Embed Size (px)
Citation preview

~ C T E R I Z A T I O N OF THE PROMOTER FOR 'ITE HUMAN FACTOR I
(C3bl C4b INACTLVATOR) GENE.
Bamini Paramaswara
Thesis submitted in conformity with the requirements for the Degree of Master of Science,
Graduate Department of Cellular and Molecular Pathology, in the University of Toronto
8 Copyright by Bamini Paramaswara 1998
1

National Library 191 ofCanada BibSihque nationale du Canada
Acquisitions and Acquisitions et Bibliographie SeMces services bibliographiques
395 Wellington Sîreet 395, nie Wellington OttawaON K 1 A W Ottawa ON K1A 0134 Canada Cariada
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, districbute or sell reproduire, prêter, distri'buer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/£ilm, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantiai extracts from it Ni la thèse ni des extraits substantiels rnay be printed or othemise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

Characterization of the promoter for the humin complement factor 1 (C3bl C4b inactivato r) gene.
Factor 1 is an 88 kDa serine protease which regulates the complernent system by
cleaving the C3b and C4b components of C3 and CS convertases. To map the upstream
regdatory regions and the basai promoter of the factor 1 gene, a series of 5' deletion
hgrnents were fused to a CAT reporter gene and tmmientiy expressed in HepG2 cells.
The regions -13 14 to -1 127 and -71 to -46 upstream of the transcription start site (+1)
possessed a silencer activity, while sequences fiom -869 to -3 1 9 positively regulated
factor 1 transcription. The basal promoter was localized to a 74 bp region (-46 to +26).
This region lacks a TATA box, but contains a TdT-like initiator (Inr) element that
overlies the major cap site (+1). CoIlStructs containing the uir (-12 to +9), Inr and
upstrearn region (-63 to +12), and the downstream TATA-like sequence (+26 to +160)
were inactive in CAT assays. Hence, factor I promoter appears to be a TATA-less Lnr-
dependent class II promoter which requires the sequences upstream and downstream for
expression. Oligonucleotides to the Inr, P2 (-12 to +9) and to the TATA-like sequence,
P3 (+89 to +130) interacted with HepG2 nuclear extract in a mobility shift assay to
generate specific complexes with similar mobilities. These complexes could be cross
competed by unlabeled P2 and P3. An oligonucleotide containing the TATA-like
sequence aione (P4) also inhibited the specific band fomed by P3. P2, P3 and P4 share a
common CTGGA sequence which may be responsible for generating the complex. The
functional role of this pentamer in constitutive expression of factor 1 needs to be
investigated.

As 1 move on to the next stage of my career, 1 would like to thank the people who
have contributed to the completion of this thesis. 1 am very grateful to Dr. Minta, my
supervisor, for giving me an oppominity to work in his lab, and for his patience, guidance
and support in completing this project. 1 would also like to thank the members of my
advisory committee, Dr. D.S.R Sarma, Dr. D. Isenrnan, Dr. J. Squire and Dr. B. Ngan for
their constructive criticisms, help and support. 1 am most indebted to Dr. P. Hamel and
his students for their technical and scientific advice.
A very special thanks to Aroon Yusuf for instilhg confidence, moral support and
most of dl, being there for me in t h e s of need. 1 wili always be grateful! Special thanks
to Soma Mondal, O'Neil Waggon, Rhea Hudson, Kathy Boras, Monty Gill, Aiko Sidle
and Subo Perampalam for seeing me tbrough those difficult moments and making this an
enjoyable expenence.
Last, but not least 1 would like to thank my family for their love, support and
encouragement. Everythmg that 1 am today, 1 owe to my Mom!

Abstract Acknowledgements Table of Contents List of Figures List of Abbreviations
OveMew of the complement system Historical perspectives The classical pathway The lectin pathway The alternative pathway The centrai role of C3 The formation of the MAC Control of the Complement System Control at the C 1 level Control at the MAC formation Pathway Control at the level of Convertases MRP of the Complement Activation Fluid phase regdators Biological significance of factor 1 Deficiency of Complement Factor 1 Transcriptional regulation of eukaryotic genes Transcription of complement genes Rationaie and Objectives
VI
vii
Preparation of pBh1474, pBh1287 and pBhl029CAT Ligation Transformation Miniprep of plasmid DNA Preparation of pBh479, pBh23 1 CAT Preparation of pBh132, pBh74CAT Preparation of pBh206CAT Preparation of pBh2 1 CAT Analysis of constructs by DNA sequencing Large scale preparation of plasmid DNA Transient transfections of HepG2 cells

CAT assay 50 p Galactosidase assay 51 Preparation of nuclear extract fiom HepG2 cells 52 Grneration of oligonucleotides for labeling 53 Labeiing of oligonucleotides 53 Electrophoretic Mobility Shift Assay 54
RESULTS Deletion mutagenesis of BI11474 & expression of CAT geme 56 Andysisofpromoteractivityinthesubh~entsofBh231 59 Analysis of ch-acting sequences by EMSA 61

Complement activation pathways
Control of the activation pathway
Degradation of C3b by factor 1
Degradation of C4b by factor I
Processing of factor 1
Promoter sequence of factor 1
Map of restriction sites used in generating 5' deletion constructs in Bh 1474
Promoter activity of 5' deletion mutants
Sequence of the basal promoter
Promoter activity in the sub fragments of the basal promoter
OligonucIeotides used in EMSA (P 1, P2, P3, P4)
EMSA on Pl, P2
EMSA on E2F, and cornpetition of P2
EMSA cornpetition on P3
Aiignment of P2Q3, and P4 sequences to show similarities
PAGE
3
4
18
24
26
29
40

Arg
AP
BBS
BSA
CHSAM
CP
CS
C4bp
ClINH
'=Pm
CR1
CR2
CR3
CYS
DAF
D m !
m
d m
DTT
EDTA
EGTA
ELISA
- Arginine
Alternative pathway
BES buffered saline
Bovine serum albumin
Chloro form: lsoarnylalcohol(24: 1)
Classicd pathway
Complement system
C4 bindiag protein
Cl inhibitor
Counts per minute
Complement receptor type 1
Complement receptor type 2
Complement receptor type 3
C ysteine
Decay accelerating factor
Dulbecco's modified Eagle's medium
Deoxynucleotide triphosphate
Dideoxynucleotide triphosphate
Dithiothreitol
E thy lenediaminetetraacetic acid
Ethylene glycol-bis-N,N,N',N' tetraacetic acid
Enzyme linked immunosorbant assay
vii

GTE
HANE
IL
Inr
kD
LDL
LPS
MAC
MBP
MASP
MCP
MRP
MW
ONPG
PBS
PMSF
PCR
RACE
SCR
SLE
I x TBE
TE
Glucose-Tris.HC1-EDTA solution
Heredi tary angionemtic oedema
Interleukin
Initiator
Kilodalton
Low density lipoprotein
Lipopolysaccharides
Membrane attack complex
Mannose binding protein
MBP associated serine protease
Membrane cofactor protein
Membrane regdatory protein
Molecular weight
O-nitrophenyl B-D galactopyranoside
Phosphate buffered saline
Phenylmethylsulfonyl fluoride
Polyrnerase Chain Reaction
Rapid amplification of cDNA ends
Short consensus repeat
Systemic lupus erythromatosus
0.089M Tris base, 0.089 bonc acid, 2.5mM EDTA, pH 8.0
1 ûmM Tris-HC1, 1 m M EDTA, pH 8 .O

INTRODUCTION

The complement system (CS) is an important humoral effector mechanism of
inflammation and immune response. The system consists of more than thirty plasma and
membrane bound proteins that interact in a highly specific sequence to generate host
defense against bacterial and Wal infections and aid in the clearance of immune
complexes fiom the circulation (Muiler-Eberhard, 1988; Nelson et al., 1966). The CS
may be initiated by three distinct activation pathways, (namely, the Classical, Lectin and
Alternative pathways) which share a common tenainal sequence (Fig. 1). Activation of
the proteins or components of complement may be effected by limited proteolysis or
protein-protein interactions. The activated products may be endowed with one or more
biological functions, such as lysis of microbial organisms, enhancement of vascular
permeability, contraction of smooth muscle, chemotaxis, opsonization, clearance of
immune complexes, modulation of immune responses and neutralization of vinises
(MulIer-Eberhard., 1988).
The biological importance of the CS is underscored in patients in which inherited
complement deficiency states are found in association with increased susceptibility to
bacterial infections and immune complex disorders. The complement system like many
other efEector systems is a "double - edged mord", since in addition to playing an
important role in the maintenance of health, it rnay also contribute to tissue damage if
complement activation is uncontrolled or inappropnately initiated. The complement
cascade is carefully controlled at different levels by several fiuid phase and membrane
bound regulatory proteins (Fig. 2). In this thesis, the mechanism of transcription of a
central regulator of CS, factor 1, was investigated. Factor I is an 88 kDa serine proteinase

FIG. 1 : Complement activation pathways. The classical pathway (CP) of complement
activation may be initiated by both antibody-dependent and antibody-independent
mechanisms and involves the complement components Cl, C4 and C2. The lectin
pathway invo lves maman binding pro tein (MBP), MBP associated serine protease
(MASP), C4 and C2 and the pathway is activated in an antibody-independent marner.
The alternative pathway (AP) is initiated in the presence of complex polysaccharides and
involves factor D, factor B and C3. The classical, altemative and lectin pathways al1
converge on C3 and follow a cornmon terminal pathway leading to the formation of a
Membrane Attack Complex (MAC) which is responsible for target cell lysis.

Fig 2: Regdation of the complement cascade. The membrane bomd regulatory proteins hclude the
decay accelerating factor @Al?), membrane cofactor protein (MCP), cornpiement receptor 1 ((CR) and
CD59. MCP, DAF and CR1 regdate the formation of C3 and CS convertase enzymes. CD59 reguiates the
assembly of the membrane atîack complex (C5b-9). The fluid phase regdators include the Cl inhi'bitor
(Clinh), C4 binding protein (C4bp), factor H 0, factor 1 (Fi), S protein and clusterin, Cl-INH inbibits
activated Cl; C4bp, factor H and I are involveci in cleavage of C4b and C3b; S protein and clusterin
prevent C5b-8 complex formation. h o w s indicate the stages influenced by each reguiator. Diagram
adapted fiom Morgan-, 1995.

which, in the presence of appropriate cofactorç, cleaves the major activation products of
the third and fourth components of complement and thereby regulate the assembly of the
terminal complement sequence and the generation of complement mediated bioiogical
products.
U HISTORICALEEPsPECmS
Complement was f h t described over a century ago as a heat labile senim factor(s)
required for the bactericidal activity of immune senim (Nuttal, 1888; Buchner, 1889;
Ehrlich, 1899). The nature of the complement factor(s) was not known until improved
techniques of protein purification which became available in the late 1950's were applied
to the isolation and characterization of the constituent components. Such studies showed
that antibody-mediated cornplement activation was composed of eleven distinct plasma
proteins (Muller-Eberhard, 1975; Nelson et al., 1966). By the mid-19603, al1 of the
components of the Classical Pathway of complement (CP) had been described and their
interactions established. The existence of a second activation pathway, the Alternative
Pathway (AP), which is independent of antibodies, was described by Pillemer and
coworkers in 1954 (Pillemer et al, 1954). In 19703, detailed structural, functional and
biosynthetic studies on the various components of the CS as well a s the proteins which
regulate the cascade were conducted. With the advent of molecular techniques, cloning at
the cDNA level of all the complement components, control proteins, and receptors
involved in the pathways became possible. Within the last decade an antibody-
independent route of activation of the CP, via a serum lectin [mannau binding protein

(MBP)], and mannan binding protein associated semm proteases (MASP) has been
described and designated as the Lectin pathway.
M M
The fint component of the cornpiement system, Cl, is a large
heteroligomeric complex consisting of a single molecule of Clq (460 kDa), two
molecules each of Clr (85 kDa) and Cls (85 kDa), all held together noncovalently in a
calcium-dependent complex (C 1 q(C 1r.C 1 s)J (Tschopp et al., 1980). C 1 q is composed of
six copies of each of three distinct polypeptide chahs (A, B and C), which are wound
around each other in a triple helix containhg an extended, collagen-like tail and a large
globular head (Ziccardi et al., 1977). C l r and Cls are serine proteases (Reid and Porter.,
198 1). The activation of the classical pathway (CP) is initiated prirnarily by the binding
of the coUagen heads of the C lq moiety of Cl to the Fc portion of IgG or IgM immune
complexes. Activation of Cl cm also be achieved by its direct interaction with a wide
variety of molecules such as polyanions (bacterial lipopolysaccharides, DNA and RNA),
certain small polysaccharides, viral membranes, C-reactive protein, but the physiological
importance of this type of activation is not clear (Law and Reid., 1995). The binding of
Clq to activators induce confornational changes in Cl q and this leads to autoactivation
of a single Clr molecule, which cleaves and activates the second Clr molecule in the
C lq(C l r.C 1 s), complex (Dodds et al., 1978). The activated C lr subsequently activates
C 1s by proteolytic cleavage at a single site (Colomb et al., 1984). Cls cleaves C4 at a
single site nea. the N-terminus of the a chah to release a 9 kDa anaphylatoxin, C4a, and

a large hgrnent, C4b. This results in the activation of the thioester bond in C4b which
allows the transacylation of the C4b hgment to nucleophilic acceptors via an ester or a .
amide linkage (Harrison et al., 1981). The half-Iife of the reactive thioester bond is only
a few seconds, such that the failure of C4b to bind to a suitable d a c e acceptor results in
the hydrolysis of the thioester bond (Campbell et al., 198 1). Hence, the binding of C4b to
surfaces is a very inefficient process and the binding is also restricted to the immediate
vicinity of the activating C 1 complex.
In the presence of magnesium ion, the next component of the CP, C2, a single-
chah plasma protein, associates with fluid phase as well as membrane bound C4b. The
C2 bond to C4b is cleaved by C 1 s with the release of a of mal1 hgment, C2b, into the
fluid phase. The Iarger hgment, C2a, remains associated with C4b to form the CP C3
convertase (C4b2a) (Polley and Muller-Eberhard., 1967). The CP C3 convertase
catalyzes the cleavage of C3 into C3a and C3b.
L A M
Recently, an antibody-independent mechanism for the activation of C4 and C2
which bypasses the C1 complex has been described (Lu et al., 1990; Reid., 1993). The
components of this pathway include serum maman binding protein (MBP) and two
serum MBP associated serine proteases (MASP-1 and MASP-2) (Fig. 1).
MBP is a high molecular weight senun lectin made up of many copies of a single
32 kDa chah (Reid., 1993; Lu et al., 1990). It possess a carbohydrate binding module
whose role in vivo is to bind mannose and N-acetyl glucosamine residues in bacterial ce11
walls. MBP also contaias a triple helical collagen-like regions that has an overall

structural similarity to Clq. Furthemore, MASP-1 and MASP-2 enzymes are similar in
structure to C 1 r and C 1 s respectively and hence there are sîructural as well as fimctional
similarities between Clq-Cl %-C lr, and MBP-MASP complex. Because of the
similarity between MASP-1 and 2 and Clr and Cls, MBP can also interact with Ch,-
C 1 s, and bring about activation of C 1 s (Lu et al., 1990). Activated MBP-MASP complex
cleaves C4 and C2 to generate C3 convertase. The lectin pathway thus provides a rapid
antibody-independent mechanism for complement activation.
fi V A r n r n A Y
The alternative pathway (AP) of the complement system c m be Uiitiated by
complex lipopolysaccharides activators, (such as bacteria, yeast and other fungi) vimes
and virally infected cells as well as by complexes of IgG, IgA and IgE (Pillemer., 1954;
Pangburn and Muller-Eberhard., 1984; Cooper and Nemerow., 1983). The constituents
of the AP include C3, factor B, factor D and properdin (Fig. 1). Factor B is a single chain
plasma serine protein (93 kDa), that is stnicturally and functionally homologous with C2.
The activation of factor B is accomplished by factor D, a 24 kDa s e ~ e protease present
in an active form in plasma Properdin is a 220 kDa protein composed of three or four
identical noncovalently linked subunits (Minta and Lepow., 1974; Gotze., 1975). The
activation of the AP is initiated by two activated forms of C3, C3b, that &ses from the
activation of CP or C3(H20) that arises h m the hydrolysis of the thioester bond of C3 by
water. C3 can be continuously activated at a slow rate in fluid phase in the foilowing
ways: 1) C3 can be cleaved by senun proteases into C3b. 2) mail nucleophiles or H,O
can gain access and react with the thiolester (Pangburn and Muller-Eberhard., 1980). 3).

C3 can also be subjected to non specific pemirbations that leads to conformational
changes and the exposure and hydrolysis of the thiolester bond (Law., 1983). C3 with a
hydrolysed thiolester without the loss of its C3a hgment is referred to as C3i or
C3m20), and its conformation resembles that of C3b. Fluid phase C3(H20) as well as
C3b bound to foreign surfaces interacts with factor B in the presence of magnesium ions
to form C3(H20)B or C3bB complexes (Pangbum and Muller-Eberhard., 1980). Factor D
cleaves factor B that is associated with C3b or C3(H,O) at a single site releasing a 30 kDa
fkgment, Ba, into the surrounding fluid h m a 60 kDa fkgment Bb, which remains
associated with C3b or C3(H20). The resulting complexes C3(H20)Bb and C3bBb are the
AP C3 convertases (Gotze and Muller-Eberhard., 1971; 1976). These enzymes are quite
labile (half-life of approxirnately 3-4 min) and rapidly undergo spontaneous dissociation.
The half-Iife of the AP C3 convertase enzyme is considerably Uicreased (to 10-30 min) by
the binding of properdin (Fearon and Austen., 1975). To date properdin is the only known
protein which stabilizes the AP C3 convertase in contrast to senun factor H, membrane
bound decay accelerating factor (DM), complement receptor 1 (CRI) which dissociates
the convertases. C3 convertase catalyses the cleavage of C3 to C3a and C3b. The C3b
molecules bind to the activating surfaces via thiolester bonds. Each molecule then
interacts with factor B to form additional C3 convertase and thereby establishing a
positive feedback amplification loop.

Lé TIJECllENTRAL oIw3
C3 is the pivotal protein in the CS since it is at this step that the classical, lectin
and alternative pathways converge. C3 is an abundant glycoprotein in normal s e m
(approximately 1.3 mg/ ml). It is a 185 kDa heterodimer composed of a 1 15 kDa a chah
linked to a 75 kDa P chah by a single disulfide bond (Muller-Eberhard and Nilsson.,
1960). The activation of Cf (Fig. 3) is the most critical event of the complement cascade,
because it permits the interactions of the terminal components to fom the membrane
attack complex (MAC) which is responsible for target ceU lysis and it generates C3a and
C3b which mediate many of the important host-defense fùnctiom of the CS.
The cleavage of C3 between amino acid residues 726 and 727 (Arg-Ser) by either
CP (C4b2a) or AP (C3bBb) C3 convertase leads to the release of a 9 kDa hgment (C3a)
and the activation of an intrarnolecular thioester bond in the large fragment, C3b (Law
and Levine., 1977). The exposed thioester bond of C3b can associate with factor B and P
to fom additional AP convertase and with CP C3 convertase (C4b2a) or to AP C3
convertase (C3bBb) to form CS convertase (C4b2a3b or C3bBb3b) (Medicus et al.,
1976a; Kim et al., 1992). The binding of C3b molecules to the Fd portion of antibodies
can aid in the clearance of immune complexes by the cells of the reticuloendothelial
system. The deposition of C3b on the surfaces of pathogenic microorganisrns results in
their opsonization and enhancernent of phagocytosis. Deposition of C3b on virus
particles also facilitates their neutraikation (Cooper et al., 1988). C3a, is an
anaphylatoxin (Lepow et al., 1970). It can bind to membrane specific C3a receptors on
mast cells, basophils, eosinophils and neutrophils to trigger release andior formation of an

array of inflammatory agents (including histamine, serotonin, platelet activating factor,
prostaglanduis, leukotrienes, lysosomal enzymes and cytokines) that are responsible for
smooth muscle contraction, increased vascular permeability, edema, hypermia, pain and
fever (Hugli., 1975; 86). C3a has been shown to inhibit B ce11 responses via the
production of prostaglandins (Law and Reid., 1995).
tz I3ULEm-oN OF -- C o -
The assembly of the membrane attack complex is initiateci by the cleavage of the
fifth component of complement, C5, by the CP CS convertase (C4b2a3b) or the AP CS
convertase (C3bBb3b) to generate C5a and CSb (Cooper and Muller-Eberhard., 1970).
CS is a heterodimer consisting of a covalently linked a chah (130 ma) and P chah (80
kDa) (Nilsson et al., 1972, Tack et al., 1976). CS convertase cleaves near the amino
terminal of the a chab to release CSa, a 15 kDa peptide with anaphylatoxin and
chemotactic properties. The anaphylatoxin activity of C5a is a hundred and a thousand
fold more potent than that of C3a and C4a respectively. This anaphylatoxin efFects of
C3a, C4a and C5a are regulated by carboxypeptidase N, which cleaves the carboxy
terminal arginine residue to yield the "desarg" product (Hugli., 1 986). C 5 b retains
residual chemotactic activity and the ability to bind to receptors and activate basophils.
This suggests that CS& is a long lived inducer of inflammation (Burgi et al., 1994).
Binding of C5a to specific membrane receptors on neutrophils augments oxidative bunt
activity, arming these cells with reactive oxygen radical for killing phagocytosed bac teria.
C5b transiently expresses binding sites for C6 and C7 (Muller-Eberhard., 1986). The

bindiig of C6 (a 125 kDa single chah protein) to C5b results in the formation of a Cm6
complex which has a capacity to bind to C7. The attachment of C7 causes
conformational changes in the C5b6 complex exposing a hydrophobic membrane binding
site, such that the complex c m now insert into the lipid bilayers of adjacent membranes.
The C5b67 complex hc t ions as an inte@ membrane receptor for C8.
CS is composed of three polypeptide chahs- a (64 kDa), P (64 kDa) and y (22
kDa) ch&. The a and y c h a h are covalently linked whereas the P chah is
noncovalently associated with the a chah (Kolb and Muller-Eberhard., 1976). The P
chab of C8 binds to C7 in C5b67 complex and the resulting complex C5b678 becomes
deeply buried in the membrane and may cause some membrane leakiness (Podack et al.,
1979; Shin et al., 1977) but has limited ability to initiate lysis. The h a l component in
the complement cascade, C9 is a 71 kDa single chab plasma protein. C9 molecule
interacts with the C5b-8 complex via C8a, and unfolds fiorn its globular, hydrophilic
form to an elongated amphipathic form that transverses the membrane and exacerbates
membrane leakiness (Mayer., 1980). Unfolding of C9 also exposes binding sites that
enable additional C9 molecules to bind, unfold and insert into the membranes. With the
recruitment of additional C9 molecules, up to 18 per complex, the transmembrane pore
(MAC) with a hydrophilic interior and hydrophobic exterior is formed (Podack and
Tschopp., 1982; Tschopp et al., 1982). The transrnembrane channel allows influx of
water and ions into the target cells, leading to osmotic lysis.

tB CDNTROL S Y S ~
The activation of the complement systern is tightly controlled at multiple stages of
the pathway by Buid phase a s well as membrane bound proteins. These regulaton
prevent the depletion of complement components, the overproduction of biologically
active fragments and the destruction of host cells (Fig. 2).
1.8.1 C O N T R O O
The macromolecular C 1 complex is regulated by C 1 inhibitor (C 1 INH) present in
plasma and body fluids. Cl INH is a 105 kDa single chain glycoprotein and a member
of the serpin farnily of proteins (Davies et al., 1986). C 1 INH rapidly foms a covalent 1 : 1
complex with both activated C 1 r and C 1 s, resulting in the generation of a (C 1 -INH)-C 1 r-
C 1 s-(C 1 -INH) compIex (Ziccardi and Cooper., 1979). This efficiently removes C 1 r and
Cls nom the activated Cl complex.
1.8.2 ~ ~ W W J d OFTHE F ~ ~ A T T O N OF MAC
The temiinal complement sequence may be controlled in three ways. Firstly the
bindiag of C5b67 complex to ce11 membrane is prevented by the rapid decay of the
membrane binding site in the Cm67 complex in the fluid phase. Secondly, the
attachment of the Cm67 complex to membranes is M e r prevented by the binding of
plasma vitronecth and clusterin to the C5b67 complex. Vitronectin (or S-protein) is a
sticky, acidic senun glycoprotein which binds to C5b67 and prevents its association with
membranes (Dahlback and Podack., 1 985). Clusterin (SP40,40) (83 ma) is ano ther
sticky plasma protein that inhibits C5b67 in a non specific manner (Jeme and Tschopp.,

1992). The most efficient fluid phase inhibitor of C5b67 is Cg. The binding of CS to
fluid phase nascent C5b-7 complex prevents its subsequent insertion into the membranes.
(Nemerow et al., 1979). Finally, there are two membrane proteins (Cg binding protein
and CD 59) present on host cells that inhibit MAC formation. The 65 kDa C8 binding
protein, binds to C8 and intaferes with the subsequent assembly of MAC (Zalman et al.,
1986). CD59 inhibits the incorporation of C9 molecules into the CS-8 complex and
prevents the development of the poly C9 lytic lesions (Davies et al., 1989; Meri et al.,
1990). As a consequence of naturd decay and presence of these inhibitory proteins, the
bulk of C5b-7 formed fails to attach to membranes and decays in the fluid phase as large,
soluble terminal complement complexes (TCC) containing al1 of the terminal
components, S protein and clusterin (Podack., 1977; Bhakdi et al., 1988).
1.8.1 C O ~ O L A T T C O N V E R T A S E S
Regulation at the level of the C3 and C5 convertases of the CP and AP is
mediated by Buid phase proteins (factor 1, factor H, C4bp, properdin) and by membrane
bound proteins (MCP, DAF and CRI). The activity of the convertase enzymes are
controlled in three ways. Firstly, the convertases are labile and the proteolytic subunits
(C2a and Bb) dissociate irreversibly kom C4b and C3b. However, the binding of
properdin to C3b stabilizes the AP C3 and CS convertases and protects the complexes
nom decay-dissociation (Smith et al., 1984). Secondly, the dissociation of the complexes
are accelerated by the binding of fztor H, C4bp, MCP, CR1 and DAF to the C3b or C4b
moiety. Thirdly, once the proteolytic subunits have been dissociated, factor H, C4bp,
MCP, and CR1 can act as cofacton for factor 1 mediated cleavage of bond C3b and C4b.

DAF however does not serve as a cofactor for factor 1 mediated cleavage of C3b or C4b.
These mechanisrns Limit the formation of C3 and CS convertases enzymes and the
activation of the terminal complement sequence.
L E - H B G w O R S OF AC"TWATT0N
The membrane proteins, DAF, MCP and CR1 restrict complement activation on
host cells at the Ievel of C3 and CS convertases. These proteins contain variable numbers
of a short consensus repeat (SCR). each of which is about 60 residues in length and is
characterized by a fkmework of highly conserved residues including four cysteines that
form two intrachain disulfide bonded loops. Factor B, C4bp and CR2 also contain the
SCR motifs. The genes encoding these SCR contalliing proteins are al1 clustered on
chromosome 1 band q23 of man (Klickstein et al., 1985) and are referred to as the
Regulators of Comptement Activation ( K A ) cluster.
DAF is a 70 kDa protein expressed on ail peripheral blood cells, endothelium and
various rnucosal epithelial cells (Medof et al., 1986). DAF is anchored to the outer leaflet
of the membrane bilayer through a glycosyl phosphatidylinositol (GPI) anchor (Davitz et
al., 1986; Medof et al., 1986). The amino terminal extracellular part of DAF consists of
four SCR domains (Nakano et al., 1992) which is followed by a 70 amino acid long
serine-threonine-proline nch (STP-nch) sequence (Lublin et al., 1 98 8). The
postranscnptional processing of DAF results in the removal of a 24 amino acid sequence
nom the carboxy tenninus and the addition of a GPI anchor (Morgan et al., 1991). DAF
binds to accelerate the dissociation C2a and Bb fiom the CP and AP convertases
(Nicholson., 1992). The activity of DAF is abolished by the removal of SCR 2-4 and the

STP-rich region, but not by the rernovai of SCR 1 (Coyne a al., 1992). The DAI? gene is
40 kb in length, contains 1 1 exons (Post et al., 1990) and is located in the RCA cluster on
chromosome lq23. Genetic deficiency of DAF does not cause paroxysmal nochunal
hemoglobinuna as suggested before, but the erythrocytes of such individuals are slightly
more susceptible to lysis than nomals, in vitro. Most patients however develop
gastrointestinal problems, such as, protein-losing enteropathy and Crohn's disease
(Asghar 1 995).
MCP (reviewed by Lublin and Atkinson., 1989) is a 45-70 kDa widely distributed
protein which is expressed b y leukocytes, lymphocytes, epithelial cells and fibrob lasts
(Seya et al., 1988; McNeaniey et al., 1989). MCP displays an unusual pattern of
migration on SDS-PAGE. The protein expressed on nomal cells resolve as two diffuse
bands of approximately 56 and 66 kDa in molecular weight. Two other forms, 76 and 35
kDa are found on certain ce11 types. The amino terminus extracellular portion of MCP is
composed of four SCR and contains the binding site for C3b and C4b (Adams et al.,
199 1). The SCR's are followed by STP (Morgan and Caras 199 l), and a track of twelve
amino acid sequence of unknown significance. The carboxyl terminus is made up of the
trammembrane region, intracytoplasmic anchor and one of the two altematively spliced
cytoplasmic tails. Like DAF, MCP is processed (removal of the 34 amino acid
hydrophobie signal sequence) to foxm the mature protein. MCP can interact with bound
C4b and C3b (Seya et al., 1986) but has greater &ty for C3b. The binding of MCP to
C3b results in the dissociation of AP C3 convertase. MCP also acts as a cofactor in the
cleavage of C4b and C3b by factor 1 (Seya and Akinson., 1989; Seya et al., 1990; Lubiin
and Coyne., 1991). The binding site for C3b in MCP resides in SCR 3 and 4. The MCP

gene is 43 kb in size and contains 14 exons and 13 introns (Bora et al., 1989). Inherited
deficiency of MCP has not yet been reported.
CRI (CD 35) (reviewed by Femn and Aheam., 1989) is not as widely expressed
as is DAF or MCP, but it is present on erythrocytes, monocytes, neutrophils, B
lymphocytes, some T lymphocytes, and podocytes. It is a single chah membrane protein
which exists in four polymorphic foms (160, 190, 220 and 250 ma). CR1 consist of a
signal peptide, an extracellular domain, trammembrane region and a cytoplasmic
domain. Contiguous blocks of seven SCRs form a larger highly homologous repeating
array called a long homologous repeat (LHR). The most common form of CR1 is
composed of 30 SCRs. CR1 has three binding sites for C4b and two binding sites for
C3b which are located in each of the f k t three LHRs. The presence of these distinct
ligand recognition sites suggests that each receptor rnolecule cm interact multivalently
with complexes containing multiple C4b and C3b molecules. CR1 regulates the
complement cascade by acceierating the dissociation of both CP and AP C3 convertases
and by acting as a cofactor in the factor 1 mediated cleavage of C3b into iC3b and
subsequently to C3dg and C4b to C4c and C4d (Fearon., 1977; Pangbum et al., 1977;
Iida and Nussenzweig 1981). The binding site for interaction of CR1 to C3b has been
mapped by deletion and site directed mutagenesis. The deletion of SCR 8-9 and 15-16
resulted in the loss of C3b binding activity and deletion of SCR 1-2 ablated C4 binding
activïty (Krych et al., 1994). CR1 on erythrocytes plays an important role in the
clearance of immune complexes and enhancement of phagocytosis of C3b and C4b
coated particles (Petersen et al., 1985).

HOOC+ $ chain (75 kDa)
O C3a HS CC
1 a-chain ( 1 10 kDa) I I I I s-7 ç --....--....-.-. S
1 B chain 1
l Factor I + cofactors
1 6 chain 1
1 0 chain 1
Fig. 3: Degradation of third component of complement C3. C3 is cleaved by C3 convertase to praduce C3a and C3b. Factor 1 in the presence of cofactors,factor H, MCP, or CR1 cleaves the a' chain of C3b to refease a 3 kDa peptide, C3f and to produce iC3b. iC3b is further degraded by factor I into C3dg and C3c mainly in the presence of C R I . Adapted from Sidle., t 995.

Lu THE A-AmoN
Factor H is a 150 kDa single chain s e m protein consistirtg entirely of 20 SCR
(Ripoche et al., 1988). It interacts with C3b through three different binding sites located
within SCR 1-4, 6-1 0 and 16-20 (Sharma and Pangbum 1996). By binding to C3b, it
dissociates Bb fiom C3b in the AP C3 convertase (Pangbum and Muller-Eberhard., 1978)
and prevents the interaction of B with C3b to form C3 convertase. Factor H also acts as a
cofactor in the cleavage of C3b by factor 1 (Weiler et al., 1976; Pangbum et al., 1977) and
exhibits a weak cofactor activity in the factor 1 mediated cleavage of C4b and C4b2a
(Pangburn., 1986). The rnechanism by which factor H fulfills these functions are
presently unclear. It has been reported that factors B and H compete for their substrate
C3b. Whether the bound C3b has higher afnnity for factor H or factor B depends on the
presence of neutral and anionic polysaccharides on the ce11 surfaces. On human cells,
factor H recognizes polyanions such as sialoglycoproteins and glycosaminoglycans and
binds to C3b with greater affinity. This binding causes conformational changes in C3b
which increases the affinity of factor 1 to C3b (Carreno et al., 1989). The factor H gene is
105 kb in length and is composed of 22 exons. Patients with homozygous factor H
deficiency have undetectable CP and AP activities, and exhibits low levels of C3 and
factor B. C5-C9 levels are also greatly depressed. The cluiical manifestations of factor H
deficiency are glomedonephritis, recment lung infections, hemolytic urernia syndrome,
meningitis and sepsis.
The predominant form of C4bp is composed of seven identical 75 kDa a chain
and a 45 kDa P chah linked together by disuifide bonds near their carboxy tennini

(Chung et al., 1985) and it assembles into a a, f3, cornplex (Hillarp and Dahback., 1988).
The C4bp a chain amino terminus is composed of 549 amino acid which contains eight
SCR and a 54 residue carboxy terminus which lacks SCRs (Chung et al., 1985). SCR 1-3
in the amino terminus are critical for murine C4bp binding (Ogata et al., 1993). The P
chain, composed of 3 SCR (Hillarp and Dahlback., 1990), does not interact with C4b.
The primary role of C4bp is to regulate the CP C3 convertase by accelerating the
dissociation of C2a h m C4b and senring as a cofactor in factor 1 mediated cleavage of
C4b to C4d and C4c (Nagasawa et al., 1980). The gene encoding the a and P chahs are
linked closely in a head to tail anangement in the RCA cluster (Pardo-Manuel et al.,
1990). The a chain is encoded by 12 exons (Rodriguez-de-Cordoba et al., 1991),
whereas the p chah is encoded by five exons (Hillarp et al., 1993). The a and P chah
genes probably arose by gene duplication.

Table 1: Complement gene size, chromosomal locations and deficiencies. Table adapted from Morgan, 1995.
C 1 q (A&B chahs) Clr, Cls
- Factor B
Prop erdin - MBP
22 kb (41 ex) 22/16 kb 20 kb (18 ex)
6 kb (18 ex)
6.5 kb (4 ex)
42 kb (41 ex)
GENE LOCATION
SLE in majority pyogenic infections incl. meningitis
As above; SLE, many are healthy
Neisserial infections; Rarely other pyogenic infections
Pyogenic infections Glomedonephritis, SLE
Nisseriai infections; Rarely SLE
Nisseriai infections

ml!
FI FH
ClINH
C4bpa C4bp B S protein Clusterin
imP
DAF
MCP CRI CD59
63 kb (12 ex) 105 kb (22 ex)
16 kb (8 ex)
40 kb (12 ex) 10 kb(8 ex)
GENE LOCATION DEFICIENCIES
As C3 deficiency Glomdonephritis Hemolytic uremia, meningitis and sepsis
Gastrointestinal pro b lems protein losing enteropathy Crohn's disease
Note: Abbreviations: AP, alternative pathway; CP, classicai pathway; ex, exons HANE, hereditary angioneurotic edema; MRP, membrane regulatory proteins; PNH, paroxysmal nocturnal hemoglobulinuria; PRP, plasma regulatory proteins; SLE, systemic lupus erythromatosus.

1.21 BIQLOGICAL S I m I F m ! w m OF CO- FACTOR 1
Factor I is a central regulator of the complement cascade. It is present in nomal
human plasma in an active form at a concentration of 35 ug/ml. Factor I activity is aiso
present in the sera of lower vertebrates (amphibians, reptiles and fishes) (Kaidoh and
Gigli., 1987). The activity of factor I is modulated by its substrates and cofactors. In the
presence of cofactors, factor H, CRI, MCP, factor 1 cleaves the a' chah of C3b to
generate C3f and iC3b (Fig 3) (Ross et al., 1982; Law et al, 1979, Harrison and
Lachmam, 1980) (Fig 3). The inactivation of C3b prevents the formation of the AP
C3K5 convertases and the positive feedback amplification of the AP. iC3b is further
cleaved by factor 1, mainly in the presence of CR1 to generate C3c and C3dg (Medicus
etal., 1983). Factor 1 has been show to play an important role in preventing spontaneous
activation of the complement via the AP by cleaving C3(H20) in the presence of factor H.
In the presence of cofactors CRI, MCP and C4bp factor 1 is also capable of cleaving C4b
to generate C4c and C4d (Nagasawa et al., 1980) (Fig 4).
The degradation products of C3b (iCfb, C3f, Cfdg, and C3c) mediate several
important biological fûnctions upon binding to specific ce11 surface recepton. The
binding of C3b or iC3b coated particles to CR1 receptors on phagocytes result in
opsonization. iC3b. C3dg and C3d are important ligands for complement receptor 2
(CR2), a 140 kDa glycoproteh expressed on B cells, some T cells and follicular
dendritic cells (Ross and Polly., 1975; Fischer et al., 1991). The interactions of iC3b,
C3dg and C3d with CR2 has been implicated in the regulation of B ceus humoral
immune responses. The binding of the polyvalent C3dK3dg to CR2 has been show to

- - - - -
a' chain 93 kDa
B chain 1
C4a
1 Y chain 1
F I
- Fig 4: Degradation of fourth component of complement C4. Cleavage of C4 by C l s results in generation of C4a and C4b. C4b is cieaved by factor I in the presence of cofactors C4bp. CR1 or MCP into C4c and C4d. Adapted frorn Ebanks., 1995.
dchain 84kDa 1 1 , 1
Factor I and cofactors

enhance the proliferation of preactivated B celis and this effect can be inhibited by
monovalent C3dC3dg (Bohnsack and Cooper., 1988). Polyvalent C3dg is also capable
of priming human B lymphocytes for anti-IgM-induced proliferation. iC3b can bind to
CR3 which is a member of integrin family composed of a 170 kDa a chain and a 95 kDa
p chain. This interaction facilitates the opsonization and phagocytosis of foreign
substances. iC3b is dso a ligand for CR4 which is a member of the family of leukocyte
adhesion receptor molecules expressed on rnyeloid cells and some activated lymphocytes.
The physiological role of CR4 is not clear, but seems to be simiIar to that of CR3. C3f is
an anaphylatoxin which exerts its biological effects by binding to C3a receptors.
Factor I is synthesized in hepatocytes, primary cultures of blood monocytes,
endothelial cells, fibroblasts, synoviocytes, Raji lymphoblastoid cells and transfected
COS4 cells (Goldberger et al., 1984; Julen et al., 1992; Gulati et al., 1994; Wong et al.,
1995). The protein is synthesized as a single polypeptide chah precursor with a linear
organization of NH,-signal peptide-heavy chain-linking peptide-light chah-COOH. The
precursor protein undergoes N-linked glycosylation and two step proteolytic cleavage to
become mature factor 1 (Goldberger et al., 1984). The fust cleavage results in the
removal of the signal peptide which is 18 amino acid in length (Fig 5). The second
cleavage results in the removal of the M e r peptide by paired amino acid cleaving
enzyme (PACE) and this converts the proprotein 1 to mature factor 1 (Wong et al., 1995).
The mature factor 1 is a heterodimer consisting of disulphide linked heavy and light
c h a h with molecular weights of 50 000 and 38 000 respectively. The heavy chah
contains 3 1 8 amino acid residues and is composed of a linear arrangement of three


domains [factor 1 module (FIM), C D 5 and LDLr] that has been found in other proteins.
The amino terminal domain (FIM) is also found as a tandem repeat at the carboxyl
terminus of the sixth and seventh component of complement (C6 and C7) (DiScipio et
al., 1984; DiScipio et al., 1989; Reid and Day., 1989). The middle CD5 domain is
structuraily similar to the CD5/CD6 domain found in membrane proteins, such as, the
differentiation antigens on human (CD5, CD6), mouse (Ly-1) T and specialized B
lymphocytes (3 copies each) (de Bniijn and Fey., 1985; Huang et al., 1987; A d o et al.,
1991; Freeman et al., 1990); as the scavenger receptor cysteine-rich (SRCR) module
found in the macrophage scavenger receptor (1 copy) (Dangott et al., 1989) and in sea
urchin sperm peptide "sperac?" receptor (4 copies) (Yamamoto et al., 1985). The C-
temiinal domain is a tandem repeat of LDL receptor (LDLr) type A domains (Al and
A2). This domain is also found in the LDL receptor (7 tandem copies) (Sudhof et al.,
1985a; Stanley et al., 1985), C6, C7, C8a, C8P and C9 (single copies) (Stanley et al.,
1986; Rao et al., 1987; Howard et al., 1987), in rat kidney membrane glycoprotein GP330
(Raychowdhury et al., 1989) and in human LDL-receptor related protein, LRP/P2
macroglobulin receptor (Herz et al., 1988) as different cluster repeats. Factor 1 is the only
known serine protease with these domains and to date the function of these domains in
factor I are unknown.
The active site of factor 1 resides in the light ch&. This 243 amino acid residue
polypeptide shows sequence homology with the catalytic subunits of other serine
proteases. The catalytic triad in factor 1 is made of histidine (362), aspartate (41 1) and
serine (507) (Davies et al., 1 98 1 ; Catterall et al., 1 987).

The size of the human factor 1 transcript is approximately 2.4 kb (Goldberger et
ai., 1984). The cDNA is 1971 bp in size and contains the sequence coding for the primary
translation product of factor 1 mRNA (Goldberger et al., 1987; Catterail et al.,I987). In
addition, it contains a 14 bp 5' untranslated sequence and a 200 bp 3' untranslated region,
including two polyadenylation signals. Alignrnent of the cDNA derived prirnary
structure of hurnan (Goldberger et al, 1987., Catterall et al 1987). Xenopus
(Kunnath-Muglia et al, 1993)] chicken and mouse factor 1 (Minta et ai, 1996) has
revealed an overd1 conservation of primary structure and domain organization of factor 1
in each species with the exception of the region between LDLR A2 and the linker peptide
(Minta et ai., 1996). This region in the mouse factor 1 cDNA contains 27 bp and 21 bp
sequences which are not present in human factor I cDNA. In Xenopus the inserts are
present as 66 bp and 21 bp respectively. These variable regions in factor 1 have been
designated as the "divergent" or D segments, there being two D segments in human m l ,
H2), four in Xenopus (X 1 -X4), four in mouse (M 1 -M4) and one in chicken.
Human factor I gene spans 63 kb of DNA and comprises of 13 exons and 12
introns (Vyse et al, 1994). The gene has been mapped to chromosome 4q25 (Goldberger
et al., 1987; Shiang et al., 1989). Exon 1 encodes the S'UT and the signal peptide. Exons
2-9 codes for the heavy chain and exons 9-13 codes for the light chain. The serine in the
catalytic triad is encoded by exon 13, histidine by exon 11 and aspartate by exon 12
(Vyse et al, 1994).
Recently, a 4 kb 5' flanking region of human factor 1 gene has been cloned f?om a
human hepatoma genomic library and the 3' 1474 bp (Bh1474) region has been
sequenced (Minta et al., 1998). This sequence contains several putative consensus

AP-1 11-6 RE
l C W K ~ T l R X t X T A ~ T A r r r C A T A A A T P I 3 i A T A TATA box
INV RPR 3b
INV RPT 3a
c r r c A A c c c r r A A r n ~ T T ~ T A 7 - - m ~ m HPR-I b HPR-1 a
T A T ~ T ~ ~ ~ I I A P L P * * X . INV RPT 2b INV RPT 2a AP-1
I S E AP-2
I S E AP-1 HXR-2c - HXR-Pb GAS IFN-GR€ Inr -
CCAAT T E CCAAT T E L + 1 - - - - CTF-NF1
HXR-2a - INV RPT 1 b INV RPT l a
~mcAGmwum---
inv NF-kB HXR-1 b
TCE
Hind III I
Fig 6:Putative consensus sequences in Bh1474. The major cap site is designated as +1 and the minor cap site at -27 and -45 are denoted by: 1

CCAAT box CCAAT box CF-NF1
AP- 1 ISE
CCAAT box CTF-NF1
CCAAT box
TATA box

sequences (including CAAT boxes, GC rich sequences, AP 1 sites, IL-1 response
element, I L 6 response element and an AP-2 site (Fig 6). Primer extension analysis has
revealed a major cap site at the position indicated as +1 in Fig 6 (152 bp upstream of
ATG) and two minor cap sites at -27 bp and -45 bp h m the major cap site (Minta et al.,
1998). Vyse et al (1994) has also reported a 426 bp 5' flankg sequence of the factor 1
gene. By RACE-polymerase chah reaction, they have detexmined the cap site to be
located 28 bp upstream of the ATG start codon. Bh1474 cloned upstrearn of pCAT
enhancer reporter plasmid has been shown to direct the expression of the CAT reporter
gene in HepG2 cells (Minta et al., 1998).
2.12 DEFICIENCY OF C O M P J , E . N T FACTOR 1
Inherited deficiency of factor 1 has been reported in 23 individuals kom 19
different pedigrees and shown to follow an autosomal recessive pattern of inheritance
(Ross and Densen., 1984; Rasmussen., 1988; Bo- et al., 1993; Vyse et al., 1994). The
deficiency is associated with uncontrolled activation of the amplification loop of the
alternative pathway leading to consurnption of C3, and factor B. The concentration of C3
and factor B in factor 1 deficient patients are 13-34% and 4-9% of normal respectively.
Factor 1-deficient s e m is incapable of solubilizing prefomed immune complexes and
exhibits reduced opsonic and chemotactic activities (Rasmussen., 1 988). The clinical
manifestations of factor 1 deficiency are similar to C3 deficiency. Factor 1 affected
individuals have propensity to suffer recurrent pyogenic infections by N. meninetides
(Bonnin et al., 1993) and S. pnewnonia (Abramson et al., 1971; Teisener et al ., 1984)

iocreased incidence of glomemlonephritis and systemic lupus erythromatosus-like illness
(West., 1989) due to impaired complement mediateci fiuictions.
The molecular basis of hereditary complement factor 1 deficiency is at present not
completely understood. Vyse et al (1995) has recently studied the molecuiar basis of
factor 1 deficiency. In one pedigree, two siblings were found to be homoygous for the
same tramversions (adenine to thymine) at nucleotide 1282 in the cDNA. This mutation
caused histidine 400, a semi-conserved residue in the serine protease family, to be
replaced by leucine. This alteration prevented the secretion of factor 1. In the second
pedigree, one allele was found to cary the same mutation as seen in the fint pedigree, the
second allele had a donor splice site mutation that caused a deletion of mRNA encoded
by the fifth exon. This deletion resulted in the premature termination of the factor 1
mscript.
1-13 TWSCBlPTlON OF EUKARYOTIC GWES
Transcription of protein encoding genes is a finely tuned and highly controlled
process. In eukaryotes, initiation of messenger RNA synthesis by RNA polymerase II
(Pol II) is governed by two classes of DNA sequence elements: the core promoter which
contains the binding site for Pol II and controls the location of the site of transcription
initiation and upstream and downstream elements which regulate the rate at which Pol II
initiates new rounds of transcription fiom the core promoter. These DNA sequences
direct the action of two classes of transcription factors: the general transcription factors,
GTFs, (TFIIA, TFIIB, TRID, TFIE, TFDF, TFW) and DNA binding transcription
activators (such as the glutamine-rich activator Spl, and the proline-rich activator

CTF/NFl) which are not essential for basal transcription but rnediate the action of
upstream promoter elements. The binding of speci fic activators to promoter elements
stimulate the recruitment of basal transcription factors in a stepwise manner to form the
preinitiation complex. According to Choy and Green, the activators function at two
stages. First, to recruit the general transcription factor TFIIB, and at a later stage after the
entry of TFIIB (Choy and Green 1993). A different model of transcriptional activation
has been proposed based on the existence of a pre-assembled Pol II transcription complex
(holoenzyme) in vivo which is composed of Pol II and a number of accessory factors
(Koleske and Young., 1994). According to this model, the transactivators interact with
different surfaces of the holoenzyme complex and brings the Pol II transcription
machinery to the template in one-step. By influencing and stabilizing the formation of
the activated initiation complex, the activators enhance the rate at which each round of
transcription is initiated (Emili and Ingles., 1995).
The core promoter of most mammalian genes transcribed by Pol II contains a
TATA box at -25-30 bp upstream of the transcription initiation site which can
independently speciS the location and the direction of transcription initiation
(Breathnach and Chambon., 1981). The TATA box is specifically recognized by TATA-
binding protein (TBP) of TFID which initiates the formation of the preinitaition
complex. TFIID is a multisubunit complex (reviewed by Burley and Roeder., 1996)
composed of TBP and seven tightly associated factors refmed to as TBP-associated
factors (TAFs) (Dynlacht et al., 1991; Pugh and Tijian., 1991 ; Tanese et al., 199 1). TBP
is one of the most highly conserved proteins in eukaryotic evolution (reviewed by
Hernandez., 1993). The binding of TBP to the minor groove of the TATA element

causes striking distortion in DNA which d o w s the stepwise assembly of other GTFs to
fom the preinitiation cornplex. TAFs are responsible for relaying signais fiom activators
to the basal rnachinery.
A series of experiments perfomed by Smale and Baltimore in 1989 led to the
identification of a second core promoter element, the initiator. This element was uiitially
identified in the TATA-less terminal deoxynucleotidyl transferase (TdT) promoter.
Initiators (Inr) are weakly conserved elements that usually encompass the transcription
start site. They are functionally analogous to the TATA box in that they can detemnine
the location of the transcription start site, direct basal transcription by Pol II and
communicate with at least some upstream activators to initiate high levels of accurate
transcription (Smale., 1994). Inr elements have been classified into different groups
based on the factors they bind and their sequence similarity to the ones that are well
characterized, such as the TdT Inr, adeno-associated virus p5-lnr and the ribosornai
protein Inr (reviewed by Weis and Reinberg., 1992).
The transcription factors which interact with the Inr have not been cteady defined.
Two proteins have been proposed: Pol II itself and TAFs of TFIID. TAF,lSO and
TAF,,250 have been shown to participate in the recognition of the Inr element and
downstream core elements (Verrijzer et al., 1994; 1995). Thus, the core eiements, the
TATA box and the Inr element seem to be recognized by TFD[D. In some genes however,
the Inr element is also recognized by sequence specific DNA bliding proteins, such a s
TFII-1, E2F, and ml, but these proteins do not appear to be respomible for basai uir
activity (KoIimar and Famham., 1 993).Although a large number of genes contain either a
TATA box or an Inr element, some genes have been reporteci which contain both

elernents, such as, the adenovins major late ptomoter. Some of the TATA-less genes
however contain A+T rich sequences that, although differing h m the original TATAAA
consensus, still appear to function as a TATA box. Several genes have also been reported
to lack both a TATA box and an Inr element, but yet retains the ability to direct Pol II to
initiate transcription f?om a single start site.
1,14 W S C R I P T I O N C ~ M E I J m E W G E N S
To date the promoters of only a few complement gens have been characterized.
The mouse C4 gene promoter has been shown to belong to an expanding group of RNA
polymerase II promoters that lacks a canonical TATA box at the -30 region. It contains a
typical Inr element at position -1 to +12 and an additional downstream element (three
direct AGAC repeats) which appears to be important for transcription. An upstream E-
box (-73 relative to transcription start site +1) has been reported to enhance transcription
(Miyagoe et al., 1994).
The prornoter of human C2 gene has ody been partially characterized. The
activity was shown to be contained in a 228 nucleotide sequence which lacked a TATA
box (Sullivan et al., 1994).
The promoter of the mouse C3 gene has many structurai similarities to that of the
human C3 promoter. Both promoters contain a TATA box located at position -30 bp that
is essential for its constitutive expression (Vik et al., 1991).
Deletion analysis of the 5' flanking region of the human factor B gene indicated
the importance of sequences located between 178 and 260 bp upstream of the

transcriptional initiation site. A typical TATA box was located at position -22 relative to
the transcriptionai start site (Wu et al., 1987).
The gene encoding the human maman binding protein contains four exons
spanning 6.5 kb of DNA. Putative TATA and CCAAT boxes are located at -41 and -82
bp upstream of the transcriptional start site (Sastry et al.. 1989).
The promoters of the genes encoduig the regulatory complement proteins have not
been fully characterized. Arnong the ones studied, the Cl-INH gene promoter lacks a
typical TATA box, but contains a potential Inr element at position -3 to +5 (Zahedi.,
1994). Whereas the human C4BPa gene promoter appears to be TATA-less, the murine
gene possess a putative TATA box at position -26. Both promoters have sequences
matching the consensus motif for liver enriched transcription factor, HNF-1 (Courtois et
al., 1988). The promoter of the human factor H gene also appears to lack canonical
TATA and CCAAT boxes, but contains an Inr element which is simila. to the CR1 uir (6
out of 8 match) (William and Vik., 1997). The 5' flanking region of the human DAF
gene lacks both a TATA box and a CCAAT box at their typical locations. However, a
TATA variant-TTAA is located at position -31. This sequence occurs in a sirnilar
position in the adenosine deaminase gene promoter, where it is speculated to act as a
TATA box variant (Thomas and Lublin., 1993). The MCP promoter also lacks a TATA
box, but has several putative ch-acting regulatory elements (Cui et al., 1993).

To date, the basai pmmoter of the factor I gene and the cis-acting elements and
cognate DNA binding proteins which regulate the constitutive and inducible expression
of the gene have not been fùlly characterized. Mutations in the regulatory regions of
genes have been shown to represent an important class of lesions causing human genetic
diseases. Such mutations have been associated with either an increase or decrease in the
transcriptionai activity of the affected gene mediated by the aitered binding of transacting
factors to specific DNA sequences in the promoter region. In this study, the technique of
deletion mutagenesis was used to map the basal promoter sequences required for the
constitutive expression of the factor 1 gene. Electrophoretic Mobility Shift Assay was
then used to localize cis-acting sequences which interact with HepG2 nuclear factors to
regulate the transcription of the factor 1 gene.

MATERIAL AND METHODS

PREPARATION OF pBh1474CAT, pBh1287CAT, pBhl029CAT
CONSTRUCTS BY RESTRICTION ENZYMES
The 5' 1474 bp flanking region of the human factor 1 gene (Bh1474) was obtained
by digesting 20 pg of the pBh1474 CAT enhancer constxuct with 40 U of HindlD
ovemight at 3 7 ' ~ . pBh1474CAT enhancer was kindly provided by Ms. May Fung in our
laboratory. 5' tnincated fragments of Bh1474 were prepared by deletion mutagenesis
using restriction endonucleases (Fig 7). A 1287 bp fï-agment was generated by cleaving
the pBh 1474CAT enhancer reporter plamid with Banll and HindlII. A A 029 bp fragment
was obtained by restricting the pBh1474CAT enhancer construct with BgM and HindII
(Fig 7) . The digested products were electrophoresed on a 0.8% low melt agarose (Sigma)
in 1xTA.E dong with 1 kb DNA markers (Gibco BRL) for two hours at 65 volts. The gel
strips containing the desired DNA bands (1474, 1287, and 1029 bp) were excised, melted
and passed through NACS columns (Pharmacia) that have been equilibrated with 1 ml of
2 M and 5M NaCl solutions. The columns were washed with 10 ml of 0.5 M NaCl
solution and the bound DNA was eluted with 0.4 mi of 2 M NaCl solution. The eluted
hgments were precipitated oveniight with two volumes of 100% ethanol. The DNA was
pelleted by centrifugation at 15,000 rpm for 15 min at 4OC, washed with 70% ethanol, air
dried and dissolved in 10 pl of Tris-EDTA buffer (TE), pH 8.0 (10 rnM Tris-HC1, pH
8.0, 1 mM EDTA). The concentration of the DNA was estirnated by agarose gel
electrophoresis in the presence of ethidium bromide by comparing the fluorescence of the
DNA with that of a known standard DNA (Sambrook et al., 1989).

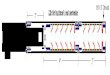
Fig 7: Map of restriction sites in pBh 1474 that were used in generating 5' deletion construcl.

To facilitate subcloning, the 3' overhang of 1287 bp hgment was rernoved by
incubating 2.5 yg of DNA with 3 pl of 10x T4 polymemse b a e r , 3 pl of 0.5 mM dNTP
mixture (dCTP, dATP, dGTP, and dTTP) and 2 U of T4 polymerase (New-England Bio
Labs) in a total volume of 30 pl at 370C for 15 min as described by Davies et al. (1 986).
The reaction mixture was extracted with phenol: chisam (chlorofonn: isoamy ldcohol,
24: 1 1 : 1 The DNA was precipitated ovemight with one-tenth volume of 3 M sodium
acetate (pH 5.2) and two volumes of 100% etbanol. The DNA was pelleted, washed, air
dried and dissolved in TE to a final concentration of 0.25 &pl.
The 5' protmding ends of 1287 bp and 1029 bp hgments were blunted by a fil1
in reaction with Klenow fi-agment DNA polymerase 1 (Gibco BRL). Approximately, 2.5
yg of each hgrnent was incubated with 3 pl of 10 x Klenow buffer, 3 U of Klenow, 3 pl
of 0.5 mM dNTP mixture and 3 pl of 10 m M DTT in a total reaction volume of 30 pl at
37'C for 15 min as described by Sambrook et al. (1989). The enzyme was heat
inactivated and the mixture was extracted with phenol: chisam. DNA was precipitated
with ethanol, washed, air dried and dissolved in 10 pl of TE.
2.1.1 LIGATION: 10 pg of pCAT basic reporter plasmid (Promega) was linearized
with 20 U of HindIII to allow subcloning of the 1474 bp hgment. To facilitate the
subcloning of the other two hgrnents, 1287 bp and 1029 bp, pCAT was linearized with
Sul1 and the 5' protruding ends were blunted as described above. Two micrograms of
1474 bp insert was incubated with 0.5 pg of pCAT basic vector linearized with H . d m
in a 20 pl ligation mixture containing l x ligation buffer, 2.5 U of T4 ligase. The 1287 bp

and 1029 bp inserts were incubated with 0.5 pg of pCAT basic vector Iinearized with
Sa11 in the same reaction buffer. Ligation was carried out for 16 hrs at 14OC.
TRANSFORMATION: Ten microliters of ligation mixture was incubated
with 100 pl of competent HB 101 cells on ice for 30 min. Then 0.9 rnls of Lauria-Bertani
broth (LB) (without ampicillin) was added to each mixture and the cells were grown at
37°C on a rotating dnun for one hour. Aliquots of the reaction mixture were plated on
1.5% agar plates containing 100 pg/ml ampicillin and incubated overnight at 37%
2.1.3 SMALL SCALE PREPARATION OF PLASMID DNA FROM E B l O l
CELLS: Muiipreparation of plasmid DNA was performed by the aikali lysis
method (Sambrook et ai., 1989). Essentially, ampicillin resistant colonies of transformed
HI3 101 cells were picked and innoculated into a 3 ml LB broth containhg 100 pg/ml
ampicillin and grown overnight at 37°C. 1.5 ml of the bacterial culture was spun briefly
and the pelleted cells were resuspended in 100 ul of GTE solution (50 mM glucose, 25
mM Tns.HCL (pH &O), 10 rnM EDTA (pH 8.0) and incubated for 5 min at room
temperature. The cells were lysed with 200 pl of aikaline lysis solution (0.2 N NaOH,
1% SDS) at room temperature for 5 min. Fhally, 150 pl of 3.5 M potassium acetate, pH
7.5 was added to the mixture and incubated on ice for 5 min to aUow plasmid DNA to
reanneal. The mixture was centrifugecl at 15,000 rpm for 5 min at 4OC . The supernatant
was extracted with phenol:chisam, and DNA was precipitated with two volumes of
ethanol. The DNA was recovered by centrifugation and the pellet was washed with 70%

of ethanol, air dried and resuspended in 30 pl of TE containing DNase fiee pancreatic
RNase (20 pg/ml). Al1 three constmcts were analyzed for the presence of insert by
restricting with HindIlI and the orientations were determined by restricting with A f l .
and HindlU.
PREPARATION OF pBh479CAT AND pBh231CAT CONSTRUCTS
pBh479CAT constnict was generated by cleaving 2 pg of purified pBh1287
CAT with 8 U of Nsil and Pstl. This released an 808 bp hgment. The vector still
containing a 479 bp 3' end of Bhl287 was eluted fiom agarose gel, the ends were blunted
and then religated to generate pBh479CAT.
pBh231CAT constnict was also derived fiom Bh1287CAT by cleaving with
BstEII and Pst l . This released a 5' 1 O56 bp fiagrnent and the vector containing the 23 1
bp 3'end of Bhl287 was isolated and religated by blunt-end ligation.
a PREPARATION OF pBhl32, pBh74CAT CONSTRUCTS BY PCR
AMPLIFICATION
A 132 bp hgment (+26 to +160) and a 74 bp hgment (-63 to +IO) were
amplified by PCR and subcloned into pCAT basic reporter plasmid to create pBhl32CAT
and pBh74 CAT constructs respectively. PCR reactions were camied out in 500 pl
microfbge tubes and contained 1 pg of template @Bh1474CAT), 0.1 FM of the sense
primer (SP), 0.1 p.M of the antisense primer (ASP) in a total reaction volume of 50 pl
containing 10 mM Tris-HC1 (pH 8.0), 50 mM KC 1, 1.5 mM MgCl, 20 pM of each d N ï P

(dATP, dTTP, dGTP, and dCTP) and 2 U of Taq polymerase (Perkin Elmer Cetus). The
primers were tagged with restriction sites (underliued) to facilitate subclonhg. The
p h e r s used for PCR amplification of the 132 bp fragment were:
SP: 5' GGA TCCAAGCTlLTCTGCAGCCAAGCTCTTTAGGAGG 3'
ASP: 5'
BamRI HittdlZI Psti
GATGAGTCGACAAGCTTCATGTTGGAGGTGTTCG 3 '
Saf I HindïU
The primers used for PCR amplification of the 74 bp hgment were :
SP: 5' GAGAAGCTTCTGCAGATGCATCCCTCAGCTCT?TAATGG 3'
ASP: 5' GTCGACTCTAGAGAAAGAATTTGGCTGAAACCT 3'
SafI XbaI
The reactions were layered with 50 pl of mineral oil and PCR amplification was
carried out in a Perkin Elmer Cetus Themai Cycler through 35 cycles of denaturation (2
min at 94OC), a~eal ing (2 min at So°C) and polymerization (3 min 72OC). The mixture
was electrophoresed on an agarose gel and the amplified hgment was gel purified,
digested with the appropriate restriction enzyme and subcloned into pCAT basic vector.
The constructs were transfomed into HBlOl cells and colonies containing the
recombinant plasmids with the inserts in the nght orientation were assessed by
sequencing from the 3' end using an antisense primer complementary to 1297-2362 5'
end of the CAT gene (5' CCATT'ïTAGCTTCCTTAGCTC 3').

PREPARATION OF pBh206CAT CONSTRUCT
A pBh206CAT construct containhg nucleotide -46 to +160 of the factor I
promoter and lacking the Spl/AP-2 Iike sites was generated by 3 3 ' exonuclease III
digestion of linearized pBh231CAT. Forty micrograms of pBh23 1CAT construct was
digested with 80 U of BstEU and 80 U of Sphl to generate a hearized vector with a 5'
and a 3' overhang. The 3' recessed end of the vector generated by BstEII was then
digested at a unifonn rate by exonuclease III whereas the 3' overhang was protected from
exonuclease III digestion. The BstELI/Sphl digested vector was gel purified and the DNA
was dissolved in stede water to give a concentration of 0.1 pg /pl. Forty microliters of
the linearized plasmid DNA was incubated with 160 U of exonuclease III (Pharmacia).
Five microliters aliquots were removed every 20 sec and mixed with 15 pl of S1 nuclease
mixture [IO p1 of 2x SI nuclease buffer, 2.5 51 of S1 nuclease (Boehringer Mannheim)
and 25 pl of water] and M e r incubated for 30 min at room temperature. The S1
nuclease action was stopped with S1 nuclease stop buffer [0.3 M Tris base and 0.05 M
EDTA (pH £?.O)] and the samples were heated to 70°C for 10 min. To facilitate iigation,
the ends were flushed by the addition of 1 pl of Klenow mixture [20 rnM Tris-HCI (pH
&O), 100 mM MgCl,] and incubation at 37OC for 5 min. In the absence of (INTP's, the
3 '-5 ' exonuclease activity of Klenow predominates. One rnicroliter of 0.5 mM cNTP mù<
was then added and the mixture was M e r incubated for 5 min at 3 7 C for the Klenow
polymerase activity to predominate. DNA was religated by the addition of 6 U of T4
ligase and Ix T4 ligase bufTer.

PREPARATION OF pBh21CAT CONSTRUCT
Twenty one base pair sense and antisense oligonucleotides (-12 to +9), containing the
major cap site, TdT-üke Inr and CTF/NFl elements w e ~ e synthesized (as s h o w beiow)
with restriction site add-ons to facilitate unidirectional cloning into pCAT basic vector.
SP: 21 bp sense primer:
HindDI Pstl Nsil mal S U
21 bp anti sense primer:
5' GTCGAmCTAGACAAAmGGCTGAAATCCATGCATCTGCAGAAGmGG 3' - Nsil Psti HindLII
Complementary sense and antisense oligonucleotides (25 pg in 50 pl of 150 mM NaCI)
were mixed and heated to 80°C for 5 min and allowed to anneal by slowly coohg to
room temperature. The DNA was precipitated overnight in two volumes of ethanol and
recovered by centrifbgation. The DNA was digested with Pst1 and f i l , extracted with
pheno1:chisa.m and precipitated with ethanol to remove the liberated oligonucleotides.
The double stranded 21 bp oligonucleotide was then subcloned into pCAT basic vector
that had been cut with Pst1 and fial.
ANALYSIS OF CONSTRUCT BY DNA SEQUENCING
Bh206, Bh132. Bh74 and Bh21 CAT constnicts were sequenced nom the 3' end
to verifjr their identity and orientation. DNA sequencing was performed by the dideoxy
method (Sanger et al.. 1977) using a DNA sequencing kit (USB corporation, Cleveland,

Ohio). An antisense primer (5' CCATM'TAGCTTCCTTAGCTC 3') complementary to
1297-2362 5' end of the CAT gene of the pCAT basic vector was used to prime the
reaction. Three to five micrograms of DNA to be sequenced was denatured with 2 N
NaOH, the primer was then annealed to the ternplate. Using sequenase enzyme, the
DNA was labeled for 5 min with 10 pCi of [a3%] dATP (>IO00 uCi/mmol, Amersham)
and d N T P ' s ( d m , dCTP and dGTP). The labeling reaction was terminated with the
addition of dideoxy nucleotide triphosphates ( d m ) and the mixture was
electrophoresed on a 24 cm x 40 cm 8% polyacrylamide gel containing 7 M urea in
lxTBE (10.8 g Tris base, 5.5 g bonc acid, 4 ml 0.5 M EDTA, pH 8.0) for 2-4 hours. The
gel was fixed in 10% acetic acid/10% methanol, dned and subjected to autoradiography.
LARGE SCALE PREPARATION OF PLASMID DNA
Maxiprep of DNA was carried out according to Sambrook et al. (1989)
Transfonned HBlOl cells containing recombinant pCAT basic reporter vector with the
promoter inserted in the nght orientation was grown ovemight at 37°C with agitation in
one liter LB broth containing 100 pg/ml ampicillin. The cells were pelleted by
centrifugation at 5,000 rpm for 10 min at 4OC and resuspended in cold GTE b a e r (8
dSOO ml culture) and incubated on ice for 10 min. 10 ml of keshly made alkaline lysis
buffer was added and the mixture was M e r incubated on ice for 10 min. Then 9.5 ml
of cold 3M potassium acetate, pH 7.5 was added and the mixture was incubated on ice for
another 10 min. The cellular debns and bacterial DNA were removed by cemtrifiging the
mixture at 15,000 rpm for 25 min at 4OC in an SS 34 rotor. The supernatant was fiItered

through cheese cloth and DNA was precipitated with 0.6 volume of isopropanol at room
temperature for 30 min. DNA was recovered by cenmgation at 10,000 rpm for 25 min
at room temperature, washed with 70% ethanol, air dned and dissolved in 4 ml of TE per
500 ml of original culture.
Cesium chloride (4.95 g/500 ml of original culture) and 450 p1 of ethidum
bromide (10 mg/& stock solution) were added to the DNA solution in TE, dissolved, and
the mixture was then centrifbged at 3,000 rpm for 30 min at room temperature. The
supernatant was tramferreci into 5 ml quick seal polyallomer VTi 80 tubes (Beckman)
and centrifbged overnight at 50,000 rpm in a Beckman VTi 65 rotor at room temperature.
The plasmid DNA band fiom each tube was removed with a hypodennic needle and
placed into a 50 ml centrifuge tube, diluted with 3 volumes of TE and extracted 3-4 tirnes
with equal volumes of n-butanol (until the top butanol layer was clear) to remove the
ethidium bromide. The aqueous phase containhg the plasmid DNA was then precipitated
with two volumes of ethanol. The DNA was recovered by centrifugation at 3,000 rpm
for 30 min at 4'C, air dned and dissolved in 500 pl of TE buffér. The solution was
extracted twice with phenol: chiasm, once with chisam and the plasmid DNA was
precipitated with 3M sodium acetate, pH 5.2 (one-tenth the volume) and two volumes of
ethanol. DNA was recovered by cenhfugation at 15,000 rpm for 10 minutes at 4OC. The
pellet was washed with 80% ethanol, air dried and dissolved in TE buffer. The purity and
the concentration of the DNA was determined by spectrophotometry. Ten microliters of
the purified plasmid DNA was diluted in 990 pl water and the O D at 260 nm and 280 nm

were measured (1 O.D. 260 = 50 pg/ml). The 260 nrn to 280 nm ratio was used to
estimate the purity of the DNA samples.
TRANSIENT TRANSF'EXTIONS OF HEPG2 CELLS
pC AT p lasrnids containhg B h sequences were transient1 y tram fected into HepG2
cells using the calcium phosphate coprecipitation technique (Sambrook et ai., 1989).
HepG2 cells were grown in Dulbecco's modified Eagie's medium (DMEM),
supplemented with 10-20% fetal calf serum and 500 U penicillin, in a humidified
incubator at 37°C in the presence of 95% air/5% CO, Twenty-four hr pnor to
transfection, HepG2 cells were seeded in 60 mm plates at a density of 3 x 106 to obtain a
50-70% confluent plate. Fifieen rnicrograms of cesium purified plasmid DNA was
mixed with 4 pg of pRSV J.3 galactosidase (Promega) and 50 pl of 2.5 M calcium chloride
in a total volume of 500 pl. Five hundred microliters of 2x BBS (N,N-bis[2-
hydroxy ethyl]-2 aminoethanesu1 fonic acid (B ES) buffered saline), pH 7.5 was p laced in
15 ml round bottom stenle tubes, and bubbled gently with a pipette while adding the
DNA-calcium chloride mixture. The tubes were then vortexed and allowed to incubate at
room temperature for 20 min for microprecipitates to form. The adherent HepG2 cells
were fed with 2 ml of nesh media The DNA precipitate was gently distributed over the
ceUs and the cultures were incubated at 3?C, in the presence of 3% CO,. After 5 hr
incubation, the ceils were shocked with 10% glycerol in medium for 3 min, washed three
times with phosphate buffered saline (PBS) (8 g NaCI, 0.2 g KCI, 1.4 g NaHPO,, and 0.2
g KH2P04 per Mer) and then cultured for 48 hrs in a 37OC incubator in the presence of

5% CO,. The cells were scraped with a rubber policeman into 1 ml TEN buffer (40 mM
Tris-Cl, pH 7.4, 1 m M EDTA, pH 8.0, and 150 mM NaCl) and microfigeci at 12,000 rpm
for 30 sec at 4OC. The ceii pellets were resuspended in 100 ul of 0.25 M Tris-HCl (pH
8.0) and subjected to three rounds of rapid fieeze and thaw cycles to lyse the cells. The
tubes were microfbged at 12,000 rpm at 4OC for two min to pellet cellular debns. niuty
microliters of the supernatant was saved for B galactosidase assay, and the remaining 60
pl of the ce11 extract was heated for 5 minutes at 60°C to degrade endogenous
deacetylases and then used for the CAT assay.
CATASSAY
CAT assays were performed according to Gorman et al., 1982. Briefly, 60 pl of
the ce11 extracts fiom transfected HepG2 cells were incubated at 370C for 3 hr with 3 pl of
"C chloramphenicol, 5 pl of n-butyryl CoA and 0.25 M Tris-HCI (pH 8.0) in a total
volume of 125 pl. The reaction mixture was extracteci with 500 pl of ethyl acetate and
vacuum dried. The material was resuspended in 20 pl of ethyl acetate and spotted on a
20 x 20 cm Baker-flex silica Gel 1B2 coated sheet for thin layer chromatography (TLC)
in a chamber containing chloroform:methanoi (97:3). The plates were air dried and
subjected to autoradiography ovemight at -80°C with intensifjmg screens. The
autoradiogram spots corresponding to the butrylated and unbutrylated chloramphenicol
products were then scraped and counted. In certain experiments, the butrylation of
chloramphenicol was quantified by extracting the reaction mixture with 300 pl of mixed
xylenes followed by scintillation counting. The n-butyryl chloramphenicol partitions into

the xylene phase, while the unmodified chloramphenicol remains predominantly in the
aqueous phase. The top xylene phase was tramferrecl into a fksh tube and back extracted
by the addition of 100 pl of kesh 0.25 M Tris-HCI (pH 8.0). After separating the two
layers by centrifugation, 200 pl of the top xylene layer was added to 5 ml of scintillation
cocktail fluid and counted in a liquid scintillation counter. The counts per minute (cpm)
obtained is a measure of the butyrylated chloramphenicol products.
2Ja P GALACTOSIDASE ASSAY
B gal activity in ce11 lysatu of traDsfected HepG2 cells was assayed according to
Sambrook et al. (1989) and used to correct for transfection efficiency. The assay is based
on the hydrolysis of the glycosidic bond of O-nitrophenyl P-D galactopyranoside
(ONPG) to release P galactose and O-nitrophmol (Kennedy., 1967). The O-nitrophenol
is responsible for the appearance of the fauit yellow color in the reaction mixture.
Essentially, 30 pl of ce11 extract was mixed with 3 pl of lOOx magnesium solution (0.1 M
MgCl,, 4.5 M P rnetacaptoethanol) and 66 pl of a solution of ONPG (Sigma) (4 mg/ml
of 0.1 M sodium phosphate pH 7.5) in a microtitre plate well. The mixture was incubated
at 370C for 45-60 min or until a faint yellow color developed and reaction was stopped
with the addition of 100 pl of 1 M sodium carbonate. The optical density was measured
at 405 nrn using an ELISA plate reader (Molecular Devices). Cell extracts from mock
transfection or buffer was used to blank the ELISA plate reader.

P R E P M O N OF NUCLEAR EXTRACT FROM HEP G2 CELLS
Nuclear extract was prepared fkom HepG2 cens according to Schreiber et al.,
(1989). A 100 mm confluent culture of HepG2 cells was ûypsinized and the cells were
transferred into a microfùge tube and sedimented. The ce11 pellet was washed twice with
PBS, resuspended in 400 pl of hypotonie buffer A (10 mM Hepes (pH 7.9). 10 mM KCI,
0.1 mM EDTA, 0.1 rnM EGTA, 1 m M DTT and 1 mM PMSF) and incubated on ice for
15 min. Twenty five microliters of cold 10% NP-40 was added to the cells and the cells
were vortexed for 10 sec to break the ce11 membrane* The nuclei were sedimented at 4OC
for 30 sec at 10,000 rpm, resuspended in 50 pl of ice cold buffer C (20 m M Hepes (pH
7.9), 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and 1 rnM PMSF) and
rocked for 15 min at 4'C. The tube was centrifuged at 4*C for 5 min at 10,000 rpm and
the supernatant (nuclear extract) was stored in aliquots at -80°C.
Protein concentration of the extract was deteRnitled using Bio Rad Protein Assay
Kit (BioRad Laboratones). The Bio Rad dye reagent was diluted with 4 parts of distilled
water and filtered through Whatman filter papa to remove parliculate matter. Bovine
Semm Albumin (BSA) (1.45 rng/ml, H,O) was used to establish a standard curve.
Dilutions of nuclear extract in buffer C were mixed with 5 ml of the diluted dye reagent,
vortexed and allowed to incubate at room temperature for 30 min. The absorbance of the
mixture was measured at 595 nm. The protein concentration of nuclear extract was
estimated from the BSA standard curve, which was linear usudy between 3-8 &pl.

GENERATION OF OLIGONUCLEOTIDES FOR LABELING
Sense and antisense oligonucleotides (Pl, P3 and P4) were custom synthesized
(AGCT, Toronto) with 5' overhangs to facilitate labeling with Klenow and [u-~*P]~ATP
(3000 Ci/mmol, Amersha.). Oligonucleotide Pl is a 38 bp sequence spanning Bh1474
fiom -53 to -9 and contains the two CCAAT boxes, AAVp5-Inr, a pyrimidine rich
sequence and two minor cap sites. P2 is a 21 bp sequence (-9 to +12) containing the
TdT-like Inr element, the major cap site and the CTFMF1-like consensus. P3 is a 45 bp
sequence (+89 to +130) that contains a TATA-üke sequence located 53 bp upstream of
the ATG codon and a weak cap site. P4 is a 23 bp oligonucleotide (+84 to +107) that
contains the TATA-like sequence alone. Ten micrograms of complementary sense and
antisense oligonucleotides were dissolved in 150 mM NaCl, heated at 80°C for 5 min and
allowed to reanneal by leaving the mixture to cool to room temperature. The double
stranded DNA was then precipitated with two volumes of ethanol and recovered by
centrifugation. P2 was digested with Xbal and Pst1 to generate a 5' overhang. Double
stranded 27 bp E2F oligonucleotide (GATTTAAGTTTCGCGCCCTTTCTCAA) was
kindly provided by Dr. Hamel and used as control in electrophoretic mobility shift assays.
,?J3 LABELING OF OLIGONUCLEOTIDES
Double stranded oligonucleotides were labeled at the 5' end with Klenow and [a-
32~]dATP as described by Sambrook et al., (1989). Pl, P2, P3 and E2F (200-500 ng)
were incubated with 3U of Klenow, 1.5 pl of 10x Klenow bae r , 10 m M of each dNTP
(dCTP, dGTP and dTTP) and 30 pCi of [ a - 3 ? ] d ~ ~ ~ in a total volume of 15 p1 for 30

min at room temperature. The labeled oligonucleotides were separated nom
unincorporated [ ~ J ' P ~ ~ A T P by spin column chromatography on Nick Columns
(Pharmacia) equilibrated with 10 mM Tris-HC1, pH 8.0, containing ImM EDTA. One
microliter of the labeled probe was mixed with scintillation fluid for counting in a (LKB
12 19 Rack Beta Liquid Scintillation Counter) spectrophotometer. Each labeied probe
was diluted to contain 20,000 cpm/pl.
2.24 ELECTROPHORETIC MOBILITY SHIFI' ASSAY (EMSA)
EMSA was perfomed accorduig to the method describecl by Zhang and
Lichtenheld., 1997. Twenty thousand counts per minute of radiolabeled double stranded
oligonucleotide was incubated with a known concentration of HepG2 nuclear extract, 1
pg41 poly dI-dC (Pharmacia) in either buffer Z (8 mM Hepes (pH 8.0), 20 mM MgC12,
100 mM KCI, 1 mM EDTA, 2 mM DTT, 200 mM ZnSO,, 100 pg/ml BSA and 20%
glycerol, in a final volume of 20 pl) (Zhang and Lichtenheld., 1997) or buffer M (60 mM
Hepes, pH 7.9, 6 mM EDTA, 1 mM DTT, 750 @ml, 10% glycerol, 40 rnM KCI, 2.5
rnM MgCl, 20 mM NaCl in a final volume of 21 pl) (GU et al., 1994). Mer 20 min
incubation at room temperature, the DNA-protein complexes were separated by
electrophoresis on a 5% polyacryIamide gel in OSxTBE buffer- The gel was dried and
visudized b y autoraciiography . Unlabeled double stranded oligonucleo tides at 1 0-50
molar excess were used in cornpetitive experirnents. The 132 bp (+26 to 1160) hgment
used as a cornpetitor was excised f?om Bh132CAT construct with Hindlll.


To map the basal promoter and define functionally important cis-acting DNA
elements in the 5' flanking region of the human factor 1 gene, a series of 5' tnincated
fragments were subcloned in the right orientation upstream of the CAT gene in the pCAT
basic reporter plasmid. Al1 the deletion mutants have a variable 5' end but a cornmon 3'
end, e n h g at +160 (with respect to the transcriptional start site designateci as +1) (Fig 8,
left panel). The constnicts were transiently transfected into HepG2 cells and assayed for
CAT activity. As shown on Fig 8, right panel, the pBh1474CAT constmct exhibited an
eighteen fold increase in CAT activity relative to promoterless pCAT and was also 2.7
times as active as the pSV40CAT positive control. pBhl287CAT, containing Bh1474
with a 187 bp 5' deletion, was thirty five times as active as pCAT basic and thus twice as
active as pBh1474CAT. The promoter strength of pBhl029CAT was similar to that of
pBh1287CAT. Removal of the 5' 550 bp fkom pBh1029 to generate Bh479 however
resulted in about 42% loss of promoter activity. The activities of pBh479CAT, and
pBh231CAT were similar to that of pBh1474CAT. The activity of pBh132CAT was
comparable to that of the promoterless pCAT constnict. This suggests that some of the
critical elements of the core promoter resides in a 99 bp region spanning -71 to +28 (Fig
9). This sequence contains two putative CCAAT boxes at -12 and -35, an Spl/AP-2 like
sequence at -62, a TdT-initiator-Like element (Inr) overlying the major cap site (+1), a
pyrimidine rich sequence overlying a minor cap site (-28). a sequence resernbling an
adeno-associated virus p5 (AAVp5)-Inr (-36) and a putative CTF/NF-1 located very close
to the TdT like-Inr element.

pCAT basic D
Rcliiivc Pmmoicr Activiiy (Fold Above pCAT Basic)
Fig 8: Left panel: Deletion construcs of pBh1474 were subcionrd upstrcam of the CAT gene of pCAT basic vector. pBhl2aICAT and pBhl029CAT were genented by cleaving pBhl474 with Ban I I Bçl I I respectively. pBh479 and pBh231CAT were obtained from pBh1287CAT by cleevinç with Nsi 1Pst 1 and BstEIUPst 1 and re-ligating the vector. pBhl32CAT was PCR amplified and subcloned. Right panel: Shows the relative pornoter activity of each construct expressed relative to pCAT basic, a promoterless CAT v-r. HepG2 cells were co-transfected with reporter plesmid and pRSV &Ga1 by calcium phosphate CO-
pcecipitation. To adjust for transfection eniciency, CAT expression obtained by LSC was normalizcd tu O- Ga1 activity. The data represents the average, SEM of three independent experiments.

-7 1 Sp-1 CCAAT BOX - 2 0
TGG~-CTC~AATGGW GGCCA i i i ~~ITGGTTTCAGIT ----- - 0 - -----
AAVp5-lnr Pyrimidine rich sequence AP-2
CCAAT BOX
TdT llke Inr CTF-NF1
Fig 9: Location of putative elements in the 99bp region (-71 to +28) of Bh231. The removal of this region frorn Bh231 resulted in loss of prom- oter activity. Adapted from Bh1474 sequence (fig 6).

In an attempt to M e r characterize the 99 bp sequence to determine the location
of regdatory elements that are essential for factor I expression in hepatocytes, three
subfi-agments of Bh23 1 (Bh206, Bh74 and Bh21) were prepared (Fig 10, left panel),
ligated to pCAT basic vector and assayed for promoter activity (Fig IO right panel).
BU06 (-46 to +160) lacking the 5' 25 bp sequence of Bh23 1 was generated using
Exonuclease III digestion to determine if the SplIAP-2 sequence at the 5' end of Bh23 1
plays a role in factor 1 transcription. A 74 bp hgment (-63 to + 10) was PCR amplified,
subcloned into pCAT and assayed to determine whether the sequence containhg the TdT-
like Inr, AAVp5-Inr, a putative CTF/NE: 1 and two CCAAT box consensus could serve as
factor 1 basal promoter. A double stranded 21 bp oligonucleotide (-12 to +9) containing
the major cap site, TdT-like In. and the CTF/NFl site was synthesized, subcloned into
pCAT and assayed for CAT activity to determine if the sequences surroundhg the
initiator element were sufficient in directhg factor 1 gene expression.
As shown in Fig 10 right panel, the CAT activity of pBh206CAT was seventy six
fold greater than that of pCAT basic control and five fold higher than that of pBh23 1 CAT
indicating the presence of silencer element(s) between -71 to -46. These results taken
together with those in Fig 9 indicate that some cntical elements of the core promoter
resides in a 74bp sequence nom 4 6 to +28. In addition, pBh74CAT and pBh21CAT
were inactive in the CAT assays, suggesting that sequences in Bh206, upstream of Inr
alone, are not sufncient to exhibit promoter activity. Hence, the sequences dowflstream
of the Inr are required for the transcription of the factor 1 gene.

pCAT basic. TU- fike Inr
TATA4 ike S q - CAT
+t60
RcIarwc Promotcr Activilv t FolJ Abovc pCAT Basic)
Fig 10: Left panel: CAT coiistnicrs of p B h l 3 1. pBh206CAT was gcnernted froiii pBh23 LCAT by exo III nuclease digestion. Bh74 was PCR miplilied and subcloned unidircctionally into pCAT basic vector. A 21 bp complimentary oligonucleoiidc was synthesizcd annealed and subcloned into pCAT basic vector to generatc pBhSlCAT. Right panel: The celntivc pro~iioter activity of each construci is expresxd as fold increase above pCAT basic. HepG2 cdls were CO-tnnsfected with reporter constructs and pRSV beta Ga1 by calcium phosphate precipitation CAT e ~ ~ c e s s i o n wvs mmlized to beta Ga1 activity to conect for trandection eficienq. The chta stiown is the mean + SEM of three independent ex'periments. f romter activity was ana1ysed by TLC and LSC. Data show is obtained by LSC.

Control of eukqotic gene expression at the transcriptional level involves the
binding of nuclear proteins to sequence specific sites on the promoter and other
regdatory regions of the gene. In this study, cornpetitive electrophoretic mobility shift
assay (EMSA) was used to localize cis-acting elements upstream and downstream of the
transcription start site in Bh206 that may be important for factor 1 expression in HepG2
cells. Three double stranded oligonucleotides (P 1 -P3) spannllig the BI1206 factor 1
promoter (Fig 11) were interacted with HepG2 nuclear extract and assessed for DNA-
protein band formation in mobility shift gel .
EMSA of Pl, P2 and P3 with HepG2 nuclear extract were performed
using two different buffers, buffet Z (Zhang and Lichtenheld, 1997) and bufFer M (Gill et
ai., 1994). As shown in Fig. 12, P2 and P3 both interacted with HepG2 nuclear extract to
form two distinct bands, a slow migrating band @and 1) and a fast migrating band @and
2). In contrast, Pl interacted with nuclear extract to generate only the fast migrating band
(band 2). Band 1 and band 2 formed with P2 and P3 had simila. electrophoretic
mobilities, but the intensity of band 1 was much stronger with P2 than with P3. Since,
the intensites of band 1 and 2 were greater in experiments using b-er Z than bufFer M
(Fig 12), funue experiments were conducted using Buffer 2. The mobility of band 2
fonned with P 1, P2, and P3 were similar. The specificity of these bands were assessed by
using an

n 1 m
- 4 6 TdT-ltke In! Inverted repeats TATA-like sequence +160 CTF-NF1
Fig II: Location of oligonucleotides used for Mobility Shift Assays. A 38bp (Pl), 21bp (P2), 45bp (P3) and 23bp (P4) seme and axaisense oiigonucleotides were synthesized and annealed to generate double stranded oligonucleotides. Pl (-53 to -9) vans Lhe two CCAAT boses found upstream of the Mtiator element P2 (-9 to + 12), contains the TdT Inr-like element AAVpS-Inr. pyrimidine rich sequence and tk CTF- NF1 site which overiies the major cap site. P3 (+89 to 134) contains the TATA like sequerice located 53bp upsvearn of the ATG codon and minor cap site. P4 (+84 to + 107) contains the dorvnstream TATA-like sequence.

Band 1
Band 2
Fig 12: EMSA of the interactions of labeled double stranded oligonucleotide P l (-53 to - 9). P2 (-9 to +12) and P3 (+89 to +134) with HepG2 nuclear exaact (NE). Pl and P2 were synthesized and amealed to generate double s m d e d oligonucleotides with 5 . overhang. P2 was also synthesized, annealed and digested with Xba 1 and Pst 1 to generate a 5' overhang. Pl , P2 and P3 were labeled with Klenow and a3'p ATP. Each double stranded Iabeled oligonucleohde (20,000 cpm) was incubated with 3 ug of HepGZ NE and 1 pg/$ poly dI-dC for 20 min at room temperature in two different buffers (Z&M) and electrophoresed on a 5% polyacrylamide gel. The gels tvere dned and autondiopaphed overnight. Buffer M contained 60 mM Hepes (pH 7.9). 6 mM EDTA. ImM Dm. 750 pgiml BSA. 10% glycerol, 40 rnM KC1, 2.5 m.!! MgCl,. 20 mM NaCl. Buffer Z contained 8 rnM Hepes (pH 8). 1 mM EDTA. 2 mM DTT. 100 &ml BSA. 20°h glycerol. 100 mM KCl. 20 mM MgCl,. 200 mM ZnSO,. Lane 1: PZ+NE+buffer Z: lane 2 : PZ+NE+buffer M; lane 3: Pl+NE+buffer 2; lane 4: Pl+NE+buffer M; lane j:

64
unrelated oiigonucleotide, E2F. Radiolabeled E2F interacted with HepG2 nuclear extract
to generate a DNA-protein complex whose mobility was similar to band 2 (Fig I3a).
Thus, band 2 may probably be due to nonspecific binding.
in cornpetitive EMS& '*P labeled P2 and 32P labeled P3 probes were competed
with 10 molar excess of Bhl32 and unlabeled double stranded oligonucleotides. As seen
in Fig 13b, the formation of band 1 by labeled P2 was unaffected by 10 molar excess of
P l but completely inhibited by IO molar excess of unlabeled PZ, P3 and the downstrearn
fragment Bh132. Band 2 was only pda l ly inhibited by unlabeled P2, P3, and Bh132,
but not by Pl. To determine whether unlabeled P2 could also inhibit the formation of
band 1 in P3, radiolabeled P3 was interacted with nuclear extract and competed with
unlabeled P2. As shown in Fig 14, band 1, DNA-protein complex formed by the
interaction of HepG2 nuclear extract with labeled P3, was completely inhibited by 10
molar excess of unlabeled P2, P3, P4 ( s u b h p e n t of P3) and Bh132, but not by P l .
Band 2 was partially inhibited by unlabeled P2, P3, P4 and Bh132 but not by P l .
Alignment of the nucleotide sequences of P2, P3 and P4 showed that TCAGC
pentamer was present in both P2 and P3 and a CTGGA motif was present in P2, P3 and
P4 (Fig. 15).

Fig 13: (A) Interaction of E X wiih HepG2 nuclear e m c c (NE). Twenty ihousand CPM of 3 2 ~ lakled E2F (lane 1) was incubated witli HepG2 NE, 0.36 ug (lane 2) and 0.18ug (lane 3) for 20 min at rwm temperature. The mixtures were eleciroplioresed on a 5% polyacrylarnide gel for 2 lus. The gel ws drid and autoradiographed ovemiglit. (B). Cornpetitive EMSA of P2. "P labeled P2 (20,000 CPM) was incubated at m m temperature for 25 min witli 0.18 ug of HepG2 NE in the absence and presence of 10 molnr escess of Bh132 and double m d e d Pl, PZ, and P3. Lane 4: P2 probe; lane 5 : PZ probe + NE; lane 6: PZ probe + NE + unlabeled Pl; lane 7: P2 probe + NE + unlabeled P2; lane 8: PZ + NE + Bli 132; lane 9: P2 + NE + P3. The reactions tvere elatrophoresed on a 5% polyacxylamide gel for 2 hrs, dried and autoradiographed ovemight.

Fig 14: Coinpeiiiivc EMSA of P3. Twcnty il~ousand C P M of "P lnbclcd P3 (P3 probe) $vas incubated ~vith 0.18 ug of HepGZ nuclear estract (NE) in ihc absence and prcscnce of 10 molnr escess of unlakled Bh132 and double sirandcd P3 and P4. Lanc 1: P3 probc; Iane 2: P3 probc + NE; lanc 3: P3 probe + NE + unlabeled P3; lanc 1: P3 probe + NE +unl:ibcled P 1: Innc 5: P3 probe + NE+ unlabcled Bh132: lane 6: P j probe + NE + unlabeled P-l: lane 7: P3 probc + NE+ unlabclcd P?.

P4 AAATTTCAAAAGAATACCTGGAG - +84 +1 07
Fig 15: Alignment of P2. P3 and P4 oligonucleotides used in Mobiiity Shift Assays. CTGGA motif is common to al1 the nucleotides.

DISCUSSION

Complement factor 1 is a plasma serine protease which plays an essential role in
the regulation of both the classical and altemative complement pathways by cleaving C3b
and C4b and preventing the assembly of C3 and CS convertase enzymes, the terminal
sequence and resultant cell darnage. In this study, transcriptional control elernents that
are essential for factor 1 expression in hepatocytes were investigated. As discussed
before (page 32), the transcriptional control regions of eukaryotic protein encoding genes
can be grouped into two categories: the upstream and downstream regulatory elements
and the core promoter. Each gene carries an array of proximal and distal regulatory
elements that can bind sequence specinc DNA binding proteins to activate or repress
transcription initiation (Tijian and Maniatis, 1994). The core promoter on the other hand
nucleates the assembly of an initiation complex containing RNA polyrnerase II and an
array of accessory proteins (T'FILA, TFIIB, TFIID, TFIIE, TFIIF, TFEH). To identiQ
sequences in the 5' flanking region of the factor I gene involved in its expression, 5'
tnuications of the factor 1 promoter region were linked to a CAT reporter plasmici,
transfected into HepG2 cells and assayed for CAT actwity. The 1474 bp 5' flanking
region of the factor 1 gene (Bh1474) was 18 fold more active in CAT activity relative to
pCAT basic controi and also 2.7-fold more active than pSV40CAT in driving the
expression of the CAT gene in HepG2 cells. Deletion mutagenesis experiments indicated
that the elernents essential for basal transcription raide in a 74 bp sequence from -46 to
+28. In addition, three upstream regions were identified which may participate in the
regulation of the expression of the gene. Removal of the h t 179 bp region from the 5'
end of Bh1474 resulted in a two-fold increase in the promoter activity, suggesting the

70
presence of negative controlling element(s) in this region. A closer examination of the
179 bp region shows a sequence resembling the rat collagen silencer consensus
(CACCTCC) (Chou et al., 1991) at position -1304 and -1250 (Y8 match). When the
sequences between -869 and -3 19 were deleted, a two fold decrease in promoter strength
was observed. This suggests the presence of an enhancer elcment(s) in this region. A
putative CCAAT box motif (7/9) located in this region between -665 to -672 may be a
likely candidate. Finaily, the removal of a 25 bp sequence between -71 to -46 resulted in
5 fold increase in promoter activity, suggesting the presence of another negative
regulatory elernent(s). This region contains an AP-2 element which partially overlaps an
Spl site. Since the removal of this region resulted in an upregulation of factor 1
transcription, it is unlikely that the members of the Spl family of proteins are involved.
AP-2 transcription factors have been shown to exert inhibitory effects. However, since
AP-2 itself is not expressed in HepG2 cells (Cunming and Clemmons., 1995), it is
possible that other members of the AP-2 family of proteins may be involved. The
identity of the regulatory elements in these three regions and their role in the
transcriptional regulation of factor 1 gene remains to be M e r investigated.
The deletion mutagenesis analysis also demonstrated that elements essential for
core promoter activity reside in a 74 bp region located between -46 and +28. This region
contains (a) two CCAAT boxes located at - 12 and -35 (8/9 and 7/9 match respectively),
(b) a TdT-like Inr element, [5/8 match with the TdT Inr element (CTCANTCT) (Smale et
al., 1990)l overlying the major transcription star t site, (c) AAVpS-Inr-iike sequence
(CTCCATTTT) (Shi et al., 1991) located at 4 0 (719 match), and (d) a pyrimidine rich
sequences (TTTCAGTTA) at position -30 which is homologous to eukaryotic consensus

71
transcription initiator sequence (YYCAYTY) (6/7 match) (Smale and Baltimore., 1989).
Since this region lacks a TATA box, but contains a TdT-like Inr located at the major
transcription start site, it is possible that factor 1 promoter is a TATA-less Inr dependent
class 11 promoter.
Initiators are weakly conserved elements that overlap the transcription start site
and are hctionally analogous to the TATA box (Reviewed by Weis and Reinberg.,
1992; Smale., 1994). The mitical nucleotides in the Inr elernent are the A at +1 and the T
at +3 and significant Inr activity is exhibited when the 5'-ANT-3' (N-is any nucleotide)
is surrounded by two to three pyrimidines at position -1 to -5, +4 and +S. The
pyrimidines at +4, +5,4 and -2 appear to be more important than the ones at -3.4, and -5
(Srnale., 1994). An examination of sequence surroundhg the major cap site (-5
ATTTCAGCCA +5) of the factor I gene shows an A at +1, a C instead of a T at +3, (both
of which are pyrimidines) and pyrimidines located at +4, -1 and -2. Thus, the factor 1
Lnr is quite homologous to other functional initiators (Smale., 1989, Kollmar and
Fadam. , 1993; Means and Famham., 1990).
Several studies have shown that sequences upstream a d o r downstream of the tnr
may be crucial for promoter activity. In this shidy, a CAT constmct containing the
sequences surromding the major transcription initiation site (Bh21) and a construct
containing the major cap site and two upstream CCAAT-like sequences (Bh74) were both
unable to support transcription. Furthemore, the Bh 132CAT constnict that contained the
region from +26 to +160 domtream of the major start site was ais0 unable to support
transcription. This suggests that sequences both upstream of +26 and downstream of the
cap site are required for factor 1 promoter activity in HepG2 ceus.

72
To detemine the location of proximal regulatory elements in the Bh206 factor 1
promoter sequence that may be involved in factor 1 expression, three double stranded
oligonucleotides complernentary to (a) the two CCAAT box consensus elements
upstream of the Inr (-53 to -9) Pl; (b) the Inr element (-12 to +9), P2 and (c) the
downstream TATA-Like sequence and a minor cap site (+89 to +130), P3 were
radiolabeled and incubated with HepG2 nuclear extract and assessed for DNA-protein
binding in a mobility shift assay. PZ and P3 both formed two DNA-protein complexes
@and 1 and band 2) with sirnilar mobilities. Pt only formed a singe band whose
migration pattern was simiIar to band 2. An unrelated probe, EZF, aIso generated a single
complex when interacted with HepG2 nuclear extract with migration pattern similar to
band 2. Since a11 the probes as well as E2F exhibited this band, it is possible that the
complex in band 2 may be due to nonspecific interactions. Band 1 in P2 and P3 appeared
to be specific, since they could be competed by their unlabeled oligonucleotides and the
band was not evident with E2F. The complex in band 1 formed with P2 could be
Uihibited by unlabeled P3 and the band 1 complex formed with P3 could also be cross-
competed with unlabeled P2. This raises the possibility that P2 and P3 may be
interacting with a common nuclear factor to generate band 1. Alignment of P2 and P3
oligonucleotides showed two pentamers -TCAGC- and -CTGGA- in both sequences. An
unlabeled double stranded O ligonucleo tide containhg the TATA-like sequence and the
CTGGA motif (P4) but not the TCAGC pentarner inhibited the formation of the
interactions of P2 or P3 with nuclear extracts. This observation tends to suggest that the
CTGGA motif may be responsible for the generation of band 1. The -CTGGA- motif,
found downstream of the transcription start site, has been shown to be important in the

73
transcriptionai regdation of two TATA-less genes, the TdT gene (+2
TTCTGGACAC+Il) and human porphobilinogen deaminase gene (Beaupain et al..
1990; Weis and Reinberg., 1992). This CTGGA motif is present upstream of the Inr in
P2, but the same motif is also present downstream of the In. in P3 and P4. The
possibility that the transcription factor may interact with multiple motifs on the factor 1
promoter cannot be at present eliminated.
The identity of the nuclear factor in band 1 is at present unknom. To date a
number of sequence specific DNA binding proteins have been recognized to bind to the
Inr element and to aid in the assembly of the preiaitiation complex. These include RNA
polymerase II itself (Carcarno et al.,1991), E2F (Means et al., 1992), YY1 (Usheva and
Shenk, 1994), upstream stimdating factor (USF) (Du et al., 1993) and initiator factor,
TFII-1 (Roy et a1.,1993). YY1, TFII-1 and USF have been shown to stimulate
transcription through the Inr element in those TATA-less initiator dependent class II
promoters where the factors were initially recognized (Du et al., 1993). TFII-1, 120 kD
protein, has been shown to bind to Inr element of the adenovirus major late promoter
(Ad-ML), the TdT and the human immunodeficiency virus promoter (Roy et al., 199 1).
Since factor 1 contains an initiator element resembling the TdT h, it is possible that band
1 may be a complex of P2 and TFII-1. TFII-1 has been shown to recognize the USF
binding site in the major late promoter with greater affinity than USF itself and this
interaction is thought to be through a different motifs (Roy et ai., 1991). Since TFII-1
contains multiple binding motif, it is possible that this protein codd recognize other
consensus sequences. YY1 has been reported to bind to the AAVpS-lnr element
(CTCCATm). A sequence homologous to the AAVpS-In. is present at -35 close to

74
one of the rninor cap sites (Fig 9). In this study, a stable DNA-protein complex was not
detected between nuclear extract and P 1, an oiigonuc leotide containing this motif.
The Ets family of transcription factors have also been show to bind specifically
to a sequence (UA GGA A/T) which is similar to the common motif CTGGA. These
factors are important for transcription from certain TATA-less promoters. Ets proteins
have been implicated in regdation of gene expression during a variety of biological
processes, such as growth control, transformation, T-ce11 activation and developmental
prograrns in many organisms (Wasylyk et al., 1993).
Foo t printing analysis on various Inr-dependent promot ers have estab lished the
role of TBP-associated factors (TAF), components of TFIID, as the uIr recognition
factors. TAFI50 and TAF60, in addition to binding to the Inr element, also contact the
downstream regions (Gilrnour et al., 1990). Several studies have also shown that TATA-
like elements located downstream of the transcriptional start site can bind TBP and play a
role in the assembly of the pre-initiation complex of Inr dependent class II promoters
(Carcamo et al., 1990). It is possible that the inverted repeat localized between the Inr
element and the TATA-like sequence in the factor 1 promoter region may serve to place
the Inr-like and TATA-like elements in close proximity such that the TBP binds to the
TATA-like sequence while the TAFs contact the Inr. The possible cooperation of the Inr
and TATA-like sequence in the assembly of the preinitiation complex should therefore be
explored in the factor 1 promoter. The role of T F D in Inr-dependent gene transcription
may offer an explanation for the requirernent of the downstream region in factor 1
expression.

c This study has identified the core pmmoter and three regulatory regions in the
S'flanking region of the factor 1 gene. Further studies employing Linker scanning
mutagenesis should be used to M e r id&@ other fiuictionally relevant sequences in
the BU06 plasmid as well as in the three upstream regulatory regions.
Site-directed mutagenesis of putative cis sequences in Bh206, such as the two
CCAAT boxes, Inr, TATA-Like sequence, the CTGGA and TCAGC pentamer repeats,
should also be used to m e r delineate the functional relevance of these sequences in the
constitutive expression factor 1. Identified functionai cis-sequences shodd be assessed
for their ability to bind nuclear extract in mobility shifi assays. Super shift assays can
then be employed to identie the transcription factor(s) involved in factor 1 expression.
CAT assays as well as mobility shift assays should also be used to analyze the tissue-
specific expression of factor 1.
Since the Bh1474 promoter contains potential consensus elements such as
glucocorticoid response element (GRE), y-IFN response element, y-IFN activating
sequence (GAS), interferon stimulating response element (ISRE), IL4 response element,
IL-6 response element and several AP 1 sites (Fig 6), the potential effects of
glucocorticoids, and idammatory cytokines (including y-UN, IL- 1, IL-6, TNF and
retinoic acid) in the regulation of expression of the factor 1 gene should be investigated.

REFERENCES

Abramson N., Alper C.A., Lachrnann P.J., Rosen F.S. and Jandl J.H. (1971).
Deficiency of C3 inactivator in man. J. Imrnunol. 107: 19.
Adams E.M., Brown MC, Nunge M., Krych M. and Atkinson S.P. (199 1).
Contribution of the repeating domains of membrane cofactor protein (CD46) of
the complement system to ligand binding and cofactor activity. I. Immunol. 147:
3005.
Aruffio B.A., Melnick M.B., Linsley P.S., and Seed B. (1991). The lymphocyte
glycoprotein CD6 contains a repeat domain structure and characteristic of a new
farnily of ce11 surface and secreted proteins. J. Exp. Med. 1 74: 949.
Asghar S.S. (1995). Membrane regdators of complement activation and their
aberrant expression in diseases. Laboratory Investigation 72: 254.
Beaupain D., Eleouet J., Romeo P. (1990). Initiation of transcription of the
erythroid promoter of the porphobilinogen deaminase gene is regulated by a cis-
acting sequence around the cap site. Nucleic Acid Res. 18: 6509.
Bhakdi S., Fassbender W., Hugo F., Carreno M.P., Berstecher C., Maiasit P. and
Kazatchkine M..D. (1 98 8). Relative inefficiency of terminal cornplement
activation. J. h u n o l . 141 : 3 1 17.
Bohnsack J.F., and Cooper N.R. (1988). CR2 ligands modulate human B ce11
activation. J. Tmmunol. 141 : 2569.
Bonnin A.J., Howard J., Zeitz and Gewurz A. (1993) Complement factor 1
deficiency with recurrent aseptic meningitis. Arch. Intem. Med. 1 53 : 1380.
Buchner H. (1 8 89). Ueber di nahere Naturder bektenentodtendon substanz im
blutsem. Central Bakter Parasitol. 6: 561

10, Bora N.S., Lubin D.M., Kumar B.V., Hockett R.D., Holers V.M. and Atkinson
J.P. (1989). Stmchiral gene for human membrane cofactor protein (MCP) of
complement maps to within 100 base pair of the 3' end of C3blC4b receptor gene.
J. Exp. Med. 169: 597.
11. Breathnach R. and Chambon P. (1981). Urganization and expression of
eukaryotic split genes coding for proteins. Ann. Rev. Biochem. 50: 349.
12. Burley and Roeder. (1996). Transcription factor IID. Am. Rev. Biochem. 65:
769.
13. Burgi B., Brunner T., Dahinder C. A. (1994). The degradation products of the
C5a anaphylatoxin C5a desarg retains basophil activating properties. Eur. J.
Immunol. 24: 1583.
14. Campbell R.D., Gagnon J., Porter R.R. (1981). Amino acid sequence around the
thiol and reactive acyl groups of human complement component C4. Biochem. J.
199: 359.
15. Catterall CF., Lyons A.L., Sim RB., Day A.J., Harris T.J.R (1957).
Characterization of primary amino acid sequence of human complement control
protein factor 1 nom an analysis of cDNA clone. Biochem. J 242: 849.
16. Carcamo J., Maldonado E., Ahu M., Ha I., Kasai Y.. Flint J. and Reinberg D.
(1 990). A TATA like-sequence located downstream of transcription initiation site
is required for expression of an RNA polymerase II transcnbed gene. Genes and
Dev. 4: 1611.

17. Carcamo J., Buckbinder L., and Reinberg D. (1991). The initiator directs the
assembly of a transcription factor 1 ID-dependent transcription complex. Roc.
Natl. Acad. Sci. USA 88: 8052.
18. Carreno M.P., Labarre D., Maillet F., Jozenfowicz M., and Kazatchkine. (1989).
Regdation of the human alternative complement pathway: Formation of a ternary
complex between factor H, surface-bomd C3b and chernical groups on
nonactivating d a c e s . Eur. J. unmMol. 19: 2145.
19. Chou J.L., Romahel Rey and Tsui L. (1991). Characterization of the promoter
region of the cystic fibrosis transmembrane conductance regulator gene. I. Biol.
Chm. 266: 24471.
20. Chung L.P., Bentley D.R., and Reid K.B.M. (1985). Molecula. cloning and
characterization of the cDNA coding for C4-biading protein, a regdatory protein
of the classicai pathway of human complement system. Biochern. 1.230: 133.
2 1. Choy B. and Green M. (1 993). Eukaryotic activator function during multiple
steps of preinitiation cornplex assembly. Nature 366: 53 1.
22. Colomb M.G., Arlaud G.J., Villiers CL. (1984). Activation of C 1. Philos.
Trans. R. Soc. London 306: 283.
23. Cooper N.R-, and Muller-Eberhard HJ. (1970). The reaction mechanism of
human C5 in immune hemolysis. J. Exp Med. 132: 775
24. Cooper N.R., Moore MD., and Nenerow G.R. (1988). Tmmunobiology of CR2,
the B lymphocyter receptor for Epstein-Barr virus and the C3d complement
hgment. Annual Review of Immunology 6: 85.

Cooper N.R., Nemerow G.R. (1983). Complernent, h s e s and virus infected
cells. Sprhger Semin. Immunopathol. 6: 327.
Courtois G., Baumhueter S., Crabtree G.R. (1988). Punfied hepatocyte nuclear
factor 1 interacts with a family of hepatocyte-specific promoters. Roc. Natl.
Acad. Sci. USA. 85: 7937.
Coyne K.E., Hall S.E., Thompson S., Arce M.A., Kinoshita T., Fujita T., Anstee
D.J., Rosse and Lublin D.M. (1992). Mapping of epitopes, glycosylation sites
and complement regdatory domains in human decay accelerating factor. J.
Lmmunol. 149: 2906.
Cui W., Hourcade D., Post T.W., Greenlund A.C., Atkinson J.P. and Kumar V.
(1 993). Characterization of the promoter region of the membrane CO factor protein
(CD46) gene of the human complernent system and cornparison to a membrane
CO factor protein-like genetic element. J. h u n o l . 1 5 1 : 4 137.
Cunming D. and Clemmons D. (1995). Transcription factor AP-2 regulates
human Insulin like growth factor binding protein 5 gene expression. J. Biol.
Chem. 270: 24844.
Dahlback B., and Podack E.R. (1985). Characterization of human S protein, an
inhib itor of the membrane attack complex. Biochemistry 25 : 6740.
Dangott L.J, Jodan J.E., Bellet RA. and Garberg D.L. (1989). Cloning of the
mRNA for the protein that crosslinks to the egg peptide speract. Proc. Natl Acad.
Sci. USA- 86: 2128.
Davies A.E. (1981). The C3b inactivation of the human cornpiement system:
homology with serine proteases. FEBs Lett. 134: 147.

33. Davies A.E., Whitehead A.S., Hanison R.A, Dauphinais A.. Bruns G.A.P.,
Cicardi M. and Rosen F.S. (1986). Human inhibitor of the fïrst cornponent of
complement, C 1 : Characterization of cDNA clones and localization of the gene
to chromosome 1 1 . Proc-Natl. Acad-Sci. USA. 83: 3 16 1.
34. Davies M.A., Simmons DL., Hale G., Harrison R.A., Tighe H., Lachmaun P.J
and Waldrnann H. (1 989). CD59, an LY-6 like protein expressed in human
lymphoid cells regulates the action of the membrane attack complex on
homologous ceils. J.Exp. Med. 170: 637.
35. Davies M.A., Low M.G and Nussenzweig V. (1986). Release of decay-
accelerating factor PAF) fiom the cell membrane by phosphatidylinositol-
specific phospholipase C (PPLC). Selective modification of a complement
regdatory protein. J. Exp. Med. 163: 1 150.
36. de Bruijin M.H., and Fey G.H. (1985). Human complement component C3.
cDNA coding sequence derived primary structure. Proc. Natl. Acad. Sci. USA.
83: 708.
37. DiScipio R G., Gehring M.R., Podack E.R, Kan C.C., Hugli T.E. and Fey G.H.
(1984). Nucleotide sequence and derived amino acid sequence of human
complement component. Proc. Natl. Acad. Sci. USA. 81: 7298.
38. DiScipio R. G., and Hugli T.E. (1989) The molecular architecture of Human
complement component C6. J. Biol. Chem. 264: 16197.
39. Dodds A.W., Sim R.B., Porter RA. and Kerr M.A. (1978). Activation of the k t
cornponent of human complement (Cl) by antibody-antigen aggregates.
Biochem. J. 175: 383.

Du, H., Roy A. L., and Roeder R. G. (1993). Human transcription factor USF
stimulates transcription through the initiator elements of the HIV-1 and the AdML
promoters. EMBO J. 12: 501.
Dynlacht B.D., Hoey T. and Tijian R. (1991). Isolation of CO-activators
associated with the TATA-binding protein that mediates transcriptional activation.
Ce11 66: 563.
Ebanks R. (1995). Studies on the localization and characterization of the CS and
C2 binding sites in the fourth component of complement. Thesis. U of T.
Ehrlich P., Morgenoth J. (1899). Zur theorie der lysenwirkung. Berlin Klin
Wschr 36: 6.
Emili A. and Ingles C.J. (1995). The RNA polymerase II carboxy-tenninal
domain links to a bigger and better holoenzyme. Current Opinion in Genetics and
Development. 5: 204.
Fearon D.T. (1 977). Purification of C3b inactivator and demonstration of its two
polypeptide chah structure. J. Immunol. 119: 1248.
Fearon D.T., Austen K.F. (1975). Properdin bhding of C3b and stabilization of
the C3b- dependent C3 convertase. J. Exp. Med. 142: 856.
Fearon D.T., Ahearn J.M. (1989). Complement receptor 1 (C3bK4b receptor;
CD39 and complement receptor 2 (C3d/Epstein-Barr vins receptor, CD21)
Curr. Topics Microbiol. Immunol. 153: 83.
Fischer E., Delibrias C., Kazatchkine M.D. (1991). Expression of CR2 (the
C3dgEBV receptor, CD21) on normal human peripherai blood T lymphocytes.
J. Immunol. 146: 865.

Freeman M., Ashkenas J., Rees D.J.G., Kingsley D.M., Copeland N.G-, Jenkins
N.A., and Krieger M. (1990). An ancient, highly conserveci family of cysteine-
rich protein domains revealed by cloning type 1 and type II murine macrophages
receptors. Proc. Natl. Acad. Sci. USA. 87: 8810.
Gill R.M., Hamel P.A., Zhe J., Zacksenhaus E., Gallie B.L. and Phillips R.A.
( 1994). Characterization of the human RB 1 promoter and of elements involved in
transcriptional regdation. Ce11 growth and differentiation 5: 467.
Gilmour D. S., Dietz T. J., and Elgin S. C. R. (1990). UV crosslinking identifies
four polypeptides that require the TATA box to bind to the Drosophila hsp7O
promoter. Mol. Cell. Biol. 10,4233.
Goldberger G., Arnaout M.A., Kay R., Rita M., and Colten H.R. (1984)
Biosynthesis and postsynthetic processing of Human C3bK4b inactivator (Factor
1) in three hepatoma ce11 lines. J. Biol. Chem. 259: 6492.
Goldberger (3.9 Bruns G.A.P, Rita M., Edge M.D. and Kwiatkowski D.J. (1987)
Human complement Factor 1. J. Biol. Chem. 282: 1.
Gonzalez S., and Lopez-Larrea C. (1996). Characterization of the human C6
promoter. J. Immunol. 157: 228.
Gotze O., and Muiler-Eberhard H.J. (1976). The alternative pathway of
complement activation. Adv. Immunol. 24: 1.
Gotze O., and Muller-Eberhard H.J. (1971). The C3 activation system: An
alternative pathway of complement activation. J. Exp. Med. 1 34: 90s.

Gotze 0. (1975). Proteases of properdin system. In proteases and biological
control. Edited by E. Reich, D.B Rifier and E. Shaw. pp 155-272. Cold Spring
Habor Labomtory, Cold Spring Harbor, New York.
Gulati P., Guc D., Lemercier C., Lappin D. and Whaiey K. (1994). Expression of
the components and regulatory proteins of the classical pathway of complement in
normal and diseased synovium. Rhematol. Int. 14: 13.
Harrison R.A., Thomas M.L., and Tack B.F. (1981). Sequence detennination of
the thiol ester site of the fourth component of human complement. Roc. Natl.
Acad. Sci. USA. 78:7388.
Harrison RA. and Lachmann P.J. (1980). The physiological breakdown of the
third component of hurnan complement. Mol. Immunol. 17: 9.
Hemandez N. (1993). TBP, a universai eukaryotic transcription factor? Genes
Dev. 7: 1291.
Herz J., Hamann U., Rogne S., Myklebost O., Gausepohl H. and Stanley K.K.
(1988). Surface location and high &ty for calcium of a 500 kD liver
membrane protein closely related to the LDL-receptor suggest a physiological role
as lipoprotein receptor. EMBO. 7: 41 19.
Hillarp A., Pardo-Manuel F., Ruiz RR, Rodriguezde-Cordoba S. and Dahlback
B. (1 993). The human C4b-binding protein beta-chah gene. J. Biol. Chem. 268:
15017.
Hillarp A. and Dahlback B. (1988). Novel subunit in C4b-binding protein
required for proteins binding. J. Biol. Chem. 263: 12759.

65. HiUarp A. and Dahlback B. (1990). Cloning of cDNA coding for the f3 chain of
human complement component C4b-binding protein. Sequence homology with
the a chah Proc. Natl. Acad. Sci. USA. 87: 1 183.
66. Howard O.M.Z., Rao A.G. and Sodetz J.M. (1987). Complementary DNA and
derived amino acid sequence of the beta subunit of human complement protein
C8. Identification of a close structura1 and a n c e s a reiationship to the alpha
subunit and C9. Biochemistry 26: 3565.
67. Huang H.J.S., Jones N.H., Strominger J.L., and Herzenberg. (1987). Molecular
cloning of LY-1, a membrane glycoprotein of mouse T-lymphocyte and a subset
of B cells: molecular homology to its human counterpart Leu-1/T1 (CD5). hoc.
Natl. Acad- USA. 84: 204.
68. Hugli T. E. (1 986). Biochemistry and biology of anaphlylatoxin. Complement 3:
111.
69. Hugli T.E. (1975). Human anaphylatoxin (C3a) f?om the third component of
complement: primary structure. J. Biol. Chem. 250: 8293.
70. Iida K., and Nussenzweig V. (1981). Complement receptor is an inhibitor of the
complement cascade. J. Exp. Med. 153: 1 138.
71. Jeme D., and Tschopp J. (1992). Clusterin: the intriguing guises of a widely
expressed glycoprotein. Trends Biochem. Sci. 17: 154.
72. Julen H., Dauchel C., Lamercier C., Sim R.B., Fontaine M., and Ripoche J.
(1992). In vitro biosynthesis of complement factor I by human endothelid cells.
Eur. J. Tmmunol. 22: 213.

Kaidoh T. and Gigli 1. (1987). Phylogeny of C4b-C3b cleaving activity. Similar
bgmentation patterns of human C4b and C3b produced by lower animal. J.
Immunol. 139: 194.
Kennedy E.P. (1967). Mechanism of hydrolysis of O-nitrophenyl P galactoside
of staphylacocus aueus and its significance for theories of sugar transport.
Proc. Natl. Acad. Sci. USA. 58:225.
Kim Y.U., Carroll M.C., Isenman D.E., Nonka M., Pranoon J., Ago P., Takeda J.,
houe K., Kinoshita T. (1992). Covalent binding of C3b and C4b within the
classical complement pathway C5 convertase. Determination of amino acid
residues involved in ester-Mage formation. J. Biol. Chem. 267: 4 1 7 1.
Klickstein L.B., Wong W.W., Smith LA., Morton C., Fearon D.T., Weis J.H.
(1985). Identification of long homologue repeat in human CRI. Complement
2: 44.
Kolb W.P., Muller-Eberhard H.J. (1976). The membrane attack complex
mechanism of complement: the three polypeptide chab structure of the eighth
component. J.Exp. Med. 143: 1 13 1.
Koleske A.J. and Young RA. (1994). An RNA polymerase Ki holoenzyme
responsive to activators. Nature 368: 466.
Koliman R., Famham P.J. (1993). Site specific initiation of transcription by
RNA polymerase II. Proc. Soc. Exp. Biol. Med. 203: 127.
Kxych M.L., Clemenza D., Howdeshell R, Hauhart D., Hourcade and Atkinson
J.P. (1994). Analysis of the fiuictional domains of complement receptor type 1

(C3b/C4b receptor; CD35) by substitution mutagenesis. J. Biol. Chem. 269:
13273.
81. Kunnath Muglia L.M., Chang G.H., Day A.J., Ezekowitz R.A.B. (1993)
Characterization of Xenopus laevis complement factor 1 structure - conservation
of modular structure except for an unusual insert not present in human factor 1.
Mol. Imrnunol. 30: 1249.
82. Law S.K., and Levine RP. (1977). Interaction between the third cornplement
protein and ce11 surfaces. Proc. Natl. Acad. Sci. USA. 74: 270 1 .
83. Law S.K. (1983). Non-enzymatic activation of the covalent binding reaction of
the complement protein C3. Biochernical J. 2 1 1 : 3 8 1.
84. Law S.K., Lichtenberg N.A., Levine R.P. (1979). Action of the C3b inactivator
on the ce11 bound C3b. J. Immiuiol. 123: 1388.
85. Law S.K., and Reid K.B.M. (1995) Cornplment. EU press, Oxford UK.
86. Lepow I.H., Willms-Kretschmerk, Patrick R.A. and Rosen F.S. (1970). Gross and
ultrastruchual observations of lesions produced by intradermal injection of human
C3a in man. Amencan Journal of Pathology 6 1 : 13.
87. Lublin D.M., and Coyne ICE. (1991). Phospholipid anchored and transmembrane
forms of either decay accelerating factor or membrane cofactor protein show
equal efficiency in protection from complement-mediated ce11 damage. J. Exp.
Med. 174: 35.

Lublin D.M., Liszewski M.K., Post T.W., Arce M.A., LeBeau M.M., Rebentisch
M.B. (1 988). Molccular cloning and chromosomal localization of human
membrane cofactor protein (MCP). J. Exp. Med. 168: 1 88
Lublin D.M. and Atkinson J.P. (1989). Decay-accelerating factor: biochemisûy,
molecular biology and bctions. Ann. Rev. Immunol. 7: 35
Lu J.H., Thiel S., Wiedemann H., Timpl R and Reid K.B.M. (1990). Binding of
the pentamerhexamer forms of mannan binding protein to zymogen activities the
proenzyme Clr,C 1 s, complex of the complement pathway of complement without
the involvement of Clq. J. Immunol. 144: 2287.
Mayer M.M. (1980). Transmembrane channels produced by complement
proteins. Am. NY. Acad. Sci. 358:43.
McNearney T., Ballard L., Seya T., and Atkuison. (1989). Membrane cofactor
protein of complement is present of on human fibroblast, epithelial, and
endotheiial cells. J. Clin. Invest. 84: 538.
Means A. and Famham P. (1990). Transcription initiation fiom the dihydrofolate
reductase promoter positioned by HlP 1 binding at the initiation site. Mol. Ce11
Biol. 10: 652.
Means A.L., Slansky J.E., McMahon S.L., Knuth M.W and Farnham P.J. (1 992).
The HIP-1 binding site is required for growth regulation of the dihydrofolate
reductase gene promoter. Mol. Celi Biol. 12: 1054.
Medicus R.G., Melamed J., h o u t M.A. (1983). Role of human factor 1 and
C3b receptor in the cleavage of surface-bound C3bi molecules. Eur. J. of
h u n o l . 13(6): 465.

Medicus R.G., Scheiber RD., Gotze 0. and Muller-Eberhard H.J. (1976a). A
molecular concept of the properdin pathway . Proc. Natl. Acad. Sci. USA 73 : 6 1 2.
Medof M.E., Walter E.I., Roberts W.L., Haas R. and Rosenberry T.L. (1986).
Decay accelerating factor of complement is anchoreci to cells by a C-temiinal
glycolipid. Biochern. 25: 6740.
Meri S., Morgan B.P., Davies A., Daniels R.H., Olavesen M.G., Waldmann H.,
Lachmann P.J. (1990). Human protectin (CD59) an 18,000- 20,000 molecular
weight complement lysis restricting factor, inhibits CS-8 catalysed insertion of
C9 into bilayer. hnmunol. 71 : 1.
Minta J.O. and Lepow I.H. (1974). Shidies on the subunit structure of human
properdin. Immunochemisûy 1 1 : 36 1-8.
&ta J.O., Wong M., Kozak CA, Kunnath-Muglia and Goldberger G. (1996)
cDNA cloning, sequencing and chromosomal assignment of the gene for mouse
complement factor I (C3bIC4b inactivator): Identificatim of a species specific
divergent segment in factor 1. Molecula. Immmology 33: 10 1.
Minta J.O., Fung M., Turner S., Eren R, Zemach L., Rits M., Goldberger G..
(1 998). Cloning and characterization of the promoter for the human complement
factor 1 (C3bX4b Inactivator) gene. In Press.
Miyagoe Y., Galibert M., Georgatsou E., Fourel G. and Meo T. (1994). Promoter
elements of mouse complement C4 gene cntical for trancriptional activation and
start site location. J. Biol. Chern. 269: 8268.
Morgan P. (1 995). Physiology and pathophysiology of complement progress and
trends. Critical reviews in Clinical Lab. Sciences 32: 265.

Morgan P., Raab H., Kohr W.J. and Caras LW. (1991). Glycophospholipid
membrane anchor attachment. Molecular anaiysis of the cleavage/attachment site.
J. Biol. Chem. 266: 1250.
Morgan P. and Caras LW. (1991). Fusion of sequence elements h m non-
anchored proteins to generate a hilly functional signal for
glycophosphatidylinisitol membrane anchor attachment. J. Ce11 Biol. 1 1 5 : 1 595.
Muller-Eberhard H.J. (1975). Complement. Annu. Rev. Biochem. 44: 697.
Muller-Eberhard H.J., Nilsson U. (1960). Relation of a P glycoprotein of human
senun of the complement systern. J.Exp. Med. 1 1 1 : 2 1 7.
Muller-Eberhard H.J. (1988) Molecular organization and function of the
cornplement syçtem. AM. Rev. Biochem. 57: 32 1.
Muller-Eberhard H.J. (1986). The membrane attack complex of cornplement.
Ann. Rev. h u n o l . 4: 503.
Nakano Y., Sumida K., Kikuta N., Muira H., Toba T., and Tomita. (1992).
Complete determination of disulfide bonds localized within the short consensus
repeat units of decay accelerating factor (CD55 antigen). Biochem. Biophy. Acta
11 16: 235.
Nagasawa S., Ichihara C., and Stroud R (1980) Cleavage of C4b and C3b
inactivator: production of a nicked form of C3b and C4b, as an intemediate
cleavage product of C3b by C3b inactivator. J. Immunol. 125578.

1 12. Nelson R.A., Jensen J, Gigli 1, and Tamura N. (1 966). Methods for the
separation, purification and measurement of nuie cornponents of hemolytic
complement in guinea-pig senun. Immunochemistry 3: 1 1 1.
113. Nemerow G.R., Yamamoto K., Lint T.F. (1979). Restriction of C-mediated
membrane damage by the eigth component of complement: a dud role for C8 in
the complement attachment sequence. J.Immuno1. 123 : 1 245.
1 14. Nicholson-Weller A. (1 992). Decay accelerating factor (CD55). In Parker CM
ed. Membrane defenses against attack by complement and perforins. pp 7-30.
Berlin Springer.
115. Nilsson U.R., Tomar R.H., Taylor F.B. Jr. (1972). Additional shidies of human
CS development of a modified purification method and characterization of the
purified product by polyaczylamide gel electrophoresis. Immunochemisîry 9:
709.
116. Nuttal G. (1888). Experimente Uber die bakterienferdilichen einflusse des
thienschen korpers. Z Hyginfektionskr 4: 3 54.
117. Ogata R.T., Mathias P., Bradt B..M., and Cooper N.R. (1993). Murine C4b-
binding protein. Mapping of the ligand binding site and the N-terminal of the pre-
protein. J. Lmmunol. 150: 2273.
118. Pangburn M.K., Schreiber R.D. and Muller-Eberhard H.J. (1977) Human C3b
inactivator: isolation, characterization and demonstration of an absolute
requirement for s e m protein for cleavage of C3b and C4b in solution. J. Exp.
Med. 144: 257.

119. Pangbum M.K., and Muller-Eberhard H.J. (1978). Complement C3 convertase:
ceU surface Cestriction of B1H control and generation of restriction of
neuroaminidase treated cells. Proc. Na& Acad. Sci. USA 75: 2416.
120. Pangbum M.K., Muller-Eberhard H.J. (1984). The alternative pathway of
complement. Springer Semin. Immunopathol. 7: 163.
12 1. Pangbum M.K. (1 986). Differences between the binding sites of the complement
regulatory proteins DAF, CRI and factor H on C3 convertases. J. Immunol. 136:
2216.
122. Pangbum MK, Muller-Eberhard H.J. (1 980). Relation of a putative thioester
bond in C3 to activation of the alternative pathway and binding of C3b to
biological targets of complement. J.Exp. Med. 152: 1 102.
1 23. Pardo-Manuel F., Rey-Campos J., Hillarp A., Dahlback B. and Rodriguez-de-
Cordoba. (1 990). Human genes for the alpha and beta c h a h of complement
C4b-binding protein are closely linked in a head-to-tail mgement. Proc. Natl.
Acad. Sci. USA. 87: 4529.
124. Petenen F., Baatnip G., Jepsen H.H. and Svehag S.E. (1985). Complement
mediated solubilization of immune complexes and their interaction with
complement C3 receptors. Complement 2: 97.
125. Pillemer L., Blim L., Lepow, IH., Ross O.A., Todd E.W., Wardlaw A.C. (1954).
The properdine system and immunity. 1. DemoIlStration of a new serum protein
and its role in immune phenornena Science 120: 279.

126. Polley M.J., Muller-Eberhard H.J. (1 967). Enhancement of the hemolytic
activity of the second component of human complement by oxidation. J. Exp.
Med. 126: 1013.
127. Podack E.R., Biesecker G., Muller-Eberhard H.J., Membrane attack cornplex of
complement: generation of high affinity phospholipid binding sites by fusion of 5
hydrophilic plasma protek. Roc. Natl. Acad. Sci. USA. 76: 897.
128. Podack E.R., and Tschopp J. (1982). Polymerization of the ninth component of
complement (Cg) with a tubula. ultrastructure resembling the MAC of
complement. Proc. Natl. Acad. Sci. USA. 79: 574.
129. Podack E.R. (1977). The C5b-9 cornplex: formation, isolation properties and
subunit composition. J. Immunol. 1 19: 2024.
130. Post T.W., Arce M..A., Liszewski M.K., Thornpson ES., Atkinson J.P. and
Lublin D.M. (1990). Structure of the gene for human cornplanent protein decay
accelerating factor. J. Immunol. 144: 740.
13 1. Pugh B.F. and Tijian R (1991). Transcription nom a TATA-less prornoter
requires a multisubunit TFIID cornplex. Genes and Dev. 5: 1935.
132. Rao A.G., Howard O.M., Ng S C , Whitehead A.S., Colten H.R. and Sodetz J.M.
(1987). Complementary DNA and derived amino acid sequence of the alpha
subunit of human complement protein C8: evidence for the existence of a separate
alpha subunit messager RNA. Biochem. 26: 3556.
133. Rasmussen M, Teisner B., Jepsen H., Svehag S., Knudsen F., Kirstein H. and
Buhl M. (1988). Three cases of factor 1 deficiency the effect of treatment with
plasma. Clin. Exp. Immunol. 74: 13 1.

134. Raychowdhwy R, Niles J.L., McCluskey RT. and Smith J.A. (1989).
Autoimmune target in Hegmann nephritis is a glycoprotien with homology to the
LDL receptor. Science 244: 1 163.
135. Reid K.B.M., and Porter. (1981). The proteolytic activation systems of
complement. AMU. Rev. Biochem. 50: 433.
136. Reid K.B.M., and Day A.J. (1989). Structure-function relaîionships of the
complement cornponents. Irnmunol. Today 10: 1 77.
137. Reid K.B.M. (1993). Structure-function relationships in the collectins. Biochem.
Soc. Trans. 21: 464.
138. Ripoche I., Day AL, Harris T.J.R. and Sim RB. (1988). The complete amino
acid sequence of huma. complement factor H. Biochem. 1.249: 593.
1 39. Rodriguez-de-Cordoba S., Sanchez-Corral P. and Rey-Compos J. (1 99 1).
Structure of the gene coding for the alpha polypeptide chain of the huma.
complement component C4b-binding protein. J. Exp. Med. 173 : 1073.
140. Ross S.C. and Densen P. (1984). Complement deficiency states and infection:
epidemology, pathogenesis and consequences of Neissenal and other infections in
an immune deficiency. Medicine 63: 243.
141. Ross G.D., Lambris J.D., Cain J.A., and Newman S.C. (1982). Generatioo of
three different fragments of bound C3 with purified factor 1 or serum 1.
Requirernent for factor H vs CR1 cofactor activity. J. Immunol. 129: 205 1.
142. Ross G.D., and Polley M.J. (1975). Specincity of human Lymphocyte
complement receptors. J. Exp. Med. 145: 1 163.

Roy A.L., Meisterernst M., Pognonec P., Roeder R.G. (1991). Cooperative
interaction of an hitiator- binding transcription intiation factor and the helix-loop-
helix activation USF. Nature 354: 245.
Roy A. L., Malik S., Meisterenst M., and Roeder R. G. (1993). An alternative
pathway for transcription initiation involving TF 1 1 - 1. Nature 365 : 3 55.
Sambrook J., Fritsch E.F., and Maniatis. (1989). Molecular cloning. A
Laboratory manual. Second edition. Cold Spring Harbor Laboratory Ress, Cold
Spring Harbor, NY.
Sanger F., Nicklen S., and Coulson A. R. (1977). DNA sequencing with chain-
tenninating inhibitors. Proc. Natl. Acad. Sci. USA. 74: 5463.
Sharma A. and Pangbum M.K. (1996). Identification of three physicaly and
functionally distinct binding sites for C3b in human complement factor H by
deletion mutagenesis. hoc. Natl. Acad. Sci. 93: 10996.
Sastry K., Heman G.A., Day L., Deignam E., Bruns G., Morton C.C., Ezekowitz
R.A.B. (1989). The human mannose binding protein gene. Exon structure
reveals its evolutionary relationship to a human pulmonary surfactant gene and
locakation to chromosome 10. J. ExpMed. 170: 1 175.
Schreiber E., Manhias M., Muiler M.M., Schaflker W. (1989). Rapid detection
of octamer binding protein with "mini-extract" prepared from a mal1 number of
cells. Vol 17 (15): 6419.
Scott V., Clark A. and Docherty K. (1994). Methods in molecular biology. Vol.
3 1. Protocols for gene analyçis. Edited by Harwood A.J. Humania Press
Inc. Totowa N.J.

Seya T., and Akinson I.P. (1989). Functional properties of membrane cofactor
protein of complement. Biochem. J. 264: 58 1.
Seya T., Turner J.R., Atkinson J.P. (1986). Purification and characterization of a
membrane protein (gp 45-70) that is a cofactor for cleavage of C3b and C4b. J.
Exp. Med. 163: 837.
Seya T., Hara T., Matsumoto M., Sugita Y. and Akedo H. (1990). Complement
mediated tumor ce11 damage induced by antibodies against membrane cofactor
protein (MCP, CD 46). 1. Exp. Med. 172: 1673.
Seya T., Ballard L.L., Bora N. S., Kumar V., Cui W. and Atkinson J. P. (1988).
Distribution of membrane cofactor protein of the complement of human
peripheral blood cells. An altered form is found on granulocytes. Eur. J.
Immunol. 139: 1260.
Shi Y., Seto E., Chang L.S., and Shrenk T. (1 99 1). Transcriptional repression by
W 1 , a human GLI-Kruppel rdated protein and relief of repression by
Adenovirus El A protein. Ce11 67: 377.
Shin M.L., Paznekas W..A., Abramovitz AS. (1977) On the mechanisrn of
membrane damage by complement: exposture of hydrophobie sites on activated
complernent proteins. J. Imnunol. 1 19: 13 58.
Shiang R., Murray J.C., Morton C., Buetow K., Wasmuth J., Olney A.H., Sanger
W., and Goldberger G. (1989). Mapping of the Human complernent factor 1 gene
to 4qZ. Genomics 4: 82.
Sidle A. (1995). Huma. complernent component C3: Mapping of contact
residues for C3 binding proteins by site-directed mutagenesis. Thesis. U of T

Smale, S. T. (1994). Core promoter architecture for eukaryotic protein-coding
genes. In Transcription: Mechanisms and Regdation, R C. Conaway and J. W.
Conaway, eds. (New York: Raven Press), pp. 63-8 1.
Smale S.T., Schmidt M., Berk A. and Baltimore D. (1990). Transcriptional
activation by Spl as directed through TATA or Initiator: specific requirment for
mammalian transcription factor IID. Roc. Natl. Acad. Sci. USA. 87: 4509.
Smale, S.T. and Baltimore D. (1989). The initiator as transcriptional control
element. Ce11 5 7: 1 03.
Smith C.A., Pangbum M.K., Vogel C. W., and Muller-Eberhard H.J. (1984).
Molecular architecture of human properdin, a positive regulator of the alternative
pathway of complement. J. Biol. Chem. 259: 4282.
StanIey K.K., Kocher H.P., Lu50 J.P., Jackson P., Tschopp L (1985). The
sequence and topology of human complement component Cg. EMBO 4: 375.
Stanley K.K., Page M., Campbell A.K., and Luzio J.P. (1986). A mechanism for
the insertion of the complement component C9 into target membranes. Mol.
Immmol. 23: 45 1.
Sudhof T.C., Goldstein J.L., Brown M.S., and Russel D.W. (1985a). The LDL
receptor gene: A mosaic of exons shared with different proteins. Science 228:
8 15.
Sullivan K-E., Wu L.C., Campbell R.D., Vdle D. (1994). Transcriptional
regulation of the gene for the second complement component of human
complement: promoter analysis. Eur. J. Immunol. 24: 393.

167. Tack B.F. and Prahl J.W. (1976). Third component of human complement
purification h m plasma and physiochemical characerization. Biochemist. 15:
4513,
168. Tanese N., Pugh B.F. and Tijan R. (1991). Coactivators for a proline rich
activator purified fiom the multisubunit human TRID complex. Genes and Dev.
5 : 2212.
169. Teisner B., Brandslund I., Foikersen J., Rasmussen LM., Poulsen L.O., Svehag
S.E. (1984). Factor 1 deficiency and C3 nepbritic factor: Immunochemical
findings and association with Nisseria meaingitides infection in two patients.
Scand. J. Immunol. 20: 291.
1 70. Thomas D., and Lubin D. (1 993). Identification of 5' flanking regions affecthg
the expression of the huma. decay accelerating factor gene and th& role in tissue
specific expression. J. Immunol. 150: 15 1.
171. Tijian R., and Maniatis T. (1994). Transcriptional activation: a complex puzzle
with few easy pieces. Cell77: 5.
172. Tshcopp J., Muller-Eberhard H.J. and Podack E.R. (1982). Formation of
transmembrane tubules by spontaneous polymenzation of the hydrophilic
complement component protein Cg. Nature 298: 534.
173. Usheva A., and Shenk T. (1994). TATA-binding protein independent initiation:
W1, TFIlB and RNA polymerase II direct basal transcription on supercoiled
template DNA. Cell76: 11 15.

174. Venijzer C.P., Yokomori K., Chen J-L and Tijian R (1994). Drosophila TAF,
150: similarity to yeast gene TSM-1 and specific binding to core promoter DNA.
Science 264: 93 3.
175. Vemjzer C.P., Yokomon K., Chen T.L. and Tijian R. (1 995). Binding of TAFs
to core elements directs promoter selectivity by RNA polymerase II. Ce11 81:
1115.
176. Vik D.P., Amiguet P., Moffat G.J., Fey M., Amiguet-Barras F.,Wetsel R.A., Tack
B.F. (1 99 1). Structural features of the human C3 gene: htrordexon organization,
transcriptional start site and promoter region sequence. Biochem. 30: 1 080.
177. Vyse T.J., Spath P.J., Davies KA., Morley B.J., Philipp P., Athanassiou P., Giles
C., and Walport M.J. (1994) Hereditary complement factor 1 deficiency. QJM
385.
178. Vyse T.J., Bates G.P., Walport M.J., and Morley B.J. (1994) The organization of
the Human complement factor 1 gene (IF): A member of the serine protease gene
family. Genomics 24: 90.
179. Weiler J.M., Daha M.R., Austen KF. and Fearon D.T. (1976). Control of the
amplification convertase of cornplement by the two plasma protein BM. hoc.
Natl. Acad. Sci. USA. 80: 3461.
180. Weis and Reinberg. (1992). Transcription by RNA polymerase II: Initiator
directed formation of transcription competent complexes. FASEB, Vol. 16: 3300.
181. West C.D. (1989) The complement profile in Clinical Medicine. Complement
Inflamm. 6: 49.

William S.A. and Vik D.P. (1997). Characterization of the 5' flanking region of
the human complement factor H gene. Scand. J. h u n o l . 45: 7.
Wong M., Goldberger G., Isenman D. and Minta J.O. (1995) Processing of
Human factor I in COS-1 ceils CO-transfected with factor 1 and paired basic amino
acid cleaving enzyme (PACE) cDNA. Mol. Immunol. 32: 379.
Zahedi K., Prada A.E., Davies A.E. (1 994). Transcriptional regdation of the C 1 -
inhibitor gene by gamma interferon. J. Biol. Chem. 269 (13): 9669.
Wasylyk B., Hahn S.L., and Giovane A. (1993). The Ets family of transcription
factors. Eur. J. Biochem. 21 1: 7.
Wu L.C., Morley B.J. and Campbell RD. (1987). Cell-specific expression of
the human complement protein factor B gene: Evidence for the role of two
distinct 5'-flanking elements. Celi 48: 33 1.
Yamamoto T., Davies CG., Brown M.S., Schneider W.J., Casey M.L., Goldstein
J.L. and Russel D. W. (1 985). The human LDL receptor: a cysteine-rich protein
with multiple Alu sequences in its mRNA. Ceil 39: 27.
Zalman L. S., Wood L. M., and Muller Eberhard H. J. (1986). Isolation of a
human erythrocyte membrane protein capable of inhibithg expression of
homologous complement trammembrane channnels. Froc. Natl. Acad. Sci.
USA 83: 6975.
Zhang Y ., Lichtenheld M.G. (1 997). Non kikr ceII-specific transcription factors
silence the perforin promoter. Journal of Tmmunol. 158: 1734.
Ziccardi RJ., and Cooper N.R. (1979). Active disassembly of the first
complement component, C 1, by C 1 inactivator. J. Tmmunol. 123: 788.

9 1 Ziccardi R.J., and Cooper N.R (1977). The subunit composition and
sedimentation properties of human C 1. J. h u n o l . I 1 8: 2047.