Embed Size (px)
Citation preview

Journal of the Iranian Chemical Society, Vol. 2, No. 3, September 2005, pp. 176-188.
JOURNAL OF THE Iranian Chemical Society
GPCRs as Therapeutic Targets: a View on Adenosine Receptors Structure and Functions, and Molecular Modeling Support
D. Dal Ben, C. Lambertucci, S. Vittori, R. Volpini and G. Cristalli* Dipartimento di Scienze Chimiche,Via S. Agostino, 1; Università di Camerino, 62032 Camerino, Italy
(Received 30 July 2005, Accepted 8 August 2005)
G-protein-coupled receptors (GPCRs) represent the largest known family of signal-transducing proteins and transmit signals
for light and many extracellular regulatory molecules. GPCRs are dysfunctional or dysregulated in several human diseases and are
estimated to be the targets of ~40% of the drugs used in clinical medicine today. Receptors for adenosine belong to this family,
and so far four subtypes, the A1, A2A, A2B, and A3, have been recognized. The activation of adenosine receptors (ARs) is largely
responsible for the variety of effects produced by adenosine throughout several organ systems. Based on the wide (and often
beneficial) effects attributed to the accumulation of endogenously released adenosine, it has long been considered that regulation
of ARs has considerable therapeutic potential. In this review, we focus on recent work on adenosine receptors as therapeutic
targets and, in particular, on molecular modelling support to adenosine receptors targeting.
Keywords: G-protein-coupled receptors, Signal transduction, Adenosine, Adenosine receptors, Homology modeling
INTRODUCTION
G-protein-coupled receptors (GPCRs) compose a big set of
sensory proteins that mediate a wide variety of physiological
processes by responding to different external stimuli such as
hormones, paracrines (local hormones), neurotransmitters,
neuromodulators, odours, and light [1-3]. These
transmembrane receptors transduce the external signals mostly
by interacting with regulatory G-proteins [4-6]. Such proteins
transmit the signal to effector enzymes, such as cytoplasmic
proteins and ion channels, by causing rapid changes in the
concentration of intracellular signalling molecules, cAMP,
cGMP, diacylglycerol, arachidonic acid, inositol phosphates,
cytosolic ions. As some GPCRs may use heterotrimeric G-
proteins and even other cytoplasmic enzymes as transducers in
* Corresponding author.
E-mail: [email protected]
their signalling and others may interact with non-G-protein
transducers only, some authors prefer alternative names for
this protein superfamily such as 7-transmembrane receptors,
heptahelical receptors, or serpentine-like receptors [7,8].
Although the GPCR families do not share sequence
similarity with each other, specific features exist in all GPCR
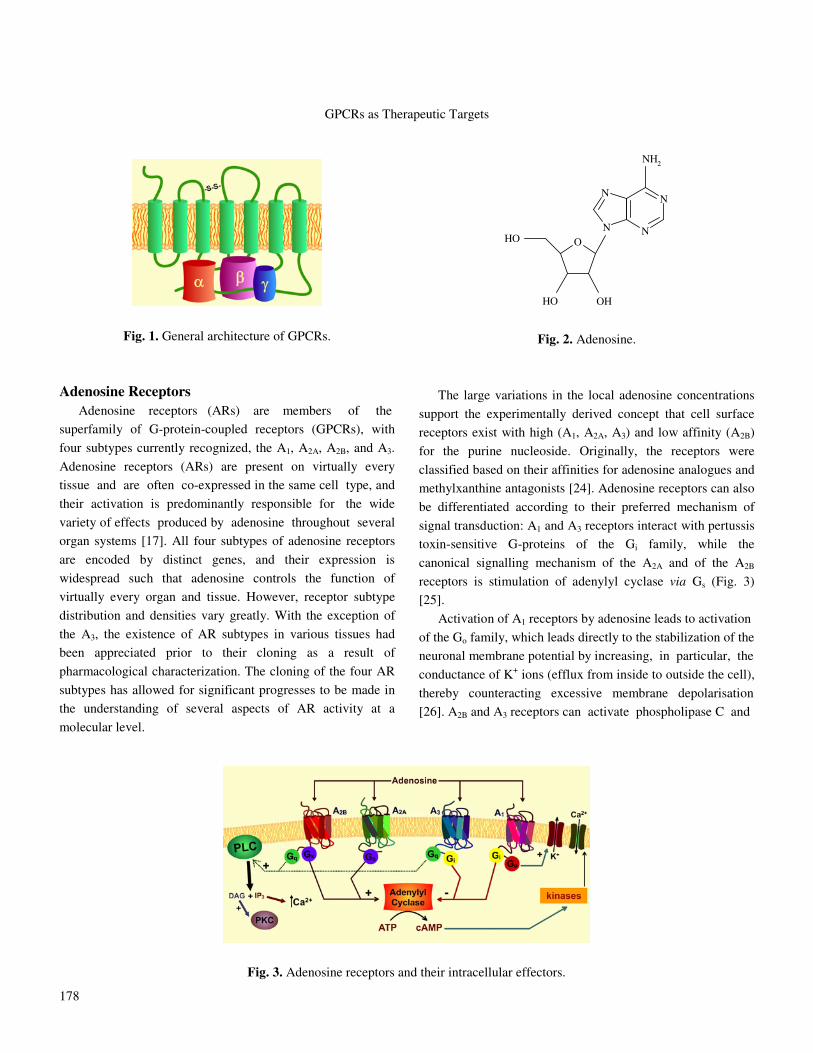
classes [9]. For example, all these receptors have in common a
central core domain that consists of seven transmembrane
(TM1-7) helices connected by three intracellular loops (IL1,
IL2, and IL3) and three extracellular loops (EL1, EL2, and
EL3). Two cysteine residues, one in TM3/EL1 interface and
one in EL2, which are conserved in almost all GPCRs, form
an essential disulfide link responsible for the packing and
stabilization of a restricted number of conformations of these
seven TM domains (Fig. 1). Beside sequence variations,
GPCRs differ in the length and function of their N-terminal
extracellular domain, their C-terminal intracellular domain and
their intracellular loops. Each of these domains confers

Dal Ben et al.
177
specific properties to these receptor proteins.
The superfamily of GPCRs can be subdivided into seven
families of receptors whose protein sequences share
significant similarity [10]. The main family (Family A), which
comprises rhodopsin and adrenoceptors, is composed by the
majority of GPCRs identified to date, and is the best studied
GPCR family both structurally and functionally. Receptors
that belong to this family are activated by a variety of stimuli,
including photons, odorants, and hormones and
neurotransmitters with molecular structures ranging from
small biogenic amines (e.g., catecholamines) to peptides (e.g.,
gonadotropin-releasing hormone) and complex glycoproteins
(e.g., luteinizing hormone). The other subfamilies are the
secretin-vasointestinal peptide (VIP) family (Family B),
members of which bind neuropeptides and peptide hormones,
the metabotropic glutamate receptor family (Family C),
comprising at least eight strongly related subtypes of
glutamate binding receptors, the fungal pheromone P family
(Family D), the fungal pheromone A receptors (Family E), and
the cAMP receptors of dictyostelium discoideum (Family F).
GPCRs can exist as dimers or as part of larger oligomeric
complexes, and many studies have intimated the existence of
heterodimeric GPCR pairings. Determination of the
prevalence and relevance of these complexes in native tissues
and the implications of heterodimerization for pharmacology
and potentially for drug design is still a key issue [11,12].
The superfamily of GPCRs comprises receptors for many
substances with important physiological functions. For this
reason their dysfunction leads to human diseases, and in fact
many GPCRs are targets for pharmaceuticals and drugs of
abuse [13-15]. It has been estimated that ~80% of known
hormones and neurotransmitters activate cellular signal
transduction mechanisms by activating GPCRs, and it has
been estimated that GPCRs represent ~40% of current drug
targets.
ADENOSINE RECEPTORS
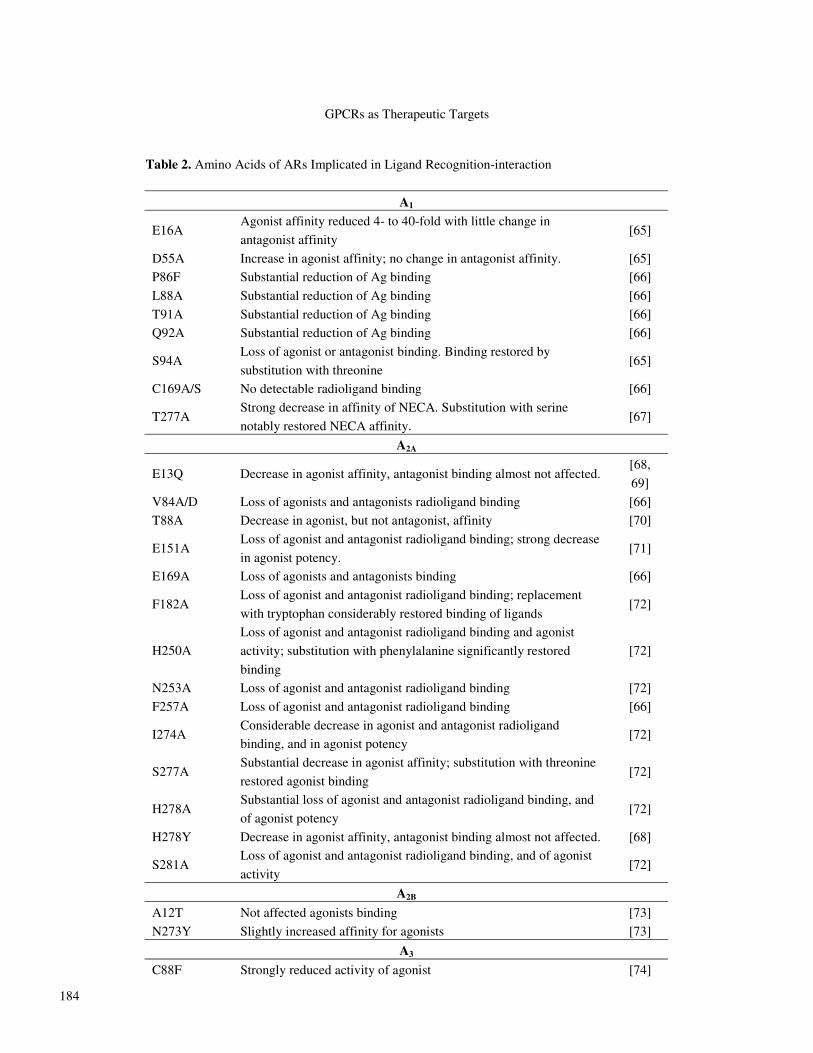
Adenosine The purine nucleoside adenosine (Fig. 2) is a mediator
playing an important role in modulating many
(patho)physiological conditions in the CNS as well as in
peripheral organs and tissues [16,17].
Adenosine is ubiquitously released in the extracellular
space from metabolically active cells and is generated by the
degradation of released ATP. As adenosine is unstable and has
a short half-life because of deamination or cellular reuptake,
hypoxia-induced increases typically bear only on local
adenosine receptor signalling. Hence, adenosine fulfils the
criteria of an autacoid. In the nervous system adenosine is
“omnipresent”, i.e. exists in all cells, and is released from
apparently all cells, including neurones and glia [18,19].
While ATP may function as a neurotransmitter in some brain
areas, adenosine is neither stored nor released as a classical
neurotransmitter since it does not accumulate in synaptic
vesicles, being released from the cytoplasm into the
extracellular space through a nucleoside transporter. The
adenosine transporters also mediate adenosine reuptake, and
the direction of the transport is dependent upon the
concentration gradient at both sides of the membrane. There
are two sources of extracellular adenosine: release of
adenosine by facilitated diffusion and extracellular conversion
of released adenine nucleotides into adenosine through a series
of ectoenzymes. Interestingly, extracellular adenosine levels
may be under control of adenosine A1 receptor since blockade
of A1 receptors increases the extracellular concentration of
adenosine in cardiac fibroblasts [20]. However, as the
extracellular formation of adenosine from released adenine
nucleotides represents a second source of adenosine, which is
not affected by transport inhibition, the transport inhibitors
usually cause an increase in the extracellular adenosine levels
[21].
Adenosine extracellular concentration is calculated to be in
the range of 50-200 nM [22], but under several adverse
conditions, including ischemia, trauma, stress and
inflammation, extracellular levels of adenosine are increased
due to increased energy demands and ATP metabolism. Under
hypoxic and ischaemic conditions there is a marked increase
in cytoplasmatic adenosine leading to an intense release of
adenosine, which is inhibited by adenosine uptake inhibitors
[16]. In the isolated heart hypoxia raises the extracellular
concentration by more than 20-fold, reaching concentrations
of > 1 μM [23].
GPCRs as Therapeutic Targets
178
Fig. 1. General architecture of GPCRs.
Adenosine Receptors Adenosine receptors (ARs) are members of the
superfamily of G-protein-coupled receptors (GPCRs), with
four subtypes currently recognized, the A1, A2A, A2B, and A3.
Adenosine receptors (ARs) are present on virtually every
tissue and are often co-expressed in the same cell type, and
their activation is predominantly responsible for the wide
variety of effects produced by adenosine throughout several
organ systems [17]. All four subtypes of adenosine receptors
are encoded by distinct genes, and their expression is
widespread such that adenosine controls the function of
virtually every organ and tissue. However, receptor subtype
distribution and densities vary greatly. With the exception of
the A3, the existence of AR subtypes in various tissues had
been appreciated prior to their cloning as a result of
pharmacological characterization. The cloning of the four AR
subtypes has allowed for significant progresses to be made in
the understanding of several aspects of AR activity at a
molecular level.
N
N
NH2
N
NO
OHHO
HO
Fig. 2. Adenosine.
The large variations in the local adenosine concentrations
support the experimentally derived concept that cell surface
receptors exist with high (A1, A2A, A3) and low affinity (A2B)
for the purine nucleoside. Originally, the receptors were
classified based on their affinities for adenosine analogues and
methylxanthine antagonists [24]. Adenosine receptors can also
be differentiated according to their preferred mechanism of
signal transduction: A1 and A3 receptors interact with pertussis
toxin-sensitive G-proteins of the Gi family, while the
canonical signalling mechanism of the A2A and of the A2B
receptors is stimulation of adenylyl cyclase via Gs (Fig. 3)
[25].
Activation of A1 receptors by adenosine leads to activation
of the Go family, which leads directly to the stabilization of the
neuronal membrane potential by increasing, in particular, the
conductance of K+ ions (efflux from inside to outside the cell),
thereby counteracting excessive membrane depolarisation
[26]. A2B and A3 receptors can activate phospholipase C and
Fig. 3. Adenosine receptors and their intracellular effectors.

Dal Ben et al.
179
this is thought to be mediated by activation of Gq.
Diacylglycerol (DAG) and inositol (1,4,5)-trisphosphate (IP3)
are implicated in the regulation of protein kinase C (PKC)
activity and the intracellular concentration of Ca2+ ions,
respectively [27]. In the brain, by the inhibition of neuronal
Ca2+ influx (through inhibition of adenylyl cyclase), adenosine
counteracts the presynaptic release of the potentially
excitotoxic neurotransmitters glutamate and aspartate, which
can impair intracellular Ca2+ homeostasis through
metabotropic glutamate receptors or the induction of
membrane depolarization through ion channel-linked
glutamate receptors [28,29]. Other G-proteins can also be
activated following adenosine receptor activation even though
the physiological significance of this is unknown.
Adenosine Receptors Structure ARs display the topology typical of GPCRs. These
receptors have in common a central core domain of seven
transmembrane helices, with each TM composed of 20-25
amino acids, connected by three intracellular (IL1, IL2 and
IL3) and three extracellular (EL1, EL2 and EL3) loops. Two
cysteine residues (one in TM3/EL1 interface and one in EL2)
which are conserved in most GPCRs, form a disulfide bond
which is crucial for the folding and for the stabilization of a
limited number of conformations of these seven TMs (Fig. 4)
[17,30].
Adenosine receptors differ in the length and function of
their N-terminal extracellular domain, their C-terminal
intracellular domain and their intracellular loops. Each of
these domains provides very specific properties to these
receptor proteins. Particularly, consensus sites for N-linked
glycosylation exist on the extracellular regions of ARs,
although the precise location of the sites for this post-
translational modification varies among the AR subtypes. The
carboxyl-terminal tails of the A1, A2B, and A3, but not the A2A,
possess a conserved cysteine residue that may putatively serve
as a site for receptor palmitoylation and permit the formation
of a fourth intracellular loop. However, site-directed
mutagenesis of this residue has not been performed for any
AR subtype, and no role for putative AR palmitoylation has
been described.
The A1, A2B, and A3 receptors are very similar in regard to
the number of amino acids composing their primary structure,
Fig. 4. Schematic view of adenosine A3 receptor structure.
and overall, these AR subtypes are among the smallest
members of the GPCR family. For example, the human
homologues of A1, A2B, and A3 consist of 326, 328, and 318
amino acid residues, respectively. On the contrary, the human
A2A is composed of 409 amino acids. All cloned species
homologues of the A2A are of similar mass, and this relatively
large size is manifested in the carboxyl-terminal tail of the
receptor, which is much longer than that of the other AR
subtypes. It should be noted that the size of ARs deduced from
their primary amino acid structure frequently is not consistent
with the mass estimated by polyacrylamide gel electrophoresis
of the expressed proteins. This is probably due to the post-
translational glycosylation of ARs which may vary in a cell
type-dependent fashion. The human A1 and A3 receptors
display about 49% overall sequence identity at the amino acid
level, while the human A2A and human A2B receptors are 45%
identical (see Table 1).
Adenosine Receptors as Therapeutic Targets Research on adenosine receptors during the past 20 years
has resulted in considerable progress in identifying selective
agonists and antagonists and an increased understanding of the
particular roles adenosine receptor subtypes play in
physiological processes [31].
Indeed, several protective effects of adenosine have been
widely reported: a) adenosine has protective effects in the

GPCRs as Therapeutic Targets
180
ischemic and reperfused myocardium by minimizing the
workload of the heart and its oxygen demand during ischemia
(through adenosine A1 receptor pathways [32]) and by causing
coronary vasodilatation and increasing myocardial oxygen
supply through the activation of adenosine A2A receptors on
vascular smooth muscle and endothelial cells; beside that,
adenosine inhibits platelet aggregation, protecting endothelial
cell, integrity and limiting the vascular injury. b) Adenosine
exhibits protecting effects against cerebral ischemia. It has
been reported that stimulation of adenosine A1 receptor
decreases the mortality associated with focal or global brain
ischemia. Additionally, by acting through adenosine A2A
receptor pathways, adenosine ameliorates post-ischemic
cerebral blood flow and decreases post-ischemic cerebral
damage. c) Adenosine has been reported to be beneficial in
organ transplantation, acting by mechanisms similar to those
for its action against ischemia and reperfusion injury. d)
Adenosine is an endogenous neuroprotective agent and
exhibits anticonvulsant effects. The anticonvulsant effects of
adenosine appear to be mediated primarily by adenosine A1
receptors [33]. e) Adenosine inhibits platelet aggregation and
attenuates tissue factor expression on endothelial cells induced
by various stimuli, acting mainly through adenosine A2A
receptor pathways. For this reason, the inhibitory effects of
adenosine on platelet and endothelial cell activation are
thought to be beneficial for the treatment of thrombosis. f)
Adenosine exerts multiple effects on pain transmission both at
spinal and peripheral sites. In the spinal cord, adenosine
provides pain relief through adenosine A1 receptor pathways
in acute nociceptive, inflammatory, and neuropathic pain tests
[34]. g) Adenosine is released at sites of inflammation and acts
as an endogenous anti-inflammatory agent, and adenosine A2A
receptor pathways seem to have an important role in the anti-
inflammatory effects of adenosine. h) In asthmatic conditions,
the extracellular concentration of adenosine increases in the
airway tissue. It inhibits eosinophil or lymphocyte function
and modulates reactive oxygen species generation in
neutrophils; anyway, many aspects of this mechanisms are still
unclear [35-37].
This knowledge has continued to stimulate considerable
interest in pursuing adenosine receptors as therapeutic targets.
For example, a number of selective agonists for A1, A2A, A2B,
and A3 [38] adenosine receptors have been developed, and
several such compounds are undergoing clinical trials for the
treatment of cardiovascular diseases (A1 and A2A) [31,39-41],
pain (A1), wound healing (A2A), diabetic foot ulcers (A2A),
colorectal cancer (A3) [42] and rheumatoid arthritis (A3). In
general, adenosine receptor agonists are derivatives of the
physiological agonist adenosine.
The development of adenosine receptor agonists has been
limited by an essential requirement for retention of the ribose
moiety for agonist activity. Despite this restriction, significant
progress has been made in the identification of potent and
selective adenosine receptor agonists. On the other hand, the
clinical evaluation of some A1 and A2A adenosine receptor
agonists has been discontinued, and the major problems
comprise: side effects due to the wide distribution of
adenosine receptors; low brain penetration, which is important
for the targeting of CNS diseases; short half-lives of
compounds; lack of effects, in some cases perhaps due to
receptor desensitisation or to low receptor density in the
targeted tissue [43,44].
Partial agonists, inhibitors of adenosine metabolism
(adenosine kinase and deaminase inhibitors) or allosteric
activators of adenosine receptors may be advantageous for
certain indications, as they may exhibit fewer side effects [45].
Adenosine itself is used as a therapeutic agent for the
treatment of supraventricular paroxysmal tachycardia and
arrhythmias and, as a vasodilatatory agent, in cardiac imaging
[46,47]. Two adenosine products, Adenocard® and
Adenoscan®, have been approved by FDA and are currently
available for cardiac arrhythmias treatment and for cardiac
imaging, respectively [48,49].
Recent focus has been on the cardioprotective and
neuroprotective effects associated with AR activation during
periods of cardiac and cerebral ischemia, respectively. On the
other hand, it has been recently proposed that antagonists of
distinct AR subtypes may be used in the treatment of certain
neurological diseases such as Parkinson's disease[50-52] .
MOLECULAR MODELING SUPPORT IN ADENOSINE RECEPTORS TARGETING The 3D-structure Problem The evolution of the field of computer-aided design of ligands (both agonists and antagonists) for GPCRs, including

Dal Ben et al.
181
adenosine receptors, has depended on the availability of
suitable molecular receptor templates. In fact, due to technical
difficulties, which complicate experimental X-ray diffraction
and NMR structure determination of GPCRs, the 3D structure
of most GPCRs is still unknown [1,9]. In fact, while it is quite
simple to deduce the amino acid sequence of a protein from
the DNA sequence of the gene encoding it, determining the
three-dimensional molecular structure of proteins has proven
to be more complex, especially for membrane proteins that
represent about half of all currently exploited drug targets. The
usual time needed to solve an eukaryotic protein target from
clone to three-dimensional structure has been one to three
years for soluble proteins and even longer, with a higher risk
of failure, for membrane proteins.
Indeed, atomic resolution crystal structures of soluble
proteins have been reported in a rapidly increasing number
over the last decade, while such improvement has not been
made in terms of membrane proteins, which have proven
extremely difficult to crystallize for two main factors: the
amphipathic nature of their surface, with a hydrophobic area in
contact with membrane phospholipids and polar surface areas
in contact with the aqueous phases on both sides of the
membrane; the very low concentrations in tissues of the
majority of medically relevant membrane proteins.
Early structural data of G-protein-coupled receptors came
from low-resolution projection maps from cryo-electron
microscopy experiments. In a paper published in 1975,
Henderson and Unwin demonstrated that bacteriorhodopsin, a
protein from halobacterium halobium that works as a light-
driven proton pump, has seven membrane spanning domains
that were interpreted as α-helices [53]. The idea of a seven
transmembrane α-helical (TMH) architecture of GPCRs,
initially based on bacteriorhodopsin which does not act
through a G-protein, was well established and had already
been proposed in lots of scientific papers suggesting GPCR
models, when strong support for the 7 TMH architecture of
GPCRs came from Henderson’s group in 1993.
An electron projection map of visual rhodopsin, a
chromoprotein in the retina that acts via a G-protein,
transducin, showed seven membrane spanning domains that
appeared to be α-helices, arranged in a slightly different way
respect those in bacteriorhodopsin [54]. New suppositions
about the structure of these receptors came from
cryomicroscopic studies of rhodopsin that indicated the
existence of seven transmembrane segments and gave an
indication of the relative disposition of these TMs [55]. The
final proof of the 7 TMH architecture of rhodopsin came when
an atomic-resolution crystal structure of bovine rhodopsin was
published a few years later [56].
Adenosine Receptors Structure-based Drug Design Molecular modeling provides a useful methodological
alternative to experimental structure determination, especially
for membrane proteins. Homology modeling of a protein
structure is based on three factors: a three-dimensional
structural template, an amino acid sequence alignment of the
modelled molecule with the template molecule (and other
related molecules), and particularly adapted computer
software. The accuracy and consistency of the protein model
depends on the accuracy of the structural data template, and
the similarity of the modelled structure with the one or those
used as a template for the initial protein model. Compared to
modeling of drug targets, which usually are macromolecules
and in most cases proteins with several hundred amino acids,
modeling of drug molecules is relatively straightforward. The
challenge in modeling of drug-receptor interactions lies in
modeling of the target molecule, and in the docking of ligand
molecules into postulated binding sites.
An early model of the TM segments of an adenosine A3
receptor (cloned from rat) was proposed by van Galen [57].
This model was based on the primary sequence and structural
homology with bacteriorhodopsin, in analogy to the method
proposed by Hibert [58]. A putative ligand binding region was
proposed and explored by docking several antagonists into the
TM domain model. In particular, this model suggested that
TM3, TM6 and TM7 might be involved in ligand binding.
However, as reported above, bacteriorhodopsin lacks any
functional or sequence homology with GPCRs. The original
model of van Galen was improved in the description of
ligand/receptor interactions by introduction of a simple way to
simulate the reorganization of the native receptor structure
induced by ligand coordination (cross-docking methodology)
[59,60]. This method gives good results in predicting local
structural changes induced by ligand in the receptor binding
site. The presence of both agonist and antagonist within the
receptor can produce a simultaneous adjustment in the

GPCRs as Therapeutic Targets
182
Table 1. Sequence Alignment of Human Adenosine A1, A2A, A2B ,and A3 Receptors.
The Respective Regions Containing Transmembrane (TM) Segments Have
been Aligned with ClustalW (http://www.ebi.ac.uk/clustalw). The Residues
Color Scheme is According to ClustalW: Color Red Corresponds to Small and
Hydrophobic Residues (AVFPMILW); Color Blue is for Acidic Residues
(DE); Color Magenta is for Basic Residues (RK) and Color Green
Corresponds to the Remaining Residues (STYHCNGQ)
TM1
AA1R_HUMAN AAYIGIEVLIALVSVPGNVLVIWAVK 35
AA2AR_HUMAN SVYITVELAIAVLAILGNVLVCWAVW 32
AA2BR_HUMAN ALYVALELVIAALSVAGNVLVCAAVG 33
AA3R_HUMAN VTYITMEIFIGLCAIVGNVLVICVVK 38
TM2
AA1R_HUMAN DATFCFIVSLAVADVAVGALVIPLAI 67
AA2AR_HUMAN NVTNYFVVSLAAADIAVGVLAIPFAI 64
AA2BR_HUMAN TPTNYFLVSLAAADVAVGLFAIPFAI 65
AA3R_HUMAN TTTFYFIVSLALADIAVGVLVMPLAI 70
TM3
AA1R_HUMAN LMVACPVLILTQSSILALLAIAVDRY 106
AA2AR_HUMAN LFIACFVLVLTQSSIFSLLAIAIDRY 103
AA2BR_HUMAN LFLACFVLVLTQSSIFSLLAVAVDRY 104
AA3R_HUMAN LFMTCLLLIFTHASIMSLLAIAVDRY 109
TM4
AA1R_HUMAN RRAAVAIAGCWILSFVVGLTPMFGWN 147
AA2AR_HUMAN TRAKGIIAICWVLSFAIGLTPMLGWN 144
AA2BR_HUMAN TRARGVIAVLWVLAFGIGLTPFLGWN 145
AA3R_HUMAN RRIWLALGLCWLVSFLVGLTPMFGWN 150
TM5
AA1R_HUMAN YMVYFNFFVWVLPPLLLMVLIYLEVF 204
AA2AR_HUMAN YMVYFNFFACVLVPLLLMLGVYLRIF 201
AA2BR_HUMAN YMVYFNFFGCVLPPLLIMLVIYIKIF 206
AA3R_HUMAN YMVYFSFLTWIFIPLVVMCAIYLDIF 201
TM6
AA1R_HUMAN IAKSLALILFLFALSWLPLHILNCIT 257
AA2AR_HUMAN AAKSLAIIVGLFALCWLPLHIINCFT 256
AA2BR_HUMAN AAKSLAMIVGIFALCWLPVHAVNCVT 257
AA3R_HUMAN TAKSLFLVLFLFALSWLPLSIINCII 253
TM7
AA1R_HUMAN ILTYIAIFLTHGNSAMNPIVYAFRIQ 293
AA2AR_HUMAN WLMYLAIVLSHTNSVVNPFIYAYRIR 293
AA2BR_HUMAN WAMNMAILLSHANSVVNPIVYAYRNR 295
AA3R_HUMAN LVLYMGILLSHANSMMNPIVYAYKIK 287

Dal Ben et al.
183
Fig. 5. Crystal structure of bovine Rhodopsin (pdb code:
1L9H). Disulfide bond and Retinal are shown. orientation of TM3, TM5, TM6, and TM7 [60].
The rhodopsin crystal structure represented a breakthrough
both in that it demonstrated beyond any doubt the 7 TMH
structure of a G-protein-coupled receptor, assumed several
years earlier, and in providing a much more accurate template
for molecular modeling of adenosine receptors than the
previous projection maps (Fig. 5). Since 2000, other 3D
structures of bovine rhodopsin have been solved through both
X-ray diffraction (pdb 1HZX, 1L9H, and 1U19) and NMR
structure determination (pdb 1JFP, 1LN6), and at the present
the rhodopsin crystal structures are the generally used
templates for building of ARs structures through homology
modeling techniques [61-63].
As first, multiple-sequence alignments of selected G-
protein-coupled receptors are used to collect data about
patterns or motifs present in these sequences, in particular in
the TM regions. The aim is to identify the same patterns into
AR sequences and correctly align them to rhodopsin crystal
structure. A guide for the alignment is the presence of some
key highly conserved amino acid residues, including the DRY
motif (in TM3) and three Pro residues (in TM4, 6 and 7) in the
TM segments of GPCRs. Once aligned the sequences, the
same boundaries are applied for the TM helices of the AR as
are identified from the X-ray crystal structure for the
corresponding sequences of bovine rhodopsin, the Cα
coordinates are used to build the seven TM helices for the
human A3 receptor. Special caution is needed for the second
extracellular (E2) loop, which is described in bovine
rhodopsin to fold back over transmembrane helices, and,
therefore, it limits the size of the active site. Hence, amino
acids of this loop could be involved in direct interactions with
the ligands. The presence of a disulfide bridge between two
cysteine residues in TM3 and E2 may be the driving force to
this peculiar fold of the E2 loop [64].
However, a structure-based approach to adenosine
receptors drug discovery in the absence, but maybe also in the
presence, of the real structures requires a multidisciplinary
approach, where molecular models represent a structural basis
to powerfully integrate experimental data derived from
molecular biological, biophysical, bioinformatics, and
pharmacological methods [13,15]. Indeed, the identification of
discrete ARs regions, or even single amino acids that are
critical for ligand recognition and are responsible for
discriminating between agonist and antagonist ligands, has
been an area of extensive study. In addition to a basic
understanding of receptor activation, it has been hoped that a
delineation of ligand-receptor interaction at a molecular level
may provide the basis for rational drug design. Both TMs and
extracellular regions of ARs have been implicated as playing a
role in the formation of the ligand-binding pocket. Site
directed mutagenesis studies in parallel with different
molecular modeling approaches have been recently used as
powerful strategy to design potent and selective ARs ligands,
and the mutagenesis data are summarized in Table 2. These
data can be very helpful, and indeed recently Jacobson and
collaborators created a “neoceptor” and several constitutively
active mutant human A3 adenosine receptors by site-directed
GPCRs as Therapeutic Targets
184
Table 2. Amino Acids of ARs Implicated in Ligand Recognition-interaction
A1
E16A Agonist affinity reduced 4- to 40-fold with little change in
antagonist affinity [65]
D55A Increase in agonist affinity; no change in antagonist affinity. [65]
P86F Substantial reduction of Ag binding [66]
L88A Substantial reduction of Ag binding [66]
T91A Substantial reduction of Ag binding [66]
Q92A Substantial reduction of Ag binding [66]
S94A Loss of agonist or antagonist binding. Binding restored by
substitution with threonine [65]
C169A/S No detectable radioligand binding [66]
T277A Strong decrease in affinity of NECA. Substitution with serine
notably restored NECA affinity. [67]
A2A
E13Q Decrease in agonist affinity, antagonist binding almost not affected. [68,
69]
V84A/D Loss of agonists and antagonists radioligand binding [66]
T88A Decrease in agonist, but not antagonist, affinity [70]
E151A Loss of agonist and antagonist radioligand binding; strong decrease
in agonist potency. [71]
E169A Loss of agonists and antagonists binding [66]
F182A Loss of agonist and antagonist radioligand binding; replacement
with tryptophan considerably restored binding of ligands [72]
H250A
Loss of agonist and antagonist radioligand binding and agonist
activity; substitution with phenylalanine significantly restored
binding
[72]
N253A Loss of agonist and antagonist radioligand binding [72]
F257A Loss of agonist and antagonist radioligand binding [66]
I274A Considerable decrease in agonist and antagonist radioligand
binding, and in agonist potency [72]
S277A Substantial decrease in agonist affinity; substitution with threonine
restored agonist binding [72]
H278A Substantial loss of agonist and antagonist radioligand binding, and
of agonist potency [72]
H278Y Decrease in agonist affinity, antagonist binding almost not affected. [68]
S281A Loss of agonist and antagonist radioligand binding, and of agonist
activity [72]
A2B
A12T Not affected agonists binding [73]
N273Y Slightly increased affinity for agonists [73]
A3
C88F Strongly reduced activity of agonist [74]

Dal Ben et al.
185
mutagenesis [76,77], which provided new insight into the
molecular recognition in the A3 receptor.
In order to provide additional insights into ligand-A3 AR
interactions, site-directed mutagenesis was used to study the
role of a number of residues in the TM domains and in the
second extracellular loop (EL2) [75]. Several residues of this
receptor within TM domains 3 and 6 and the EL2, which have
been predicted by previous molecular modeling to be involved
in the ligand recognition, were mutated. The N250A mutant
receptor lost the ability to bind both radiolabeled agonist and
antagonist [75]. The H95A mutation considerably reduced the
affinity of both agonists and antagonists. In contrast, the
K152A (EL2), W243A and W243F (TM6) mutations did not
significantly change the agonist binding but decreased
antagonist affinity, suggesting that these residues were critical
for the high affinity of A3 AR antagonists [75]. Although not
important for agonist binding, Trp243 was critical for receptor
activation. The results were successfully interpreted using a
rhodopsin based model of ligand-A3 receptor interactions [78].
Some attempts to set up a high-throughput tools to model
GPCRs have been recently reported. One of these,
“GPCRmod”, firstly proposes a reliable alignment of the
seven transmembrane domains based on pattern/motif
recognition f or each of the 7 TMs that are considered
independently. It then converts the alignment into knowledge-
based three-dimensional (3-D) models starting from a set of 3-
D backbone templates and two separate rotamer libraries for
side chain positioning [79]. Another software, “PREDICT”,
tries to model the 3D structure of GPCRs without relying on
homology to rhodopsin. This algorithm starts from the primary
sequence of the receptor, simultaneously optimizing multiple
conformations of the protein in order to find its most stable
structure, culminating in a virtual receptor-ligand complex
[80].
Combined Target-based and Ligand-based Drug Design Strategy In a recent work, Moro proposes a combined target-based
and ligand-based drug design approach to define a
pharmacophore model of the human A3 receptor antagonists
[81]. This strategy is composed by two different steps; in a
first phase a high throughput molecular docking study is
carried out, and information about the putative binding site
and the binding conformations of some A3 inhibitors are
collected. Secondly, docking based structure superimposition
is used to perform a quantitative study of the structure activity
relationships for binding of these inhibitors to adenosine A3
receptor using a comparative molecular field analysis
(CoMFA). The results are combined to assemble a new target
based pharmacophore model. This method seems to give good
Table 2. Continued.
H95A Considerable decrease in both agonist and antagonist affinity [75]
R108A/K Enhanced potency of agonist [74]
Y109F Strongly reduced activity of agonist [74]
D107K/R108K/E
Complete loss of agonist activity [74]
K152A Agonist binding not affected but rather decreased antagonist affinity [75]
A229E Enhanced potency of agonist [74]
W243A/F Agonist binding not affected but rather decreased antagonist affinity [75]
N250A Loss of agonist and antagonist radioligand binding [75]
H272E Decrease in both agonist and antagonist affinity [76]
Y282F Strongly reduced activity of agonist [74]

GPCRs as Therapeutic Targets
186
results, as the predicted Ki values of new A3 inhibitors were in
agreement with the experimental values.
CONCLUDING REMARKS
G-protein-coupled receptors (GPCRs) compose a big
family of sensory proteins that mediate a wide variety of
physiological processes by responding to different external
stimuli such as hormones, paracrines (local hormones),
neurotransmitters, neuromodulators, odours, and light. These
transmembrane receptors transduce the external signals mostly
(but not only) by interacting with regulatory G-proteins. The
superfamily of GPCRs comprises receptors for many
substances with important physiological functions. For this
reason their dysfunction leads to human diseases, and in fact
many GPCRs are targets for pharmaceuticals and drugs of
abuse. The purine nucleoside adenosine is a mediator playing
an important role in modulating many pathophysiological
conditions in the CNS as well as in peripheral organs and
tissues. Four adenosine receptor subtypes have been identified
by molecular cloning; they belong to the family of GPCRs,
and represent a promising drug target. This is due to the fact
that the receptors are expressed in a large variety of cells, and
there is a large number of ligands, which have been generated
by introducing several modifications in the structure of the
lead compound adenosine some of them highly specific.
Currently, adenosine receptors drug discovery is focused
on the development of more selective and/or potent molecules
that act at the orthosteric site (i.e. the binding site of the
endogenous agonist) of the receptor. Knowledge and mapping
of the structural determinants for optimal receptor function are
essential for understanding the molecular basis of ligand
action and receptor function in normal and abnormal
conditions. Moreover, deciphering structure-function
relationships in adenosine receptors will promote computer-
aided drug discovery by elucidating the binding mode (s) of
known ligands in their receptor binding sites.
REFERENCES
[1] K. Kristiansen, Pharmacol. Ther. 103 (2004) 21.
[2] R.J. Lefkowitz, Trends Pharmacol. Sci. 25 (2004) 413.
[3] J. Bockaert, J.P. Pin, Embo J. 18 (1999) 1723.
[4] T.B. Patel, Pharmacol. Rev. 56 (2004) 371.
[5] J. Bockaert, P. Marin, A. Dumuis, L. Fagni. FEBS Lett.
546 (2003) 65.
[6] T. Schoneberg, G. Schultz, T. Gudermann. Mol. Cell.
Endocrinol. 151 (1999) 181.
[7] K.L. Pierce, R.T. Premont, R.J. Lefkowitz, Nat. Rev.
Mol. Cell. Biol. 3 (2002) 639.
[8] O. El Far, H. Betz, Biochem. J. 365 (2002) 329.
[9] A.L. Lomize, I.D. Pogozheva, H.I. Mosberg, J.
Comput. Aided Mol. Des. 13 (1999) 325.
[10] R. Fredriksson, M.C. Lagerstrom, L.G. Lundin, H.B.
Schioth, Mol. Pharmacol. 63 (2003) 1256.
[11] G. Milligan, Mol. Pharmacol. 66 (2004) 1.
[12] I. Gomes, B.A. Jordan, A. Gupta, C. Rios, N. Trapaidze,
L.A. Devi, J. Mol. Med. 79 (2001) 226.
[13] J. Ballesteros, K. Palczewski, Curr. Opin. Drug Discov.
Devel. 4 (2001) 561.
[14] G. Milligan, G.J. Feng, R.J. Ward, N. Sartania, D.
Ramsay, A.J. McLean, J.J. Carrillo, Curr. Pharm.
Des. 10 (2004) 1989.
[15] O.M. Becker, Y. Marantz, S. Shacham, B. Inbal, A.
Heifetz, O. Kalid, S. Bar-Haim, D. Warshaviak, M.
Fichman, S. Noiman, Proc. Natl. Acad. Sci. USA 101
(2004) 11304.
[16] T. Noji, A. Karasawa, H. Kusaka, Eur. J. Pharmacol.
495 (2004) 1.
[17] a) P.G. Baraldi, B. Cacciari, G. Cristalli, S. Vittori, in:
C. Melchiorre (Ed.), I Recettori Purinergici, Klueb
Editrice, Bologna, 2000; b) M. Klinger, M. Freissmuth,
C. Nanoff, Cell Signal. 14 (2002) 99.
[18] J.A. Ribeiro, A.M. Sebastiao, A. de Mendonca, Prog.
Neurobiol. 68 (2002) 377.
[19] P. Meghji, J.B. Tuttle, R. Rubio, J. Neurochem. 53
(1989) 1852.
[20] B.T. Andresen, D.G. Gillespie, Z. Mi, R.K. Dubey,
E.K. Jackson, J. Pharmacol. Exp. Ther. 291 (1999) 76.
[21] K.K. Kalsi, R.T. Smolenski, M.H. Yacoub, Mol. Cell.
Biochem. 180 (1998) 193.
[22] S. Latini, F. Pedata, J. Neurochem. 79 (2001) 463.
[23] U.K. Decking, G. Schlieper, K. Kroll, J. Schrader, Circ.
Res. 81 (1997) 154.
[24] B.B. Fredholm, M.P. Abbracchio, G. Burnstock, J.W.
Daly, T.K. Harden, K.A. Jacobson, P. Leff, M.

Dal Ben et al.
187
Williams, Pharmacol. Rev. 46 (1994) 143.
[25] I. Feoktistov, I. Biaggioni, Pharmacol. Rev. 49 (1997)
381.
[26] G.F. Baxter, D.M. Yellon, J. Mol. Cell. Cardiol. 31
(1999) 981.
[27] T.C. Zhao, R.C. Kukreja, Am. J. Physiol. Heart Circ.
Physiol. 285 (2003) H434.
[28] K.A. Rudolphi, P. Schubert, F.E. Parkinson, B.B.
Fredholm, Cerebrovasc. Brain Metab. Rev. 4 (1992)
346.
[29] P. Schubert, T. Ogata, C. Marchini, S. Ferroni, K.
Rudolphi, Ann. NY Acad. Sci. 825 (1997) 1.
[30] M.E. Olah, G.L. Stiles, Annu. Rev. Pharmacol. Toxicol.
35 (1995) 581.
[31] a) G. Cristalli, R. Volpini (Eds.), Adenosine Receptors:
Chemistry and Pharmacology, Curr. Top. Med. Chem.
3 (2003); b) G. Cristalli, S. Costanzi, C. Lambertucci,
S. Taffi, S. Vittori, R. Volpini, I1 Farmaco 58 (2003)
193.
[32] C.F. Neely, F.V. Dipierro, M. Kong, J.P. Greelish, T.J.
Gardner, Circulation 94 (1996) II376.
[33] R.F. Bruns, J.J. Katims, Z. Annau, S.H. Snyder,
J.W. Daly, Neuropharmacology 22 (1983) 1523.
[34] J. Sawynok, Eur. J. Pharmacol. 347 (1998) 1.
[35] D. Marx, C.I. Ezeamuzie, K. Nieber, I. Szelenyi, Drug
News Perspect. 14 (2001) 89.
[36] P. Forsythe, L.P. McGarvey, L.G. Heaney, J.
MacMahon, M. Ennis. Clin. Sci. (Lond) 96 (1999) 349.
[37] P.J. Oldenburg, S.J. Mustafa, J. Pharmacol. Exp. Ther.
313 (2005) 319.
[38] a) B.B. Fredholm, Drug News Perspect. 16 (2003) 283;
b) G. Cristalli, C. Lambertucci, S. Taffi, S. Vittori, R.
Volpini, Curr. Top. Med. Chem. 3 (2003) 387; c) R.
Volpini, S. Costanzi, S. Vittori, G. Cristalli, K.N. Klotz,
Curr. Top. Med. Chem. 3 (2003) 427; d) R. Volpini, S.
Costanzi, C. Lambertucci, S. Vittori, K.-N. Klotz, G.
Cristalli, J. Med. Chem. 45 (2002) 3271; e) R. Volpini,
S. Costanzi, C. Lambertucci, S. Vittori, G. Cristalli,
Curr. Pharm. Des. 8 (2002) 2285.
[39] C.F. Neely, I.M. Keith, Am. J. Physiol. 268 (1995)
L1036.
[40] A. Conti, A. Monopoli, M. Gamba, P.A. Borea, E.
Ongini, Naunyn Schmiedebergs Arch. Pharmacol. 348
(1993) 108.
[41] L. Yan, J.C. Burbiel, A. Maass, C.E. Muller, Expert
Opin. Emerg. Drugs 8 (2003) 537.
[42] S. Merighi, P. Mirandola, K. Varani, S. Gessi, E.
Leung, P.G. Baraldi, M.A. Tabrizi, P.A. Borea,
Pharmacol. Ther. 100 (2003) 31.
[43] K.N. Klotz, Naunyn Schmiedebergs Arch. Pharmacol.
362 (2000) 382.
[44] C.E. Muller,T. Scior. Pharm. Acta Helv. 68 (1993) 77.
[45] G. Cristalli, S. Costanzi, C. Lambertucci, G. Lupidi, S.
Vittori, R. Volpini, E. Camaioni. Med. Res. Rev. 21
(2001) 105.
[46] R.M. Berne, Cardiovasc. Res. 27 (1993) 2.
[47] M. Quintana, T. Kahan, P. Hjemdahl, Am. J.
Cardiovasc. Drugs 4 (2004) 159.
[48] A.C. Roach, Crit. Care Nurse 11 (1991) 78.
[49] M.M. LaManna, R. Mohama, I.L. Slavich, 3rd, F.J.
Lumia, S.D. Cha, N. Rambaran, V. Maranhao, Clin.
Nucl. Med. 15 (1990) 804.
[50] P.J. Richardson, A.K. Gubitz, T.C. Freeman, A.K.
Dixon, Adv. Neurol. 80 (1999) 111.
[51] W. Bara-Jimenez, A. Sherzai, T. Dimitrova, A. Favit, F.
Bibbiani, M. Gillespie, M.J. Morris, M.M. Mouradian,
T.N. Chase, Neurology 61 (2003) 293.
[52] A. Pinna, R. Volpini, G. Cristalli, M. Morelli. Eur. J.
Pharmacol. 512 (2005) 157.
[53] R. Henderson, P.N. Unwin, Nature 257 (1975) 28.
[54] G.F. Schertler, C. Villa, R. Henderson, Nature 362
(1993) 770.
[55] V.M. Unger, P.A. Hargrave, J.M. Baldwin, G.F.
Schertler, Nature 389 (1997) 203.
[56] K. Palczewski, T. Kumasaka, T. Hori, C.A. Behnke, H.
Motoshima, B.A. Fox, I. Le Trong, D.C. Teller, T.
Okada, R.E. Stenkamp, M. Yamamoto, M. Miyano,
Science 289 (2000) 739.
[57] P.J. van Galen, A.H. van Bergen, C. Gallo-Rodriguez,
N. Melman, M.E. Olah, I.J. AP, G.L. Stiles, K.A.
Jacobson, Mol. Pharmacol. 45 (1994) 1101.
[58] M.F. Hibert, S. Trumpp-Kallmeyer, A. Bruinvels, J.
Hoflack, Mol. Pharmacol. 40 (1991) 8.
[59] S. Moro, D. Guo, E. Camaioni, J.L. Boyer, T.K. Harden,
K.A. Jacobson, J. Med. Chem. 41 (1998) 1456.
[60] S. Moro, A.H. Li, K.A. Jacobson, J. Chem. Inf.

GPCRs as Therapeutic Targets
188
Comput. Sci. 38 (1998) 1239.
[61] J. Saunders, Drug Discov. Today 6 (2001) 288.
[62] E. Archer, B. Maigret, C. Escrieut, L. Pradayrol, D.
Fourmy, Trends Pharmacol. Sci. 24 (2003) 36.
[63] R.E. Stenkamp, D.C. Teller, K. Palczewski, Arch.
Pharm. (Weinheim) 338 (2005) 209.
[64] a) S. Moro, F. Deflorian, G. Spalluto, G. Pastorin, B.
Cacciari, S.K. Kim, K.A. Jacobson, Chem. Commun.
(Camb) (2003) 2949; b) S. Costanzi, C. Lambertucci, S.
Vittori, R. Volpini, G. Cristalli, J. Mol. Graph. Model.
21 (2003) 253.
[65] H. Barbhaiya, R. McClain, A. Ijzerman, S.A. Rivkees,
Mol. Pharmacol. 50 (1996) 1635.
[66] S.K. Kim, Z.G. Gao, P. Van Rompaey, A.S. Gross,
A. Chen, S. Van Calenbergh, K.A. Jacobson, J. Med.
Chem. 46 (2003) 4847.
[67] A. Townsend-Nicholson, P.R. Schofield, J. Biol. Chem.
269 (1994) 2373.
[68] Z.G. Gao, Q. Jiang, K.A. Jacobson, A.P. Ijzerman,
Biochem. Pharmacol. 60 (2000) 661.
[69] I.J. AP, J.K. Von Frijtag Drabbe Kunzel, J. Kim, Q.
Jiang, K.A. Jacobson, Eur. J. Pharmacol. 310 (1996)
269.
[70] Q. Jiang, A.M. Van Rhee, J. Kim, S. Yehle, J. Wess, K.
A. Jacobson, Mol. Pharmacol. 50 (1996) 512.
[71] J. Kim, Q. Jiang, M. Glashofer, S. Yehle, J. Wess, K.A.
Jacobson, Mol. Pharmacol. 49 (1996) 683.
[72] J. Kim, J. Wess, A.M. van Rhee, T. Schoneberg, K.A.
Jacobson, J. Biol. Chem. 270 (1995) 13987.
[73] M.W. Beukers, H. den Dulk, E.W. van Tilburg, J.
Brouwer, A.P. Ijzerman, Mol. Pharmacol. 58 (2000)
1349.
[74] A. Chen, Z.G. Gao, D. Barak, B.T. Liang, K.A.
Jacobson, Biochem. Biophys. Res. Commun. 284
(2001) 596.
[75] Z.G. Gao, A. Chen, D. Barak, S.K. Kim, C.E. Muller,
K.A. Jacobson, J. Biol. Chem. 277 (2002) 19056.
[76] K.A. Jacobson, Z.G. Gao, A. Chen, D. Barak, S.A.
Kim, K. Lee, A. Link, P.V. Rompaey, S. van
Calenbergh, B.T. Liang, J. Med. Chem. 44 (2001)
4125.
[77] K.A. Jacobson, M. Ohno, H.T. Duong, S.K. Kim,
S. Tchilibon, M. Cesnek, A. Holy, Z.G. Gao, Chem.
Biol. 12 (2005) 237.
[78] Z.G. Gao, S.K. Kim, T. Biadatti, W. Chen, K. Lee,
D. Barak, S.G. Kim, C.R. Johnson, K.A. Jacobson, J.
Med. Chem. 45 (2002) 4471.
[79] C. Bissantz, A. Logean, D. Rognan, J. Chem.
Inf. Comput. Sci. 44 (2004) 1162.
[80] S. Shacham, Y. Marantz, S. Bar-Haim, O. Kalid,
D. Warshaviak, N. Avisar, B. Inbal, A. Heifetz, M.
Fichman, M. Topf, Z. Naor, S. Noiman, O.M. Becker,
Proteins 57 (2004) 51.
[81] S. Moro, P. Braiuca, F. Deflorian, C. Ferrari, G.
Pastorin, B. Cacciari, P.G. Baraldi, K. Varani, P.A.
Borea, G. Spalluto, J. Med. Chem. 48 (2005) 152.





























