Embed Size (px)
Citation preview

Glomerulonephritis
FRACP LECTURE SERIES 2014
Amelia Le Page
Nephrologist Monash Children’s

Glomerulonephritis
• GLOMERULAR INJURY WITH INFLAMMATION
• Clinically: Haematuria, Proteinuria +/-Hypertension, Oedema and Renal Impairment

Overall Pathogenesis
• Trigger = Humoral Immune response (most)
Immune complexes
Pre-formed circulating complexes Native Intraglomerular Ags Deposited Antigens
In situ Immune complex formation
• Secondary Complement Activation
• Secondary Coagulation cascasde
• Secondary Cell mediated immune response
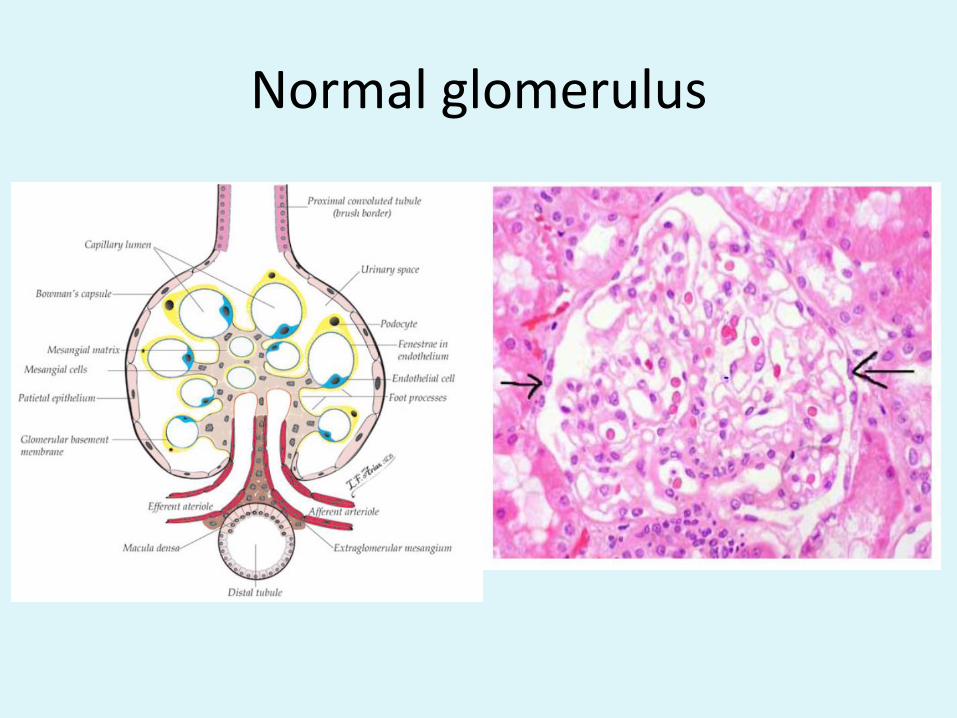
Normal glomerulus

Clinical Presentation: • Acute (Subacute) Nephritis
Macroscopic haematuria, Oedema, HypertensionProteinuriaRenal Impairment
Common causes• Post-Streptococcal/Infectious Nephritis• IgA/HSP• MPGN• Lupus Nephritis• (Endocarditis associated nephritis)
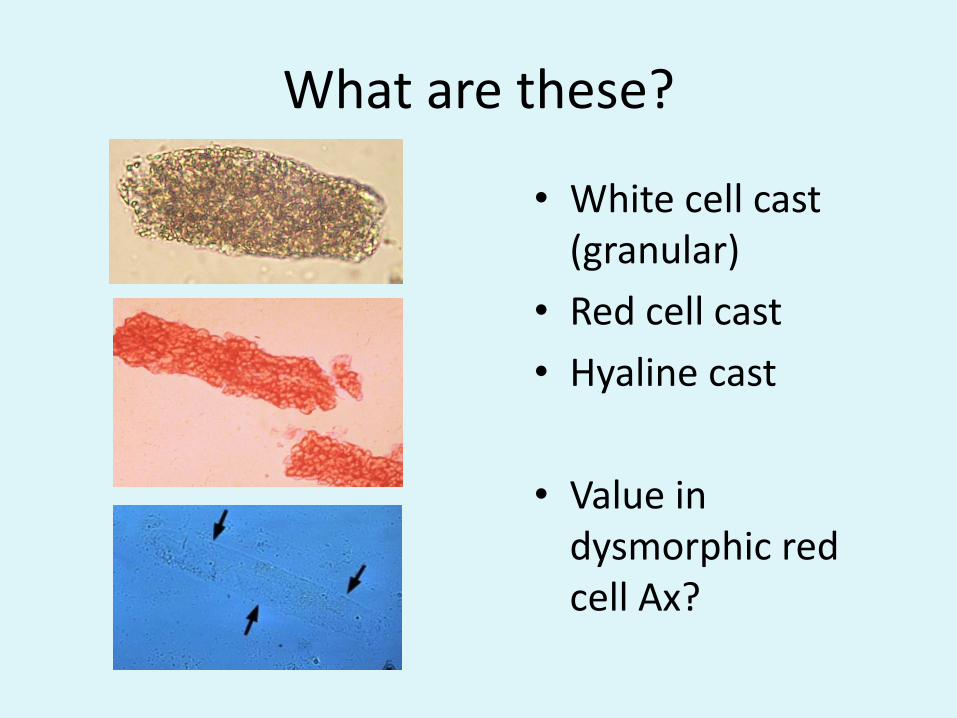
What are these?
• White cell cast (granular)
• Red cell cast
• Hyaline cast
• Value in dysmorphic red cell Ax?

Clinical presentation:
• RPGN
Acute nephritis with rapid progression to ARF
• Common causes
• ANCA associated vasculitis
• Goodpasture’s
• All of the causes of acute

Clinical Presentation:
• Chronic Nephritis
Subacute/Asymptomatic presentation
• Main causes:
• Primary GN: IgA, MPGN,Membranous
• Secondary GN: ANCA, Lupus, Membranous

Clinical Presentation
• Nephrotic
• Ie. Non-Minimal Change, Non-FSGS
• Membranous: Primary, Secondary
• Lupus

The Renal biopsy (LM/IP or IF/EM)
3 Broad GN patterns on LM:
• Proliferative vs Non-Proliferative• Diffuse vs focal proliferative• Segmental vs global proliferative • Crescenteric : cellular vs fibrous type
Isolate abnormal glomerulus :
1. Mesangial Cellularity2. Capillaries3. Basement membranes4. Check other gloms: sclerosed, crescents5. Check tubulointerstitium

Light Microscopy Histology do not define disease but help

Specific biopsy patterns to recognise
• Crescenteric:
more likely with 1) ANCA associated GN 2)antiGBM
but can occur with most cases of GN
• Tram tracking: MPGN LM
• Wire-loops: Lupus Class III/IV LM LM
• Spikes on special stains: Membranous
• Starry Sky: IF of APSGN
• Subepithelial humps: EM of APSGN

The IF/IP
What and whereC3, IgA, IgM, IgG, C1q
Typical patterns:• Granular= immune complex• Nil or very low grade = pauci-immune (ANCA associated)• Linear IgG at GBM= antiGBM• Full house pattern = all present. Main ddx= Lupus
• C3/IgG granular deposition = PSGN (subepithelial), SLE, MPGN types I and III(GBM/mesangial), Infective Endocarditis
• C3 only at GBM, bowmans capsule, tubular= MPGN II (DDD) • IgA +/- C3/IgG mesangial granular= IgA/HSP

The EM
• Information on SITE of immune complexes
• Helpful esp with ddx MPGN versus APSGN.
• Subepithelial humps=APSGN• Subendothelial/mesangial deposits= MPGN I• GBM deposits = MPGN II (DDD)• Subepithelial +subendothelial/mesangial
deposits= MPGN III

Acute GN Ix
• C3,C4
• ANA
• Post Streptococcal Abs
• And then consider others depending on results to date/clinical progression

RPGN Ix
• C3,C4• ANA• Post Streptococcal Abs• ANCA• Anti-GBM Abs
• Biopsy: Hallmark= crescents (fibrous = more advanced versus cellular) IF (Anti-GBM, Full house nephropathy, C3 deposition patterns of APSGN, pauci-immune)

Acute PostStrep (Infectious) GN
• Other infectious causes (EBV,CMV,HepB, Endocarditis)
• GAS: specific M protein serotypes
• Age 2-12
• Pathophysiologic trigger theories: immune complex trapping, molecular mimicry, in situ immune complex formation: strep antigen deposition and complement activation.

APSGN
• Clinically: acute GN, RPGN, incidental microhaematuria/proteinuria
• C3 low, C4 normal
• Antibody titres
• Follow-up: hypertension & macro haematuria resolve within a week, proteinuria/microhaematuria within 6 months. Complement normalises by 6-8 weeks.
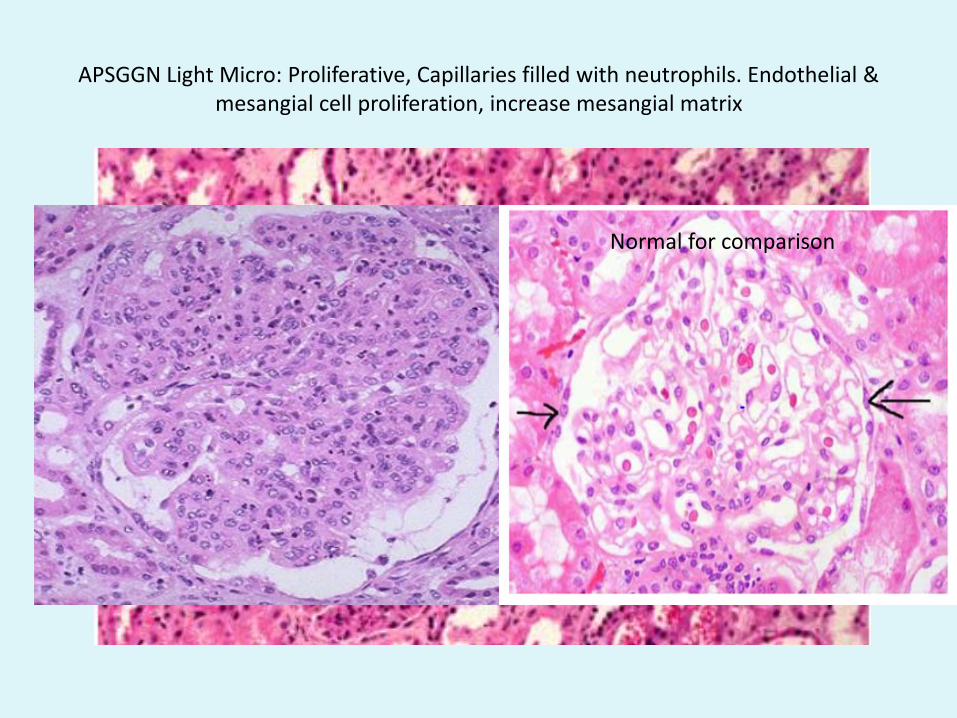
APSGGN Light Micro: Proliferative, Capillaries filled with neutrophils. Endothelial & mesangial cell proliferation, increase mesangial matrix
Normal for comparison
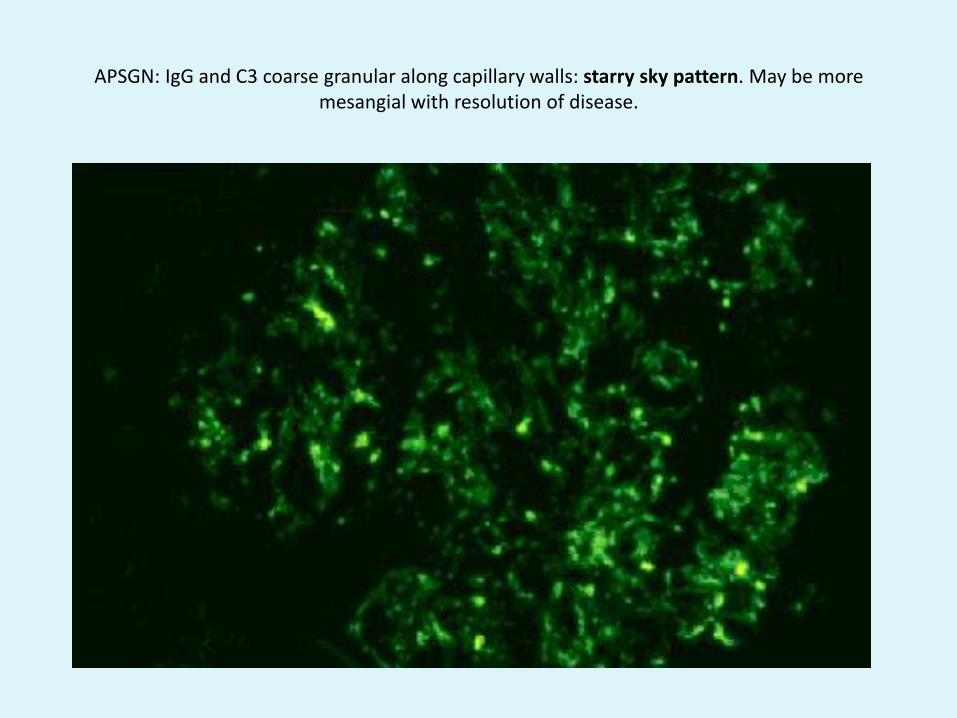
APSGN: IgG and C3 coarse granular along capillary walls: starry sky pattern. May be more mesangial with resolution of disease.
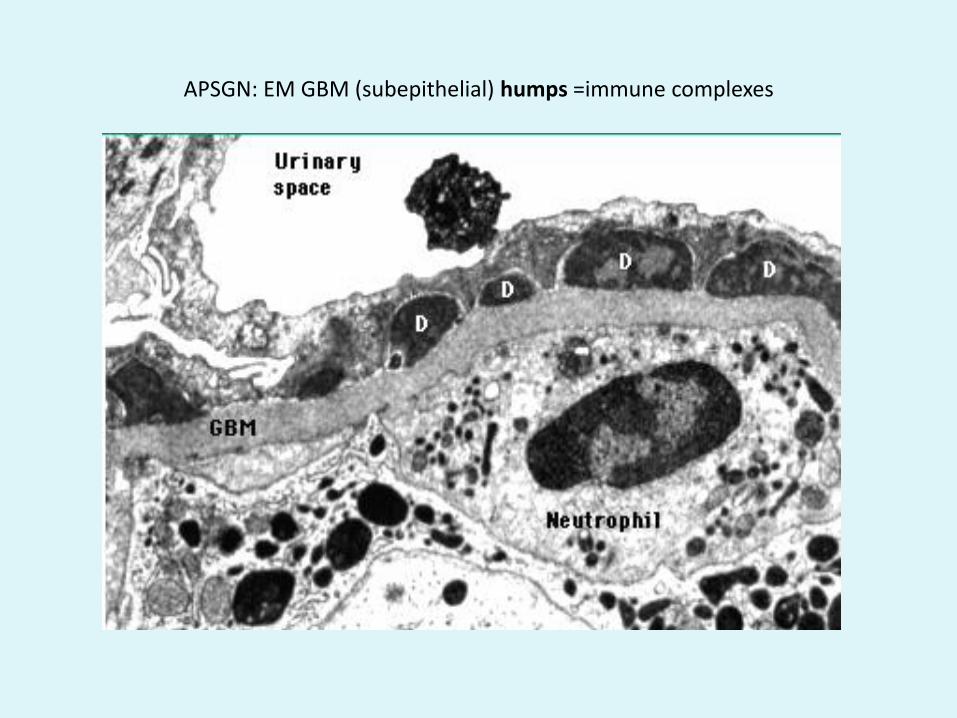
APSGN: EM GBM (subepithelial) humps =immune complexes

APSGN Management
• With Acute nephritis:
Salt and fluid restriction
Diuretics for HeT
• Treat only active infections
• RPGN & where dialysis requiring alternative therapies (e.g. methylpred) maybe considered although little evidence.

IgA Nephropathy
• Pathogenesis:
• Mesangial deposition of IgA complexes.
• Why?
• Dysregulated synthesis or metabolism of mucosal IgA
• Poor galactosylation of IgA1 (genetic predisposition)
• Polygenic/Environmental
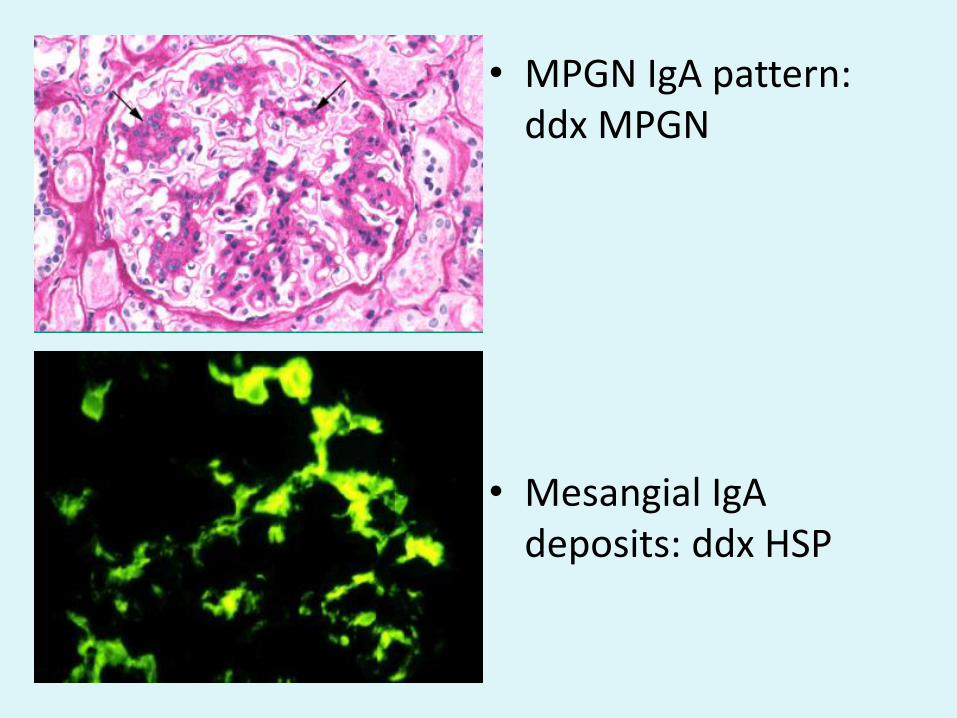
• MPGN IgA pattern: ddx MPGN
• Mesangial IgA deposits: ddx HSP

IgA Nephropathy
CFs: 1.Synpharyngitc macro haematuria 2.Acute nephritis3.RPGN4. Chronic nephritis5.Nephrotic
Those presenting with CFs 2-5----diagnosis on biopsy
Rx:Immunosuppressive:Active biopsy/disease: clinically -persistent high grade proteinuria or increased CrGlucocorticoids+/- steroid sparing agents (azathioprine/CPA)
Non-Immunosuppressive:ACEI/ARBBP optimisation

HSP nephritis
• Prevention: 2009 Cochrane metanalysis: no evidence to support Prednisolone use to reduce nephritis.
• 20-54% evidence of renal involvement (varying from haematuria to nephrotic syndrome to nephritis)—more severe in older
• Prognosis worse with initial nephrotic state, more signficant acute nephritis – refer
• 97% with nephritis present within 6 mo—therefore monitor for 6 mo – persistent proteinuria - refer
• Biopsy: as per IgA• Rx: as per IgA

Membranoproliferative GN (AKA mesangiocapillary)
• Disease defined on biopsy
• Clinically presents as Acute GN, Chronic GN, Nephrotic syndrome with microhaematuria, RPGN
• Low C3 +/- Low C4.

MPGN Pathogenic Classification (another is based on EM)
Immune complex mediated : • Hep C, Hep B, Autoimmune (including lupus). • Subendothelial and mesangial depositis on EM. IF: full
house pattern in SLE, IgM and C3 in HepC.
Complement mediated:
• Dysregulated persistent alternative complement pathway activation: C3 nephropathy, dense deposit disease.
• C3 staining of capillary walls and mesangium. EM deposits: subendothelial, mesangial +/- subepithelial, GBM
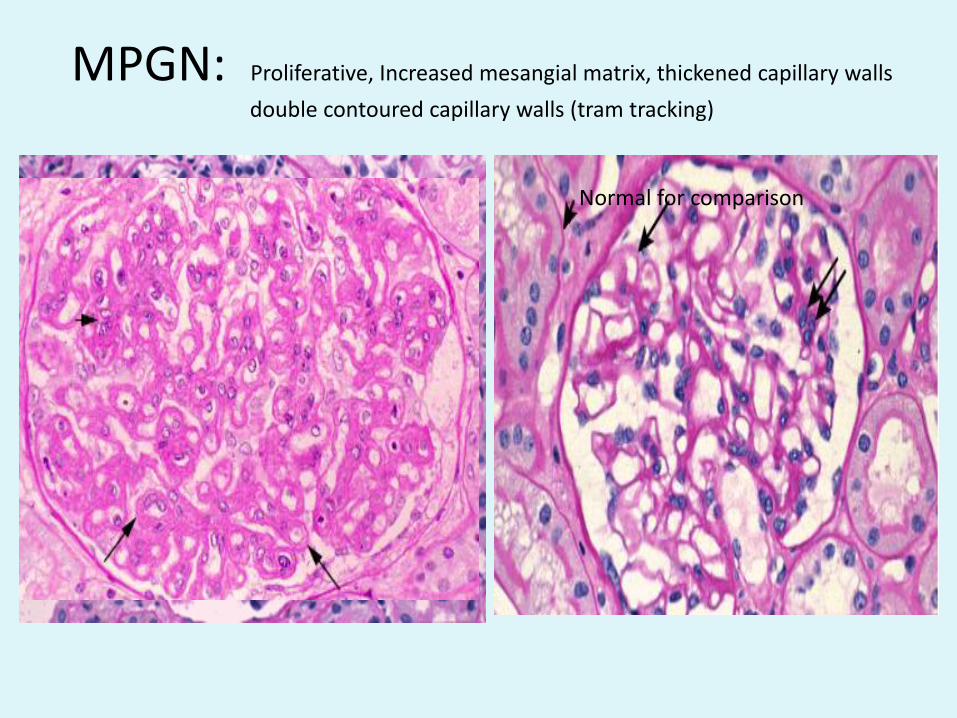
MPGN: Proliferative, Increased mesangial matrix, thickened capillary walls
double contoured capillary walls (tram tracking)
Normal for comparison

MPGN cntd• Consider as a ddx to post-strep GN esp. where hypocomplementemia persists.
• Clinical Features: acute, subacute, chronic, RPGNLook for other non-renal clinical features in type II MPGN (DDD): drusen, lipodystrophy
• Test CH50, AH50, factors I,H, C3 nephritic factor, Factor H mutation screening etc.
• Complement mediated MPGN (dense deposit disease) poor prognosis with high recurrence rate post transplant.
• Therapy: tailored to cause, clinical severity.
– Idiopathic immune complex: steroids +/- seroid sparing, CPA if more severe.
– C3 nephropathy/dense deposit disease: plasma exchange, rituximab (eg. Antibody
associated), complement inhibitors

SLE nephritis• 60-75% with SLE will develop nephritis
• CFs: Acute GN, Chronic GN, RPGN, nephrotic, asymptomatic.
• Pathophysiology: Pattern of glomerular injury depends on site of deposition of immune complex. Site dependent on type of antibody/charge.
• Mesangial/Subendothelial deposits- acute proliferative (nephritis) disease
• Subepithelial deposits- less acute inflammatory disease-membranous type with nephrotic features.
• Antibody deposition and secondary local response

SLE nephritis• Regular urine screening for evidence of
nephritis/disease activity.
• Any evidence of proteinuria should lead to biopsy.
• Lupus Biopsy classification (ISN based)I normal LM, mesangial deposits on IF/EM only
II MPGN LM
III Focal proliferative -
IV Diffuse – Acute Nephritis
V Membranous - Nephrotic
VI Advanced sclerosing – ESKD
• Chronicity : TIS changes
Good prognosis, zero or minimal urine sediment
CFs may overlap, disease may evolve between classes
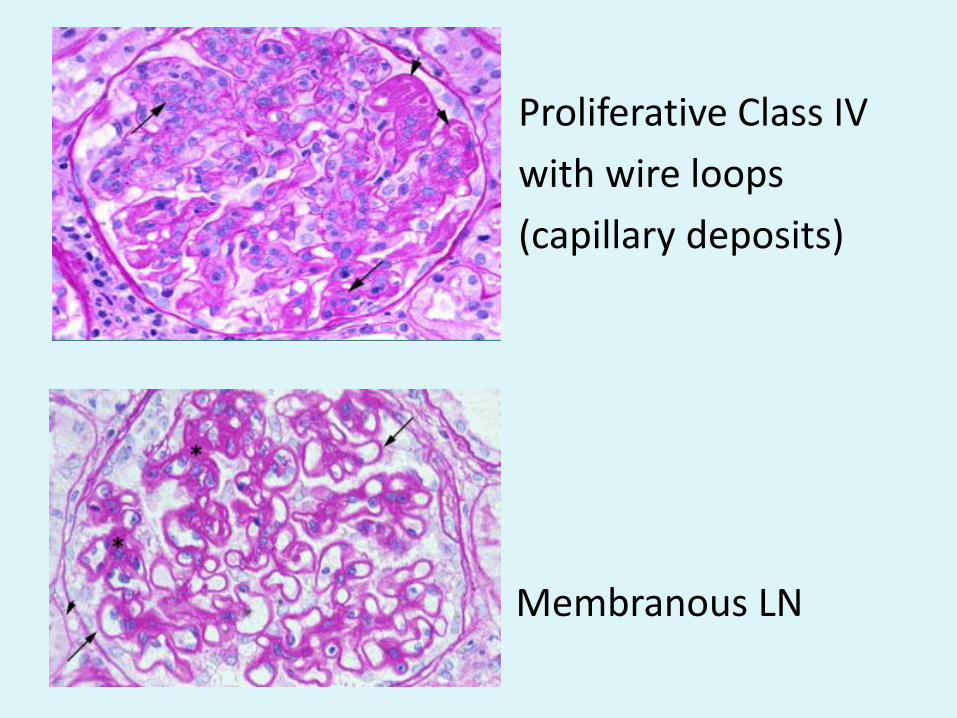
Proliferative Class IV
with wire loops
(capillary deposits)
• Membranous LN

Lupus Nephritis Rx
• Indicated for class III +• Immunosuppressive:• Induction:
Class III/IV: Glucocorticoids + CPA or MMF (max 3g/day)If crescents, severe nephritis-methlypred pulse x3Class V: Glucocorticoids + CPA or cyclosporin (insufficient evidence to date to use MMF)
• Maintenance:• Class III/IV: MMF or azathioprine and low dose prednisolone for 2-3 years
Class V: as above for 1 year.
• Relapsing/resistant disease: consider the other agent (MMF or CPA) or rituximab
• Non Immunosuppressive: ACEI, Statin

ANCA associated nephritis and other renal vasculitis
• Includes Granulomatous Polyangiitis (GPA), microscopic polyangiitis (MPA), Churg Strauss, renal limited vasculitis.
• Classification according to type of ANCA (PR3, MPO, or seronegative) also used and may have more prognostic significance.
• PR3 more associated with GPA• MPO more associated with MPA
• Renal CFs: may develop in up to 85%RPGNProteinuriaHaematuriaAcute Nephritis
• Non renalENT: GPA>MPA (relapsing polychondritis-saddle nose deformity)Pulmonary: GPA + Churg Strauss > MPASkin: vasculitis: purpura/uritcarial/erythema nodosum/livedo reticularisOther sites of vasculitis
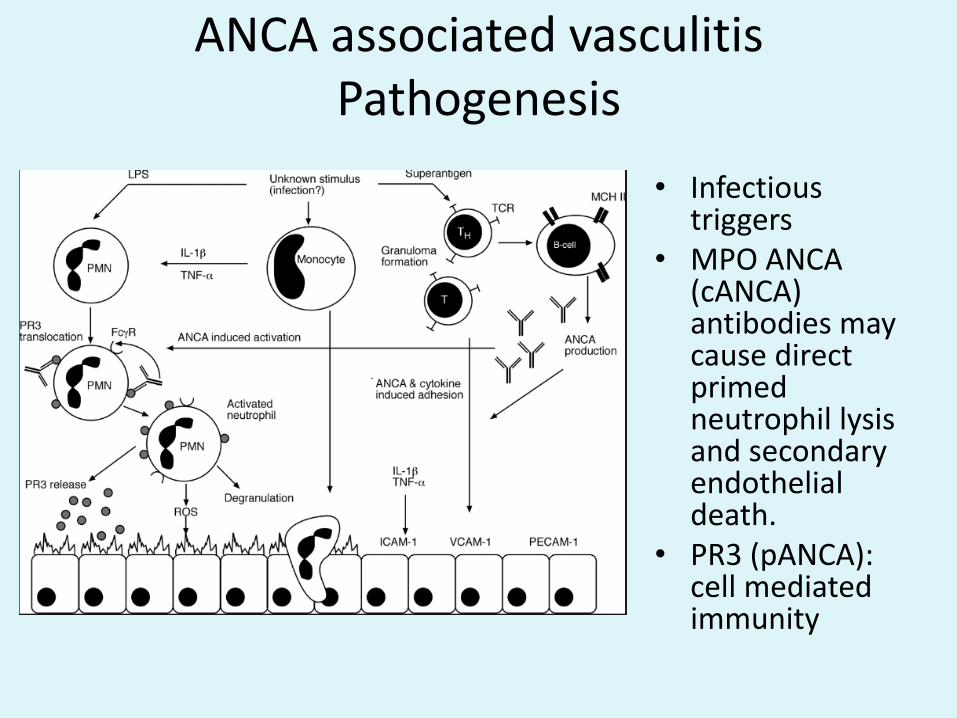
ANCA associated vasculitisPathogenesis
• Infectious triggers
• MPO ANCA (cANCA) antibodies may cause direct primed neutrophil lysisand secondary endothelial death.
• PR3 (pANCA): cell mediated immunity
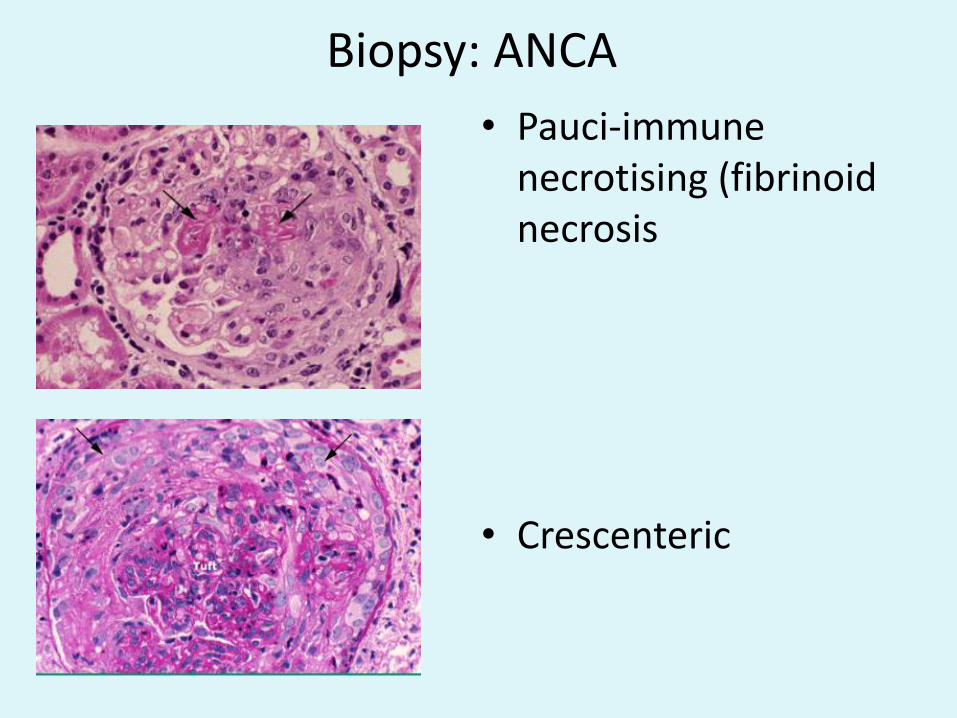
Biopsy: ANCA
• Pauci-immune necrotising (fibrinoidnecrosis
• Crescenteric

Rx ANCA associated nephritis
• Induction: according to severity
Dialysis requiring: plasma exchange/CPA/GC
CPA & glucocorticoids: remission in >80%
Ritux & glucocorticoids: Equivalent
• Maintenance:
Azathioprine, glucocorticoids
Duration according to risk of relapse
(GPA>MPA)

Goodpastures
• AKA anti-GBM antibody-mediated disease
• Abs bind to alpha-3(IV) collagen chain of GBM and alveolar membrane
• ? Initiated by autoreactive T cells
• Genetic (HLA associations) & environmental triggers (smoking)
• Clinical Features: RPGN (rarely subacute nephritis)
30-40% pulmonary features
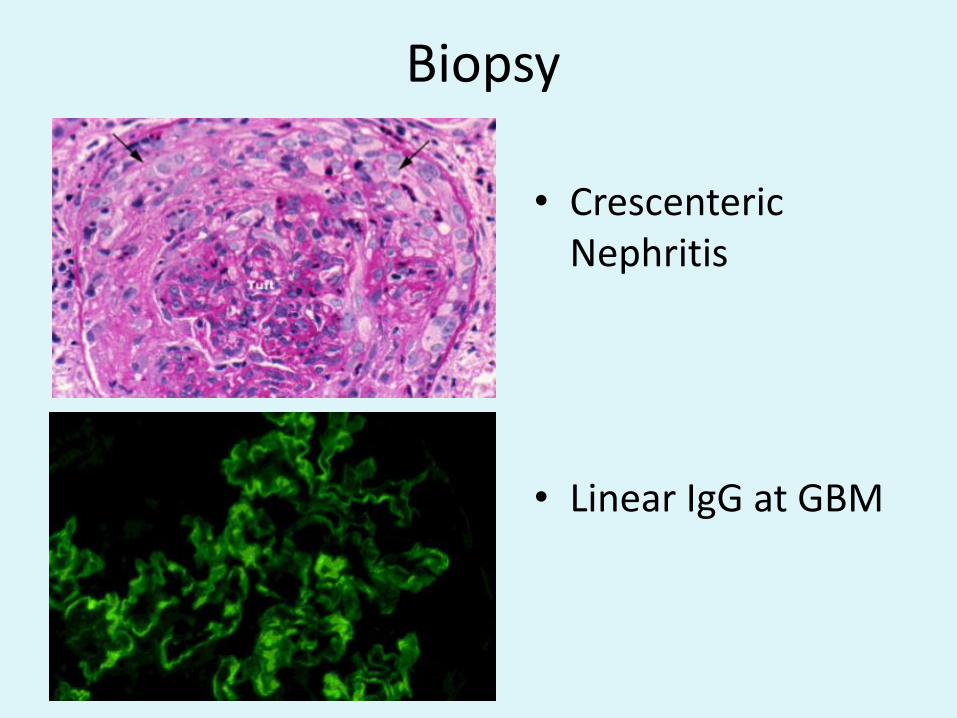
Biopsy
• CrescentericNephritis
• Linear IgG at GBM

GoodPastures Rx
• Prednisolone
• Cyclophosphamide
• Plasma Exchange
• High risk of ESKD

Membranous Nephropathy
• Idiopathic v secondary (hepB, tumors,SLE)
• Diffuse thickening of GBM without hypercellularity
• Special stains: spikes= expansion of GBM beside deposits
• IF: c3,IgG on GBM
• EM: subepithelial deposits outside GBM, foot process effacement
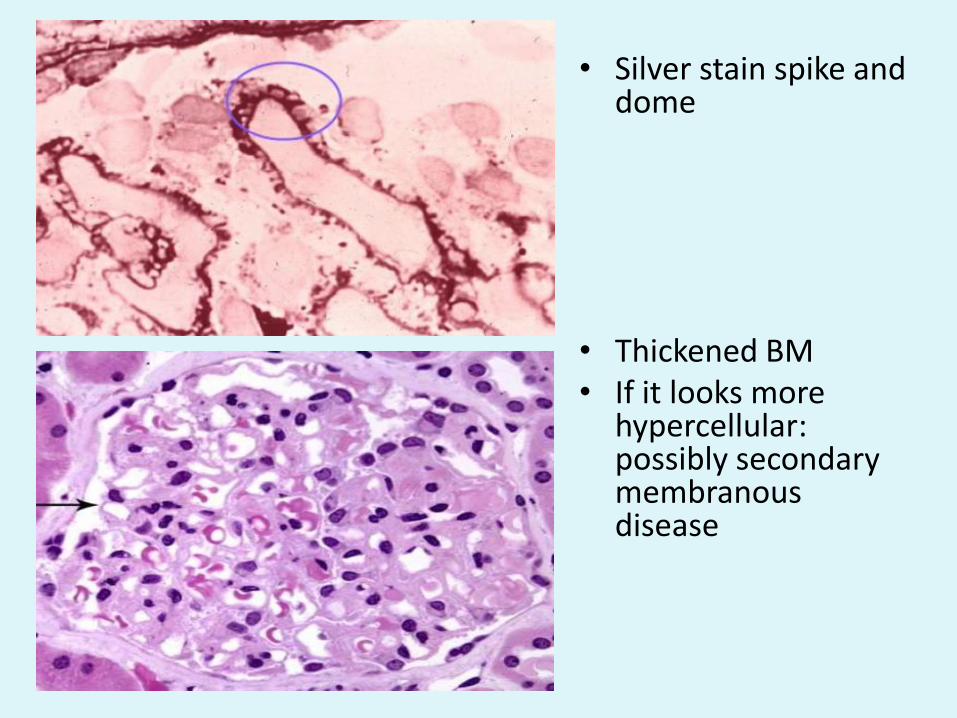
• Silver stain spike and dome
• Thickened BM• If it looks more
hypercellular: possibly secondary membranous disease
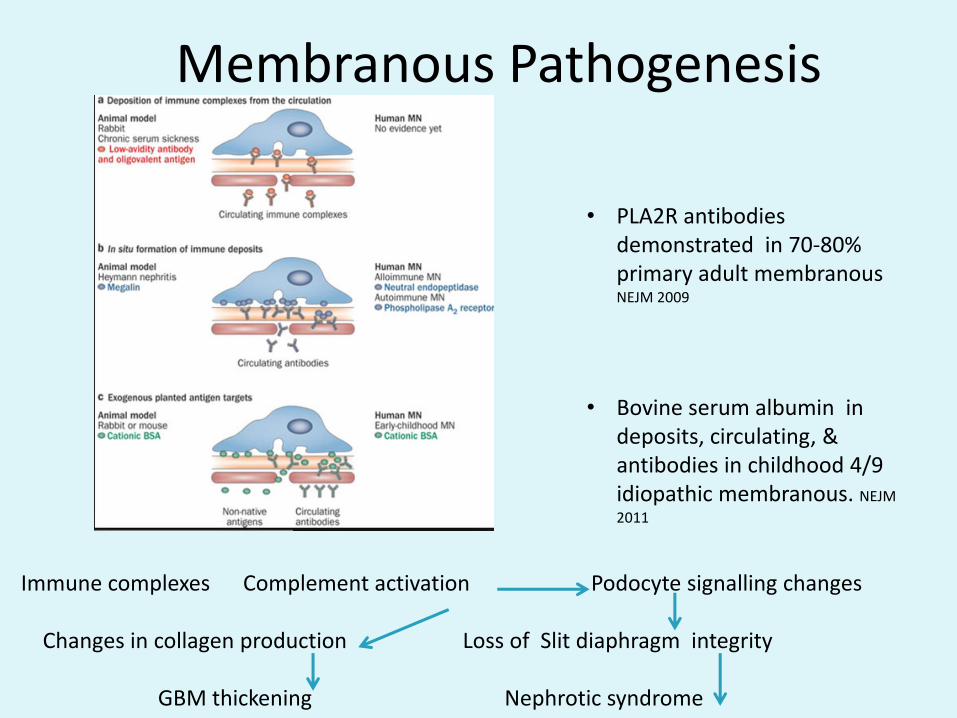
Membranous Pathogenesis
• PLA2R antibodies demonstrated in 70-80% primary adult membranous NEJM 2009
• Bovine serum albumin in deposits, circulating, & antibodies in childhood 4/9 idiopathic membranous. NEJM
2011
Immune complexes Complement activation Podocyte signalling changes
Changes in collagen production Loss of Slit diaphragm integrity
GBM thickening Nephrotic syndrome

Membranous Rx
• Primary:Determine severity based degree of proteinuria/biopsy signs of chronicity.
If nephrotic-Corticosteroids-With Other Immunosuppression (CSA/CPA)-Remember other risks: clottting
Non nephrotic-ACEI therapy
• Overall prognosis in childhood unclear: ? 25% progression to CKD. Increased progression where have resistant disease.
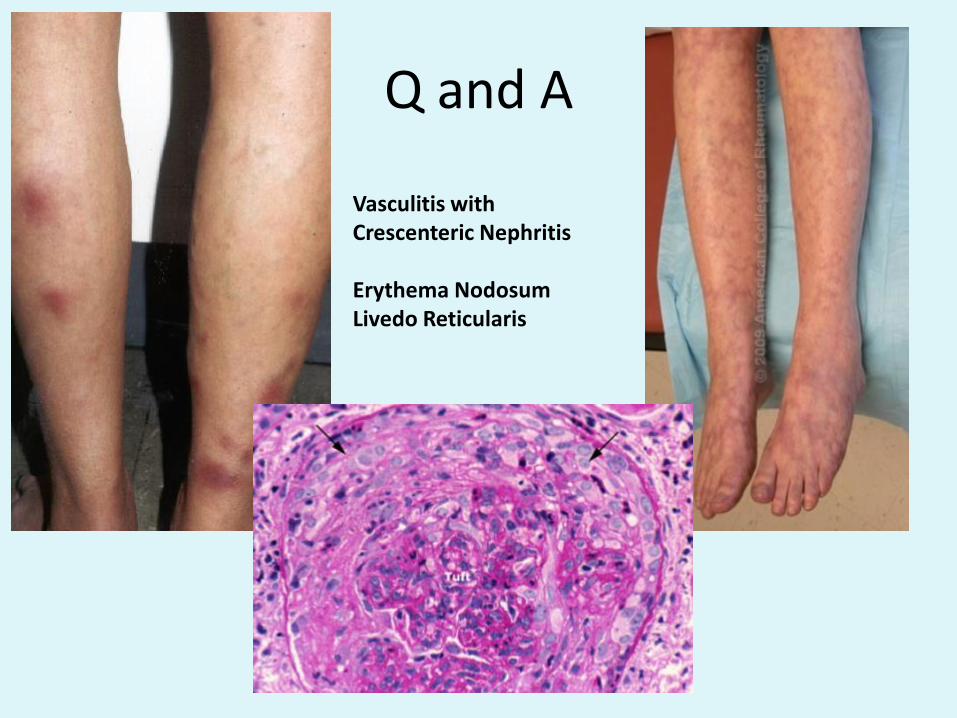
Q and A
Vasculitis with Crescenteric Nephritis
Erythema NodosumLivedo Reticularis
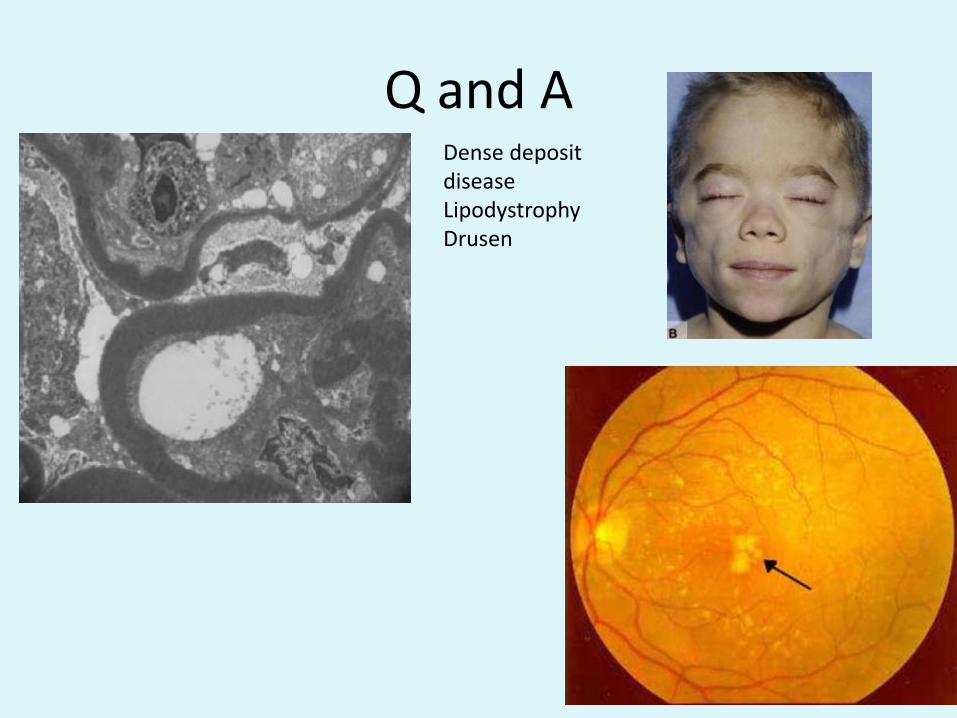
Q and ADense deposit diseaseLipodystrophyDrusen


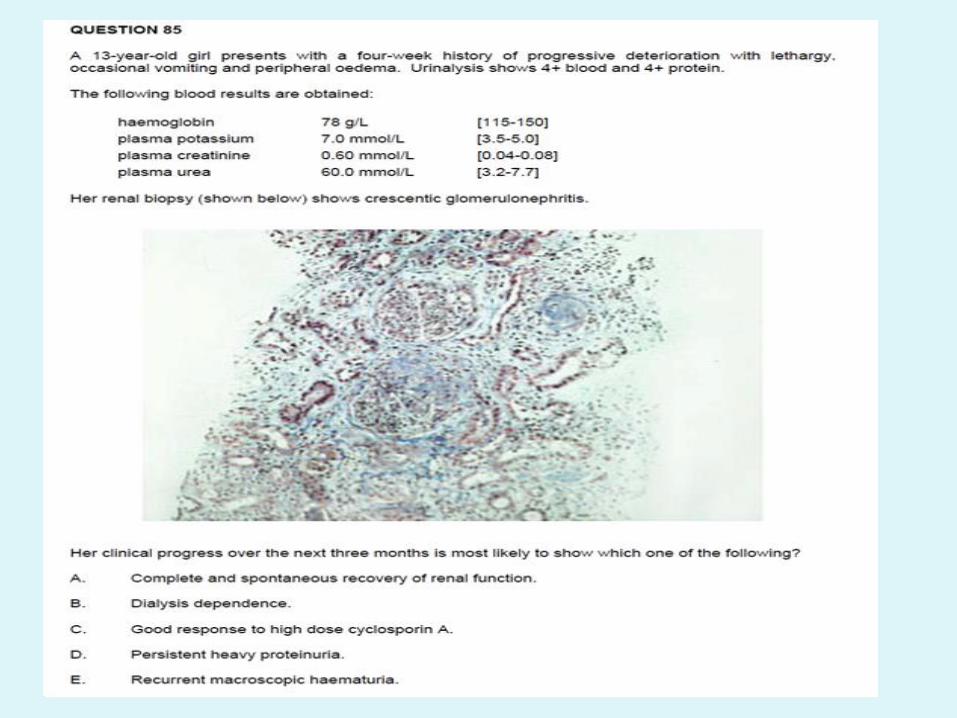

• QUESTION 96
A ten-year-old boy presents with facial swelling, hypertension, proteinuria and haematuria.
Complement levels are 0.7 mg/dL [0.8-1.8]; antistreptolysin-O titre (ASOT) and anti-DNase are positive. He is excreting 500 mg of urinary protein in 24 hours [<200]. Anti-nuclear antibodies are negative, and he has no other symptoms or signs suggesting autoimmune phenomena. After initial treatment with fluid restriction, diet and antihypertensives, his urinary sediment improves, and blood pressure returns to normal. At one month he is reassessed.
• Which of the following would most strongly indicate the need for renal biopsy?
• A. Creatinine of 0.11 mmol/L [0.05 – 0.10].
• B. Haematuria.
• C. Hypertension.
• D. Hypocomplementaemia.
• E. Proteinuria of > 200 mg/24 hours.





























