Embed Size (px)
DESCRIPTION
Glimepiride Nano
Citation preview

ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
15
ABSTRACT
S.Vidyadhara*, J.Ramesh Babu, RLC.Sasidhar, A.Ramu, S.Siva Prasad and M.TejasreeChebrolu Hanumaiah Institute of Pharmaceutical Sciences,Chowdavaram,Guntur-19, A.P, India.
FORMULATION AND EVALUATION OF GLIMEPIRIDE SOLID DISPERSIONS ANDTHEIR TABLET FORMULATIONS FOR ENHANCED BIOAVAILABILITY
Solid dispersions of Glimepiride with sodiumstarch glycolate(SSG) were prepared and further compressed astablets by using diluents such as lactose, dicalcium phosphate and microcrystalline cellulose. The solid dispersionsof Glimepiride with SSG at different ratios were prepared by physical mixing, solvent evaporation and kneadingmethods. The rapid release of poorly soluble Glimepiride from solid dispersions was influenced by the proportionof polymer and the method employed for its preparation. Among the three methods employed solvent evaporationand kneading methods were found to be suitable for improving the dissolution rate of Glimepiride. The releasewas found to follow the first order kinetics. Some of the dispersions prepared by the solvent evaporation methodand kneading method were formulated into tablets with diluents such as lactose, DCP and MCC. All the tabletpreparations containing diluents were found to release the drug in the order of DCP> MCC > Lactose. The dissolutionrate of such tablet formulations were found to release the drug at a faster rate than that of tablets prepared withplain drug.
Key Words: Glimepiride, Sodium starch glycolate, Solid Dispersions, Solid dispersionTablets.
INTRODUCTIONOral drug delivery is the simplest and easiest way ofadministering drugs [1, 2]. Because of the greater stability,smaller bulk, accurate dosage and easy production solidoral dosage forms have many advantages over othertypes of dosage forms. Therefore, most of the newchemical entities (NCE) under development these daysare intended to be used as a solid dosage form thatoriginate an effective and reproducible in vivo plasmaconcentration after oral administration[3,4].In fact, mostNCEs are poorly water soluble drugs, not well-absorbedafter oral administration which can detract from the drugsinherent efficacy[5,6].Moreover, most promising NCEs,despite their high permeability, are generally only absorbedin the upper small intestine, absorption being reducedsignificantly after the ileum, showing that there is a smallabsorption window[7,8]. Consequently, if these drugs arenot completely released in the gastro intestinal area, theymay have a low bioavailability. Therefore, one of the majorcurrent challenges in the pharmaceutical industry is relatedto strategies that improve the water solubility of drugs[9].Drug release is a crucial and limiting step for oral drugbioavailability, particularly for drugs with lowgastrointestinal solubility and high permeability. Byimproving the drug release profile of these drugs, it ispossible to enhance their bioavailability and reduce theirside effects. Solid dispersions are one of the mostsuccessful strategies to improve the drug release of poorlysoluble drugs[10]. Presenting the compound as themolecular dispersion combining the benefits of a localincrease in the solubility (within the solid solution) andmaximizing the surface area of the compound that comesin contact with the dissolution medium as the carrierdissolves. When a mixture with composition E, consistingof a slightly soluble drug and an inert, highly water solublecarrier when dissolved in aqueous medium, the carrier
will dissolve rapidly, releasing very fine crystals of thedrug[11]. The large surface area of the resulting suspensionshould result in an enhanced dissolution rate and therebyimproved bioavailability[12]. The advantage of soliddispersions over other approaches is that many of thecarriers that can be applied are already extensively usedin the pharmaceutical industry as excipients, so additionaltoxicity studies above and beyond what is required forthe drug itself should not be required. The possibility ofcombining several carriers to produce an optimizedproduct further extends the range of possibilities forformulation[13]. Glimepiride is one of the third generationsulphonylurea,antidiabetic drug which stimulates insulinrelease. It is used for treatment of non-insulin-dependentdiabetesmellitus[14,16]. Glimepiride is classified under classII according to biopharmaceutical classification system[15].The drugshows low, pH dependent solubility. In acidic andneutral aqueous media, glimepiride exhibits very poorsolubility at 370C (<0.004 mg/ml). In media pH>7, solubilityof drug is slightly increased to 0.02 mg/ml. This poorsolubility may cause poor dissolution and unpredictedbioavailability[15,16]. Glimepiride is rapidly absorbed by theliver after oral administration. It undergoes extensive firstpass metabolism in the liver. It is practically insoluble inwater and other aqueous media. The very poor aqueoussolubility and wettability of Glimepiride give rise todifficulties in the design of pharmaceutical formulationsand led to variable oral bioavailability. A few reports areavailable on the enhancement of solubility, dissolution rateof Glimepiride . Rate of absorption and/or extent ofbioavailability for such insoluble drug is controlled byrate of dissolution in gastrointestinal fluids. The peakplasma concentration (Cmax) and the (tmax) depend uponextent and rate of dissolution of drug respectively.Hence the present investigation was aimed to increasethe rate of dissolution of Glimepiride.

ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
16
MATERIALS AND METHODSGlimepiride was a gift sample from Life line formulations,Vijayawada , Sodiumstarch glycolate(SSG),Microcrystalline cellulose (MCC), Dicalciumphosphate(DCP), Lactose were gift samples obtainedfrom Pellets Pharma Ltd.,Hyderabad. Methanol, Sodiumhydroxide, Hydrochloric acid, Potassium hydrogenphosphate (S.D.Fine chemicals, Mumbai) was procuredfrom commercial sources. All other materials used wereof pharmacopoeial grade.
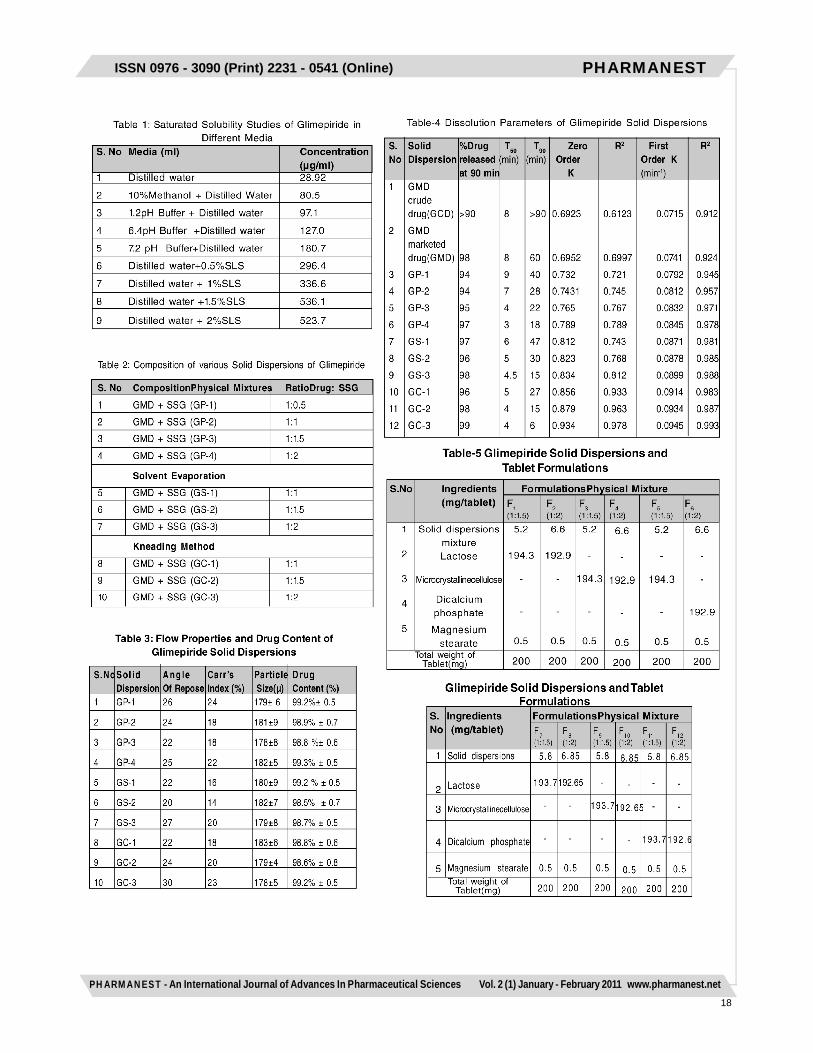
Saturated solubility studies1g of Glimepiride was weighed and transferred in conicalflasks containing 100ml of different dissolution media.These flasks were hermatically sealed and incubated at370C in a incubator shaker, rotated at 50rpm for 24hrs.Then the samples were filtered and subsequently dilutedwith same media. The corresponding absorbance valueswere noted at 228nm.
Preparation of solid dispersionsGlimepiride solid dispersions with SSG were preparedby employing three methods such as:
1. Physical Mixing2. Solvent Evaporation
3. Co-grinding (Kneading Method)
Physical Mixing MethodKnown quantity of drug (20mg) Glimepiride and SSG wereweighed separately and passed through sieve no: 80. Thematerials passed through sieve no: 80 were collected andtransferred into a clean and dry glass mortar, and weretriturated together for 5min. Then the blended mixture waspassed through sieve no: 80 and it is collected and packedin a wide mouthed amber colored glass containers andwere hermatically sealed
Solvent Evaporation MethodGlimepiride was taken in a china dish and was dissolvedin few ml of methanol. To the methanolic solution, specifiedamount of SSG was added and the mixture was heatedto 500C on a mantle with continuous stirring until thesolvent is evaporated. Then the mixture was collected andpacked in a amber colored glass container and washermatically sealed. Then the mixture was stored atambient conditions .
Kneading MethodGlimepiride and SSG were taken in a glass mortar andfew ml of water was added and triturated vigorously untilthe damp granular mass was obtained. The mixture wasthen dried in a hot air oven to form dry granules. Then the
mixture was taken and passed through sieve no: 80 andthe granules were collected which were packed insidewide mouthed amber colored glass container andhermatically sealed for storage .Various compositions ofsolid dispersions are shown in table-2.
Characterization and Evaluation of SolidDispersionsThe solid dispersions prepared by various methods werecharacterized by particle size determination, and flowproperties such as angle of repose and Carr’s index.
Estimation of Glimepiride in Solid Dispersions20mg of solid dispersion was dissolved in methanol byvigorous shaking in the solvent. The solutions were filteredand filtrate was diluted suitably with 6.4pH Phosphatebuffer containing 0.5% Sodium lauryl sulphate (SLS). Drugcontent of samples were determined by measuringabsorbance at 228nm.
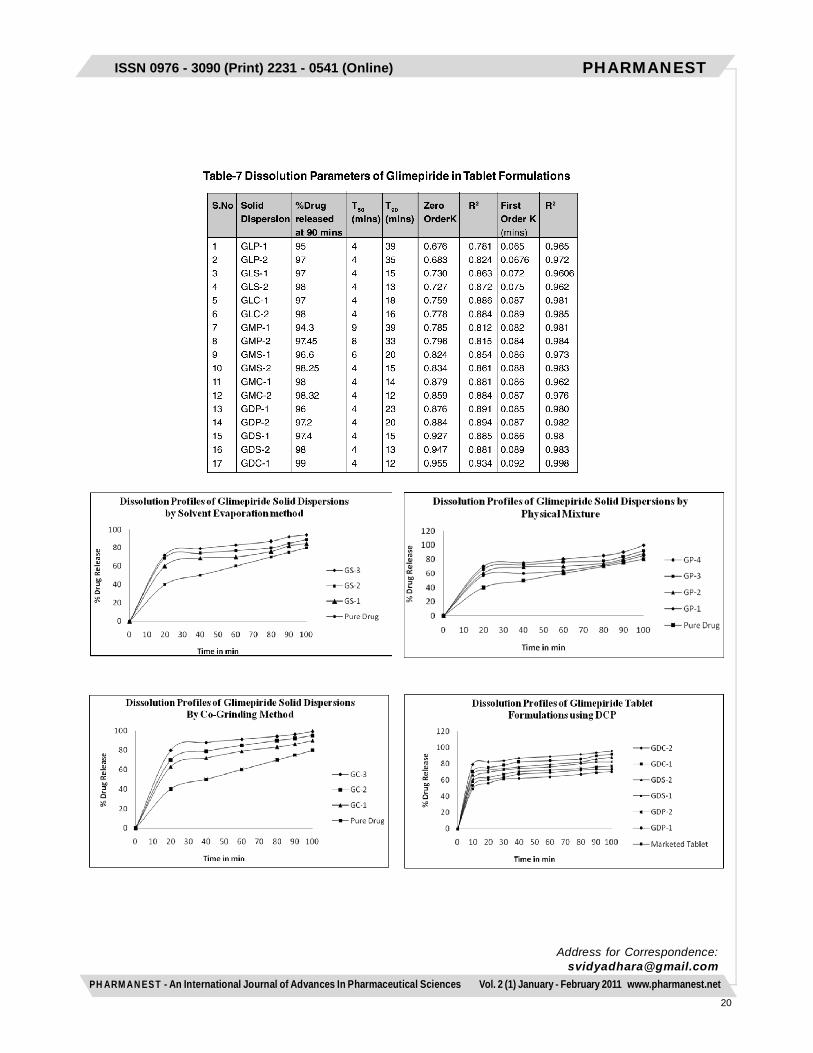
Dissolution rate studies on Glimepiride SolidDispersionsDissolution rate studies of pure Glimepiride and soliddispersions were performed in 8 stage Toshibadissolution test apparatus with rotating paddle methodat 50rpm using 900ml of 6.4pH buffer containing 0.5%SLS. The temperature of the bath was maintained at37±0.50C throughout the experiment. 5ml of sampleswere withdrawn at various time intervals and werefurther diluted with 6.4pH Phosphate buffer containing0.5% SLS medium. The absorbance of the sampleswas measured at 228nm for determining the amountof drug released at various time intervals. Each timethe same volume of buffer was added to the dissolutionmedia for maintaining the sink conditions. Thedissolution studies were carried out in triplicate. Basedupon the data obtained from the dissolution studiesvarious parameters such as T50, T90, zero order andfirst order release rate constants were estimated.The dissolution parameters such as T50 and T90 weremeasured directly from the dissolution profile curves.The zero order constant (K value) was obtained bycalculating the slope value from the percentage drugreleased versus time profile curve. The first orderconstant was calculated by multiplying the slope valueobtained from log percent drug undissolved versustime plot with 2.303.

ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
17
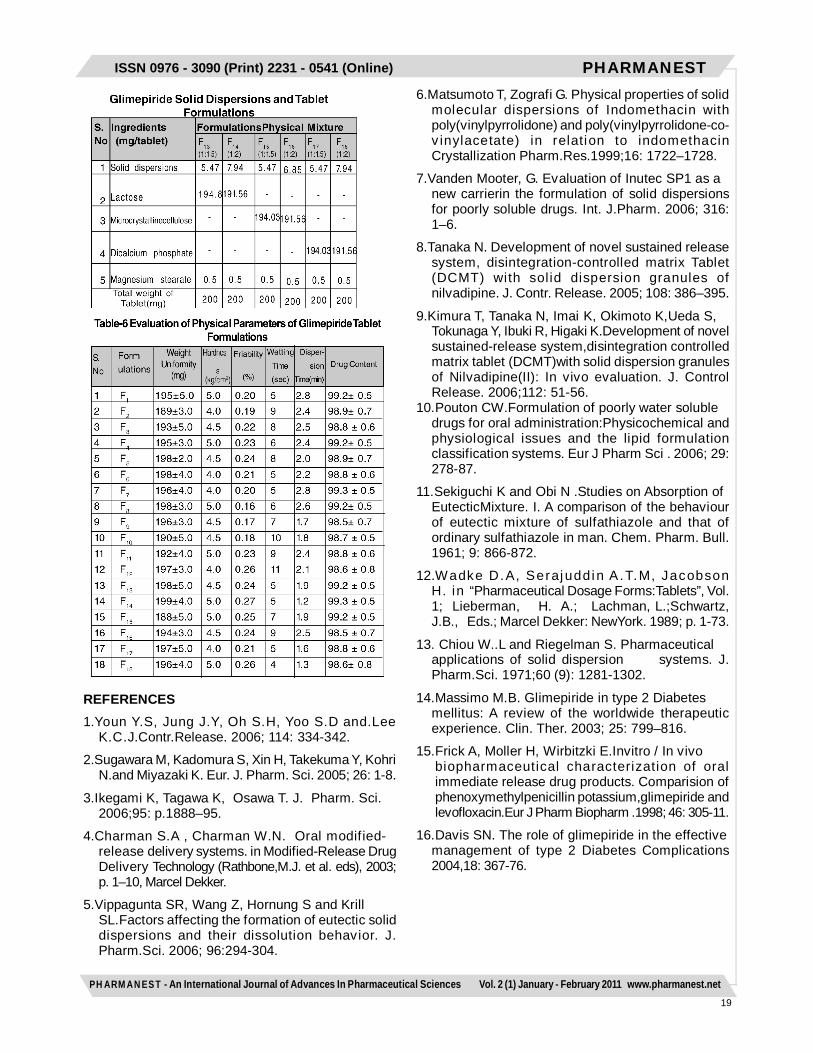
Preparation of Glimepiride Tablets with SolidDispersionsAmong the solid dispersions prepared and based uponthe dissolution studies performed, two optimizeddispersions were selected for preparation of tablets. Theselected solid dispersions were blended with diluents likelactose, DCP, MCC and 0.5% of lubricant and then directlycompressed by using 16 station rotary punching machinewith 3mm flat surface punches with a compression forceof 3-5kg/cm2. The compositions of various tabletformulations were given in Table.5.Evaluation of Physical Parameters for GlimepirideTabletsThe physical parameters such as weight uniformity,hardness, friability, drug content and disintegration timewere evaluated for the prepared tablets as perI.P.standards .Dissolution rate studies on Glimepiride TabletsDissolution rate studies of Glimepiride tablets wereperformed in 8 stage Toshiba dissolution test apparatusas per the procedure described earlier.RESULTS AND DISCUSSIONSaturated solubility studies revealed that Glimepirideexhibit maximum solubility in the 6.4pH phosphate buffercontaining 0.5% SLS as medium among the differentmedia used (Table 1). Hence 6.4pH phosphate buffercontaining 0.5% SLS was used as dissolution mediumfor further studies. The drug concentration was measuredat an absorption maximum of 228nm using UVspectrophotometer for all the dissolution media. The soliddispersions were prepared with a novel super disintegrantsuch as SSG by physical mixing, solvent evaporation andkneading methods as per the compositions shown in Table2. All the dispersions were prepared under similarconditions to avoid batch to batch variations. Thedispersions were found to be uniform in theircharacteristics. All the solid dispersions were in the sizerange of 178±10µm. The angle of repose and Carr’s indexvalues of all the dispersions prepared indicated the goodand free flowing characteristics (Table 3). The drug contentestimated in all the solid dispersions were highly uniformand in the size range of 98±2%, indicated the uniformity(Table 3).The dissolution studies of Glimepiride as pure drug andits solid dispersions prepared was performed in 6.4pHphosphate buffer containing 0.5% SLS as medium byusing paddle method. The dissolution rate of all the soliddispersions were found to be rapid than compared to itspure drug Glimepiride. The T50, T90 values of thedispersions indicated their rapid drug dissolution than theirrespective counterpart Glimepiride pure drug. The kineticsof drug release from all the dispersions followed firstorder. The R2 values obtained for all the dispersions werelinear for the first order plots. Among the solid dispersionsprepared, solvent evaporation and kneading methodswere found to be suitable in increasing the dissolution
rates of poorly soluble Glimepiride. It was observed thatas the concentration of SSG increases the rate ofdissolution of the drug was also increased. Soliddispersions prepared by solvent evaporation and kneadingmethods at a drug to super disintegrant ratio of 1:2 werefound to undergo rapid dissolution rates.Hence selected solid dispersion, were further directlycompressed as tablets by using lactose, MCC and DCPas diluents. The compositions are shown in Table 5. Allthe tablets were compressed under identical conditionsto avoid processing variables. The physical parameterssuch as weight uniformity, hardness, friability, drug contentand wetting time and dispersion time were evaluated forthe prepared tablets. The physical parameters evaluatedhighly uniform ad all the tablets were found to be withinthe Pharmacopoeial limits . The dissolution studies on theGlimepiride marketed tablet and all the tablet formulationswere performed in 6.4pH Phosphate buffer containing0.5% SLS medium using paddle method. The rate ofdissolution of tablet formulations was rapid whencompared to the marketed tablet of Glimepiride. The rateof drug release from all the tablets followed first orderkinetics. Among the tablets prepared with the lactose,MCC and DCP as diluents, tablets with DCP tend to exhibitrapid dissolution. The rate of rapid release is in the orderof DCP > MCC > Lactose in the tablet formulations.The disintegration time for the DCP containing tabletswere found to be much faster than its respective counterparts. The rapid disintegration may be due to increaseduptake of water by both diluents and super disintegrantwhich lead to faster dissolution of the tablets gaveimproved dissolution profiles of poorly soluble Glimepiride.CONCLUSIONThe present study has shown that it is possible toincrease the dissolution rate of poorly soluble drugGlimepiride by preparing solid dispersions withsuperdisintegrant like Sodiumstarch glycolate. The soliddispersions exhibit faster dissolution characteristics ascompared to plain drug. This was due to solubilisingeffect of carrier or crystallization of drug entrapped inmolecular state by the carrier. A higher dissolution ratewas obtained with solid dispersions prepared by solventevaporation method and kneading method in the ratioof 1:2 for the drug and SSG. Based on the study it maybe concluded that Glimepiride tablets prepared by soliddispersions with DCP as a diluent was found to be idealfor rapid disintegration and for improving the dissolutionrate and bioavailability.
ACKNOWLEDGEMENTSThe authors express their gratitude to Life lineformulations., and Pellets Pharma Ltd., Hyderabad forproviding the gift samples. The authors are thankfulto the management of Chebrolu Hanumaiah Instituteof Pharmaceutical Sciences, Guntur for providing thefacilities to carry out the research work.
ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
18
ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
19
REFERENCES1.Youn Y.S, Jung J.Y, Oh S.H, Yoo S.D and.Lee
K.C.J.Contr.Release. 2006; 114: 334-342.
2.Sugawara M, Kadomura S, Xin H, Takekuma Y, KohriN.and Miyazaki K. Eur. J. Pharm. Sci. 2005; 26: 1-8.
3.Ikegami K, Tagawa K, Osawa T. J. Pharm. Sci.2006;95: p.1888–95.
4.Charman S.A , Charman W.N. Oral modified-release delivery systems. in Modified-Release DrugDelivery Technology (Rathbone,M.J. et al. eds), 2003;p. 1–10, Marcel Dekker.
5.Vippagunta SR, Wang Z, Hornung S and KrillSL.Factors affecting the formation of eutectic soliddispersions and their dissolution behavior. J.Pharm.Sci. 2006; 96:294-304.
6.Matsumoto T, Zografi G. Physical properties of solidmolecular dispersions of Indomethacin withpoly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-v inylacetate) in relation to indomethacinCrystallization Pharm.Res.1999;16: 1722–1728.
7.Vanden Mooter, G. Evaluation of Inutec SP1 as anew carrierin the formulation of solid dispersionsfor poorly soluble drugs. Int. J.Pharm. 2006; 316:1–6.
8.Tanaka N. Development of novel sustained releasesystem, disintegration-controlled matrix Tablet(DCMT) with sol id dispersion granules ofnilvadipine. J. Contr. Release. 2005; 108: 386–395.
9.Kimura T, Tanaka N, Imai K, Okimoto K,Ueda S,Tokunaga Y, Ibuki R, Higaki K.Development of novelsustained-release system,disintegration controlledmatrix tablet (DCMT)with solid dispersion granulesof Nilvadipine(II): In vivo evaluation. J. ControlRelease. 2006;112: 51-56.
10.Pouton CW.Formulation of poorly water solubledrugs for oral administration:Physicochemical andphysiological issues and the lipid formulationclassification systems. Eur J Pharm Sci . 2006; 29:278-87.
11.Sekiguchi K and Obi N .Studies on Absorption ofEutecticMixture. I. A comparison of the behaviourof eutectic mixture of sulfathiazole and that ofordinary sulfathiazole in man. Chem. Pharm. Bull.1961; 9: 866-872.
12.Wadke D.A, Serajuddin A .T.M, JacobsonH. in “Pharmaceutical Dosage Forms:Tablets”, Vol.1; Lieberman, H. A.; Lachman, L.;Schwartz,J.B., Eds.; Marcel Dekker: NewYork. 1989; p. 1-73.
13. Chiou W..L and Riegelman S. Pharmaceuticalapplications of solid dispersion systems. J.Pharm.Sci. 1971;60 (9): 1281-1302.
14.Massimo M.B. Glimepiride in type 2 Diabetesmellitus: A review of the worldwide therapeuticexperience. Clin. Ther. 2003; 25: 799–816.
15.Frick A, Moller H, Wirbitzki E.Invitro / In vivobiopharmaceutical characterization of oralimmediate release drug products. Comparision ofphenoxymethylpenicillin potassium,glimepiride andlevofloxacin.Eur J Pharm Biopharm .1998; 46: 305-11.
16.Davis SN. The role of glimepiride in the effectivemanagement of type 2 Diabetes Complications2004,18: 367-76.
ISSN 0976 - 3090 (Print) 2231 - 0541 (Online)
PHARMANEST - An International Journal of Advances In Pharmaceutical Sciences Vol. 2 (1) January - February 2011 www.pharmanest.net
PHARMANEST
20
Address for Correspondence:[email protected]





























