European Commission to approve products covered
by the Medical Devices Directive. All medical
device manufacturers must designate a notified
body to certify and register their products. For
all classes of devices, a detailed technical file must
be submitted providing objective evidence demon-
strating compliance with the Medical Device
Directive’s essential requirements and with appro-
priate harmonized standards which include ISO
13485:2003 and ISO 10993 standards [8].
Products shipped must bear the CE marking to
show compliance with the directive (Figure 14.1).
If a Notified Body is involved in the approval, the
number of the Notified Body must also appear adja-
cent to the CE marking.
Additionally, the product must be shipped with a
Declaration of Conformity, an example of which is
shown in Figure 14.2.
Documentation can include the following:
• Evidence demonstrating compliance with
essential requirements detailed in the directive
for the particular product’s end use;
• Demonstration of design verification and
validation;
• Risk assessment and analysis;
• Clinical evidence demonstrating effectiveness
of the device;
• Procedures for post-market surveillance;
• Complete declaration of conformity;
• Technical information of the finished device—
including toxicity and biocompatibility
studies;
Figure 14.1 The CE mark.
EC Declaration of Conformity
Council Directive 93/42/EEC concerningmedical devices
We (Name and address of manufacturer)
Certify that the product described is in conformitywith the applicable provisions of Council Directive
93/42/EEC concerning medical devices.
(Name, type or model, lot, batch or serial no. etc.)
(Description)
(Name of Responsible Person) (Signature of Responsible Person)
(Date)
Figure 14.2 CE marking declaration of conformity.
342 HANDBOOK OF POLYMER APPLICATIONS IN MEDICINE AND MEDICAL DEVICES

• Accurate product identification, labels, proce-
dures, and user instructions; and
• CE mark or label on product or packaging.
14.4 Other Countries
14.4.1 Japan
The Japanese government, through the Ministry
of Health, Labor, and Welfare (MHLW), regulates
all medical devices, whether manufactured in Japan
or imported from other countries. In Japan, the
term “medical device” is used for any instrument,
apparatus, or material as designated by the
Japanese government that is used in diagnosing,
treating, and/or preventing diseases in humans or
animals and which can be used to affect the struc-
ture and functions of humans or animals. The
Pharmaceutical Affairs Law (PAL) is the primary
governing law for medical devices in Japan.
Medical devices must undergo thorough safety
examinations and demonstrate medical efficacy
before they are granted approval, or “shonin,” to be
sold in Japan. PAL regulations specify very
detailed requirements for companies that manufac-
ture or import medical devices for sale in Japan,
ranging from infrastructure and facilities to person-
nel and processes. For new medical devices for
which there are no equivalent products already
approved in Japan or for devices that have been
improved or modified that might affect device safety
and efficacy, clinical trials are required. Clinical trials
must be conducted to demonstrate the safety and
efficacy of the product under strict Good Clinical
Practice (GCP) standards, and must be followed by
standard Post-Marketing Assessment (PMA) reporting
and a follow-up program.
In April 2004, the Pharmaceuticals and Medical
Devices Agency (PMDA) was established in an
effort to create a more efficient and transparent
medical device registration review process. The
PMDA was formed by merging three already exist-
ing organizations: (1) the Pharmaceuticals and
Medical Devices Evaluation Center (PMDEC), (2)
the Organization of Pharmaceutical Safety and
Research (OPSR), and (3) the Japan Association for
the Advancement of Medical Equipment (JAAME).
Two of these three agencies (PMDEC and JAAME)
were previously involved in the medical device
approval process, including the review of product
registration applications and clinical trial consulta-
tions. Prior to the creation of the PMDA, the appli-
cation and review process for new devices could
take as long as 2 years. Over the next several years,
the PMDA intends to shorten this process, although
it has not had success in doing so thus far.
Under the New PAL, the Quality Assurance
Controller will be responsible for ensuring compli-
ance with the new Good Manufacturing Practice
(GMP) requirement, based on Japan’s own adapta-
tion of ISO 13845:2003, as well as Good Quality
Practice Ordinance (GQP) standards [15]. The
Standard Operating Procedures (SOPs) for GQP
include product storage controls, the release of pro-
ducts into the market, quality control at local
offices, ensuring the maintenance of all quality
assurance documents and reports, the handling of
product recalls, and audits.
Necessary Governmental Authorizations (for sale
of devices into Japan):
• Manufacturing (or import) approval (“Shonin”)
which guarantees the safety and efficacy of the
device, obligatory for every product;
• Manufacturing (or import) license (“Kyoka”) of
a device, which the Japanese manufacturer and
importer hold, renewable every 5 years; and
• Reimbursement listing approval.
14.4.2 China
There are two main agencies in China that regu-
late medical devices, the State Food and Drug
Administration (SFDA) and the Department of
Medical Devices. The State Food and Drug
Administration (SFDA) is the Chinese equivalent
of the FDA in the United States. All imported
medical devices must be registered with the
SFDA. The Department of Medical Devices under
the SFDA is responsible for the standardization,
product registration, safety, and supervision of all
imported devices into China. Some of the stan-
dards used by the agency are ISO 10993 (Biologic
Evaluation of Materials and Medical Devices),
ISO 14971 (Risk Management), and ISO 13485
(Medical Devices—Quality Management Systems).
14.4.3 Australia
The medical device legislation has been estab-
lished by the Therapeutic Goods Act 1989 as
34314: REGULATIONS FOR MEDICAL DEVICES AND APPLICATION TO PLASTICS SUPPLIERS

amended by the Therapeutic Goods Amendment
(Medical Devices) Bill 2002 and the Therapeutic
Goods (Medical Devices) Regulations 2002. The
new framework also adopts the philosophies of the
Global Harmonization Task Force on medical
devices.
The new regulatory system has the following
features:
• a device classification scheme based on differ-
ent levels of risk for each class of device;
• essential principles for the quality, safety, and
performance of the medical device that must
be complied with before the product can be
supplied;
• options as to how compliance with the essen-
tial principles can be satisfied and assessed;
• manufacturer quality systems, type testing, and
design evaluation;
• the use of recognized standards to satisfy the
requirements of the essential principles;
• a comprehensive post-market surveillance and
adverse incident reporting program;
• appropriate regulatory controls for the
manufacturing processes of medical devices;
• the continued use of the Australian Register of
Therapeutic Goods as the central point of con-
trol for the legal supply of medical devices in
Australia; and
• chemical, physical, and biological properties.
14.4.4 India
The Central Drugs Standards Control
Organization (CDSCO) under the Ministry of
Health and Family Welfare regulates the licensing,
import, manufacture, and sale of medical devices
into the country. Approvals can be facilitated by
evidence of approval from the US FDA, the EU
MDD (CE certificate), and approvals from
Australia, Canada, Japan, and other countries. ISO
certification for specific manufacturing facilities
(ISO 13485) is also accepted. Device master files
must contain details of good manufacturing prac-
tices including components and materials used
in the device. It must also include the manufactur-
ing and quality assurance processes, risk assess-
ment, design verification, sterilization, stability,
biocompatibility, and toxicological data associated
with the materials and production of the finished
device.
14.4.5 South America
For most companies the access point to South
America is Brazil. Brazil has the second largest
healthcare market in the Americas (bigger than
Canada and second only to the United States). It is
a member of Mercosur—the South American Free
Trade Area that includes Brazil, Argentina,
Uruguay, and Paraguay. Separate submissions
have to be made in each country. Registration of
products or product families must contain informa-
tion on the manufacturer, the materials and com-
position used, and the intended use.
Mexico has patterned its regulations after the US
FDA and ISO requirements under the Secretarıa
de Salud.
14.5 Global Harmonization TaskForce (GHTF)
The GHTF was conceived in 1992 and is an
informal grouping that was formed to respond to
the growing need for the international harmoniza-
tion of regulations in medical devices. The mem-
bers of the GHTF include government and industry
officials from the European Union, Japan, Canada,
Australia, and the United States. These representa-
tives working with medical device manufacturers
and other organizations related to medical devices
try to harmonize global approaches to the safety,
efficacy, clinical performance, and quality of medi-
cal devices with the goal of protecting public
health, promoting innovation, and facilitating inter-
national trade. Global harmonization is the aligning
of the different regulatory systems of the world
making them globally on par with each other to
manufacture and sell safe and effective devices.
The GHTF is committed to developing guidelines
accepted in all GHTF countries and gives technical
guidance toward a more coherent approach on the
interpretation of technical and quality requirements
for medical devices. It has four study groups, deal-
ing with product approval-related issues, post-
market surveillance, quality system requirements,
and audits of quality systems.
344 HANDBOOK OF POLYMER APPLICATIONS IN MEDICINE AND MEDICAL DEVICES

14.6 Applicability of theRegulations to Material Suppliers
The regulations (FDA 21 CFR Parts 820 and
ISO 13485:2003) are applicable to the manufac-
turers of “finished devices.” Suppliers of raw mate-
rials are not expected to comply with these
regulations but must meet acceptable material
requirements set forth by the device manufacturers
(as per their purchasing controls). Finished device
or component manufacturers expect their material
suppliers to have consistently good quality and pro-
cess control in their facilities.
In July 1998 in the United States, the Biomaterials
Access Assurance Act—BAAA (HR 872) was signed
into law by President Bill Clinton. The purpose of
the act was to “establish rules governing product lia-
bility actions against raw materials and bulk compo-
nent suppliers to medical device manufacturers, and
for other purposes” [16]. This was a very important
bill, as it protects the suppliers of biomaterials or
components of implanted devices from liability if an
entire device results in injury or death, provided it
was not the fault of the material or component. This
act was in response to a very serious concern
expressed by suppliers following many expensive
lawsuits where it was found that the eventual cause
of the problem was not with the material but with
the finished device itself. Many plastics suppliers are
willing to supply materials as long as their materials
are used in devices that are in contact with the
human body for less than 29 days (minimal contact
with and minimal residence time within the body).
A few plastics suppliers are willing to recommend
their products for implants and devices that are in the
body for more than 29 days (implantable devices)
based on the extensive studies and data that show
their materials pass all physical, chemical, biocom-
patibility, hemocompatibility, and toxicity tests
required for implantable devices.
Finished device manufacturers are expected to
establish purchasing controls [17], providing mate-
rial suppliers with acceptance criteria and material
specifications and requirements needed for their
specific devices and applications. Such require-
ments might include the following:
• Raw material performance specifications,
• Biocompatibility,
• Sterilization requirements,
• Material purity,
• Chemical resistance,
• Toxicity requirements,
• Product quality and consistency,
• Notification of formula changes,
• Adherence to good manufacturing practices,
and
• Assurance of supply.
14.7 Conclusion
The purpose of regulations for medical devices
is to ensure that the products are consistent, safe,
and effective for their intended use. The two major
regulations are the 21 CFR Parts 820 Quality
Systems Regulations enforced by the Food and
Drug Administration in the United States and the
global standard by the International Organization of
Standards ISO 13485:2003 “Medical devices—
Quality management systems—Requirements for
regulatory purposes” enforced by the European
Union. Most countries have adopted modified ver-
sions of the ISO 13485 and/or the FDA regulations.
Finished device manufacturers need to comply with
the regulations. Suppliers of raw materials and
components do not need to comply with the regula-
tions, but are subject to the purchasing controls of
the finished device manufacturers. Finished device
manufacturers must have stringent supplier qualifi-
cation procedures that include supplier audits,
incoming raw material and component specifica-
tions, and quality metrics. Plastic material suppliers
must provide appropriate data and information
about their products that the regulatory bodies and
the finished device manufacturers can use to assess
the performance and viability of the raw materials
for their specific devices. This is only required for
high-risk devices. Such information includes the
formulation, the performance specifications, the
test methods and release criteria, the quality
metrics, material characteristics (physical, chemi-
cal, biocompatibility, and toxicity), the assurance of
supply, and the notification of any formulation
changes. This information is typically maintained
by the regulatory bodies in master files, is kept con-
fidential, and is accessible only to the regulatory
bodies but not to the finished device manufacturers
or the public at large.
34514: REGULATIONS FOR MEDICAL DEVICES AND APPLICATION TO PLASTICS SUPPLIERS

References
[1] Estrin NF. The medical device industry. New
York: CRC Press: Marcel Dekker Inc; 1990.
[2] Squibb ER. The collected papers of Edward
Robinson Squibb, M.D., 1819�1900.
In: Porter D, Earl R, editors. Food labeling:
toward national uniformity. Washington DC,
USA: National Academies Press; 1992. p. 39.
[3] Federal food and drugs act of 1906 (The
“Wiley Act”) Public Law Number 59�384 34
Stat. 768 (1906) 21 U.S.C. Sec 1�15 (1934)
(Repealed in 1938 by 21 U.S.C. Sec 329 (a)).
[4] Study Group on Medical Devices. Medical
devices: a legislative plan. Washington, D.C.:
Department of Health Education and Welfare;
1970.
[5] FDA office of compliance and surveillance.
Device recalls: a study of quality problems.
HHS Publication FDA-90-4235: Washington
DC, USA; 1990.
[6] FDA medical device regulation from premarket
approval to recall—department of health and
human services inspector general’s study; 1990.
[7] 21 CFR Part 820—Quality Systems Regulation.
[8] 21 CFR Parts 808, 812, 820 Medical Devices;
Current Good Manufacturing Practices
(CGMP); Final Rule; October 7, 1996,
p. 52606, response #7.
[9] Premarket Approval (PMA)—is the FDA pro-
cess of scientific and regulatory review of
devices “that support or sustain human life,
are of substantial importance in preventing
impairment of human health, or which present
a potential, unreasonable risk of illness or
injury.”
[10] 510(k) application and submission—is the
FDA scientific and regulatory approval pro-
cess of devices that a manufacturer thinks is
“substantially equivalent” to a similar device
that was on the market prior to May 28, 1976.
This is less involved than the premarket
approval defined in reference 9.
[11] Investigational Device Exemption (IDE) is
issued by the FDA to allow the use of investi-
gational devices in human subjects for clinical
trials and investigation in order to evaluate the
safety and effectiveness of the investigational
medical device.
[12] ISO 13485:2003, Medical devices—Quality
management systems—Requirements for regu-
latory purposes.
[13] ISO 14971:2007, Medical devices—Application
of risk management to medical devices.
[14] ISO/TR 14969:2004, Medical devices—Quality
management systems—Guidance to the appli-
cation of ISO 13485:2003.
[15] Ministerial Ordinance on Standards for
Quality Assurance for Drugs, Quasi-drugs,
Cosmetics, and Medical Devices MHLW.
Ordinance Number 136; September 22, 2004.
[16] Public Law 105�230, sect. 1, 112 Stat. 1519
codified in 21 U.S.C. 1601�1606, 1999.
[17] Quality Management System—Medical
Devices—Guidance on the Control of Products
and Services Obtained from Suppliers.
346 HANDBOOK OF POLYMER APPLICATIONS IN MEDICINE AND MEDICAL DEVICES


![[ MEDICAL DEVICES ] - Global Affairs Canadainternational.gc.ca/.../pdfs/download/medical_devices.pdf · 2015-06-02 · CANADA’S MEDICAL DEVICES SECTOR Canada’s medical devices](https://img.pdfslide.us/doc/110x75/5f9f36e00d70d4306d589c5a/-medical-devices-global-affairs-2015-06-02-canadaas-medical-devices-sector.jpg)


























