Embed Size (px)
Citation preview

J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
104
Case Report h
Multisystem Langerhans Cell Histiocytosis in high-risk group: A case series of two infants
Anggun Putri Yuniaswan1, Putri Rachma Safitri1, Diah Prabawati Retnani2
1. Department of Dermatology and Venereology, Faculty of Medicine, Universitas Brawijaya
Dr. Saiful Anwar Regional General Hospital, Malang, Indonesia 2.Department of Pathology Anatomy, Faculty of Medicine, Universitas Brawijaya
Dr. Saiful Anwar Regional General Hospital, Malang, Indonesia h
Email: [email protected]
Abstract Background: Langerhans Cell Histiocytosis (LCH) has diverse manifestations, from asymptomatic to aggressive, which involves many organs. Histopathological examination plays a crucial role as a basic diagnostic standard for LCH. Writing Group of the Histiocyte Society proposes a guideline for diagnosing LCH, divided into presumptive, designated, and definitive diagnosis. Case Illustration: Two cases of a 14 month-old girl and an 18 month-old girl presented similar clinical manifestation and multi-organ involvement. Dermatological examination revealed red papules and plaques covered by brownish scales and crusts on the scalp and body, erosion in some folds of the body. Histopathological examination of the first case revealed an early purpuric phase. S100 immunostaining just revealed hyperplasia of Langerhans cell but still could not support the diagnosis of LCH. Fine Needle Aspiration Biopsy of the enlarged submandibular lymph node after two months of observation suggested LCH. In the second case, histopathological examination revealed proliferation of round-oval nucleated cells, pleomorphic, some reniform nuclei, with amphophilic cytoplasm. S100 and CD1a immunostaining revealed a positive reaction in the proliferative cells. Discussion: Patients aged 14 and 18 months old indicated almost similar clinical manifestations leading to LCH diagnosis, with different histopathological pictures. The first patient was presumptively diagnosed as high-risk multisystem LCH, but the initial histopathology results did not support LCH diagnosis. On the other hand, the second patient was definitively diagnosed with high-risk multisystem LCH. Conclusion: Patients with clinically suspected LCH without histopathological confirmation should be observed at least six months to reassess the necessity of a follow-up biopsy. Keywords: Langerhans Cell Histiocytosis, multisystem organ, histopathology, S100 Background A histiocyte is a group of various disorders with major signs of accumulation and infiltration of monocytes, macrophages, and dendritic cells to the target tissues.1 Langerhans cells histiocytosis (LCH) is characterized by clonal proliferation and reactive accumulation of dendritic cells.2 This disease is a rare one with less than one incidence in 200,000 children.1 Patient with LCH demonstrate a variety of clinical presentation depending on associated involved in several organ systems,
indicates the different possible outcomes.3 Tissue biopsy is required as the basis for the LCH diagnosis.4 This case series present two cases of high-risk multisystem type LCH in patients aged 14 and 18 months Case Illustration 1 A 14-month-old girl patient was admitted to our hospital with a complaint of weakness, pallor, fever, and liquid diarrhea. The patient was consulted to Dermatology and Venereology Department due to brownish thick scales on the scalp, noted 7 months

J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
105
previously. Multiple red spots also had appeared on the forehead and extended to the back, hands, legs and feet at the age of 1 month. In addition, reddish spots and abrasions occasionally appeared behind the ears, folds of the neck, folds of the armpits, groin and genital during the last 7 months. Whitish plaque was also found in one of the patients' tongue. Since 2 months, there was brownish yellow discharge from patient's ears. Patients also experienced a decrease in appetite and weight. There was no abnormality in the patients' growth and development history. General examination of the patient showed vital signs within normal limits. Anthropometry status defined the patient in malnourished condition (marasmus), also looked anemic. Her abdomen was distended and hard. Spleen was scored as Schuffner III. There were also enlarged lymph nodes on both sides of the neck. Dermatological examination revealed red papules covered with brownish crust and scales on the scalp (Figure 1.A).On the tongue and lips inspection, a white plaque was demonstrated. In the anterior and posterior trunk, brownish-red papules and macules are noted (Figure 1.B). There was an erythematous erosion in the retro auricular region, neck, armpit, and inguinal folds (Figure 1.C). During genital examination, thick whitish vaginal discharge was demonstrated. Haematological examination suggested anemia (Haemoglobin level 3,7 g/dL). Erythrocytes decreased to 2.28 x 106 cells/µL, leucocytosis with a level of 14.580 cells/µL also occurred. Physiology of hemostasis examination showed increased PTT INR to 1.62, and APTT of the patients reached 53 (Control: 26). Blood smear revealed normochromic erythrocytes, anisopoicilocytosis, teardrop cells, normoblasts, neutrophilia, monocytosis, giant platelets, and platelet clumping. Coombs test was negative. There was a decrease in iron levels to 10 ug/dL, a
reduction in TIBC to 98 ug/dL, and a reduction in transferrin saturation of -10%. Liver function, kidney function, and blood glucose were within normal limits. A gram smear examination of white plaque on the tongue and vaginal discharge showed candidiasis infection. In the bone marrow examination, no foreign cells were found (a), suggesting megaloblastic anemia. Histopathological examination revealed initial purpura phase (Figure 1.D), and immunohistochemistry with S100 staining showed slight Langerhans cell hyperplasia, thus did not support the LCH diagnosis, and it was not continued with checking CD1a (Figure 1.E). The patient was diagnosed with LCH (presumptive diagnosis) involving the skin, lymph nodes, haematopoietic system, liver, and spleen. Therefore, this patient was classified into high-risk multisystem LCH and received PRC transfusion, albumin transfusion, normal saline dressing 3x10 minutes on erosion, desoximetasone 0.05% cream twice per day on the scalp, and sodium fusidate cream twice per day in the skin and genitalia erosions, and ear drop of ofloxacin 3 mg/ml from otorhinolaryngologist. The patient was observed for six months, but in the second month, the patient's condition deteriorated, and she complained of pallor and enlarged lumps in the neck. From the physical examination, anemia was suggested, and enlarged lymph nodes in the left submandibular and left inguinal region were found. The FNAB examination on enlarged submandibular lymph node revealed proliferation of round nucleated cells with nuclear grooves, suggested LCH (Figure 1.F). The patient was admitted to the hospital and received a transfusion of PRC and therapy of PO prednisone 40 mg/m2/day and chemotherapy in the form of IV vinblastine 6 mg/m2, IV leucovorin 12 mg/m2 for six circles. The patient passed away after receiving some chemotherapy due to septic shock.

J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
106
Figure 1. Case 1. (a) papules and erythema covered with brownish-yellow scales (b) brownish-red papule (c) erosion with erythema base (d) First histopathological examination revealed erythrocyte in
the dermis, suggesting initial purpura (e) S100 examination was positive in several cell groups, showing Langerhans cell hyperplasia (f) Fine needle aspiration biopsy (FNAB) examination revealed
proliferation of round nucleated cells with nuclear groves.
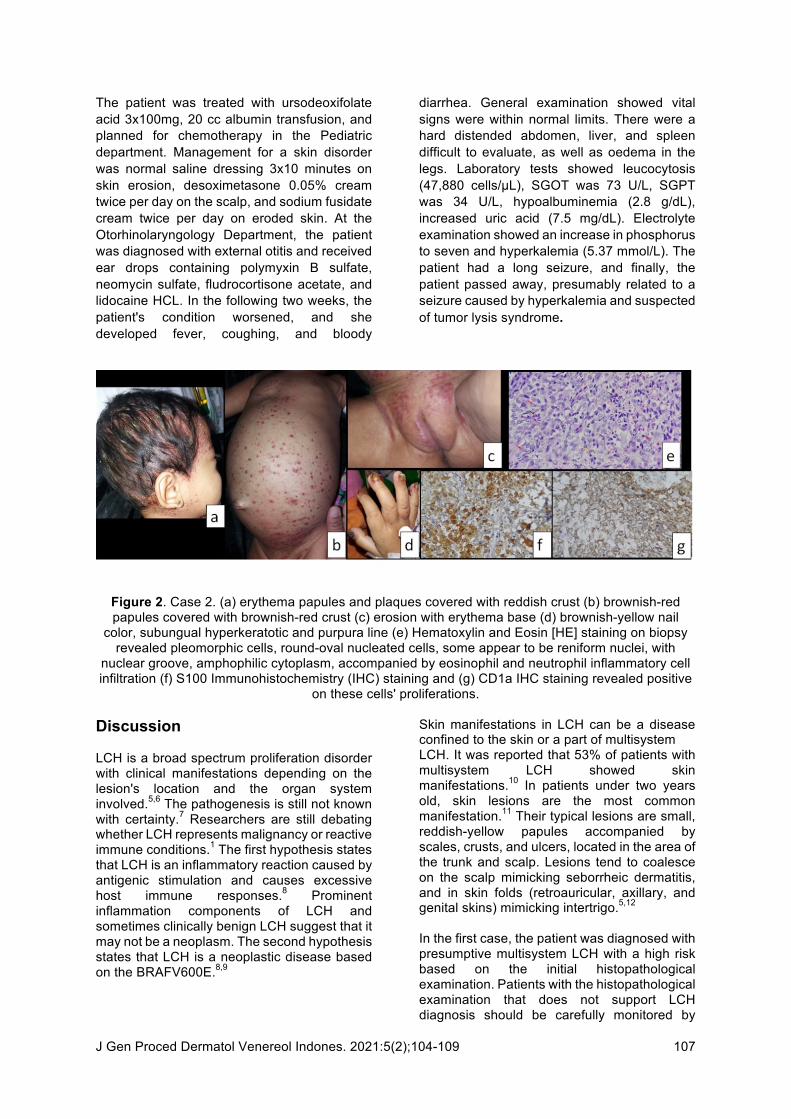
Case Illustration 2 An 18-month-old girl patient was admitted to our hospital with complaint of weakness, nausea, and vomiting. The patient was referred to Dermatology and Venereology Department due to brownish-yellow thick scales on the scalp noted ten months ago. Red spots and brownish scales were also scattered on the forehead, nose, and back of the ear. Red spots covered by crust also had appeared on the abdomen, back, hands, legs, feet, and thigh folds for the last five months. In addition, red spots and abrasions appeared on the neck and armpit folds for these ten months. The patient's nails also turned brownish in color and thickened for four months. In the patient's ear, sometimes clear liquid comes out during the past four months. The patient had a low appetite, hence low weight. In the last month, the patient had a recurrent fever, and her eyes gradually became yellowish. Initially, the patient was diagnosed with seborrheic dermatitis. The patient visited the otorhinolaryngologist and was diagnosed with ear infections. The patient's growth and development history were within normal limits. General examination revealed vital signs were of normal value. The patient was weighed 10 kg, jaundiced, had a hard distended abdomen. The liver and spleen were difficult to evaluate. There was edema in the lower limbs and enlarged lymph nodes in the neck and left inguinal region. Dermatological status revealed red-brownish papules and plaque covered by yellow-brownish crusts and scales (Figure 2.A&B). On
the folds of the neck and both armpit, erythema-based erosion was demonstrated (Figure 2.C). Both hands' fingernails showed brownish yellow, subungual hyperkeratotic, and purpuric lines (Figure 2.D). On hematological examination, hemoglobin was found within normal limits. There was leukocytosis with 22,040 cells/µL. The total bilirubin and direct bilirubin level were above normal value (5.1 and 5.01, respectively), while indirect bilirubin was within normal limits at 0.09. SGOT level was 76 U/L, SGPT level was 39 U/L, hypoalbuminemia was found (2.31 g/dL). Alkaline phosphatase and ɤ-GT were high (1019 and 915 respectively), while total protein was low (5.69). Ultrasound examination showed hepatomegaly with diffuse parenchymal liver disease, suggesting hepatoblastoma, with Gall Bladder sludge. Thus, we suspected subileus to be present. There was no clear visualization of an intra-abdominal mass. Histopathological examination revealed pleomorphic cells, round-oval nucleated cells, some appear to be reniform nuclei, nuclear groove, amphophilic cytoplasm, accompanied by eosinophilic and neutrophilic inflammatory cell infiltration (Figure 2.E). IHC S100 and CD1a staining revealed a positive proliferation of these cells (Figure 2.F&G). Thus the patient was diagnosed with LCH (Definitive Diagnosis). Based on examination, it was found that there were organs involvement of the skin, lymph nodes, and liver. The patient was then classified into high-risk multisystem LCH.
J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
107
The patient was treated with ursodeoxifolate acid 3x100mg, 20 cc albumin transfusion, and planned for chemotherapy in the Pediatric department. Management for a skin disorder was normal saline dressing 3x10 minutes on skin erosion, desoximetasone 0.05% cream twice per day on the scalp, and sodium fusidate cream twice per day on eroded skin. At the Otorhinolaryngology Department, the patient was diagnosed with external otitis and received ear drops containing polymyxin B sulfate, neomycin sulfate, fludrocortisone acetate, and lidocaine HCL. In the following two weeks, the patient's condition worsened, and she developed fever, coughing, and bloody
diarrhea. General examination showed vital signs were within normal limits. There were a hard distended abdomen, liver, and spleen difficult to evaluate, as well as oedema in the legs. Laboratory tests showed leucocytosis (47,880 cells/µL), SGOT was 73 U/L, SGPT was 34 U/L, hypoalbuminemia (2.8 g/dL), increased uric acid (7.5 mg/dL). Electrolyte examination showed an increase in phosphorus to seven and hyperkalemia (5.37 mmol/L). The patient had a long seizure, and finally, the patient passed away, presumably related to a seizure caused by hyperkalemia and suspected of tumor lysis syndrome.
Figure 2. Case 2. (a) erythema papules and plaques covered with reddish crust (b) brownish-red papules covered with brownish-red crust (c) erosion with erythema base (d) brownish-yellow nail
color, subungual hyperkeratotic and purpura line (e) Hematoxylin and Eosin [HE] staining on biopsy revealed pleomorphic cells, round-oval nucleated cells, some appear to be reniform nuclei, with
nuclear groove, amphophilic cytoplasm, accompanied by eosinophil and neutrophil inflammatory cell infiltration (f) S100 Immunohistochemistry (IHC) staining and (g) CD1a IHC staining revealed positive
on these cells' proliferations. Discussion LCH is a broad spectrum proliferation disorder with clinical manifestations depending on the lesion's location and the organ system involved.5,6 The pathogenesis is still not known with certainty.7 Researchers are still debating whether LCH represents malignancy or reactive immune conditions.1 The first hypothesis states that LCH is an inflammatory reaction caused by antigenic stimulation and causes excessive host immune responses.8 Prominent inflammation components of LCH and sometimes clinically benign LCH suggest that it may not be a neoplasm. The second hypothesis states that LCH is a neoplastic disease based on the BRAFV600E.8,9
Skin manifestations in LCH can be a disease confined to the skin or a part of multisystem LCH. It was reported that 53% of patients with multisystem LCH showed skin manifestations.10 In patients under two years old, skin lesions are the most common manifestation.11 Their typical lesions are small, reddish-yellow papules accompanied by scales, crusts, and ulcers, located in the area of the trunk and scalp. Lesions tend to coalesce on the scalp mimicking seborrheic dermatitis, and in skin folds (retroauricular, axillary, and genital skins) mimicking intertrigo.5,12 In the first case, the patient was diagnosed with presumptive multisystem LCH with a high risk based on the initial histopathological examination. Patients with the histopathological examination that does not support LCH diagnosis should be carefully monitored by

J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
108
proper imaging for at least six months to reassess the necessity for re-biopsy and monitor the possibility of developed malignancy.6,13 During the observation period, our first patient had a lymph node enlargement. Thus, FNAB was performed. FNAB's examination revealed proliferation of round nucleated cells with nuclear grooves, suggested LCH. Patients receiving prednisone 40 mg/m2/day and chemotherapy in the form of IV vinblastine 6 mg/m2, IV leucovorin 12 mg/m2 for six circles. In the second case, the patient is definitively diagnosed as a high-risk multisystem LCH. Both patients, in this case, passed away from the disease. Writing Group of the Histiocyte Society proposes guidelines for identifying the level of trust for diagnosing LCH. These criteria include a presumptive diagnosis if the morphological characteristics are striking. It will be a designated diagnosis if the morphological features are striking, plus two or more additional positive results for adenosine triphosphatase, S100 protein, α-d-mannosidase, and peanut lectin. A definitive diagnosis is if the morphological characteristics are striking plus Birbeck granule in the lesion cell seen with Langerin CD270 stained and confirmed with an electron microscope and/or positive results in CD1a antigen staining in the lesion.5,6
Based on the organs affected, LCH is divided into several groups. It is called a single LCH system if it only involves one organ system and is said to be a multisystem LCH if it involves more than one organ. Single-system LCH is divided into local and multiple, whereas multisystem LCH is divided into low-risk groups when dealing with the skin, bone, lymph nodes, pituitary organs, whereas high-risk groups is when dealing with one or more high-risk organs such as the hematopoietic system, lungs, liver and spleen.5,6
Management of LCH depends on the patient's age, the disease's severity, and the lesions' location. Therapy in patients with multisystem involvement has several treatment options. One of them is monochemotherapy using vinblastine with or glucocorticoids. When monochemotherapy is ineffective, a multiagent regimen may be considered. Multisystem LCH has an unpredictable journey, especially when there is a high-risk organ involvement, often associated with a poor prognosis.2,5,14
Conclusion Patients with clinically suspected LCH, while the histopathological examination results do not support the diagnosis, need to be confirmed with immunohistochemistry stain or observed at least six months to reassess the need for re-biopsy to exclude malignancy.6,13 Since LCH has an unpredictable course, and it requires treatment options, varying depending on the risk and the extent of organ involvement. 2,5,14
References 1. Grana N. Langerhans cell histiocytosis.
Cancer Control. 2014 Oct;21(4):328-34. Available from: https://journals.sagepub.com/doi/abs/10.1177/107327481402100409. [Accessed on 26th December 2019]
2. Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multi-system Langerhans cell histiocytosis. Blood. 2013 Jan 1:blood-2012. Available from: https://pdfs.semanticscholar.org/4b1c/c4528d10513f4b0c9f2e5c5fe1e94ca730e7.pdf. [Accessed on 26th December 2019]
3. Donadieu J, Bernard F, Van Noesel M, Barkaoui M, Bardet O, Mura R, et al. Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis (LCH): Results of an international phase II study. Blood. 2015 Jan 1:blood-2015. Available from: https://pdfs.semanticscholar.org/c7bc/fc6cc8e76a14dd2cfed00e4234e3381020a0.pdf. [Accessed on 26th December 2019]
4. DiCaprio MR, Roberts TT. Diagnosis and management of Langerhans cell histiocytosis. JAAOS-Journal of the American Academy of Orthopaedic Surgeons. 2014 Oct 1;22(10):643-52. Available from: https://journals.lww.com/jaaos/Abstract/2014/10000/Diagnosis_and_Management_of_Langerhans_Cell.5.aspx. [Accessed on 26th December 2019]
5. Gelmetti C. ‘Cutaneous Langerhans Cell Histiocytosis’ in Goldsmith LA, Fitzpatrick’s Dermatology in General Medicine, 8thed., Fizpatrick’s dermatology in general medicine. 7th ed. New York: Mc Graw Hill Co; 2008. p 1782-95.

J Gen Proced Dermatol Venereol Indones. 2021:5(2);104-109
109
6. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatric blood & cancer. 2013 Feb;60(2):175-84. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/pbc.24367. [Accessed on 26th
December 2019] 7. Battistella M, Fraitag S, Teillac DH,
Brousse N, de Prost Y, et al. Neonatal and Early Infantile Cutaneous Langerhans Cell Histiocytosis: Comparison of Self-regressive and Non–self-regressive Forms. Arch Dermatol. 2010 Feb 1;146(2):149-56. Available from: https://jamanetwork.com/journals/jamadermatology/article-abstract/209621. [Accessed on 26th December 2019]
8. Morren MA, Vanden Broecke K, Vangeebergen L, Sillevis-Smitt JH, Van Den Berghe P, Hauben E, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: A Retrospective cohort study. Pediatric blood & cancer. 2016 Mar;63(3):486-92. Availble from: https://onlinelibrary.wiley.com/doi/abs/10.1002/pbc.25834. [Accessed on 26th
December 2019] 9. Badalian-Very G, Vergilio JA, Fleming M,
Rollins BJ. Pathogenesis of Langerhans cell histiocytosis. Annu. Rev. Pathol. 2013 Jan 24;8:1-20. Available from: https://www.annualreviews.org/doi/abs/10.1146/annurev-pathol-020712-163959. [Accessed on 26th December 2019]
10. Simko SJ, Garmezy B, Abhyankar H, Lupo PJ, Chakraborty R, Lim KP, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J. Pediatr. 2014 Nov 1;165(5):990-6. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0022347614007380. [Accessed on 26th December 2019]
11. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: History, classification, pathobiology,
clinical manifestations, and prognosis. J Am Acad Dermatol. 2018 Jun 30;78(6):1035-44. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0190962217319308. [Accessed on 26th December 2019]
12. Reddy IS, Gowrishankar S, Somani VK, Murthy DB. Adult onset Langerhans cell histiocytosis: Report of two patients. IJDVL. 2014 Nov 1;80(6):560. Available from: http://www.ijdvl.com/article.asp?issn=0378-6323;year=2014;volume=80;issue=6;spage=560;epage=562;aulast=Reddy. [Accessed on 26th December 2019]
13. Micic D, Rodriguez-Galindo C. Langerhans Cell Histiocytosis (LCH): Guidelines for diagnosis, clinical work-up and treatment for patients until the age of 18 years. Available from: https://s3.amazonaws.com/academia.edu.documents/44212353/Langerhans_cell_histiocytosis_LCH_Guid20160330-22847-nqfiva.pdf?response-content-disposition=inline%3B%20filename%3DLangerhans_cell_histiocytosis_LCH_Guidel.pdf&X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAIWOWYYGZ2Y53UL3A%2F20191226%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20191226T071131Z&X-Amz-Expires=3600&X-Amz-SignedHeaders=host&X-Amz-Signature=ab97b9cd4164a3f12703b08ca1e3f3ed5445c2b950ec6ce1ee6b5e06758f1281. [Accessed on 26th December 2019]
14. Morimoto A, Shioda Y, Imamura T, Kanegane H, Sato T, Kudo K, et al. Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr. Blood Cancer. 2011 Jan;56(1):110-5. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/pbc.22703. [Accessed on 26th
December 2019





























