Embed Size (px)
Citation preview

ARTHRITIS & RHEUMATISMVol. 58, No. 4, April 2008, pp 1010–1019DOI 10.1002/art.23482© 2008, American College of Rheumatology
Genetic Association of Vasoactive Intestinal Peptide ReceptorWith Rheumatoid Arthritis
Altered Expression and Signal in Immune Cells
Mario Delgado,1 Gema Robledo,1 Blanca Rueda,1 Nieves Varela,1
Francisco O’Valle,2 Pedro Hernandez-Cortes,3 Marta Caro,1 Gisela Orozco,1
Elena Gonzalez-Rey,4 and Javier Martin1
Objective. Vasoactive intestinal peptide (VIP) hasbeen shown to be one of the endogenous factors involvedin the maintenance of immune tolerance. Administra-tion of VIP ameliorates clinical signs in various exper-imental autoimmune disorders. This study was un-dertaken to investigate whether the exacerbatedinflammatory autoimmune response in rheumatoid ar-thritis (RA) might result directly from altered expres-sion and/or signaling of VIP receptors in immune cells.
Methods. The effect of specific agonists of differ-ent VIP receptors on collagen-induced arthritis in micewas investigated by clinical and histologic assessmentand measurement of cytokine and chemokine produc-tion. Expression of VIP receptor type 1 (VPAC1) insynovial cells and monocytes from RA patients wasdetermined by flow cytometry. Potential associations ofVPAC1 genetic polymorphisms with RA susceptibilitywere investigated.
Results. A VPAC1 agonist was very efficient in thetreatment of experimental arthritis, and deficient ex-pression of VPAC1 in immune cells of RA patients was
associated with the predominant proinflammatory Th1milieu found in this disease. Immune cells derived fromRA patients were less responsive to VIP signaling thanwere cells from healthy individuals and showed reducedVIP-mediated immunosuppressive activity, renderingleukocytes and synovial cells more proinflammatory inRA. A significant association between multiple-markerhaplotypes of VPAC1 and susceptibility to RA wasfound, suggesting that the reduced VPAC1 expression inRA-derived immune cells is associated with the de-scribed VPAC1 genetic polymorphism.
Conclusion. These findings are highly relevant tothe understanding of RA pathogenesis. They suggestthat VIP signaling through VPAC1 is critical to main-taining immune tolerance in RA. In addition, the resultsindicate that VPAC1 may be a novel therapeutic targetin RA.
Rheumatoid arthritis (RA), a chronic auto-immune disease characterized by inflammation of syno-vial joints, affects up to 1% of the white population. RAis a complex disease, with both genetic and environmen-tal factors contributing to its etiology. The contributionof HLA genes, particularly HLA–DRB1, to RA geneticpredisposition was the first described, and remains as thebest characterized, single genetic risk factor contributingto RA. However, it has been estimated that only 30% ofthe genetic contribution to RA can be attributed to HLAgenes, and it is suggested that other non-HLA genes mayplay a relevant role in RA susceptibility (1). Identifica-tion of non-HLA RA susceptibility genes remains achallenge. The search for additional RA susceptibilitygenes has been facilitated by the performance of whole-genome linkage scans.
Supported by the Junta de Andalucıa (Proyectos de Excelen-cia, CTS-870) and the Plan Nacional de I�D (grant SAF06-00398).
1Mario Delgado, PhD, Gema Robledo, BS, Blanca Rueda,PhD, Nieves Varela, PhD, Marta Caro, BS, Gisela Orozco, PhD, JavierMartin MD, PhD: Instituto de Parasitologıa y Biomedicina LopezNeyra, CSIC, Granada, Spain; 2Francisco O’Valle, MD, PhD: Univer-sity of Granada, Granada, Spain; 3Pedro Hernandez-Cortes, MD,PhD: San Cecilio University Hospital, Granada, Spain; 4ElenaGonzalez-Rey, PhD: University of Seville, Seville, Spain.
Address correspondence and reprint requests to Mario Del-gado, PhD, Instituto de Parasitologia y Biomedicina, CSIC, Avenidadel Conocimiento s/n, PT Ciencias de la Salud, Granada 18100, Spain.E-mail: [email protected].
Submitted for publication August 28, 2007; accepted inrevised form December 7, 2007.
1010

Vasoactive intestinal peptide (VIP) is a 28–amino acid peptide that, in addition to being a neu-ropeptide, is produced by immune cells in response toantigen stimulation and under inflammatory/auto-immune conditions (2). VIP elicits a broad spectrum ofbiologic actions, including immunomodulatory func-tions, acting predominantly as a potent antiinflamma-tory factor (3). In addition, VIP inhibits Th1 responses,promotes immune tolerance by inducing generation ofTreg cells, and has emerged as a promising factor in thetreatment of autoimmune/inflammatory diseases (3,4).Indeed, VIP is very efficient at ameliorating the pathol-ogy of several experimental autoimmune disorders, in-cluding RA, ulcerative colitis, multiple sclerosis, type 1diabetes, and uveoretinitis (2,3,5). The therapeutic ef-fect of VIP is associated with reduction in the severity ofthe 2 main phases of these immune disorders: VIPimpairs early events that are associated with the initia-tion and establishment of autoimmunity to self tissuecomponents, as well as later phases that are associatedwith the evolving immune and destructive inflammatoryresponses (3).
It is therefore tempting to speculate that theproinflammatory Th1 trait commonly seen in patientswith various autoimmune disorders might be associatedwith altered responsiveness of immune cells to VIP.Although studies have been undertaken in an effort tocorrelate altered VIP levels, presence of VIP autoanti-bodies, or VIP genetic variants with severity and inci-dence of autoimmunity, no association has been conclu-sively demonstrated (6–9). We hypothesized that theexacerbated inflammatory autoimmune response in RAmight result directly from altered expression and/orsignaling of VIP receptors in immune cells. VIP exertsits biologic activities by binding to 2 closely related classII G protein–coupled receptors, VIP receptor type 1(VPAC1) and VIP receptor type 2 (VPAC2), that havesimilar affinities for the peptide (2). VPAC1 is constitu-tively expressed by various immune cells, including Tcells, macrophages, and dendritic cells, while VPAC2 isexpressed marginally and its expression is induced byimmune stimulation (2,3). Therefore, the aim of thiswork was to determine which of the VIP receptors areinvolved in the therapeutic effect of VIP in arthritis, andwhether altered expression of VIP receptors in immunecells might render inflammatory cells less responsive toendogenously produced VIP and exogenously addedVIP. In addition, we investigated the possible associa-tion of VIP receptor polymorphism with RA suscepti-bility or with VIP receptor levels.
MATERIALS AND METHODS
Arthritis induction and treatment. Animal experimen-tal protocols were reviewed and approved by the EthicalCommittee of the Spanish Council of Scientific Research. Toinduce collagen-induced arthritis (CIA), DBA/1J mice (7–10weeks old; The Jackson Laboratory, Bar Harbor, ME) wereinjected subcutaneously with 200 �g of type II collagen (CII;Sigma, St. Louis, MO) emulsified in Freund’s complete adju-vant (CFA) containing 200 �g Mycobacterium tuberculosisH37Ra (Difco, Detroit, MI). On day 21 after primary immu-nization, mice were boosted by subcutaneous injection of 100�g of CII in CFA. Treatment with VPAC agonists consisted ofintraperitoneal administration of 3 nmoles/day of [K15, R16,L27] VIP (1-7)-GRF (8-27) (a VPAC1 agonist) or of Ac-[Glu8,Lys12, Nle17,Ala19, Asp25, Leu26, Lys27,28, Gly29,30, Thr31]-VIPcyclo (21-25) (Ro 25-1553; a VPAC2 agonist) on 5 consecutivedays starting 26 days postimmunization, when all mice exhib-ited established arthritis (clinical score �2). In each experi-ment, a control group of mice was injected intraperitoneallywith phosphate buffered saline.
Mice were assessed under blinded conditions everyother day by 2 independent examiners, and scored for signs ofarthritis as follows: grade 0 � no swelling; grade 1 � slightswelling and erythema; grade 2 � moderate swelling andedema; grade 3 � extreme swelling and pronounced edema;grade 4 � joint rigidity. Each limb was graded, for a maximumpossible score of 16 per animal. For histologic analysis, micewere killed on day 45 after primary immunization and the pawsof 4–6 animals were randomly collected, fixed in 4% bufferedformaldehyde, decalcified in Bouin’s fixative, embedded inparaffin, sectioned, and stained with Masson’s-Goldnertrichrome stain. Histopathologic changes were scored in ablinded manner based on cell infiltration, cartilage destruction,and bone erosion parameters as previously described (10). Fordetermination of cytokine levels in joints, protein extracts wereisolated by homogenization of joints (50 mg tissue/ml) in 50mM Tris HCl (pH 7.4) with 0.5 mM dithiothreitol and protein-ase inhibitor cocktail (10 �g/ml; Sigma). Serum samples werecollected at the peak of disease (day 35), and the levels of IgG,IgG1, and IgG2a anti-CII antibody were measured by enzyme-linked immunosorbent assay (ELISA) as previously described(10). Cytokine and chemokine levels in joint protein extractsprepared on day 35 were determined with specific sandwichELISAs using capture/biotinylated detection antibodies ac-cording to the recommendations of the manufacturer (BDPharMingen, San Diego, CA).
Assessment of T cell autoreactive response. Single-cellsuspensions (106 cells/ml) from draining lymph nodes wereobtained 32 days postimmunization. Cells were stimulated incomplete medium (RPMI 1640 containing 10% fetal calfserum [FCS], 2 mM L-glutamine, 100 units/ml penicillin, and100 �g/ml streptomycin) with various concentrations of heat-inactivated CII for 48 hours (for cytokine determination) or for72 hours (for proliferative response assays) (10). Cell prolifer-ation was evaluated using a bromodeoxyuridine-based prolif-eration assay (Roche Diagnostics, Mannheim, Germany). Cy-tokine content in culture supernatants was determined byspecific sandwich ELISA as described above.
To determine the number of Treg cells in mice with
VASOACTIVE INTESTINAL PEPTIDE SIGNALING IN RA 1011
CIA, draining lymph node cells isolated on day 30 postimmu-nization were incubated with fluorescein isothiocyanate(FITC)–conjugated anti-CD25 and peridin chlorophyllprotein–conjugated anti-CD4 monoclonal antibodies (mAb)(2.5 �g/ml) for 1 hour at 4°C. After extensive washing, cellswere fixed/saponin permeabilized, incubated for 45 minutesat 4°C with phycoerythrin-conjugated anti–forkhead box P3(anti-FoxP3) mAb (0.5 �g/sample), diluted in 0.5% saponin,and analyzed on a FACSCalibur flow cytometer.
Alternatively, synovial cells (106/ml) isolated frommice with CIA on day 32 postimmunization were stimulatedwith inactivated CII (10 �g/ml) in the absence or presence ofdifferent concentrations of VPAC1 or VPAC2 agonists. Cyto-kine levels were determined in supernatants after 48 hours ofculture.
Subjects and tissue samples. A case–control associa-tion study in a cohort of 381 RA patients and 355 healthycontrols recruited at Hospital Virgen de las Nieves (Granada,Spain) was designed to analyze the role of the VPAC1 gene inRA genetic predisposition. All RA patients were of Spanishwhite origin and met the American College of Rheumatology(formerly, the American Rheumatism Association) 1987 re-vised classification criteria for RA (11). Seventy-seven percentof the RA patients were women, the mean � SD age at theonset of the disease was 51.5 � 13 years, 56.4% carried the RAshared epitope, 77.2% were positive for rheumatoid factor,25% had extraarticular manifestations, and 19% had nodulardisease. The mean � SD age of the controls at the time ofanalysis was 48 � 11 years, and 78% were women. Peripheralblood mononuclear cells (PBMCs) were freshly isolated byFicoll-Histopaque density gradient centrifugation. Synovialtissue was obtained from 6 patients with RA at time of kneereplacement surgery. Fibroblast-like synoviocyte (FLS) cul-tures were established from homogenized synovium in 10%FCS/Dulbecco’s modified Eagle’s medium (12). FLS cultureswere used between passages 3 and 6. Approval of the institu-tional human studies committee and individual informed con-sent from each subject were obtained before the initiation ofthe study.
VPAC1 single-nucleotide polymorphism (SNP) screen-ing and genotyping. We selected the 9 genetic variants de-scribed in the public database (http://www.ncbi.nlm.nih.gov/entrez/snp) as localized within the VPAC1 3�-untranslatedregion (3�-UTR) as genetic markers for screening and associ-ation studies. A set of primers (forward CTATCCTCTAC-TGCTTCCTC; reverse ACATCTCTGACCTAGTTAC) wasdesigned to amplify a 1,123-bp fragment of the 3�-UTR con-taining 7 SNPs (rs1061363, rs7636205, rs897, rs895, rs896,rs342511, rs14380). Polymerase chain reaction (PCR) condi-tions were as follows: an initial denaturation step of 94°C for5 minutes, followed by 35 3-step cycles of 94°C for 1 minute,58°C for 1 minute, and 72°C for 1 minute, plus a final extensionstep of 72°C for 10 minutes. The presence of SNPs wasdetermined by direct sequencing of the amplicons using theBigDye Terminator cycle sequencing kit on an ABI 3100sequencer (Applied Biosystems, Foster City, CA). DetectedSNPs were also genotyped by direct sequencing for the asso-ciation study. The genotyping of the more distal VPAC13�-UTR genetic variant (rs9677) was performed by TaqMan 5�allelic discrimination assay. Primers and probes were providedwith a TaqMan SNP genotyping assay (with part numberC_7627363_10; Applied Biosystems). After PCR, the genotype
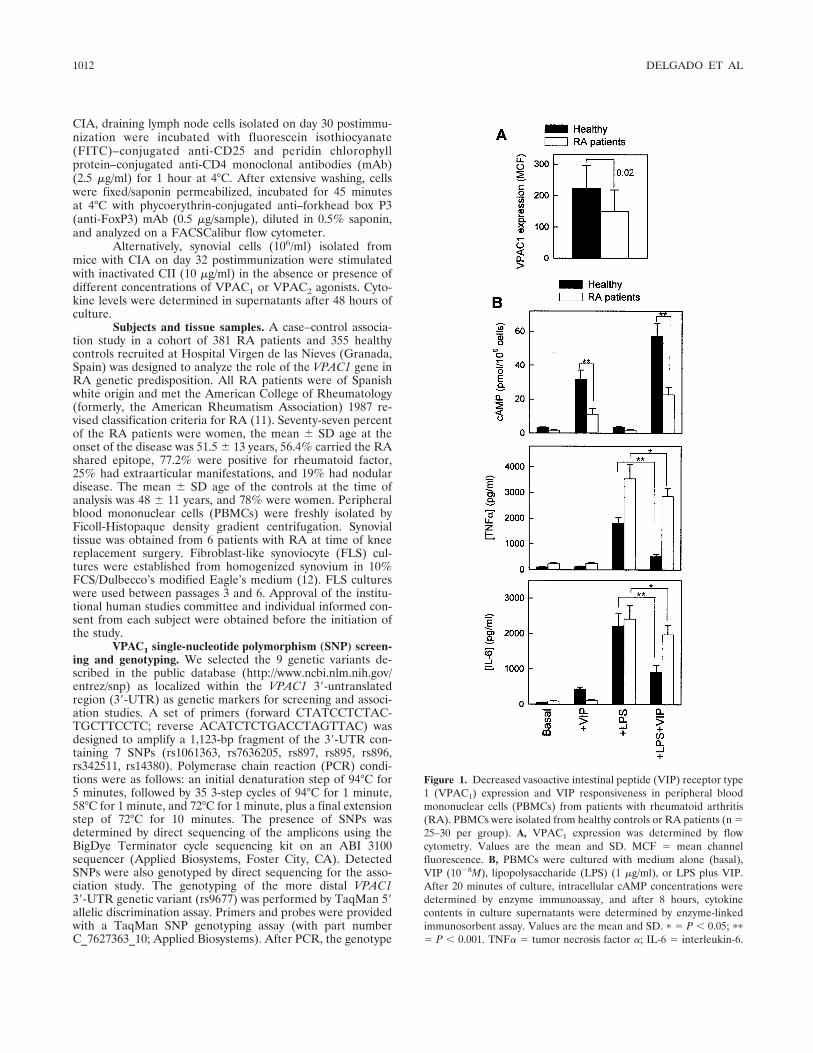
Figure 1. Decreased vasoactive intestinal peptide (VIP) receptor type1 (VPAC1) expression and VIP responsiveness in peripheral bloodmononuclear cells (PBMCs) from patients with rheumatoid arthritis(RA). PBMCs were isolated from healthy controls or RA patients (n �25–30 per group). A, VPAC1 expression was determined by flowcytometry. Values are the mean and SD. MCF � mean channelfluorescence. B, PBMCs were cultured with medium alone (basal),VIP (10�8M), lipopolysaccharide (LPS) (1 �g/ml), or LPS plus VIP.After 20 minutes of culture, intracellular cAMP concentrations weredetermined by enzyme immunoassay, and after 8 hours, cytokinecontents in culture supernatants were determined by enzyme-linkedimmunosorbent assay. Values are the mean and SD. � � P � 0.05; ��
� P � 0.001. TNF� � tumor necrosis factor �; IL-6 � interleukin-6.
1012 DELGADO ET AL

of each sample was attributed automatically by measuring theallele-specific fluorescence on the ABI Prism 7500 SequenceDetection System using SDS 1.3.1 software for allelic discrim-ination (Applied Biosystems). The rs8913 genotypes werededuced from rs342511 genotyping by virtue of the completelinkage disequilibrium (LD) described for these SNPs (D� � 1)(www.hapmap.org).
Determination of VPAC1 expression and cytokine andcAMP production. PBMCs and FLS (5 � 105/ml) were acti-vated with lipopolysaccharide (LPS) (1 �g/ml) or tumor ne-crosis factor � (TNF�) (10 nM), respectively, in the presenceor absence of VIP (10�8M; American Peptides, Sunnyvale,CA). Cells were incubated with rabbit anti-human VPAC1antibody (Affinity BioReagents, Golden, CO) followed byFITC-conjugated anti-rabbit mAb, and VPAC1 expression wasevaluated by flow cytometry. After 20 minutes of culture, cellswere lysed with HCl and intracellular cAMP levels determinedwith an enzyme immunoassay kit (Amersham, Piscataway, NJ).After 8 hours, cytokine contents in culture supernatants weredetermined by specific ELISA as described above.
Statistical analysis. Allele and genotype frequencieswere calculated and compared using the Unphased and Stat-calc software packages (Epi Info 2002; Centers for DiseaseControl and Prevention, Atlanta, GA). Significance was calcu-lated with 2 � 2 contingency tables and Fisher’s exact test; Pvalues, odds ratios (ORs), and 95% confidence intervals (95%CIs) were calculated. P values less than 0.05 were consideredsignificant. For haplotype analysis, pairwise LD measures wereinvestigated and haplotype blocks constructed using theexpectation-maximization algorithm implemented with Haplo-view and Unphased software. The power of the study to detecteffect of a polymorphism in disease susceptibility was esti-mated using Quanto version 0.5 software (Department ofPreventive Medicine, University of Southern California, LosAngeles). Values for clinical score, cytokine content, VPAC1expression, and cAMP level were expressed as the mean � SD,and the differences between groups were analyzed by Mann-Whitney U test and, if appropriate, by Kruskal-Wallis analysisof variance.
RESULTS
Correlation of decreased VPAC1 expression withVIP responsiveness in activated PBMCs from RA pa-tients. Previous lines of evidence support the implicationof VPAC1 in the effect of VIP on inflammatory andTh1-driven responses both in vitro and in vivo (2–4).Therefore, we first investigated whether altered expres-sion of VPAC1 in immune cells of RA patients thatmight render these cells less responsive to VIP could beassociated with the proinflammatory Th1 trait com-monly seen in RA. PBMCs isolated from RA patientsexhibited significantly reduced levels of VPAC1 (Figure1A). This altered expression of VPAC1 on immune cellsfrom RA patients had functional consequences andcorrelated with VIP responsiveness of the inflammatorycells. Thus, unstimulated and activated PBMCs isolatedfrom RA patients produced significantly lower levels ofcAMP in response to VIP, compared with PBMCs fromhealthy control subjects (Figure 1B). In addition,whereas VIP strongly inhibited the production of TNF�and interleukin-6 (IL-6) by LPS-activated PBMCs fromhealthy subjects (72% and 59% inhibition, respectively),it had only a moderate inhibitory effect on both cyto-kines in LPS-activated immune cells from RA patients(20% inhibition) (Figure 1B).
Association of VPAC1 genetic variants with RAsusceptibility. We further addressed whether VPAC1
genetic polymorphisms were potentially associated withRA susceptibility and tested whether these variants hadfunctional effects in the expression of VPAC1 in RA-derived immune cells. The human VPAC1 gene com-
Table 1. Distribution of VPAC1 3�-UTR SNPs in RA patients and healthy controls*
VPAC1 SNP,allele 1
(major allele)/allele 2
(minor allele)
RA patients (n � 381) Healthy controls (n � 355)
OR (95% CI)† P
Genotype, no. (%)
MAF
Genotype, no. (%)
MAF1/1 1/2 2/2 1/1 1/2 2/2
rs7636205, A/C 258 (67.7) 119 (31.2) 4 (1.0) 0.17 230 (64.8) 118 (33.2) 7 (2.1) 0.19 1.14 (0.86–1.50) 0.34rs897, C/T 160 (42.0) 181 (47.5) 40 (10.5) 0.34 123 (34.6) 181 (51.0) 51 (13.4) 0.40 1.27 (1.02–1.58) 0.02rs895, C/T 145 (38.1) 186 (48.8) 50 (13.1) 0.37 113 (31.8) 183 (51.5) 59 (16.6) 0.43 1.22 (0.99–1.52) 0.05rs896, C/T 145 (38.1) 186 (48.8) 50 (13.1) 0.37 113 (31.8) 183 (51.5) 59 (16.6) 0.43 1.22 (0.99–1.52) 0.05�2155, C/T‡ 361 (94.8) 20 (5.2) 0 0.03 339 (95.5) 16 (4.5) 0 0.02 1.17 (0.57–2.37) 0.64rs342511, A/G 135 (35.4)§ 190 (49.9) 56 (14.7) 0.40 97 (27.3) 196 (55.2) 62 (17.5) 0.45 1.25 (1.01–1.54) 0.03rs14380, A/T 229 (60.1) 134 (35.2) 18 (4.7) 0.22 202 (56.9) 132 (37.2) 21 (5.9) 0.24 1.13 (0.88–1.43) 0.31rs8913, G/A 135 (35.4)¶ 190 (50) 56 (14.7) 0.40 97 (27.3) 196 (55.2) 62 (17.5) 0.45 1.25 (1.01–1.54) 0.03rs9677, C/T 100 (26.2) 173 (45.4) 108 (28.3) 0.49 91 (25.6) 185 (52.1) 79 (22.3) 0.48 1.12 (0.92–1.36) 0.27
* 3�-UTR � 3�-untranslated region; SNPs � single-nucleotide polymorphisms; MAF � minor allele frequency.† Odds ratios (ORs) and 95% confidence intervals (95% CIs) refer to the genotype distribution in rheumatoid arthritis (RA) patients versus controls.‡ New genetic variant.§ OR 1.45 (95% CI 1.05–2.01), P � 0.02, A/A-homozygous RA patients versus controls.¶ OR 1.45 (95% CI 1.05–2.01), P � 0.02, G/G-homozygous RA patients versus controls.
VASOACTIVE INTESTINAL PEPTIDE SIGNALING IN RA 1013

prises 13 exons and 12 introns spanning 22 kb (13). Weinvestigated the presence of SNPs localized withinVPAC1 putative regulatory regions which might havefunctional relevance by altering promoter activity orRNA splicing and processing. Since SNPs in the pro-moter region have not been described for the VPAC1gene, we focused on the VPAC1 3�-UTR that maycontain regulatory sequences.
The VPAC1 3�-UTR was sequenced in a total of736 individuals (381 RA patients and 355 healthy con-trols). One of the 9 SNPs described in the publicdatabase for the VPAC1 3�-UTR (rs1061363) was notpolymorphic in our population and therefore was ex-
cluded for the association study. The rest of the VPAC13�-UTR SNPs (rs7636205, rs897, rs895, rs896, rs342511,rs14380, rs8913, rs9677) were polymorphic and showedallele frequencies very similar to those previously re-ported in white populations (http://www.ncbi.nlm.nih.gov/) (Table 1). Our study population exhibited anew genetic variant consisting of a C-to-T change andlocated between SNPs rs896 and rs342511 SNPs (posi-tion 2155 from the transcription start site). This newSNP (called �2155) showed very low minor allelefrequency in our population (0.03) (Table 1).
The 9 tested VPAC1 3�-UTR SNPs were ob-served to be in Hardy-Weinberg equilibrium in both theRA patient group and the control group (Table 1).Interestingly, rs897, rs342511, and rs8913 SNPs showeda significant association with susceptibility to RA, with Pvalues ranging from 0.02 to 0.03 (Table 1). However,rs342511 and rs8913 were in complete LD (r2 � 1). Inaddition, the numbers of rs342511 A/A– and rs8913G/G–homozygous individuals were significantly in-creased among RA patients compared with healthycontrols (OR 1.45 [95% CI 1.05–2.01], P � 0.02) (Table 1).
We further investigated the LD pattern of the 9tested VPAC1 3�-UTR SNPs. Six genetic markers(rs7636205, rs897, rs895, rs896, rs342511, and rs8913)showed strong pairwise LD (D� � 0.90), conforming aclearly defined haplotype block (Figure 2). Interestingly,the ACCCAC haplotype was significantly associatedwith RA predisposition (OR 1.50 [95% CI 1.20–1.86],P � 0.0002), while the ATTTGT haplotype showed aprotective effect (OR 0.78 [95% CI 0.62–0.96], P � 0.02)(Table 2). However, we did not observe significantdifferences between the 2 haplotypes in terms of VPAC1
expression or VIP-induced cAMP production in PBMCsfrom RA patients (Figures 3A and B). The possibleimplication of VPAC1 polymorphism and haplotypes inRA clinical course was also investigated, and no evi-dence of association between any of the clinical variables
Figure 2. Haplotype block estimated for the VPAC1 3�-untranslatedregion. Numbers in boxes represent linkage disequilibrium (D� values).Empty boxes indicate complete disequilibrium.
Table 2. Distribution of VPAC1 haplotypes in RA patients and healthy controls*
Haplotype(rs7636205/rs897/
rs895/rs896/rs342511/rs8913)
RA patients(2n � 760),
no. (%)
Healthy controls(2n � 690)
no. (%) OR (95% CI) P
ACCCAC 311 (40.9) 218 (31.6) 1.50 (1.20–1.86) 0.0002ATTTGT 254 (33.4) 270 (39.1) 0.78 (0.62–0.96) 0.02CCCCAT 116 (15.3) 109 (15.8) 0.96 (0.72–1.27) 0.7Other 79 (10.4) 93 (13.5) 0.7 (0.86–1.15) 0.07
* RA � rheumatoid arthritis; OR � odds ratio; 95% CI � 95% confidence interval.
1014 DELGADO ET AL

and VPAC1 SNPs or haplotypes was observed (data notshown).
Finally, we investigated the potential relationshipbetween altered expression of VPAC1 and the different
haplotypes in synovial cells isolated from RA patients.Interestingly, FLS from RA patients with the protectivehaplotype ATTTG showed increased VPAC1 expressionand an increased percentage of VPAC1-positive cellsfollowing activation (Figure 3C). In contrast, VPAC1expression was slightly reduced in activated FLS fromRA patients with the predisposition haplotypes (Figure3C). This paralleled the finding that VIP significantlyreduced the production of inflammatory cytokines inactivated synovial cells from patients with the haplotypeATTTG, but not in FLS from patients with otherhaplotypes (Figure 3C). Collectively, these findings sup-port the notion that the protective VPAC1 haplotypeATTTG is associated with increased VPAC1 expressionand VIP response by activated synovial inflammatorycells.
A specific agonist of VPAC1 decreases the sever-ity of experimental arthritis by inhibiting inflammatoryand autoimmune responses. CIA is a murine experimen-tal disease model that shares a number of clinical,histologic, and immunologic features with RA, and it isused as a model system to test potential therapeuticagents. Administration of VIP at the time of diseaseonset was previously found to be effective in reducingCIA severity and incidence (10,14). In order to investi-gate the receptor involved in the therapeutic effect ofVIP, specific agonists of VPAC1 (15) and VPAC2 (16)were administered to animals with established clinicalsigns of arthritis (clinical score �2). Treatment with theVPAC1 agonist, but not the VPAC2 agonist, progres-sively attenuated the severity of CIA and decreased thepercentage of mice with arthritis (Figure 4A). His-topathologic analysis of joints showed that treatmentwith the VPAC1 agonist significantly reduced the sever-ity of characteristic features of CIA, including chronicinflammation of synovial tissue (infiltration of inflam-matory cells [lymphocytes, plasma cells, macrophages,and neutrophils] into the joint cavity and periarticularsoft tissue), pannus formation, cartilage destruction, andbone erosion (Figure 4A).
We next investigated the mechanisms underlyingthe decrease in severity of CIA following VPAC1 agonisttreatment. We first evaluated the effect of both VPAC1and VPAC2 agonists on the production of inflammationmediators that are mechanistically linked to CIA sever-ity. Administration of the VPAC1 agonist significantlyreduced protein expression of inflammatory cytokines(TNF�, interferon-� [IFN�], IL-6, IL-1�, and IL-12) andchemokines (RANTES and macrophage inhibitory pro-tein 2) in the joints of arthritic mice (Figure 4B). Inaddition, the joints of VPAC1 agonist–treated mice
Figure 3. VPAC1 expression and VIP response by synovial cells andleukocytes from healthy controls and RA patients with differentVPAC1 gene variations. A and B, PBMCs were isolated from healthycontrols or RA patients with the VPAC1 haplotypes ACCCA orATTTG (n � 25–30 per group). VPAC1 expression was determined byflow cytometry (A), and intracellular cAMP concentrations in PBMCscultured for 20 minutes with VIP (10�8M) were determined by enzymeimmunoassay (B). Values are the mean and SD. � � P � 0.05; �� �P � 0.001. C, Fibroblast-like synoviocytes were isolated from RApatients with the haplotype ATTTG (n � 1) or the other haplotypes ofVPAC1 (n � 8). Cells were cultured for 24 hours with medium(unstimulated) or TNF� (10 nM). VPAC1 expression and the percent-age of VPAC1-expressing cells were determined by flow cytometry,and cytokine contents in culture supernatants were determined byenzyme-linked immunosorbent assay. Values for patients with otherhaplotypes of VPAC1 are the mean and SD. See Figure 1 fordefinitions.
VASOACTIVE INTESTINAL PEPTIDE SIGNALING IN RA 1015
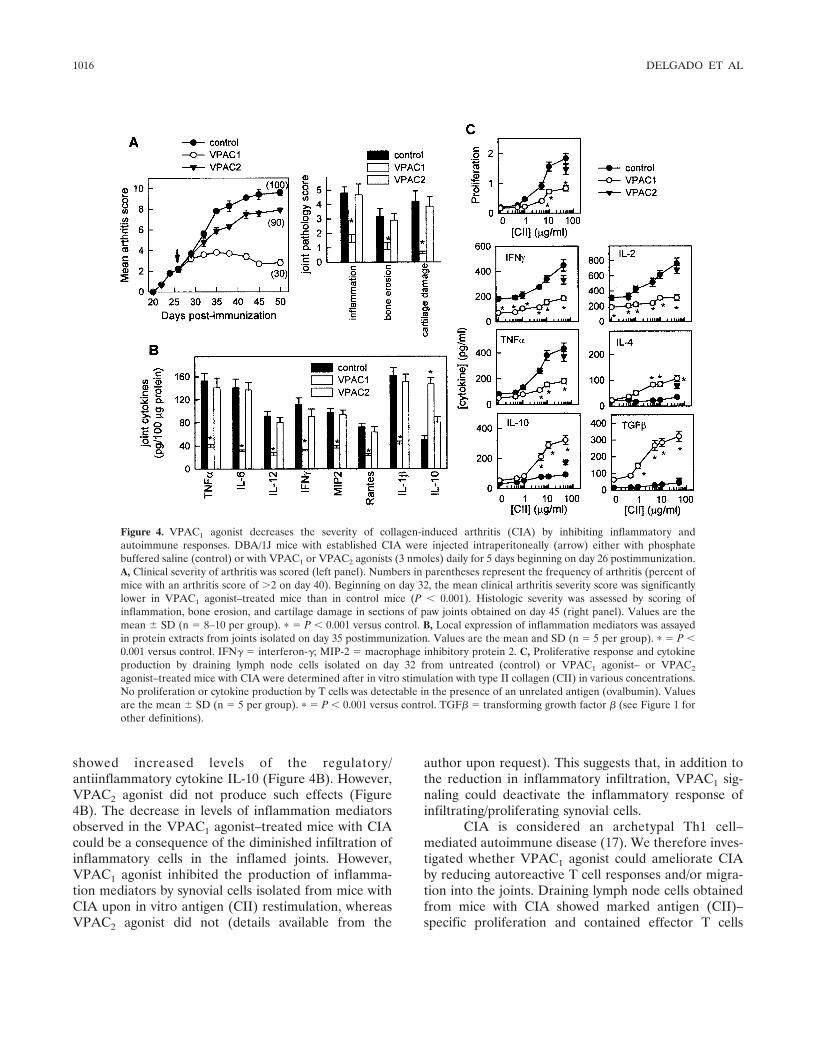
showed increased levels of the regulatory/antiinflammatory cytokine IL-10 (Figure 4B). However,VPAC2 agonist did not produce such effects (Figure4B). The decrease in levels of inflammation mediatorsobserved in the VPAC1 agonist–treated mice with CIAcould be a consequence of the diminished infiltration ofinflammatory cells in the inflamed joints. However,VPAC1 agonist inhibited the production of inflamma-tion mediators by synovial cells isolated from mice withCIA upon in vitro antigen (CII) restimulation, whereasVPAC2 agonist did not (details available from the
author upon request). This suggests that, in addition tothe reduction in inflammatory infiltration, VPAC1 sig-naling could deactivate the inflammatory response ofinfiltrating/proliferating synovial cells.
CIA is considered an archetypal Th1 cell–mediated autoimmune disease (17). We therefore inves-tigated whether VPAC1 agonist could ameliorate CIAby reducing autoreactive T cell responses and/or migra-tion into the joints. Draining lymph node cells obtainedfrom mice with CIA showed marked antigen (CII)–specific proliferation and contained effector T cells
Figure 4. VPAC1 agonist decreases the severity of collagen-induced arthritis (CIA) by inhibiting inflammatory andautoimmune responses. DBA/1J mice with established CIA were injected intraperitoneally (arrow) either with phosphatebuffered saline (control) or with VPAC1 or VPAC2 agonists (3 nmoles) daily for 5 days beginning on day 26 postimmunization.A, Clinical severity of arthritis was scored (left panel). Numbers in parentheses represent the frequency of arthritis (percent ofmice with an arthritis score of �2 on day 40). Beginning on day 32, the mean clinical arthritis severity score was significantlylower in VPAC1 agonist–treated mice than in control mice (P � 0.001). Histologic severity was assessed by scoring ofinflammation, bone erosion, and cartilage damage in sections of paw joints obtained on day 45 (right panel). Values are themean � SD (n � 8–10 per group). � � P � 0.001 versus control. B, Local expression of inflammation mediators was assayedin protein extracts from joints isolated on day 35 postimmunization. Values are the mean and SD (n � 5 per group). � � P �0.001 versus control. IFN� � interferon-�; MIP-2 � macrophage inhibitory protein 2. C, Proliferative response and cytokineproduction by draining lymph node cells isolated on day 32 from untreated (control) or VPAC1 agonist– or VPAC2
agonist–treated mice with CIA were determined after in vitro stimulation with type II collagen (CII) in various concentrations.No proliferation or cytokine production by T cells was detectable in the presence of an unrelated antigen (ovalbumin). Valuesare the mean � SD (n � 5 per group). � � P � 0.001 versus control. TGF� � transforming growth factor � (see Figure 1 forother definitions).
1016 DELGADO ET AL

producing high levels of Th1-type cytokines (IFN�, IL-2,and TNF�) and low levels of Th2-type cytokines (IL-4and IL-10) (Figure 4C). In contrast, draining lymphnode cells from VPAC1 agonist–treated mice prolifer-ated to a much lower degree and produced low levels ofTh1 cytokines and increased amounts of suppressivecytokines (IL-10 and transforming growth factor �1[TGF�1]) and the Th2-type cytokine IL-4 (Figure 4C).This suggests that VPAC1 agonist administration duringCIA progression partially inhibits CII-specific Th1 cellclonal expansion. These findings paralleled the findingthat VPAC1 agonist administration resulted in reducedserum levels of CII-specific IgG, particularly autoreac-tive IgG2a antibodies (data not shown), generally reflec-tive of Th1 activity. In contrast, the VPAC2 agonistfailed to reduce the autoreactive response in mice withCIA (data not shown).
Various lines of evidence indicate that Treg cellsconfer significant protection against CIA by decreasingthe activation and joint homing of autoreactive Th1 cells(18), and VIP has been shown to generate Treg cells(19). In addition, VPAC1 agonist treatment inhibitsevents in the inflammatory phase of CIA followingactivation of antigen-specific Th1 cells, and induces thegeneration of IL-10/TGF�1–producing T cells. There-fore, we investigated whether VPAC1 signaling inducesTreg cells with suppressive activity during the progres-sion of the disease. Mice with CIA that were treated withthe VPAC1 agonist, but not those treated with theVPAC2 agonist, had significantly higher numbers ofCD4�,CD25�,FoxP3� Treg cells in both draininglymph nodes and synovium compared with control micewith CIA (details available from the author upon re-quest).
DISCUSSION
RA is a systemic autoimmune disease that leadsto chronic inflammation in the joints and subsequentdestruction of cartilage and erosion of bone. Althoughits etiology is unknown, various lines of evidence indi-cate that the recruitment and activation of neutrophils,macrophages, and lymphocytes into joint tissue and theformation of pannus are hallmarks of RA pathogenesis.The contribution of Th1 responses in RA is not com-pletely understood, but the results of several studiesusing animal models indicate a pathogenic role of Th1-derived cytokines (17). Th1 cells react with componentsof the joint, infiltrate the synovium, release proinflam-matory cytokines and chemokines, and promote macro-phage and neutrophil infiltration and activation. Inflam-
mation mediators, such as cytokines and free radicals,produced by infiltrating inflammatory cells play a criticalrole in joint damage (17).
VIP has recently emerged as one of the endoge-nous factors involved in the maintenance of immunetolerance in RA and other autoimmune disorders (2–5).In this study, we obtained the first evidence indicatingthat deficient expression of the VIP receptor VPAC1 inimmune cells of RA patients is associated with thepredominant proinflammatory Th1 milieu found in thisdisease. Our results show that altered expression ofVPAC1 has direct functional consequences character-ized by reduced VIP-mediated antiinflammatory activityin immune cells from patients with RA. These cells areless responsive to VIP signaling than are PBMCs fromhealthy individuals, as evidenced by production ofcAMP, which is the major VPAC1-coupled intracellularmediator described for VIP, and a well-known immuno-suppressive agent (20). These findings are of importanceand highly relevant to the understanding of the patho-genesis of RA in that they provide a possible explanationfor the proinflammatory nature of the immune cells inthis disease.
Interestingly, when we analyzed 9 SNPs in theVPAC1 gene in a case–control cohort from Spain, wefound a significant association between multiple-markerhaplotypes of VPAC1 and susceptibility to RA. This isthe first report to describe a genetic association of VIPor of its 2 VIP receptors with any autoimmune disorder.Reduced levels of VIP and altered expression of VPAC2in T cells from patients with multiple sclerosis wererecently reported (9); however, results of 2 differentstudies have failed to support the notion of geneticpolymorphisms within the VIP and VPAC2 genes in thatdisease (8,9).
Our findings provide evidence of a critical role ofVPAC1 in immune homeostasis. This is also supportedby the fact that VPAC1 is constitutively expressed inmost immunocompetent cells; in addition, the notion ofa role of VPAC1 in VIP’s effect on the inflammatory andTh1-driven responses and the generation of Treg cellshas been supported by both in vitro and in vivo findings(2–4). However, studies using mice deficient in ortransgenic for VPAC2 have demonstrated the involve-ment of VPAC2 only in a shift toward a Th2-prominentresponse (21–23), revealing a secondary role of thisreceptor in the regulation of autoimmunity by VIP.Accordingly, the present study shows that treatment witha VPAC1 agonist, but not with a VPAC2 agonist, reducesthe frequency of arthritis, ameliorates symptoms, andprevents joint damage in an experimental model of
VASOACTIVE INTESTINAL PEPTIDE SIGNALING IN RA 1017

arthritis. Similar to VIP (10,14), the therapeutic effect ofthe VPAC1 agonist is associated with a striking reduc-tion of the 2 deleterious components of the disease, i.e.,the autoimmune and inflammatory responses. Thistreatment reduced the presence of autoreactive Th1cells in the periphery and the joints, and strongly de-creased the inflammatory response during CIA progres-sion by down-regulating the production of several in-flammation mediators in the joints, including variouscytokines and chemokines.
It is tempting to speculate that reduced expres-sion of VPAC1 in RA PBMCs is associated with thedescribed VPAC1 genetic polymorphism. All of theSNPs analyzed in our study are localized on the VPAC13�-UTR, whereas the promoter region of the VPAC1gene does not contain any SNPs. Because the 3�-UTRmay contain regulatory sequences, most of them relatedto the stability of the messenger RNA (mRNA), severalpossibilities emerge to potentially relate the VPAC1susceptibility haplotypes with altered VPAC1 expressionin RA. First, the presence of a SNP could preventbinding of a transcription factor to the gene. Thus, asubstitution of T for C in the sequence ACCCTA makesthe polymorphic sequence unable to bind the transacti-vator factor p300. Second, it has been recently reportedthat certain small noncoding RNAs (called microRNAs)recognize and bind to partially complementary sites inthe 3�-UTR of target genes in animals and, by unknownmechanisms, regulate protein production of the targettranscript (24,25). This raises the possibility that inclu-sion of a SNP could generate a target sequence in theVPAC1 3�-UTR that makes its mRNA less stable.
Further studies will determine the involvement ofthese or other mechanisms in the reduced expression ofVPAC1 in immune cells from patients with RA. Inaddition, we have identified a protective VPAC1 haplo-type, which is associated with increased expression ofVPAC1 in activated synovial cells. However, the dataremain merely suggestive because of the limited numberof synovial samples analyzed.
In summary, reduced expression of VPAC1,probably as a result of altered gene regulation in the3�-UTR of the VIP receptor, results in decreased VIPsignaling in RA. This has the functional consequence ofrendering leukocytes and synovial cells more proinflam-matory in RA. In our studies, VPAC1 has emerged ashaving a significant genetic association with RA outsidethe HLA region. Our results not only suggest that VIPsignaling through VPAC1 is critical to maintaining im-mune tolerance in RA, but also identify VPAC1 as anovel therapeutic target for RA and other chronic
autoimmune disorders. Further, they provide a powerfulrationale for assessment of the efficacy of VPAC1 ago-nists as new immunomodulatory factors with the capac-ity to deactivate the inflammatory response at multiplelevels.
ACKNOWLEDGMENT
We are grateful to the patients who participated in thisstudy for their essential collaboration.
AUTHOR CONTRIBUTIONS
Dr. Delgado had full access to all of the data in the study andtakes responsibility for the integrity of the data and the accuracy of thedata analysis.Study design. Delgado, Martin.Acquisition of data. Robledo, Rueda, Varela, O’Valle, Hernandez-Cortes, Caro, Orozco, Gonzalez-Rey.Analysis and interpretation of data. Delgado, Robledo, Gonzalez-Rey,Martin.Manuscript preparation. Delgado, Martin.Statistical analysis. Robledo, Rueda, Varela.
REFERENCES
1. Orozco G, Rueda B, Martin J. Genetic basis of rheumatoidarthritis. Biomed Pharmacother 2006;60:656–62.
2. Delgado M, Pozo D, Ganea D. The significance of vasoactiveintestinal peptide in immunomodulation. Pharmacol Rev 2004;56:249–90.
3. Gonzalez-Rey E, Chorny A, Delgado M. Regulation of immunetolerance by anti-inflammatory neuropeptides. Nat Rev Immunol2007;7:52–63.
4. Gonzalez-Rey E, Delgado M. Vasoactive intestinal peptide andregulatory T-cell induction: a new mechanism and therapeuticpotential for immune homeostasis. Trends Mol Med 2007;13:241–51.
5. Pozo D, Gonzalez-Rey E, Chorny A, Anderson P, Varela N,Delgado M. Tuning immune tolerance with vasoactive intestinalpeptide: a new therapeutic approach for immune disorders. Pep-tides 2007;28:1833–46.
6. Bangale Y, Karle S, Planque S, Zhou YX, Taguchi H, NishiyamaY, et al. VIPase autoantibodies in Fas-defective mice and patientswith autoimmune disease. FASEB J 2003;17:628–35.
7. Larsson J, Ekblom A, Henriksson K, Lundeberg T, TheodorssonE. Concentration of substance P, neurokinin A, calcitonin gene-related peptide, neuropeptide Y and vasoactive intestinal polypep-tide in synovial fluid from knee joints in patients suffering fromrheumatoid arthritis. Scand J Rheumatol 1991;20:326–35.
8. Cunningham S, O’Doherty C, Patterson C, McDonnell G,Hawkins S, Marrosu MG, et al. The neuropeptide genes TAC1,TAC3, TAC4, VIP and PACAP(ADCYAP1), and susceptibility tomultiple sclerosis. J Neuroimmunol 2007;183:207–13.
9. Sun W, Hong J, Zang YC, Liu X, Zhang JZ. Altered expression ofvasoactive intestinal peptide receptors in T lymphocytes andaberrant Th1 immunity in multiple sclerosis. Int Immunol 2006;18:1691–700.
10. Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vaso-active intestinal peptide prevents experimental arthritis by down-regulating both autoimmune and inflammatory components of thedisease. Nat Med 2001;7:563–8.
11. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF,
1018 DELGADO ET AL

Cooper NS, et al. The American Rheumatism Association 1987revised criteria for the classification of rheumatoid arthritis.Arthritis Rheum 1988;31:315–24.
12. Takeba Y, Suzuki N, Kaneko A, Asai T, Sakane T. Evidence forneural regulation of inflammatory synovial cell functions by se-creting calcitonin gene-related peptide and vasoactive intestinalpeptide in patients with rheumatoid arthritis. Arthritis Rheum1999;42:2418–29.
13. Sreedharan SP, Huang JX, Cheung MC, Goetzl EJ. Structure,expression, and chromosomal localization of the type I humanvasoactive intestinal peptide receptor gene. Proc Natl Acad SciU S A 1995;92:2939–43.
14. Williams RO. Therapeutic effect of vasoactive intestinal peptide incollagen-induced arthritis. Arthritis Rheum 2002;46:271–3.
15. Gourlet P, Vandermeers A, Vertongen P, Ratche J, De Neef P,Cnudde J, et al. Development of high affinity selective VIP1receptor agonists. Peptides 1997;18:1539–45.
16. Xia M, Sreedharan SP, Bolin DR, Gaufo GO, Goetzl EJ. Novelcyclic peptide agonist of high potency and selectivity for the typeII vasoactive intestinal peptide receptor. J Pharmacol Exp Ther1997;281:629–33.
17. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature2003;423:356–61.
18. Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ,van Ewijk W, van Laar JM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25� regulatory T cells.Arthritis Rheum 2005;52:2212–21.
19. Gonzalez-Rey E, Fernandez-Martin A, Chorny A, Delgado M.Vasoactive intestinal peptide induces CD4�,CD25� T regulatorycells with therapeutic effect in collagen-induced arthritis. ArthritisRheum 2006;54:864–76.
20. Banner KH, Trevethick MA. PDE4 inhibition: a novel approachfor the treatment of inflammatory bowel disease. Trends Pharma-col Sci 2004;25:430–6.
21. Voice JK, Dorsam G, Lee H, Kong Y, Goetzl EJ. Allergicdiathesis in transgenic mice with constitutive T cell expression ofinducible vasoactive intestinal peptide receptor. FASEB J 2001;15:2489–96.
22. Goetzl EJ, Voice JK, Shen S, Dorsam G, Kong Y, West KM, et al.Enhanced delayed-type hypersensitivity and diminished immedi-ate-type hypersensitivity in mice lacking the inducible VPAC2receptor for vasoactive intestinal peptide. Proc Natl Acad Sci U SA 2001;98:13854–9.
23. Voice J, Grinninger C, Kong Y, Bangale Y, Paul S, Goetzl EJ.Roles of vasoactive intestinal peptide (VIP) in the expression ofdifferent immune phenotypes by wild-type mice and T cell-targeted type II VIP receptor transgenic mice. J Immunol 2003;170:308–14.
24. Scherr M, Eder M. Gene silencing by small regulatory RNAs inmammalian cells. Cell Cycle 2007;6:444–9.
25. Behm-Ansmant I, Rehwinkel J, Izaurralde E. MicroRNAs silencegene expression by repressing protein expression and/or by pro-moting mRNA decay. Cold Spring Harb Symp Quant Bio 2006;71:523–30.
VASOACTIVE INTESTINAL PEPTIDE SIGNALING IN RA 1019








![Sinisa Dovat, MD, DSc, Vasoactive intestinal peptide ......Sinisa Dovat, MD, DSc, Series Editor Dorsam GP et al. VIP in human leukemia mast cells[2], neutrophils[3], eosinophils[4],](https://img.pdfslide.us/doc/110x75/60bdfd263eb7801e007d5d66/sinisa-dovat-md-dsc-vasoactive-intestinal-peptide-sinisa-dovat-md-dsc.jpg)




















