Embed Size (px)
Citation preview

Gastrointestinal Drugs Advisory CommitteeIntroductory Remarks
NDA203214Tofacitinib: treatment of adults with moderate to severely active ulcerative colitis
Tara Altepeter, MDClinical Team Leader
Division of Gastroenterology and Inborn Errors Products
Office of Drug Evaluation III
Center for Drug Evaluation and Research, FDA

2
Disease Overview• Ulcerative colitis (UC) is a chronic, inflammatory disease affecting the
colon• Currently there is no cure• More than 900,000 US adults are affected• Patients experience recurrent flares of abdominal pain and bloody
diarrhea, which may be associated with various extra-intestinal manifestations
• Available treatments are aimed at controlling symptoms, reducing complications, avoiding the need for surgery, and reducing the long term risk of colon cancer

3
Product Overview• Drug: Tofacitinib• Class: Janus Kinase (JAK) Inhibitor • Mode of Administration: oral tablet• Proposed indication:
– Treatment of adults with moderate to severely active UC who have had inadequate response, loss of response, or intolerance to: corticosteroids, azathioprine (AZA), 6-mercaptopurine (6-MP), or tumor necrosis factor (TNF) blocker therapy.

4
FDA Approved
Treatments
Year of Approval
ClassEfficacy Information
Clinical RemissionBoxed Warning
Infliximab
(Remicade®)2005
TNF blocker
Study 1: 32% -39% at Week 8
20% at Week 8, 30 and 54
Study 2: 28-34% at Week 8
Serious infections including tuberculosis (TB),malignancies, hepatitis B virus reactivation, hepatotoxicity, hypersensitivity, cytopenias, demyelinating disease, heart failure, lupus-like syndrome
Adalimumab
(Humira®)2011
TNF blocker
Study I: 18.5% at week 8
Study II: 16.5% at Week 8.
8.5% at both Weeks 8 and 52
Serious infections including TB, bacterial sepsis, invasive
fungal infections, and opportunistic infections,
malignancies, anaphylaxis or serious allergic reactions,
hepatitis B virus reactivation, demyelinating disease,
cytopenias/pancytopenia, heart failure, lupus-like
syndrome.
Golimumab
(Simponi®)2013
TNF blocker
Study I: 18% of at Week 6
Study II: 28% at both Week 30 and Week
54
Serious infections including TB, invasive fungal
infections, hepatitis B reactivation, malignancies ,
congestive heart failure, demyelinating disorders,
hematologic cytopenias, Lupus-like syndrome,
hypersensitivity reactions
Vedolizumab
(Entyvio®)2014
Anti-integrin Agent
Study I: 17% at Week 6
Study II: 42% at Week 52
21% at both Weeks 6 and 52
Serious infections including anal abscess, sepsis (some
fatal), tuberculosis, salmonella sepsis, Listeria
meningitis, giardiasis, cytomegaloviral colitis.
FDA Approved Chronic Treatments for Moderate to Severe UC

5
Regulatory History• Rheumatoid Arthritis
– Initial approval: 5mg twice daily (BID) 2012– Extended release formulation 11mg once daily (QD) subsequently
approved 2016
• Plaque Psoriasis– Complete Response 2015
• Psoriatic Arthritis – Recently approved: 5mg BID 2017
• Ulcerative Colitis – Application submitted on May 4, 2017

6
Drug Development Considerations in Inflammatory Bowel Disease
• Phase 2 – Adequate dose finding
– Exploration of time to response/remission
• Phase 3– Confirmation of efficacy and broader understanding of
safety
– Relevant subgroup analyses

7
Applicant’s Proposed DosingAdults with moderate to severely active UC who have had inadequate response, loss of response, or intolerance to: corticosteroids, azathioprine, 6-mercaptopurine, or TNF blocker therapy.
• Induction: – 10 mg twice daily for 8 weeks– For patients who do not achieve adequate therapeutic benefit (response) by Week 8, the induction dose
of 10 mg twice daily can be extended for an additional 8 weeks (16 weeks total). – Discontinue therapy in patients who do not achieve adequate therapeutic benefit by Week 16.
• Maintenance: – 5 mg twice daily– For those with an inadequate response, loss of response, or intolerance to TNF blocker therapy: 10 mg
twice daily

8
Questions to Committee (1):1) The Applicant has proposed an induction dosing regimen of 10 mg BID for a total for 16 Weeks in patients who have not achieved “adequate therapeutic benefit” by Week 8 based on exploratory analyses of trial data in patients who continued induction treatment when they had not achieved clinical response defined as a decrease from baseline in Mayo score of ≥3 points and ≥ 30%, with an accompanying decrease in the subscore of rectal bleeding of ≥ 1 point or absolute subscore for rectal bleeding of 0 or 1.
a) Discuss the adequacy of the efficacy data to support the use of the 10 mg BID dosing for extended induction therapy for a total of 16 weeks in patients who have not achieved “adequate therapeutic benefit” by Week 8.
b) Discuss the adequacy of the safety data to support the use of the 10 mg BID dosing for induction for a total of 16 weeks in patients who have not achieved “adequate therapeutic benefit” by Week 8.
c) Do you recommend the inclusion of this dosing regimen for this population in the product label?
d) If you recommend inclusion of this dosing regimen in the product label, discuss how inadequate therapeutic benefit that merits extension of induction treatment should be distinguished from inadequate therapeutic benefit that should prompt discontinuation of tofacitinib therapy.

9
Questions to Committee (2):2) For adult patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to TNF blocker therapy:
a) Discuss the adequacy of the efficacy data to support the use of the 10 mg BID dosing as continuous maintenance treatment.
b) Discuss the adequacy of the safety data to support the use of the 10 mg BID dosing as continuous maintenance treatment.
c) Do you recommend inclusion of this dosing regimen for this population in the product label?
d) Do you recommend that the Applicant conduct a post-marketing efficacy trial in this population comparing a 10 mg BID continuous dosing regimen vs a regimen of 10 mg induction and 5mg BID as maintenance?

10
Questions to Committee (3):3) For discussion (non-voting):
– Discuss if additional post-marketing evaluation of safety is warranted, and the mechanism(s) you recommend (e.g., registry, observational study, etc.) for such evaluation.
4) For discussion (non-voting):
a) Are there unique characteristics of the pediatric UC population that should be taken into account when planning the tofacitinib pediatric development program? In particular, consider the ontogeny of the immune system and the described mechanism of action of tofacitinib.
b) Given the safety concerns (malignancy and serious infections) described with long term use of 10mg BID and the severity of UC in the pediatric population, please recommend the maximum dose that should be targeted for evaluation for long term treatment in pediatric UC.
c) Discuss whether you recommend limiting enrollment in the pediatric trials (and subsequent pediatric indications) to patients who have failed other biologic therapies.


FDA Gastrointestinal Drugs Advisory Committee (GIDAC)
Introduction of FDA Speakers and Presentations
NDA 203214
Tofacitinib for the Treatment of Adult Patients with Moderately to Severely Active Ulcerative Colitis (UC)
Lesley S. Hanes, MD MSc
Division of Gastroenterology and Inborn Errors Products
Center for Drug Evaluation and Research
March 8, 2018

13
Introduction of FDA Presentations
1. Clinical Pharmacology Dose Selection: Dilara Jappar, PhD
2. Statistical Efficacy Evaluation: Sara Jimenez, PhD
3. Clinical Safety Evaluation: Lesley Hanes, MD MSc
4. Remarks About Results from Truven MarketScan® Database: Joel Weissfeld, MD MPH
5. Pediatric Considerations: Melanie Bhatnagar, MD


Clinical Pharmacology Findings of Tofacitinib for Treatment of Moderately to Severely Active
Ulcerative Colitis (UC)
NDA 203214/S018
Dilara Jappar, Ph.D.
Fang Li, Ph.D.
Office of Clinical Pharmacology
Office of Translational Sciences
Center for Drug Evaluation and Research
March 8, 2018

16
Outline
• Exposure Comparison among Disease Populations
• Dose Selection Rationale for Phase 3 Induction Studies
• Exposure–Response Analysis for Efficacy in Maintenance Trial
www.fda.gov
17
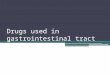
Systemic Exposure Comparison
• Similar systemic exposure between UC, PsO, and PsA at 10 mg BID dose
www.fda.gov
PK Parametersa
Geometric Mean (CV%)
5 mg BID
UC PsO PsA RA
AUC0-24,ss (ng·h/mL)423
(22.6%)
404
(22.5%)
419
(34.1%)
504
(22.0%)
a: PK parameters estimated based on population PK analysis;
UC=Ulcerative Colitis; PsO= Plaque psoriasis; PSA= psoriatic arthritis; RA=Rheumatoid Arthritis

18
Outline
• Exposure Comparison among Disease Populations
• Dose Selection Rationale for Phase 3 Induction Studies
• Exposure–Response Analysis for Efficacy in Maintenance Trial
www.fda.gov
19
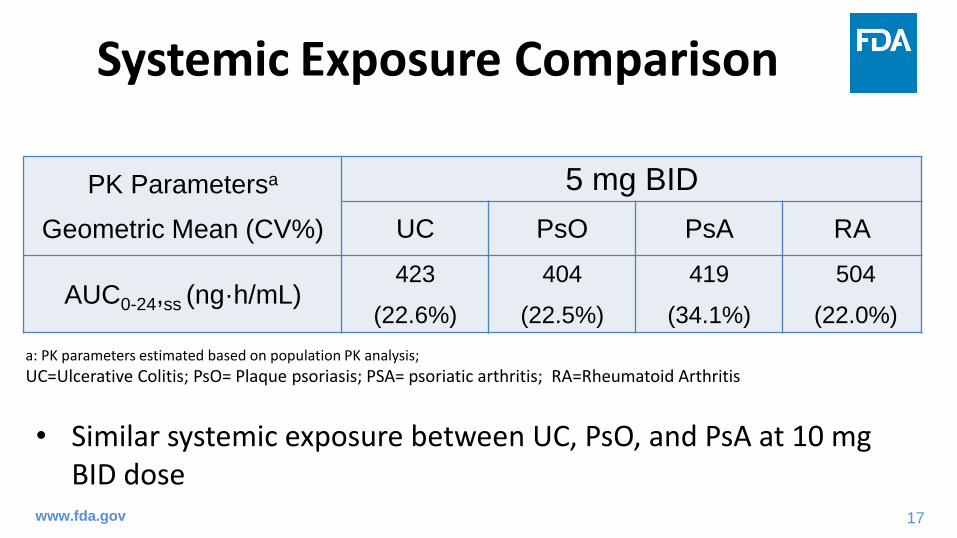
Phase 2 Dose-Ranging Trial for Induction
www.fda.gov
Ran
do
miz
atio
n 10 mg BID
3 mg BID
Placebo BID
0.5 mg BID
15 mg BID
Weeks 0 2-1-3 4 128-2
Efficacy Follow-Up
Safety
End
of
Tre
atm
en
t
Screening
Week -1 to -3
Study 1063
20
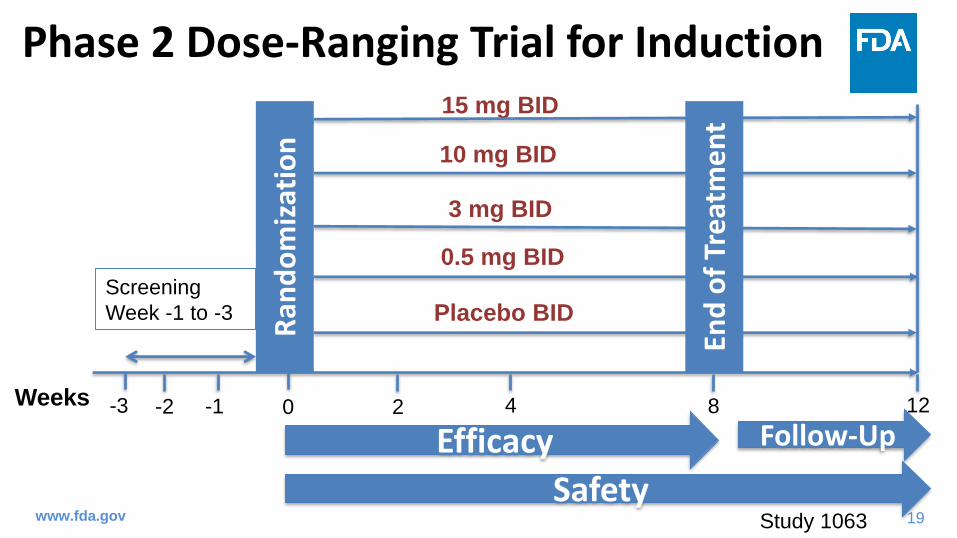
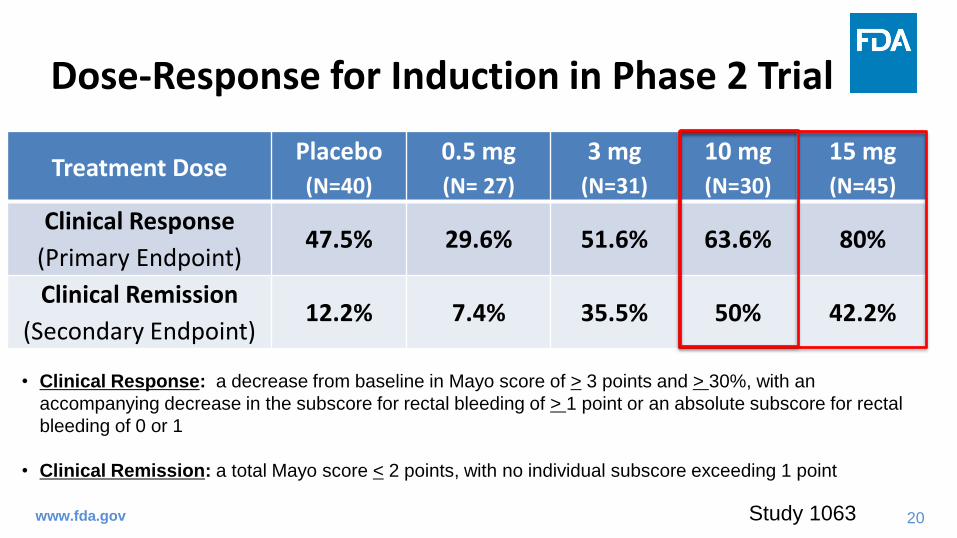
Dose-Response for Induction in Phase 2 Trial
www.fda.gov
Treatment DosePlacebo
(N=40)
0.5 mg
(N= 27)
3 mg
(N=31)
10 mg
(N=30)
15 mg
(N=45)
Clinical Response
(Primary Endpoint)47.5% 29.6% 51.6% 63.6% 80%
Clinical Remission
(Secondary Endpoint)12.2% 7.4% 35.5% 50% 42.2%
• Clinical Response: a decrease from baseline in Mayo score of > 3 points and > 30%, with an
accompanying decrease in the subscore for rectal bleeding of > 1 point or an absolute subscore for rectal
bleeding of 0 or 1
• Clinical Remission: a total Mayo score < 2 points, with no individual subscore exceeding 1 point
Study 1063
21
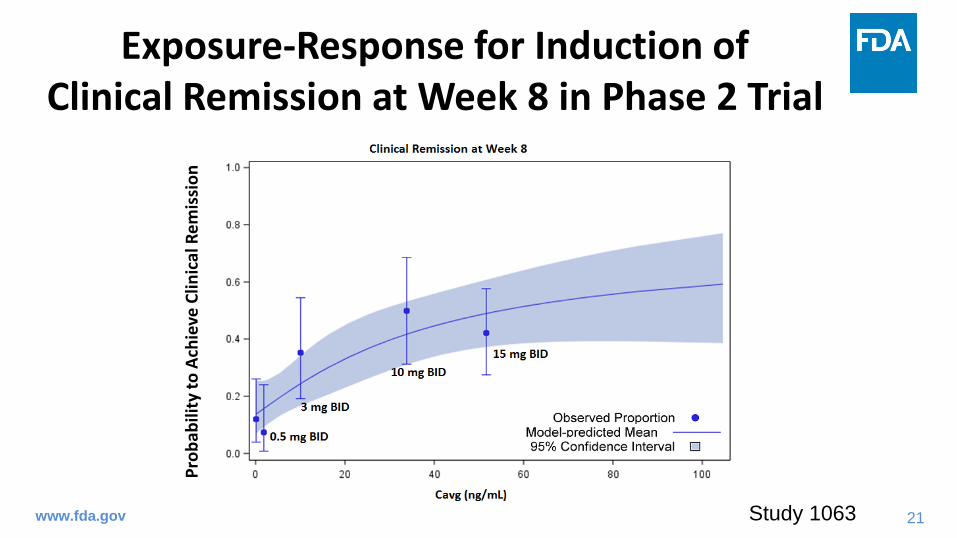
Exposure-Response for Induction of Clinical Remission at Week 8 in Phase 2 Trial
www.fda.gov
Pro
bab
ility
to
Ach
ieve
Clin
ical
Re
mis
sio
n
Study 1063

22
Dose Selection for Phase 3 Trials
• The dose-response and exposure-response relationship in phase 2 trial supported the selection of 10 mg BID dose for induction of remission in phase 3 trials.

23
Would 5 mg BID Dose be as Efficacious as 10 mg BID Dose for Induction of Remission?
www.fda.gov
24
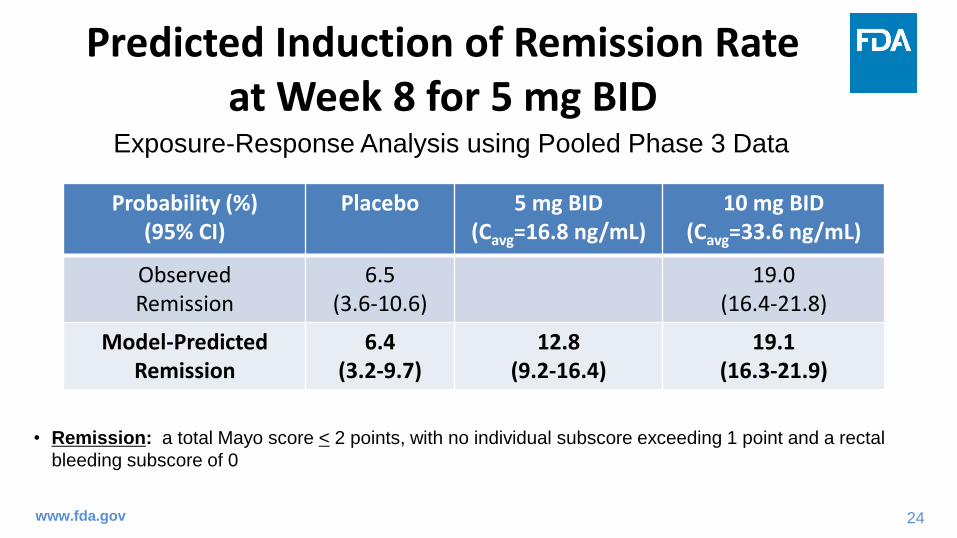
Predicted Induction of Remission Rate at Week 8 for 5 mg BID
www.fda.gov
Probability (%)(95% CI)
Placebo 5 mg BID(Cavg=16.8 ng/mL)
10 mg BID(Cavg=33.6 ng/mL)
Observed Remission
6.5(3.6-10.6)
19.0(16.4-21.8)
Model-Predicted Remission
6.4(3.2-9.7)
12.8(9.2-16.4)
19.1(16.3-21.9)
• Remission: a total Mayo score < 2 points, with no individual subscore exceeding 1 point and a rectal
bleeding subscore of 0
Exposure-Response Analysis using Pooled Phase 3 Data

25
Outline
• Exposure Comparison Among Disease Populations
• Dose Selection Rationale for Phase 3 Induction Studies
• Exposure–Response Analysis for Efficacy in Maintenance Trial
www.fda.gov
26
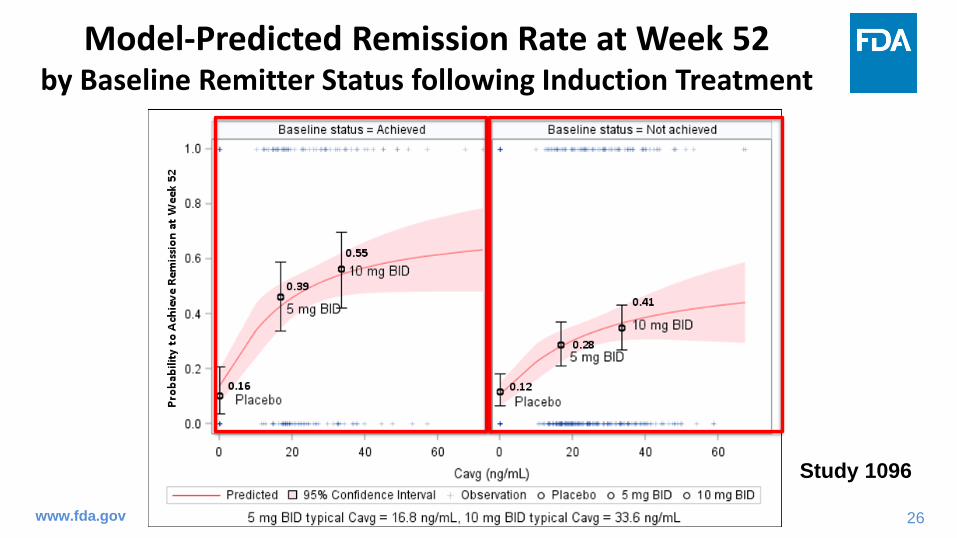
Model-Predicted Remission Rate at Week 52 by Baseline Remitter Status following Induction Treatment
www.fda.gov
Study 1096
27
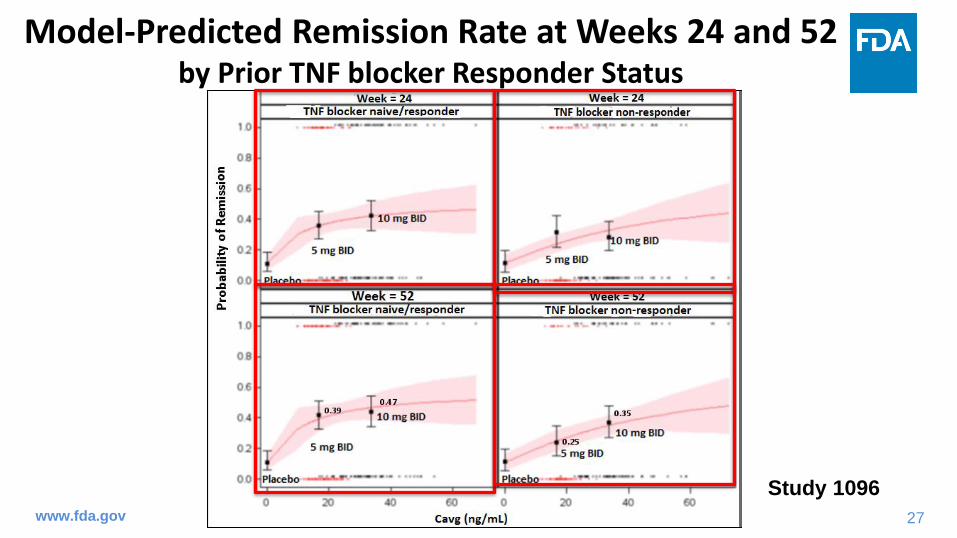
Model-Predicted Remission Rate at Weeks 24 and 52by Prior TNF blocker Responder Status
www.fda.gov
Study 1096

28
Summary• UC patients have similar systemic exposure to patients with plaque
psoriasis and psoriatic arthritis and about 20% lower exposure than rheumatoid arthritis patients at both 5 mg BID and 10 mg BID doses.
• The dose/exposure dependent increase in clinical remission rate supported the selection of 10 mg BID dose for induction treatment in phase 3 trials.
• In maintenance treatment, the 10 mg BID dose showed numerically higher remission rate at week 52 than the 5 mg BID dose regardless of baseline remitter status or prior TNF blocker responder status.
www.fda.gov


NDA 203214Tofacitinib (Xeljanz)
Analyses of Efficacy Data for Proposed Dosing Regimens
Sara Jimenez, PhDOffice of Biostatistics, CDER
March 8, 2018

31
Outline• Overview of studies in the New Drug Application (NDA)
• Dosing regimens for ulcerative colitis (UC)
• Evidence regarding the applicant’s additional dosing regimens for two subgroups
o Induction non-responders
o TNF blocker failures

32
Overview of Studies
www.fda.gov
33
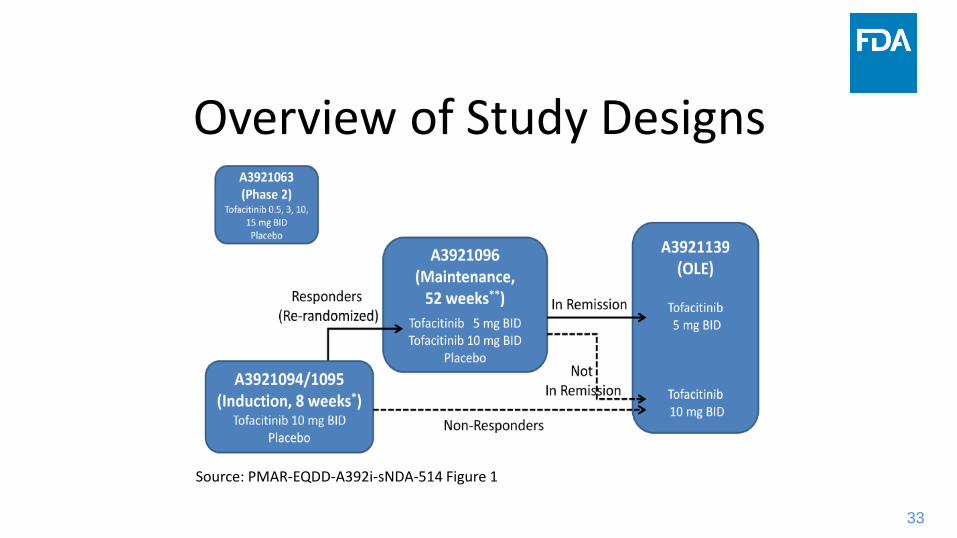
Overview of Study Designs
Source: PMAR-EQDD-A392i-sNDA-514 Figure 1
34
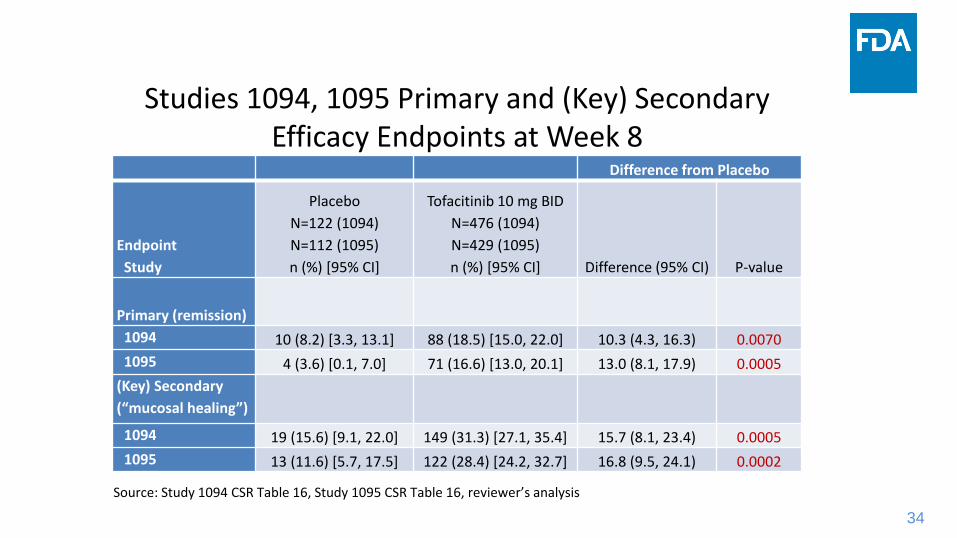
Studies 1094, 1095 Primary and (Key) Secondary Efficacy Endpoints at Week 8
Difference from Placebo
Endpoint
Study
Placebo
N=122 (1094)
N=112 (1095)
n (%) [95% CI]
Tofacitinib 10 mg BID
N=476 (1094)
N=429 (1095)
n (%) [95% CI] Difference (95% CI) P-value
Primary (remission)
1094 10 (8.2) [3.3, 13.1] 88 (18.5) [15.0, 22.0] 10.3 (4.3, 16.3) 0.0070
1095 4 (3.6) [0.1, 7.0] 71 (16.6) [13.0, 20.1] 13.0 (8.1, 17.9) 0.0005
(Key) Secondary
(“mucosal healing”)
1094 19 (15.6) [9.1, 22.0] 149 (31.3) [27.1, 35.4] 15.7 (8.1, 23.4) 0.0005
1095 13 (11.6) [5.7, 17.5] 122 (28.4) [24.2, 32.7] 16.8 (9.5, 24.1) 0.0002
Source: Study 1094 CSR Table 16, Study 1095 CSR Table 16, reviewer’s analysis
35
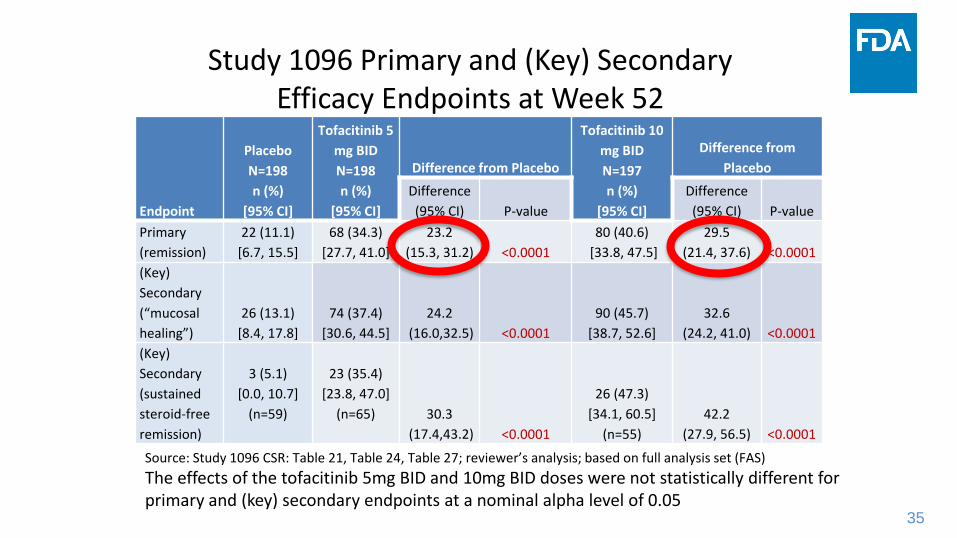
Study 1096 Primary and (Key) Secondary Efficacy Endpoints at Week 52
Endpoint
Placebo
N=198
n (%)
[95% CI]
Tofacitinib 5
mg BID
N=198
n (%)
[95% CI]
Difference from Placebo
Tofacitinib 10
mg BID
N=197
n (%)
[95% CI]
Difference from
Placebo
Difference
(95% CI) P-value
Difference
(95% CI) P-value
Primary
(remission)
22 (11.1)
[6.7, 15.5]
68 (34.3)
[27.7, 41.0]
23.2
(15.3, 31.2) <0.0001
80 (40.6)
[33.8, 47.5]
29.5
(21.4, 37.6) <0.0001
(Key)
Secondary
(“mucosal
healing”)
26 (13.1)
[8.4, 17.8]
74 (37.4)
[30.6, 44.5]
24.2
(16.0,32.5) <0.0001
90 (45.7)
[38.7, 52.6]
32.6
(24.2, 41.0) <0.0001
(Key)
Secondary
(sustained
steroid-free
remission)
3 (5.1)
[0.0, 10.7]
(n=59)
23 (35.4)
[23.8, 47.0]
(n=65) 30.3
(17.4,43.2) <0.0001
26 (47.3)
[34.1, 60.5]
(n=55)
42.2
(27.9, 56.5) <0.0001
Source: Study 1096 CSR: Table 21, Table 24, Table 27; reviewer’s analysis; based on full analysis set (FAS)
The effects of the tofacitinib 5mg BID and 10mg BID doses were not statistically different for primary and (key) secondary endpoints at a nominal alpha level of 0.05

36
Dosing Regimens
www.fda.gov

37
Applicant’s Pre-specified Tofacitinib Dosing Regimen
• For adult patients with moderate to severely active UC, 10mg twice daily (BID) for induction for 8 weeks and 5mg BID for maintenance
o Results for the pre-specified endpoints in this dosing regimen were statistically significant
www.fda.gov

38
Applicant’s Proposed Dosing Regimens for Two Subgroups
www.fda.gov

39
Additional Dosing Regimen (Induction Non-responders)
• For patients who do not achieve adequate therapeutic benefit by week 8 (induction non-responders), the induction dose of 10 mg BID can be extended for an additional 8 weeks (16 weeks total), followed by 5 mg BID for maintenance
• Discontinue therapy in patients who do not achieve adequate therapeutic benefit by week 16
www.fda.gov

40
Additional Dosing Regimen (Prior TNF Blocker Failure)
• For patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to TNF blocker therapy, 10 mg BID for maintenance may be considered in order to maintain therapeutic benefit
o Refractory patients are defined as those with a history of prior TNF blocker failure
www.fda.gov

41
Induction Non-responders• Remission results for
o Locally read endoscopies
o Centrally read endoscopies
• Focus on patients with 16 weeks (active treatment during induction) of tofacitinib 10mg BID

42
Exploratory Analyses for Induction Non-responders
• Patients in Study 1094/1095 who did not achieve clinical response by week 8, and who were then treated with an additional 8 weeks of tofacitinib 10mg BID in the open-label extension Study 1139
• Patients in this group may have received o 8 weeks (placebo during induction) of tofacitinib 10mg
BID, or o 16 weeks (active treatment during induction) of tofacitinib
10mg BID
www.fda.gov
43
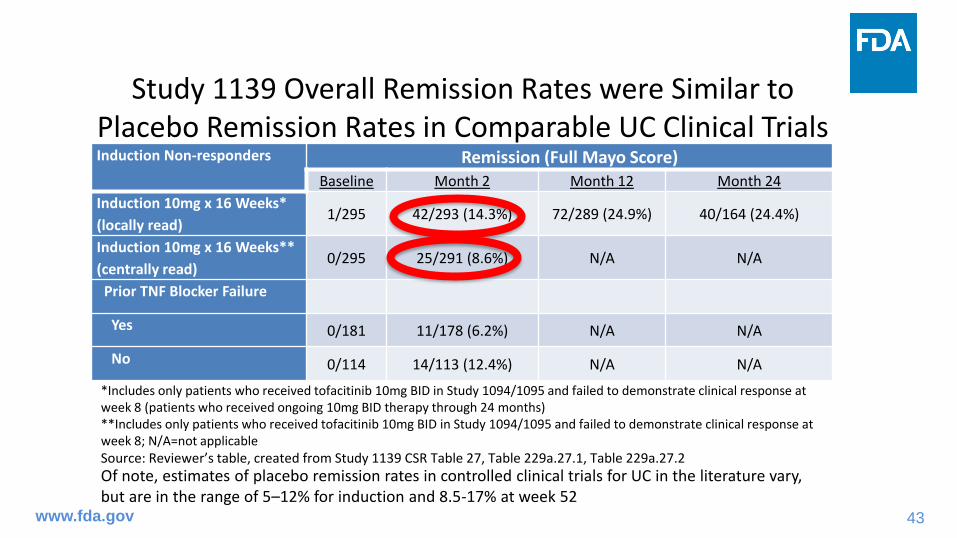
Study 1139 Overall Remission Rates were Similar to Placebo Remission Rates in Comparable UC Clinical TrialsInduction Non-responders Remission (Full Mayo Score)
Baseline Month 2 Month 12 Month 24
Induction 10mg x 16 Weeks*
(locally read)1/295 42/293 (14.3%) 72/289 (24.9%) 40/164 (24.4%)
Induction 10mg x 16 Weeks**
(centrally read)0/295 25/291 (8.6%) N/A N/A
Prior TNF Blocker Failure
Yes 0/181 11/178 (6.2%) N/A N/A
No 0/114 14/113 (12.4%) N/A N/A
www.fda.gov
*Includes only patients who received tofacitinib 10mg BID in Study 1094/1095 and failed to demonstrate clinical response at week 8 (patients who received ongoing 10mg BID therapy through 24 months)**Includes only patients who received tofacitinib 10mg BID in Study 1094/1095 and failed to demonstrate clinical response at week 8; N/A=not applicableSource: Reviewer’s table, created from Study 1139 CSR Table 27, Table 229a.27.1, Table 229a.27.2
Of note, estimates of placebo remission rates in controlled clinical trials for UC in the literature vary, but are in the range of 5–12% for induction and 8.5-17% at week 52

44
Patients with Prior TNF Blocker Failure
www.fda.gov
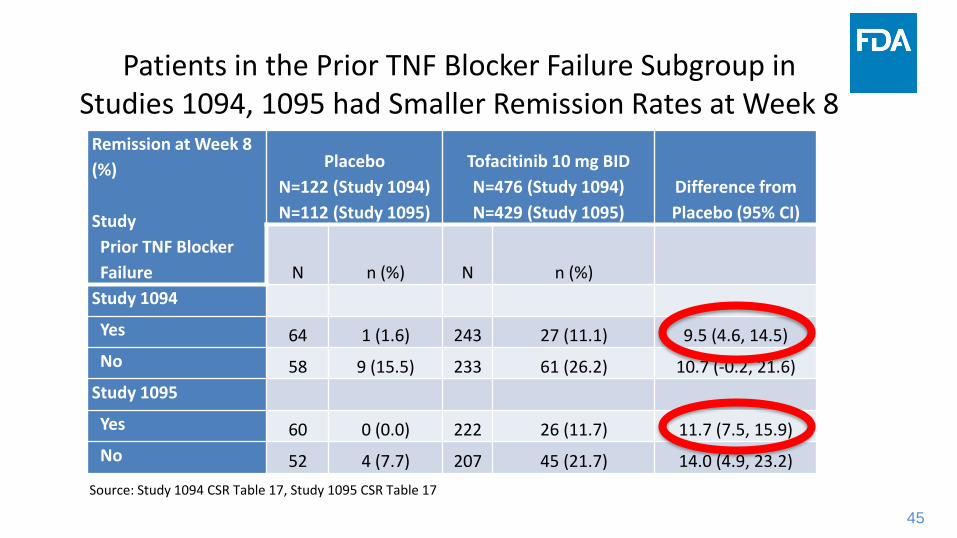
45
Remission at Week 8
(%)
Study
Prior TNF Blocker
Failure
Placebo
N=122 (Study 1094)
N=112 (Study 1095)
Tofacitinib 10 mg BID
N=476 (Study 1094)
N=429 (Study 1095)
Difference from
Placebo (95% CI)
N n (%) N n (%)
Study 1094
Yes 64 1 (1.6) 243 27 (11.1) 9.5 (4.6, 14.5)
No 58 9 (15.5) 233 61 (26.2) 10.7 (-0.2, 21.6)
Study 1095
Yes 60 0 (0.0) 222 26 (11.7) 11.7 (7.5, 15.9)
No 52 4 (7.7) 207 45 (21.7) 14.0 (4.9, 23.2)
Patients in the Prior TNF Blocker Failure Subgroup in Studies 1094, 1095 had Smaller Remission Rates at Week 8
Source: Study 1094 CSR Table 17, Study 1095 CSR Table 17
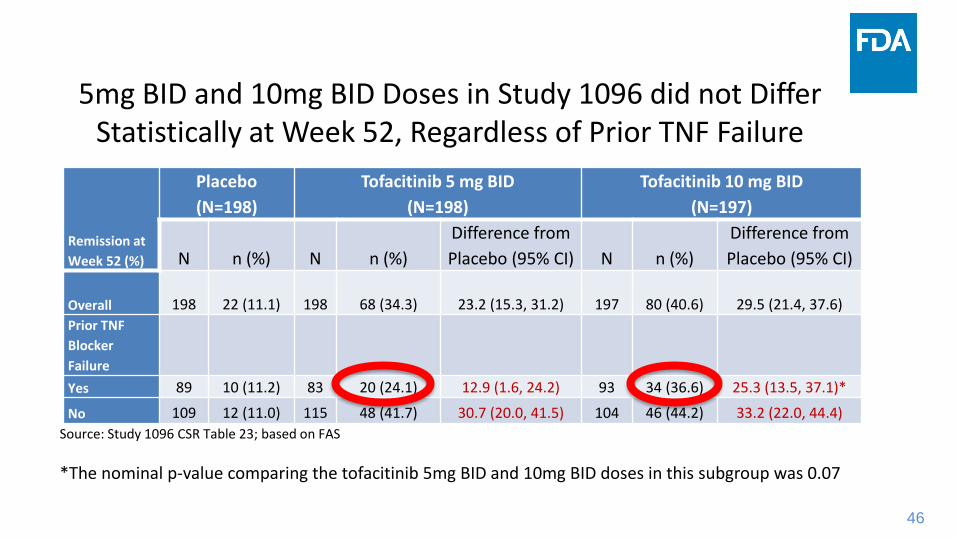
46
5mg BID and 10mg BID Doses in Study 1096 did not Differ Statistically at Week 52, Regardless of Prior TNF Failure
Remission at
Week 52 (%)
Placebo
(N=198)
Tofacitinib 5 mg BID
(N=198)
Tofacitinib 10 mg BID
(N=197)
N n (%) N n (%)
Difference from
Placebo (95% CI) N n (%)
Difference from
Placebo (95% CI)
Overall 198 22 (11.1) 198 68 (34.3) 23.2 (15.3, 31.2) 197 80 (40.6) 29.5 (21.4, 37.6)
Prior TNF
Blocker
Failure
Yes 89 10 (11.2) 83 20 (24.1) 12.9 (1.6, 24.2) 93 34 (36.6) 25.3 (13.5, 37.1)*
No 109 12 (11.0) 115 48 (41.7) 30.7 (20.0, 41.5) 104 46 (44.2) 33.2 (22.0, 44.4)
Source: Study 1096 CSR Table 23; based on FAS
*The nominal p-value comparing the tofacitinib 5mg BID and 10mg BID doses in this subgroup was 0.07

47
Summary• The maintenance efficacy of tofacitinib 5 mg BID in treating
adult patients with moderate to severely active UC after 8 weeks induction of tofacitinib 10 mg BID has been demonstrated
• The benefit of the long-term use of tofacitinib 10 mg BID compared to tofacitinib 5 mg BID has not been demonstrated
• The applicant’s exploratory analyses were not adequate to support the two additional dosing regimens
www.fda.gov


FDA Gastrointestinal Drugs Advisory Committee (GIDAC)
Focused Safety Evaluation
NDA 203214
Focused Tofacitinib UC Program Safety Evaluation
Lesley S. Hanes, MD MSc
Division of Gastroenterology and Inborn Errors Products
Center for Drug Evaluation and Research
March 8, 2018

50
Benefit and Risk Assessment
Benefit: Both the 5 mg and 10 mg dose of tofacitinib are efficacious for long-term UC treatment
Risk: Evident and potentially, dose-dependent safety risks exist with the use of the higher 10 mg dose in comparison to the lower 5 mg dose

51
Xeljanz® (tofacitinib) 5 mg BID
Source: Xeljanz labeling dated 12/17

52
Xeljanz® (tofacitinib) 5 mg BIDLabelling Warnings and Precautions
• Avoid use of XELJANZ/ XELJANZ XR during an active serious infection, including localized infections
• Gastrointestinal Perforations – Use with caution in patients that may be at increased risk
• Laboratory Monitoring – Recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids
• Immunizations – Live vaccines: Avoid use with XELJANZ/ XELJANZ XR

53
Discussion• History of Tofacitinib Dosage
• 8-Week Induction and 16-Week Extended Induction Treatment
• Limitations to Safety Analyses
• Adverse Events of Special Interest (AESIs):
Higher safety risks observed with 10 mg dose treatment
• Summary and Benefit-Risk Considerations

54
History of Dosage Safety
Tofacitinib 10 mg BID dose is not approved
Tofacitinib 5mg BID is approved for the treatment for patients with rheumatoid arthritis (RA) and psoriatic arthritis (PsA)
RA program: > 7000 patients, with ~22,000 patient years (PYs)
• RA safety risks: dose-dependent
• “Over-immunosuppression” concern for 10 mg dose
Long-term, post-approval safety outcome trial in RA patients for particular adverse events of special interest (AESIs)

55
Discussion
• History of Tofacitinib Dosage
• 8-Week Induction and 16-Week Extended Induction
Treatment
• Limitations to Safety Analyses
• Adverse Events of Special Interest (AESIs):
Higher safety risks observed with 10 mg dose treatment
• Summary and Benefit-Risk Considerations

56
Proposed Induction Treatment Regimens
Phase 3 Induction Trials: 8 Weeks of Tofacitinib 10mg BID
Proportion of patients with Serious Adverse Events (SAEs):• 4% in the tofacitinib 10 mg group for 8 Weeks• 6% in the placebo group
Extended Induction Treatment for 16 Weeks(Initial 8 Weeks + Additional 8 Weeks)
Proportion of patients with SAEs: • 5% in induction non-response group, 10 mg for 16 weeks • A randomized, concurrent control group did not exist

57
Discussion
• History of Tofacitinib Dosage
• 8-Week Induction and 16-Week Extended Induction Treatment
• Limitations to Safety Analyses
• Adverse Events of Special Interest (AESIs):
Higher safety risks observed with 10 mg dose treatment
• Summary and Benefit-Risk Considerations
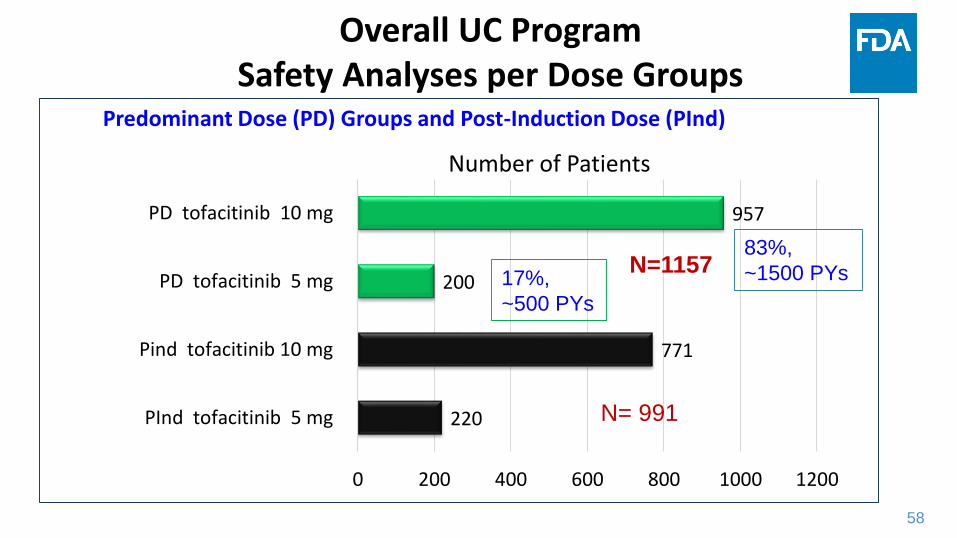
58
220
771
200
957
0 200 400 600 800 1000 1200
PInd tofacitinib 5 mg
Pind tofacitinib 10 mg
PD tofacitinib 5 mg
PD tofacitinib 10 mg
Number of Patients
N= 991
Overall UC ProgramSafety Analyses per Dose Groups
Predominant Dose (PD) Groups and Post-Induction Dose (PInd)
83%,
~1500 PYs 17%,
~500 PYs
N=1157
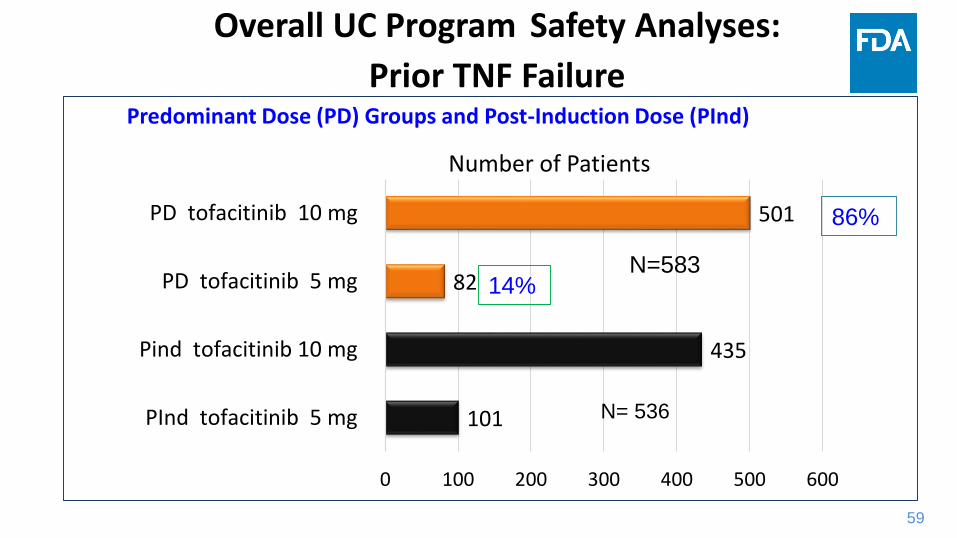
59
101
435
82
501
0 100 200 300 400 500 600
PInd tofacitinib 5 mg
Pind tofacitinib 10 mg
PD tofacitinib 5 mg
PD tofacitinib 10 mg
Number of Patients
N= 536
Overall UC Program Safety Analyses:
Prior TNF Failure Predominant Dose (PD) Groups and Post-Induction Dose (PInd)
86%
14% N=583

60
UC Maintenance Trial (1096): Limitations
• Small numbers of patients in the randomized, controlled maintenance trial
5 mg group: n= 198
10 mg group: n = 196
Placebo group: n = 198
• Patient exposure to each tofacitinib dose possibly too small to capture uncommon AESI
61
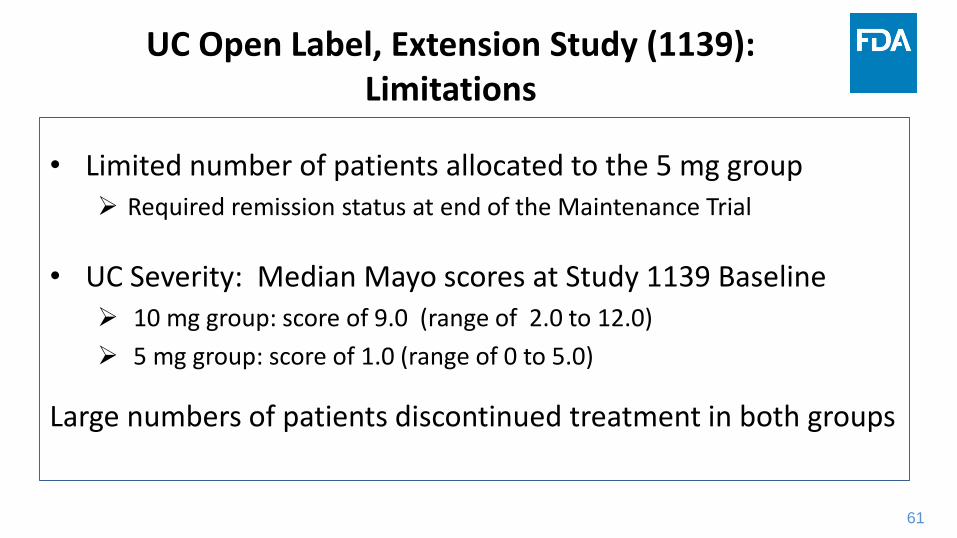
UC Open Label, Extension Study (1139):Limitations
• Limited number of patients allocated to the 5 mg group Required remission status at end of the Maintenance Trial
• UC Severity: Median Mayo scores at Study 1139 Baseline 10 mg group: score of 9.0 (range of 2.0 to 12.0)
5 mg group: score of 1.0 (range of 0 to 5.0)
Large numbers of patients discontinued treatment in both groups

62
Discussion
• History of Tofacitinib Dosage
• 8-Week Induction and 16-Week Extended Induction Treatment
• Limitations to Safety Analyses
• Adverse Events of Special Interest (AESIs):
Higher safety risks observed with 10 mg dose treatment
• Summary and Benefit-Risk Considerations

63
Dose-Dependent Risks: 10 mg vs. 5 mg
• Evident Dose-Dependent Risks: Herpes zoster infection, serious infections, and nonmelanoma skin cancer (NMSC)
• Potential, Dose-Dependent Imbalance in Risks: Deaths and Malignancies (excluding NMSC)
Herpes zoster-related opportunistic infection, “possible” likelihood of drug-induced liver injury, adverse cardiovascular and thromboembolic events
Increase in lipid levels, liver enzyme levels, creatinine, creatinine phosphokinase (CPK); and decrease in absolute lymphocyte (ALC) and neutrophil counts (ANC)

64
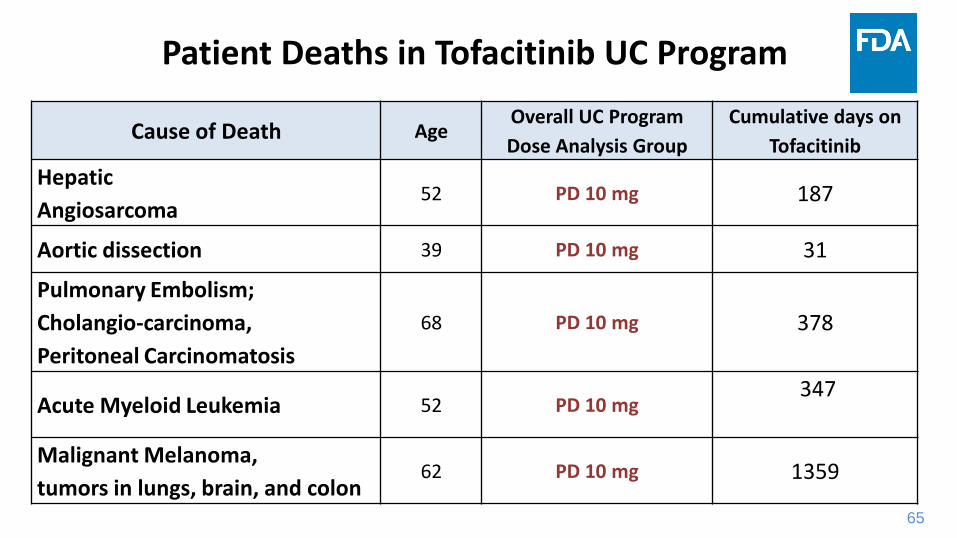
Patient Deaths in the Tofacitinib UC Program
65
Patient Deaths in Tofacitinib UC Program
Cause of Death Age Overall UC Program
Dose Analysis Group
Cumulative days on
Tofacitinib
Hepatic
Angiosarcoma52 PD 10 mg 187
Aortic dissection 39 PD 10 mg 31
Pulmonary Embolism;
Cholangio-carcinoma,
Peritoneal Carcinomatosis
68 PD 10 mg 378
Acute Myeloid Leukemia 52 PD 10 mg347
Malignant Melanoma,
tumors in lungs, brain, and colon62 PD 10 mg 1359

66
Malignancies in the Tofacitinib UC Program

67
Thirteen (13) patients had 15 adjudicated malignancies (excluding NMSC) in the UC Program (Sept 2017)
• PD 10 mg dose group and all events occurred in Open-Label, Extension study
• Incidence Rate (IR) of 0.84 per 100 Patient Years (PYs)
92% (12/13) had prior AZA/ 6MP exposure
77% (10/13) had history of prior TNF Failure
Malignancies (excluding NMSC)
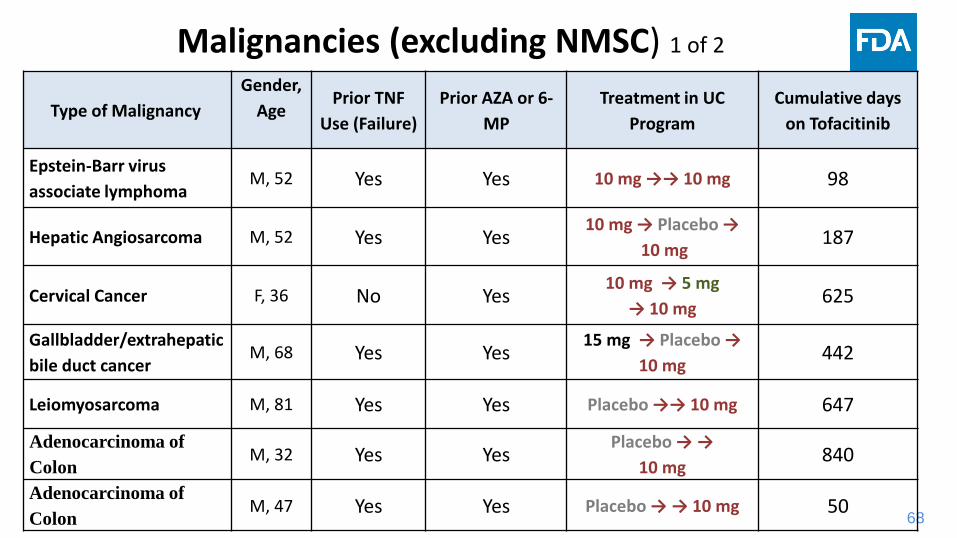
68
Malignancies (excluding NMSC) 1 of 2
Type of Malignancy
Gender,
AgePrior TNF
Use (Failure)
Prior AZA or 6-
MP
Treatment in UC
Program
Cumulative days
on Tofacitinib
Epstein-Barr virus
associate lymphomaM, 52 Yes Yes 10 mg →→ 10 mg 98
Hepatic Angiosarcoma M, 52 Yes Yes10 mg → Placebo →
10 mg 187
Cervical Cancer F, 36 No Yes10 mg → 5 mg
→ 10 mg 625
Gallbladder/extrahepatic
bile duct cancerM, 68 Yes Yes
15 mg → Placebo →
10 mg 442
Leiomyosarcoma M, 81 Yes Yes Placebo →→ 10 mg 647
Adenocarcinoma of
ColonM, 32 Yes Yes
Placebo → →
10 mg840
Adenocarcinoma of
ColonM, 47 Yes Yes Placebo → → 10 mg 50
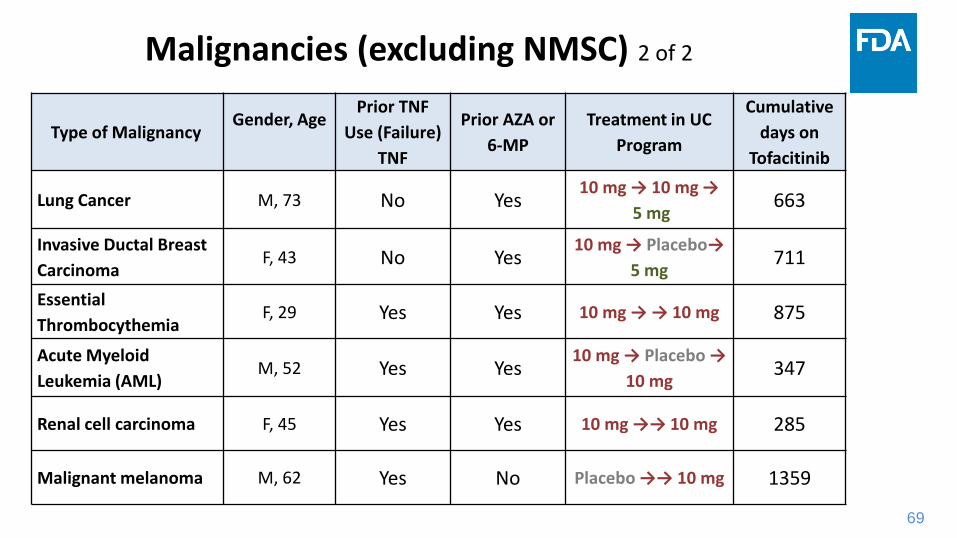
69
Type of MalignancyGender, Age
Prior TNF
Use (Failure)
TNF
Prior AZA or
6-MP
Treatment in UC
Program
Cumulative
days on
Tofacitinib
Lung Cancer M, 73 No Yes10 mg → 10 mg →
5 mg663
Invasive Ductal Breast
CarcinomaF, 43 No Yes
10 mg → Placebo→
5 mg711
Essential
ThrombocythemiaF, 29 Yes Yes 10 mg → → 10 mg 875
Acute Myeloid
Leukemia (AML)M, 52 Yes Yes
10 mg → Placebo →
10 mg347
Renal cell carcinoma F, 45 Yes Yes 10 mg →→ 10 mg 285
Malignant melanoma M, 62 Yes No Placebo →→ 10 mg 1359
Malignancies (excluding NMSC) 2 of 2
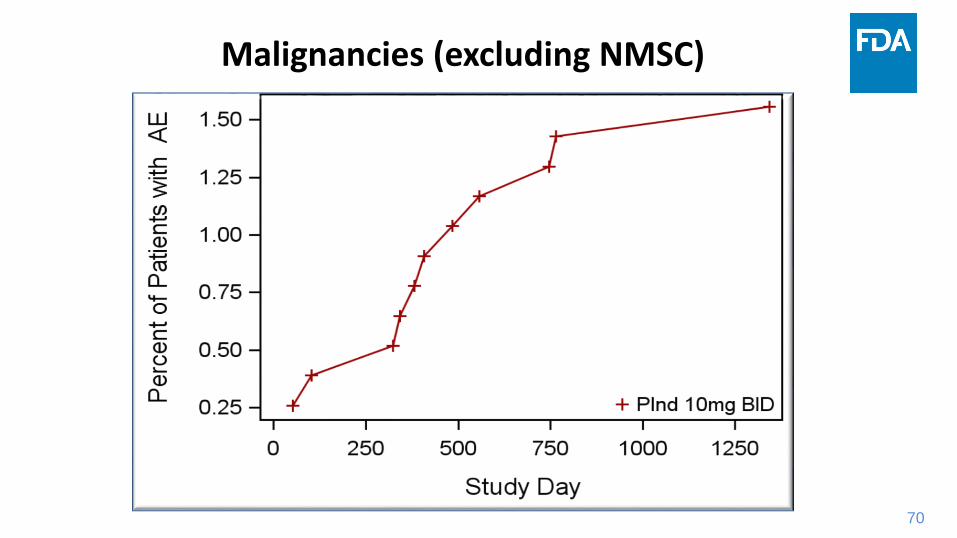
70
Malignancies (excluding NMSC)

71
Nonmelanoma Skin Cancers (NMSC)
Adjudicated NMSCs in 15 patients (September 29, 2017 data)
Dose-Dependent Risk
Incidence Rate Ratio of = 1.3
Estimated 30% increased risk
• PD 10 mg group: 0.79 per 100 PYs• PD 5 mg group: 0.61 per 100 PYs
Characteristics:
14 out of 15: prior exposure to azathioprine (AZA) or 6-MP
13 out of 15: prior TNF Failure
7 out of 15: history of NMSC
www.fda.gov
72
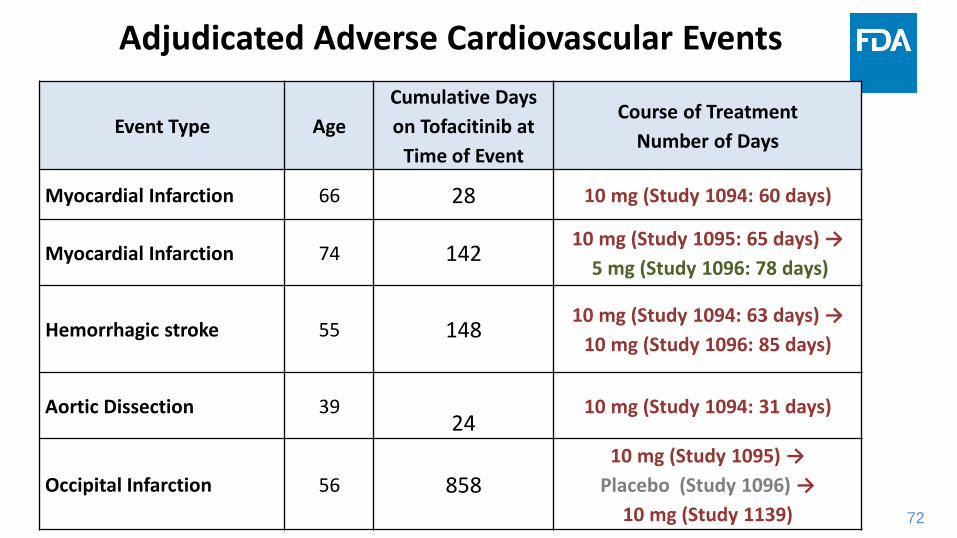
Adjudicated Adverse Cardiovascular Events
Event Type Age
Cumulative Days
on Tofacitinib at
Time of Event
Course of Treatment
Number of Days
Myocardial Infarction 66 28 10 mg (Study 1094: 60 days)
Myocardial Infarction 74 14210 mg (Study 1095: 65 days) →
5 mg (Study 1096: 78 days)
Hemorrhagic stroke 55 14810 mg (Study 1094: 63 days) →
10 mg (Study 1096: 85 days)
Aortic Dissection 3924
10 mg (Study 1094: 31 days)
Occipital Infarction 56 858
10 mg (Study 1095) →
Placebo (Study 1096) →
10 mg (Study 1139)

73
Thromboembolic Events
Four (4) patients had pulmonary embolism (PE) in the open, label extension study (1139)
PD 10 mg group
There were no occurrences of deep vein thrombosis in patients treated with tofacitinib

74
Select Laboratory Abnormalities

75
Laboratory Abnormalities: Overall UC Program
For both dose groups, laboratory abnormalities were similar to those noted in the Xeljanz label
Potentially dose-dependent:
• Decreases in Absolute lymphocyte count (ALC) and Absolute neutrophil count (ANC)
• Increases in lipids, creatinine, creatinine phosphokinase (CPK), and liver enzyme levels

76
“Possible” Likelihood ofDrug-Induced Liver Injury (DILI)
Five (5) patients with liver injury adjudicated as “possible” likelihood of drug-induced liver injury
• Did not meet Hy’s law criteria
• > 25 to 50% certainty of tofacitinib causing injury versus other risk factors
• PD 10 mg dose group

77
Absolute Lymphocyte Count (ALC)
Decreased ALC < 1.0 × 103/µL, (1000) confirmed post-baseline:
• 20% of the PD 10 mg
• 17% of the PD 5 mg
Discontinuation criteria of ALC < 0.5 × 103/µL , (500) confirmed:
• 0.9% (8/938) of the PD 10 mg
• 0.5% (1/186) of the PD 5 mg
Pattern(s) of recovery from low ALC levels are not well described in the UC patient population

78
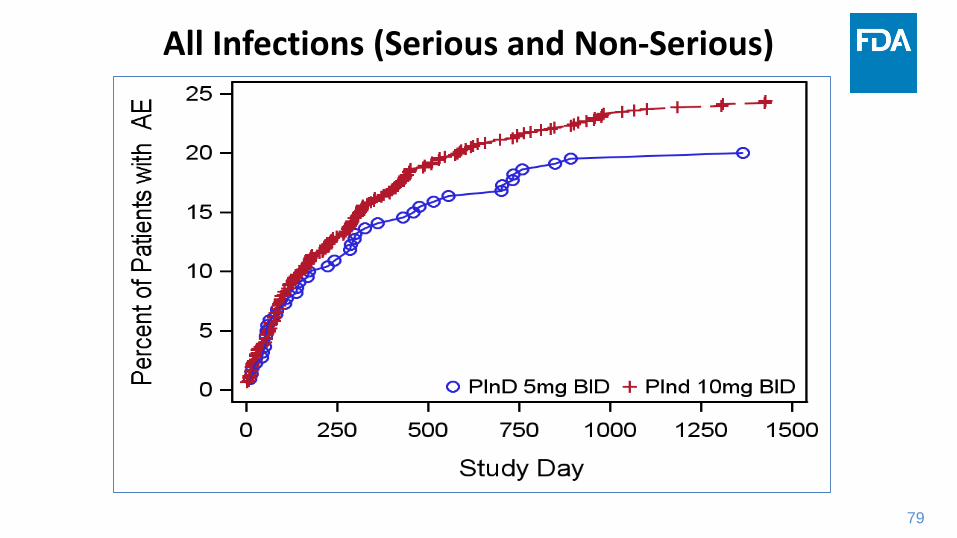
Infection Risks in the Tofacitinib UC Program
79
All Infections (Serious and Non-Serious)
80
Serious Infection Risks
Dose-dependent risk for serious infections seen in UC program
3% of patients in the UC Program developed serious infections
PD 10 mg vs. PD 5 mg group: Incidence Rate Ratio (IRR) of 1.4
Estimated, increase in risk of 40%
• PD 10 mg: Incidence Rate of 2.01 per 100 PYs• PD 5 mg : Incidence Rate of 1.41 per 100 PYs

81
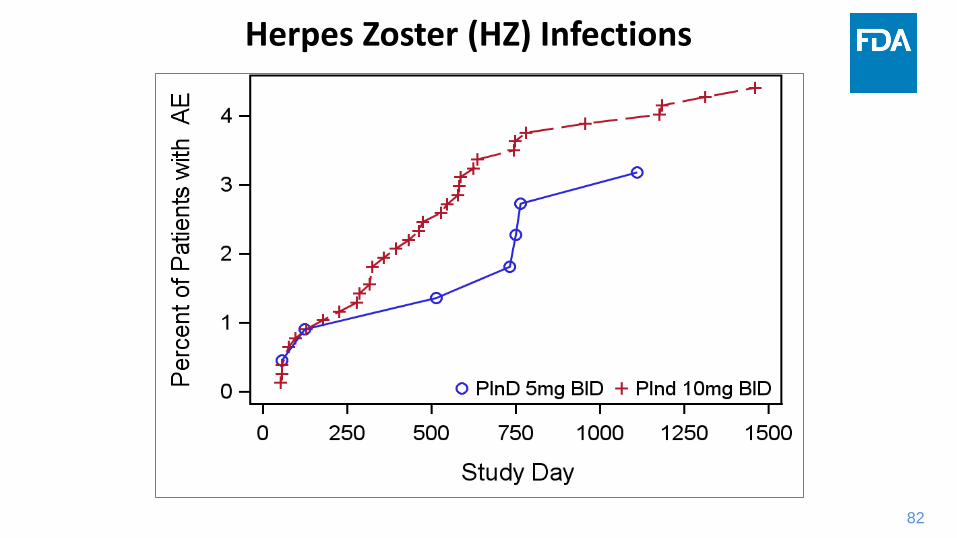
Opportunistic Infections (OI) Risks
2% (n=22) Opportunistic Infections in phase 3 program
• 18 were Herpes Zoster (HZ) - related opportunistic infections
• Six disseminated HZ cases:
one case of meningoencephalitis
two ophthalmic infections
three multi-dermatomal infection
• Non-HZ opportunistic infections occurred in four (4) patients
82
Herpes Zoster (HZ) Infections
83
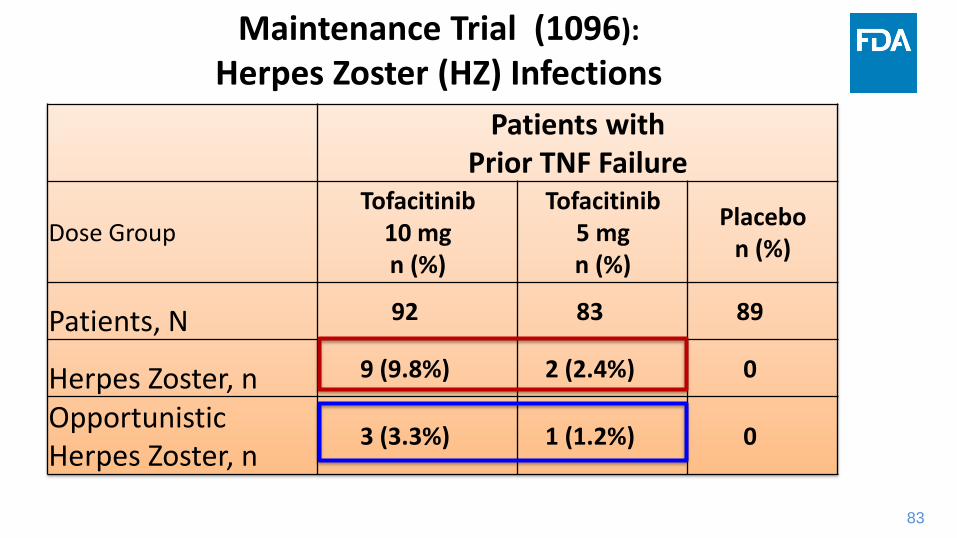
Maintenance Trial (1096):
Herpes Zoster (HZ) Infections
Patients withPrior TNF Failure
Dose GroupTofacitinib
10 mgn (%)
Tofacitinib5 mgn (%)
Placebon (%)
Patients, N 92 83 89
Herpes Zoster, n 9 (9.8%) 2 (2.4%) 0
OpportunisticHerpes Zoster, n
3 (3.3%) 1 (1.2%) 0

84
Discussion
• History of Tofacitinib Dosage
• 8-Week Induction and 16-Week Extended Induction Treatment
• Limitations to Safety Analyses
• Adverse Events of Special Interest (AESIs):
Higher safety risks observed with 10 mg dose treatment
• Summary and Benefit-Risk Considerations

85
Safety Analysis Limitations and Challenges
Multiple limitations and challenges in the safety analyses:
1. The majority of patients in PD 10 mg dose safety analyses group
as compared to the PD 5 mg dose group
2. Overall, the severity of disease was greater in those treated with
the higher versus lower dose
3. High rates of discontinuation in both dose groups

86
Benefit and Risk Assessment
Benefit: Both the 5 mg and 10 mg dose of tofacitinib are efficacious for long-term UC treatment
Risk: Evident and potentially, dose-dependent safety risks exist with the use of the higher 10 mg dose in comparison to the lower 5 mg dose


FDA Gastrointestinal Drugs Advisory Committee (GIDAC)
Tofacitinib UC Program Focused Safety Evaluation
NDA 203214
Remarks about Results from Truven MarketScan®
Joel L. Weissfeld, MD MPH
Office of Surveillance and Epidemiology (OSE)
Center for Drug Evaluation and Research
March 8, 2018

89
Truven MarketScan®
• Truven captures data submitted to U.S. health insurance companies or Medicaid.
• The Applicant used diagnosis, procedure, and drug codes in Truven.
www.fda.gov

90
Malignancy except NMSC
In 4420 patients with a drug or procedures code for a TNF-alpha blocker, the Applicant used an undisclosed code set to observe 31 malignancy events over 4895 patient-years, i.e., 0.6 per 100 patient-years.
www.fda.gov

91
Estimates of Background Risk
• Malignancy occurs in patients with moderate-to-severe UC.
• Background risks in moderate-to-severe UC might explain the number of patients observed with malignancy in the tofacitinib UC safety population.
www.fda.gov

92
Consideration of the Differences
• Real World vs. Clinical Research.
• Other challenges to interpretation,
– Inadequate controls for confounding.
– Markedly different methods for defining, ascertain-ing, and validating malignancy outcomes.
– Statistical uncertainty.
www.fda.gov


Tofacitinib Development Program:Pediatric Ulcerative Colitis
Melanie E. Bhatnagar, MD
Medical Officer
Division of Pediatric and Maternal Health
Center for Drug Evaluation and Research
March 8, 2018

95
Outline
• Pediatric Drug Legislation
• JAK Inhibitors for Pediatric Use
• Applicant’s Proposed Pediatric UC Study Plan
• FDA’s Current Thinking on Pediatric UC
www.fda.gov

96
Outline
• Pediatric Drug Legislation
• JAK Inhibitors for Pediatric Use
• Applicant’s Proposed Pediatric UC Study Plan
• FDA’s Current Thinking on Pediatric UC
www.fda.gov

97
Pediatric Research Equity Act (PREA)
• Gives FDA authority to require pediatric dosing, safety, and efficacy data for the same indication(s) being sought in adults
• Applies to drug or biological products for which applicants are proposing a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration
• Requires an initial Pediatric Study Plan (iPSP)
www.fda.gov

98
PREA + Janus Kinase (JAK) Inhibitors• JAK inhibitors approved by FDA for use in adults:
– Ruxolitinib• Myelofibrosis• Polycythemia Vera
– Tofacitinib• Psoriatic Arthritis• Rheumatoid Arthritis• Ulcerative Colitis (UC)*
www.fda.gov
PREA did not apply due to orphan designation
PREA waiver
Subject to PREA
*proposed indication

99
Outline
• Pediatric Drug Legislation
• JAK Inhibitors for Pediatric Use
• Applicant’s Proposed Pediatric UC Study Plan
• FDA’s Current Thinking on Pediatric UC
www.fda.gov
100
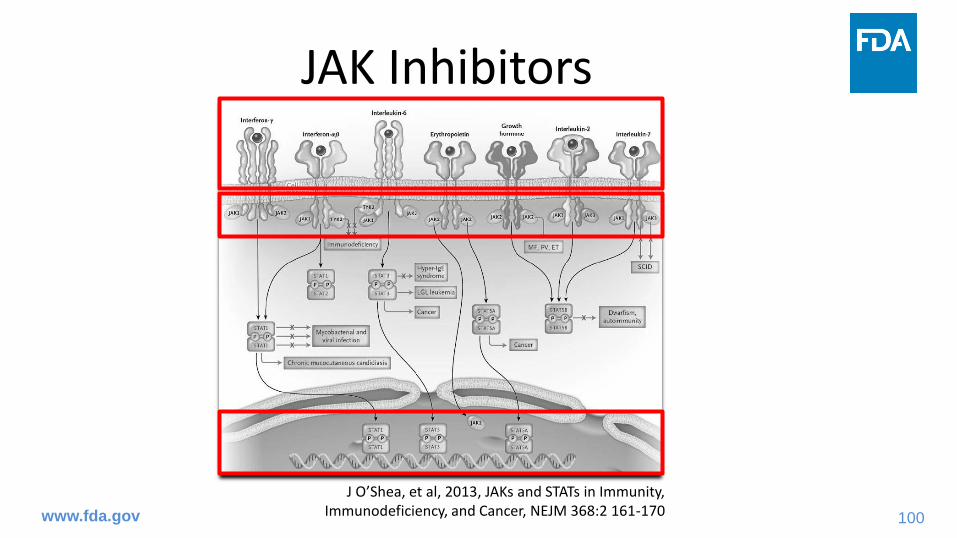
JAK Inhibitors
www.fda.gov
J O’Shea, et al, 2013, JAKs and STATs in Immunity, Immunodeficiency, and Cancer, NEJM 368:2 161-170
101
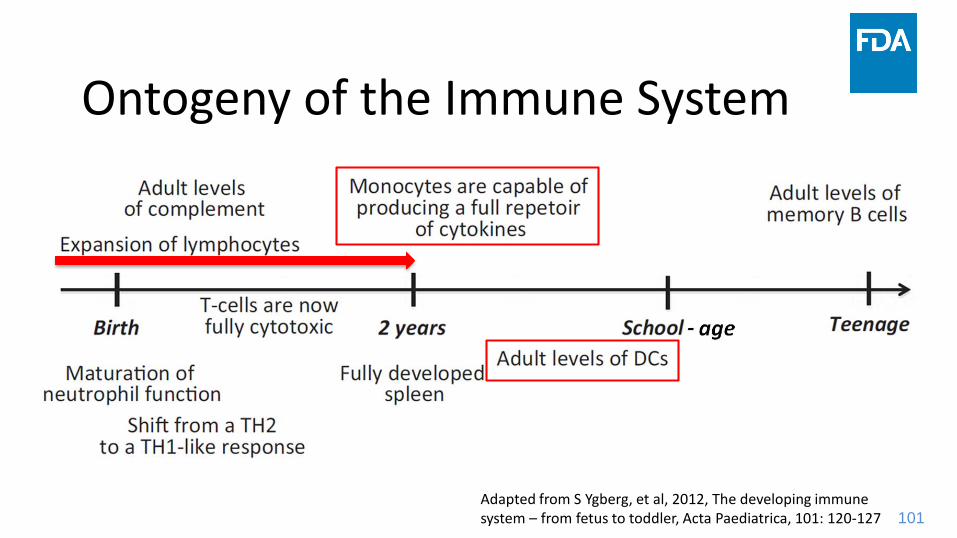
Ontogeny of the Immune System
Adapted from S Ygberg, et al, 2012, The developing immune system – from fetus to toddler, Acta Paediatrica, 101: 120-127

102
Outline
• Pediatric Drug Legislation
• JAK Inhibitors for Pediatric Use
• Applicant’s Proposed Pediatric UC Study Plan
• FDA’s Current Thinking on Pediatric UC
www.fda.gov

103
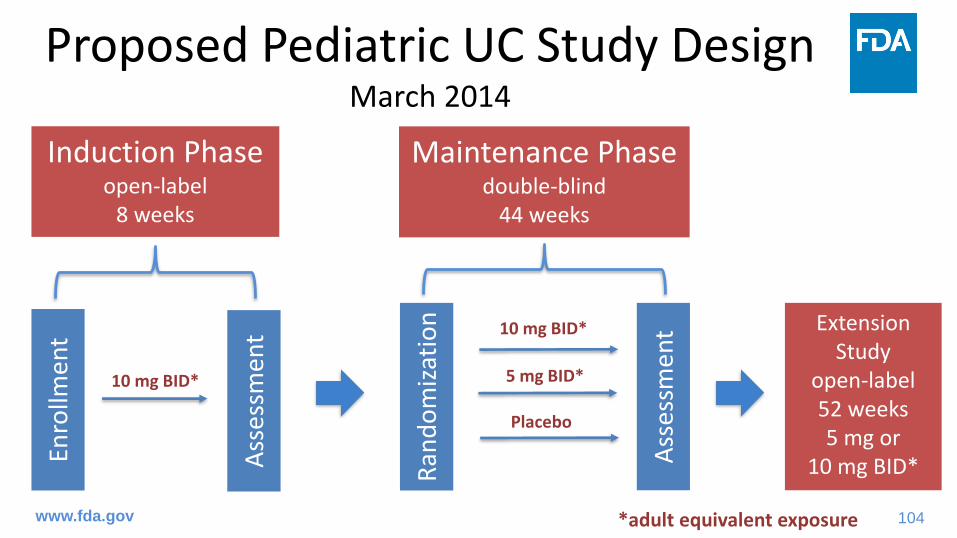
Proposed Pediatric UC Study Plan• Initial Pediatric Study Plan (March 18, 2014 )
– Single study of the safety and effectiveness of tofacitinib in pediatric patients 4 years to less than 18 years of age with moderate to severe UC who have had an inadequate response to prior therapies
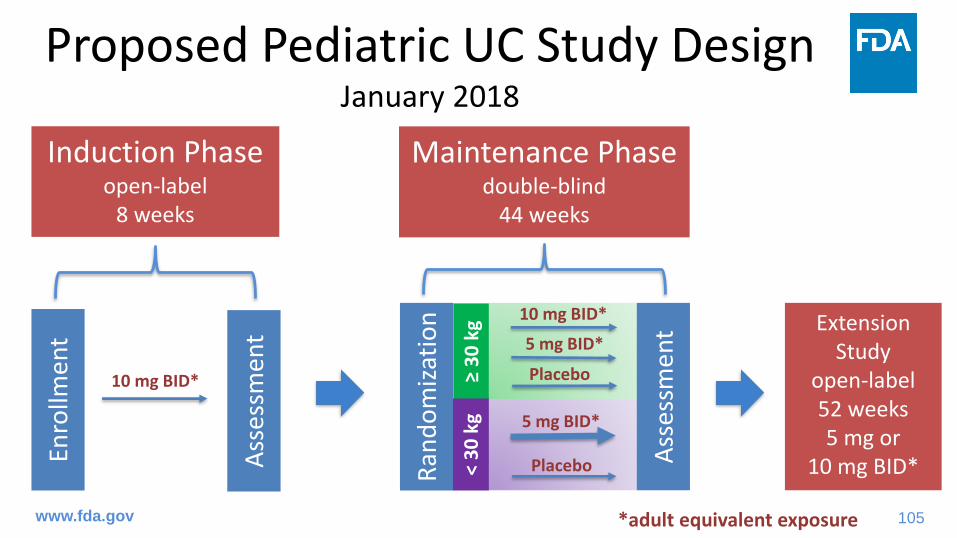
• Pediatric Study Protocol (January 19, 2018)– Revised maintenance dosing regimen based on weight
stratification at randomization– Extended lower age limit for study enrollment from 4 years
to 2 years
www.fda.gov
104
Proposed Pediatric UC Study DesignMarch 2014
Enro
llmen
t
Induction Phaseopen-label
8 weeks
10 mg BID*
Maintenance Phasedouble-blind
44 weeks
Extension Study
open-label52 weeks5 mg or
10 mg BID*Ass
essm
ent
Ran
do
miz
atio
n
Ass
essm
ent 10 mg BID*
5 mg BID*
Placebo
*adult equivalent exposurewww.fda.gov
105
Proposed Pediatric UC Study DesignJanuary 2018
Enro
llmen
t
Induction Phaseopen-label
8 weeks
10 mg BID*
Maintenance Phasedouble-blind
44 weeks
Extension Study
open-label52 weeks5 mg or
10 mg BID*Ass
essm
ent
Ran
do
miz
atio
n
Ass
essm
ent
10 mg BID*
5 mg BID*
Placebo
Placebo
5 mg BID*
≥ 3
0 k
g<
30
kg
*adult equivalent exposurewww.fda.gov

106
Proposed Pediatric UC Study Plan
• Initial pediatric study plan (March 2014)– Plan to request that FDA waive the PREA requirement
to study patients less than 4 years of age based on theoretical safety concerns
• Extensive and severe disease in pediatric patients with UC may increase susceptibility to bacterial translocation across intestinal mucosa
• Severe sequelae of bacteremia if taking an immunomodulator– Immature immune systems – Immune suppression related to their disease
www.fda.gov

107
Outline
• Pediatric Drug Legislation
• JAK Inhibitors for Pediatric Use
• Applicant’s Proposed Pediatric UC Study Plan
• FDA’s Current Thinking on Pediatric UC
www.fda.gov

108
FDA’s Current Thinking on Pediatric UC
• Pathogenesis of UC is similar to adults, though pediatric patients tend to present with more severe disease– Higher rates of extensive colonic involvement and increased likelihood for colectomy
• Assessment of safety and effectiveness required for pediatric UC patients ≥ 2 years of age
• One adequate, well-controlled pediatric UC study is generally acceptable if the expected response to the drug is similar between adults and pediatric patients
www.fda.gov

109
FDA’s Current Thinking on Pediatric UC
• Important need for therapies for pediatric UC
www.fda.gov *proposed

110
Summary
• Information on use of JAK inhibitors in the pediatric population is limited
• Pediatric UC tends to be more severe compared to adults• The applicant proposes studying pediatric UC patients 2
years of age and older at dosages targeting adult exposures up to 10 mg twice daily
• The apparent dose-dependent safety findings in adults, including malignancies and serious infections, raise concerns regarding the appropriate population and dosage to target in the pediatric UC study
www.fda.gov


FDA Backup Slides
113
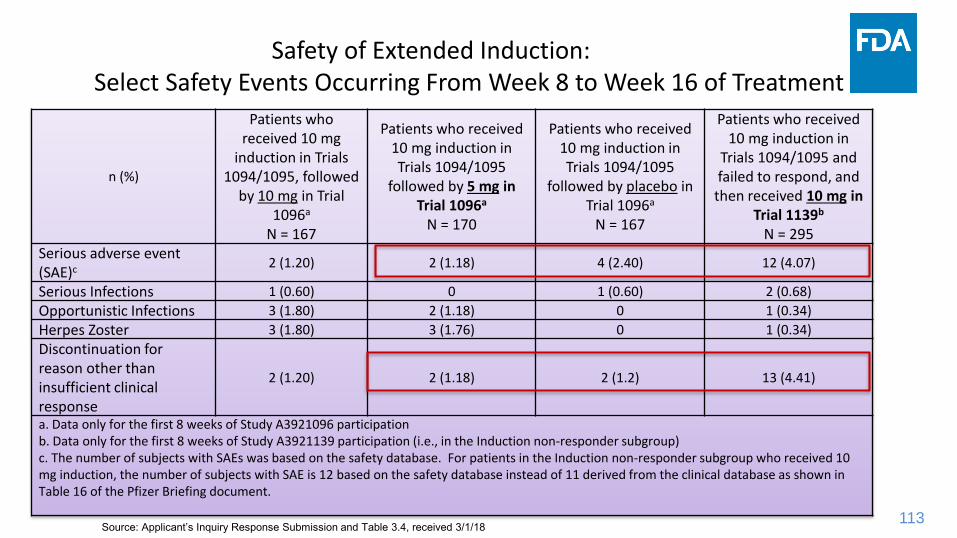
Safety of Extended Induction: Select Safety Events Occurring From Week 8 to Week 16 of Treatment
n (%)
Patients who received 10 mg
induction in Trials1094/1095, followed
by 10 mg in Trial1096a
N = 167
Patients who received 10 mg induction in Trials 1094/1095
followed by 5 mg in Trial 1096a
N = 170
Patients who received 10 mg induction in Trials 1094/1095
followed by placebo inTrial 1096a
N = 167
Patients who received 10 mg induction in
Trials 1094/1095 and failed to respond, and
then received 10 mg in Trial 1139b
N = 295Serious adverse event (SAE)c 2 (1.20) 2 (1.18) 4 (2.40) 12 (4.07)
Serious Infections 1 (0.60) 0 1 (0.60) 2 (0.68)
Opportunistic Infections 3 (1.80) 2 (1.18) 0 1 (0.34)
Herpes Zoster 3 (1.80) 3 (1.76) 0 1 (0.34)
Discontinuation for reason other than insufficient clinical response
2 (1.20) 2 (1.18) 2 (1.2) 13 (4.41)
a. Data only for the first 8 weeks of Study A3921096 participationb. Data only for the first 8 weeks of Study A3921139 participation (i.e., in the Induction non-responder subgroup)c. The number of subjects with SAEs was based on the safety database. For patients in the Induction non-responder subgroup who received 10 mg induction, the number of subjects with SAE is 12 based on the safety database instead of 11 derived from the clinical database as shown in Table 16 of the Pfizer Briefing document.
Source: Applicant’s Inquiry Response Submission and Table 3.4, received 3/1/18
114
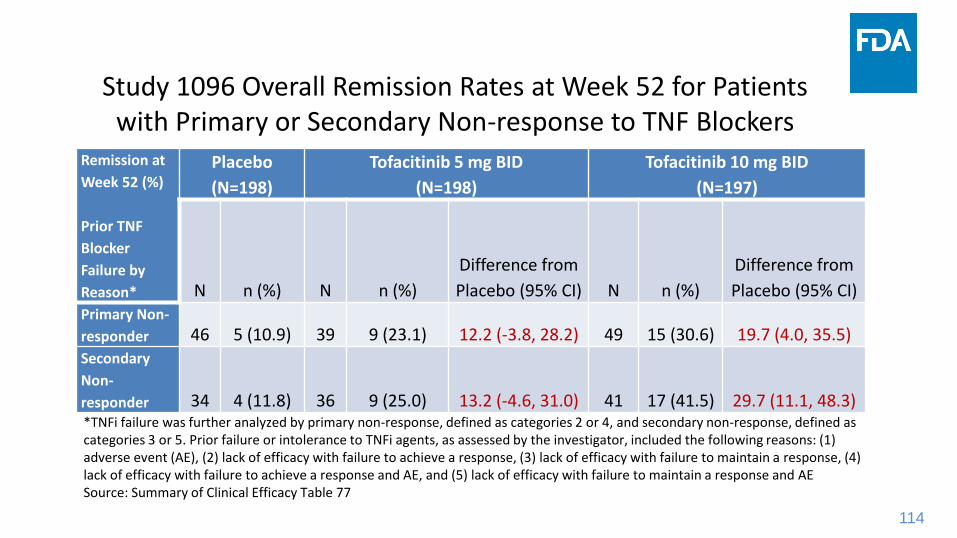
Study 1096 Overall Remission Rates at Week 52 for Patients with Primary or Secondary Non-response to TNF Blockers
Remission at
Week 52 (%)
Prior TNF
Blocker
Failure by
Reason*
Placebo
(N=198)
Tofacitinib 5 mg BID
(N=198)
Tofacitinib 10 mg BID
(N=197)
N n (%) N n (%)
Difference from
Placebo (95% CI) N n (%)
Difference from
Placebo (95% CI)Primary Non-
responder 46 5 (10.9) 39 9 (23.1) 12.2 (-3.8, 28.2) 49 15 (30.6) 19.7 (4.0, 35.5)Secondary
Non-
responder 34 4 (11.8) 36 9 (25.0) 13.2 (-4.6, 31.0) 41 17 (41.5) 29.7 (11.1, 48.3)*TNFi failure was further analyzed by primary non-response, defined as categories 2 or 4, and secondary non-response, defined as categories 3 or 5. Prior failure or intolerance to TNFi agents, as assessed by the investigator, included the following reasons: (1) adverse event (AE), (2) lack of efficacy with failure to achieve a response, (3) lack of efficacy with failure to maintain a response, (4) lack of efficacy with failure to achieve a response and AE, and (5) lack of efficacy with failure to maintain a response and AESource: Summary of Clinical Efficacy Table 77