Embed Size (px)
DESCRIPTION
hemolytic anemia
Citation preview

HAEMOLYTIC ANAEMIA
Dr Maryam Jameelah AizuddinDr Siti Nurzafira

PRESENTATION OUTLINE
Definition
Types of haemolytic disorders
Haemoglobinopathy
Enzymopathy
Membranopathy
Extrinsic Factors
Immune Haemolytic Anemia
Presentation (signs and symptoms)
Pathophysiology
Investigations
Management

HAEMOLYTIC ANAEMIA:DEFINITIONS
Haemolysis: Premature destruction of RBCs.Anaemia results => destructions exceed capacity of BM to produce RBCsNormal RBCs survival time => 110-120 days
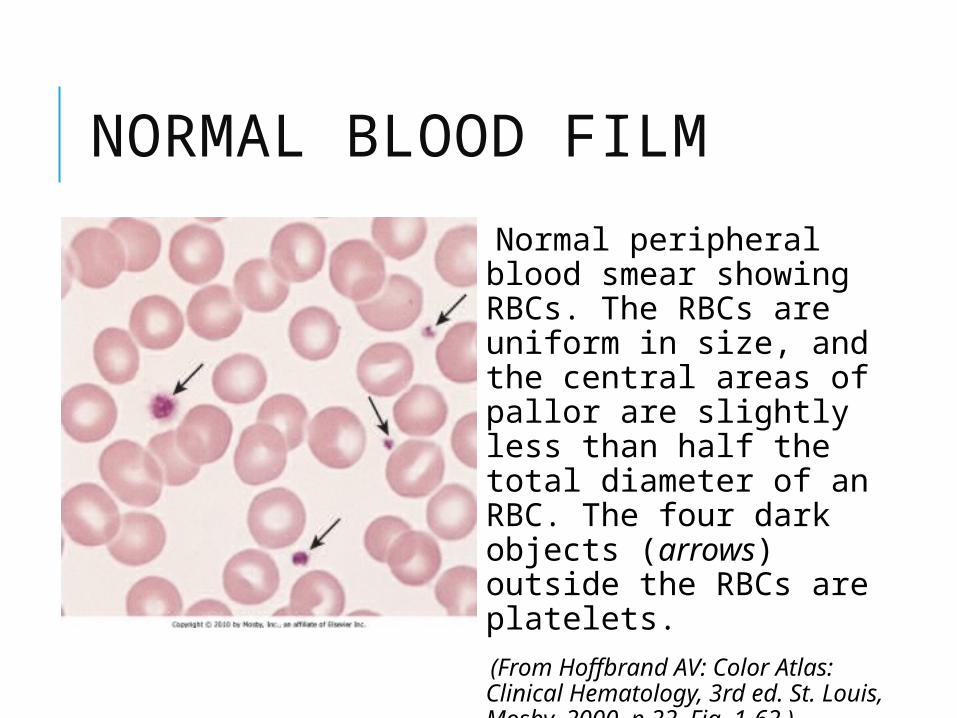
NORMAL BLOOD FILM
Normal peripheral blood smear showing RBCs. The RBCs are uniform in size, and the central areas of pallor are slightly less than half the total diameter of an RBC. The four dark objects (arrows) outside the RBCs are platelets.
(From Hoffbrand AV: Color Atlas: Clinical Hematology, 3rd ed. St. Louis, Mosby, 2000, p 22, Fig. 1-62.)



HAEMOLYTIC ANAEMIA:CLINICAL FEATURESJaundice: generally mild and often not noticed by the patient.
Anaemia: Recent onset = acquired Long-standing = possibly congenital.
Haemoglobinuria: intravascular haemolysis.Urobilinogenuria: increased Hb catabolism.
Splenic pain: spenomegaly or splenic infarction.
Leg ulcers: intrinsic red cell disorders, e.g. sickle cell disease.

WHAT HAPPENED DURING HAEMOLYIS?
RBCs survival shorthened => RBCs
EPO (stimulation in BM to RBCs)
=> Reticulocytosis (2-3)
(Ddx: acute blood loss, vit B 12 or folate deficiency)

HAEMOLYTIC ANAEMIA:LABORATORY FINDINGS
Features of increased erythrocyte breakdown:
Unconjugated bilirubinaemia.
Urobilinogenuria.
Haptoglobins decreased.

Features of increased erythrocyte production:
Reticulocytosis Polychromasia and nucleated
red cells in peripheral blood film. Erythroid hyperplasia in bone
marrow aspirate. Radiological changes, e.g. "hair
on end" appearance of cranial X-ray.

The ‘hair’ represents the accentuated trabeculae extending between the inner and outer skull tables in the expanded diploic marrow spaces. It appears to be ‘on end’ because the trabeculae are oriented perpendicular to the inner and outer tables of the skull. The term is now classically associated with the radiographic changes seen in hemolitic anemia. A. Sickle cell disease. B. Thalassemia.

Features specific to intravascular haemolysis:
• Haemoglobinaemia(haptoglobin and haemopexin
exhausted).• Methaemoglobinaemia.• Haemoglobinuria.• Haemosiderinuria.

CLASSIFICATION OF HAEMOLYTIC ANAEMIA
Cellular Defect
• Enzyme deficiencies (G6PD deficiency, Pyruvate kinase deiciency
• Haemoglobin defects• Membrane defects (hereditary
spherocytes)
Extracellula
r defects
• Autoimmune Haemolytic anaemia (warm and cold antibosy)
• Isoimmune Haemolytic anaemia

CELLULAR DEFECT:HEREDITARY SPEROCYTOSIS The most common inherited abnormality of RBCs membrane
Prevalence ~1 in 5000 people of Nothern European decsent but has been described in most ethnics group
Others:Hereditary elliptocytosisUsually no anemiaAutosomal dominant, affecting 1/2500 Europeans
Hereditary pyropoikilocytosis

CELLULAR DEFECT:HEREDITARY SPEROCYTOSIS Autosomal Dominant trait
Patients develop hemolysis and abdominal pain with even trivial infectious illnesses
Defect is in proteins of the membrane skeleton, usually spectrin or ankyrin
Lipid microvesicles are pinched off in the spleen and other RE organs, causing decreased MCV and spherocytic change.


CELLULAR DEFECT:HEREDITARY SPEROCYTOSIS
Normal
Abnormal-HS cells lyse more readily at low ionic strength
Diagnosed by Osmotic Fragility

CELLULAR DEFECT:HEREDITARY SPEROCYTOSISBLOOD FILM
Peripheral blood with spherocytes in hereditary spherocytosis. Numerous, round, dense red blood cells without central areas of pallor represent spherocytes (arrows). The mean corpuscular hemoglobin concentration is increased.
(From Damjanov I, Linder J: Pathology: A Color Atlas. St. Louis, Mosby, 2000, p 75, Fig. 5-7.)

HEREDITARY SPHEROCYTOSISHOW DO WE TREAT?
None : if Hb > 10g/dL and reticulocytes counts 10% Transfusion : if severe anaemia, poor growth, aplastic crises and age <2 years Folic acid

CELLULAR DEFECTS:G6PD DEFICIENCY

CELLULAR DEFECT: G6PD DEFICIENCYPATHOPHYSIOLOGY
G6PD activity results in NADPH and
glutathione, which are required to protect hemoglobin from oxidative damage.
Oxidizing agents converts hemoglobin to methemoglobin, then denature it, causing it to precipitate as Heinz bodies.
The spleen pinches off the Heinz body and the overlying membrane, leaving a “bite cell” or “blister cell”

GLUCOSE 6-PHOSPHATE DEHYDROGENASE
FUNCTIONS Regenerates NADPH, allowing regeneration of glutathione
Protects against oxidative stress
Lack of G6PD leads to hemolysis during oxidative stress Infection Medications Fava beans

G6PD DEFICIENCYCLINICAL FEATURES
Typically, hemolysis is triggered by drugs or infections. Anemia is maximal 7-10 d after exposure. Urine becomes dark associated with low back & abd pain Wait for 6 wk after hemolysis to do enzyme assay
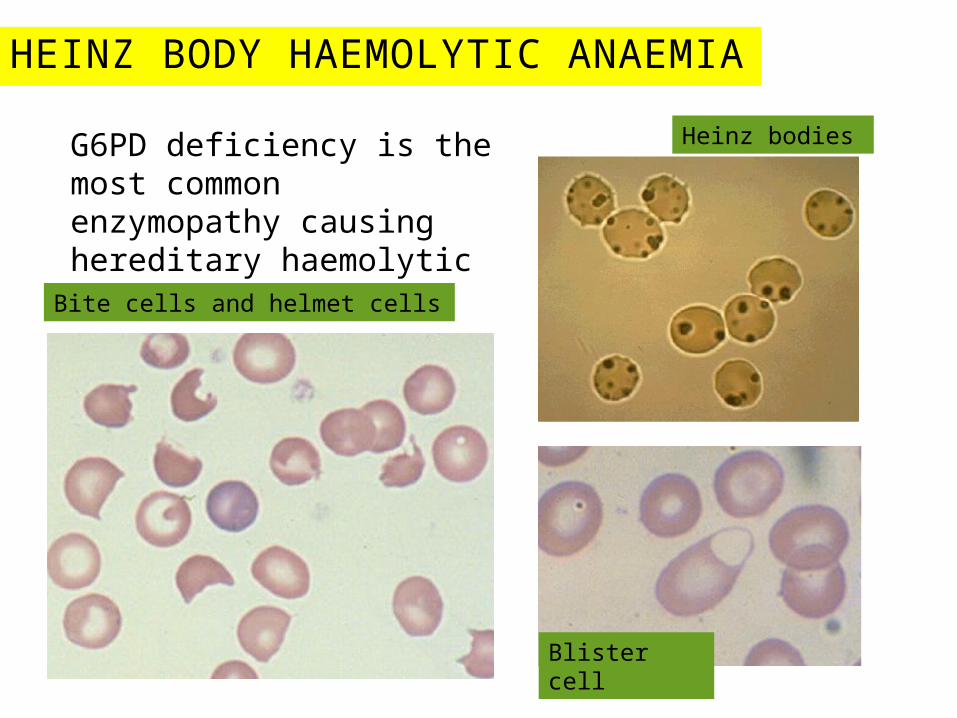
HEINZ BODY HAEMOLYTIC ANAEMIA
G6PD deficiency is the most common enzymopathy causing hereditary haemolytic anaemia.Bite cells and helmet cells
Blister cell
Heinz bodies

Peripheral blood smear with a bite cell and inset showing Heinz bodies in glucose-6-phosphate dehydrogenase deficiency. The arrowshows a bite cell with part of the red blood cell membrane removed. The inset shows a peripheral blood smear with a supravital stain visualizing punctate inclusions representing denatured hemoglobin (Heinz bodies).
(From Kumar V, Fausto N, Abbas A: Robbins and Cotran's Pathologic Basis of Disease, 7th ed. Philadelphia, WB Saunders, 2004, Fig. 13-8; inset from Wickramasinghe SN, McCullough J: Blood and Bone Marrow Pathology. London, Churchill Livingstone, 2003, Fig. 8-8.)

G6PD DEFICIENCY AGENTS TO AVOID
–Primaquine
–Aspirin
–Quinolones(cipro)
–Sulfa drugs
–Dapsone
–Fava beans(favism)
–Naphtha compounds (mothballs)

G6PD DEFICIENT: TREATMENT
Avoid oxidant drugs
Transfuse in severe cases
IV fluids to maintain good urine output
Splenectomy in severe recurrent hemolysis
Folic acid supplements

WHY 5 DAYS OF OBSERVATION?
The bilirubin levels usually are the highest during day 3-5 of life

INHERITANCE OF G6PD DEFICIENT IN FEMALE? G6PD is an x linked enzyme deficiency
Usually affected males
How female are affected?
Lyon Hypothesis : in Heterozygous female may have G6PD when there is random inactivation of normal X chromosome.
Most affected female do not have clinical hemolysis after exposure of oxidant drug
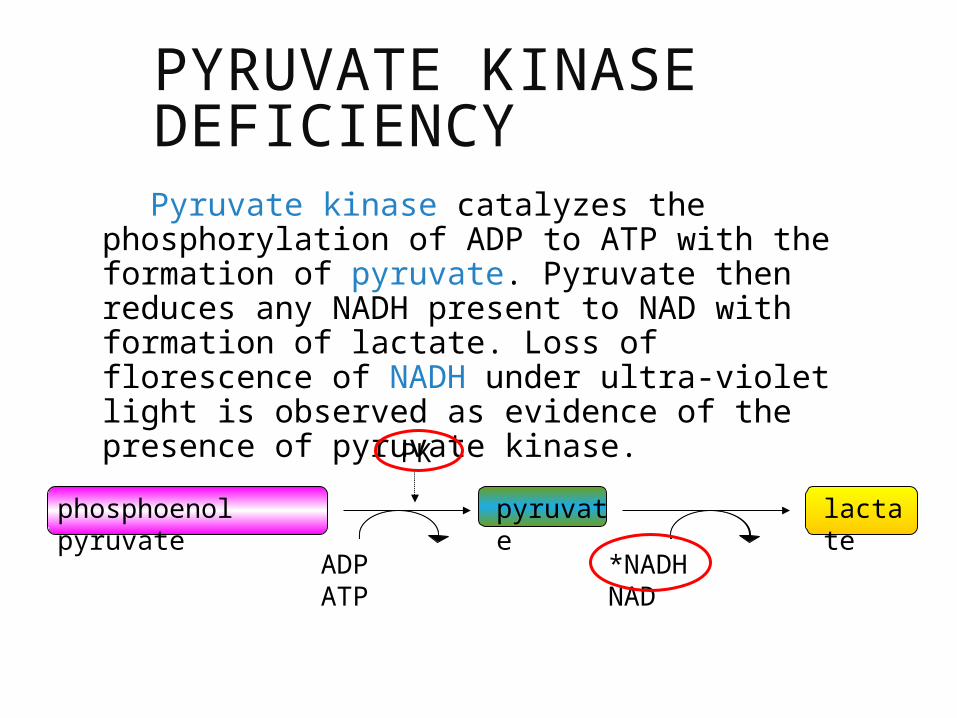
PYRUVATE KINASE DEFICIENCY
Pyruvate kinase catalyzes the phosphorylation of ADP to ATP with the formation of pyruvate. Pyruvate then reduces any NADH present to NAD with formation of lactate. Loss of florescence of NADH under ultra-violet light is observed as evidence of the presence of pyruvate kinase.
phosphoenol pyruvate
pyruvate
lactate
PK
ADP ATP *NADH NAD

PYRUVATE KINASE DEFICIENCY:CLINICAL MANIFESTATIONS In new born
Severe jaundice, kernicterus
Anaemic symptoms
Older children
Hb ranging 8-12g/dL, some pallor, jaundice,
splenomegaly (usually doesn’t need Tx)
Relative high incidence among the Amish

Peripheral blood smear in pyruvate kinase deficiency. The arrow shows one of many red blood cells with thorny projections (echinocytes) extending from the red blood cell membrane. (From Wickramasinghe SN, McCullough J: Blood and Bone Marrow Pathology. London, Churchill Livingstone, 2003, Fig. 8-10.)

PYRUVATE KINASE DEFICIENCY: THE AMISH
Autosomal recessive trait The amish - Intermarriage

PYRUVATE KINASE DEFICIENCY: DIAGNOSIS Demonstration of marked reduction in RBC PK activity Increase Michaeli-Menten dissociation constant (Km) for phophoenolpyruvate

Peripheral blood smear in pyruvate kinase deficiency. The arrow shows one of many red blood cells with thorny projections (echinocytes) extending from the red blood cell membrane.
(From Wickramasinghe SN, McCullough J: Blood and Bone Marrow Pathology. London, Churchill Livingstone, 2003, Fig. 8-10.)

PYRUVATE KINASE DEFICIENCY:TREATMENTS Exchange transfusion maybe indicated for hyperbilirubinaemia in newborn
Transfusion of PCs for severe anaemia or aplastic crises
Splenectomy after age of 5-6 years old

Structural Hemoglobin Defects
Hemoglobin SHemoglobin C
Thalassemias
Alpha thalassemiaBeta thalassemia
Haemoglobin Defect
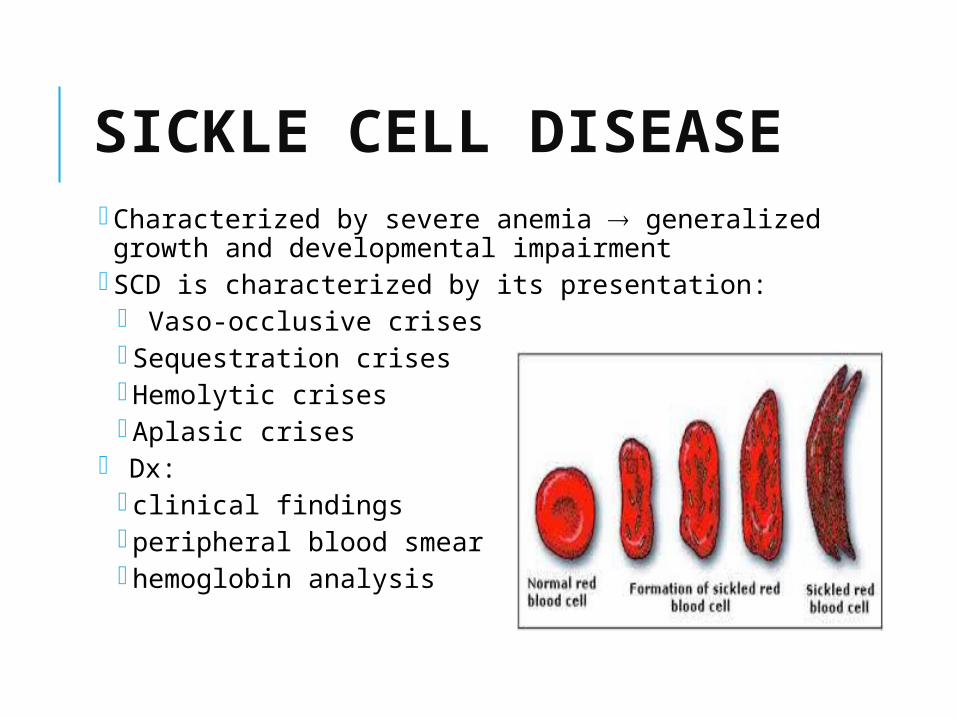
SICKLE CELL DISEASE Characterized by severe anemia generalized growth and developmental impairment
SCD is characterized by its presentation: Vaso-occlusive crises Sequestration crises Hemolytic crises Aplasic crises
Dx: clinical findings peripheral blood smear hemoglobin analysis
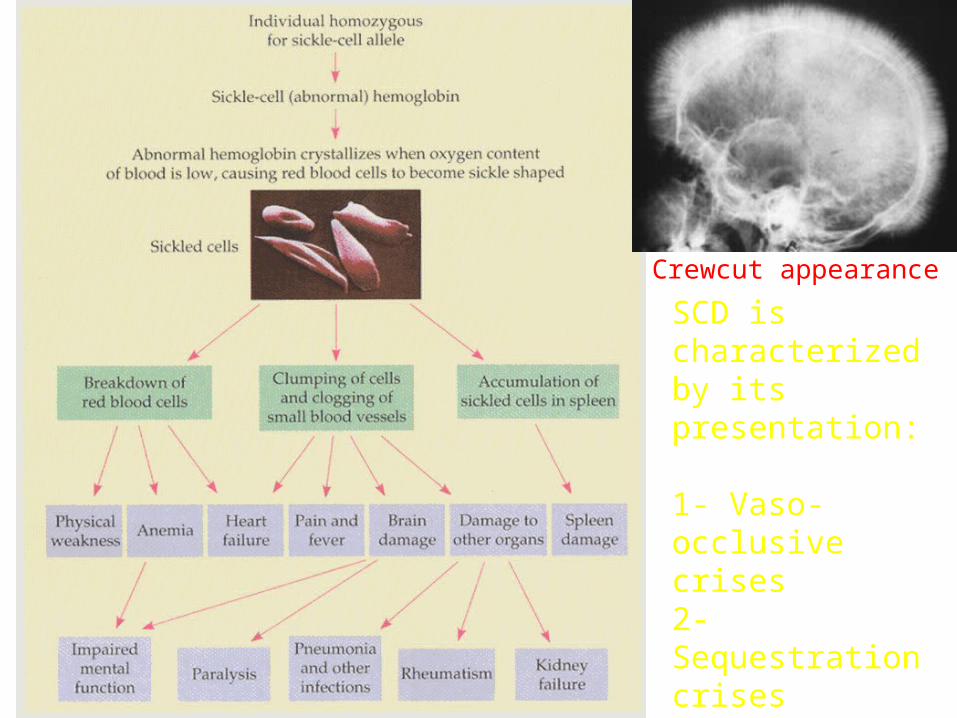
SCD is characterized by its presentation:
1- Vaso-occlusive crises2- Sequestration crises3- Hemolytic crises4- Aplasic crises
Crewcut appearance

Pathophysiology of sickle cell anemia

EXTRACELLULAR:AIHA ASSOCIATED WITH “WARM” ANTIBODY Hallmark of this disease => + direct (Coombs Test) detects coating of Ig/complements on RBCs
Antibodies are active at 35-40 oC
Most often belong to IgG class
Do not require complement for its activity
Usually do not produce agglutination in vitro
Etilology:
Abnormal Ig directed against RBCs but mechanism not clearly understood
Molecular mimicry
Infectious agents – results RBCs membrane antigenic
Primary/ idiopathic type
Secondary – associated with SLE/ immunodeficiency

CLINICAL MANIFESTATIONS Acute transient
Lasting 3-6 month
Predominant in children age 2-12 yr
Often preceded by infection usually respiratory
Pallor, jaundice, pyrexia, haemoglobinuria, fatigue
Spleen usually enlarged
Consistent response to glucocorticoids therapy
Chronic
May lasts from months to years
Frequent in infants and children older than 12yr
Response to glucocorticoids variable and inconsistent

LABORATORY FINDINGS
Profound anaemia Hb < 6g/dL
High retics
Leucocytosis common
High LDH
Platlet usually normal but concomitant ITP sometimes occur
Direct Coombs => POSITIVE
Indirect Coombs sometimes can be positive (free antibody in the serum)

BLOOD FILM
Spherocytes and polychromasia

Polychromasia. The arrow depicts a blue discolored red blood cell without a central area of pallor. (From Naeim F: Atlas of Bone Marrow and Blood Pathology. Philadelphia, WB Saunders, 2001, Fig. 1-15A.)
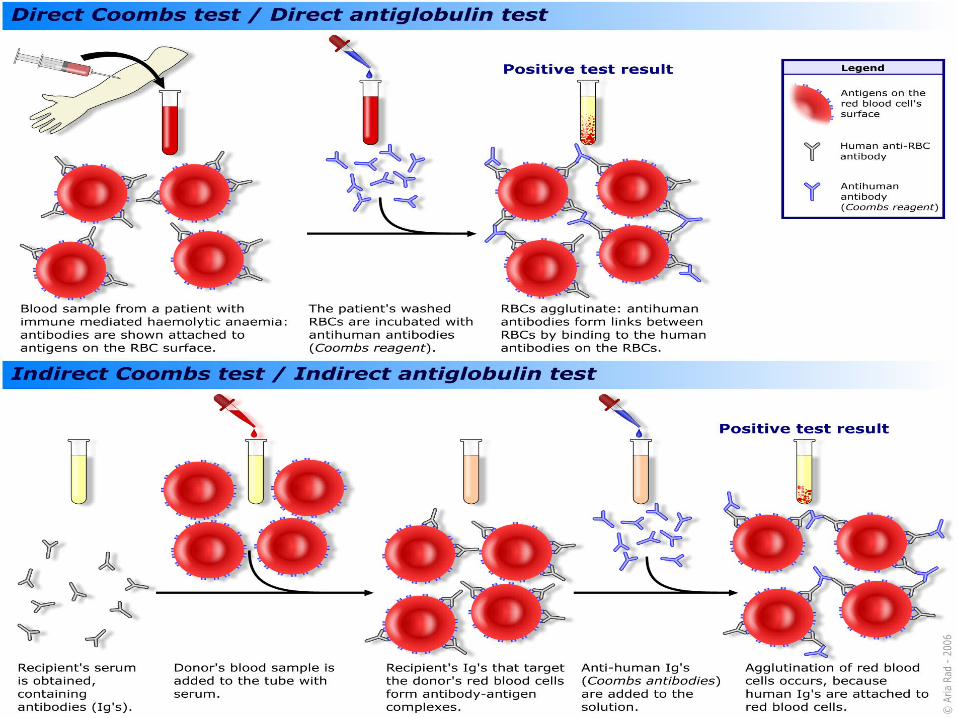

TREATMENT
Transfusion may be required initially (May be EXTREMELY difficult to find compatible blood: blood in which RBCS give least positive in vitro reaction by Coombs technique)
Mild disease may not require any treatment
More severe disease => Prednisolone 2-6mg/kg/day ( rate of haemolysis by blocking macrophages, production of autoantibody)
Tx should be continued until haemolysis rate , gradually tappering down dose)
If haemolytic anaemia still remains => higher dose of glucocorticoids , IViG maybe tried , Rituximab
Refractory case => plasmapheresis
Splenectomy may be beneficial

AIHA ASSOCIATED WITH COLD ANTIBODIES Cold antibodies => RBCs antibodies more active at T and agglutinates RBCs at T <37oC
Primarily IgM and require complements for activity
Primary/Idiopathic
Secondary to infection Mycoplasma pneumoniae, EBV
Secondary to lymphoproliferative disorder

LABORATORY FINDINGS
Haemoglobinaemia
Haemoglobinurea
Spontaneous RBCs agglutination => RBCs agglutination seen in blood film
MCV

Red blood cell agglutination in a patient with a cold (IgM) immune hemolytic anemia. (Courtesy of Jean Schafer.)

TREATMENT
Self limiting
? transfusion
Glucocorticoids much less effective
Patient should avoid exposure to cold (warm blood products, fluids)
Treat for underlying disease
Rarely, in severe haemolytic anaemia => immunosuppression and plasmapheresis
Steroids and splenectomy are usually ineffective

EXTRACELLULAR:ISOIMMUNE HAEMOLYTIC ANAEMIAHAEMOLYTIC DISEASE OF THE NEWBORN(ERYTHROBLASTOSIS FETALIS)
Transpacental passage of maternal Ig active against paternal RBC antigens of infant
=> RBCs destruction
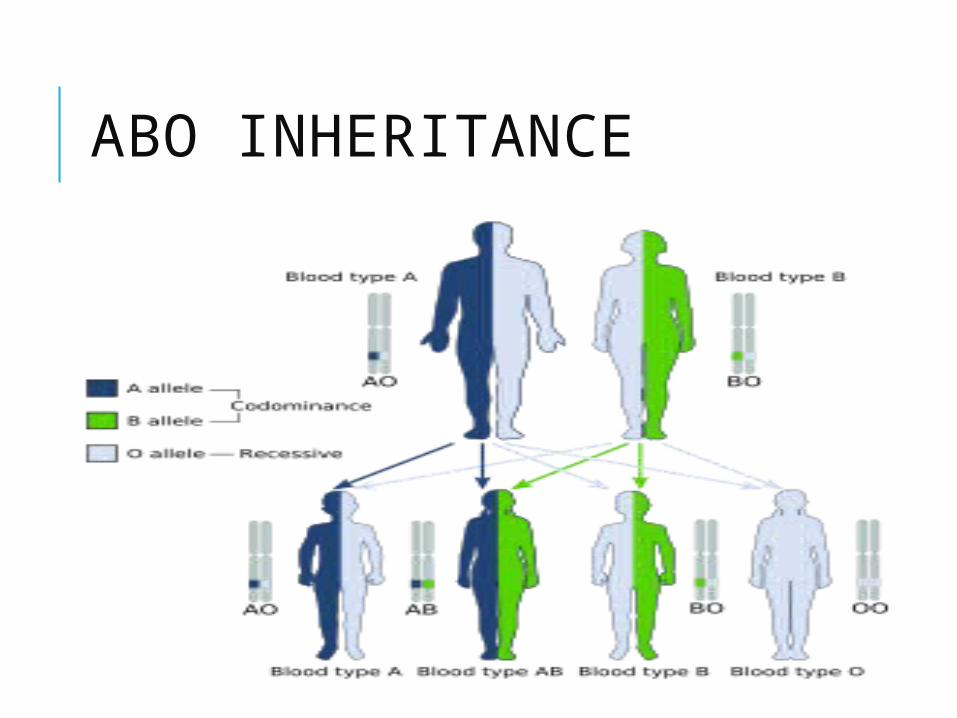
ABO INHERITANCE


RH INCOMPATIBILITY
Rh antigenic => genetically transmitted by each parents
Pathogenesis:
Rh negative mother
Rh positive infant’s blood (D antigen) infused into mother’s system
Ig formation against D antigen occur in unsensitised Rh negative mother
Once senstisation has occurred => small dose of antigen Ig titre
Initially IgM antibody, later takes place by IgG=> readily cross placenta

WHY DOES IT RARELY OCCUR DURING FIRST PREGNANCY? Transfusion occur near to time of delivery => too late for mother to become sensitized and trasmit Ig
Ability of mother to produce antibody => produce only low titre

CLINICAL MANIFESTATION Laboratory evidence of mild haemolysis (15%)
Severe anaemia with compensatory hyperplasia or erythropoetic tissue => hepatosplenomegaly
Anaemic S&S => when compensatory capacity of haemopoetic system is exceeded
Profound anaemia (pallor, signs of cardiac decompensation)
Massive anasarca
Hydrops fetalis
Jaundice first day of life => massive haemolysis
(Risk of bilirubin enchephalopathy/ kenicterus)
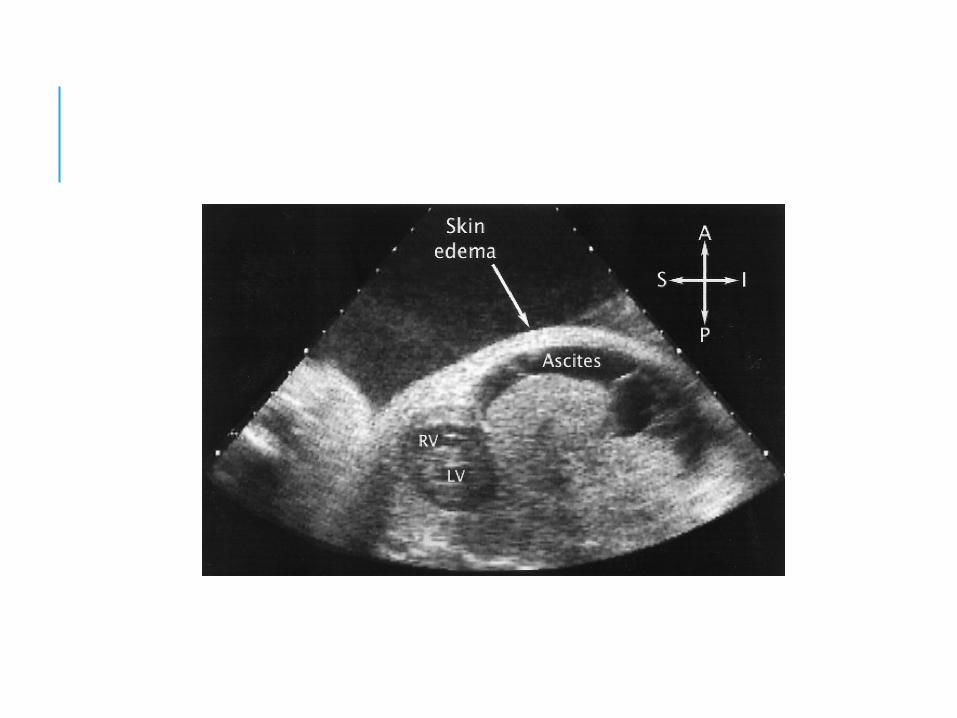


DIAGNOSISANTENATAL DX:For Rh negative mother: Hx of previous transfusion, abortion, pregnancy => possible sensitisation, prev affected infant, stillbirth
parents blood types should be tested => incompatibility
Maternal titre Ig towards D antigens=> 12-26 wk, 28-32wk and 38 wk
Monitoring severity of fetal disease=> TAS (evidence of hydrops fetalis), doppler US( evidence of increase vascular resistance in middle cerebral artery), amniocentesis/ cordocentesis??, PUBS is the goal standard

DIAGNOSIS:POSTNATAL Definitive: demonstration of blood group incompatibilty and corresponding Ig bound to infant’s RBCs, Rh type
Anaemia (in hydrop fetalis may be as low as 3-4g/dL)
WCC may be normal or elevated
Thrombocytopenia in severe cases
Serum bilirubin
Retics increase
Direct coombs test : POSITIVE
Blood film : polychromasia and marked increase in nucleated RBCs

TREATMENT
Unborn infant: Hydrops fetal anaemia (Hct 30%) => intravascular fetal transfusion
Liveborn infant: Fresh, low-titre, group O, leucoreduced and irradiated Rh-neg blood should be available
Evidence of hydrops fetalis
immediate resuscitation, supportive treatment, haemodynamic stabilisation before proceeding with ET
ET => previous kernicterus, retic >15%, prematurity
Monitoring => FBC and serum bilirubin 4-6 hr interval

PREVENTION OF RH SENSITISATION RhoGAM (human anti D globulin)
IM 300 microgramwithin 72 hrs of delivery of Rh postive infant, ectopic pregnancy, abdominal trauma in pregnancy, abortion
RhoGAm also more effective if administered at 28-32 week than again at birth than given as a single dose

BLOOD GROUP A AND B INCOMPATIBILITY Most common cause of incompatibility
15% of birth are at risk, manifestation 0.3-2.2%
IgM antibodies do not cross placenta, but mother who have develop IgM antibody towards A/B from previous sensitisation => haemolytic anameia
Presentation (milder than Rh incompatibility)
Treatment: phototherapy or rarely ET (post discharge, monitoring of Hg/Hct is essential)



![Retrospective study of canine infectious haemolytic …...Haemolytic anaemia develops as a consequence of red blood cell lysis due to infectious or non-infectious causes [1]. Infectious](https://img.pdfslide.us/doc/110x75/5f3a35eedf03db47f4785f1b/retrospective-study-of-canine-infectious-haemolytic-haemolytic-anaemia-develops.jpg)


























