Embed Size (px)
Citation preview

Neuza dos Prazeres Lima TeixeiraNeuza dos Prazeres Lima Teixeira
Dissertation presented to obtain the Ph.D degree in BiologyInstituto de Tecnologia Química e Biológica | Universidade Nova de Lisboa
Oeiras,March, 2014
FSR QUORUM SENSING:Role in Enterococcus faecalis Biology & Host Infection

Auth
or:
Neu
za T
eixei
raFS
R Q
UO
RU
M S
ENSI
NG
: R
ole
in E
nter
ococ
cus
faec
alis
Bi
olog
y &
H
ost
Infe
ctio
nO
eira
s, M
arch
, 2014


Neuza dos Prazeres Lima Teixeira
Dissertation presented to obtain the Ph.D degree in BiologyInstituto de Tecnologia Química e Biológica | Universidade Nova de Lisboa
Oeiras, March, 2014
FSR QUORUM SENSING:
Role in Enterococcus faecalis
Biology & Host Infection

From the left to the right: Luís Paulo Rebelo, Maria de Fátima Lopes,
Neuza Teixeira, Constança Pomba, António Jacinto, Miguel Prudêncio
and Francisco Dionísio.
14nd March 2014
Second Edition, March 2014
Stress by Antibiotics and Virulence of Enterococci Laboratory
Instituto de Tecnologia Química e Biológica (ITQB)
Universidade Nova de Lisboa
Avenida da Republica (EAN)

Financial Support from Fundação para a Ciência e Tecnologia (FCT)
Ph.D. Grant: SFRH/BD/65750/2009

ii

iii
Supervisor:
PhD Maria de Fátima Gonçalves Ribeiro dos Santos Silva Lopes
Auxiliary investigator at Instituto de Tecnologia Química e Biológica, Oeiras.
Co-supervisor: PhD Michael S. Gilmore
Sir William Osler Professor at Harvard Medical School, USA
Examining Committee PhD Miguel Prudêncio (Principal Examiner)
Investigator/ Group Leader at Instituto de Medicina Molecular (IMM), Lisboa.
PhD Constança Pomba (Principal Examiner)
Associate Professor at Faculdade de Medicina Veterinária da Universidade Técnica
de Lisboa.
PhD António Jacinto
Principal investigator at Chronic Diseases FCM Nova (CEDOC), Lisboa.
PhD Francisco Díonisio
Auxiliary investigator at Faculdade de Ciências da Universidade de Lisboa.

iv

v
To my supervisor
Fátima Lopes

vi

vii
ACKNOWLEDGMENTS
I would like to express my sincere gratitude to everyone who directly or indirectly
helped me through the development of this thesis. I would also like to acknowledge
the institutes where I worked: ITQB, Shepens Research Eye Institute of Harvard
Medical School and CEDOC.
Special thanks to my great supervisor, Maria de Fátima Silva Lopes. This thesis is
the result of great team work, without you I would never have developed this thesis,
that´s why I dedicate this thesis to you, Fátima. I admire you professionally and
personally, I learned a lot with you. During the 8 years I worked with you, you never
let me down, you were always a great supervisor. Even when you were crossing
difficult times you have never put me aside. You are a special person, a special
friend, thanks for all our discussions about science, live conversations and all the
laughter we had. For me, this PhD is not the end of our team work, it´s the beginning!
Thanks to my co-supervisor, Michael S. Gilmore for having received me in his
laboratory and for having accepted to be my co-supervisor. It was a pleasure working
in your lab and with your team. Thanks for believing in my abilities/skills and in my
work and for helping me during the last four years. Thanks for the advice and wise
words that you have always directed me.
Thanks to my thesis committee, António Jacinto and Francisco Dionísio who
accepted to be part of this work. Special thanks to António for having received me in
his lab at IMM and, later, at CEDOC. António I admire and respect your work and
how you manage your team and now the institute, CEDOC. Thanks for always
saying to me: “You are a part of this team” and for always including me in
congresses and lab retreats, this was very special to me. Thanks for the interesting

viii
discussions about science and for sharing your experiences. I´ll never forget what
you did for me.
Thanks to my collaborators, Kelli Palmer, Lynn Hancock, Jiro Nakayama and
Anna Zaidman-Remy who accepted to work with me and Fátima. We had
interesting discussions about my PhD work.
Thanks to all past colleagues of SAVE laboratory, especially to Sofia Santos,
Paulo Marujo and Teresa Braga for the discussions about science and for being my
friends during that time. From the lab next door to: Beatriz, Marta and Rusa for the
incredible moments and good laughter we had. Special thanks to my friends Paula
Alves and Filipa Silva. Thanks for the nice lunches, dinners and happy moments! I
believe our friendship will persist!
A special thanks to all present and past colleges from António Jacinto lab´s. All of
you contributed for the success of my PhD. I spent great times with you and I learned
a lot about Drosophila and Zebrafish. Thanks to Ana Roberto and Fernanda for
always being ready to help me! Thanks to Lara for the great moments while sharing
the desk, I really spend wonderful moments with you! Thanks for the help that you
gave me in understanding the “Drosophila world”! Thanks to Ana Sofia for helping
me with Drosophila protocols and for always being available to help. To Mariana,
Maria, Angela, Carolina and Telmo for the great time we spent together in fly room
and CEDOC´s sofas! Thanks to Marta Carapuço and Virginia for being great
friends and giving me very wise advices, Thanks!!!
Thanks to all my PhD collegues for the nice moments during the PhD program
classes, our dinners and parties. We spent great times together!! I made very good
friends! Thanks to Margarida Saramago for being my friend and partner in
congresses, we spent a great time together in Greece, I´ll never forget! Thanks to
Claudia Queiroga for being such a good friend, since our first PhD group we never

ix
separated and we built a real friendship. Thanks for all our discussions about
science, life and business Thanks for being by my side in the most difficult
moments of my PhD and of my personal life, you are a really good friend!
Thanks to all past and present InteraQB colleagues, we spent great times organizing
the parties and SunSetSessions! A special thanks to João Damas, Barbara, Lia,
Fábio, Rui, Joana and both Filipa, through InteraQB we built an incredible team and
we built a solid project that is a success in ITQB!
Um grande Obrigado às minhas amigas Ana Margarida Pardelha, Cláudia Xavier e
Rita Fidalgo por terem tido paciência para ouvir todas as minhas preocupações,
reclamações e fúrias! Sem o vosso apoio seria difícil ultrapassar as fases menos
boas do doutoramento. Sempre serão um grande suporte da minha vida, obrigado!
Um grande obrigado à minha irmã, cunhado e sobrinhos por ajudarem a não me
sentir sozinha durante o meu doutoramento e me incluírem sempre nos seus planos!
Um especial obrigado aos meus Pais, por SEMPRE acreditarem em mim e
SEMPRE me apoiarem, mesmo não percebendo bem o que eu fazia no laboratório.
São os meus heróis, admiro-vos muito por serem tão especiais. Obrigado!
Um obrigado doce ao Ricardo Cesário por ter sido SEMPRE o meu suporte, o meu
porto seguro! Pelo amor e carinho incondicional que sempre me deu e por SEMPRE
acreditar em mim. Mesmo nos momentos em que estivemos longe, foste sempre a
pessoa em que me deu a palavra e a força que mais precisava. És a pessoa que
mais me entende, Amo-te!

x

xi
THESIS OUTLINE
The present thesis dissertation is the result of more than four years of research at the
Stress by Antibiotics and Virulence of Enterococci (SAVE) laboratory from Instituto
de Tecnologia Química e Biologica (Oeiras, Portugal); Tissue Morphogenesis and
Repair laboratory from Chronic Diseases FCM Nova (Oeiras, Portugal) and
Departments of Ophthalmology and Microbiology and Immunobiology from Harvard
Medical School (Boston, USA), under supervision of Maria de Fátima Silva Lopes
and Michael S. Gilmore.
The thesis is divided in six chapters. In Chapter I some general concepts of quorum
sensing and Enterococcus genus are introduced. Particular attention is given to
Enterococcus faecalis pathogenesis correlated with quorum sensing Fsr system and
virulence factors it regulates. Additionally, the advantages of using Drosophila
melanogaster as a model organism to study host-pathogen interaction are described.
The Chapters II and III focus on an interesting phenomenon, which is the shutting off
of the QS under certain circumstances. Chapter II focus on a particular diary strain,
E. faecalis LN68, previously reported to show incongruence between gelatinase
genotype and phenotype. We report all experiments performed to explain the reason
of this incongruence. From this work a manuscript was published, which the author of
this dissertation played a major contribution and is the first author. In Chapter III we
describe work produced in order to understand the antagonistic effect of vancomycin,
a cell-wall active antibiotic, on expression of fsr, gelE and sprE genes. This work
resulted in a publication in which the author of this thesis is a co-author.
In Chapter IV we report experiments made to identify all genes regulated, directly
and indirectly, by the Fsr system. To complete the study we also established
Drosophila as a good model to study Fsr virulence. This approach allowed the
identification of new virulence genes in E. faecalis. This work is published in which
the author of this thesis made the majority of the experimental work and is the first
author. The Chapter V described the follow-up work of Chapter IV, and reports the

xii
influence of Fsr system on the Drosophila humoral and cellular immune system
responses. The publication of this work is in preparation.
This PhD thesis is finalized with Chapter VI, a general discussion that correlates all
findings described in the previous chapters, and provides future perspectives for
research on E. faecalis infectivity.

xiii
THESIS PUBLICATIONS
- Teixeira N, Santos S., Marujo P, Yokohata R.,Iyer V. , Nakayama J., Hancock LE,
Serror P. and Maria de Fátima Silva Lopes (2012); The incongruent gelatinase
genotype and phenotype in Enterococcus faecalis are due to shutting off the
ability to respond to the gelatinase biosynthesis-activating pheromone (GBAP)
quorum-sensing signal; Microbiology,158, 519–528 (Doi: 10.1099/mic.0.055574-0).
- Teixeira N., Varahan S., Gorman Matthew J., Palmer L. K., Zaidman-Remy A.,
Yokohata R., Nakayama J., Hancock E. L., Jacinto A., Gilmore M. S., Maria de
Fátima Silva Lopes (2013); Drosophila host model reveals new Enterococcus
faecalis quorum-sensing associated virulence factors; PLoS One 8(5): e64740
(Doi: 10.1371/journal.pone.0064740).
- Ribeiro T., Teixeira N., Yokohata R., Nakayama J., Gilmore M.S. and Maria de
Fátima Silva Lopes (2013); Transcriptomic study Reveals new pathways and
genes involved in Enterococcus faecalis V583 response to a therapeutic dose
of vancomycin, Archives of Microbiology, 4(5:3).(Doi: 10.3823/274).
- Teixeira N., Jacinto A. and Maria de Fátima Silva Lopes; Contribution of
melanization to Drosophila survival changes with E. faecalis V583 genomic
content; (in preparation).

xiv

xv
ABSTRACT
When Quorum Sensing (QS) was discovered it was realized that bacteria have a
kind of “social life” and they cooperate and coordinate their activities on the
bodies/environments they infect/live. Many bacteria only become dangerous to us
when they sense that their numbers are high enough to overwhelm human defences.
Only then they release their toxins and cause illness and death. Since this important
discovery, many bacteria (both Gram negative and Gram positive) were identified as
QS participants. In Gram positive bacteria, the Fsr system (Enterococcus faecalis
system regulator) is one example of QS that controls the expression of two virulence
factors, gelatinase and serine protease, important for the prevalence and survival of
E. faecalis during infection. Enterococcus is a peculiar and controversial genus of
Gram-positive lactic acid bacteria. It includes commensal species inhabiting the
gastro-intestinal tracts of humans and animals. However, they are also capable of
causing opportunistic infections including bacteraemia, endocarditis, meningitis,
wound, urinary tract and nosocomial bloodstream infections. E. faecalis is the
predominant species in human/animal associated environments, and therefore the
most studied species of this genus. Recent data indicates that E. faecalis is the third
most commonly isolated nosocomial pathogen (12% of all hospital infections). Over
representation of E. faecalis among clinical isolates may be related to its natural
abundance, to the presence of virulence factors and/or the ability to acquire easily
antibiotic resistances. The two most studied virulence factors are gelE (encoding
gelatinase, GelE) and sprE (encoding a serine protease, SprE). These genes are
present in clinical and diary enterorococal strains but are not always phenotypically
positive. Fsr system and GeE-SprE have been proven important for E. faecalis
virulence but their role in E. faecalis biology and during the infectious process is
poorly understood. Moreover, the opportunistic nature of E. faecalis makes it difficult
to find the perfect animal model to study host-pathogen interactions. This thesis work
was thus designed to fill these knowledge gaps.

xvi
Under certain conditions, E. faecalis shuts down Fsr QS. In order to investigate the
main reasons for this, we studied E. faecalis LN68 strain that has all fsr and gelE-
sprE genes but a negative gelatinase phenotype (Chapter II). The fsr and gelE-sprE
operons were sequenced, and the negative gelatinase phenotype was attributed to a
nonsense mutation (a premature STOP codon). This mutation in the fsrC gene is
translated into a deficient ATPase sensor domain, responsible for sensing and
transducing the signal from the quorum-sensing molecule. This mutation was found
in other enterococcal strains revealing that this is a natural way to shutdown the QS-
associated production of GelE and SprE and suggesting that some benefits may
come from silencing the QS.
In a previous microarray study from our lab in strain E. faecalis V583, fsr-gelE-sprE
genes were found to be down-regulated by vancomycin, a cell-wall active antibiotic,
(Chapter III). In order to check the hypothesis of QS shutdown by vancomycin, we
used E. faecalis V583ΔfsrB, which is unable to produce GBAP but is able to sense it.
Cells from this mutant were collected after incubation (0 min, 10 and 20 min) with
GBAP or with GBAP and vancomycin .Expression levels of gelE, sprE and vanB
genes were evaluated by semi-quantitative RT-PCR. When GBAP was added after
vancomycin none of the Fsr regulated genes, gelE and sprE, was induced,
suggesting that this antibiotic turns FsrC sensor blind to the QS molecule. It was the
first time Fsr system activity was associated with an antibiotic. These two previous
studies indicate that Fsr associated QS is not essential for growth as it is repressed
in some environments.
Previous studies have suggested that Fsr is a global regulator in E. faecalis. To
complete the study of Fsr biology it is therefore essential to know which other genes
are regulated by the Fsr system and to investigate their role in E. faecalis virulence.
To achieve this goal, we did a transcriptomic analysis using isogenic mutants of
V583 variously defective in either Fsr QS or protease expression to identify the
genes directly and indirectly regulated by Fsr (Chapter IV). QS was artificially
induced by addition of the quorum signal, GBAP, exogenously and in a controlled
manner. The Fsr QS system was found to regulate five genes (gelE, sprE, ef1097

xvii
and ef1351-52) and twelve additional genes were found to be dependent on the
presence of the QS-induced proteases. Additionally, the induction of gelE and sprE
resulted in up-regulation of two genes important in cell autolysis – lrgAB – that were
confirmed to be regulated by LytRS.
Drosophila melanogaster has proven useful as a tool to study host-pathogen
interactions, both for bacterial pathogens and human commensals. We therefore
chose this model organism to gather clues on the role of Fsr, and of the genes it
regulates, on host disease and death inflicted by E. faecalis infections. To study the
mechanism of pathogenesis associated with Fsr, proteases and new genes,
Drosophila was first established as an infection model. We then infected the fruit fly
with V583 mutants on the newly found genes. Two new Fsr - associated virulence
factors were found, namely, lrgAB operon and the bacteriocin coding gene ef1097.
We also found that inactivation of Fsr and the two proteases increased fly´s tolerance
to E. faecalis, whereas flies showed similar resistance towards all studied E. faecalis
strains. These results suggest that future approaches to combat the E. faecalis
infection can be through improving host tolerance, providing an alternative, or a
complement, to the use of antibiotics.
These new findings lead us to further investigate the role of Fsr in the cross-talk with
the Drosophila immune system (Chapter V). We measured the expression of
drosomycin; analyzed the phagocytosis and the melanization during Drosophila
infection (control W1118, W1118HmlΔ>GFP/UAS-Bax and W1118 PPO1Δ, PPO2Δ) with
V583 and V583ΔfsrBΔgelEΔsprE strains. We found that Fsr interfered with the
melanization process. Moreover, Drosophila was only able to survive in the absence
of both Fsr-GelE-SprE factors and melanization. Melanization is used by Drosophila
to combat the pathogens but when exacerbated is also able to cause host injury. We
believe that fly death, caused by E. faecalis carrying Frs-GelE-SprE, is due to
exacerbated host injury by host´s own immune response.
Overall, the work presented in this thesis gives us important clues on the role of Fsr
and QS both in the E. faecalis biology, in its struggle in the environment it inhabits,
as well as in the cross-talk with the host. For future, the data reported in this thesis

xviii
can be further explored to find new therapies to control the E. faecalis infections and
decrease their impact on host death numbers.

xix
RESUMO
Desde a descoberta do Quorum Sensing (QS), percebeu-se que as bactérias têm
uma espécie de “vida social” e que cooperam e coordenam as suas actividades no
hospedeiro/ambiente que infectam/habitam. Muitas bactérias só se tornam perigosas
quando sentem que estão em número suficiente para sobrecarregar a defesa
humana. Em seguida, libertam toxinas e causam doença e, eventualmente, morte. A
partir do momento em que foi feita esta importante descoberta, muitas bactérias
(Gram negativas e Gram positivas) foram identificadas como participantes no QS.
Em bactérias Gram positivas, o sistema Fsr (Enterococcus faecalis system regulator)
é um exemplo de QS que tem como molécula sinal a GBAP (Gelatinase biosynthesis
activating pheromone).O Fsr controla a expressão de dois factores de virulência,
gelatinase e proteinase sérica, importantes para a persistência e sobrevivência de E.
faecalis durante a infecção. Enterococcus é um género peculiar e controverso que
pertence ao grupo de bactérias lácticas Gram-positivas. Este género inclui espécies
comensais que habitam no trato gastrointestinal de humanos e animais. Contudo,
são capazes de causar infecções oportunistas como bacteremias, endocardites,
meningites, feridas, infecções urinárias e infecções nosocomiais da corrente
sanguínea. E. faecalis é a espécie predominante em humanos/animais, e também a
mais estudada. Estudos recentes indicam que E. faecalis é o terceiro patogénico
nosocomial mais comum (12% das infecções hospitalares). A abundante presença
de E. faecalis em isolados clínicos poderá estar relacionada com a sua natureza de
produzirem factores de virulência e/ou com a sua facilidade de adquirirem
resistência a antibióticos. Os factores de virulência mais estudados são gelE
(gelatinase, GelE) e sprE (proteinase sérica, SprE). Embora estes genes estejam
presentes em estirpes clínicas e ambientais de Enterococcus, nem sempre
produzem um fenótipo positivo. O sistema Fsr e as proteases GelE-SprE têm vindo
a ser referenciados como importantes para a virulência de E. faecalis, mas a sua
função na biologia e durante o processo de infecção de E. faecalis é ainda

xx
desconhecida. Além disso, a natureza oportunista de E. faecalis tem tornado difícil a
escolha de um modelo animal perfeito para o estudo da interacção entre o
hospedeiro e o patogénico. O trabalho desta tese foi desenhado para preencher
estas lacunas do conhecimento.
Em certas condições, E. faecalis desliga o QS Fsr. É o caso da estirpe E. faecalis
LN68 que tem todos os genes fsr e gelE-sprE mas apresenta um fenótipo gelatinase
negativo (Capítulo II). Os operões fsr e gelE-sprE foram sequenciados e o fenótipo
gelatinase negativo foi atribuído à presença de uma mutação sem sentido (um
codão STOP prematuro). Esta mutação no gene fsrC afecta o domínio sensor
ATPase, responsável por sentir e traduzir a GBAP. Esta mutação foi também
identificada noutras estirpes de Enterococcus revelando ser uma forma natural de
desligar a produção de GelE e SprE associada ao QS, e sugerindo que esta bactéria
poderá ter benefícios energéticos com o silenciamento do QS.
Num estudo anterior de transcriptómica em E. faecalis V583, os genes fsr-gelE-sprE
foram identificados como serem negativamente regulados pela vancomicina,
antibiótico que inibe a síntese da parede celular, (Capítulo III). Para testar a
hipótese de que o QS é desligado pela presença da vancomicina, usou-se a estirpe
mutante E. faecalis V583ΔfsrB, que é incapaz de produzir GBAP mas sente o seu
sinal e activa a síntese das proteases. Células deste mutante foram recolhidas após
incubação (0 minutos, 10 e 20 minutos) com GBAP ou com GBAP e vancomicina.
Os níveis de expressão dos genes gelE, sprE e vanB foram quantificados por RT-
PCR semi-quantitativo. Quando a GBAP foi adicionada depois da vancomicina
nenhum dos genes regulados pelo Fsr, nomeadamente gelE e sprE, foram induzidos
sugerindo que este antibiótico torna o sensor FsrC cego à molécula QS. Esta
constitui a primeira vez em que a actividade do sistema Fsr foi correlacionada com
um antibiótico. Estes dois estudos previamente descritos demonstraram que o QS
associado ao Fsr não é essencial ao crescimento da bactéria mas é reprimido em
alguns ambientes/condições.
Estudos anteriormente realizados têm sugerido que o Fsr é um regulador global em
E. faecalis. Para completar o estudo da biologia do Fsr é essencial identificar os

xxi
outros genes que o sistema Fsr regula, e investigar o seu contributo para a
virulência de E. faecalis. Para atingir este objectivo, procedeu-se à análise
transcriptómica usando mutantes isogénicos de V583 no Fsr e nas proteases, para
identificar os genes directamente e indirectamente regulados por este sistema de
QS (Capítulo IV). O QS foi artificialmente induzido pela GBAP, adicionada
exogenamente e de uma forma controlada. O sistema Fsr foi identificado como
regulador de cinco genes (gelE, sprE, ef1097 e ef1351-52). Doze genes adicionais
foram identificados como sendo dependentes da presença das proteases induzidas
pelo QS. Adicionalmente, a indução do gelE e sprE resultou na regulação positiva de
dois genes importantes para a autólise celular – lrgAB- tendo-se confirmado que são
regulados pelo sistema de dois componentes LytRS.
Drosophila melanogaster tem vindo a demostrar ser uma ferramenta útil para o
estudo da interacção do hospedeiro – patogénico, em particular, no estudo de
bactérias patogénicas e comensais humanos. Foi por essa razão que se escolheu
como modelo para estudar a função do Fsr e dos genes que este regula, no
desenvolvimento da doença do hospedeiro e na morte inerente à infecção causada
por E. faecalis. D. melanogaster foi primeiro estabelecida como modelo de infecção
para estudar o mecanismo patogénico associado ao Fsr, proteases e os novos
genes. Em seguida, a mosca da fruta foi infectada com mutantes de V583 nos genes
novos anteriormente identificados. Dois novos factores de virulência associados ao
Fsr foram assim identificados, nomeadamente lrgAB e a bacteriocina ef1097.
Também foi observado que a inactivação do Fsr e das duas proteases aumenta a
tolerância da mosca relativamente a E. faecalis, sido demonstrada resistência similar
para todas as estirpes E. faecalis testadas. Este resultado sugere que o combate de
infecções por E. faecalis poderá passar por beneficiar a tolerância do hospedeiro,
proporcionando uma alternativa ou um complemento ao uso de antibióticos.
Estas novas descobertas levaram-nos a investigar a função do Fsr na comunicação
com o sistema imunitário da Drosophila (Capítulo V). Durante a infecção de
Drosophila (controlo W1118, W1118HmlΔ>GFP/UAS-Bax and W1118 PPO1Δ, PPO2Δ)
com as estipes V583 e V583ΔfsrBΔgelEΔsprE foi medida a expressão da

xxii
drosomicina, analisada a fagocitose e a melanização. Observou-se que o Fsr
interfere com o processo de melanização. Além disso, Drosophila só foi capaz de
sobreviver quando, em simultâneo, não estão presentes nem o Fsr e as proteases
GelE-SprE, nem a melanização. Esta é um mecanismo utilizado pela Drosophila
para combater os patogénicos mas quando exacerbada é capaz de provocar danos
no próprio hospedeiro. É possível que a morte da mosca da fruta causada por E.
faecalis com Fsr-GelE-SprE seja devida às lesões decorrentes de uma exacerbada
resposta imune do hospedeiro.
No geral, o trabalho apresentado nesta tese fornece pistas importantes sobre o
papel do Fsr - QS, tanto na biologia de E. faecalis, na sua forma de sobreviver nos
seus habitats, bem como no cross-talk com o hospedeiro. Para futuro, os resultados
apresentados nesta tese podem ser explorados para a procura de novas terapias
para o controlo de infecções provocadas por E. faecalis de forma a diminuir o seu
impacto sobre a morte do hospedeiro.

xxiii
ABREVIATIONS
Abbreviation Full form
Δ Deletion
ΔCt Cycling threshold
Ace Adhesion to collagen
agr Accessory gene regulator
AIP Autoinducer peptide
AHL Acyl-homoserine lactone
AIs Autoinducers
AMPs Antimicrobial peptides
A. thaliana Arabidopsis thaliana
AS protein Aggregation substance
Bee Biofilm enhancer
BHI Brain heart infusion
bp Base pairs
°C degree Celsius
C. elegans Caenorhabditis elegans
cDNA Complementary DNA
CFUs Colony forming units
cps cluster Capsular polysaccharides
Cyl Cytolysin
ddl D-alanine-D-alanine ligase
D-Lac D-lactate
D-Ser D-serine
DNA Deoxyribonucleic acid
Drosophila Drosophila melanogaster
Ebp Endocarditis and biofilm associated pili
(e)DNA Extracellular DNA
ElrA Surface protein
epa cluster Enterococcal polysaccharide antigen
EPS Extracellular polymeric substances
E. casseliflavus Enterococcus casseliflavus

xxiv
E. coli Escherichia coli
E. durans Enterococcus durans
E. gallinarum Enterococcus gallinarum
E. faecalis Enterococcus faecalis
E. faecium Enterococcus faecium
Esp Surface protein
fsr E. faecalis regulator
iRNA RNA interference
GBAP Gelatinase biosynthesis activating pheromone
GelE Gelatinase
G. melonella Galleria melonella
GRAS Generally Recognized As Safe
h hours
HK Histidine Kinase
hld virulence factor δ-lysin
JAK/STAT Janus kinase/signal transducer and activator of transcription
LB Luria Bertani Broth
LN68 Strain isolated from Niza milk
LSE4 Strain isolated from Serra da Estrela milk
MDR Multi-Drug Resistance
mg milligram
min minute
ml millilitre
M. luteus Micrococcus luteus
mRNA messenger RNA
M. sexta Manduca sexta
NaCl Sodium Chloride
NCBI National Center for Biotecnology Information
nl nanoliters
ng nanogram
nM nanoMolar
nt Nucleotides
mM milliMolar
OD Optical density
OG1RF E. faecalis OG1RF
PCR Polymerase chain reaction

xxv
PPO Pro-phenoloxidase
PO Phenoloxidase
PRR Pattern recognition receptor
PG Peptidoglycan
(PGRP)-LC Peptidoglycan recognition protein
QA29b Strain isolated from Azeitão cheese
qRT-PCR Quantitative real-time polymerase chain reaction
QS Quorum sensing
S. aureus Staphylococcus aureus
SPHs Serine protease homologues
SprE Serine protease
StrA Sortase
TM Melting temperature
VRE Vancomycin resistant Enterococci
V583 E. faecalis V583
RHK Receptor histidine kinase
RNA Ribonucleic Acid
rDNA Ribossomal RNA
ROS Reactive oxygen species
RR Response Regulator
RT-PCR Reverse Transcriptase-polimerase chain reaction
V. fischeri Vibrio fischeri
V. harveyi Vibrio harveyi
wt Wild-type
XIP Double-tryptophan peptide pheromone

xxvi

xxvii
TABLE OF CONTENTS
Acknowledgments ..................................................................................................... vii
Thesis outline ............................................................................................................. xi
Thesis publications ....................................................................................................xiii
Abstract ..................................................................................................................... xv
Resumo .................................................................................................................... xix
Abreviations ............................................................................................................ xxiii
Table of contents .................................................................................................... xxvii
INTRODUCTION
1. QUORUM SENSING
A Way to Communicate………………………………..………………………………….5
1.1 Different QS Systems Among Bacteria……………………………………………….6
1.2 Enterococcus faecalis Fsr Quorum Sensing System………………………………..8
2. ENTEROCOCCUS GENUS
General Characteristics…………………………………………….……………………12
2.1 Enterococcus spp. - An Opportunistic Pathogen…………………………………...13
3. ENTEROCOCAL VIRULENCE
The Role of Fsr, Gelatinase and Serine Protease………………………………......16
3.1 Animal Models to Study Fsr and Proteases………………………………………...20
4. DROSOPHILA MELANOGASTER
A model to Study Host-pathogen Interaction………………..………………………23
5. AIMs AND SCOPE OF THESE THESIS…………………………………...………...30
6. Bibliography……..…...…………………………………..………………………………32
CHAPTER I

xxviii
SILENCING FSR SYSTEM: A Way to Survive
1. Summary………………………………...……………………………………………….51
2. Introduction………………………………………………………………………………52
3. Material and methods…………………………………………………………………...55
4. Results and Discussion…………………………………………………………………59
5. Acknowledgements……………………………………………………………………..67
6. Bibliography……...………………………………………………………………………68
7. Supplementary data…………………………………………………………………….74
FSR AND VANCOMYCIN:
The antagonistic relation
1. Summary…………………………………………………………………………………81
2. Introduction………………………………………………………………………………82
3. Material and methods…………………………………………………………………...84
4. Results and Discussion………………………………………………………………...86
5. Bibliography……………………………………………………………………………...90
NEW FINDINGS ON FSR SYSTEM: New
virulence genes and their impact during Drosophila infection
1.Summary………………………………………………………………………………….99
2. introduction……………………………………………………………………………..100
3. material and methods………………………………………………………………….103
4. Results…………………………………………………………………………………..110
5. Discussion………………………………………………………………………………118
6. Acknowledgements……………………………………………………………………124
7. Bibliography…………………………………………………………………………….125
8. Supplementary data……………………………………………………………………135
CHAPTER III
CHAPTER IV
CHAPTER II

xxix
FSR SYSTEM AND DROSOPHILA:
The collapse of the immune system
1.Summary………………………………………………………………………………...145
2. Introduction………………………………………………………………………...…...146
3. Material and methods………………………………………………………………….149
4. Results ………………………………………………………………………………….152
5. Discussion………………………………………………………………………………159
6. Acknowledgements……………………………………………………………………162
7. Bibliography…………………………………………………………….………………163
GENERAL DISCUSSION
1. FSR QUORUM SENSING SYSTEM
- Different environments lead different ways to persist………………………….……173
2. FSR REGULON
- New genes and potential virulence factors…………………………………………...177
3. FUTURE PRESPECTIVES
- Fsr system can be a future target for therapy………………………………………..183
4. Bibliography………………...…………………………………………………………..185
CHAPTER V
CHAPTER VI

xxx

INTRODUCTION


CONTENTS
1. QUORUM SENSING
A Way to Communicate ............................................................................................ 5
1.1 Different QS Systems Among Bacteria ............................................................. 6
1.2 Enterococcus faecalis Fsr Quorum Sensing System ........................................ 8
2. ENTEROCOCCUS GENUS
General Characteristics .......................................................................................... 12
2.1 Enterococcus spp. - An Opportunistic Pathogen ............................................ 13
3. ENTEROCOCAL VIRULENCE
The Role of Fsr, Gelatinase and Serine Protease ................................................. 16
3.1 Animal Models to Study Fsr and Proteases .................................................... 20
4. DROSOPHILA MELANOGASTER ....................................................................... 23
A model to Study Host-pathogen Interaction
5. AIMs AND SCOPE OF THESE THESIS ............................................................... 30
6. BLIBLYOGRAPHY ............................................................................................... 32

Chapter I
4
Neuza Teixeira has written the whole chapter based on the referred bibliography.

INTRODUCTION
5
Cha
pter
I
1. QUORUM SENSING
A Way to Communicate
Many bacteria are known to regulate their cooperative activities and physiological
processes trough a mechanism called quorum sensing (QS), in which bacterial cells
communicate with each other by releasing, sensing and responding to small
diffusible signal molecules. QS as a concept grew out of the work of JW Hastings in
the 1960’s (Hastings & Greenberg, 1999). QS describes a process of cell-to-cell
communication used by bacteria to understand and adapt in their environment and
consequently to apply specific strategies that allow adaptation to environmental
stress in space and time (Fuqua et al., 1994; Skandamis & Nychas, 2012).
QS cell-to-cell signaling results from production of small, diffusible signal molecules
called autoinducers that are secreted at a basal level during bacterial growth by
emitter cells. This signal accumulates in the surrounding environment. When the
concentration of these signal molecules reaches a threshold level (the quorum level),
the signal molecule binds to receptors on or in the bacteria cell and alter gene
expression (Figure 1) (Skandamis & Nychas, 2012).
A B
Figure 1 - Diagram of quorum sensing. (A) At low cell density, the concentration of the autoinducer (blue
dots) is relatively low and the expression of a regulated product (red dots) is restricted. (B) At high cell density, the
concentration of the autoinducer is high and expression of the regulated bacterial product is induced or derepressed
(http://commons.wikimedia.org/wiki/File:Quorum_sensing_diagram.png).

Chapter I
6
QS control genes that are beneficial when expressed by groups of bacteria acting in
synchrony. The list of processes that bacteria coordinate by QS is extensive. These
activities are generally unproductive when carried out by small number of cells.
Bacteria use quorum sensing communication circuits to regulate a diverse array of
physiological activities that include symbiosis, virulence, competence, conjugation,
antibiotic production, motility, sporulation, and biofilm formation (Rutherford &
Bassler, 2012).
One classical example of QS is the production of bioluminescence by the marine
bacteria Vibrio fischeri, a symbiont of Hawaiian bobtail squid Euprymna scolopes
(Nealson & Hastings, 1979; Reading & Sperandio, 2006). V. fischeri lives in squid
light organ (nutrient-rich environment) and is only beneficial to the bacteria to
synthesize the light producing enzymes (luciferase) when certain number of
autoinducers inside the squid light organ are detect. In contrast, autoinducers do not
accumulate to any significant level in free ocean and V. fischeri does not make light.
On the other hand, light production by the bacteria enables the squid to eliminate its
shadow in the ocean and thus the light is used as squid in a strategy to evade
predators (Bassler, 1999; Reading & Sperandio, 2006).
1.1 Different QS Systems Among Bacteria
QS systems in bacteria have been generally divided into at least three classes: (1)
LuxI/LuxR-type QS in Gram negative bacteria which use acyl-homoserine lactones
(AHL) as signal molecules; (2) oligopeptides-two-component-type QS in Gram
positive bacteria, which use small, often post-translationally modified peptides as a
signal molecule; and (3) luxS-encoded autoinducer 2 (AI-2) QS in both Gram
negative and Gram positive bacteria (Li & Tian, 2012).
Gram negative bacterial, typically use AHL molecules as autoinducers, and each
species has distinct AHL to communicate with members of its own species. AHLs are
produced by LuxI-type proteins and AHLs diffuse freely across cell membrane. At

INTRODUCTION
7
Cha
pter
I
high concentration, the autoinducer binds to a transcription factor of the LuxR type in
the cytoplasm, and this LuxR-AHL complex binds to a specific promoter thereby
activating gene expression for group-specific processes, like bioluminescence (lux)
(Figure 2) (Bassler & Vogel, 2013; Bassler, 1999).
In Gram positive bacteria there are two types of QS systems. The first type uses
autoinducer peptides (AIP), called peptide pheromones. Gram positive bacteria
normally produce a signal peptide precursor, which is cleaved and the active AIP is
then exported through a peptide-specific ABC transporter into their environment. AIP
is recognized by the input domain of a typical sensor element of a histidine kinase
(HK) two-component signal transduction system. HK phosphorylates the response
regulator (RR) which binds to the promoter of genes of interest (Figure 2).
Figure 2 - Quorum-sensing signalling pathways in Gram negative (A) and Gram positive bacteria
(B). (A) LuxI/LuxR in a Gram-negative bacteria. The autoinducer (black dots) synthesized through pathways
involving LuxI, is released, and then reenters into bacteria and binds to receptors (LuxR) that alter cellular response
elements. (B) QS in Gram positive bacteria. Amino acids or short peptides (wavy lines) are exported and then bind
to cell surface-bound sites that activate phosphorylation cascades, leading to transcriptional changes (Raffa et al.,
2005) (http://www.accessscience.com).
Another common feature of many QS systems is the involvement of a dedicated
ATP-binding cassette (ABC) exporter in the secretion of the peptide pheromone.
Very often, the genes encoding the precursor of the peptide pheromone and the
genes encoding the proteins involved its detection through a the two-component

Chapter I
8
sensing system, as well as those involved in the secretion of peptide, are
transcriptionally linked, and the synthesis of the peptide signal forms an
autoregulatory loop (Figure 2) (Waters & Bassler, 2005).
In recent years, a second type of QS system has been identified in several groups of
Gram-positive streptococci. This system is called ComRS, and uses a small double-
tryptophan peptide pheromone (XIP) as a signal molecule. XIP is internalized by an
oligopeptide ABC transporter typified by Opp/Ami, and interacts with transcriptional
regulator ComR, a proximal regulator of sigX that encodes a master regulator or
alternative sigma factor SigX (ComX). Later , the competence genes for genetic
transformation are activated (Li & Tian, 2012).
In additional to these QS systems, another QS type called autoinducer 2 (AI-2) has
been described in both Gram negative and Gram positive organisms. This type of QS
system enables inter-species signaling. AI-2 was first characterized in marine
bacterium Vibrio harveyi and contributes to regulation of cell-density-dependent
bioluminescence. The synthesis of AI-2 depends on a luxS encoded synthase, which
is a metabolic enzyme involved primarily in the conversion of ribosyl-homocysteine
into homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), the precursor of AI-2.
The LuxR protein is a cytoplasmic receptor and transcription activator. Homologues
of luxS have been found in many species of bacteria, suggesting that AI-2 QS is
widely spread among prokaryotes (Li & Tian, 2012).
1.2 Enterococcus faecalis Fsr Quorum Sensing System One example of QS with a cell density-dependent two-component regulatory system
mechanism in Gram positive bacteria is the Fsr system in Enterococcus faecalis (E.
faecalis regulator). This QS system, first described by Qin et al, controls the
expression of pathogenicity-related extracellular proteases, gelatinase (gelE) and
serine protease (sprE), and has been suggested to also regulate biofilms formation
and other genes important for virulence (Nakayama et al., 2001a; Nakayama et al.,
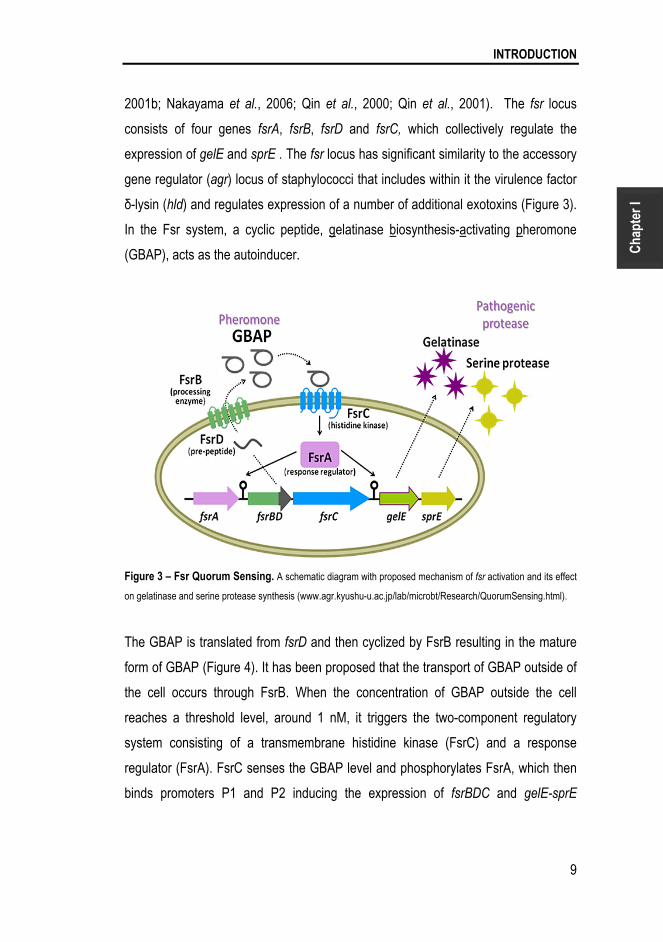
INTRODUCTION
9
Cha
pter
I
2001b; Nakayama et al., 2006; Qin et al., 2000; Qin et al., 2001). The fsr locus
consists of four genes fsrA, fsrB, fsrD and fsrC, which collectively regulate the
expression of gelE and sprE . The fsr locus has significant similarity to the accessory
gene regulator (agr) locus of staphylococci that includes within it the virulence factor
δ-lysin (hld) and regulates expression of a number of additional exotoxins (Figure 3).
In the Fsr system, a cyclic peptide, gelatinase biosynthesis-activating pheromone
(GBAP), acts as the autoinducer.
Figure 3 – Fsr Quorum Sensing. A schematic diagram with proposed mechanism of fsr activation and its effect
on gelatinase and serine protease synthesis (www.agr.kyushu-u.ac.jp/lab/microbt/Research/QuorumSensing.html).
The GBAP is translated from fsrD and then cyclized by FsrB resulting in the mature
form of GBAP (Figure 4). It has been proposed that the transport of GBAP outside of
the cell occurs through FsrB. When the concentration of GBAP outside the cell
reaches a threshold level, around 1 nM, it triggers the two-component regulatory
system consisting of a transmembrane histidine kinase (FsrC) and a response
regulator (FsrA). FsrC senses the GBAP level and phosphorylates FsrA, which then
binds promoters P1 and P2 inducing the expression of fsrBDC and gelE-sprE

Chapter I
10
transcripts (Figure 3) (Nakayama et al., 2001a; Nakayama et al., 2001b; Nakayama
et al., 2006; Qin et al., 2000; Qin et al., 2001).
Qin et al (2000) reported that the gelatinase phenotype requires the concomitant
presence of the fsr and gelE (Qin et al., 2000). In the years following this finding,
several studies reported the loss of the gelatinase phenotype in different
Enterococcus strains. In some cases, this phenomenon was found to be associated
with a deletion of part of Fsr operon, but in other cases incongruence between the
genotype and the phenotype was reported. All these reports indicate that the loss of
GelE phenotype, in the presence of an apparently complete fsr operon, occurs both
in natural and laboratory subcultured E. faecalis isolates (Eaton & Gasson, 2001;
Galloway-Pena et al., 2011; Lopes Mde et al., 2006; Nakayama et al., 2002).
Figure 4 – Structure and properties of GBAP (Gelatinase Biosynthesis Activating Pheromone).
GBAP is a cyclic peptide pheromone in E. faecalis with 11 aminoacid residues and a cyclic peptide containing a
lactone linkage. The lactone ring is indispensable for the activity (Nakayama et al., 2001b).
In 2006, Bourgogne et al. made a transcriptomic study comparing OG1-RF (a
clinical E. faecalis isolate) with its isogenic fsrB deletion mutant and suggested that
fsr is more than a regulator of the gelE and sprE protease genes (Bourgogne et al.,
2006). Moreover, the effect of fsrB mutation had different effects on overall
transcription depending on the growth phase, which points to fsr as a major regulator
of many functions in the cell. This study revealed that besides the proteases, Fsr also
regulates directly a bacteriocin ef1097, and ef0954-0957 (bopABCD) a transcriptional

INTRODUCTION
11
Cha
pter
I
regulator of biofilm formation in plastic surface. The detailed mechanism of gene
regulation and pathogenesis associated with Fsr system and proteases are currently
incomplete (Bourgogne et al., 2006).
Recent transcription studies have also shown that fsr and gelE–sprE expression is
modulated during some stress conditions, namely in blood (Vebo et al., 2009), urine
(Vebo et al., 2010) and vancomycin (unpublished data from our Lab).

Chapter I
12
2. ENTEROCOCCUS GENUS
General Characteristics
The name “entérocoque” was first used in 1899 by Thiercelin to identify a new
species of Gram positive coccus found in the human gut (Thiercelin, 1899).
Enterococci are lactic acid Gram positive bacteria, with ovoid shape (coccus) that
grows in short chains, pairs or as single cells. They are facultative anaerobic
bacteria, catalase negative and can grow between 10-45ºC, although their optimal
temperature is 35-37ºC. Most enterococcal species are able to grow in the presence
of 6, 5% of NaCl, at pH 9, 6 and 40% of bile salts. They are homo-fermentative; and
produce lactic acid from glucose (Mundt, 1986).
The identification of species from the genus Enterococcus by physiological tests
has always been problematic because of their considerable phenotypic diversity.
Furthermore, identification of species by conventional tests often requires long
incubation times. Genotypic identification methods using the 16S and 23S rDNA
genes are more accurate; although they cannot differentiate between all
Enterococcus species (e.g. Enterococcus gallinarum and Enterococcus casseliflavus
show 99.8% homology in their 16S rDNA). Alternative methods have been
successfully applied using amplification of specific genes, for example, the ddl gene
with encodes D-alanine-D-alanine ligase (Ogier & Serror, 2008). Nowadays, the
genus Enterococcus is composed of forty-five species, with the most common
species studied being Enterocccus faecalis and Enterococcus faecium
(http://old.dsmz.de). Historically E. faecalis has been the most intensively studied
due to its prominence in the nosocomial setting.
The first sequenced genome available was that of E. faecalis V583 (Paulsen et al.,
2003), which was isolated from a patient suffering from a persistent bloodstream
infection and was the first reported vancomycin resistant clinical isolate in the United
States (Sahm et al., 1989). E. faecalis V583 has served as a model clinical strain
causing human infections. In 2008, the genome of another E. faecalis strain, OG1RF,

INTRODUCTION
13
Cha
pter
I
(Bourgogne et al., 2008) was reported and in 2010 the genome of 28 other
enterococcal strains (including E. faecalis, E. faecium, E. casseliflavus and E.
gallinarum species) became available (Palmer et al., 2010).
E. faecalis and E. faecium are natural members of the gastrointestinal microbiota in
humans, varying in abundance among individuals along the gastro-intestinal tract.
Enterococci are commonly isolated from foods, plants, water and soils, because of
their use in fermentations, and also as a result of dissemination from fecal sources
combined with their natural tolerance to adverse environmental conditions (Lopes et
al., 1999; Ogier & Serror, 2008). Unlike many other lactic acid bacteria, enterococci
are not considered as “Generally Recognized As Safe” (GRAS) and their detection in
water is regarded as an indicator of fecal contamination (Godfree et al., 1997).
Enterococci therefore have an ambiguous status concerning assessment of
enterococci food containing safety. On the one hand, enterococci are used in cheese
making, in the development of flavors, aroma and contributing to the ripening of
cheeses such Cheddar, Feta and Mozarella. On the other hand, their ability to
produce biogenic amines in cheese and fermented sausages and their propensity for
genetic exchange constitute negative aspects for their utilization as probiotic
(Foulquie Moreno et al., 2006; Giraffa, 2003; Ogier & Serror, 2008).
2.1 Enterococcus spp. - An Opportunistic Pathogen
Enterococci are commensal bacteria of the gastro-intestinal tracts of humans,
animals and insects. Although harmless in healthy individuals, enterococcal clinical
isolates become pathogenic in patients in intensive care units, in hospitalized
patients with impaired immune systems and elderly people. As opportunistic
pathogens, they are prevalent in the nosocomial environment, causing infections of
the urinary tract, bloodstream, intra-abdominal and pelvic regions, surgical sites, and
rarely the central nervous system (Foulquie Moreno et al., 2006). Recent data

Chapter I
14
indicate that E. faecalis is the third most commonly isolated nosocomial pathogen
(12% of all hospital infections) (Hollenbeck & Rice, 2012).
Currently, in the United States and Europe, infections caused by E. faecium are
much more frequently resistant to vancomycin and ampicillin than those caused
by E. faecalis. E. faecium is now almost as common a cause of nosocomial infection
as E. faecalis. This change in species is of paramount clinical importance, as E.
faecium is by far the more difficult of the two species to treat (Arias & Murray, 2012).
Antibiotics interrupt vital cellular functions through different modes of action.
Inhibition of growth is usually achieved by attacking the cell-wall and cell membrane
integrity, or by interfering with DNA, RNA or protein synthesis. Antibiotics can be
either bactericidal or bacteriostatic depending on their mechanism of action.
Bactericidal antibiotics effectively kill the target bacteria; and bacteriostatic halt cell
growth and replication. From a clinical perspective, infections caused by multidrug
resistant enterococci are difficult to treat due to limited therapeutic options.
Enterococci have been shown to possess a broad range of intrinsic antibiotic
resistances (Leclercq et al., 1992; Moellering, 1992; Murray, 1990) and are able to
acquire high-level drug resistance to certain antibiotics. Resistances may arise by
point mutations in the drug binding site, like in quinolones (Onodera et al., 2002) and
ampicillin, or by acquisition of resistance genes, as in the case of aminoglycosides,
macrolides, chloramphenicol, tetracycline and glycopeptides, of which vancomycin
resistance is the most relevant clinically (Leclercq et al., 1992; Moellering, 1992;
Murray, 1990; Onodera et al., 2002; Saurina et al., 1997). Enterococal success as
nosocomial pathogens is also related to their ability to survive for long periods on
environmental surfaces, including medical equipment, bed rails and doorknobs. They
are tolerant to heat, chlorine and some alcohol preparations (Arias & Murray, 2012;
Braga et al., 2011). Dissemination of resistance to different antibiotics is a problem
among clinical, dairy and veterinary Enterococcus strains (de Fatima Silva Lopes et
al., 2005; Lopes Mde et al., 2003). Enterococcal infections that result in human
disease can be fatal, particularly those caused by vancomycin-resistant enterococci
(VRE). In 1986, the first VRE strains appeared in Europe and, in 1989 the first case
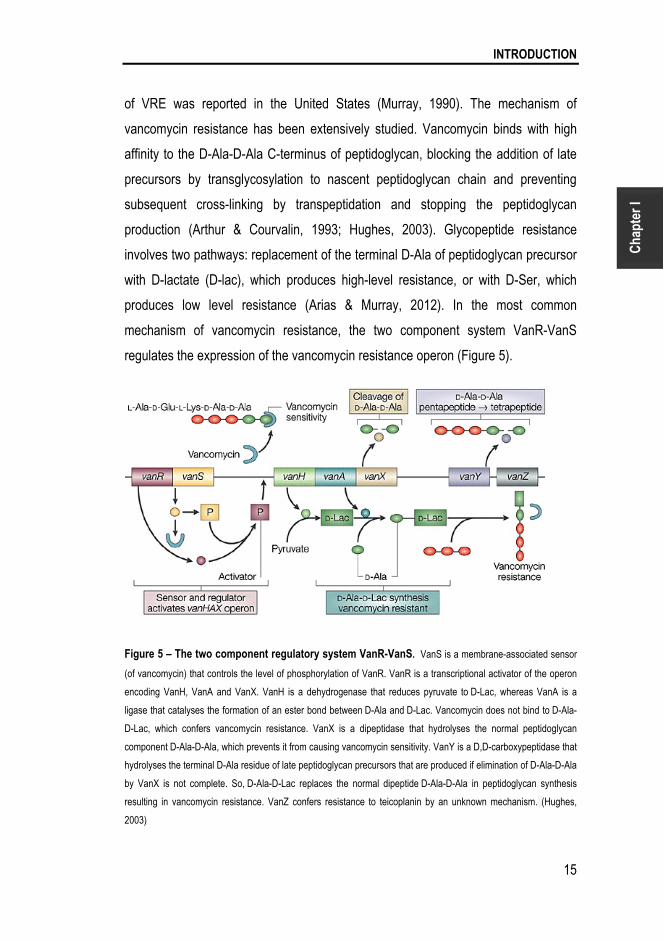
INTRODUCTION
15
Cha
pter
I
of VRE was reported in the United States (Murray, 1990). The mechanism of
vancomycin resistance has been extensively studied. Vancomycin binds with high
affinity to the D-Ala-D-Ala C-terminus of peptidoglycan, blocking the addition of late
precursors by transglycosylation to nascent peptidoglycan chain and preventing
subsequent cross-linking by transpeptidation and stopping the peptidoglycan
production (Arthur & Courvalin, 1993; Hughes, 2003). Glycopeptide resistance
involves two pathways: replacement of the terminal D-Ala of peptidoglycan precursor
with D-lactate (D-lac), which produces high-level resistance, or with D-Ser, which
produces low level resistance (Arias & Murray, 2012). In the most common
mechanism of vancomycin resistance, the two component system VanR-VanS
regulates the expression of the vancomycin resistance operon (Figure 5).
Figure 5 – The two component regulatory system VanR-VanS. VanS is a membrane-associated sensor
(of vancomycin) that controls the level of phosphorylation of VanR. VanR is a transcriptional activator of the operon
encoding VanH, VanA and VanX. VanH is a dehydrogenase that reduces pyruvate to D-Lac, whereas VanA is a
ligase that catalyses the formation of an ester bond between D-Ala and D-Lac. Vancomycin does not bind to D-Ala-
D-Lac, which confers vancomycin resistance. VanX is a dipeptidase that hydrolyses the normal peptidoglycan
component D-Ala-D-Ala, which prevents it from causing vancomycin sensitivity. VanY is a D,D-carboxypeptidase that
hydrolyses the terminal D-Ala residue of late peptidoglycan precursors that are produced if elimination of D-Ala-D-Ala
by VanX is not complete. So, D-Ala-D-Lac replaces the normal dipeptide D-Ala-D-Ala in peptidoglycan synthesis
resulting in vancomycin resistance. VanZ confers resistance to teicoplanin by an unknown mechanism. (Hughes,
2003)

Chapter I
16
3. ENTEROCOCAL VIRULENCE
The Role of Fsr, Gelatinase and Serine Protease
For enterococci to act as pathogen they must first adhere to host tissues. During
infection of sterile tissues, enterococci encounter an environment vastly different than
the gut, with high redox potentials, limited nutrients, phagocytic leukocytes, and other
host defenses. Enterococci express factors – virulence factors - that permit
adherence to host cell and extracellular matrix, facilitate tissue invasion, effect
immunomodulation and cause toxin-mediated damage (Gilmore, 2002).
The first examination of enterococal virulence was reported in 1899, the same year
this organism was discovered. MacCalum and Hasting described a fatal case of
endocarditis caused by an organism that they termed Micrococcus zymogenes. The
bacteria expressed cytolitic (or hemolytic) and protease (gelatinase) activities and
likely represented E. feacalis (MacCallum & Hastings, 1899). Since then
Enterococcus virulence has been extensively studied. About a dozen putative
virulence factors have been reported from virulence analysis in various animal
models (Table 1). They are involved in attachment both to host cells and to
extracellular matrix proteins (AS, Esp, EfaA), in resistance to macrophages (AS,
HypR), in cell and tissue damage (Cyl, GelE, SprE) and in immune system evasion
(capsular polysaccharides) (Gilmore, 2002; Tendolkar et al., 2003).
Some of these virulence factors are encoded by conjugative plasmid genes (AS and
Cyl) or rearranged chromosomal regions such as i) the fsr locus (GelE, SprE and Fsr
(Nakayama et al., 2001a; Qin et al., 2000)), ii) the large chromosomal region
described as the pathogenicity island (Esp, Cyl, AS and Gls24-like (Shankar et al.,
2002)) and iii) the cps locus (Hancock & Gilmore, 2002). Plasmid encoded virulence
factors have been shown to be transmissible by gene transfer mechanisms (Chow et
al., 1993; Wirth, 1994) (Table 1).

INTRODUCTION
17
Cha
pter
I
Table 1 – E. faecalis virulence factors and their putative role (Arias & Murray, 2012; Jett et al., 1994;
Ogier & Serror, 2008).
Gene Name Putative role Reference(s)
Cell surface determinants AS protein Aggregation
substance
Adhesion, tissue colonization (Schlievert et al., 1998)
(Waters et al., 2004)
Esp Surface protein Biofilm formation (Shankar et al., 1999)
Ace Adhesion
to collagen
Adhesion to ECM (Nallapareddy et al., 2000)
(Nallapareddy et al., 2011b)
Bee Biofilm enhancer Biofilm formation (Tendolkar et al., 2006)
Ebp Endocarditis and
biofilm associated
pili
Biofilm formation and
adhesion to human platelets
(Nallapareddy et al., 2006)
(Nallapareddy et al., 2011a)
(Nallapareddy et al., 2011b)
ElrA Surface protein Role in experimental
peritonitis, resistance to host
defenses
(Brinster et al., 2007)
StrA Sortase Biofilm formation, role in
catheter-associated UTIs
(Guiton et al., 2009)
(Guiton et al., 2010)
Exopolysaccharides
cps cluster Capsular
polysaccharides
Resistance to host defenses (Hancock & Gilmore, 2002)
epa cluster Enterococcal
polysaccharide
antigen
Resistance to host defenses (Teng et al., 2002)
(Teng et al., 2009)
Secreted factors
GelE Gelatinase Tissue damage, formation of
biofilms, immune evasion
(Singh et al., 2005)
SprE Serine Protease Tissue damage, formation of
biofilms, immune evasion
(Kawalec et al., 2005)
CylA-M Cytolysin Tissue damage (Jett et al., 1992)
Regulators
FsrA Fsr System gelE, sprE and ace regulation (Nakayama et al., 2001a; Qin
et al., 2001)
CylR1-R2 Cyl operon
regulator
Cytolysin regulation (Jett et al., 1994)

Chapter I
18
E. faecalis is an example of an opportunistic pathogen that uses QS system to
produce virulence factors to succeed during infection. One of the most studied is the
fsr QS (see below) that regulates the virulence factors serine protease and
gelatinase. The serine protease has high similarity to the Staphylococcus glutamil-
endopeptidases but has not been purified or characterized. Some studies have
reported this protease to have some role in biofilm formation, but its exact role is still
unknown (Kawalec et al., 2005). The gelatinase has been largely described as an
important virulence factor. This protease was first described in 1975 by Gold et al.
who identified a protease in E. faecalis OG1-10 responsible for human gelatin oral
degradation, suggesting that it was a virulence factor (Gold et al., 1975). This protein
is secreted as a zinc metalloprotease (thermolysin-M4 protease) capable to
hydrolyze numerous subtracts, including gelatin, collagen, casein, fibrin, hemoglobin
and other small bioactive peptides. The protein gelatinase produced by E. faecalis
OG1-10 was isolated by Makinen et al. in 1989 (Makinen et al., 1989). The gene was
subsequently identified (gelE) and its sequence determined (Su et al., 1991).
Gelatinase activity was detected in enterococal clinical strains (Lopes Mde et al.,
2006; Singh et al., 2005).
Fsr is the only QS system known to contribute to E. faecalis biofilm formation
(Mohamed & Huang, 2007). Biofilms are important for enterococcal infections
because they protect bacteria against antibiotics and phagocytosis (Paganelli et al.,
2012). The formation of multilayer biofilms involves a complex process from
attachment of single cells to the development of a 3D biofilm structure. Under optimal
conditions a mature biofilm develops consisting of loosely packed microcolonies held
together with extracellular polymeric substances (EPS) interspersed with water
channels through which nutrients reach deeper parts of the biofilm. During biofilm
formation, autolysins contribute to different aspects. They can act as adhesins, but
when released by proteases they can lyse cells and thereby generate extracellular
(e)DNA, which is necessary to stabilize the EPS biofilm in microcolonies. Autolysis
regulation is very important for bacterial growth and division. Enzymes involved in
autolysis are peptidoglycan (PG) hydrolases which play a role in cell wall growth and

INTRODUCTION
19
Cha
pter
I
turnover, cell separation, recycling of muropeptides, lysis by cell-wall synthesis
inhibitors, competence, sporulation, flagellum formation and pathogenicity (Bayles,
2007; Mohamed & Huang, 2007). Autolysis control may also be involved in tolerance
to cell-wall active antibiotics, as previously demonstrated for the homologous Agr
system of Staphylococcus aureus (Antignac et al., 2007; Bayles, 2007). Knowing that
PG degradation products are also a major elicitor of the host immune response it is
obvious to assume/speculate a relation between autolysis and host immune
recognition and response. Therefore, autolysis control is crucial for virulence, stress
response and host immune system modulation (Antignac et al., 2007; Bayles, 2007;
Thomas et al., 2008).
Different studies described that gelatinase has a critical role in biofilm formation
(Hancock & Perego, 2004; Kristich et al., 2004; Mohamed & Murray, 2005; Mohamed
& Murray, 2006). In 2008, Thomas et al invoked the fratricidal model for E. faecalis
biofilm development. In this model, GelE activated the lysis of a subpopulation of
bacteria and thereby catalyzes the release of genomic DNA (e)DNA, as originally
proposed for S. pneumoniae autolysis (Gilmore & Haas, 2005). SprE negatively
affects autolysis, (e)DNA release and early biofilm maturation by negatively
regulating GelE activity, and thereby acts as an immunity protein against lysis. GelE
and SprE execute their characteristic effects following downstream interactions with
the primary autolysin, AtlA, by modifying the cell-wall affinity of proteolytically
processed AtlA. The interplay of the two secreted and co-regulated proteases seems
to be tightly regulated. A minority subpopulation of quorum non-responders (GelE–
SprE–) act as prey cells, for targeted fratricide mediated by the quorum-responsive
predator cells that form the majority in the biofilm. In response to the GBAP peptide,
predator cells secrete GelE and SprE proteases. Prey cells are susceptible cells that
have not (yet) responded to GBAP. If GelE reaches the cells before SprE, this results
in lysis via release of AtlA from their surface, and this in turn can also lyse
neighboring cells (Thomas et al., 2008; Thomas et al., 2009).
In 2011, Pinkston et al, demonstrated that Fsr modulates Ace surface levels through
its regulation of GelE which directly cleaves Ace, subsequently impacting on the

Chapter I
20
ability of cells to adhere to collagen. The bacterial surface protein has an important
role in E. faecalis virulence by mediating adherence and colonization to host tissue
which is an early step toward clinical infection (Pinkston et al., 2011). Another study
indicated that, the absence of gelatinase (in E. faecalis OG1RF) leads to high levels
of secreted antigenic SalB, in the exoproteome. The relation between GelE and SalB
it still not clear but it is known that the absence of SalB increases autolysis and cell
morphological changes (Shankar et al., 2012). In addition to these studies, GelE and
SprE have also been reported to have an important role in translocation across
intestinal T84 cells and in degradation of antimicrobial peptides (AMPs) from immune
system of Galleria mellonella (Cytrynska et al., 2007).
All these studies indicate that Fsr-GelE-SprE has an important role in E. faecalis
virulence and place these traits as promising targets to combat the E. faecalis
infection. Nakayama et al., discovered two secondary metabolites, siamycin and
ambuic acid, which act as QS inhibitors. Siamycin inhibits GBAP-induced
phosphorylation of receptor histidine kinase FsrC and ambuic acid inhibits the
proteolytic processing of FsrD, the propeptide of GBAP. However, none of these
compounds influence E. faecalis growth (Nakayama et al., 2007; Nakayama et al.,
2009).
3.1 Animal Models to Study Fsr and Proteases
Several studies provided evidence that both Fsr and the proteases independently
contribute to the pathogenicity of E. faecalis in different infection models, (Table 2)
but their exact contributions to E. faecalis infection are still unknown. The use of
animal models is important to elucidate the pathogen actions in the host.

INTRODUCTION
21
Cha
pter
I
Table 2 – Host models used to study virulence associated to the Fsr and/or gelatinase.
Animal model used E. faecalis mutants used for
the study
Enterococcus strains References
Rabbit endophthalmitis OG1RFΔfsrB2 E. faecalis OG1RF (Mylonakis et al., 2002)
Rat endocarditis
OG1RFΔfsrB1
OG1RFΔfsrB2
OG1RFΔgelE1
E. faecalis OG1RF (Singh et al., 2005)
Galleria mellonella
QA29bΔfsrB2
QA29bΔgelE2
LSE4aΔfsrB2
LSE4aΔgelE2
E. faecalis OG1RF
E. faecalis QA29B
E. faecalis LSE4a
E. faecalis LN68
E. faecium QSE32
E. durans QN1
E. faecalis ATCC 51299
(Gaspar et al., 2009;
Park et al., 2007)
Arabidopsis thaliana
OG1RFΔfsrA,
OG1RFΔfsrB1,
OG1RFΔfsrC1,
OG1RFΔgelE1,
OG1RFΔsprE1
E. faecalis FA-2-2,
E. faecalis V583,
E. faecalis OG1RF
(Jha et al., 2005)
Caenorhabditis elegans OG1RFΔfsrB1
OG1RFΔgelE1
E. faecalis OG1RF (Sifri et al., 2002)
Zebrafish OG1RFΔfsrB1
OG1RFΔgelE1
OG1RFΔsprE1
OG1RFΔgelEΔsprE1
E. faecalis OG1RF
(Prajsnar et al., 2013)
Different outcomes have been observed in different assays when fsrABC or gelE-
sprE mutants were compared with the parental strain. In rat endocarditis a greater
decrease in endocarditis severity was observed when the proteases were deleted
comparing with deletion of fsrB (Singh et al., 2005). In other studies the observation
was the opposite - in rabbit endophtalmitis (Engelbert et al., 2004; Mylonakis et al.,
2002), murine and C. elegans infection (Garsin et al., 2001; Sifri et al., 2002), G.
mellonella infection (Gaspar et al., 2009) - a greater attenuation was observed when
1 insertional mutant 2 in-frame deletion mutant

Chapter I
22
fsrB was deleted than when proteases were absent. These results highlight the
complexity of interaction between this system and the host, and the importance of
finding a highly tractable animal model that will allow precise determination of the
role of Fsr-GelE-SprE in the infection.

INTRODUCTION
23
Cha
pter
I
4. DROSOPHILA MELANOGASTER
A model to Study Host-pathogen Interaction
The use of invertebrate animal models has provided tremendous insights into
pathogen-host interaction of many human pathogens, and has revealed that many
aspects of these interactions in higher host organisms are conserved in
invertebrates. The fruit fly Drosophila melanogaster (Drosophila) is one of the most
used for studying host-pathogen interaction of bacteria, fungal and viral pathogens.
The life cycle of Drosophila has different stages: the embryo, 1st instar larva, 2nd
instar larva, 3rd instar larva, prepupa, pupa and adult (Igboin et al., 2012) (Figure 6).
Figure 6 – The life cycle of Drosophila. The life cycle is divided in six stages: embryo, 1st instar larva, 2nd
instar larva, 3rd instar larva, prepupa, pupa and adult. (http://www.immortalhumans.com/longevity-research-on-fruit-
flies-providing-promising-hope-for-longer-human-lifespan/).

Chapter I
24
Drosophila is a model organism with many advantages: small size, short generation
time (depending on the ambient temperature, from being an egg to become an adult
it takes 7 days), a fully sequenced genome and pre-existing libraries of genetic
mutants. Studies often use a clear endpoint (death), and this model host can be used
in large quantities to facilitate statistical analysis. Numerous studies have revealed
significant parallels between the Drosophila immune response and mammalian
innate response. The absence of an adaptive immune response permits the study of
interactions between pathogens and the host innate immune response in isolation.
Drosophila loss-of-function immune response mutants have been used to examine
the roles of the genes in the response to infection with various pathogens.
Transgenic Drosophila has been used to monitor the activation of immune response
pathways upon infection and to examine the effects of transgenically expressed
pathogen proteins on the host. Drosophila rely solely on an innate immune system to
combat infecting microbes and, like mammals, they detect the presence of invading
microbes using pattern recognition receptors, which recognize conserved microbial
motifs and activate a response that is specific for the type of invading microbes
(Igboin et al., 2012) (Figure 7).
Figure 7 – Scheme of humoral immune system
inside Drosophila. A systemic infection induces the
transcription of antimicrobial peptides, mainly in the fat
body of the fruit fly (blue), which is analogous to the liver.
These peptides are transported into the haemolymph
(blood), where they accumulate to high concentrations and
circulate throughout the body. Some tissues respond
directly to localized sites of infection, such the trachea
(orange) .The cellular immune response is characterized by
the presence of haemocytes (blood cells), which circulate
or attach themselves to organs. A systemic infection can be
instigated in the laboratory by puncture with a septic needle
(as indicated in the figure) or in nature by a septic wound.
In both cases, the site of wounding clots containing a
melanin- seal (Kimbrell & Beutler, 2001).

INTRODUCTION
25
Cha
pter
I
The innate immune system consists of both cellular and humoral components. The
cellular response involves specialized hemocytes (blood cells), which engage in
phagocytosis and encapsulation of foreign microbes. The body cavity of Drosophila
is filled with circulating hemolymph that contains free-floating hemocytes (Figure 7
and 8). Drosophila larvae contain several thousand hemocytes, which can be divided
into the following three cell types on the basis of their structural and functional
features: plasmocytes, crystal cells and lamellocytes. Plasmocytes represent 90%-
95% of all mature larval hemocytes and function in phagocytic removal of dead cells
and microbial pathogens. Lamellocytes are relatively large, flat, and adherent cells
that primarily function in encapsulation and neutralization of objects too large to be
phagocytized. Crystal cells constitute 5% of the larval hemocytes and are
nonphagocytic cells involved in the melanization process (Figure 8).
Figure 8 – Scheme of an overview of Drosophila host defense – cellular and humoral responses.
These overview demonstrated that the two response types are connected (Lemaitre & Hoffmann, 2007)
Melanization is characterized by the blackening reaction (deposition of melanin) at
the site of cuticular injury, or on surface of parasites invading the hemocoel, and
plays an important role in reactions such as wound healing, encapsulation,

Chapter I
26
sequestration of microorganisms and production of toxic intermediates that kill the
pathogens. Melanization requires the activation of proPhenoloxidase, an enzyme that
catalyzes the oxidation of phenols to quinones, which polymerize melanin (Figure 8
and 9) (Lemaitre & Hoffmann, 2007; Meister & Lagueux, 2003). During this process
reactive oxygen species are produced, which can harm the host in addition to
harming the pathogen (Chambers et al., 2012).
Figure 9 - Model for melanization activation upon microbial Infection. Upon the recognition of a
microorganism, a pattern recognition receptor (PRR) is presumed to trigger a protease cascade involving the
proteases MP1 and MP2/Sp7/PAE1, which culminates in the cleavage of prophenol oxidase (PPO) to phenol oxidase
(PO). The serpin Spn28D controls the release and availability of PO by inhibiting its activation, possibly in crystal
cells. Spn27A acts in the hemolymph to inhibit the MP1/MP2 cascade and prevent excessive melanization. Spn77Ba
regulates melanization in the tracheal epithelium by inhibiting the same protease cascade. Other studies revealed the
involvement of serine protease homologues (SPHs) in activating PO. PO inhibitors limit melanization by directly
inhibiting the enzymatic activity of PO. Melanization reaction it also involved in others immune responses such as
blood coagulation, AMP expression, wound healing and phagocytosis (Tang, 2009).
The humoral response is mediated by three signaling pathways: the Imd (immune
deficiency), Toll and JAK/STAT (janus kinase/signal transducer and activator of

INTRODUCTION
27
Cha
pter
I
transcription) pathways (Figure 8 and 10). The humoral response involves the
production of antimicrobial peptides (AMPs) through Toll or Imd pathway.
Figure 10 – Drosophila humoral Immune system model – Toll and Imd pathways. These models
represent the cascade of events for the activation of Toll and Imd pathways. The Toll pathway is activated by fungi
and Gram positive bacteria and Imd is activated only by Gram negative bacteria. Toll pathway: The Toll receptor is
activated upon binding with a cleaved form of Spätzle that is processed by proteolytic cascade activated by bacteria
secreted molecules. After the activation of Toll receptor the AMPs are produced through a cascade of events. Imd
Pathway: The bacteria components bind directly to receptor and are recruit the adaptor Imd. The Imd interacts with
dFADD and the cascade of events is activated and the AMPs are produced(Lemaitre & Hoffmann, 2007).

Chapter I
28
Produced AMPs can be classified in seven groups, with differential effectiveness,
against fungi (Drosomycin and Metchnikowin), Gram positive bacteria (Defensin) and
Gram negative bacteria (Diptericin, Drosocin, Attacin and Cecropin) (Lemaitre &
Hoffmann, 2007)The Imd signaling pathway is homologous to the mammalian tumor
necrosis factor receptor 1 signaling pathway, and only differs at the level of
detection/activation. This pathway regulates the production of AMPs by fat body cells
and gut epithelial cells, respectively, in response to primarily Gram negative bacterial
infections. The Imd pathway, activated by its receptor peptidoglycan recognition
protein (PGRP)-LC and the Imd protein, acts through TAK1, signaling the IkB kinase
complex to activate the NF-kB transcription factor Rel, which responds to infection by
Gram-negative bacteria (Figure 8 and 10) (Davis & Engstrom, 2012; Kounatidis &
Ligoxygakis, 2012; Lemaitre & Hoffmann, 2007).
The Toll signaling pathway is homologous to the mammalian Toll/IL-1 receptor
signaling pathway, although unlike the mammalian receptors Drosophila Toll does
not directly recognize bacterial components. Like Imd, this pathway regulates
systemic AMP expression in fat body, primarily in response to fungal and Gram-
positive bacterial infection. The Toll pathway relies on cleavage of the extracellular
ligand, Spätzle, followed by signaling through the Toll receptor and its intracellular
adaptor protein complex, which contains MyD88, Tube and the Pelle kinase. The Toll
pathway is activated in response to Gram positive bacteria, fungi and yeast (Figure
10) (Davis & Engstrom, 2012; Kounatidis & Ligoxygakis, 2012; Lemaitre & Hoffmann,
2007).
The Drosophila JAK-STAT signaling pathway comprises the same components as
the mammalian pathway, although they differ in the number of JAKs (one in
Drosophila and four in mammalians) and STATs (one in Drosophila and seven in
mammalians) they possess. The JAK-STAT pathway regulates numerous
physiological processes including immune response; however its contribution to the
immune response is not as well studied as Imd and Toll pathways. The JAK-STAT
pathway is involved in the immune response in the Drosophila gut, where it helps

INTRODUCTION
29
Cha
pter
I
maintain epithelial cell homeostasis by regulating stem cell proliferation (Igboin et al.,
2012; Lemaitre & Hoffmann, 2007).
In 2005, Cox and Gilmore characterized the Drosophila microflora and examined the
occurrence of enterococci in the gastrointestinal consortium of Drosophila (Cox &
Gilmore, 2007). They found that Drosophila was naturally colonized by
representatives of five bacterial phyla. Among these were several species of
enterococci, including E. faecalis, E. faecium, E. gallinaraum, and E. durans, as well
as a previously detected but uncultured Enterococcus species. These strains were
found only in the GI tract of Drosophila, as occurs normally in humans. In this study
they also proved that Drosophila is a good model to study the virulence factor
cytolysin (Cox & Gilmore, 2007).

Chapter I
30
5. AIMS AND SCOPE OF THESE THESIS
The discovery of communication between bacteria via quorum-sensing, which
orchestrate important temporal events during the infection process, provides a novel
opportunity to combat bacterial infection. E. faecalis infections constitute a serious
problem in the hospital environment and its ability to acquire/transfer antibiotic
resistance and virulence factors is limiting the use of antibiotics. New therapeutics
are urgently needed, and these may be based on a better understanding of the
factors that are important for E. faecalis infection.
Since the Fsr system was described, numerous studies have shown its importance in
the infections process. These studies have demonstrated that fsr is not only involved
during the E. faecalis infection, but also contributes to enterococcal survival in
different environments. However, the exact mechanism by which Fsr contributes to
virulence during infection is not entirely clear. Moreover, many strains have been
reported to carry the entire fsr and gelE-sprE operons, despite their inability to
phenotypically show gelatinase activity isolates (Eaton & Gasson, 2001; Galloway-
Pena et al., 2011; Lopes Mde et al., 2006; Nakayama et al., 2002).
This PhD thesis aims at clarifying the contribution Fsr regulon to E. faecalis biology
and to host injury using different approaches. Three main questions were addressed
in four chapters of this thesis:
- Under which conditions the Fsr system is shut down? (Chapters II and III)
- Which other genes, if any, do Fsr and/or GelE-SprE regulate? (Chapter IV)
- How does Fsr contribute to E. faecalis infection in Drosophila? (Chapters IV and V)
Chapters II and III describe two different conditions in which fsr is shutdown. Chapter
II focuses on a particular strain, E. faecalis LN68 (already described by (Lopes Mde
et al., 2006)), that has all fsr-gelE-sprE genes but does not exhibit the gelatinase

INTRODUCTION
31
Cha
pter
I
phenotype. In a previous transcriptomic study made in our laboratory, the VRE strain
V583 was subjected to a therapeutic vancomycin dose. fsr-gelE-sprE genes were
found to be affected by that cell wall active antibiotic. In Chapter III we looked for the
reason for vancomycin interference with Fsr activity.
In Chapter IV we looked for genes directly and indirectly regulated by Fsr and
attributed a role in E. faecalis virulence. To study these genes, we first established
Drosophila as a model to study Fsr virulence. In Chapter V we investigated the
influence that Fsr system has on Drosophila humoral and cellular immune system.
The final chapter of this thesis is a general discussion (Chapter VI) correlating all
chapters discussing the importance of the new findings presented, and proposing
future advances in controlling E. faecalis infection.

Chapter I
32
6. BLIBLYOGRAPHY
Antignac, A., Sieradzki, K. & Tomasz, A. (2007). Perturbation of cell wall synthesis
suppresses autolysis in Staphylococcus aureus: evidence for coregulation of cell wall
synthetic and hydrolytic enzymes. J Bacteriol 189, 7573-7580.
Arias, C. A. & Murray, B. E. (2012). The rise of the Enterococcus: beyond
vancomycin resistance. Nat Rev Microbiol 10, 266-278.
Arthur, M. & Courvalin, P. (1993). Genetics and mechanisms of glycopeptide
resistance in enterococci. Antimicrob Agents Chemother 37, 1563-1571.
Bassler, B. & Vogel, J. (2013). Bacterial regulatory mechanisms: the gene and
beyond. Curr Opin Microbiol 16, 109-111.
Bassler, B. L. (1999). How bacteria talk to each other: regulation of gene expression
by quorum sensing. Curr Opin Microbiol 2, 582-587.
Bayles, K. W. (2007). The biological role of death and lysis in biofilm development.
Nat Rev Microbiol 5, 721-726.
Bourgogne, A., Hilsenbeck, S. G., Dunny, G. M. & Murray, B. E. (2006).
Comparison of OG1RF and an isogenic fsrB deletion mutant by transcriptional
analysis: the Fsr system of Enterococcus faecalis is more than the activator of
gelatinase and serine protease. J Bacteriol 188, 2875-2884.
Bourgogne, A., Garsin, D. A., Qin, X. & other authors (2008). Large scale
variation in Enterococcus faecalis illustrated by the genome analysis of strain
OG1RF. Genome Biol 9, R110.

INTRODUCTION
33
Cha
pter
I
Braga, T. M., Marujo, P. E., Pomba, C. & Lopes, M. F. (2011). Involvement, and
dissemination, of the enterococcal small multidrug resistance transporter QacZ in
resistance to quaternary ammonium compounds. J Antimicrob Chemother 66, 283-
286.
Brinster, S., Posteraro, B., Bierne, H., Alberti, A., Makhzami, S., Sanguinetti, M.
& Serror, P. (2007). Enterococcal leucine-rich repeat-containing protein involved in
virulence and host inflammatory response. Infect Immun 75, 4463-4471.
Chambers, M. C., Lightfield, K. L. & Schneider, D. S. (2012). How the fly balances
its ability to combat different pathogens. PLoS Pathog 8, e1002970.
Chow, J. W., Thal, L. A., Perri, M. B., Vazquez, J. A., Donabedian, S. M., Clewell,
D. B. & Zervos, M. J. (1993). Plasmid-associated hemolysin and aggregation
substance production contribute to virulence in experimental enterococcal
endocarditis. Antimicrob Agents Chemother 37, 2474-2477.
Cox, C. R. & Gilmore, M. S. (2007). Native microbial colonization of Drosophila
melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect
Immun 75, 1565-1576.
Cytrynska, M., Mak, P., Zdybicka-Barabas, A., Suder, P. & Jakubowicz, T.
(2007). Purification and characterization of eight peptides from Galleria mellonella
immune hemolymph. Peptides 28, 533-546.
Davis, M. M. & Engstrom, Y. (2012). Immune response in the barrier epithelia:
lessons from the fruit fly Drosophila melanogaster. J Innate Immun 4, 273-283.
de Fatima Silva Lopes, M., Ribeiro, T., Abrantes, M., Figueiredo Marques, J. J.,
Tenreiro, R. & Crespo, M. T. (2005). Antimicrobial resistance profiles of dairy and
clinical isolates and type strains of enterococci. Int J Food Microbiol 103, 191-198.

Chapter I
34
Eaton, T. J. & Gasson, M. J. (2001). Molecular screening of Enterococcus virulence
determinants and potential for genetic exchange between food and medical isolates.
Appl Environ Microbiol 67, 1628-1635.
Engelbert, M., Mylonakis, E., Ausubel, F. M., Calderwood, S. B. & Gilmore, M. S.
(2004). Contribution of gelatinase, serine protease, and fsr to the pathogenesis of
Enterococcus faecalis endophthalmitis. Infect Immun 72, 3628-3633.
Foulquie Moreno, M. R., Sarantinopoulos, P., Tsakalidou, E. & De Vuyst, L.
(2006). The role and application of enterococci in food and health. Int J Food
Microbiol 106, 1-24.
Fuqua, W. C., Winans, S. C. & Greenberg, E. P. (1994). Quorum sensing in
bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J
Bacteriol 176, 269-275.
Galloway-Pena, J. R., Bourgogne, A., Qin, X. & Murray, B. E. (2011). Diversity of
the fsr-gelE region of the Enterococcus faecalis genome but conservation in strains
with partial deletions of the fsr operon. Appl Environ Microbiol 77, 442-451.
Garsin, D. A., Sifri, C. D., Mylonakis, E., Qin, X., Singh, K. V., Murray, B. E.,
Calderwood, S. B. & Ausubel, F. M. (2001). A simple model host for identifying
Gram-positive virulence factors. Proc Natl Acad Sci U S A 98, 10892-10897.
Gaspar, F., Teixeira, N., Rigottier-Gois, L., Marujo, P., Nielsen-LeRoux, C.,
Crespo, M. T., Lopes Mde, F. & Serror, P. (2009). Virulence of Enterococcus
faecalis dairy strains in an insect model: the role of fsrB and gelE. Microbiology 155,
3564-3571.

INTRODUCTION
35
Cha
pter
I
Gilmore, M. S. & Haas, W. (2005). The selective advantage of microbial fratricide.
Proc Natl Acad Sci U S A 102, 8401-8402.
Gilmore, M. S., Phillip S. Coburn, Sreedhar R. Nallapareddy, Barbara E. Murray
(2002). Enterococcal Virulence. In The Enterococci Pathogenesis, Molecular Biology,
and Antibiotic Resistance, pp. 301-354. Edited by M. S. Gilmore, Clewell DB,
Courvalin P, Dunny GM, Murray BE, Rice LB. Washington D.C.: American Society
for Microbiology.
Giraffa, G. (2003). Functionality of enterococci in dairy products. Int J Food Microbiol
88, 215-222.
Godfree, A. F., Kay, D. & Wyer, M. D. (1997). Faecal streptococci as indicators of
faecal contamination in water. Soc Appl Bacteriol Symp Ser 26, 110S-119S.
Gold, O. G., Jordan, H. V. & van Houte, J. (1975). The prevalence of enterococci in
the human mouth and their pathogenicity in animal models. Arch Oral Biol 20, 473-
477.
Guiton, P. S., Hung, C. S., Kline, K. A. & other authors (2009). Contribution of
autolysin and Sortase a during Enterococcus faecalis DNA-dependent biofilm
development. Infect Immun 77, 3626-3638.
Guiton, P. S., Hung, C. S., Hancock, L. E., Caparon, M. G. & Hultgren, S. J.
(2010). Enterococcal biofilm formation and virulence in an optimized murine model of
foreign body-associated urinary tract infections. Infect Immun 78, 4166-4175.
Hancock, L. E. & Gilmore, M. S. (2002). The capsular polysaccharide of
Enterococcus faecalis and its relationship to other polysaccharides in the cell wall.
Proc Natl Acad Sci U S A 99, 1574-1579.

Chapter I
36
Hancock, L. E. & Perego, M. (2004). The Enterococcus faecalis fsr two-component
system controls biofilm development through production of gelatinase. J Bacteriol
186, 5629-5639.
Hastings, J. W. & Greenberg, E. P. (1999). Quorum sensing: the explanation of a
curious phenomenon reveals a common characteristic of bacteria. J Bacteriol 181,
2667-2668.
Hollenbeck, B. L. & Rice, L. B. (2012). Intrinsic and acquired resistance
mechanisms in enterococcus. Virulence 3, 421-433.
Hughes, D. (2003). Exploiting genomics, genetics and chemistry to combat antibiotic
resistance. Nature reviews Genetics 4, 432-441.
Igboin, C. O., Griffen, A. L. & Leys, E. J. (2012). The Drosophila melanogaster host
model. J Oral Microbiol 4.
Jett, B. D., Jensen, H. G., Nordquist, R. E. & Gilmore, M. S. (1992). Contribution
of the pAD1-encoded cytolysin to the severity of experimental Enterococcus faecalis
endophthalmitis. Infect Immun 60, 2445-2452.
Jett, B. D., Huycke, M. M. & Gilmore, M. S. (1994). Virulence of enterococci. Clin
Microbiol Rev 7, 462-478.
Jha, A. K., Bais, H. P. & Vivanco, J. M. (2005). Enterococcus faecalis mammalian
virulence-related factors exhibit potent pathogenicity in the Arabidopsis thaliana plant
model. Infect Immun 73, 464-475.
Kawalec, M., Potempa, J., Moon, J. L., Travis, J. & Murray, B. E. (2005).
Molecular diversity of a putative virulence factor: purification and characterization of

INTRODUCTION
37
Cha
pter
I
isoforms of an extracellular serine glutamyl endopeptidase of Enterococcus faecalis
with different enzymatic activities. J Bacteriol 187, 266-275.
Kimbrell, D. A. & Beutler, B. (2001). The evolution and genetics of innate immunity.
Nature reviews Genetics 2, 256-267.
Kounatidis, I. & Ligoxygakis, P. (2012). Drosophila as a model system to unravel
the layers of innate immunity to infection. Open Biol 2, 120075.
Kristich, C. J., Li, Y. H., Cvitkovitch, D. G. & Dunny, G. M. (2004). Esp-
independent biofilm formation by Enterococcus faecalis. J Bacteriol 186, 154-163.
Leclercq, R., Dutka-Malen, S., Brisson-Noel, A., Molinas, C., Derlot, E., Arthur,
M., Duval, J. & Courvalin, P. (1992). Resistance of enterococci to aminoglycosides
and glycopeptides. Clin Infect Dis 15, 495-501.
Lemaitre, B. & Hoffmann, J. (2007). The host defense of Drosophila melanogaster.
Annu Rev Immunol 25, 697-743.
Li, Y. H. & Tian, X. (2012). Quorum sensing and bacterial social interactions in
biofilms. Sensors (Basel) 12, 2519-2538.
Lopes Mde, F., Ribeiro, T., Martins, M. P., Tenreiro, R. & Crespo, M. T. (2003).
Gentamicin resistance in dairy and clinical enterococcal isolates and in reference
strains. J Antimicrob Chemother 52, 214-219.
Lopes Mde, F., Simoes, A. P., Tenreiro, R., Marques, J. J. & Crespo, M. T.
(2006). Activity and expression of a virulence factor, gelatinase, in dairy enterococci.
Int J Food Microbiol 112, 208-214.

Chapter I
38
Lopes, M. F., Pereira, C. I., Rodrigues, F. M. & other authors (1999). Registered
designation of origin areas of fermented food products defined by microbial
phenotypes and artificial neural networks. Appl Environ Microbiol 65, 4484-4489.
MacCallum, W. G. & Hastings, T. W. (1899). A case of acute endocarditis caused
by Micrococcus Zymogenes (Nov. spec), with description of the microorganism
Journal of Experimental Medicine 4, 521-534.
Makinen, P. L., Clewell, D. B., An, F. & Makinen, K. K. (1989). Purification and
substrate specificity of a strongly hydrophobic extracellular metalloendopeptidase
("gelatinase") from Streptococcus faecalis (strain 0G1-10). J Biol Chem 264, 3325-
3334.
Meister, M. & Lagueux, M. (2003). Drosophila blood cells. Cell Microbiol 5, 573-580.
Moellering, R. C., Jr. (1992). Emergence of Enterococcus as a significant pathogen.
Clin Infect Dis 14, 1173-1176.
Mohamed, J. A. & Murray, B. E. (2005). Lack of correlation of gelatinase production
and biofilm formation in a large collection of Enterococcus faecalis isolates. J Clin
Microbiol 43, 5405-5407.
Mohamed, J. A. & Murray, B. E. (2006). Influence of the fsr locus on biofilm
formation by Enterococcus faecalis lacking gelE. J Med Microbiol 55, 1747-1750.
Mohamed, J. A. & Huang, D. B. (2007). Biofilm formation by enterococci. J Med
Microbiol 56, 1581-1588.

INTRODUCTION
39
Cha
pter
I
Mundt, J. O. (1986). Enterococci. In Bergey’s Manual of Systematic Bacteriology pp.
1063-1065. Edited by N. S. M. P. H. A. Sneath, M. E. Sharpe & J. G. Holt:
Baltimore:Williams & Wilkins.
Murray, B. E. (1990). The life and times of the Enterococcus. Clin Microbiol Rev 3,
46-65.
Mylonakis, E., Engelbert, M., Qin, X., Sifri, C. D., Murray, B. E., Ausubel, F. M.,
Gilmore, M. S. & Calderwood, S. B. (2002). The Enterococcus faecalis fsrB gene, a
key component of the fsr quorum-sensing system, is associated with virulence in the
rabbit endophthalmitis model. Infect Immun 70, 4678-4681.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S., Akkermans, A. D., de Vos, W. M. &
Nagasawa, H. (2001a). Gelatinase biosynthesis-activating pheromone: a peptide
lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 41,
145-154.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S. & Nagasawa, H. (2001b). Chemical
synthesis and biological activity of the gelatinase biosynthesis-activating pheromone
of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem 65, 2322-2325.
Nakayama, J., Kariyama, R. & Kumon, H. (2002). Description of a 23.9-kilobase
chromosomal deletion containing a region encoding fsr genes which mainly
determines the gelatinase-negative phenotype of clinical isolates of Enterococcus
faecalis in urine. Appl Environ Microbiol 68, 3152-3155.
Nakayama, J., Chen, S., Oyama, N., Nishiguchi, K., Azab, E. A., Tanaka, E.,
Kariyama, R. & Sonomoto, K. (2006). Revised model for Enterococcus faecalis fsr
quorum-sensing system: the small open reading frame fsrD encodes the gelatinase

Chapter I
40
biosynthesis-activating pheromone propeptide corresponding to staphylococcal agrd.
J Bacteriol 188, 8321-8326.
Nakayama, J., Tanaka, E., Kariyama, R. & other authors (2007). Siamycin
attenuates fsr quorum sensing mediated by a gelatinase biosynthesis-activating
pheromone in Enterococcus faecalis. J Bacteriol 189, 1358-1365.
Nakayama, J., Uemura, Y., Nishiguchi, K., Yoshimura, N., Igarashi, Y. &
Sonomoto, K. (2009). Ambuic acid inhibits the biosynthesis of cyclic peptide
quormones in gram-positive bacteria. Antimicrob Agents Chemother 53, 580-586.
Nallapareddy, S. R., Qin, X., Weinstock, G. M., Hook, M. & Murray, B. E. (2000).
Enterococcus faecalis adhesin, ace, mediates attachment to extracellular matrix
proteins collagen type IV and laminin as well as collagen type I. Infect Immun 68,
5218-5224.
Nallapareddy, S. R., Singh, K. V., Sillanpaa, J., Garsin, D. A., Hook, M.,
Erlandsen, S. L. & Murray, B. E. (2006). Endocarditis and biofilm-associated pili of
Enterococcus faecalis. J Clin Invest 116, 2799-2807.
Nallapareddy, S. R., Sillanpaa, J., Mitchell, J., Singh, K. V., Chowdhury, S. A.,
Weinstock, G. M., Sullam, P. M. & Murray, B. E. (2011a). Conservation of Ebp-type
pilus genes among Enterococci and demonstration of their role in adherence of
Enterococcus faecalis to human platelets. Infect Immun 79, 2911-2920.
Nallapareddy, S. R., Singh, K. V., Sillanpaa, J., Zhao, M. & Murray, B. E. (2011b).
Relative contributions of Ebp Pili and the collagen adhesin ace to host extracellular
matrix protein adherence and experimental urinary tract infection by Enterococcus
faecalis OG1RF. Infect Immun 79, 2901-2910.

INTRODUCTION
41
Cha
pter
I
Nealson, K. H. & Hastings, J. W. (1979). Bacterial bioluminescence: its control and
ecological significance. Microbiological reviews 43, 496-518.
Ogier, J. C. & Serror, P. (2008). Safety assessment of dairy microorganisms: the
Enterococcus genus. Int J Food Microbiol 126, 291-301.
Onodera, Y., Okuda, J., Tanaka, M. & Sato, K. (2002). Inhibitory activities of
quinolones against DNA gyrase and topoisomerase IV of Enterococcus faecalis.
Antimicrob Agents Chemother 46, 1800-1804.
Paganelli, F. L., Willems, R. J. & Leavis, H. L. (2012). Optimizing future treatment
of enterococcal infections: attacking the biofilm? Trends Microbiol 20, 40-49.
Palmer, K. L., Carniol, K., Manson, J. M. & other authors (2010). High-quality
draft genome sequences of 28 Enterococcus sp. isolates. J Bacteriol 192, 2469-
2470.
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Paulsen, I. T., Banerjei, L., Myers, G. S. & other authors (2003). Role of mobile
DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299,
2071-2074.
Pinkston, K. L., Gao, P., Diaz-Garcia, D., Sillanpaa, J., Nallapareddy, S. R.,
Murray, B. E. & Harvey, B. R. (2011). The Fsr quorum-sensing system of
Enterococcus faecalis modulates surface display of the collagen-binding MSCRAMM
Ace through regulation of gelE. J Bacteriol 193, 4317-4325.

Chapter I
42
Prajsnar, T. K., Renshaw, S. A., Ogryzko, N. V., Foster, S. J., Serror, P. &
Mesnage, S. (2013). Zebrafish as a novel vertebrate model to dissect enterococcal
pathogenesis. Infect Immun 81, 4271-4279.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2000). Effects of
Enterococcus faecalis fsr genes on production of gelatinase and a serine protease
and virulence. Infect Immun 68, 2579-2586.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2001). Characterization of
fsr, a regulator controlling expression of gelatinase and serine protease in
Enterococcus faecalis OG1RF. J Bacteriol 183, 3372-3382.
Raffa, R. B., Iannuzzo, J. R., Levine, D. R., Saeid, K. K., Schwartz, R. C., Sucic,
N. T., Terleckyj, O. D. & Young, J. M. (2005). Bacterial communication ("quorum
sensing") via ligands and receptors: a novel pharmacologic target for the design of
antibiotic drugs. The Journal of pharmacology and experimental therapeutics 312,
417-423.
Reading, N. C. & Sperandio, V. (2006). Quorum sensing: the many languages of
bacteria. FEMS Microbiol Lett 254, 1-11.
Rutherford, S. T. & Bassler, B. L. (2012). Bacterial quorum sensing: its role in
virulence and possibilities for its control. Cold Spring Harb Perspect Med 2.
Sahm, D. F., Kissinger, J., Gilmore, M. S., Murray, P. R., Mulder, R., Solliday, J.
& Clarke, B. (1989). In vitro susceptibility studies of vancomycin-resistant
Enterococcus faecalis. Antimicrob Agents Chemother 33, 1588-1591.
Saurina, G., Landman, D. & Quale, J. M. (1997). Activity of disinfectants against
vancomycin-resistant Enterococcus faecium. Infect Control Hosp Epidemiol 18, 345-
347.

INTRODUCTION
43
Cha
pter
I
Schlievert, P. M., Gahr, P. J., Assimacopoulos, A. P., Dinges, M. M., Stoehr, J.
A., Harmala, J. W., Hirt, H. & Dunny, G. M. (1998). Aggregation and binding
substances enhance pathogenicity in rabbit models of Enterococcus faecalis
endocarditis. Infect Immun 66, 218-223.
Shankar, J., Walker, R. G., Wilkinson, M. C., Ward, D. & Horsburgh, M. J. (2012).
SalB inactivation modulates culture supernatant exoproteins and affects autolysis
and viability in Enterococcus faecalis OG1RF. J Bacteriol 194, 3569-3578.
Shankar, N., Baghdayan, A. S. & Gilmore, M. S. (2002). Modulation of virulence
within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature
417, 746-750.
Shankar, V., Baghdayan, A. S., Huycke, M. M., Lindahl, G. & Gilmore, M. S.
(1999). Infection-derived Enterococcus faecalis strains are enriched in esp, a gene
encoding a novel surface protein. Infect Immun 67, 193-200.
Sifri, C. D., Mylonakis, E., Singh, K. V., Qin, X., Garsin, D. A., Murray, B. E.,
Ausubel, F. M. & Calderwood, S. B. (2002). Virulence effect of Enterococcus
faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans
and mice. Infect Immun 70, 5647-5650.
Singh, K. V., Nallapareddy, S. R., Nannini, E. C. & Murray, B. E. (2005). Fsr-
independent production of protease(s) may explain the lack of attenuation of an
Enterococcus faecalis fsr mutant versus a gelE-sprE mutant in induction of
endocarditis. Infect Immun 73, 4888-4894.
Skandamis, P. N. & Nychas, G. J. (2012). Quorum sensing in the context of food
microbiology. Appl Environ Microbiol 78, 5473-5482.

Chapter I
44
Su, Y. A., Sulavik, M. C., He, P., Makinen, K. K., Makinen, P. L., Fiedler, S.,
Wirth, R. & Clewell, D. B. (1991). Nucleotide sequence of the gelatinase gene
(gelE) from Enterococcus faecalis subsp. liquefaciens. Infect Immun 59, 415-420.
Tang, H. (2009). Regulation and function of the melanization reaction in Drosophila.
Fly 3, 105-111.
Tendolkar, P. M., Baghdayan, A. S. & Shankar, N. (2003). Pathogenic enterococci:
new developments in the 21st century. Cell Mol Life Sci 60, 2622-2636.
Tendolkar, P. M., Baghdayan, A. S. & Shankar, N. (2006). Putative surface
proteins encoded within a novel transferable locus confer a high-biofilm phenotype to
Enterococcus faecalis. J Bacteriol 188, 2063-2072.
Teng, F., Jacques-Palaz, K. D., Weinstock, G. M. & Murray, B. E. (2002).
Evidence that the enterococcal polysaccharide antigen gene (epa) cluster is
widespread in Enterococcus faecalis and influences resistance to phagocytic killing
of E. faecalis. Infect Immun 70, 2010-2015.
Teng, F., Singh, K. V., Bourgogne, A., Zeng, J. & Murray, B. E. (2009). Further
characterization of the epa gene cluster and Epa polysaccharides of Enterococcus
faecalis. Infect Immun 77, 3759-3767.
Thiercelin, M. E. (1899). Sur un diplocoque saprophyte de l'intestin susceptible de
devenir pathogen. C R Soc Biol 5, 269-271.
Thomas, V. C., Thurlow, L. R., Boyle, D. & Hancock, L. E. (2008). Regulation of
autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular
proteases influences biofilm development. J Bacteriol 190, 5690-5698.

INTRODUCTION
45
Cha
pter
I
Thomas, V. C., Hiromasa, Y., Harms, N., Thurlow, L., Tomich, J. & Hancock, L.
E. (2009). A fratricidal mechanism is responsible for eDNA release and contributes to
biofilm development of Enterococcus faecalis. Mol Microbiol 72, 1022-1036.
Vebo, H. C., Snipen, L., Nes, I. F. & Brede, D. A. (2009). The transcriptome of the
nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to
growth in blood. PLoS One 4, e7660.
Vebo, H. C., Solheim, M., Snipen, L., Nes, I. F. & Brede, D. A. (2010).
Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis
isolates, and their transcriptional responses to growth in human urine. PLoS One 5,
e12489.
Waters, C. M., Hirt, H., McCormick, J. K., Schlievert, P. M., Wells, C. L. & Dunny,
G. M. (2004). An amino-terminal domain of Enterococcus faecalis aggregation
substance is required for aggregation, bacterial internalization by epithelial cells and
binding to lipoteichoic acid. Mol Microbiol 52, 1159-1171.
Waters, C. M. & Bassler, B. L. (2005). Quorum sensing: cell-to-cell communication
in bacteria. Annu Rev Cell Dev Biol 21, 319-346.
Wirth, R. (1994). The sex pheromone system of Enterococcus faecalis. More than
just a plasmid-collection mechanism? Eur J Biochem 222, 235-246.

Chapter I
46

SILENCING FSR SYSTEM:
A Way to Survive
This chapter is based on the following manuscript:
The incongruent gelatinase genotype and phenotype in Enterococcus faecalis are due to shutting off the ability to respond to the gelatinase biosynthesis-activating pheromone
(GBAP) quorum-sensing signal
Neuza Teixeira, Sofia Santos, Paulo Marujo, Ryoji Yokohata, Vijayalakshmi S. Lyer, Jiro Nakayama, Lynn E. Hancock,
Pascale Serror and Maria de Fátima Silva Lopes (2012), Microbiology, 158, 519-528.


CONTENTS
1. SUMMARY ........................................................................................................... 51
2. INTRODUCTION ................................................................................................... 52
3. MATERIAL AND METHODS ................................................................................ 55
3.1 Bacterial strains and plasmids ........................................................................ 55
3.2 General DNA techniques ................................................................................ 55
3.3 Sequence analysis of the fsr and gelE–sprE regions of LN68 ........................ 55
3.4 Mutant construction ........................................................................................ 56
3.5 E. faecalis LN68 complementation with the V583 fsrC gene .......................... 57
3.6 RNA extraction and semiquantitative RT-PCR ............................................... 57
4. RESULTS AND DISCUSSION .............................................................................. 59
5. ACKNOWLEDGEMENTS ..................................................................................... 67
6. BIBLIOGRAPHY ................................................................................................... 68
7. SUPPLEMENTARY DATA .................................................................................... 74

Chapter II
50
The author of this thesis performed the majority of the experiments. Experimental
design, data analysis and manuscript preparation were done by the author of this
thesis and the supervisor Maria de Fatima Silva Lopes. Sofia Santos and
Vijayalakshmi S. Iyer helped with mutant construction and Ryoji Yokohata produced
the GBAP.

Silencing Fsr System: A way to Survive
51
Cha
pter
II
1. SUMMARY
The concomitant presence of a complete fsr quorum-sensing system and gelE–sprE
operons in Enterococcus faecalis is known to be essential for the detection of
gelatinase activity. However, there are reports of the absence of gelatinase activity
despite the presence of complete fsr and gelE loci. In order to understand this
incongruence between genotype and phenotype we sequenced fsr and gelE loci of
the E. faecalis LN68 strain, which was previously found to carry both operons but to
lack gelatinase activity. Of the 59 nucleotide differences detected compared with the
gelatinase-positive V583 strain, we found a nonsense mutation (a premature STOP
codon) predicted to truncate the ATPase sensor domain of the FsrC protein,
responsible for sensing and transducing the signal from the quorum-sensing
molecule. Strain LN68 was highly affected in the expression of the gelE and sprE
genes, further supporting the lack of Fsr dependent gelE induction. When we
constructed a V583 mutant with the same premature stop mutation in the fsrC gene
the resulting strain was no longer able to degrade gelatin. We conclude that the
reduced ability to transduce the quorum-sensing signal of the prematurely truncated
FsrC protein is sufficient to explain the negative gelatinase phenotype. As the
incongruent genotype and phenotype is detected in natural isolates, we believe that
the silencing of the quorum-sensing system Fsr may be beneficial for some E.
faecalis strains.

Chapter II
52
2. INTRODUCTION
Enterococci are natural inhabitants of the oral cavity, intestinal tract and female
genital tract of both humans and animals, and are also among the predominant
microbiota of traditionally fermented dairy products (Lopes et al., 1999) In contrast to
their beneficial role in intestinal homeostasis, these micro-organisms are becoming
increasingly important to human health as leading causes of nosocomial infections,
namely of the urinary tract, bloodstream, intra-abdominal and pelvic regions, and
surgical sites. To do so, they rely on several mechanisms, including the fsr operon of
Enterococcus faecalis, the species most frequently associated with nosocomial
infections (Gilmore, 2002; Mundt, 1986; Ogier & Serror, 2008). The fsr (Enterococcus
faecalis Regulator) two-component system, a homologue of the agr system in
Staphylococcus aureus, is a quorum sensing dependent regulatory system. The fsr
operon comprises four genes: fsrA, fsrB, fsrC and fsrD. The last encodes an
autoinducing cyclic peptide named Gelatinase Biosynthesis Activating Pheromone
(GBAP) that is processed and exported out of the cell by the FsrB protein.
Accumulation of GBAP outside cells is sensed by the FsrC histidine kinase, leading
to the activation of the response regulator FsrA. Activated FsrA induces expression
of the fsrBDC genes. These genes are involved in an autoregulatory circuit that
results in a boost of GBAP signalling and in induction of the Fsr regulon, among
which the gelE–sprE operon is the most induced (Bourgogne et al., 2006). This
operon encodes gelatinase (GelE), an extracellular zinc metalloprotease (Makinen et
al., 1989; Su et al., 1991), and SprE, a serine protease (Nakayama et al., 2001a;
Nakayama et al., 2001b; Qin et al., 2000).
Although the secreted protease SprE has been implicated in disease in animal
models, the role of SprE is still unknown. GelE is known to contribute to biofilm
formation, and contributes also to virulence through degradation of a broad range of
host proteinaceous substrates (Hancock & Perego, 2004; Park et al., 2007; Steck et
al., 2011). The role of the gelE and fsr loci in E. faecalis virulence has been
demonstrated in different mammalian infection models (Mohamed & Murray, 2006),

Silencing Fsr System: A way to Survive
53
Cha
pter
II
in the Caenorhabditis elegans nematode model (Gaspar et al., 2009; Sifri et al.,
2002) and in the Arabidopsis thaliana plant model (Jha et al., 2005). Gelatinase has
also been implicated in evasion of the immune system of the insect Galleria
mellonella (Park et al., 2007). Recent transcription studies have also shown that fsr
and gelE–sprE expression is modulated during some stress conditions, namely in
blood (Vebo et al., 2009) and urine (Vebo et al., 2010). Despite their evident
importance for E. faecalis virulence and stress responses inside the host, many
reports have shown that both loci are present in enterococcal isolates from different
environments. This suggests that both the Fsr system and the GelE and SprE
proteins may play a role, not associated with virulence, in the biology of E. faecalis
(Thomas et al., 2009). The presence of the gelE genetic locus has often been
complemented by the search for the phenotype, i.e. detection of gelatinase activity
on plates containing gelatin. Qin et al. (2000) reported that the gelatinase phenotype
requires the concomitant presence of the fsr and gelE genes. Soon after, Eaton &
Gasson (2001) reported the loss by subculturing of the gelatinase phenotype in 20%
of the analysed strains. Later, Nakayama et al. (2002) suggested that the loss of the
gelatinase phenotype after subculturing might be due to a 23.9 kb chromosomal
deletion, which was found in urine isolates with silent gelE genes. This deletion
included the fsrA, fsrB and partially the fsrC gene. However, it has recently been
shown that the 23.9 kb chromosomal deletion does not occur spontaneously by
subculturing strains in the laboratory, but likely results from horizontal transfer and
recombination (Galloway-Pena et al., 2011). We and others have reported the loss of
the gelatinase phenotype by subculturing, but also the existence of natural
gelatinase-negative strains carrying gelE and an apparently complete fsr operon
(Lopes Mde et al., 2006; Nakayama et al., 2002). Altogether, these reports indicate
that silent gelE genes, in the presence of an apparently complete fsr operon, occur
both in natural and laboratory subcultured E. faecalis isolates. However, no further
demonstration of the involvement of mutations in either the fsr or the gelE–sprE
operon in the absence of gelatinase activity has been reported or demonstrated so
far.

Chapter II
54
In the present study we investigated the reason for the incongruence between the fsr
and gelE genotype and the gelatinase phenotype in E. faecalis strain LN68 (Lopes
Mde et al., 2006). We demonstrate that this incongruence, in our strains with
apparently complete fsr and gelE loci, is correlated with a specific nonsense codon in
the FsrC protein. We further demonstrate that this codon change originates a
truncated FsrC protein, preventing E. faecalis from sensing GBAP.

Silencing Fsr System: A way to Survive
55
Cha
pter
II
3. MATERIAL AND METHODS
3.1 Bacterial strains and plasmids Strains and plasmids used in this study are listed in Table 1. Enterococcal strains
were grown in BHI (brain heart infusion) medium at 37ºC, and Escherichia coli
strains were grown in LB medium at 37ºC with agitation.
3.2 General DNA techniques General molecular biology techniques were performed by standard methods
(Sambrook, 1989). Restriction enzymes, polymerases and T4 DNA ligase were used
according to manufacturers’ instructions. PCR amplification was performed using a
Biometra thermocycler. When necessary, PCR products and DNA restriction
fragments were purified with purification kits (Macherey-Nagel). Plasmids were
purified using the Miniprep kit (Macherey-Nagel). Electrotransformation of
Escherichia coli and E. faecalis was carried out as described by Dower et al. (1988)
and Dunny et al. (1991), using a Gene Pulser apparatus (Bio-Rad) (Dower et al.,
1988; Dunny et al., 1991). Plasmid inserts were sequenced at BaseClear (The
Netherlands).
3.3 Sequence analysis of the fsr and gelE–sprE regions of LN68 PCR amplification of overlapping fragments of the fsr and gelE–sprE regions was
carried out using Expand High FidelityPLUS DNA polymerase (Roche) and primers
from Gaspar et al. (2009) (Gaspar et al., 2009). Amplicons were sequenced by
BaseClear and DNA sequence analysis was accomplished using the Vector NTI
10.3.0 program (Invitrogen). The results were compared with the V583 genomic DNA
sequence available at the J. Craig Venter Institute website (http://www.jcvi.org/).

Chapter II
56
3.4 Mutant construction Markerless fsrC mutants of E. faecalis LN68 and of a derivative erythromycin-
susceptible V583 strain (kindly provided by Axel Hartke, Université de Caen, France)
were constructed essentially as described by Brinster et al. (2007) (Brinster et al.,
2007)(Figure 1). In this procedure we changed the nucleotide guanine for adenine
(position 1764864 in the V583 genome), which leads to the substitution of a
tryptophan by a STOP codon (in V583). In LN68, we did the opposite, i.e. we
substituted the STOP codon by a tryptophan at position 403. Briefly, flanking regions
of fsrC were amplified from chromosomal DNA by PCR with primers LN68_fsrC-
PGMT5 and LN68_mfsrC_M, LN68_fsrC_PGMT3 and LN68_fsrC_M, for strain
LN68; and primers V583_mfsrC_M and LN68_fsrC-PGMT5, LN68_fsrC_PGMT3 and
V583_fsrC_M, for strain V583 (Table 1). The two cognate PCR fragments were fused
by PCR using the external primers LN68_fsrCPGMT5 and LN68_fsrC_PGMT3, for
both strains, and the resulting products were cloned into pGEM-T (Promega). After
being checked by sequencing, the inserted PCR fragment was removed from its
cloning vector by restriction enzymes and subsequently cloned into plasmid
pG+host9 (Maguin et al., 1996), which was then electroporated into the respective E.
faecalis strain. The fsrC single- and double-crossover mutants were selected as
described by Brinster et al. (2007) (Brinster et al., 2007). Successful targeted
mutations of fsrC in strains LN68 and V583 were first identified by PCR screening
and then confirmed by sequencing. The cognate phenotypes were confirmed by
gelatinase activity assay. An isogenic in-frame deletion of fsrB in E. faecalis V583
was generated using an Escherichia coli–enterococcal temperature-sensitive cloning
vector, pLT06 (Thurlow et al., 2009). Upstream regions flanking fsrB (ef1821) in the
V583 genome were PCR-amplified using the primer pair FsrBP1 and FsrBP2, and
the downstream region of ef1821 was amplified using primers FsrBP3 and FsrBP4
(Table 1). The resultant PCR products were digested with BamHI followed by
ligation, and the resulting product was reamplified with primers FsrBP1 and FsrBP4.
For the construction of the fsrB deletion vector pVI02, the amplified product was

Silencing Fsr System: A way to Survive
57
Cha
pter
II
digested with EcoRI and PstI, followed by ligation to similarly digested plasmid vector
pLT06. The ligated product was electroporated into Escherichia coli EC1000 for
propagation, and blue colonies were selected for by culture at 37 ºC on LB agar
containing chloramphenicol (10 mg ml-1) and X-Gal (80 mg ml-1). Positive clones
were identified by PCR using primers OriF and SeqR (Thurlow et al., 2009). Plasmid
pVI02 was confirmed by restriction mapping and electroporated into E. faecalis V583
cells. To generate E. faecalis strain VI13 possessing an in-frame deletion of fsrB, a
published protocol was used (Thurlow et al., 2009). Primers FsrAP1 and GelEP2
were employed to confirm the presence of the deletion.
3.5 E. faecalis LN68 complementation with the V583 fsrC gene
To complement strain LN68 with the fsrC gene from V583, a 1595 bp BamHI–PstI
fragment from V583 was recovered from the PCR amplicon obtained by using
primers fsrC39PstI and fsrC59BamHI (Gaspar et al., 2009). The fragment was
cloned into the vector pOri23 (Que et al., 2000) cut with BamHI and PstI, resulting in
plasmid pOri23–fsrC. Primers pOri23_fw and pOri23_rv (Braga et al., 2011) were
used to confirm cloning. pOri23–fsrC was then introduced into LN68 cells by
electroporation. Gelatinase activity was evaluated as described below, on gelatin
plates supplemented with 500 mg erythromycin ml-1.
3.6 RNA extraction and semi-quantitative RT-PCR
RNA was extracted from cells grown in BHI broth at 37 ºC. Briefly, overnight cultured
cells were diluted 1:100 and growth was monitored by following OD600. Following
exponential, early stationary and late stationary growth phases, cells were collected
for RNA isolation. Total RNA was extracted and purified with an RNeasy Mini kit
(Qiagen). RNA integrity was checked by electrophoresis on a 1% agarose gel
(RNase free), and DNA contamination was checked using primers for 23S
(Supplementary Table S1). The cDNA was synthesized using random primers
Chapter II
58
(Roche Diagnostics), 3 mg total RNA and a Transcriptor High Fidelity cDNA
Synthesis kit (Roche Diagnostics). Two serial dilutions (1:10 and 1:100) of cDNA
were used for PCR in order to amplify cDNA of fsrA (primers: mfsrA, fsrA), fsrB
(primers: fsrB, mfsrB), fsrC (primers: fsrC_2, mfsrC), gelE (primers: mgelE_2, gelE)
and sprE (primers: sprE_4, msrpE_4) transcripts (Supplementary Table S1). 23S
rRNA was used as a control.
Table 1 – Strains and plasmids used in this study.
Strains Relevant characteristics Reference
E. coli DH5α F- Ø80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1
hsdR17(rK - mK- ) phoA supE44 λ- thi-1 gyrA96 relA1
(Grant et al., 1990)
E. coli TG1 supE hsdD5 thi (Δlac-proAB) F- (traD36 proAB-lacZΔM15)
repA
(Law et al., 1995)
E. faecalis V583 Clinical isolate, TIGR sequence strain; VanR (Sahm et al., 1989)
E. faecalis V583ermS derivative of E. faecalis V583, susceptible to erythromycin due
to deletion of the erm(B)
supplied by Axel
Hartke,Caen,France
E. faecalis LN68 Wild-type E. faecalis; isolated from milk, GelE- (Lopes Mde et al., 2006)
E. faecalis LN66 Wild-type E. faecalis; isolated from milk, GelE- This Study
E. faecalis QSE15 Wild-type E. faecalis isolated from cheese, GelE- This Study
E. durans QN8 Wild-type E. durans; isolated from cheese, GelE- This study
E. faecalis EF_SAVE3 E. faecalis V583ermS W403STOP, Gel- This Study
E.faecalis EF_SAVE 5 E. faecalis LN68 (pSAVE7), EryR, GelE+ This study
Plasmids
pORI23 E. coli – E. faecalis shuttle plasmid, EryR (Que et al., 2000)
pGEM-T E. coli replicating plasmid, AmpR Promega
pG+host9 E. faecalis thermosensitive plasmid, EryR (Maguin et al., 1996)
pSAVE3 pGEM-T derivative carrying fsrC_W403STOP from E. faecalis
V583
This study
pSAVE4 pGEM-T derivative carrying fsrC_W403STOP This study
pSAVE5 pGhost9 derivative carrying fsrC_W403STOP This study
pSAVE6 pGhost9 derivative carrying fsrC_STOP403W This study
pSAVE7 pORI23 derivative carrying a 1595 pb BamHI/PstI fsrC
fragment from E. faecalis V583
This study

Silencing Fsr System: A way to Survive
59
Cha
pter
II
4. RESULTS AND DISCUSSION
The importance of E. faecalis gelatinase to virulence and biofilm-forming ability has
promoted the detection of the fsr and gelE–sprE operons as a presumption of
pathogenicity. However, an incongruent genotype and phenotype may lead to false-
positive assumptions on the virulence potential of strains, since the detection of the
gene does not necessarily equate to gelatinase activity (Eaton & Gasson, 2001). In a
previous study, the E. faecalis LN68 dairy strain was found to lack gelatinase activity
on plates, despite apparently carrying the entire fsr and gelE–sprE operons,
suggesting a defect in the functionality of the fsr and gelE– sprE operons or in their
expression (Lopes Mde et al., 2006).
We first sequenced 6106 bp, including the promoter and terminator regions, of both
the fsr and gelE–sprE operons of LN68 strain. Comparison of the LN68 sequence
with that of E. faecalis V583 strain revealed 59 differences. Thirty-one, 18 and one
corresponded to silent, missense and nonsense mutations, respectively. We found
also nine differences located in the intergenic regions (Table 2).
Table 2 - Mutations identified in E. faecalis LN68.
Mutation
type
Gene/IRa
IR
fsrA IR fsrB IR fsrC IR gelE IR sprE IR
Silent - 2 - 4 - 5 - 12 - 8 -
Missense - 2 - 3 - 5 - 6 - 3 -
Nonsense 3 - 4 - - 1 - - - - 1
Deletions - - - - - - - - - - -
Insertions - - 1 - - - - - - - -
a IR, intergenic region.

Chapter II
60
Even if all of these differences could potentially influence the gelatinase phenotype,
either by disturbing the regulation of gene expression through changed transcript
stability for silent mutations, or by promoting a change in protein structure for
missense mutations, the most drastic effect was predicted to result from the
nonsense codon in the fsrC gene responsible for the substitution of tryptophan 403
by a STOP codon (Figure 1). This leads to FsrC402, a truncated histidine kinase
shortened by the last 45 aa compared with the FsrC of V583 strain (447 aa).
Figure 1 -Schematic representation of fsr and gelE genetic loci in LN68 and V583 strains. The
nonsense mutation W423STOP is also identified.
To further investigate how the detected differences in the LN68 sequence would
affect the structure of FsrC we used TopPred software (http://mobyle.pasteur.fr/cgi-
bin/portal.py?#forms::toppred). FsrC proteins of both the V583 and the LN68 strain
are predicted to have six transmembrane segments and three extracellular loops
(Figure 2). The major difference in LN68 FsrC is a truncated kinase domain due to
the nonsense mutation in the C-terminal region. These results support our prediction
that substitution of tryptophan 403 by a STOP codon promotes a structural change in
the FsrC protein and most likely has an impact on its activity. Noticeably, the GXG
triplet highly conserved in the ATPase domain of many histidine kinases is
interrupted in LN68 FsrC (Figure 2). This triplet is important for nucleotide binding
and histidine phosphorylation of the cognate response regulators (Parkinson &
Kofoid, 1992; Zhu & Inouye, 2002). In response to GBAP, the ATPase domain of

Silencing Fsr System: A way to Survive
61
Cha
pter
II
W G
COOH447
cytoplasmicmembrane
ES
IS
FV
I
LF
V
FS
S VL
L VN
T AL
L SL
I
WL
I
GY
S
YA
L TV
V IF
T GF
L LI
H
FS
V
YN
I
ML
L TF
V SL
F IA
K FI
S
IA
S
LL
W
NV
L LL
L IN
I WI
A LK
I
FV
F
SI
L
LL
L LI
L LL
L LC
G FF
I
LI
S
LE
L
VG
L FL
I LI
G LV
I EA
Y
22
2
53
73
98
78
116
136
173
153
192
212M
H2
N
1
RV
TLIKIE
CKIP
LL S
L LI
AA
VINL C
ML
FS
L
K K R F
TI
S
QE
Y
FQQI
Q
L
T KN
FV
NS
F
R
L
SN
LV
E
P
V
L R NR
I
P
F
EP
V
FG
VT
N
D
S
RM
SS
K
ECL-1
ECL-2ECL-3
RQEL E
I
H245
G402K385
Y 252
cytoplasmicmembrane
ES
IS
FV
I
LF
V
FS
S VL
L VN
T AL
L SL
I
SW
L
LG
C
IY
A LT
V VI
F TG
F LL
I
FS
V
YN
I
ML
L TF
V SL
F IA
K FI
S
IA
S
LL
W
NV
I LL
L IN
I WI
A LK
I
FV
F
SI
L
LL
L LI
L LL
L LC
G FF
I
LI
S
LE
L
VG
L FL
I LI
G LV
I EA
Y
22
2
52
72
98
78
116
136
173
153
192
212M
H2
N
1
RV
TLIKIE
CKIP
LL S
L LI
AA
VINLC
ML
FS
HK K R
F
TI
S
QE
Y
FQQI
Q
L
T KN
FV
NS
F
R
L
SN
LV
E
P
V
L R NR
I
P
F
EP
V
FG
VT
N
D
S
RM
SS
K
ECL-1
ECL-2ECL-3
RQEL E
I
H245
G402
COOH
R385
N 252
FsrC phosphorylates the FsrA response regulator, which activates transcription from
PfsrB and PgelE promoters, increasing expression of the fsrBDC, gelE and sprE
genes. If phosphorylation of FsrA does not occur, the response regulator will not
activate transcription, and expression of the proteases will not increase. Thus, we
hypothesize that a truncated ATPase domain of FsrC impairs its ability to transduce
GBAP signalling and consequently it cannot induce the expression of the GelE and
SprE proteases.
Figure 2 - Intramembrane
structures of FsrC proteins
from V583 and LN68,
predicted using TopPred
software. Mutations found in the
fsrC gene of LN68 are shown:
tyrosine (Y) 54 to cysteine (C);
leucine (L) 124 to isoleucine (I);
tyrosine 252 to asparagine (N);
lysine (K) 385 to arginine (R);
tryptophan (W) 403 to STOP. ES,
extracellular space; IS, intracellular
space; ECL, extracellular loop.
E. faecalis V583
E. faecalis LN68

Chapter II
62
The low transcript levels of the gelE and sprE genes, in LN68 strain (Supplementary
Figure S1), are in accordance with the absence of gelatinase activity on gelatin
plates. In order to prove that LN68 has a negative gelatinase phenotype because it is
a GBAP non-responder (derived from its functionally impaired FsrC) and not because
of any other mutation detected in the fsrA, fsrB and gelE genes (Table 2), we
constructed two mutants: EF_SAVE3 in V583, where tryptophan 403 was substituted
by a STOP codon, and EF_SAVE4 in LN68, where the 403STOP codon was
substituted by a tryptophan (Figure 1), and we checked for induction of gelatinase
activity of LN68, V583 and their derived mutants by exogenous GBAP. The activity of
FsrC was indirectly detected by testing gelatinase activity on plates. Compared with
the V583 wild-type strain, EF_SAVE3 showed no gelatinase activity, indicating that
the nonsense mutation is sufficient to explain loss of FsrC activity (Figure 3).
Unexpectedly, we observed that the E. faecalis EF_SAVE4 mutant was unstable, as
it reverted easily to the wild-type sequence of the LN68 fsrC gene. In fact, if
gelatinase activity was tested right after constructing and confirming strain
EF_SAVE4, the strain behaved as a gelatinase producer. However, if we repeated
the test with EF_SAVE4 subcultures, it behaved as a gelatinase non-producer, and
sequencing of fsrC showed that the strain reverted to the STOP codon of the wild-
type strain. We thus decided to complement strain LN68 with fsrC from strain V583,
using plasmid pORI23. The resulting strain, E. faecalis EF_SAVE5, exhibited
gelatinase activity, indicating that expression of fsrC from V583 was sufficient to
restore gelatinase expression in strain LN68 (Figure 3). Then, to investigate whether
LN68 and EF_SAVE3 were GBAP non-responders and GBAP producers, we
performed two experiments, whose results are presented in Figure 4. We observed
no gelatinase activity when purified GBAP was added to LN68 and EF_SAVE3 cells
(Figure 4a). These results indicate that neither LN68 nor EF_SAVE3 is a GBAP
responder, showing that the W403STOP mutation in the FsrC histidine kinase is
enough to turn E. faecalis into a GBAP non-responder. We then wondered whether
these two strains were still producing sufficient amounts of GBAP to induce
detectable gelatinase. The results are presented in Figure 4b.
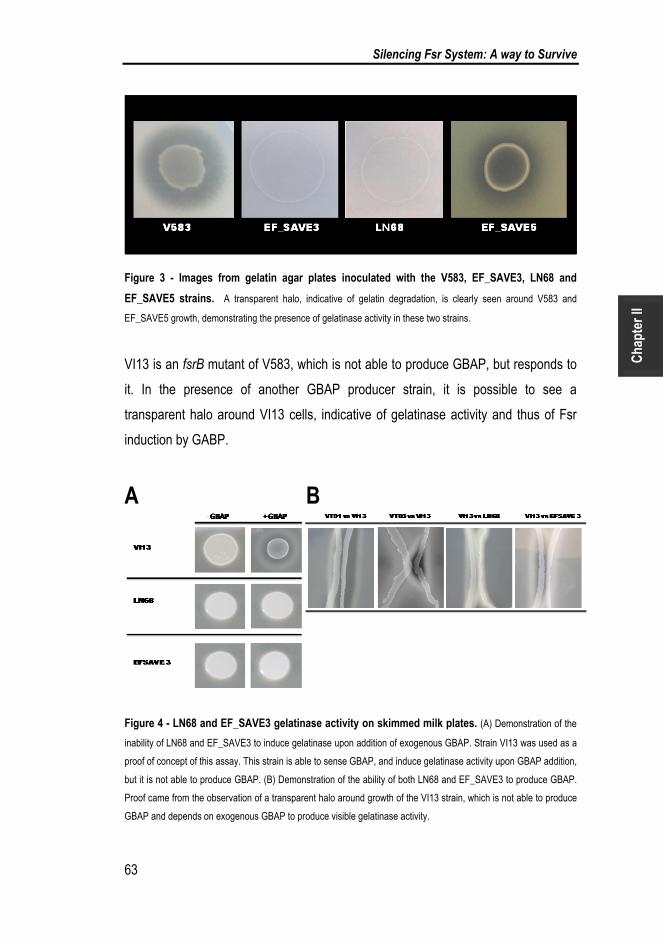
Silencing Fsr System: A way to Survive
63
Cha
pter
II
Figure 3 - Images from gelatin agar plates inoculated with the V583, EF_SAVE3, LN68 and
EF_SAVE5 strains. A transparent halo, indicative of gelatin degradation, is clearly seen around V583 and
EF_SAVE5 growth, demonstrating the presence of gelatinase activity in these two strains.
VI13 is an fsrB mutant of V583, which is not able to produce GBAP, but responds to
it. In the presence of another GBAP producer strain, it is possible to see a
transparent halo around VI13 cells, indicative of gelatinase activity and thus of Fsr
induction by GABP.
A B
Figure 4 - LN68 and EF_SAVE3 gelatinase activity on skimmed milk plates. (A) Demonstration of the
inability of LN68 and EF_SAVE3 to induce gelatinase upon addition of exogenous GBAP. Strain VI13 was used as a
proof of concept of this assay. This strain is able to sense GBAP, and induce gelatinase activity upon GBAP addition,
but it is not able to produce GBAP. (B) Demonstration of the ability of both LN68 and EF_SAVE3 to produce GBAP.
Proof came from the observation of a transparent halo around growth of the VI13 strain, which is not able to produce
GBAP and depends on exogenous GBAP to produce visible gelatinase activity.

Chapter II
64
When VI13 was allowed to grow in the proximity of either VT01 or VT03, GBAP
producer strains but with a negative gelatinase phenotype, the GBAP produced by
VT01 and VT03 was able to induce gelatinase activity in VI13. Similarly, both LN68
and EF_SAVE3 were able to induce gelatinase activity in VI13, although to different
extents, and at much lower levels than those seen with VT01 or VT03. This is
because both LN68 and EF_SAVE3 have a truncated FsrC, which allows these
strains to produce only basal levels of GBAP to which VI13 responded. Our results
thus demonstrate that both LN68 and EF_SAVE3 are GBAP producers.
Recently, Thomas et al. (2009) have reported the existence, among planktonic E.
faecalis cells, of GBAP non-responders, i.e. cheaters, which, at the end of stationary
phase, reach approximately 12% of the population (Thomas et al., 2009). In a
population where individuals work cooperatively, cheaters are individual cells that
benefit from the cooperation with others, but do not cooperate. In light of our data, it
is possible that E. faecalis strains accumulate mutations in the fsr locus, in particular
in fsrC, and upon selection, due to still unknown factors, the loss of the ability to
transduce the GBAP signal is selected, as if the strains no longer need high levels of
GelE and SprE for survival, and therefore do not require a fully efficient quorum-
sensing Fsr system. As fsr has also been suggested to regulate other genes in the
genome of E. faecalis (Bourgogne et al., 2006), it is also plausible that the advantage
of the loss of quorum sensing is selected by other functions in the cell which are
shutdown. This raises interesting future prospects for further investigating the role of
Fsr, GelE and SprE in the biology of E. faecalis.
In order to establish whether the GBAP non-responder behaviour in other E. faecalis
isolates was due to the same W403STOP mutation or to other codon variations, we
searched for this nonsense codon in seven genetically unrelated enterococcal
isolates from our culture collection, which also carried the apparently complete fsr
and gelE loci, but lacked gelatinase activity (Figure 5). The same nonsense codon
was detected in only two E. faecalis tested strains, suggesting that it is likely that
other variations in the FsrC sequence lead to the same GBAP non-responder
behaviour.

Silencing Fsr System: A way to Survive
65
Cha
pter
II
Figure 5 - Enterococcal strains with an incongruent gelatinase genotype and phenotype. The fsrC
gene of these strains was sequenced and was found to carry the W403STOP mutation, indicated by a star.
The detection of this nonsense codon in Enterococcus durans suggests either an
ancient event that was disseminated in the genus or a mutation which frequently
occurs due to unknown specific conditions. We have also detected other codon
variations in the fsrC gene of strain LN68. It is possible that these, though not
responsible for the loss of FsrC activity, are needed for stabilization of the
STOP403W substitution that seems to be deleterious in the LN68 genetic
background. It is thus likely that different sets of fsrC codon variations will be
responsible for negative gelatinase phenotypes in different genomes. In fact, when
we searched the E. faecalis sequenced genomes
(http://www.broadinstitute.org/annotation/genome/enterococcus_faecalis/MultiHome.html), we found different
sets of codon variations in fsrC among strains with a negative gelatinase phenotype
and positive genotype (Galloway-Pena et al., 2011). This fact, together with the
inability of LN68 to displace the W403STOP mutation, suggests that each genome
may accommodate different sets of codon variations in fsrC in order to achieve a
gelatinase negative phenotype.
In summary, previous work suggested that, besides deletion, mutations in any of the
fsr or gelE genes could explain the negative gelatinase phenotype observed in
natural E. faecalis isolates. However, no proof was available. In the present work we
demonstrate that a single nonsense codon in fsrC is enough to explain the
incongruence between a gelE genotype and a gelatinase-negative phenotype. The
substitution of tryptophan 403 by a STOP codon in strain LN68 was shown to
generate a truncated FsrC that was most likely impaired in the ATPase activity

Chapter II
66
needed for GBAP signal transduction. By demonstrating the hypothesized cause–
effect relationship between codon variations in the fsr–gelE loci and the gelatinase-
negative phenotype, we came across interesting findings which may prove very
important for future research on E. faecalis biology and host–pathogen relationships.
First, it is clear that the missense mutations that we detected in other fsr genes and
in gelE were not important for the negative gelatinase phenotype, as strain
EF_SAVE5, despite still carrying those mutations, was able to produce gelatinase.
Therefore, E. faecalis achieves the gelatinase-negative phenotype by accumulating
mutations in the fsrC gene. Accordingly, and despite the fact that each isolate may
accumulate different sets of codon variations in fsrC, the important fact here is that
some E. faecalis isolates appear to stabilize a quorum-sensing non-responder
behaviour. This is worth studying in future research on E. faecalis, as it could shed
some light on the role of Fsr and gelatinase in the biology and pathogenicity of this
important nosocomial pathogen.

Silencing Fsr System: A way to Survive
67
Cha
pter
II
5. ACKNOWLEDGEMENTS
The authors are grateful to Axel Hartke (Universite´ de Caen, France) for kindly
providing the E. faecalis V583ermS. This work was supported by Fundação para a
Ciência e a Tecnologia (FCT) through project grant PDC/CVT/67270/2006, co-
financed through FEDER, and grant PEst-OE/EQB/LA0004/2011, and by the bilateral
cooperation project Portugal/France (GRICES/EGIDE, Pessoa program, 2006/07). N.
T. is grateful to FCT for grant SFRH/BD/65750/2009. P. M. is grateful to FCT for
grant SFRH/BPD/14595/2003.

Chapter II
68
6. BIBLIOGRAPHY
Bourgogne, A., Hilsenbeck, S. G., Dunny, G. M. & Murray, B. E. (2006).
Comparison of OG1RF and an isogenic fsrB deletion mutant by transcriptional
analysis: the Fsr system of Enterococcus faecalis is more than the activator of
gelatinase and serine protease. J Bacteriol 188, 2875-2884.
Braga, T. M., Marujo, P. E., Pomba, C. & Lopes, M. F. (2011). Involvement, and
dissemination, of the enterococcal small multidrug resistance transporter QacZ in
resistance to quaternary ammonium compounds. J Antimicrob Chemother 66, 283-
286.
Brinster, S., Furlan, S. & Serror, P. (2007). C-terminal WxL domain mediates cell
wall binding in Enterococcus faecalis and other gram-positive bacteria. J Bacteriol
189, 1244-1253.
Dower, W. J., Miller, J. F. & Ragsdale, C. W. (1988). High efficiency transformation
of E. coli by high voltage electroporation. Nucleic Acids Res 16, 6127-6145.
Dunny, G. M., Lee, L. N. & LeBlanc, D. J. (1991). Improved electroporation and
cloning vector system for gram-positive bacteria. Appl Environ Microbiol 57, 1194-
1201.
Eaton, T. J. & Gasson, M. J. (2001). Molecular screening of Enterococcus virulence
determinants and potential for genetic exchange between food and medical isolates.
Appl Environ Microbiol 67, 1628-1635.

Silencing Fsr System: A way to Survive
69
Cha
pter
II
Galloway-Pena, J. R., Bourgogne, A., Qin, X. & Murray, B. E. (2011). Diversity of
the fsr-gelE region of the Enterococcus faecalis genome but conservation in strains
with partial deletions of the fsr operon. Appl Environ Microbiol 77, 442-451.
Gaspar, F., Teixeira, N., Rigottier-Gois, L., Marujo, P., Nielsen-LeRoux, C.,
Crespo, M. T., Lopes Mde, F. & Serror, P. (2009). Virulence of Enterococcus
faecalis dairy strains in an insect model: the role of fsrB and gelE. Microbiology 155,
3564-3571.
Gilmore, M. S., Phillip S. Coburn, Sreedhar R. Nallapareddy, Barbara E. Murray
(2002). Enterococcal Virulence. In The Enterococci Pathogenesis, Molecular Biology,
and Antibiotic Resistance, pp. 301-354. Edited by M. S. Gilmore, Clewell DB,
Courvalin P, Dunny GM, Murray BE, Rice LB. Washington D.C.: American Society
for Microbiology.
Grant, S. G., Jessee, J., Bloom, F. R. & Hanahan, D. (1990). Differential plasmid
rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction
mutants. Proc Natl Acad Sci U S A 87, 4645-4649.
Hancock, L. E. & Perego, M. (2004). The Enterococcus faecalis fsr two-component
system controls biofilm development through production of gelatinase. J Bacteriol
186, 5629-5639.
Jha, A. K., Bais, H. P. & Vivanco, J. M. (2005). Enterococcus faecalis mammalian
virulence-related factors exhibit potent pathogenicity in the Arabidopsis thaliana plant
model. Infect Immun 73, 464-475.
Law, J., Buist, G., Haandrikman, A., Kok, J., Venema, G. & Leenhouts, K. (1995).
A system to generate chromosomal mutations in Lactococcus lactis which allows fast
analysis of targeted genes. J Bacteriol 177, 7011-7018.

Chapter II
70
Lopes Mde, F., Simoes, A. P., Tenreiro, R., Marques, J. J. & Crespo, M. T.
(2006). Activity and expression of a virulence factor, gelatinase, in dairy enterococci.
Int J Food Microbiol 112, 208-214.
Lopes, M. F., Pereira, C. I., Rodrigues, F. M. & other authors (1999). Registered
designation of origin areas of fermented food products defined by microbial
phenotypes and artificial neural networks. Appl Environ Microbiol 65, 4484-4489.
Maguin, E., Prevost, H., Ehrlich, S. D. & Gruss, A. (1996). Efficient insertional
mutagenesis in lactococci and other gram-positive bacteria. J Bacteriol 178, 931-935.
Makinen, P. L., Clewell, D. B., An, F. & Makinen, K. K. (1989). Purification and
substrate specificity of a strongly hydrophobic extracellular metalloendopeptidase
("gelatinase") from Streptococcus faecalis (strain 0G1-10). J Biol Chem 264, 3325-
3334.
Mohamed, J. A. & Murray, B. E. (2006). Influence of the fsr locus on biofilm
formation by Enterococcus faecalis lacking gelE. J Med Microbiol 55, 1747-1750.
Mundt, J. O. (1986). Enterococci. In Bergey’s Manual of Systematic Bacteriology pp.
1063-1065. Edited by N. S. M. P. H. A. Sneath, M. E. Sharpe & J. G. Holt:
Baltimore:Williams & Wilkins.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S., Akkermans, A. D., de Vos, W. M. &
Nagasawa, H. (2001a). Gelatinase biosynthesis-activating pheromone: a peptide
lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 41,
145-154.

Silencing Fsr System: A way to Survive
71
Cha
pter
II
Nakayama, J., Cao, Y., Horii, T., Sakuda, S. & Nagasawa, H. (2001b). Chemical
synthesis and biological activity of the gelatinase biosynthesis-activating pheromone
of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem 65, 2322-2325.
Nakayama, J., Kariyama, R. & Kumon, H. (2002). Description of a 23.9-kilobase
chromosomal deletion containing a region encoding fsr genes which mainly
determines the gelatinase-negative phenotype of clinical isolates of Enterococcus
faecalis in urine. Appl Environ Microbiol 68, 3152-3155.
Ogier, J. C. & Serror, P. (2008). Safety assessment of dairy microorganisms: the
Enterococcus genus. Int J Food Microbiol 126, 291-301.
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Parkinson, J. S. & Kofoid, E. C. (1992). Communication modules in bacterial
signaling proteins. Annu Rev Genet 26, 71-112.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2000). Effects of
Enterococcus faecalis fsr genes on production of gelatinase and a serine protease
and virulence. Infect Immun 68, 2579-2586.
Que, Y. A., Haefliger, J. A., Francioli, P. & Moreillon, P. (2000). Expression of
Staphylococcus aureus clumping factor A in Lactococcus lactis subsp. cremoris
using a new shuttle vector. Infect Immun 68, 3516-3522.
Sahm, D. F., Kissinger, J., Gilmore, M. S., Murray, P. R., Mulder, R., Solliday, J.
& Clarke, B. (1989). In vitro susceptibility studies of vancomycin-resistant
Enterococcus faecalis. Antimicrob Agents Chemother 33, 1588-1591.

Chapter II
72
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular Cloning: a Laboratory
Manual., Cold Spring Harbor Laboratory edn. New York: Cold Spring Harbor.
Sifri, C. D., Mylonakis, E., Singh, K. V., Qin, X., Garsin, D. A., Murray, B. E.,
Ausubel, F. M. & Calderwood, S. B. (2002). Virulence effect of Enterococcus
faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans
and mice. Infect Immun 70, 5647-5650.
Steck, N., Hoffmann, M., Sava, I. G. & other authors (2011). Enterococcus faecalis
metalloprotease compromises epithelial barrier and contributes to intestinal
inflammation. Gastroenterology 141, 959-971.
Su, Y. A., Sulavik, M. C., He, P., Makinen, K. K., Makinen, P. L., Fiedler, S.,
Wirth, R. & Clewell, D. B. (1991). Nucleotide sequence of the gelatinase gene
(gelE) from Enterococcus faecalis subsp. liquefaciens. Infect Immun 59, 415-420.
Thomas, V. C., Hiromasa, Y., Harms, N., Thurlow, L., Tomich, J. & Hancock, L.
E. (2009). A fratricidal mechanism is responsible for eDNA release and contributes to
biofilm development of Enterococcus faecalis. Mol Microbiol 72, 1022-1036.
Thurlow, L. R., Thomas, V. C. & Hancock, L. E. (2009). Capsular polysaccharide
production in Enterococcus faecalis and contribution of CpsF to capsule
serospecificity. J Bacteriol 191, 6203-6210.
Vebo, H. C., Snipen, L., Nes, I. F. & Brede, D. A. (2009). The transcriptome of the
nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to
growth in blood. PLoS One 4, e7660.

Silencing Fsr System: A way to Survive
73
Cha
pter
II
Vebo, H. C., Solheim, M., Snipen, L., Nes, I. F. & Brede, D. A. (2010).
Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis
isolates, and their transcriptional responses to growth in human urine. PLoS One 5,
e12489.
Zhu, Y. & Inouye, M. (2002). The role of the G2 box, a conserved motif in the
histidine kinase superfamily, in modulating the function of EnvZ. Mol Microbiol 45,
653-663.

Chapter II
74
7. SUPPLEMENTARY DATA
Figure S1 - (A) Growth curves of V583 (squares) and LN68 (lozenges) strains in BHI. 1, 2 and 3
mark the three time points for RNA extraction. (B) Analysis of transcription of fsr and gelE–sprE operon
genes by RT‐PCR for V583 and LN68 strains at three different growth stages.

Silencing Fsr System: A way to Survive
75
Cha
pter
II
Table S1 - Primers used in this study.
primer Sequence (5´-3´) MT (ºC) Reference
fsrA5´EcoRI GAA TCG AAT TCG TTT TTG TTT GCA GG 60,1 (Gaspar et al., 2009)
fsrA3´PstI GAA TCC TGC AGT TCG CTT AAC GTC CC 66,4 (Gaspar et al., 2009)
fsrB5´EcoRI GAA TCG AAT TCT TAC TTA GGG AGG G 61,3 (Gaspar et al., 2009)
fsrB3´PstI GAA TCC TGC AGA ACA TTA ATG CCG C 63,0 (Gaspar et al., 2009)
fsrC5´EcoRI GAA TCG AAT TCG ACA ATG GAT GGG AC 63,2 (Gaspar et al., 2009)
fsrC3´PstI GAA TCC TGC AGT TGC TTT ATC CTC CC 64,8 (Gaspar et al., 2009)
gelE5´EcoRI GAA TCG AAT TCT TGA GTT ATG AGG GG 61,6 (Gaspar et al., 2009)
gelE3´PstI GAA TCC TGC AGC AAG CTA AAA CCG GC 66,4 (Gaspar et al., 2009)
sprE5´EcoRI GAA TCG AAT TCT GAA TCT GTT CTG GTC 61,9 (Gaspar et al., 2009)
mfsrA ATG AGT GAA CAA ATG GC 47,9 (Gaspar et al., 2009)
T7fsrA T7 - GT AAG AAA TAG TGC C 65,9 (Gaspar et al., 2009)
fsrA_2 CTA GGA AAA AGA TAT TTA GTT GGG C 58,1 (Gaspar et al., 2009)
mfsrA_2 CAA GAA CAG TTT GGC GGT TG 57,3 (Gaspar et al., 2009)
mfsrB TTG AAG AGG AGG GCG 50,6 (Gaspar et al., 2009)
T7fsrB T7 - GT CCA AAT ATA TTG GGC 67,2 (Gaspar et al., 2009)
T7mfsrB T7 - GC AAT ACT TGA AGA GGA GGG 70,5 (Gaspar et al., 2009)
fsrB GTC CAA ATA TAT TGG GC 47,9 (Gaspar et al., 2009)
fsrC_2 TTT ATA ATC ATG ACG AAA CAT CGC 55,9 (Gaspar et al., 2009)
mfsrC_2 TGA AGA AAC GAT TGC ACC AAC C 58,4 (Gaspar et al., 2009)
fsrC_3 TTT CTT TTT ACA TAT AAC AAT CCC C 54,8 (Gaspar et al., 2009)
mfsrC_3 TTT TTG TGT TTT TGA TTT CGC C 52,8 (Gaspar et al., 2009)
mfsrC TTT GTT CGT TTG CGG C 49,2 (Gaspar et al., 2009)
T7fsrC T7 - GT TGA GTG ACC GCT CC 70,6 (Gaspar et al., 2009)
mgelE AAT ATT TAC GCA GGG 42,4 (Gaspar et al., 2009)
T7gelE T7 - GT TTA CCT GAA TGT CC 67,1 (Gaspar et al., 2009)
T7mgelE T7 -GC AAC AAA TAT TTA CGC AGG G 69,5 (Gaspar et al., 2009)
gelE TCA TTC ATT GAC CAG 42,4 (Gaspar et al., 2009)
gelE_2 GTG TAA AGC AAT TCC CG 50,4 (Gaspar et al., 2009)
mgelE_2 AAC GGA TAA CAC AGG GG 52,8 (Gaspar et al., 2009)
gelE_3 CAA CAC TCT GAG TAT CCG CAC C 62,1 (Gaspar et al., 2009)
mgelE_3 TCT TCG CCA ACT GGT GAC C 58,8 (Gaspar et al., 2009)
msprE TAA CTT TGA TCG CCG G 49,2 (Gaspar et al., 2009)
T7sprE T7 - GC TGC TGG CAC AGC GG 73,0 (Gaspar et al., 2009)
sprE_2 TCA AAC AAA CGA AAC TGG C 52,4 (Gaspar et al., 2009)
msprE_2 TTT GTT TAG TAA AAG TAC TCT GG 53,5 (Gaspar et al., 2009)
sprE_3 CAT TCT TAA AAC TTT CAG CCA C 54,7 (Gaspar et al., 2009)
T7mfsrD T7 GA AAT TTG GTA AAA A 62,2 (Gaspar et al., 2009)
sprE_4 TTT CCT GTT TGC TTA ATG CCG C 58,4 This study

Chapter II
76
msrpE_4 GGC TGA AAG TTT TAA GAA TGC CAA A 58,1 This study
LN68_fsrC-PGMT5 TTA TAA ACC AAT GAT ACG GG 51,2 This study
LN68_fsrC_PGMT3 AAA ATA AAT TAT TAT GGA TTG CC 50 This study
LN68_mfsrC_M CCC AGA A GAG CAC GGT TGG GGA TTG TTA TAT GTA AAA G 70,5 This study
LN68_fsrC_M CTT TTA CAT ATA ACA ATC CCC AAC CGT GCT CTT CTG GG 70,5 This study
V583_mfsrC_M CCA GAA GAG CAC GGT TGA GGA TTG TTA TAT GTA AAA G 68,4 This study
V583_fsrC_M CTT TTA CAT ATA ACA ATC CTC AAC CGT GCT CTT CTG G 68,4 This study
fsrC5´_BamHI GAA TCG GAT CCG ACA ATG GAT GGG AC 66,4 This study
pOri23_fw GGA TTG GAT TAG TTC TTG TGG 55,9 (Braga et al.)
Pori23_rv TTG AGT GAG CTG ATA CCG C 56,7 (Braga et al.)

FSR AND VANCOMYCIN:
The antagonistic relation
This chapter complements a previous PhD work and is published in the following manuscript:
Transcriptomic study Reveals new pathways and genes Involved in Enterococcus faecalis V583 response to a therapeutic dose of vancomycin
Tânia Ribeiro, Neuza Teixeira, Ryoji Yokohata, Jiro Nakayama, Michael S. Gilmore
and Maria de Fátima S. Lopes (2013), Archives of Clinical Microbiology, Vol. 4 (5:3).


CONTENTS
1. SUMMARY ........................................................................................................... 81
2. INTRODUCTION ................................................................................................... 82
3. MATERIALS AND METHODS .............................................................................. 84
3.1 Bacterial strains and RNA extraction conditions ............................................. 84
3.1 Semi-quantitative RT-PCR ............................................................................. 85
4. RESULTS AND DISCUSSION .............................................................................. 86
6. BIBLIOGRAPHY ................................................................................................... 90

Chapter III
80
Neuza Teixeira performed the experiments correlating vancomycin with the Fsr
system. Ryoji Yokohata produced the GBAP.

Fsr and Vancomycin: The antagonistic relation
81
Cha
pter
III
1. SUMMARY
An enterococcal strain carrying the VanB resistance type can become susceptible if
impaired in other genes unrelated to the vanB operon. This fact alone illustrates the
lack of knowledge on the vancomycin mode of action. This antibiotic is still usable to
treat serious infections caused by multi-resistant enterococcal strains, but may not be
so for long. In a previous PhD work from our lab, microarrays were used to detect the
genetic response of the VanB carrying strain Enterococcus faecalis V583 to a
therapeutic dose (10 µg/ ml) of vancomycin. This therapeutic dose of vancomycin
was found to act as an anti-virulence agent, by down-regulating fsr-gelE-sprE genes.
We wondered if vancomycin was playing a role in preventing FsrC from sensing
GBAP molecule. In order to elucidate on this E. faecalis V583 and its isogenic mutant
fsrB cells were collected 0 min, 10 min and 20 min after GBAP addition, in the
presence and absence of vancomycin. RNA was extracted and semi-quantitative RT-
PCR was performed. We concluded that E. faecalis V583 strain responds to
vancomycin therapy by becoming blind to the quorum sensing molecule GBAP.

Chapter III
82
2. INTRODUCTION
During the five decades of vancomycin usage, eight genotypes of resistance to this
antibiotic have been described in only one genus, Enterococcus. Vancomycin, a cell-
wall active glycopeptide antibiotic, was first introduced to clinical practice in the 60’s
and in 1986 the first resistant enterococcal strain was isolated (Leclercq et al., 1988).
Since then, vancomycin resistant enterococci (VRE) have been isolated from
endocarditis, bacteraemia, urinary tract infections and wound infections, and have
emerged as one of the major nosocomial agents in hospitals (Arias & Murray, 2012).
Among the eight resistance types, VanA and VanB constitute the two most widely
disseminated, both conferring resistance by the same mechanism and encoding
related enzymes (Reynolds et al., 1994). In both cases, resistance is due to
synthesis of peptidoglycan precursors ending in the depsipeptide D-alanyl–D-lactate
(D-Ala–D-Lac) that binds glycopeptides with reduced affinity (Evers & Courvalin,
1996). The vanB operon contains the vanYBWHBBXBV resistance genes (Ribeiro et
al., 2011), and vanH, vanB and vanX are essential for resistance phenotype. vanHB
encodes a dehydrogenase that reduces pyruvate to D-Lac; vanB encodes a ligase
that synthetizes the depsipeptide D-Ala-D-Lac; and VanXB hydrolyses the D-Ala-D-
Ala dipeptide synthetized by the native Enterococcus Ddl ligase. Expression of the
resistance genes is regulated by the vanRBSB two-component system, which is
composed of a membrane-associated sensor kinase (VanSB), and a cytoplasmic
response regulator (VanRB) that acts as a transcriptional activator (Arthur et al.,
1994; Evers & Courvalin, 1996). The regulatory and resistance genes are transcribed
from distinct promoters that appear to be co-ordinately regulated (Aslangul et al.,
1997).
The Fsr (Enterococcus faecalis sensor regulator) is a quorum sensing regulatory
system and is similar to agr. The Fsr operon comprises four genes: fsrA, fsrB, fsrC
and fsrD. The last encodes an auto-inducing cyclic peptide named gelatinase
biosynthesis- activating pheromone (GBAP) and this peptide is processed and
exported out of the cell by FsrB. Accumulation of GBAP outside cells is sensed by

Fsr and Vancomycin: The antagonistic relation
83
Cha
pter
III
the FsrC histidine kinase, leading to the activation of the response regulator FsrA.
Activated FsrA induces expression of the fsrBDC genes, which are involved in an
auto regulatory circuit that results in a boost of GBAP signalling and the induction of
another operon, the gelE-sprE operon (Nakayama et al., 2001a; Nakayama et al.,
2001b; Nakayama et al., 2006; Qin et al., 2000; Qin et al., 2001). The proteases
have been shown to be involved in biofilm formation, in translocation across intestinal
T84 cells, in degradation of antimicrobial peptides (AMPs) from the immune system
of Galleria mellonella, in autolysis regulation and as regulators of Ace surface protein
exposure on the surface of E. faecalis cells (Hancock & Perego, 2004; Park et al.,
2007; Pinkston et al., 2011; Singh et al., 2010; Thomas et al., 2009; Zeng et al.,
2005).
However, in a recent work from our lab (PhD thesis), we found that Fsr quorum-
sensing system, and the genes directly regulated by it, are repressed by the cell-wall
active antibiotic. This result came from a transcriptomic study in which we subjected
E. faecalis V583, the first VRE genome to be sequenced, to vancomycin, and
examined the transcriptional response by microarray to identify the cellular pathways
affected. A therapeutic dose of vancomycin (10 µg/ml) was chosen to represent the
range between peak levels of 20-40 µg/ml and trough serum levels of 5-10 µg/ml
achieved in therapy (Rybak et al., 2009).
The purpose of the work presented in this chapter was thus to elucidate on the
reason/mechanism responsible for Fsr shutdown by vancomycin.

Chapter III
84
3. MATERIAL AND METHODS
3.1 Bacterial strains and RNA extraction conditions
Strains used in this study are described in Table 1. The effect of vancomycin on the
ability of E. faecalis V583 to sense GBAP, was directly evaluated by measuring
changes in expression of fsr regulated genes gelE and sprE. To control the timing of
the response of this operon, we used a V583 fsrB mutant (Teixeira et al., 2013),
which is unable to produce GBAP by itself, but is able to sense and activate Fsr upon
GBAP addition. This strain, termed V583ΔfsrB, was grown in BHI to an OD ~0.4,
and the culture was then split. To half, only GBAP (10nM), which was chemically
synthesized (Nakayama et al., 2001b), was added. To the other portion, both GBAP
(10 nM) and vancomycin (10 µg/ml) were added, and after 10 or 20 minutes, cells
were collected for RNA extraction and expression levels of gelE, sprE and vanB
genes were evaluated. Total RNA was extracted and purified with an RNeasy Mini kit
(Qiagen).
Table 1 - Strains and primers used in this study.
Strains Relevant characteristics Reference
V583 Clinical isolate, VaR, with VanB type of resistance (Aslangul et al., 1997)
VI13 E. faecalis V583 ∆fsrB, GelE-, GBAP- (Teixeira et al., 2012)
Primers
Gene Sequence (5´-3´)
gelE TCATTCATTGACCAG (forward)
AACGGATAACACAGGGG (reverse) (Gaspar et al., 2009)
sprE CATTCTTAAAACTTTCAGCCAC (forward)
TAACTTTGATCGCCGG (reverse) (Gaspar et al., 2009)
vanB CCTACCCTGTCTTTGTGAAGC (forward)
ATTGTCCTGCTGCTTCTATCG (reverse) This study

Fsr and Vancomycin: The antagonistic relation
85
Cha
pter
III
3.1 Semi-quantitative RT-PCR
cDNA was synthesized using random primers (Roche Diagnostics), 3 mg total RNA
and a Transcriptor High Fidelity cDNA Synthesis kit (Roche Diagnostics). Three
serial dilutions (1:1; 1:10 and 1:100) of cDNA were used for PCR in order to amplify
cDNA of gelE, sprE and vanB transcripts (see the primers in Table 1). vanB RNA
was used as a control of vancomycin activity

Chapter III
86
4. RESULTS AND DISCUSSION
In a previous work from our lab (PhD Thesis), the expression profile of E. faecalis
V583 exposed to a therapeutic dose of vancomycin was compared with the
expression profile of V583 cells in the absence of the antibiotic. This allowed the
identification of genes which respond either to vancomycin, or to the changes
introduced by the induction of the vanB operon genes in response to the antibiotic. In
this study Fsr system was repressed following the addition of vancomycin (Table 2)
and message corresponding to genes known to be regulated by this system, namely
gelE and sprE, was less abundant. At the late exponential growth phase when the
cells were collected, we would have expected that GBAP was accumulating outside
the cells and thus activating the Fsr system (Nakayama et al., 2001a).
Table 2 - Fsr and vancomycin resistance genes differentially expressed at both time points (t10
and t30 min) under exposure to10 µg/ml of vancomycin, with fold-change values higher than 2.
Fold change values are given for each gene, as well as its putative function and role, according to NCBI.
Locus Gene Descriptions Role Fold change
t10 t30
EF1097 - putative bacteriocin Unknown function -7.1 -12.5
EF1817 sprE serine proteinase, V8 family Protein fate -5.0 -9.6
EF1818 gelE Coccolysin Cell envelope/Protein fate/Cellular
processes -3.6 -8.0
EF1820 fsrC histidine kinase, putative Signal transduction -2.7 -2.9
EF1821 fsrB agrBfs protein Unknown function -2.3 -2.9
EF2292 - hypothetical protein Unknown function 131.5 176.9
EF2293 vanX D-alanyl-D-alanine dipeptidase Protein fate 129.5 123.9
EF2294 vanB D-alanine-D-lactate ligase Protein fate 188.0 132.9
EF2295 vanH D-specific alpha-keto acid
dehydrogenase Cell envelope/Cellular processes 203.5 217.8
EF2296 vanW vancomycin B-type resistance protein
VanW Cellular processes/Signal transduction 219.1 241.1
EF2297 vanYB D-alanyl-D-alanine carboxypeptidase Cell envelope/Cellular processes 145.6 170.5
EF2298 vanSB sensor histidine kinase VanSB Signal transduction 2.8 3.0
EF2299 vanRB DNA-binding response regulator VanRB Regulatory functions/Signal transduction 3.0 3.2

Fsr and Vancomycin: The antagonistic relation
87
Cha
pter
III
The reduction in mRNA for the Fsr system was unexpected. One explanation would
be that vancomycin affected the ability of FsrC to sense the external GBAP, and
interfering with Fsr auto-induction, resulting in down-regulation of the system and the
genes it controls. To test this hypothesis, we used the V583ΔfsrB strain, which can
sense GBAP and activate fsr and gelE-sprE transcription, but is unable to produce it
(Teixeira et al., 2012). We added chemically synthesized GBAP to cultures of this
strain, with and without vancomycin, and looked for induction of gelE and sprE gene
expression. As shown in Figure 1B, and as expected, expression of both genes was
increased upon GBAP addition to V583ΔfsrB cells. However, when vancomycin was
added, neither gelE nor sprE were induced (Figure 1), demonstrating that
vancomycin somehow prevents the Fsr system from responding to its quorum signal.
In this experiment, vanB gene was, as expected, induced by vancomycin,
corroborating the microarray data and also validating our results with GBAP and the
V583ΔfsrB strain.
Figure 1 - Effect of GBAP and vancomycin on the expression of gelE, sprE and vanB genes in
strain V583fsrB. A, vancomycin was added at T0, prior to addition of GBAP; B, GBAP was added alone at T0.
The immediate repression of the Fsr system upon exposure to vancomycin indicates
that for reasons unknown, FsrC becomes blind to the quorum signal GBAP. The

Chapter III
88
quorum-sensing disrupting activity of vancomycin would be predicted to limit the
contributions of GelE and SprE proteases to bacterial virulence. It is possible that
other cell-wall active antibiotics may produce the same effect. Possibly, vancomycin
prevents FsrC phosphorylation, as is the case for the antimicrobial peptide
antimicrobial siamycin I. Siamycin I, active against E. faecalis and previously found to
inhibit both gelatinase and GBAP production (Nakayama et al., 2007), directly inhibits
autophosphorylation of FsrC kinase (Ma et al., 2011).
The observed negative effect of vancomycin on Fsr induction may be related to the
fitness cost that Fsr activation can have on E. faecalis. When under antibiotic stress
bacteria need to reduce the energy waste in general ant that used for Fsr activation
in particular and apply this energy to other resources, namely the vancomycin
resistance operon activity, which helps the bacteria to survive. In Staphylococcus
aureus it was described that sub-inhibitory concentrations of antibiotics are known to
modulate virulence gene expression in a process likely involving a quorum sensing
called agr, that is similar to E. faecalis fsr (Joo et al., 2010). Agr-negative isolates
frequently arise in hospital infections (Traber et al., 2008). Even though virulence
gene expression is compromised in agr-deficient isolates, they still give rise to
concern. In terms of resistance to antimicrobials, agr-negative strains are known to
display intermediate resistance or heteroresistance to glycopeptides such as
vancomycin, glycopeptide intermediate-level resistant S. aureus [GISA] and hetero-
GISA) (Paulander et al., 2013). This fact can be correlated with the fitness cost of
carrying agr, which is enhanced by the presence of some antibiotics and that
treatment with those antibiotics will select for agr-deficient mutants. Like in S. aureus,
the fitness cost in E. faecalis can be one of the main reasons of the Fsr shutdown in
vancomycin presence.
When vancomycin is added to E. faecalis V583, it stops growing after 30 minutes
and enterers earlier into stationary phase (Figure S1). This drastic change on
bacterial growth phase could be also correlated with changes in cell autolysis trough
Fsr shutdown. As already described, the proteases GelE and SprE are correlated
with cell autolysis (Teixeira et al., 2013; Thomas et al., 2009). During bacterial growth

Fsr and Vancomycin: The antagonistic relation
89
Cha
pter
III
autolysis plays a crucial role in cell-division and is important for peptidoglycan
construction and renovation. In 2009, Thomas et al, show that autolysin AtlA is a
target of both GelE and SprE and this interaction is critical to the regulation of
enterococcal fratricide and biofilm development (Thomas et al., 2009). Our results
indicated the hypothesis that in the absence of the proteases GelE-SprE autolysin is
repress and bacteria stop growing. More studies are needed to prove test these two
hypotheses.
This quorum-sensing disrupting ability of vancomycin found in this work, is an
important step to finding news targets to combat E. faecalis infections.

Chapter III
90
6. BIBLIOGRAPHY
Arias, C. A. & Murray, B. E. (2012). The rise of the Enterococcus: beyond
vancomycin resistance. Nat Rev Microbiol 10, 266-278.
Arthur, M., Depardieu, F., Snaith, H. A., Reynolds, P. E. & Courvalin, P. (1994).
Contribution of VanY D,D-carboxypeptidase to glycopeptide resistance in
Enterococcus faecalis by hydrolysis of peptidoglycan precursors. Antimicrob Agents
Chemother 38, 1899-1903.
Aslangul, E., Baptista, M., Fantin, B., Depardieu, F., Arthur, M., Courvalin, P. &
Carbon, C. (1997). Selection of glycopeptide-resistant mutants of VanB-type
Enterococcus faecalis BM4281 in vitro and in experimental endocarditis. J Infect Dis
175, 598-605.
Evers, S. & Courvalin, P. (1996). Regulation of VanB-type vancomycin resistance
gene expression by the VanS(B)-VanR (B) two-component regulatory system in
Enterococcus faecalis V583. J Bacteriol 178, 1302-1309.
Gaspar, F., Teixeira, N., Rigottier-Gois, L., Marujo, P., Nielsen-LeRoux, C.,
Crespo, M. T., Lopes Mde, F. & Serror, P. (2009). Virulence of Enterococcus
faecalis dairy strains in an insect model: the role of fsrB and gelE. Microbiology 155,
3564-3571.
Hancock, L. E. & Perego, M. (2004). The Enterococcus faecalis fsr two-component
system controls biofilm development through production of gelatinase. J Bacteriol
186, 5629-5639.
Joo, H. S., Chan, J. L., Cheung, G. Y. & Otto, M. (2010). Subinhibitory
concentrations of protein synthesis-inhibiting antibiotics promote increased

Fsr and Vancomycin: The antagonistic relation
91
Cha
pter
III
expression of the agr virulence regulator and production of phenol-soluble modulin
cytolysins in community-associated methicillin-resistant Staphylococcus aureus.
Antimicrob Agents Chemother 54, 4942-4944.
Leclercq, R., Derlot, E., Duval, J. & Courvalin, P. (1988). Plasmid-mediated
resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med
319, 157-161.
Ma, P., Nishiguchi, K., Yuille, H. M., Davis, L. M., Nakayama, J. & Phillips-Jones,
M. K. (2011). Anti-HIV siamycin I directly inhibits autophosphorylation activity of the
bacterial FsrC quorum sensor and other ATP-dependent enzyme activities. FEBS
Lett 585, 2660-2664.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S., Akkermans, A. D., de Vos, W. M. &
Nagasawa, H. (2001a). Gelatinase biosynthesis-activating pheromone: a peptide
lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 41,
145-154.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S. & Nagasawa, H. (2001b). Chemical
synthesis and biological activity of the gelatinase biosynthesis-activating pheromone
of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem 65, 2322-2325.
Nakayama, J., Chen, S., Oyama, N., Nishiguchi, K., Azab, E. A., Tanaka, E.,
Kariyama, R. & Sonomoto, K. (2006). Revised model for Enterococcus faecalis fsr
quorum-sensing system: the small open reading frame fsrD encodes the gelatinase
biosynthesis-activating pheromone propeptide corresponding to staphylococcal agrd.
J Bacteriol 188, 8321-8326.

Chapter III
92
Nakayama, J., Tanaka, E., Kariyama, R. & other authors (2007). Siamycin
attenuates fsr quorum sensing mediated by a gelatinase biosynthesis-activating
pheromone in Enterococcus faecalis. J Bacteriol 189, 1358-1365.
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Paulander, W., Nissen Varming, A., Baek, K. T., Haaber, J., Frees, D. & Ingmer,
H. (2013). Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus
aureus. MBio 3, e00459-00412.
Pinkston, K. L., Gao, P., Diaz-Garcia, D., Sillanpaa, J., Nallapareddy, S. R.,
Murray, B. E. & Harvey, B. R. (2011). The Fsr quorum-sensing system of
Enterococcus faecalis modulates surface display of the collagen-binding MSCRAMM
Ace through regulation of gelE. J Bacteriol 193, 4317-4325.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2000). Effects of
Enterococcus faecalis fsr genes on production of gelatinase and a serine protease
and virulence. Infect Immun 68, 2579-2586.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2001). Characterization of
fsr, a regulator controlling expression of gelatinase and serine protease in
Enterococcus faecalis OG1RF. J Bacteriol 183, 3372-3382.
Reynolds, P. E., Snaith, H. A., Maguire, A. J., Dutka-Malen, S. & Courvalin, P.
(1994). Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus
gallinarum BM4174. Biochem J 301 ( Pt 1), 5-8.

Fsr and Vancomycin: The antagonistic relation
93
Cha
pter
III
Ribeiro, T., Santos, S., Marques, M. I., Gilmore, M. & de Fatima Silva Lopes, M.
(2011). Identification of a new gene, vanV, in vanB operons of Enterococcus faecalis.
Int J Antimicrob Agents 37, 554-557.
Rybak, M. J., Lomaestro, B. M., Rotschafer, J. C., Moellering, R. C., Jr., Craig,
W. A., Billeter, M., Dalovisio, J. R. & Levine, D. P. (2009). Therapeutic monitoring
of vancomycin in adults summary of consensus recommendations from the American
Society of Health-System Pharmacists, the Infectious Diseases Society of America,
and the Society of Infectious Diseases Pharmacists. Pharmacotherapy 29, 1275-
1279.
Singh, K. V., Nallapareddy, S. R., Sillanpaa, J. & Murray, B. E. (2010). Importance
of the collagen adhesin ace in pathogenesis and protection against Enterococcus
faecalis experimental endocarditis. PLoS Pathog 6, e1000716.
Teixeira, N., Santos, S., Marujo, P., Yokohata, R., Iyer, V. S., Nakayama, J.,
Hancock, L. E., Serror, P. & Silva Lopes Mde, F. (2012). The incongruent
gelatinase genotype and phenotype in Enterococcus faecalis are due to shutting off
the ability to respond to the gelatinase biosynthesis-activating pheromone (GBAP)
quorum-sensing signal. Microbiology 158, 519-528.
Teixeira, N., Varahan, S., Gorman, M. J. & other authors (2013). Drosophila host
model reveals new enterococcus faecalis quorum-sensing associated virulence
factors. PLoS One 8, e64740.
Thomas, V. C., Hiromasa, Y., Harms, N., Thurlow, L., Tomich, J. & Hancock, L.
E. (2009). A fratricidal mechanism is responsible for eDNA release and contributes to
biofilm development of Enterococcus faecalis. Mol Microbiol 72, 1022-1036.

Chapter III
94
Traber, K. E., Lee, E., Benson, S., Corrigan, R., Cantera, M., Shopsin, B. &
Novick, R. P. (2008). agr function in clinical Staphylococcus aureus isolates.
Microbiology 154, 2265-2274.
Zeng, J., Teng, F. & Murray, B. E. (2005). Gelatinase is important for translocation
of Enterococcus faecalis across polarized human enterocyte-like T84 cells. Infect
Immun 73, 1606-1612.

NEW FINDINGS ON FSR SYSTEM:
New virulence genes and their impact during Drosophila
infection
This chapter is based on the following manuscript:
Drosophila Host Model Reveals New Enterococcus faecalis Quorum-Sensing Associated Virulence Factors
Neuza Teixeira, Sriram Varahan, Matthew J. Gorman, Kelli L. Palmer, Anna Zaidman-Remy,
Ryoji Yokohata, Jiro Nakayama, Lynn E. Hancock, António Jacinto, Michael S. Gilmore and
Maria de Fátima Silva Lopes (2013), PLoS One 8, e64740.


CONTENTS 1. SUMMARY ........................................................................................................... 99
2. INTRODUCTION ................................................................................................. 100
3. MATERIAL AND METHODS .............................................................................. 103
3.1 Bacterial Strains and Plasmids ..................................................................... 103
3.2 Antibiotic Resistance Assay ......................................................................... 103
3.3 General DNA Techniques ............................................................................. 103
3.4 Mutant Construction ..................................................................................... 105
3.5 RNA Extraction and cDNA Synthesis for Microarrays .................................. 107
3.6 Semiquantitative RT-PCR ............................................................................ 108
3.7 D. melanogaster Infection ............................................................................. 108
3.8 Percentage of Similarities between V583 Genome and Other Genomes
Published ............................................................................................................ 109
3.9 Statistical Analysis ........................................................................................ 109
4. RESULTS ............................................................................................................ 110
4.1 Fsr dependent genes .................................................................................... 110
4.2 Genes dependent on simultaneous Fsr and Proteases activation ................ 112
4.3 LytRS system is required for GBAP induction of lrgAB genes ...................... 113
4.4 Fsr and the proteases affect D. melanogaster tolerance to E. faecalis infection
........................................................................................................................... 114
4.5 ef1097 contributes to toxicity in D. melanogaster infection ........................... 115
4.6 LrgAB and LytRS contribute differently to death of D. melanogaster ........... 116
5. DISCUSSION ...................................................................................................... 118
6. ACKNOWLEDGEMENTS ................................................................................... 124
7. BIBLIOGRAPHY ................................................................................................. 125
8. SUPPLEMENTARY DATA .................................................................................. 135

Chapter IV
98
The author of this thesis performed the majority of the experiments. Experimental
design, data analysis and manuscript preparation were done by Neuza Teixeira and
the supervisor Maria de Fatima Silva Lopes. Matthew J. Gorman and Vijayalakshmi
S. Iyer did the Fsr mutants. Kelli L. Palmer did the microarray analysis. Anna
Zaidman-Remy helped with the Drosophila experiments and Ryoji Yokohata
produced the GBAP.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
99
Cha
pter
IV
1. SUMMARY
Enterococcus faecalis V583 is a vancomycin-resistant clinical isolate which belongs
to the hospital-adapted clade, CC2. This strain harbours several factors that have
been associated with virulence, including the fsr quorum-sensing regulatory system
that is known to control the expression of GelE and SprE proteases. To discriminate
between genes directly regulated by Fsr, and those indirectly regulated as the result
of protease expression or activity, we compared gene expression in isogenic mutants
of V583 variously defective in either Fsr quorum sensing or protease expression.
Quorum sensing was artificially induced by addition of the quorum signal, GBAP,
exogenously in a controlled manner. The Fsr regulon was found to be restricted to
five genes, gelE, sprE, ef1097, ef1351 and ef1352. Twelve additional genes were
found to be dependent on the presence of GBAP-induced proteases. Induction of
GelE and SprE by GBAP via Fsr resulted in accumulation of mRNA encoding lrgAB,
and this induction was found to be lytRS dependent. Drosophila infection was used
to discern varying levels of toxicity stemming from mutations in the fsr quorum
regulatory system and the genes that it regulates, highlighting the contribution of
LrgAB and bacteriocin EF1097 to infection toxicity. A contribution of SprE to infection
toxicity was also detected. This work brought to light new players in E. faecalis
success as a pathogen and paves the way for future studies on host tolerance
mechanisms to infections caused by this important nosocomial pathogen.

Chapter IV
100
2. INTRODUCTION
Drosophila melanogaster is used increasingly as a model for identifying virulence
factors of pathogenic microbes, and for elucidating their effects on the host (Boyer et
al., 2012). The fruit fly presents several advantages, such as small size, short life
cycle, short generation time, a fully sequenced genome and pre-existing libraries of
genetic mutants. In addition, its immune system shares similarities with the
mammalian immune system, including genes and pathways. In particular, the Toll
and Imd pathways in D. melanogaster have parallels in the mammalian Toll-like
(TLR) and interlleukin-1 (IL-1) receptor families, and the mammalian tumour necrosis
factor signalling pathway (Glavis-Bloom et al., 2012). In 2007, Cox and Gilmore
characterized the microbiome of this host and showed that Enterococcus sp. and
naturally colonize its alimentary canal; and that cytolysin, a toxin expressed by some
strains of Enterococcus faecalis, contributes to death of the flies when colonized
(Cox & Gilmore, 2007). It is also known that E. faecalis are able to kill the flies and
induce the Toll pathway after infection by septic injury, and that haemocytes
(Drosophila circulating cells that function as phagocytes) also play a role in fly’s
defense against these bacteria (Nehme et al., 2011; Schneider et al., 2007).
Enterococci are Gram-positive bacteria commonly found in gastrointestinal tract
consortia, but are also adapted to survive and persist in the environment. In contrast
to their benign role as members of the gut flora, select lineages of several
enterococcal species have become leading causes of antibiotic resistant nosocomial
infection, causing infections of the urinary tract, bloodstream, intra-abdominal and
pelvic regions, and surgical sites (Gilmore, 2002). E. faecalis, the species most
frequently associated with nosocomial infections (Qin et al., 2000), possesses a
number of traits that exacerbate the effects of infection. Fsr (Enterococcus faecalis
sensor regulator) a two-component, quorum sensing regulatory system, was first
described in 2000 by Qin et al. as a paralog of the Agr system in Staphylococcus
aureus (Qin et al., 2000). Despite similarities, Agr is functionally distinct from Fsr as it
uses the RNAIII riboregulator (Novick et al., 1993). The fsr operon comprises four

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
101
Cha
pter
IV
genes: fsrA, fsrB, fsrC and fsrD (Nakayama et al., 2001a). The last encodes an auto-
inducing cyclic peptide named gelatinase biosynthesis-activating pheromone
(GBAP), and this peptide is processed and exported out of the cell by FsrB.
Accumulation of GBAP outside cells is sensed by the FsrC histidine kinase, leading
to the activation of the response regulator FsrA. Activated FsrA induces expression
of the fsrBDC genes forming an auto regulatory circuit that results in a rapid,
exponential increase in GBAP signalling. Expression of a second operon is induced
by FsrA consisting of two cistrons gelE-sprE. The first cistron, gelE, encodes
gelatinase, an extracellular zinc metalloprotease, and the second, sprE, encodes a
serine protease (Qin et al., 2000; Qin et al., 2001). Several studies provided
evidence that both Fsr and the proteases independently contribute to the
pathogenicity of E. faecalis in different infection models (Engelbert et al., 2004;
Garsin et al., 2001; Gaspar et al., 2009; Jha et al., 2005; Mylonakis et al., 2002; Sifri
et al., 2002; Singh et al., 2005). The proteases have also been shown to be involved
in biofilm formation (Hancock & Perego, 2004), in translocation across intestinal T84
cells (Zeng et al., 2005), in degradation of antimicrobial peptides (AMPs) from the
immune system of Galleria mellonella (Park et al., 2007), in autolysis regulation
(Thomas et al., 2009) and as regulators of Ace surface protein exposure on the
surface of E. faecalis cells (Pinkston et al., 2011; Singh et al., 2010). The exact
mechanisms by which Fsr and its regulated proteases contribute to toxicity of
infection are not known. This has been confounded in part by unexplained variation
in experimental results. In 2005, Singh et al. tested fsrB and gelE mutants in E.
faecalis strain OG1RF in a rat endocarditis model. Deletion of the proteases led to a
greater decrease in endocarditis severity than deletion of fsrB. In the absence of
fsrB, the gelE expression was reduced, and the authors postulated that was the
reason for the smaller attenuation of fsrB mutant (Singh et al., 2005). In contrast,
studies examining the role of these traits in rabbit endophtalmitis (Engelbert et al.,
2004; Mylonakis et al., 2002), murine and C. elegans infection (Garsin et al., 2001;
Sifri et al., 2002), and in a G. mellonella infection model (Gaspar et al., 2009) all
found that fsrB deletion led to a greater attenuation than deletion of the proteases.

Chapter IV
102
These last results raised the possibility that Fsr could be affecting directly or
indirectly more genes or their products than just the proteases. Bourgogne et al.
compared gene expression in OG1RF with an isogenic fsrB deletion mutant, and
provided some evidence that Fsr regulates more than gelE and sprE protease genes
(Bourgogne et al., 2006). While it is known that host substrates, such as complement
components C3, C3a and C5a are targeted by GelE (Park et al., 2007; Park et al.,
2008; Thurlow et al., 2010), little is known regarding a functional role for SprE in
production of host injury and death. To decipher the role of Fsr-regulated genes in
virulence, we used a clonal-complex (CC) 2 strain (McBride et al., 2007), E. faecalis
V583, the first vancomycin-resistant enterococcal isolate in the US, which was
obtained from a chronic bloodstream infection (Sahm et al., 1989). E. faecalis CC2 is
the leading multidrug resistant hospital adapted clade (McBride et al., 2007; Willems
et al., 2011). To rigorously characterize the Fsr regulon, we compared gene
expression in isogenic mutants in Fsr genes and each of the Fsr-regulated protease
genes using microarrays and purified GBAP. D. melanogaster was used to examine
the individual contribution to virulence of SprE protease and other genes found to be
part of the Fsr regulon (or related to it, including EF1097, LrgAB and the two-
component system LytRS).

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
103
Cha
pter
IV
3. MATERIAL AND METHODS
3.1 Bacterial Strains and Plasmids
Strains and plasmids used in this study are listed in Table 1. E. faecalis strains were
grown either in BHI, M17 broth/agar (Oxoid) or Enterococcel Agar (Quilaban) at
37ºC, unless a different growth temperature is specified. Escherichia coli strains
were grown in LB medium (Sigma) at 37ºC with agitation. The following antibiotic
concentrations were used: with E. faecalis, tetracycline 30 mg/ml; with E. coli,
ampicillin 150 mg/ml and tetracycline 150 mg/ml.
3.2 Antibiotic Resistance Assay
Resistance to different antibiotics (Ciprofloxacin, Penicillin, Sulphamethoxazole,
Vancomycin, Nitrofurantoin, Ofloxacin, Ampicillin, and Ceftriaxone) was determined
according to the recommendations of the disk providers (Oxoid) (Lopes Mde et al.,
2003), and results were interpreted according to the recommendations of the Clinical
and Laboratory Standards Institute (CLSI, formerly NCCLS) (http: //www.clsi.org/).
3.3 General DNA Techniques
General molecular biology techniques were performed by standard methods.
Restriction enzymes, polymerases and T4 DNA ligase were used according to
manufacturers’ instructions. PCR amplification was performed using a Biometra
thermocycler. When necessary, PCR products and DNA restriction fragments were
purified with purification kits (Macherey-Nagel). Plasmids were purified using the
Miniprep kit (Macherey-Nagel). Electro- transformation of E. coli and E. faecalis was
carried out as described by Dower et al. (1988) and Dunny et al. (1991), using a
Gene Pulser apparatus (Bio-Rad) (Dower et al., 1988; Dunny et al., 1991). Plasmid
inserts and mutant sequence were confirmed by sequencing at StabVida (Portugal).

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
104
Table 1 - Strains, plasmids and primers used in this study.
Strains Relevant characteristics Reference
E. coli
DH5α F- Ø80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK - mK- ) phoA supE44 λ- thi-1 gyrA96 relA1
(Grant et al., 1990)
TG1 RepA supE hsdD5 thi (Δlac-proAB) F- (traD36 proAB-lacZΔM15) repA
(Law et al., 1995)
VE14188 GM1674 (dam- dcm- repA+) (Rigottier-Gois et al., 2011)
E. faecalis
V583 Clinical isolate, TIGR sequence strain; VnR (Sahm et al., 1989)
VE14089 V583 free of replicating plasmids
(Rigottier-Gois et al., 2011)
VI13 E. faecalis V583∆fsrB, GelE-, SprE-, GBAP-
(Teixeira et al., 2012)
MG01 E. faecalis V583ΔfsrB∆gelE; GelE-, SprE-, GBAP-
This study
MG02 E. faecalis V583ΔfsrB∆sprE; GelE-,SprE-, GBAP- This Study
MG03 E. faecalis V583ΔfsrB∆gelEΔsprE ; GelE-, SprE-, GBAP-
This Study
VT01 E. faecalis V583∆gelE, GelE-, GBAP+
(Thomas et al., 2008)
VT02 E. faecalis V583∆sprE, SprE-, GBAP+
(Thomas et al., 2008)
VT03
E. faecalis V583∆gelE∆sprE, GelE-, SprE-, GBAP+ (Thomas et al., 2008)
KS17 E. faecalis V583∆lytRS, GelE+, SprE+, GBAP+
This study
KS18 E. faecalis V583∆lrgAB, GelE+, SprE+, GBAP+
This study
KS19 E. faecalis V583∆fsrB∆lytRS, GelE-, SprE-, GBAP- This study
SAVE38 E. faecalis VE14089∆ef1097, GelE+, SprE+, GBAP+ This study
Plasmids
pGEM-T High copy plasmid, AmpR Promega
pG+host9 E. faecalis thermosensitive plasmid, EryR
(Maguin et al., 1996)
pLT06 Temperature-sensitive cloning vector, CmR
(Thurlow et al., 2009a)
pVI02 pLT06 containing engineered fsrB deletion (Teixeira et al., 2012)
pVT01
pLT06 containing engineered gelE deletion (Thomas et al., 2008)
pVT02
pLT06 containing engineered sprE deletion (Thomas et al., 2008)
pVT03
pLT06 containing engineered gelEsprE deletion (Thomas et al., 2008)
pKS103
pLT06 containing engineered lytSR deletion This study
pKS104 pLT06 containing engineered lrgAB deletion
This study

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
105
Cha
pter
VI
pSAVE37 pGEM-T containing engineered EF1097 deletion This study
pSAVE38 pG+host9 containing engineered EF1097 deletion This study
Primers
EF1097_1 AAG ACA ACA CGGGATAACACTCG
This study
EF1097_2 GCTTAGCCCACATTGAACTGCTGTCATTAGTAATGCCATCGCC
This study
EF1097_3 GCAGTTCAATGTGGGCTAAGC This study
EF1097_4 CTGAGTTACGGTCCATCCTTCTTCC This study
LytP1
GAGAGAATTCGCTTGGGAACTTCATTGC
This study
LytP2
CTCTGGATCCGACCACACCGGCACCTCC
This study
LytP3
GAGAGGATCCGTTAGCCGTTCATACGTC
This study
LytP4
CTCTCTGCAGGGTACGGCAATCGCTGTTG
This study
LytUp
GTATCAACGGTATGAATACGG
This study
LytDown
AATGCAATTCGACCCAAGGC
This study
LrgP1 GAGAGAATTCGGAAAGACGACAGTGACTTC This study
LrgP2
CTCTGGATCCTTCCATTCTTCTTCGCTCCCT
This study
LrgP3
GAGAGGATCCGCAACGGTCATTGGTCTATAA
This study
LrgP4
CTCTCTGCAGGCCTGCGAATAACTGGTTGA
This study
LrgUp
CCATCAAGCATGCATTTGGC
This study
LrgDown
TGGTACCGCTTGTTTTGACG
This study
mgelE_2 AAC GGA TAA CAC AGG GG
(Gaspar et al., 2009)
gelE TCA TTC ATT GAC CAG
(Gaspar et al., 2009)
lrgA_fw GGGCTTGTTCATTTCCCC
This study
lrgA_rv AAGGCGCCCGTCCAACCAG
This study
lrgB TTCTATGCCAACTGCCACAC
This study
mlrgB AAGGTTTCTTCTTATTTACGCC
This study
gls24_f TGCGTGGTAGAATACGGCAAAG
This study
gls24_rv GTCCATATGTCGCATGTTGC This study
3.4 Mutant Construction
E. faecalis V583 mutants (MG01[V583ΔfsrBΔgelE]; MG02 [V583ΔfsrBΔsprE]; and
MG03[V583ΔfsrBΔgelEΔsprE] were constructed by introducing pVT01(ΔgelE),

Chapter IV
106
pVT02(ΔsprE), and pVT03(ΔgelEΔsprE), respectively into the VI13[V583ΔfsrB] strain
and selecting for protease gene deletions essentially as described by Thomas et al.
2009 (Thomas et al., 2009). These strains are still responsive to external GBAP, but
are not able to produce the QS molecule, as is the case of VI13[V583ΔfsrB] (Teixeira
et al., 2012). Construction of KS17 [V583ΔlytSR] and KS18 [V583ΔlrgAB] mutants
was done similarly to the method described by Thurlow et al. using the marker less
deletion vector pLT06 (Thurlow et al., 2009b). In brief, flanking regions of lytSR and
lrgAB were amplified from E. faecalis V583 chromosomal DNA by PCR with primers
LytP1, LytP2, LytP3, LytP4 and LrgP1, LrgP2, LrgP3, LrgP4 respectively (Table 1).
The flanking PCR fragments were ligated together following BamHI digestion and
reamplified by PCR using the external primers P1 and P4, for both the lytSR and
lrgAB deletion constructs. The resulting amplicons were digested with EcoRI and PstI
and cloned into similarly digested pLT06 to create pKS103 (ΔlytSR) and pKS104
(ΔlrgAB). The resulting plasmids were confirmed by restriction analysis and
sequenced. Plasmids were introduced into E. faecalis V583 by electroporation and
selection of the desired mutant was performed as described (Thurlow et al., 2009b).
To create KS19 [V583ΔfsrBΔlytSR], VI13 was transformed with pKS103 (ΔlytSR)
and selection for deletion of lytSR was performed as described (Thurlow et al.,
2009b). E. faecalis V583Δef1097 was constructed essentially as described by
Brinster et al. (2007) (Brinster et al., 2007) in strain VE14089 (Rigottier-Gois et al.,
2011). Briefly, flanking regions of EF1097 were amplified from chromosomal DNA of
V583 by PCR with primers EF1097_1, EF1097_2, EF1097_3 and EF1097_4
respectively (Table 1). The two cognate PCR fragments were fused by PCR using
the external primers EF1097_1 and EF1097_4 for EF1097, respectively, and the
resulting product was cloned into pGEM-T (Promega). The inserted PCR fragment
was removed from its cloning vector by restriction enzymes and subsequently cloned
into pG+host9 plasmid (Maguin et al., 1996), which was then electroporated into E.
faecalis VE14089. The ef1097 single- and double crossover mutants were selected
as described by Brinster et al. (2007) (Brinster et al., 2007; Maguin et al., 1996).
Successful targeted mutations of ef1097 were first identified by PCR screening and

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
107
Cha
pter
VI
were confirmed by sequencing (StabVida, Portugal), and analysed by Vector NTI
program (Invitrogen).
3.5 RNA Extraction and cDNA Synthesis for Microarrays
E. faecalis strains were grown in BHI, at 37ºC, until 0.4 OD (600 nm). At this point,
purified GBAP, prepared as previously described (Nakayama et al., 2001b), was
added to a final concentration of 10 nM in the culture. This concentration was
previously shown to be able to induce the Fsr system (Nakayama et al., 2001a;
Nakayama et al., 2001b). In order to determine the effect of GBAP induction at a time
in growth when we knew, from previous work (Nakayama et al., 2001b), that the Fsr
system was not yet fully activated, we chose 0.4 OD to add GBAP. The quorum-
sensing molecule was added to induce the Fsr quorum-sensing system in strains
which lack the ability to produce the GBAP molecule, but are still able to sense it. At
time zero (immediately after GBAP addition) and after 10 min post-GBAP addition,
RNA was extracted from cells and used to synthesize cDNA and perform microarray
transcriptional analysis. Experiments without GBAP were also performed. To prepare
samples for Affymetrix GeneChip analysis, a previously published protocol was used
with few modifications (Schuster et al., 2003). Briefly, RNA was stabilized with RNA
protect (Qiagen) and RNA was isolated with RNeasy columns per the manufacturer’s
instructions (Qiagen). Samples were treated with RNase-free DNase I (Roche) to
remove contaminating DNA, and the absence of contaminating DNA was confirmed
by PCR. RNA integrity was verified using agarose gel electrophoresis of glyoxylated
samples (Ambion). cDNA was prepared from RNA using Superscript II Reverse
Transcriptase (Invitrogen) with random (N6) priming. cDNA was fragmented with
dilute DNase I (Roche) and fragments were biotinylated with the BioArray Terminal
Labeling Kit (Enzo Life Sciences) prior to hybridization. Affymetrix GeneChip
Analysis Samples were hybridized to a previously described custom E. faecalis
Affymetrix GeneChip (McBride et al., 2007) and scanned at the University of Iowa
DNA Core Facility. All microarray experiments were performed in duplicate. Data was

Chapter IV
108
analysed using Affymetrix GeneChip Operating Software, which identifies probe sets
with statistically significant hybridization over background (i.e. presence versus
absence calls) and among those, identifies probe sets for which hybridization is
significantly increased or decreased in pairwise comparisons of microarray
experiments. Signal log ratios for differentially expressed probe sets were averaged
and converted to fold change values. Only genes with ≥3-fold differential expression
were considered. The data discussed in this publication have been deposited in
NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through
GEO Series accession number GSE42036 (http:
//www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc =GSE42036).
3.6 Semiquantitative RT-PCR
RNA was extracted from strains V583ΔlytRS and V583ΔfsrB grown in BHI broth at
37ºC. Briefly, overnight cultured cells were diluted 1:100 and growth was monitored
by following OD600. Cells were collected in the same conditions as those used for
RNA extraction for microarrays. Total RNA was extracted and purified with an
RNeasy Mini kit (Qiagen). RNA integrity was checked by electrophoresis on a 1%
agarose gel (RNase free). cDNA was synthesized using random primers (Roche
Diagnostics), 3 mg total RNA and a Transcriptor High Fidelity cDNA Synthesis kit
(Roche Diagnostics). Serial dilutions of V583ΔlytRS and V583ΔfsrB cDNA were used
for PCR in order to amplify cDNA of lrgA (primers: lrgA, mlrgA), lrgB (primers: lrgB,
mlrgB) and gelE (primers: mgel_2, gelE) (Table 1).
3.7 D. melanogaster Infection
Oregon R male flies were injected with 50 nl of bacteria at 0.02 OD (600 nm) from
one of the strains: V583, V583ΔfsrBΔgelE, V583ΔfsrBΔsprE,
V583ΔfsrBΔgelEΔsprE, V583ΔlytRS, V583ΔlrgAB, VE14089 and VE14089Δef1097.
As control, flies were injected with the same volume of BHI medium. Male flies were
anesthetized with CO2 and the injections were carried out with a pulled glass

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
109
Cha
pter
VI
capillary needle using a nano-injector (Nanoliter 2000, World Precision Instruments).
Reproducibility was measured by determining the number of bacteria injected at time
zero. Injected flies were placed at 29ºC, 65% humidity. Seventy-five flies were
assayed for each survival curve, and they were placed in three vials of 25 flies each.
Each experiment was repeated three times, making a total of 225 flies tested per
strain in each set of three replicates, to ensure high confidence results. Death was
recorded at 0, 4, 6, 8, 10, 12, 14 and 24 h hours post-injection. All experiments were
performed at least three times. Following challenge with bacteria, six individual flies
were collected (at 0 h, 4 h, 8 h, 12 h and 24 h), homogenized, diluted serially, and
plated onto Enterococcel agar (Quilaban). E. faecalis CFUs (colony forming units)
were determined by testing three groups of six flies for each time point.
3.8 Percentage of Similarities between V583 Genome and Other Genomes
Published
The percentage of similarities was made with blast program (http:
//blast.ncbi.nlm.nih.gov/). The genomes that were used on this analysis were from Broad
Institute page (http: //www.broadinstitute.org/annotation/genome/enterococcus_faecalis/Multi
Home.html) and compared with V583 genome (http://www.ncbi.
nlm.nih.gov/nuccore/NC_004668.1).
3.9 Statistical Analysis
Statistical analysis of Drosophila survival was performed using GraphPad Prism
software version 5.03. Survival curves were compared using Log-rank and Gehan-
Breslow-Wilcoxon tests. Statistical analysis of Drosophila survival was performed
using t-test.

Chapter IV
110
4. RESULTS
In order to precisely identify genes for which expression is altered when GBAP
reaches effective quorum sensing concentration, we used a fsrB mutant, which is
unable to produce GBAP, but is able to sense it (Teixeira et al., 2012). We also used
single and double protease mutants in the fsrB mutant background in order to identify
any genes for which expression is indirectly controlled by Fsr through its regulation of
protease levels. Table 2 shows key changes in gene expression in V583ΔfsrB,
V583ΔfsrBΔgelE, V583ΔfsrBΔsprE and V583ΔfsrBΔgelEΔsprE after 10 min of GBAP
exposure. Besides genes previously known, or predicted, to be regulated by Fsr
through GBAP (gelE, sprE and ef1097) (Bourgogne et al., 2006; Qin et al., 2000; Qin
et al., 2001), 15 additional genes were differentially regulated by GBAP addition
collectively in all four mutants (Table 2). In contrast to previous results using oligo-
array study (Bourgogne et al., 2006), the current approach employed a statistically
more robust technology (Woo et al., 2004) and isolated the effects of only Fsr
quorum sensing through the use of mutants and the exogenous quorum molecule.
4.1 Fsr dependent genes
As expected, V583ΔfsrB responded to GBAP by substantially increasing the
expression of gelE (ef1818) (fold change 63) and sprE (ef1817) (fold change 59). To
a lesser extent, fsrC (ef1820) (fold change 3) transcript abundance was also
increased. As shown in Table 2, mutation of each protease gene did not affect the
expression of the other genes in the fsr or gelE-sprE operons, showing that the
presence of the deletions in these operons did not have polar effects on transcript
abundance of the remaining protease gene (V583ΔfsrBΔgelE expresses wild type
levels of sprE, and V583ΔfsrBΔsprE expresses wild type levels of gelE). In
accordance to previous results by others (Bourgogne et al., 2006), fsrA expression
was not affected by GBAP.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
111
Cha
pter
VI
Table 1- Genes differentially expressed upon addition of GBAP to V583ΔfsrB, V583ΔfsrBΔgelE,
V583ΔfsrBΔsprE and V583ΔfsrBΔgelEΔsprE strains. Fold-change values were obtained by comparing
gene expression at 10 min against 0 min post-GBAP addition, by microarray analysis.
Locus Putative function Fold Change1
V583ΔfsrB V583ΔfsrBΔgelE V583ΔfsrBΔsprE V583ΔfsrBΔgelEΔsprE
EF04113 PTS system mannitol-specific IIBC - - - - 3
EF04684 LemA family protein - + 3 - -
EF05635 Hypothetical protein - - - + 3
EF07766 Hypothetical protein - + 11 - - EF08917 Aspartate aminotransferase
putative - - - - 4
EF0892 Aminoacid ABC transporter ,ATP-binding protein
- - - - 3
EF0893 Aminoacid ABC transporter/ permease
- 3 - 3 - 3
EF1097 Putative Bacteriocin + 31 + 23 + 30 + 47
EF12188 spermidine/putrescine ABC transporter,permease
- - - - 3
EF1351 Hypothetical protein - + 6 + 8 + 4 EF1352 Magnesium-translocating, P-type
ATPase + 5 + 7 + 5 + 3
EF18159 Transcriptional regulator, LysR family putative
- - + 12 + 11
EF1816 Hypothetical protein , with domain β-lactamase
- - + 4
+ 3
EF1817 Serine protease – SprE + 60 + 90 - -
EF1818 Gelatinase – GelE + 63 - + 42 -
EF1820 Histidine Kinase – FsrC + 3 + 4 + 3 + 4 EF31932 Antiholin-like protein LrgB + 34 - - - EF31942 Murein hydrolase regulator LrgA + 79 - - -
1 (+) up-regulated (-) down-regulated; 2 These two genes were up-regulated in the experiments done without GBAP, only in
the V583ΔfsrB strain with a fold change of + 7 for E3193 and + 6 for EF3194; 3 ef0411 is part of the predicted operon
ef0411-0412-0413, which encodes a mannitol specific PTS-system; 4 LemA-like protein likely involved in cell wall
metabolism. LemA proteins contain a predicted amino terminal transmembrane helix and a short extracellular amino
terminus. The exact molecular function of this protein is uncertain; 5 Has two predicted transmembrane helixes and a Blast
search does not reveal similarity to proteins of known function. Upstream is a putative operon encoding the potassium-
transporting ATPase KdpABC (EF0567-EF0569) and the two-component system KdpED (EF0570-EF0571) (TCS12)
(Hancock & Perego, 2002); 6 It has a predicted transmembrane domain at its N-terminus (residues 4 to 20) and the rest of
the protein is located outside the cell. It has a predicted thioredoxin fold domain similar to bacteriocin accessory proteins
((http://www.genome.jp/dbget-bin/www_bget?efa:EF0776); 7 Predicted to facilitate the conversion of aspartate and alpha-
ketoglutarate to oxaloacetate and glutamate; 8 Part of the predicted operon ef1218-ef1224, which codes for a
spermidine/putrescine ABC transporter; 9 EF1815 has 25% amino acid sequence similarity to CidR from S. aureus
(http://blast.ncbi.nlm.nih.gov/); EF1816 is a hypothetical protein with a -lactamase domain, has no transmembrane domain,
and is orthologous to PhnP, which is involved in phosphonate metabolism. EF1815 and EF1816 are located upstream of
SprE (EF1817), but only EF1816 is located in the positive DNA strand.

Chapter IV
112
Genes for which expression was affected by GBAP in all the four mutants are
therefore under the direct control of FsrA and not influenced by indirect activities of
the proteases on secondary regulators. In addition to Fsr and protease genes,
ef1097 was induced by GBAP addition showing transcript abundance changes (fold
change 31) similar to those observed for the protease genes. Transcripts of the
ef1352 gene where more abundant upon GBAP induction, but exhibited an increase
of a lower magnitude (fold change 5).
To determine whether a specific promoter motif could be identified upstream of
genes found to be regulated by Fsr through its quorum sensing, we compared known
(Del Papa & Perego, 2011) and putative promoter regions. The V583 promotor
regions of ef1097, gelE and fsrB possess a predicted FsrA binding motif (Del Papa &
Perego, 2011). However, this motif does not occur upstream of ef1351. This raises
the possibility that induction of ef1351-ef1352 in our experiments may be related to
increased expression of the only gene which was also induced in the four mutants,
but not independently controlled, ef1097. Alternatively, direct FsrA regulation
mechanisms may be more complex than previously suspected.
4.2 Genes dependent on simultaneous Fsr and Proteases activation
Some genes were found to be affected by the presence or absence of proteases,
indicating an indirect regulatory pathway. Those only affected if sprE was absent
(ef1815, ef1816); those affected only if either one of the proteases was absent
(ef0893); those for which mRNA levels were altered only when both proteases were
absent (ef0411, ef0563, ef0891, ef0892, ef1218); those for which mRNA
accumulated only in the presence of both proteases (ef3193 and ef3194) and those
affected in the absence of only the gelE gene (ef0468, ef0776). These last two genes
might respond to the high expression levels of sprE in a way yet to be determined.
Overall, the twelve genes affected by the combined activation of Fsr and the
proteases are putatively involved in different cellular processes, such as regulation,
cell-wall metabolism and transport, and some are even of unknown function.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
113
Cha
pter
VI
Currently available data does not allow us to further clarify the connection between
these genes and the Fsr-GelE-SprE system.
4.3 LytRS system is required for GBAP induction of lrgAB genes
EF3193-EF3194 correspond to the lrgAB genes which, in S. aureus, are described to
be involved in repression of murein hydrolase activity, decreased autolysis and
increased tolerance to penicillin (Groicher et al., 2000). In S. aureus these genes are
regulated by the LytRS two-component regulatory system, located immediately
upstream of the lrgAB genes (Sharma-Kuinkel et al., 2009). There is no data about
the function of lrgAB genes in E. faecalis but it is known that they are also located
downstream of lytRS homologs, which suggests that in V583 lrgAB are regulated by
LytRS. In our experiments, ef3193-3194 mRNA was more abundant upon GBAP
induction only in the fsrB mutant, suggesting that these genes are not responding
directly to FsrA activation, but probably to increased protease GelE and SprE
expression, which only occurs when GBAP is added to the fsrB mutant. In order to
test the hypothesis that the large increase in lrgAB abundance was the result of
GBAP induction via the LytRS system, we deleted this two-component system from
the fsrB mutant strain and compared the expression of lrgAB genes in the
ΔfsrBΔlytRS and fsrB mutants (Figure 1). We found that GBAP is only able to induce
lrgAB genes if LytRS is functional. These results were not observed in previous
studies of fsr regulation in OG1RF (Bourgogne et al., 2006). None of the E. faecalis
∆lytRS or ∆lrgAB mutant strains showed different antibiotic resistance profiles (Table
S1) nor gelatinase activities when compared to the wild-type strain (data not shown).
Low level expression of lrgAB genes was observed in the ΔfsrBΔlytRS mutant
(Figure S1), which points either to a low constitutive expression of those genes or to
the existence of another regulator(s) able to modulate their expression.

Chapter IV
114
4.4 Fsr and the proteases affect D. melanogaster tolerance to E. faecalis
infection
To test the functional importance of genes found to be directly and indirectly
dependent on Fsr, we then tested the virulence of the fsr-related mutants in a D.
melanogaster injection model. We first compared the ability of the triple mutant
V583ΔfsrBΔgelEΔsprE, to the single V583ΔfsrB mutant, and the V583 parental
strain, to kill Drosophila. The fate of both the host (percentage of survival) and the
bacteria (number of CFU) was followed for 24 h. In our assay, 50% of the flies were
killed by the wild type strain 10 hours post-injection and after 14 h nearly all flies
were dead (Figure 2A). For the same period of infection, the triple mutant
V583ΔfsrBΔgelEΔsprE strain only killed 15% of the infected flies. 24 h post-injection,
the triple mutant V583ΔfsrBΔgelEΔsprE was significantly attenuated (see Table S2
for detailed statistical analysis). These results show that the Fsr system and the
proteases it regulates contribute measurably to toxicity in this model.
Figure 1 - LytRS is required for GBAP induction of lrgAB genes. The semi-quantitative RT-PCR shows
expression of lrgAB genes in the VI13 (∆fsrB mutant) and KS19 (∆fsrB∆lytRS mutant), in the presence of GBAP.
Expression of gelE and gdh were used as positive and negative controls, respectively, of Fsr induction by GBAP and
of RNA concentration, respectively. The RNA used for this analysis was previously treated with RNase-free DNase I
to remove contaminating DNA.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
115
Cha
pter
VI
The survival curve of flies infected with the wild type strain shows two different killing
rates: until 8h, V583 strain is able to kill around 3 flies/ hour; after this time, and until
12h, V583 kills flies at a much higher rate, 15 flies/ hour. At 8h post infection, V583
cells reach the cell density considered to be able to induce the activation of the Fsr
system in broth culture (Nakayama et al., 2001a; Nakayama et al., 2001b). Although
there is no data on the in vivo Fsr expression during E. faecalis growth inside the
host, we cannot exclude the possibility that the increased killing rate after 8h is due
to induced expression of the proteases.
In order to dissect the contribution of fsr-regulated genes to the lethality of infection,
we tested these genes separately by infecting the flies with single deletion mutants
(Figure 2B). Deletion of both proteases, either in the double protease mutant or in the
triple mutant, led to a greater attenuation of virulence then deletion of fsrB
(p0.0001, Table S2). Consistent with previous demonstrations that in an fsrB
mutant strain, proteases are still expressed (Singh et al., 2005), we observed an
attenuation of the virulence in the triple mutant over that of the fsrB mutant,
suggesting that low level expression of both proteases is enough to induce increased
killing of the flies by the fsrB mutant. Absence of gelE alone produced the lowest
attenuation of E. faecalis virulence, differing significantly (pable S2) from
the effect of the absence of sprE gene alone, which was attenuated to a similar level
achieved by deletion of fsrB (Table S2). This result points to SprE as having a major
role in E. faecalis virulence in the Drosophila model. All strains grew similarly inside
Drosophila (Figure S2).
4.5 ef1097 contributes to toxicity in D. melanogaster infection
The large increase in ef1097 mRNA abundance upon GABP addition, and the fact
that it has been previously associated with Fsr system in another E. faecalis strain
(Bourgogne et al., 2006), led us to delete this gene to test its role in E. faecalis
virulence. This mutant was constructed in VE14089, a plasmid cured derivative strain
of V583, previously reported in G. mellonella to be less virulent than parental V583

Chapter IV
116
strain (Rigottier-Gois et al., 2011). Our results confirm that strain VE14089 is less
virulent than V583 in the D. melanogaster model as well (compare control in Figure
2A and 2C). Previously, we compared the toxicity of V583ΔfsrBΔgelEΔsprE and
V583ΔgelEΔsprE strains in the fly (Figure 2A and 2B). Both strains express ef1097,
and therefore, the role of this protein was not assessed. Figure 2C clearly shows that
deletion of ef1097 reduces killing of the flies by E. faecalis, therefore providing
evidence for a role of this bacteriocin in E. faecalis toxicity in the fly. As deletion of
ef1097 did not affect the gelatinase production ability of V583 strain (results not
shown), the reduction of toxicity does not appear to be due to an effect on expression
of fsr or the proteases it regulates.
4.6 LrgAB and LytRS contribute differently to death of D. melanogaster
LytRS appears to induce lrgAB expression upon addition of GBAP to the fsrB mutant
strain (Figure 1). Interestingly, lytRS was previously found to be strongly induced
during infection of G. mellonella, and proposed to contribute to E. faecalis VE14089
virulence in the same model (Hanin et al., 2010). The importance of LytRS was
therefore tested in Drosophila infection. Our results (Figure 2D) did not show a
significant difference in the fly survival (Table S2) following infection with the lytRS
mutant as compared to wild type. Our results cannot be compared to those of Hanin
et al. (Hanin et al., 2010) as both the strains and the infection protocols used were
different.
lgrAB are still expressed in the lytRS mutant. We thus wondered if complete
abolishment of its expression would have a more pronounced effect on D.
melanogaster toxicity than that of its regulator LytRS. The lrgAB mutant strain was
significantly reduced in toxicity for D. melanogaster (Figure 2D, Table S2). This result
highlights the relevance of the lrgAB operon in infection by E. faecalis and constitutes
the first report on such a role for this operon in this species.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
117
Cha
pter
VI
Figure 2 - Drosophila survival
rates upon infection with E.
faecalis strains. 75 Oregon R (5-
to 7-day-old) male adult flies,
raised at 25°C, were divided in
tubes of 25 flies each, and
infected, by septic injury onto the
thorax with a thin needle, with
V583 (A, B, D) and VE14089
derived strains (C). Data is
representative of three
independent experiments (225 flies
per strain). Curves assigned with
an are significantly different (p<
0.0001) from the respective wild-
type -infected curve, as
determined by log-rank analysis
(Table S2).

Chapter IV
118
5. DISCUSSION
Assessing the basis for virulence of an opportunistic pathogen, such as E. faecalis, is
difficult because it is invariably subtle and multifactorial. Research on this topic in
recent years has concluded that the sole presence of a gene predicted to induce
virulence in a strain does not necessarily imply that the same gene may lead to the
same host fate in a different E. faecalis strains (Gaspar et al., 2009; Gaspar et al.,
2012). Besides the genome background and the host, the manner in which the
microbe is introduced also play a roles in determining whether or not a factor
contributes to toxicity. D. melanogaster has been used as a model host to study
pathogenesis because it provides easy handling, fast results, a fully sequenced
genome, pre-existing libraries of genetic mutants, the possibility to play on the host
side and similarities with the mammal immune system. In this work, we show that it
can be used to discern varying levels of toxicity stemming from mutations in the fsr
quorum regulatory system and the genes that it regulates.
In a representative of the hospital endemic lineage CC2, V583, the Fsr regulon is
largely restricted to the five genes, namely gelE, sprE, ef1097, ef1351 and ef1352
found to be directly dependent on GBAP-induced Fsr activation, and twelve
additional genes found to be dependent on GBAP induction of the proteases. Among
these are genes coding for proteins involved in cell-wall, transport and regulatory
functions. These genes are thus candidates to link the Fsr-proteases activity with the
phenotypes known to be associated to their impairment, namely biofilm formation,
adhesion and translocation to/in host-cells, autolysis and host damage and death.
This contrasts with previous findings in the more commensal background, OG1RF,
which was tested using an X-mer based oligonucleotide array with fewer controls and
less redundancy than the Affymetrix microarrays used here. Our experiment assayed
the first ten minutes after a burst of GBAP aiming to get clear, measurable and
immediate changes in expression, whereas the study by Bourgogne et al (Bourgogne
et al., 2006) followed the changes in expression of an fsrB mutant spanning different
growth stages. Their experimental design likely allowed for further events of

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
119
Cha
pter
VI
differential expression to take place. Whether the differences in results stem from
differences in strains, or differences in techniques and experimental approaches
used, is not currently known.
In the present study, we found that induction of GelE and SprE by GBAP via the fsr
regulator resulted in accumulation of mRNA encoding lrgAB, and that this induction
was lytRS dependent, indicating a functional relationship between Fsr and LytRS
regulons. In S. aureus, autolysis is positively regulated by Agr, a paralog of Fsr, that
positively regulates LrgAB (Fujimoto et al., 2000). Unlike S. aureus, in E. faecalis
FsrA does not regulate lrgAB genes directly, but does so indirectly. Both GelE and
SprE have previously been shown to play a role in autolysis regulation in E. faecalis,
respectively promoting and repressing it (Thomas et al., 2008). GelE is known to
proteolytically activate AtlA (Thomas et al., 2009), a major autolysin. Recently, GelE
was also found to control the levels of SalB, a protein with no evident peptidoglycan
hydrolytic activity, but affecting the levels of proteins involved in cell-wall synthesis
and cell division (Shankar et al., 2012). A salB mutant in OG1-RF strain showed
anomalous cell-division and increased autolysis (Shankar et al., 2012). Given the
current knowledge, we could speculate that autolysis regulation could constitute the
functional link, found in this study, between Fsr and LytRS. Future studies should
address the mechanism behind GelE-SprE regulation of autolytic activities in E.
faecalis and how they affect the expression of lrgAB operon through LytRS
regulation.
EF1097 protein, found by Bourgogne et al. 2006 (Bourgogne et al., 2006) to be
dependent on Fsr regulation in E. faecalis OG1RF, was here confirmed to be true
also for the V583 strain. In 2007, Swe et al. (Swe et al., 2007) suggested that ef1097
gene encodes a precursor of antimicrobial proteins with similarities to the
streptococcin SA-M57 in S. aureus. EF1097 is conserved in all E. faecalis strains
(Supplementary Table S3). Finding this bacteriocin to be similarly regulated in
distinct E. faecalis strains, namely OG1RF and V583, suggests this is a common
feature in the species. QS-activated bacteriocin production may constitute a means
to kill surrounding and competing bacteria thus providing competitive advantage to E.

Chapter IV
120
faecalis when colonizing or infecting a host. The Fsr homologue in S. aureus, Agr, is
known to regulate the expression of pro-inflammatory peptides, the phenol-soluble
modulins (PSM), in a RNAIII independent way (Queck et al., 2008). Several roles in
pathogenesis have been attributed to these amphipathic peptides (Periasamy et al.,
2012a), including antimicrobial activity (Joo et al., 2011), biofilm formation,
maturation and detachment (Periasamy et al., 2012b), and cytolytic ability to
neutrophils and other human cells (Kretschmer et al., 2012). Although the role of
EF1097 is not as extensively studied as that of PSMs, their shared features, namely
quorum-sensing induction and role in virulence, should direct further studies on
EF1097 role in E. faecalis biology and interaction with the host.
Despite the inexistence of clues on the EF1097 mechanism of action, bacteriocins
have been shown to produce changes in membrane potential and affect transport of
magnesium and amino acids (Uratani & Hoshino, 1984). EF1352, which codes for a
putative magnesium-translocating P-type ATPase, was induced in all strains used in
the microarrays. However, this operon lacks the previously described FsrA binding
motif in its promoter region. It is thus licit to speculate that expression of this operon
may be dependent on expression of ef1097, as this is the only Fsr dependent gene
with the FsrA motif not deleted and tested in the microarrays assays. Further studies
are needed to understand the link between bacteriocin production and induction of
an MgtA transporter, although we could hypothesise that EF1097 could induce ion
leakage, which in turn, would induce MgtA.
Despite different mortality curves were produced upon infection of Drosophila with
the tested mutants, they all grew similarly inside the host. Hosts have two ways to
deal with an infection: resistance and tolerance (Hanin et al., 2010; Rigottier-Gois et
al., 2011). Resistance is related with pathogen load and with mechanisms used to kill
the pathogens: more resistant hosts have fewer pathogens. Tolerance is a
consequence of the host ability to overcome the fitness cost imposed upon infection
and induction of the immune system and is related to the ability of the host to remain
healthy. Tolerance can be defined and measured from the slope of the health-by
microbe curve. We plotted the fly´s survival against pathogen load, assuming host
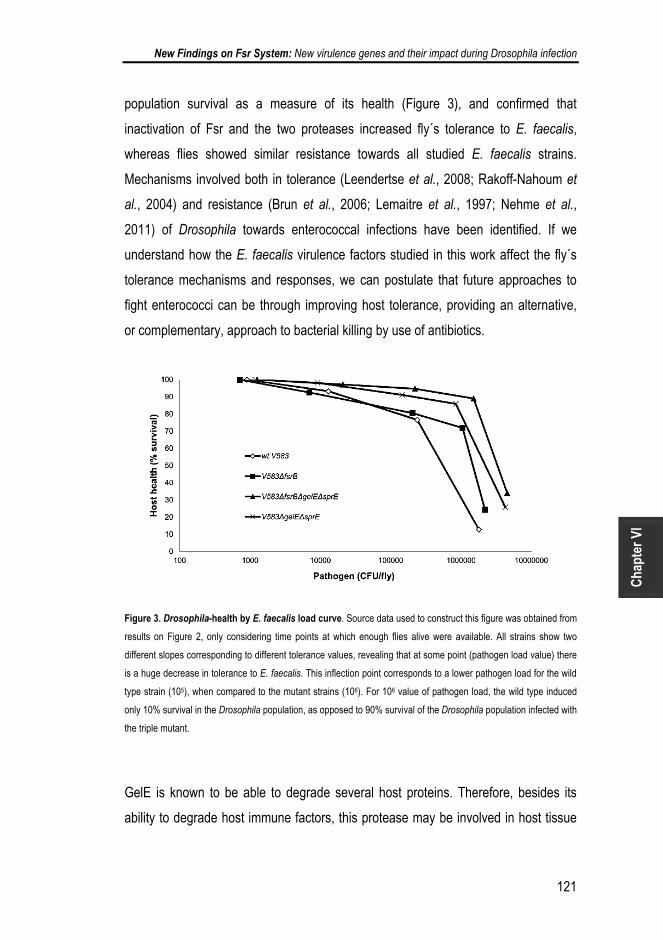
New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
121
Cha
pter
VI
population survival as a measure of its health (Figure 3), and confirmed that
inactivation of Fsr and the two proteases increased fly´s tolerance to E. faecalis,
whereas flies showed similar resistance towards all studied E. faecalis strains.
Mechanisms involved both in tolerance (Leendertse et al., 2008; Rakoff-Nahoum et
al., 2004) and resistance (Brun et al., 2006; Lemaitre et al., 1997; Nehme et al.,
2011) of Drosophila towards enterococcal infections have been identified. If we
understand how the E. faecalis virulence factors studied in this work affect the fly´s
tolerance mechanisms and responses, we can postulate that future approaches to
fight enterococci can be through improving host tolerance, providing an alternative,
or complementary, approach to bacterial killing by use of antibiotics.
Figure 3. Drosophila-health by E. faecalis load curve. Source data used to construct this figure was obtained from
results on Figure 2, only considering time points at which enough flies alive were available. All strains show two
different slopes corresponding to different tolerance values, revealing that at some point (pathogen load value) there
is a huge decrease in tolerance to E. faecalis. This inflection point corresponds to a lower pathogen load for the wild
type strain (105), when compared to the mutant strains (106). For 106 value of pathogen load, the wild type induced
only 10% survival in the Drosophila population, as opposed to 90% survival of the Drosophila population infected with
the triple mutant.
GelE is known to be able to degrade several host proteins. Therefore, besides its
ability to degrade host immune factors, this protease may be involved in host tissue

Chapter IV
122
injury. Recently, GelE has also been implicated in release of Ace protein from the
surface of E. faecalis cells in OG1RF strain (Pinkston et al., 2011). In that study,
authors showed that deletion of gelE gene increased the number of Ace proteins
bound to the surface of the bacterial cells, increasing adherence to collagen. In the
insect model G. mellonella, collagen adherence has been shown to be required for
invasion and virulence (Abranches et al., 2011). Although this remains to be proven
true for Drosophila, it is licit to speculate that the lower attenuation of the gelE mutant
in this insect host model could be due to increased adherence to host cells and
proteins. Despite considered to be cell-bound, SprE is also able to degrade host
proteins, such as insulin and fibrinogen, but not immune system elements, such as
complement from human serum or Cecropin from insect hemolymph (Park et al.,
2007). Its major contribution to host death proven in this work needs thus urgent
clarification.
This work brought to light new players (Figure 4) in Fsr role in E. faecalis, namely
LrgAB operon, which will help unravel the bacterial programmed cell death which, in
turn, may help discover new approaches to control this important nosocomial
pathogen. Moreover, Drosophila was successfully established as a model to study
virulence associated genes in E. faecalis, highlighting LrgAB and EF1097 as novel
virulence factors induced by QS. Using Drosophila as a model also allowed us to
show that SprE is, per se, a relevant player in host injury and to suggest that E.
faecalis success during septic injury is not due to GelE acting as a bacterial defence
against the flies AMPs, but that it could rather be through host injury.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
123
Cha
pter
VI
Figure 4 - GBAP-dependent regulatory network. Once the GBAP (black disks) concentration outside cells reaches
a certain threshold (upper part of the cell), the Fsr system is activated, and the FsrA regulator induces expression of
gelE, sprE and ef1097 genes. Both produce proteins which will be located to the cell membrane and cell wall.
Although GelE is loosely bound to the cell, it will also be released from it. The induced expression of ef1352, which
encodes a putative MgtA protein, by GBAP is likely due to increased amounts of EF1097, predicted to be a
bacteriocin. EF1352 could function as an auto-immunity factor against EF1097. The increased level of GelE and
SprE proteins in the cell-wall in response to GBAP are proposed to induce changes sensed by LytS protein, which in
turn, activates LytR, responsible for induction of lrgAB genes. When no GBAP is produced (lower part of the cell)
ef1097 is not expressed, but both GelE and SprE are still produced, although in lower amounts (dotted line). In this
situation, lrgAB genes are still expressed, but the increment in their expression during growth in the exponential
phase (assayed during microarrays performed without GBAP) is not due to the QS molecule. As we found that lrgAB
can still be expressed in a lytRS mutant, we propose that this is not the only regulator able to induce expression of
that operon.

Chapter IV
124
6. ACKNOWLEDGEMENTS
The authors are grateful to Isabel Marques, from IGC, for her help in enterococcal
genome comparison regarding genes directly regulated by Fsr.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
125
Cha
pter
VI
7. BIBLIOGRAPHY
Abranches, J., Miller, J. H., Martinez, A. R., Simpson-Haidaris, P. J., Burne, R.
A. & Lemos, J. A. (2011). The collagen-binding protein Cnm is required for
Streptococcus mutans adherence to and intracellular invasion of human coronary
artery endothelial cells. Infect Immun 79, 2277-2284.
Bourgogne, A., Hilsenbeck, S. G., Dunny, G. M. & Murray, B. E. (2006).
Comparison of OG1RF and an isogenic fsrB deletion mutant by transcriptional
analysis: the Fsr system of Enterococcus faecalis is more than the activator of
gelatinase and serine protease. J Bacteriol 188, 2875-2884.
Boyer, L., Paquette, N., Silverman, N. & Stuart, L. M. (2012). Bacterial effectors:
learning on the fly. Adv Exp Med Biol 710, 29-36.
Brinster, S., Furlan, S. & Serror, P. (2007). C-terminal WxL domain mediates cell
wall binding in Enterococcus faecalis and other gram-positive bacteria. J Bacteriol
189, 1244-1253.
Brun, S., Vidal, S., Spellman, P., Takahashi, K., Tricoire, H. & Lemaitre, B.
(2006). The MAPKKK Mekk1 regulates the expression of Turandot stress genes in
response to septic injury in Drosophila. Genes Cells 11, 397-407.
Cox, C. R. & Gilmore, M. S. (2007). Native microbial colonization of Drosophila
melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect
Immun 75, 1565-1576.
Del Papa, M. F. & Perego, M. (2011). Enterococcus faecalis virulence regulator
FsrA binding to target promoters. J Bacteriol 193, 1527-1532.

Chapter IV
126
Dower, W. J., Miller, J. F. & Ragsdale, C. W. (1988). High efficiency transformation
of E. coli by high voltage electroporation. Nucleic Acids Res 16, 6127-6145.
Dunny, G. M., Lee, L. N. & LeBlanc, D. J. (1991). Improved electroporation and
cloning vector system for gram-positive bacteria. Appl Environ Microbiol 57, 1194-
1201.
Edgar, R., Domrachev, M. & Lash, A. E. (2002). Gene Expression Omnibus: NCBI
gene expression and hybridization array data repository. Nucleic Acids Res 30, 207-
210.
Engelbert, M., Mylonakis, E., Ausubel, F. M., Calderwood, S. B. & Gilmore, M. S.
(2004). Contribution of gelatinase, serine protease, and fsr to the pathogenesis of
Enterococcus faecalis endophthalmitis. Infect Immun 72, 3628-3633.
Fujimoto, D. F., Brunskill, E. W. & Bayles, K. W. (2000). Analysis of genetic
elements controlling Staphylococcus aureus lrgAB expression: potential role of DNA
topology in SarA regulation. J Bacteriol 182, 4822-4828.
Garsin, D. A., Sifri, C. D., Mylonakis, E., Qin, X., Singh, K. V., Murray, B. E.,
Calderwood, S. B. & Ausubel, F. M. (2001). A simple model host for identifying
Gram-positive virulence factors. Proc Natl Acad Sci U S A 98, 10892-10897.
Gaspar, F., Teixeira, N., Rigottier-Gois, L., Marujo, P., Nielsen-LeRoux, C.,
Crespo, M. T., Lopes Mde, F. & Serror, P. (2009). Virulence of Enterococcus
faecalis dairy strains in an insect model: the role of fsrB and gelE. Microbiology 155,
3564-3571.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
127
Cha
pter
VI
Gaspar, F. B., Montero, N., Akary, E., Teixeira, N., Matos, R., Gonzalez-Zorn, B.,
Barreto Crespo, M. T., Serror, P. & Silva Lopes Mde, F. (2012). Incongruence
between the cps type 2 genotype and host-related phenotypes of an Enterococcus
faecalis food isolate. Int J Food Microbiol 158, 120-125.
Gilmore, M. S., Phillip S. Coburn, Sreedhar R. Nallapareddy, Barbara E. Murray
(2002). Enterococcal Virulence. In The Enterococci Pathogenesis, Molecular Biology,
and Antibiotic Resistance, pp. 301-354. Edited by M. S. Gilmore, Clewell DB,
Courvalin P, Dunny GM, Murray BE, Rice LB. Washington D.C.: American Society
for Microbiology.
Glavis-Bloom, J., Muhammed, M. & Mylonakis, E. (2012). Of model hosts and
man: using Caenorhabditis elegans, Drosophila melanogaster and Galleria
mellonella as model hosts for infectious disease research. Adv Exp Med Biol 710,
11-17.
Grant, S. G., Jessee, J., Bloom, F. R. & Hanahan, D. (1990). Differential plasmid
rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction
mutants. Proc Natl Acad Sci U S A 87, 4645-4649.
Groicher, K. H., Firek, B. A., Fujimoto, D. F. & Bayles, K. W. (2000). The
Staphylococcus aureus lrgAB operon modulates murein hydrolase activity and
penicillin tolerance. J Bacteriol 182, 1794-1801.
Hancock, L. & Perego, M. (2002). Two-component signal transduction in
Enterococcus faecalis. J Bacteriol 184, 5819-5825.
Hancock, L. E. & Perego, M. (2004). The Enterococcus faecalis fsr two-component
system controls biofilm development through production of gelatinase. J Bacteriol
186, 5629-5639.

Chapter IV
128
Hanin, A., Sava, I., Bao, Y., Huebner, J., Hartke, A., Auffray, Y. & Sauvageot, N.
(2010). Screening of in vivo activated genes in Enterococcus faecalis during insect
and mouse infections and growth in urine. PLoS One 5, e11879.
Jha, A. K., Bais, H. P. & Vivanco, J. M. (2005). Enterococcus faecalis mammalian
virulence-related factors exhibit potent pathogenicity in the Arabidopsis thaliana plant
model. Infect Immun 73, 464-475.
Joo, H. S., Cheung, G. Y. & Otto, M. (2011). Antimicrobial activity of community-
associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble
modulin derivatives. J Biol Chem 286, 8933-8940.
Kretschmer, D., Nikola, N., Durr, M., Otto, M. & Peschel, A. (2012). The virulence
regulator Agr controls the staphylococcal capacity to activate human neutrophils via
the formyl peptide receptor 2. J Innate Immun 4, 201-212.
Law, J., Buist, G., Haandrikman, A., Kok, J., Venema, G. & Leenhouts, K. (1995).
A system to generate chromosomal mutations in Lactococcus lactis which allows fast
analysis of targeted genes. J Bacteriol 177, 7011-7018.
Leendertse, M., Willems, R. J., Giebelen, I. A., van den Pangaart, P. S.,
Wiersinga, W. J., de Vos, A. F., Florquin, S., Bonten, M. J. & van der Poll, T.
(2008). TLR2-dependent MyD88 signaling contributes to early host defense in
murine Enterococcus faecium peritonitis. J Immunol 180, 4865-4874.
Lemaitre, B., Reichhart, J. M. & Hoffmann, J. A. (1997). Drosophila host defense:
differential induction of antimicrobial peptide genes after infection by various classes
of microorganisms. Proc Natl Acad Sci U S A 94, 14614-14619.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
129
Cha
pter
VI
Lopes Mde, F., Ribeiro, T., Martins, M. P., Tenreiro, R. & Crespo, M. T. (2003).
Gentamicin resistance in dairy and clinical enterococcal isolates and in reference
strains. J Antimicrob Chemother 52, 214-219.
Maguin, E., Prevost, H., Ehrlich, S. D. & Gruss, A. (1996). Efficient insertional
mutagenesis in lactococci and other gram-positive bacteria. J Bacteriol 178, 931-935.
McBride, S. M., Fischetti, V. A., Leblanc, D. J., Moellering, R. C., Jr. & Gilmore,
M. S. (2007). Genetic diversity among Enterococcus faecalis. PLoS ONE 2, e582.
Mylonakis, E., Engelbert, M., Qin, X., Sifri, C. D., Murray, B. E., Ausubel, F. M.,
Gilmore, M. S. & Calderwood, S. B. (2002). The Enterococcus faecalis fsrB gene, a
key component of the fsr quorum-sensing system, is associated with virulence in the
rabbit endophthalmitis model. Infect Immun 70, 4678-4681.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S., Akkermans, A. D., de Vos, W. M. &
Nagasawa, H. (2001a). Gelatinase biosynthesis-activating pheromone: a peptide
lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 41,
145-154.
Nakayama, J., Cao, Y., Horii, T., Sakuda, S. & Nagasawa, H. (2001b). Chemical
synthesis and biological activity of the gelatinase biosynthesis-activating pheromone
of Enterococcus faecalis and its analogs. Biosci Biotechnol Biochem 65, 2322-2325.
Nehme, N. T., Quintin, J., Cho, J. H., Lee, J., Lafarge, M. C., Kocks, C. &
Ferrandon, D. (2011). Relative roles of the cellular and humoral responses in the
Drosophila host defense against three gram-positive bacterial infections. PLoS One
6, e14743.

Chapter IV
130
Novick, R. P., Ross, H. F., Projan, S. J., Kornblum, J., Kreiswirth, B. &
Moghazeh, S. (1993). Synthesis of staphylococcal virulence factors is controlled by
a regulatory RNA molecule. Embo J 12, 3967-3975.
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Park, S. Y., Shin, Y. P., Kim, C. H., Park, H. J., Seong, Y. S., Kim, B. S., Seo, S. J.
& Lee, I. H. (2008). Immune evasion of Enterococcus faecalis by an extracellular
gelatinase that cleaves C3 and iC3b. J Immunol 181, 6328-6336.
Periasamy, S., Chatterjee, S. S., Cheung, G. Y. & Otto, M. (2012a). Phenol-soluble
modulins in staphylococci: What are they originally for? Commun Integr Biol 5, 275-
277.
Periasamy, S., Joo, H. S., Duong, A. C., Bach, T. H., Tan, V. Y., Chatterjee, S. S.,
Cheung, G. Y. & Otto, M. (2012b). How Staphylococcus aureus biofilms develop
their characteristic structure. Proc Natl Acad Sci U S A 109, 1281-1286.
Pinkston, K. L., Gao, P., Diaz-Garcia, D., Sillanpaa, J., Nallapareddy, S. R.,
Murray, B. E. & Harvey, B. R. (2011). The Fsr quorum-sensing system of
Enterococcus faecalis modulates surface display of the collagen-binding MSCRAMM
Ace through regulation of gelE. J Bacteriol 193, 4317-4325.
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2000). Effects of
Enterococcus faecalis fsr genes on production of gelatinase and a serine protease
and virulence. Infect Immun 68, 2579-2586.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
131
Cha
pter
VI
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2001). Characterization of
fsr, a regulator controlling expression of gelatinase and serine protease in
Enterococcus faecalis OG1RF. J Bacteriol 183, 3372-3382.
Queck, S. Y., Jameson-Lee, M., Villaruz, A. E., Bach, T. H., Khan, B. A.,
Sturdevant, D. E., Ricklefs, S. M., Li, M. & Otto, M. (2008). RNAIII-independent
target gene control by the agr quorum-sensing system: insight into the evolution of
virulence regulation in Staphylococcus aureus. Mol Cell 32, 150-158.
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov,
R. (2004). Recognition of commensal microflora by toll-like receptors is required for
intestinal homeostasis. Cell 118, 229-241.
Rigottier-Gois, L., Alberti, A., Houel, A. & other authors (2011). Large-scale
screening of a targeted Enterococcus faecalis mutant library identifies envelope
fitness factors. PLoS One 6, e29023.
Sahm, D. F., Kissinger, J., Gilmore, M. S., Murray, P. R., Mulder, R., Solliday, J.
& Clarke, B. (1989). In vitro susceptibility studies of vancomycin-resistant
Enterococcus faecalis. Antimicrob Agents Chemother 33, 1588-1591.
Schneider, D. S., Ayres, J. S., Brandt, S. M. & other authors (2007). Drosophila
eiger mutants are sensitive to extracellular pathogens. PLoS Pathog 3, e41.
Schuster, M., Lostroh, C. P., Ogi, T. & Greenberg, E. P. (2003). Identification,
timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes:
a transcriptome analysis. J Bacteriol 185, 2066-2079.

Chapter IV
132
Shankar, J., Walker, R. G., Wilkinson, M. C., Ward, D. & Horsburgh, M. J. (2012).
SalB inactivation modulates culture supernatant exoproteins and affects autolysis
and viability in Enterococcus faecalis OG1RF. J Bacteriol 194, 3569-3578.
Sharma-Kuinkel, B. K., Mann, E. E., Ahn, J. S., Kuechenmeister, L. J., Dunman,
P. M. & Bayles, K. W. (2009). The Staphylococcus aureus LytSR two-component
regulatory system affects biofilm formation. J Bacteriol 191, 4767-4775.
Sifri, C. D., Mylonakis, E., Singh, K. V., Qin, X., Garsin, D. A., Murray, B. E.,
Ausubel, F. M. & Calderwood, S. B. (2002). Virulence effect of Enterococcus
faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans
and mice. Infect Immun 70, 5647-5650.
Singh, K. V., Nallapareddy, S. R., Nannini, E. C. & Murray, B. E. (2005). Fsr-
independent production of protease(s) may explain the lack of attenuation of an
Enterococcus faecalis fsr mutant versus a gelE-sprE mutant in induction of
endocarditis. Infect Immun 73, 4888-4894.
Singh, K. V., Nallapareddy, S. R., Sillanpaa, J. & Murray, B. E. (2010). Importance
of the collagen adhesin ace in pathogenesis and protection against Enterococcus
faecalis experimental endocarditis. PLoS Pathog 6, e1000716.
Swe, P. M., Heng, N. C., Ting, Y. T., Baird, H. J., Carne, A., Tauch, A., Tagg, J. R.
& Jack, R. W. (2007). ef1097 and ypkK encode enterococcin V583 and corynicin JK,
members of a new family of antimicrobial proteins (bacteriocins) with modular
structure from Gram-positive bacteria. Microbiology 153, 3218-3227.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
133
Cha
pter
VI
Teixeira, N., Santos, S., Marujo, P., Yokohata, R., Iyer, V. S., Nakayama, J.,
Hancock, L. E., Serror, P. & Silva Lopes Mde, F. (2012). The incongruent
gelatinase genotype and phenotype in Enterococcus faecalis are due to shutting off
the ability to respond to the gelatinase biosynthesis-activating pheromone (GBAP)
quorum-sensing signal. Microbiology 158, 519-528.
Thomas, V. C., Thurlow, L. R., Boyle, D. & Hancock, L. E. (2008). Regulation of
autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular
proteases influences biofilm development. J Bacteriol 190, 5690-5698.
Thomas, V. C., Hiromasa, Y., Harms, N., Thurlow, L., Tomich, J. & Hancock, L.
E. (2009). A fratricidal mechanism is responsible for eDNA release and contributes to
biofilm development of Enterococcus faecalis. Mol Microbiol 72, 1022-1036.
Thurlow, L. R., Thomas, V. C., Fleming, S. D. & Hancock, L. E. (2009a).
Enterococcus faecalis capsular polysaccharide serotypes C and D and their
contributions to host innate immune evasion. Infect Immun 77, 5551-5557.
Thurlow, L. R., Thomas, V. C. & Hancock, L. E. (2009b). Capsular polysaccharide
production in Enterococcus faecalis and contribution of CpsF to capsule
serospecificity. J Bacteriol 191, 6203-6210.
Thurlow, L. R., Thomas, V. C., Narayanan, S., Olson, S., Fleming, S. D. &
Hancock, L. E. (2010). Gelatinase contributes to the pathogenesis of endocarditis
caused by Enterococcus faecalis. Infect Immun 78, 4936-4943.
Uratani, Y. & Hoshino, T. (1984). Pyocin R1 inhibits active transport in
Pseudomonas aeruginosa and depolarizes membrane potential. J Bacteriol 157,
632-636.

Chapter IV
134
Willems, R. J., Hanage, W. P., Bessen, D. E. & Feil, E. J. (2011). Population
biology of Gram-positive pathogens: high-risk clones for dissemination of antibiotic
resistance. FEMS Microbiol Rev 35, 872-900.
Woo, Y., Affourtit, J., Daigle, S., Viale, A., Johnson, K., Naggert, J. & Churchill,
G. (2004). A comparison of cDNA, oligonucleotide, and Affymetrix GeneChip gene
expression microarray platforms. J Biomol Tech 15, 276-284.
Zeng, J., Teng, F. & Murray, B. E. (2005). Gelatinase is important for translocation
of Enterococcus faecalis across polarized human enterocyte-like T84 cells. Infect
Immun 73, 1606-1612.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
135
Cha
pter
VI
8. SUPPLEMENTARY DATA
Figure S1. lrgAB expression in the absence of GBAP. The semi-quantitative RT-PCR shows expression of lrgAB
genes in the VI13 (∆fsrB mutant) and KS19 (∆fsrB∆lytRS mutant) strains, in the absence of GBAP. Expression of
gelE and gdh were used as negative and positive controls, respectively. The RNA used for this analysis was
previously treated with RNase-free DNase I to remove contaminating DNA and PCR was done in order to confirm
absence of DNA from the RNA samples analysed.

Chapter IV
136
Figure S2. E. faecalis growth curves in injected flies. Oregon R (5- to 7-day-old) male adult flies, raised at 25°C,
were divided in tubes of 25 flies each, and infected, by septic injury onto the thorax with a thin needle, with V583
mutants. Flies were collected at 0, 4, 8, 12, and 24 h. Three groups of six flies for each time point were homogenized
and plated in Enterococcel agar and E. faecalis CFUs were determined.

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
137
Cha
pter
VI
Table S1 – Antibiotic resistance profiles of E. faecalis V583 derivative mutants. Values correspond to diameter
of halos (µm) around antibiotic disks.
Strain Ciprofloxacin Penicillin Sulphamethoxazole Vancomycin Nitrofuratoin Ofloxacin Ampicilin Ceftriaxone
V583 24,03 22,02 30,09 10,06 22,09 20,08 28,05 15,02
V583∆lrgAB 24,01 21,08 29,06 10,06 23,03 20,08 28,09 16,04
V583∆lytRS 23.03 21,03 30,08 10,02 21,07 20,08 27,04 11,09
V583∆fsrB 24,09 21,03 30,00 10,04 20,06 20,01 28,04 15,0
Table S2 – Statistical analysis of figure 2 using Kaplan-Myer curves. Experimental and control populations are
compared using Log-Rank and Wilcoxon tests (ChiSquare and p-values). Analysis was performed using GraphPad
Prism statistical software.
Strain 1
Strain 2
ChiSquare Log Rank Wilcoxon
p-value Log Rank Wilcoxon
V583wt
V583∆fsrB
112,5
83,09
<0,0001
<0,0001
V583∆fsrB∆gelE∆sprE
272,0 237,9 <0,0001 <0,0001
V583∆gelE 41.32 25.85 <0,0001 <0,0001 V583∆sprE 162.9 136.1 <0,0001 <0,0001 V583∆gelE∆sprE 286.4 252.0 <0,0001 <0,0001
V583∆lytRS V583∆lrgAB
0.389 120.1
0.013 107.7
ns
<0,0001
ns
<0,0001
V583∆fsrB V583∆gelE
V583∆sprE
V583∆gelE∆sprE
V583∆fsrB∆gelE∆sprE
24.38
0,2742
36.26
28.73
19.12
1.163
39.45
32.80
<0,0001
ns
<0,0001
<0,0001
<0,0001
ns
<0,0001
<0,0001
V583∆gelE
V583∆sprE V583∆gelE∆sprE V583∆fsrB∆gelE∆sprE
40.56 128.5
120.1
39.85 126.5
119.5
<0,0001 <0,0001
<0,0001
<0,0001 <0,0001
<0,0001
V583∆sprE
V583∆gelE∆sprE V583∆fsrB∆gelE∆sprE
38.30
31.46
36.28
31.10
<0,0001
<0,0001
<0,0001
<0,0001
V583∆gelE∆sprE
V583∆fsrB∆gelE∆sprE 0,6964 0,5534 ns ns
V583∆lytRS V583∆lrgAB
95.58 95.15 <0,0001 <0,0001
V583∆ABC V583∆ABC∆ef1097 30.00 32.61 <0,0001 <0,0001

Chapter IV
138
Table S3 – Similarity (%) between E. faecalis V583 proteins (found to have their genes differentially expressed in the microarray
experiments) and sequenced genomes from Pubmed and Enterococcus group Sequencing Project, Broad Institute of Harvard and MIT
(http://www.broadinstitute.org/). Grey shade represents absence of gene coding for the protein.
Fsr-dependent proteins Fsr proteins
Genomes EF0411 EF0412 EF0413 EF1097 EF1351 EF1352 EF3193 EF3194 EF1817 EF1818 EF1820 EF1821 EF1822
E. faecalis
OG1RF 99 99 99 99 99 99 99 99 98 99 99 99 99
dg1 98 98 97 98 98 99 98 100 98 98 98 97 99
atcc_4200 98 98 97 98 98 99 98 99 98 98 98
ch188_1 98 98 97 98 100 99 99 99 98 98 98 98 98
d6_1 98 98 96 98 100 99 98 99
ds5 98 98 98 98 100 99 98 99
e1sol_1 98 98 97 97 97 99 97 99 98 98 98 97 99
fly1_1 98 98 97 97 97 99 98 99 98 97 98 97 99
hip11704_1 98 98 98 98 100 99 98 99 98 98 98
jh1_1 98 98 98 98 100 99 98 99 98 97 98 97 98
merz96_1 98 98 97 97 98 99 98 98 100 99 98 97 100
t11_1 99 97 98 98 100 100 98 99 99 98 98 97 99
t1_1 98 98 98 98 98 99 98 99 98 98 98 97 99

New Findings on Fsr System: New virulence genes and their impact during Drosophila infection
139
t2_1 98 97 98 98 100 99 98 98 98 98 98
t3_1 98 98 98 98 100 99 98 99 99 98 98 97 99
t8_1 98 98 98 98 97 99 98 99 98 98 98
x98_1 98 98 98 98 98 99 98 99 98 98 98 97 98
62 99 99 99 99 100 99 99 99 99 99 99
E. faecium
1,141,733_1 80 81
1,230,933_1 79 82 81
1,231,408_1 79 82 81
1,231,410_1 79 82 81
1,231,501_1 79 82 79
1,231,502_1 79 82 81
com12_1 80 81
com15 79 81
E. gallinarum_eg2_1
80 79 81 78
E. Casseliflavus_ec30_1
80 83 86 80

Chapter IV
140

FSR SYSTEM AND DROSOPHILA:
The collapse of the immune system
This chapter is based on the following manuscript:
Contribution of melanization to Drosophila survival changes with E. faecalis V583 genomic content
Neuza Teixeira, António Jacinto and Maria de Fátima Silva Lopes,
In preparation.


CONTENTS 1. SUMMARY ......................................................................................................... 145
2. INTRODUCTION ................................................................................................. 146
3. MATERIAL AND METHODS ............................................................................... 149
3.1 Bacterial Strains ........................................................................................... 149
3.2 RNA extraction and Real-Time PCR for AMP expression ............................ 149
3.3 Drosophila Infection ...................................................................................... 150
3.4 Drosophila Melanization Assay .................................................................... 151
3.5 Statistical Analysis ........................................................................................ 151
4. RESULTS ............................................................................................................ 152
4.1 Drosomycin expression is similar during Drosophila infection with either V583
or V583ΔfsrBΔgelEΔsprE strains ....................................................................... 152
4.2 E. faecalis Fsr, GelE and SprE do not interfere with Drosophila phagocytosis
........................................................................................................................... 153
4.3 Fsr-GelE-SprE leads to increased melanization ........................................... 155
5. DISCUSSION ...................................................................................................... 159
6. ACKNOWLEDGEMENTS ................................................................................... 162
7. BIBLIOGRAPHY ................................................................................................. 163

The author of this thesis performed all the experimental work. Neuza Teixeira also
participated in the experimental design, data analysis and manuscript preparation.

FSR System and Drosophila: The collapse of the immune system
145
Cha
pter
V
1. SUMMARY
Enterococcus faecalis is a human opportunist pathogen able to infect Drosophila,
leading to its death within 24 hours. Previous studies proved that E. faecalis carrying
the Fsr quorum sensing system are extremely virulent. Fsr is the regulator of two
important virulence factors, gelatinase and serine protease, which cause death of
Drosophila adult flies by decreasing its tolerance to infection. The exact mechanism
underlying the toxicity of these E. faecalis virulence factors is nevertheless not
known, in particular the way they interfere with the host immune response. In the
present study, we investigated the influence of Fsr-GelE-SprE bacterial factors on
different immunity responses, namely antimicrobial peptide production, phagocytosis
and melanization. Using E. faecalis V583 wild type and E. faecalis V583
ΔfsrBΔgelEΔsprE mutant we showed that both drosomycin production and
phagocytosis were activated to similar levels by the two bacterial strains. However,
fly pupae infected with the mutant strain showed less melanization and higher
survival rates when compared to pupae infected with wild type bacteria. Moreover,
Drosophila mutants in the melanization pathway infected with the mutant bacteria
showed substantially increased survival rates (80%) compared to Drosophila infected
with wild type bacteria (20%). These results suggest that the bacterial factors studied
contribute to fly death by interfering with the melanization pathway.
As melanization is involved both in resistance and tolerance to infections, this study
thus provides new insights into the mechanisms whereby the Fsr system and the
proteases it regulates in E. faecalis contribute to disease and death of the host.

Chapter V
146
2. INTRODUCTION
In order to cause disease and death, pathogens must overcome the host´s immune
defenses. Understanding how the host immune defense mechanisms react to
pathogens and how pathogens inflict disease on the host can therefore provide us
with clues to fight those more efficiently. Among the most challenging pathogens are
the opportunistic ones, namely Enterococcus faecalis, which are commensal to
humans but can cause disease in patients with impaired immune systems.
Enterococci are natural inhabitants of the oral cavity, intestinal tract and female
genital tract of both human and animals. In contrast to the beneficial role they play in
intestinal homeostasis, these organisms are becoming increasingly important to
human health as leading causes of nosocomial infections. They are prevalent in the
nosocomial environment, causing infections of the urinary tract, bloodstream, intra-
abdominal and pelvic regions, surgical sites and central nervous system (Gilmore,
2002). To do so, they rely on several mechanisms including the fsr operon in the
case of E. faecalis. The fsr (Enterococcus faecalis regulator) two component system,
a homologue of the agr system in Staphylococcus aureus, is a quorum sensing-
dependent regulatory system that regulates the expression of two other important
virulence factors, gelE and sprE. These genes encode, respectively, gelatinase
(GelE), an extracellular zinc metalloprotease, and SprE, a serine protease
(Nakayama et al., 2001; Nakayama et al., 2002; Qin et al., 2000).
Recently, our Lab provided evidence for their role, and also for Fsr function, in
Drosophila melanogaster mortality (Teixeira et al., 2013). D. melanogaster (fruit fly) is
a powerful model organism to understand both the molecular mechanisms regulating
the activation of innate immune response and to screen for bacterial effectors
involved in virulence (Lemaitre & Hoffmann, 2007). The fruit fly has a multilayered
immune system consisting of at least seven defensive mechanisms: regulation of the
native microbiota in the gut through antimicrobial peptides (AMPs) and reactive
oxygen species; the barrier epithelial response, which recognizes infections and
wounds, produces local AMPs and sends signals to the rest of the body; the clotting

FSR System and Drosophila: The collapse of the immune system
147
Cha
pter
V
response, which not only seals wounds and prevents bleeding, but can physically
trap bacteria; the phenoloxidase response, which deposits melanin at the site of an
immune reaction, releasing potentially antimicrobial reactive oxygen species; the
phagocytic response, through which phagocytes can kill microbes directly by either
encapsulation or phagocytosis, or indirectly by releasing systemic signals; the
systemic AMP response, which involves the release of massive quantities of AMPs
from the fat body (the liver analog) into the circulation (Meister et al., 1997); and the
RNAi response, which is required to fight viral infections.
The expression of AMPs, regulated by the Toll and Imd pathways (Dionne &
Schneider, 2008), can take a few hours to a few days to occur. In contrast, a more
immediate immune response, induced within a few minutes after infection, is
melanization (Tang, 2009). This is considered to be the earliest and most acute
reaction of insects against pathogens upon injury (Tang, 2009) and is used to
encapsulate and sequester pathogens too large to be phagocytized (Kounatidis &
Ligoxygakis, 2012). During melanization reaction, phenols are oxidized to
quinolones, which then polymerize to form melanin that is deposited around intruding
microorganisms to help sequester them at the wound site. The quinolone substances
and other reactive oxygen intermediates are thought to be directly toxic to
microorganisms. Melanin synthesis is the final product of the proteolytic cascade
leading to the cleavage of prophenoloxidase (proPO) to phenoloxidase (PO).
We have shown that the E. faecalis virulence factors Fsr, GelE and SprE are
necessary to cause Drosophila mortality upon infection (Teixeira et al., 2013).
However, it remains unclear how these factors control this process. In the present
study we asked how different aspects of the immune response in Drosophila were
affected upon infection with E. faecalis and how that depends on the Fsr, GelE and
SprE machinery. We found that important resistance mechanisms, such as
drosomycin expression and phagocytosis, were not altered in the absence of Fsr and
the proteases. In contrast, the melanization response was severely affected in flies
infected with wild type but not with Fsr mutant bacteria. Furthermore, we show that
the impairment in the melanization reaction seems to be a critical event for the death

Chapter V
148
of infected flies, leading us to propose this pathway as one of the key targets of the
Fsr system.

FSR System and Drosophila: The collapse of the immune system
149
Cha
pter
V
3. MATERIAL AND METHODS
3.1 Bacterial Strains
Strains used in this study are listed in Table 1. Enterococcal strains were grown in
BHI (Brain Heart Infusion) medium at 37ºC, and Micrococcus luteus strain was grown
in LB medium at 37ºC with agitation.
Table 1 – Bacterial strains and primers used in this study.
Strains Relevant Characteristics References
E. faecalis V583 Clinical Isolate, TIGR sequenced strain;
VaR
(Sahm et al., 1989)
MG03 E. faecalis V583ΔfsrBΔgelEΔsprE;
GelE-, SprE-, GBAP-
(Teixeira et al., 2013)
Micrococcus luteus Gram positive bacteria (#)
Primers for RT-PCR Sequence (5´-3´)
Drosomycin R TCCCAGGACCACCAGCAT (Pili-Floury et al., 2004)
Drosomycin F CGTGAGAACCTTTTCCAATATGATG (Pili-Floury et al., 2004)
Ribosomal protein 49 F GACGCTTCAAGGGACAGTATCTG (Pili-Floury et al., 2004)
Ribosomal protein 49 R AAACGCGGTTCTGCATGAG (Pili-Floury et al., 2004)
(#) strain provided by Luís Teixeira laboratory, IGC.
3.2 RNA extraction and Real-Time PCR for AMP expression
E. faecalis and M. luteus strains were grown in BHI and LB, at 37ºC, until OD
(600nm) 0,02. The bacterial strains were injected into W1118 flies. At 6h and 24h after
W1118 flies infections 10 flies were collected and homogenized to proceed to RNA
extraction. Total RNA extraction was prepared using a TRIzol (Life Technologies)
extraction protocol and purified with RNA Clean-up & Concentration from Zymo
Research Company. SYBR Green quantitative real-time PCR analysis was
performed using 1st Strand cDNA Synthesis kit RT-PCR (AMV) and LightCycler® 96
System from Roche Company. The primers used are listed in Table 1. The amount of
mRNA detected was normalized to control rp49 mRNA values. Normalized data were
used to quantify the relative levels of a given mRNA according to cycling threshold

Chapter V
150
analysis (ΔCt). Relative ΔCt gene/ΔCt rp49 ratios of unchallenged wild-type controls
were anchored in 1 to indicate fold induction. Graphs represent the mean and SD of
relative ratios detected in 3 independent biological repetitions.
3.3 Drosophila Infection
Drosophila male flies (Table 2) were injected with 50 nl of bacteria at OD (600 nm)
0.02 from one of the strains: V583, V583ΔfsrBΔgelEΔsprE and M. luteus. As control,
flies were injected with the same volume of BHI medium. Male flies were
anesthetized with CO2 and injections were carried out with a pulled glass capillary
needle using a nano-injector (Nanoliter 2000, World Precision Instruments).
Reproducibility was measured by determining the number of bacteria injected at time
zero. Injected flies were placed at 29ºC, 65% humidity. 75 flies were assayed for
each survival curve, and they were placed in three vials of 25 flies each. Each
experiment was repeated three times, making a total of 75 flies tested per strain in
each set of three replicates, to ensure high confidence results. Death was recorded
at 0, 4, 6, 8, 10, 12, and 24 h hours post-injection. All experiments were performed at
least three times. Following challenge with bacteria, six individual flies were collected
(at 0 h, 6 h, 12 h and 24 h), homogenized, diluted serially, and plated onto
Enterococcel agar (Quilaban). E. faecalis CFUs (colony forming units) were
determined by testing three groups of six flies for each time point.
Table 2 – Flies used in this study.
Fly’s Relevant Characteristics
W1118 Wild type fly
W1118PPO1ΔPPO2Δ Flies without PPO1 and PPO2 (*)
W1118HmlΔ>GFP/UAS W1118 flies with hemocytes labeled with GFP
W1118HmlΔ>GFP/UAS-Bax W1118 flies without hemocytes
(*)Drosophila line provided by Bruno Lemaitre.

FSR System and Drosophila: The collapse of the immune system
151
Cha
pter
V
3.4 Drosophila Melanization Assay
Drosophila W1118, in pre-pupa stage, was injected with 50 nl of bacteria at OD (600
nm) 0.02 from one of the strains: V583, V583ΔfsrBΔgelEΔsprE and M. luteus. As
control, flies were injected with the same volume of BHI medium. Injections were
carried out with a pulled glass capillary needle using a nano-injector (Nanoliter 2000,
World Precision Instruments). The melanization process was recorded at 0, 6, 24
and 48 h hour’s post-injection using the stereoscope Zeiss Lumar V12. All
experiments were performed at least three times.
3.5 Statistical Analysis
Statistical analysis of Drosophila survival was performed using GraphPad Prism
software version 5.03. Survival curves were compared using Log-rank and Gehan-
Breslow-Wilcoxon tests. Statistical analysis of Drosophila survival was performed
using Student's t-test.

Chapter V
152
4. RESULTS
In order to understand the mechanisms by which the Fsr-GelE-SprE factors in E.
faecalis induce fast death of Drosophila upon infection, we tested whether known
innate immune system pathways are differentially regulated in two E. faecalis strains,
V583 (wild type) and its isogenic mutant devoid of fsr, gelE and sprE genes.
4.1 Drosomycin expression is similar during Drosophila infection with either
V583 or V583ΔfsrBΔgelEΔsprE strains
It is known that Gram positive bacteria activate the Toll pathway and that Drosomycin
is one of the AMPs produced to kill this group of bacteria (Lemaitre & Hoffmann,
2007). One way bacteria use to hamper the immune system of the host is by
inhibiting these peptides. Indeed, Park et al demonstrated that gelatinase from E.
faecalis is able to degrade Gm cecropin, an inducible AMP in the insect Galleria
mellonela (Park et al., 2007). We were therefore interested to know whether the
presence of Fsr-GelE-SprE influenced the expression levels of AMPs. For that we
measured the expression of Drosomycin by qRT-PCR at 6h and 24h post-infection in
both V583 and V583ΔfsrBΔgelEΔsprE strains and in the control strain M. luteus.
Interestingly, we found that all strains induced Drosomycin expression to similar
levels in both time points analyzed, over the period of 24h (Figure 1). These results
suggest that the E. faecalis virulence factors tested do not regulate AMP production
in Drosophila.

FSR System and Drosophila: The collapse of the immune system
153
Cha
pter
V
Figure 1- Drosomycin relative expression (scale log10) measured by qRT-PCR. W1118 flies (wild type)
were challenged with septic injury with Gram positive bacteria: M. luteus (black), E. faecalis V583 (white) and E.
faecalis V583ΔfsrBΔgelEΔsprE (grey). Total RNAs were extracted at 6h and 24h post-infection. Results were
normalized to rp49 expression levels. M. luteus was used as a positive control of drosomycin expression and of the
Toll pathway activation. Normalized data were used to quantify the relative levels of a drosomycin according to
cycling threshold analysis (ΔCt).
4.2 E. faecalis Fsr, GelE and SprE do not interfere with Drosophila
phagocytosis
Phagocytosis is an important defense mechanism that has been conserved during
evolution. In Drosophila the circulating phagocytic cells are the plasmocytes, which
are part of the innate immune system. This complex cellular process is initiated by
the recognition of the particles or pathogens to be ingested, followed by cytoskeletal
remodeling and signaling events leading to their engulfment and destruction (Ulvila et
al., 2011). It is known that E. faecalis can survive for a prolonged period in mouse
peritoneal, human and zebrafish macrophages after being phagocyted (Gentry-
Weeks et al., 2003; Prajsnar et al., 2013; Sussmuth et al., 2000). To investigate
whether E. faecalis Fsr-GelE-SprE perturb phagocytosis in the fruit fly, we used a
1
10
6h 24h
Drosomycin
Realtive
Expression
log 1
0
hours after infection
M.luteus
V583
V583∆fsrB∆gelE∆sprE

Chapter V
154
Drosophila line genetically modified to lack all hemocytes (W1118HmlΔ>GFP/UAS-
Bax). We found that flies without hemocytes (HmlΔ>GFP/UAS-Bax) show only
slightly increased survival rates upon infection with V583 when compared with
control flies (W1118HmlΔ>GFP/UAS) (Figure 2A). The same was observed when the
two Drosophila lines were infected with the E. faecalis mutant strain
V583ΔfsrBΔgelEΔsprE (Figure 2B). The flies died at the same rate with or without
hemocytes and regardless of the presence of the E. faecalis virulence factors
studied. These data suggest that the role of the E. faecalis virulence factors tested in
host death does not seem to occur through changes in phagocytosis by the
hemocytes.
Figure 2 – Survival curves of
Drosophila, with and without
phagocytes, infected with E.
faecalis V583 and
V583ΔfsrBΔgelEΔsprE.
(A) W1118HmlΔ>GFP/UAS-Bax survival
to septic injury with V583.
(B) W1118HmlΔ>GFP/UAS-Bax survival
to septic injury with
V583ΔfsrBΔgelEΔsprE. As a control
Drosophila W1118HmlΔ>GFP/UAS flies
were used. For each survival curve, 75
male adult flies, raised at 25ºC, were
divided in tubes 25 flies each, and
infected, by septic injury onto the thorax
with a thin needle. Data are
representative of three independent
experiments (making a total of 75 flies
tested per strain). Statistical analysis
was performed using GraphPad Prism
software version 5.03. Survival curves
were compared using Log-rank and
Gehan-Breslow-Wilcoxon tests and they
were not statistically different.

FSR System and Drosophila: The collapse of the immune system
155
Cha
pter
V
4.3 Fsr-GelE-SprE leads to increased melanization
One of the key immune reactions in Drosophila is the activation of tyrosinase-type
phenoloxidases (POs), which catalyze several reactions leading to the crosslinking of
proteins, the production of reactive intermediates with potential cytotoxic activity and
ultimately to the production of melanin (Bidla et al., 2009). Melanization is the earliest
reaction against the evasion of pathogen and it is visible by the blackening of wound
site. To determine if melanization is affected by the presence of Fsr-GelE-SprE in
infecting E. faecalis, we injected wild type pre-pupae, a stage that allows the easy
detection of melanized dark spots, with E. faecalis V583 and E. faecalis
V583ΔfsrBΔgelEΔsprE strains. At 6h post-infection, melanized spots are only around
the site of injection in all strains analyzed (Figure 3).
Figure 3 – Melanization in wild type fly pre-pupae after infection. W1118 pre-pupae were infected by
septic injury with 50nl of M. luteus at 0, 02 OD; V583 and V583ΔfsrBΔgelEΔsprE at 0, 02 OD, and placed at 29ºC.
Injection with BHI medium and M. luteus are controls of this experiment. Pictures were taken with stereoscope Zeiss
Lumar V12 at 0h, 6h, 24h and 48h post-infection. This procedure was made at least in 10 pre-pupae and the results
were always the same. After 24h hours an exacerbated melanization in the pre-pupae infected with V583 was
observed. All the other pre-pupae had only the normal black dots around the injection site (indicated with an arrow).

Chapter V
156
After 24h it is clear that the pre-pupae infected with V583 strain have an exacerbated
melanization, which is observed all over the body. In pre-pupae infected with E.
faecalis triple mutant, however, melanization remains restricted to the wound site,
similar to pre-pupae infected with the M. luteus control strain. Moreover, pre-pupae
infected with wild type bacteria were dead after 24h whereas those infected with the
mutant bacteria were still alive after 48h. These results indicate that the presence of
the Fsr-GelE-SprE E. faecalis virulence factors interferes with the melanization
process during infection through which it contributes to host death.
We thus asked if the excessive melanization was responsible for the fast and
massive death of the infected hosts. To answer this, we infected flies mutant for two
prophenoloxidases (PPO1 and PPO2), which makes them unable to produce
melanin (Tang, 2009). Figure 4 shows the survival rates of W1118PPO1ΔPPO2Δ
mutant and wild type flies infected with V583 and mutant strains. When we compare
PPO mutant and control flies infected with the same wild type bacteria, survival rates
are similar. However, they have different shapes: while control flies die massively
only after 12h of infection, PPO mutant flies steadily die during the time course of the
assay (Figure 4A). Although the overall bacterial growth inside the two flies was
similar after 24h, V583 grew faster in the PPO mutant flies (Figure 5). This result
suggests that melanization may not play a role as a resistance mechanism against E.
faecalis. It also means that, in flies unable to perform the melanization reaction, and
during the first 12 hours of infection a lower number of V583 cells can cause a higher
host death rate, i.e., PPO mutants show decreased tolerance to E. faecalis. The two
bacterial proteases present in V583, GelE and SprE, are able to degrade host
structural proteins, thus causing tissue damage, which must be healed in order for
the fly to maintain its healthy status. In the absence of melanization, which
contributes to tissue healing, it is possible that the flies tolerate less the presence of
V583 carrying Fsr-GelE-SprE factors.

FSR System and Drosophila: The collapse of the immune system
157
Cha
pter
V
Figure 4 –Survival curves of
Drosophila with and without
melanization. (A) W1118 and
W1118PPO1ΔPPO2Δ survival to septic
injury with V583 strain (B) W1118 and
W1118PPO1ΔPPO2Δ survival to septic
injury with V583ΔfsrBΔgelEΔsprE. For
each survival curve, 75 male adult
flies, raised at 25ºC, were divided in
tubes 25 flies each, and infected by
septic injury onto the thorax with a thin
needle. Data are representative of
three independent experiments
(making a total of 75 flies tested per
strain). Drosophila survival was
performed using GraphPad Prism
software version 5.03. For statistical
analysis was used Student's t-test.
Asterisks (*) indicate the level of
statistical significance (*p < 0.05; **p <
0.005).
When we compare the survival rates of PPO mutant and control flies infected with
the mutant bacteria major differences were observed. 24 hours after infection, 80%
of the W1118PPO1ΔPPO2Δ mutant flies infected with V583ΔfsrBΔgelEΔsprE triple
mutant were still alive (Figure 4B). In contrast, all wild type flies were dead when
infected with the wild type bacteria (Figure 4A).
* *
**
* *
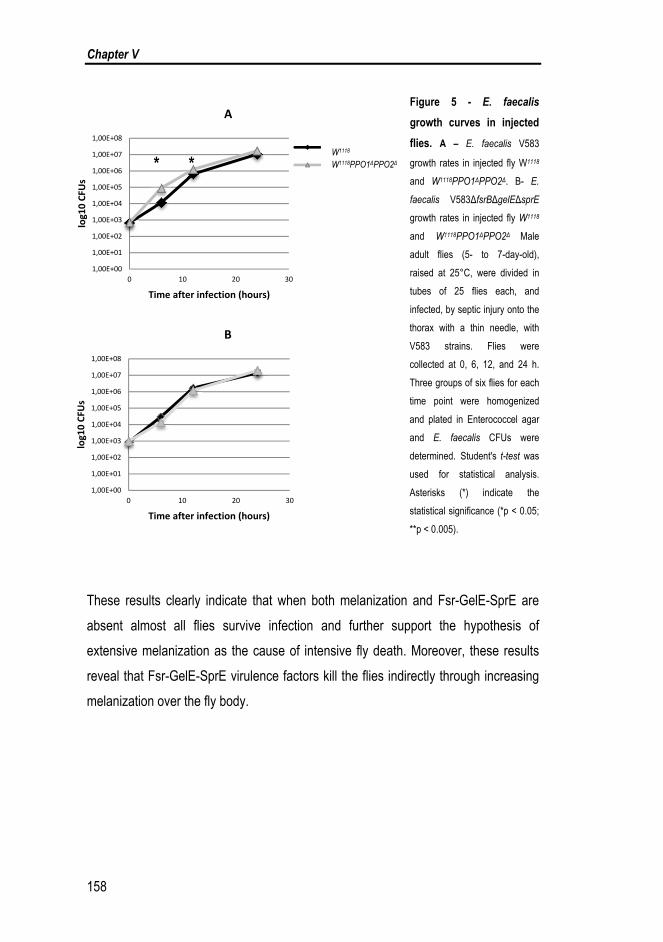
Chapter V
158
Figure 5 - E. faecalis
growth curves in injected
flies. A – E. faecalis V583
growth rates in injected fly W1118
and W1118PPO1ΔPPO2Δ. B- E.
faecalis V583ΔfsrBΔgelEΔsprE
growth rates in injected fly W1118
and W1118PPO1ΔPPO2Δ Male
adult flies (5- to 7-day-old),
raised at 25°C, were divided in
tubes of 25 flies each, and
infected, by septic injury onto the
thorax with a thin needle, with
V583 strains. Flies were
collected at 0, 6, 12, and 24 h.
Three groups of six flies for each
time point were homogenized
and plated in Enterococcel agar
and E. faecalis CFUs were
determined. Student's t-test was
used for statistical analysis.
Asterisks (*) indicate the
statistical significance (*p < 0.05;
**p < 0.005).
These results clearly indicate that when both melanization and Fsr-GelE-SprE are
absent almost all flies survive infection and further support the hypothesis of
extensive melanization as the cause of intensive fly death. Moreover, these results
reveal that Fsr-GelE-SprE virulence factors kill the flies indirectly through increasing
melanization over the fly body.
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
0 10 20 30
log10 CFU
s
Time after infection (hours)
B
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
0 10 20 30
log10 CFU
s
Time after infection (hours)
A
PPO mutant
W118 wild type
W1118 W1118PPO1ΔPPO2Δ * *

FSR System and Drosophila: The collapse of the immune system
159
Cha
pter
V
5. DISCUSSION
The fly mechanisms responsible for protection against bacterial infections are not
clearly understood yet. Drosophila has four distinct pathways implicated in regulation
of genes induced upon septic injury, namely Toll, Imd, JNK and JAK-STAT (Brun et
al., 2006). Previous studies have shown that E. faecalis induces both cellular and
humoral immune response mechanisms in Drosophila. Toll seems to be the crucial
pathway in the defense against E. faecalis (Nehme et al., 2011): whereas Toll
pathway mutants are susceptible to E. faecalis, Imd mutants are not (Brun et al.,
2006). The Toll pathway is responsible for production of several AMPs: diptericin,
cecropin, drosocin and attacin are active against Gram-negative bacteria and
drosomycin, metchnikowin and defensin to fungi and Gram-positive bacteria. Except
for defensin, E. faecalis is resistant to the bactericidal activity of all AMPs produced
by Drosophila, and even from G. mellonella (Smeianov et al., 2000). It is thus not
clear how the Toll pathway confers protection against Gram-positive bacteria, as it is
known that defensin is not necessary to mediate protection (Nehme et al., 2011).
Previous in vitro studies showing that the proteases GelE and/or SprE may degrade
insect AMPs, have led researchers suggest that E. faecalis success in insect species
could be attributed to the degradation of the host innate immune AMPs by the
proteases. However, in a previous study, our findings suggest otherwise (Teixeira et
al., 2013). In fact, as we observed no difference in growth inside the host between
any of the mutants and wild type V583, we conclude that neither the Fsr system nor
the proteases it controls affect bactericidal action by the fly. This implies that none of
the proteases provides self-protection against any AMP in the fly immune system
(Teixeira et al., 2013). In the present study we showed that the presence of Fsr-
GelE-SprE does not affect the levels of drosomycin expression, further supporting
the likely irrelevant role of Drosophila AMPs on E. faecalis infection progression.
However, this does not exclude the possibility that the presence of these proteases
in high amounts may turn the host more fragile to other bacteria due to AMPs
degradation. In fact, previous work has shown that GelE is able to degrade host

Chapter V
160
AMP´s and that this is responsible for insects getting less able to deal with
Escherichia coli strains (Park et al., 2007).
The way the fly is able to fight invading microorganisms also includes a cellular
immune response that can result in the phagocytosis of relatively small organisms
like bacteria or the encapsulation of larger parasites (Khush & Lemaitre, 2000).
However, little is known about how Drosophila phagocytes affect the course of
infections (Brandt et al., 2004). On the other hand, bacteria that are specialized in
growing inside phagocytes have developed ways to fight these cells from within.
Moreover, previous studies have demonstrated that the pathogenesis mechanisms
developed by Mycobacterium marinum and Listeria monocytogenes to fight
vertebrate phagocytes also function in the fly (Dionne et al., 2003; Mansfield et al.,
2003). In the case of extracellular pathogens, such as E. faecalis, it is known that
these bacteria are able to stand macrophages defense mechanisms for hours and
days (Gentry-Weeks et al., 1999). Although some E. faecalis defense mechanisms
have been implicated in its prolonged life inside macrophages (Abrantes et al., 2013;
Verneuil et al., 2005; Waters & Dunny, 2001), neither Fsr nor the two proteases it
regulates seem to play a role in bacterial survival inside these defensive cells.
Recently, macrophages in zebrafish were shown to phagocytize bacteria in blood
circulation being only able to engulf surface-associated microbes (Prajsnar et al.,
2013). It is also known that homolog of tumor necrosis factor (TNF) encoded by eiger
is required for innate immune responses that are effective at fighting extracellular
pathogens but are wasteful or simply ineffective when fighting intracellular pathogens
(Schneider et al., 2007). In our model, despite being phagocytized by Drosophila
hemocytes (results not shown), neither Fsr nor GelE or SprE were found to affect the
cellular immune response of Drosophila.
Melanization is another Drosophila immune response. It is visible by the blackening
of a wound site or the surface of pathogens, which results from the synthesis and
deposition of melanin. In addition to being important for wound healing, melanin can
encapsulate and sequester pathogens, and the reaction intermediates appear to be
directly toxic to microbes as well (Tang et al., 2006). Here, we evaluated/investigated

FSR System and Drosophila: The collapse of the immune system
161
Cha
pter
V
the effect of Fsr-GelE-SprE presence in melanization in pre-pupa infected with either
V83 or V583ΔfsrBΔgelEΔsprE strains. Pre-pupa infected with the mutant strain not
only survived longer but also showed normal melanization around the injection site,
suggesting an association between the extent of melanization and death and the
presence of the proteases and the Fsr. This was confirmed by measuring survival
rates of Drosophila without PPO1 and PPO2, two prophenoloxidases important for
the melanization reaction. After 24h, flies without PPOs and infected with
V583ΔfsrBΔgelEΔsprE showed 80% higher survival providing evidence that Fsr-
GelE-SprE decrease Drosophila tolerance to E. faecalis infection through
melanization and promoting the fly death. It is known the melanization reaction
produces toxic intermediates, namely reactive oxygen species that can kill the host
(Tang, 2009). If uncontrolled melanization occurs an overproduction of these toxic
intermediates can cause collateral damage in the host (Ayres & Schneider, 2012).
Our result leads us to hypothesize that Fsr-GelE-SprE interfere with melanization,
possibly by activating the proteolytic cascade responsible for the tight control of this
pathway. Host death by exacerbated immune response to infections has also been
observed for other bacteria. For example, after Drosophila infection with Salmonella
typhimurium bacterial secreted effectors cause an immune response that is
damaging both to the bacteria and to the host (Brandt et al., 2004).
In summary, the present study reveals that E. faecalis Fsr system does not interfere
with AMP expression or phagocytosis but leads to excessive melanization. Our
results constitute an important step towards the discovery of the Fsr role during E.
faecalis infection. Further studies should determine which step(s) of the melanization
cascade is/are affected by the Fsr-proteases.

Chapter V
162
6. ACKNOWLEDGEMENTS
The authors are grateful to Bruno Lemaitre from Global Health Institute, School of
Life Sciences, École Polytechnique Fédérale Lausanne (EPFL) - Switzerland for the
W1118PPO1ΔPPO2Δ Drosophila lines; and to Luis Teixeira, from Instituto Gulbenkian
de Ciência, for the M. luteus strain.
We are also grateful to Anabela Bensimon-Brito for the technical support in qRT-
PCR experiment, to Carolina Moreira and Ana Sofia Brandão for the technical
support in Drosophila phagocytosis experiments and Lara Carvalho for the
comments and revision of this paper.

FSR System and Drosophila: The collapse of the immune system
163
Cha
pter
V
7. BIBLIOGRAPHY
Abrantes, M. C., Kok, J. & Lopes Mde, F. (2013). EfaR is a major regulator of
Enterococcus faecalis manganese transporters and influences processes involved in
host colonization and infection. Infect Immun 81, 935-944.
Ayres, J. S. & Schneider, D. S. (2012). Tolerance of infections. Annu Rev Immunol
30, 271-294.
Bidla, G., Hauling, T., Dushay, M. S. & Theopold, U. (2009). Activation of insect
phenoloxidase after injury: endogenous versus foreign elicitors. J Innate Immun 1,
301-308.
Brandt, S. M., Dionne, M. S., Khush, R. S., Pham, L. N., Vigdal, T. J. &
Schneider, D. S. (2004). Secreted Bacterial Effectors and Host-Produced Eiger/TNF
Drive Death in aSalmonella-Infected Fruit Fly. PLoS Biol 2, e418.
Brun, S., Vidal, S., Spellman, P., Takahashi, K., Tricoire, H. & Lemaitre, B.
(2006). The MAPKKK Mekk1 regulates the expression of Turandot stress genes in
response to septic injury in Drosophila. Genes Cells 11, 397-407.
Dionne, M. S., Ghori, N. & Schneider, D. S. (2003). Drosophila melanogaster is a
genetically tractable model host for Mycobacterium marinum. Infect Immun 71, 3540-
3550.
Dionne, M. S. & Schneider, D. S. (2008). Models of infectious diseases in the fruit
fly Drosophila melanogaster. Disease models & mechanisms 1, 43-49.

Chapter V
164
Gentry-Weeks, C., Estay, M., Loui, C. & Baker, D. (2003). Intravenous mouse
infection model for studying the pathology of Enterococcus faecalis infections. Infect
Immun 71, 1434-1441.
Gentry-Weeks, C. R., Karkhoff-Schweizer, R., Pikis, A., Estay, M. & Keith, J. M.
(1999). Survival of Enterococcus faecalis in mouse peritoneal macrophages. Infect
Immun 67, 2160-2165.
Gilmore, M. S., Phillip S. Coburn, Sreedhar R. Nallapareddy, Barbara E. Murray
(2002). Enterococcal Virulence. In The Enterococci Pathogenesis, Molecular Biology,
and Antibiotic Resistance, pp. 301-354. Edited by M. S. Gilmore, Clewell DB,
Courvalin P, Dunny GM, Murray BE, Rice LB. Washington D.C.: American Society
for Microbiology.
Khush, R. S. & Lemaitre, B. (2000). Stop press: genes that fight infections: what the
drosophila genome says about animal immunity. Trends in genetics : TIG 16, 468.
Kounatidis, I. & Ligoxygakis, P. (2012). Drosophila as a model system to unravel
the layers of innate immunity to infection. Open Biol 2, 120075.
Lemaitre, B. & Hoffmann, J. (2007). The host defense of Drosophila melanogaster.
Annu Rev Immunol 25, 697-743.
Mansfield, B. E., Dionne, M. S., Schneider, D. S. & Freitag, N. E. (2003).
Exploration of host-pathogen interactions using Listeria monocytogenes and
Drosophila melanogaster. Cell Microbiol 5, 901-911.
Meister, M., Lemaitre, B. & Hoffmann, J. A. (1997). Antimicrobial peptide defense
in Drosophila. Bioessays 19, 1019-1026.

FSR System and Drosophila: The collapse of the immune system
165
Cha
pter
V
Nakayama, J., Cao, Y., Horii, T., Sakuda, S., Akkermans, A. D., de Vos, W. M. &
Nagasawa, H. (2001). Gelatinase biosynthesis-activating pheromone: a peptide
lactone that mediates a quorum sensing in Enterococcus faecalis. Mol Microbiol 41,
145-154.
Nakayama, J., Kariyama, R. & Kumon, H. (2002). Description of a 23.9-kilobase
chromosomal deletion containing a region encoding fsr genes which mainly
determines the gelatinase-negative phenotype of clinical isolates of Enterococcus
faecalis in urine. Appl Environ Microbiol 68, 3152-3155.
Nehme, N. T., Quintin, J., Cho, J. H., Lee, J., Lafarge, M. C., Kocks, C. &
Ferrandon, D. (2011). Relative roles of the cellular and humoral responses in the
Drosophila host defense against three gram-positive bacterial infections. PLoS One
6, e14743.
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Pili-Floury, S., Leulier, F., Takahashi, K., Saigo, K., Samain, E., Ueda, R. &
Lemaitre, B. (2004). In vivo RNA interference analysis reveals an unexpected role
for GNBP1 in the defense against Gram-positive bacterial infection in Drosophila
adults. J Biol Chem 279, 12848-12853.
Prajsnar, T. K., Renshaw, S. A., Ogryzko, N. V., Foster, S. J., Serror, P. &
Mesnage, S. (2013). Zebrafish as a novel vertebrate model to dissect enterococcal
pathogenesis. Infect Immun 81, 4271-4279.

Chapter V
166
Qin, X., Singh, K. V., Weinstock, G. M. & Murray, B. E. (2000). Effects of
Enterococcus faecalis fsr genes on production of gelatinase and a serine protease
and virulence. Infect Immun 68, 2579-2586.
Sahm, D. F., Kissinger, J., Gilmore, M. S., Murray, P. R., Mulder, R., Solliday, J.
& Clarke, B. (1989). In vitro susceptibility studies of vancomycin-resistant
Enterococcus faecalis. Antimicrob Agents Chemother 33, 1588-1591.
Schneider, D. S., Ayres, J. S., Brandt, S. M. & other authors (2007). Drosophila
eiger mutants are sensitive to extracellular pathogens. PLoS Pathog 3, e41.
Smeianov, V., Scott, K. & Reid, G. (2000). Activity of cecropin P1 and FA-LL-37
against urogenital microflora. Microbes Infect 2, 773-777.
Sussmuth, S. D., Muscholl-Silberhorn, A., Wirth, R., Susa, M., Marre, R. &
Rozdzinski, E. (2000). Aggregation substance promotes adherence, phagocytosis,
and intracellular survival of Enterococcus faecalis within human macrophages and
suppresses respiratory burst. Infect Immun 68, 4900-4906.
Tang, H., Kambris, Z., Lemaitre, B. & Hashimoto, C. (2006). Two proteases
defining a melanization cascade in the immune system of Drosophila. J Biol Chem
281, 28097-28104.
Tang, H. (2009). Regulation and function of the melanization reaction in Drosophila.
Fly 3, 105-111.
Teixeira, N., Varahan, S., Gorman, M. J. & other authors (2013). Drosophila host
model reveals new enterococcus faecalis quorum-sensing associated virulence
factors. PLoS One 8, e64740.

FSR System and Drosophila: The collapse of the immune system
167
Cha
pter
V
Ulvila, J., Vanha-Aho, L. M. & Ramet, M. (2011). Drosophila phagocytosis - still
many unknowns under the surface. APMIS : acta pathologica, microbiologica, et
immunologica Scandinavica 119, 651-662.
Verneuil, N., Rince, A., Sanguinetti, M., Auffray, Y., Hartke, A. & Giard, J. C.
(2005). Implication of hypR in the virulence and oxidative stress response of
Enterococcus faecalis. FEMS Microbiol Lett 252, 137-141.
Waters, C. M. & Dunny, G. M. (2001). Analysis of functional domains of the
Enterococcus faecalis pheromone-induced surface protein aggregation substance. J
Bacteriol 183, 5659-5667.

Chapter V
168

GENERAL DISCUSSION


CONTENTS
1. FSR QUORUM SENSING SYSTEM
- Different environments lead to different ways to persist ................................ 173
2. FSR REGULON
- New genes and potential virulence factors ...................................................... 177
3. FUTURE PRESPECTIVES
- Fsr system can be a future target for therapy .................................................. 184
4. BIBLYOGRAPHY ............................................................................................... 186

Chapter VI
172

GENERAL DISCUSSION
173
Cha
pter
VI
Among the complex molecular processes involved in the development of bacteria-
borne disease is Quorum sensing (QS), the way bacteria communicate and
coordinate collective behaviours. The study of how to inhibit QS may provide
important clues to create antibacterial pharmaceuticals.
Bacteria use QS to control a wide variety of processes, which include the
synchronized expression of genes involved in colonization, symbiosis and virulence
in host–bacterial infections. Activation of virulence genes at bacterial low population
density would result in the generation of host defence responses, thus providing the
host an early lead over the invading bacteria. A certain threshold of bacterial cell
density, and therefore autoinducer concentration, has to be accomplished in order for
the pathogen to overpower the host defence mechanisms hence, in order to
guarantee their survival and the synthesis of virulence factors until the success of the
infection process is guaranteed (Raina et al., 2009).
1. FSR QUORUM SENSING SYSTEM
- Different environments lead to different ways to persist
In this thesis we presented two examples in which Fsr QS is silenced: by mutation in
the fsrC gene (chapter II); and by the presence of a therapeutic dose of vancomycin
(chapter III). Despite the difference between these studies they share a common
feature, the Fsr repression through the same target, the histidine kinase FsrC. In
both studies we associated the QS silencing with fitness cost of activating the Fsr.
When the conditions are not favourable for growth, E. faecalis reduces the energy
costs by shutting down the genes not relevant in that specific situation.
The phenotypes regulated by QS (for example, extracellular factors) are typically
relevant and beneficial only when expressed in a concerted manner by large
populations of cells (Chong et al., 2013). When in high concentrations the QS signal
producer cells are achieved, the QS activated genes are induced, and cells work in

Chapter VI
174
a cooperative way producing the “public goods” (factors that are beneficial for the
population). At low cell densities, the action of extracellular factors would be
relatively inefficient, since they would disperse away before they could be used. Cells
that produce extracellular factors can be exploited by ‘free-riders’ or cheaters, which
avoid the costs of producing QS-regulated factors themselves by benefiting from
those produced by others in order to grow and prevail (Rumbaugh et al., 2012)
(Figure 1).
The question then is, under which conditions cheating strains will increase to such an
extent that QS breaks down as a regulatory system of cooperative behaviour – with
the consequence that the cooperative behaviour itself cannot be maintained (Czaran
& Hoekstra, 2009). Our studies presented in chapters II and III provide two examples
of cheating. In both cases, E. faecalis silence the Fsr through the modification of
histidine kinase FsrC, but maintaining the virulence factors genes intact. This can be
related with the fact that the extracellular factors regulated by Fsr can produce some
benefit for the cell itself and the Fsr genes only provide benefit for cell-
communication/cooperation.
In Chapter III, we propose that the negative effect of vancomycin on Fsr induction
may function as a survival strategy that E. faecalis developed. By decreasing the
fitness cost of Fsr activation saves energy to be used in the vancomycin resistance
operon expression and activity. In VRE, it is known that vancomycin binds to
bacteria cell-wall and replaces the terminal D-Ala of peptidoglycan precursor with D-
Lac producing a high-level of resistance (Chapter I). The cell-wall modification by
vancomycin changes the levels of autolysis and consequently the cell-division and
growth (Bisicchia et al., 2011; Sieradzki & Tomasz, 1997). When vancomycin was
added to E. faecalis V583, cells entered rapidly into the stationary phase (Chapter
III). Recent publications indicates that autolysis is also correlated with proteases
GelE and SprE (Teixeira et al., 2013; Thomas et al., 2008).

GENERAL DISCUSSION
175
Cha
pter
VI
Figure 1 – E. faecalis QS model under changing environmental conditions. Among the population
able to sense the quorum signal, there are cells that participate in the cross-talk (the “talk” cells) and cells that
do not participate (the “silence” cells). The “talk” cells are cooperators that produce GBAP, sense the GBAP and
activate the QS. Through the QS activation “public goods” such as GelE and SprE are produced and promote host
damage. This cooperation is associated with fitness cost and it is depend of cell density. The “silence” cells are non-
cooperators, do not produce GBAP, do not sense GBAP and do not activate the QS. The cell cheaters use the
effects of the public goods (from the “talk” cells) in the host to grow and proliferate, overcoming/avoiding the fitness
cost. This phenomenon is called selfishness. The “talk” cells can develop to “silence” cells and vice versa under
environmental pressure.
Altogether, these results lead us to hypothesize that this peptidoglycan alteration
promotes a conformational change of the histidine kinase FsrC and, consequently,
fsr repression. Furthermore, the cell wall modification promotes the silencing of fsr
and decreased production of GelE and SprE, reducing cell autolysis and
consequently stopping growth (Figure 2).

Chapter VI
176
Figure 2 – Model for vancomycin effect on the Fsr system. A - Without stress. When E. faecalis cells
activate the fsr, FsrC senses the GBAP signal and, through the quorum sensing, the proteases are produced. GelE
and SprE are involved in autolysis regulation (Teixeira et al., 2013; Thomas et al., 2008). The cells divide normally. B
– Under vancomycin stress. Vancomycin modifies the cell wall and the FsrC cannot sense the GBAP signal.
Production of GelE and SprE is decreased, which has an impact on autolysis. The vancomycin also interferes with
autolysis (Bisicchia et al., 2011; Sieradzki & Tomasz, 1997). E. faecalis stop growth and cell division.
Opportunist bacteria such as E. faecalis have numerous ways to survive and persist
in the different environments and these two examples of QS silencing are an
evidence of this characteristic. These two studies are important to understand why
and when E. faecalis needs to becomes a cheater, activating/silencing the QS.
However, more studies are necessary to prove our hypothesis and reveal the relation
between vancomycin and the Fsr system.

GENERAL DISCUSSION
177
Cha
pter
VI
2. FSR REGULON
- New genes and potential virulence factors
The E. faecalis Fsr QS system is known to regulate the virulence factors GelE and
SprE and a putative bacteriocin EF1097. The expression of these three genes was
found to be exponentially increased by activation of the Fsr system by GBAP in two
genetically unrelated E. faecalis strains (Bourgogne et al., 2006; Teixeira et al.,
2013). The concomitant presence of these three genes with Fsr is 98% in the 18
sequenced enterococcal genomes (Teixeira et al., 2013). These observations
suggest a mechanistic association between the proteases and the bacteriocin that
needs clarification. EF1097 was found to be important during infection in Drosophila.
When ef1097 and the genes encoding the two proteases are expressed, as a
response to high cell density, they may be seen as public goods, expected to be
produced in a cooperative population for the benefit of the entire population. We
speculate that the putative bacteriocin can have some function helping the QS to be
more efficient in killing the non-cooperators. As already referred, public goods are
sent outside the cells. Among the genes found to be up-regulated by GBAP are
ef1351 and ef1352, which are in operon and encode a putative (Mg2+)-magnesium-
translocating P-type ATPase. Mg2+ is a modulator of virulence in many bacteria and
its concentration allows bacteria to sense their surroundings. EF1352 is orthologous
to mgtA gene from S. typhimurium, MgtA is a Mg2+ transporter that, together with
MgtB, pumps Mg2+ into the cell (Tao et al., 1995). It is possible that EF1351-EF1352,
that it is not exported from the cell, constitutes an immunity factor against self-
produced EF1097 (Figure 3). In fact, it is known that bacteriocins can affect
magnesium transport (Uratani & Hoshino, 1984). More studies are needed to
investigate this correlation.
We found twelve additional genes dependent on the presence of GBAP-induced
proteases. Among these genes, lrgAB were highly induced when both protease
genes were induced in response to the quorum-sensing molecule. lrgAB were found
to be under the control of the LytRS two-component system, similar to what has been

Chapter VI
178
described in S. aureus. In this bacterium, LytRS and Agr are two important players in
the tight regulation of cell autolysis; the first positively regulates the lrgAB expression
and the second reduces the rate of autolysis. In E. faecalis, GelE was found to
proteolitically activate AtlA autolysin (Thomas et al., 2009). Altogether, these
findings suggest that, in E. faecalis, LytRS through LrgAB, and Fsr, through GelE
and SprE, are also key players in autolysis regulation (Figure 3). lrgAB genes were
highly induced only when the protease genes were induced in response to GBAP.
Assuming that both play an opposite role in autolysis, it is possible that lrgAB were
induced to protect cells form huge production of the proteases. In addition, it is
possible that those additional genes induced indirectly by the QS activation of the Fsr
system are playing a role in preventing cell damage by self or by the neighbour cells.
Once new genes were found to be part of the Fsr regulon in V583 strain, their role in
the virulence of this strain was assessed. Different infection models have been used
to study E. faecalis virulence, but with some limitations. In our study, we used the
fruit fly Drosophila to test virulence of the Fsr system and the newly identified genes.
According to our in vivo tests, we can conclude that ef1097 and lrgAB are important
to E. faecalis virulence (Chapter IV), demonstrating that Fsr virulence is not only
through the proteases. We also observed that in the absence of Fsr, the fly´s
tolerance to E. faecalis increases and the flies have a higher survival rate.
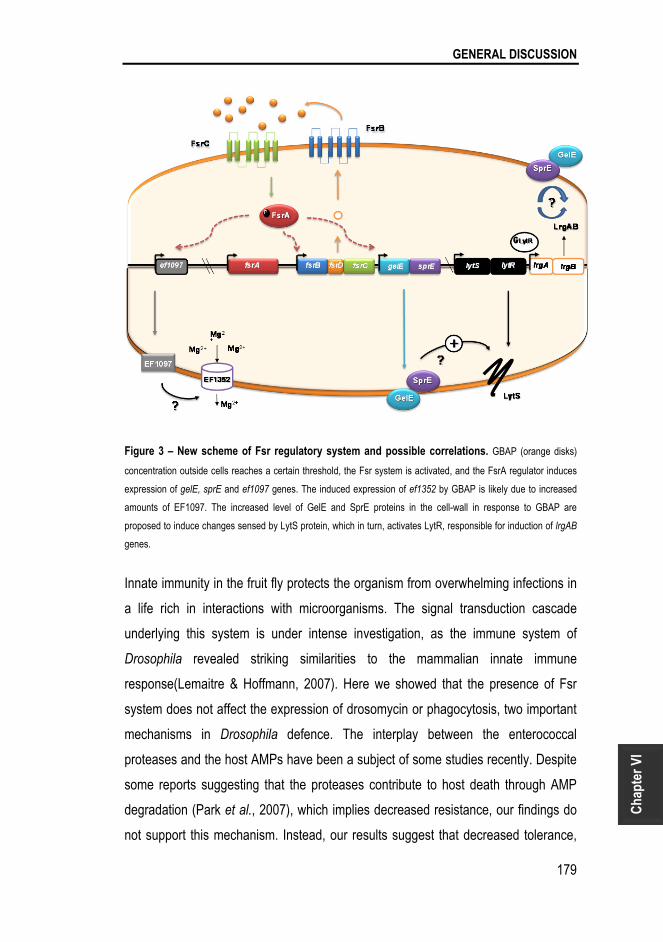
GENERAL DISCUSSION
179
Cha
pter
VI
Figure 3 – New scheme of Fsr regulatory system and possible correlations. GBAP (orange disks)
concentration outside cells reaches a certain threshold, the Fsr system is activated, and the FsrA regulator induces
expression of gelE, sprE and ef1097 genes. The induced expression of ef1352 by GBAP is likely due to increased
amounts of EF1097. The increased level of GelE and SprE proteins in the cell-wall in response to GBAP are
proposed to induce changes sensed by LytS protein, which in turn, activates LytR, responsible for induction of lrgAB
genes.
Innate immunity in the fruit fly protects the organism from overwhelming infections in
a life rich in interactions with microorganisms. The signal transduction cascade
underlying this system is under intense investigation, as the immune system of
Drosophila revealed striking similarities to the mammalian innate immune
response(Lemaitre & Hoffmann, 2007). Here we showed that the presence of Fsr
system does not affect the expression of drosomycin or phagocytosis, two important
mechanisms in Drosophila defence. The interplay between the enterococcal
proteases and the host AMPs have been a subject of some studies recently. Despite
some reports suggesting that the proteases contribute to host death through AMP
degradation (Park et al., 2007), which implies decreased resistance, our findings do
not support this mechanism. Instead, our results suggest that decreased tolerance,

Chapter VI
180
rather than decreased resistance, leads to fly´s death by E. faecalis carrying Fsr and
the proteases. After 24h of infection with E. faecalis wild type or with Fsr mutants,
Drosophila had the same E. faecalis CFUs inside its body but presented different
survival rates. This result indicates that Fsr system influences the fly´s tolerance but
not its resistance to infection. Fly´s resistance/tolerance can also be altered by the
immune system as a way to survive to the pathogen. One obvious mechanism
affecting tolerance is the intensity of an immune response; an overly exuberant
immune response can cause collateral damage through immune effectors and
because of the energy allocated away from other physiological functions (Ayres &
Schneider, 2012). We thus investigated some of the immunity functions of Drosophila
by measuring the survival rates of flies without haemocytes and without melanization
during infection with V583wt and V583 ΔfsrBΔgelEΔsprE strains (chapter V). We
found that haemocytes are able to identify and phagocyte the bacteria, but ddo not
play a role in combating E. faecalis infection (Table 1 and Figure 4). From Table 1,
we also observed that flies without melanization and infected with E. faecalis without
Fsr system have a survival rate near to 100 % (Table 1).
Table 1 – Drosophila survival results 24h after E. faecalis infection. The survival (percentage) is
identified with: (+++) more than 80%; (+) less than 30% and (-) less than 5% (Data from chapter V).
E. faecalis V583
Drosophila lines Wild type ΔfsrBΔgelEΔsprE
Wild type W1118 + +
Without haemocytes (hmlΔ>bax) - +
Without melanization (PPO1PPO2) - + + +
From the results of table 1 and visual analysis, allow us to say that, in the presence
of Fsr and proteases, an uncontrolled melanization occurs forcing pupa death (dead
pupas were completely black). Activation of melanization is strictly regulated.
Uncontrolled melanization generates excessive toxic intermediates that can kill the
host (Tang, 2009). Recognition of pathogens and injury leads to the activation of a

GENERAL DISCUSSION
181
Cha
pter
VI
serine protease cascade that culminates in proteolytic cleavage of inactive PPO to
active PO. Serine protease inhibitors, called serpins, are responsible for keeping the
melanization strictly localized at the site of injury or infection (Tang, 2009). Some
specific pathogens have developed means to prevent PPO activation by producing
serpin-type inhibitors or other factors specifically interfering with proteolytic activation
of PPO or upstream components (Cerenius et al., 2008). We therefore hypothesized
that during E. faecalis infection; one of the factors regulated by Fsr inhibits the
serpins that control the melanization. In response to infection by injury, the fly
activates melanization and, with no control of melanization reaction, an excess of
melanin and toxic intermediates are produced, promoting host death. This hypothesis
would explain the high percentage of fly survival when flies without melanization are
infected with E. faecalis ΔfsrBΔgelEΔsprE. The reactive oxygen species (ROS)
generated during melanin production cause host damage and, in high concentration
the death of the host (Ayres & Schneider, 2012). ROS affect several biological
processes such as cellular signalling and aging, and play an important role in host
defence against invading microorganisms; however, an overproduction of ROS is
cytotoxic and carcinogenic (Strickertsson et al., 2013). Recent work in human cells
found that infection by E. faecalis induces an intracellular production of ROS
suggesting that E. faecalis is associated with gastric pathogenicity by promoting the
appearance of gastric cancer cells (Strickertsson et al., 2013). They propose a
common link in bacterial induced pathogenicity by which the infection stimulates a
general activation of pro-inflammatory cytokines and ROS by the cells themselves,
making the epithelial cells harmful to themselves (Strickertsson et al., 2013). In our
study we also suspect that E. faecalis infection block the melanization control,
promoting the ROS overproduction, cell damage and inevitable death.
GelE has an important role in the development of endocarditis (Singh et al., 2005).
We also suspect that during infection, the proteases GelE and SprE cause tissue
damage in Drosophila body. This would explain the reduced survival in V583wt
infected flies mutant for PPOs.

Chapter VI
182
Melanization is an important mechanism in insects and possibly in other
invertebrates (Cerenius et al., 2008). In humans melanization does not occur,
however PPO activation mediated by a serine protease cascade is somewhat
analogous to the coagulation pathway and complement system (CS) in human
plasma (Jiang et al., 2003). Like the melanization, the fast activation of the
complement system after a microorganism infection of a potential host is a crucial
step in clearance of many pathogens. For example, anaphylatoxins like C3a and
C5a, products of the CS cascade, are commonly involved in exacerbated
inflammatory reactions that can cause direct harm to the host following infections
(Garcia et al., 2013). We know that GelE destroys the C3a complement of human
cells and AMPs of G. mellonella (Park et al., 2007; Park et al., 2008). Taking into
account that the serine protease cascade during melanization is analogous to the
complement system, we hypothesize that in humans, Fsr regulated components
promote an exacerbated complement response. Future studies should investigate
this hypothesis.
The study presented in this thesis is important for the understanding the E. faecalis
Fsr system biology and how it interferes with Drosophila innate immune system. The
conservation between the innate immune system of humans and Drosophila, it will
allow us/future studies to develop new targets to control E. faecalis infections in
humans.

GENERAL DISCUSSION
183
Cha
pter
VI
Figure 4 – Drosophila Immune response upon infection with E. faecalis V583. When Drosophila is
infected by E. faecalis, three immune responses are activated: Phagocytosis (orange panel), the melanization (green
panel) and the Toll pathway (blue panel). Orange panel: After an E. faecalis V583 infection, the haemocytes (green)
capture the bacteria (blue dots in the picture) but do not kill the bacteria. Green panel: The melanization is the first
immune response activated when Drosophila is infected. Recognition of E. faecalis V583 triggers a serine protease
cascade that activates phenol oxidase (PPO), a key enzyme in the melanin biosynthetic pathway. During this
process reactive oxygen species (ROS) are produced which are toxic to the bacteria (pathogen killing) and, in
excess, to the host (wound healing and tissue damage). Serpin-type protease inhibitors are involved in the control of
this cascade. Our results suggest that the Fsr interferes with the control of melanization and promotes an excess of
ROS and consequent death. Blue panel: Drosomycin is an AMP produced when Drosophila is infected by Gram
positive bacteria, such as E. faecalis. Toll pathway is activated and drosomycin is produced by the fatbody. It is
known that melanization and production of drosomycin are connected (Tang, 2009).

Chapter VI
184
3. FUTURE PRESPECTIVES
- Fsr system can be a future target for therapy
The QS system allows bacteria to produce, detect, and exchange signalling
molecules. This process is crucial to disease development because it ultimately
controls the way that bacteria express “virulence factors” — the molecules that
enable the bacteria to colonize, evade and compromise the host's immune response,
exit and enter cells, and obtain nutrients. Activation of virulence genes at low
population density would result in the generation of host defence responses, thus
providing the host an early lead over the invading bacteria. Therefore, in order to
guarantee their survival, bacteria synthesized virulence factors until their number
make certain their success in the infection process. The discovery of new therapeutic
approaches to target bacterial virulence is essential, owing to the increasing
emergence of bacterial strains that are acquiring resistance to antibiotics (Defoirdt et
al., 2013; Raina et al., 2009).
Blocking the Fsr QS and inhibiting virulence seems to be one of the most appealing
uses of the knowledge acquired about cell to-cell communication in E. faecalis. Jiro
Nakayama laboratory already proposed peptide antagonists of GBAP that inhibit the
Fsr QS. However, the peptides effectiveness in the host has not been tested yet
(Nakayama et al., 2007; Nakayama et al., 2009; Nakayama et al., 2013).
Our study gives us new clues on the Fsr contribution to E. faecalis biology and during
infection. Through transcription studies we discovered new genes regulated by Fsr
and involved in E. faecalis virulence. Altogether, we propose that inhibition of Fsr can
interfere with different cell processes in bacteria and in the host but does not kill the
bacteria. Adding to this, blocking the Fsr QS does not give us an immediate
decrease of E. faecalis virulence, because even though inhibition of GBAP signalling
is effective, we still have a basal expression and production of GelE and SprE. Even
in small concentration, the virulence factors regulated by Fsr are powerful. We only
had a significant decrease on virulence when we infected the host with the mutant for

GENERAL DISCUSSION
185
Cha
pter
VI
fsrB, gelE and sprE genes. A target that blocks Fsr needs to ensure that QS is shut
down and that the basal production of virulence factors is eliminated.
But the use of new therapeutics based in QS blocking is not straightforward. Studies
in S. aureus and P. aeruginosa, relevant pathogenic bacteria in hospital environment,
showed that QS-cheaters are frequently isolated during colonization of untreated
colonized patients (Kohler et al., 2010; Paulander et al., 2013). The most likely
explanation for this advantage is that the mutants exploit the wild type public goods,
without paying the metabolic cost of their production; although other direct costs of
QS in clinical contexts cannot be ruled out (Kohler et al., 2010). When QS-gene
expression is reduced by the QS inhibiting drug, this advantage of QS-cheater is lost
and virulent isolates predominate both in colonized patients and during in vitro
experiments. These results suggest that QS-blockage may increase the prevalence
of more virulent QS-responders among colonizing isolates in the hospital
environment (Kohler et al., 2010). Recently, the antibiotic azithromycin, that blocks
the QS of P. aeruginosa, was proposed for the prevention of ventilator-associated
pneumonia in patients with prolonged colonization and the results suggest that
virulence inhibition is a promising anti-microbial strategy (van Delden et al., 2012).
Taking into account these recent studies, the use of QS-blockers needs to be
carefully studied but can be a powerful tool for the control or prevention of E. faecalis
infection in the hospital environment.
Altogether, the work developed in this thesis leads us to propose a novel therapeutic
strategy against E. faecalis based on the Fsr system, by mediating the prevention or
elimination of cell communication and virulence factors that cause host damage.
Nevertheless, the Fsr role during melanization process needs to be further explored
in order to understand which part of the cascade is blocked. It will be interesting to
elucidate the role of each gene regulated by Fsr during infection and in the
melanization control. Concerning the effect of vancomycin on the Fsr QS, it would be
interesting to further exploit the possibility of using this knowledge to produce a new
and effective therapeutic strategy.

Chapter VI
186
4. BIBLYOGRAPHY
Ayres, J. S. & Schneider, D. S. (2012). Tolerance of infections. Annu Rev Immunol
30, 271-294.
Bisicchia, P., Bui, N. K., Aldridge, C., Vollmer, W. & Devine, K. M. (2011).
Acquisition of VanB-type vancomycin resistance by Bacillus subtilis: the impact on
gene expression, cell wall composition and morphology. Mol Microbiol 81, 157-178.
Bourgogne, A., Hilsenbeck, S. G., Dunny, G. M. & Murray, B. E. (2006).
Comparison of OG1RF and an isogenic fsrB deletion mutant by transcriptional
analysis: the Fsr system of Enterococcus faecalis is more than the activator of
gelatinase and serine protease. J Bacteriol 188, 2875-2884.
Cerenius, L., Lee, B. L. & Soderhall, K. (2008). The proPO-system: pros and cons
for its role in invertebrate immunity. Trends in immunology 29, 263-271.
Chong, G., Kimyon, O. & Manefield, M. (2013). Quorum Sensing Signal Synthesis
May Represent a Selective Advantage Independent of Its Role in Regulation of
Bioluminescence in. PLoS One 8, e67443.
Czaran, T. & Hoekstra, R. F. (2009). Microbial communication, cooperation and
cheating: quorum sensing drives the evolution of cooperation in bacteria. PLoS One
4, e6655.
Defoirdt, T., Brackman, G. & Coenye, T. (2013). Quorum sensing inhibitors: how
strong is the evidence? Trends Microbiol 21, 619-624.

GENERAL DISCUSSION
187
Cha
pter
VI
Garcia, C. C., Weston-Davies, W., Russo, R. C. & other authors (2013).
Complement C5 activation during influenza A infection in mice contributes to
neutrophil recruitment and lung injury. PLoS One 8, e64443.
Jiang, H., Wang, Y., Yu, X. Q., Zhu, Y. & Kanost, M. (2003). Prophenoloxidase-
activating proteinase-3 (PAP-3) from Manduca sexta hemolymph: a clip-domain
serine proteinase regulated by serpin-1J and serine proteinase homologs. Insect
Biochem Mol Biol 33, 1049-1060.
Kohler, T., Perron, G. G., Buckling, A. & van Delden, C. (2010). Quorum sensing
inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS
Pathog 6, e1000883.
Lemaitre, B. & Hoffmann, J. (2007). The host defense of Drosophila melanogaster.
Annu Rev Immunol 25, 697-743.
Nakayama, J., Tanaka, E., Kariyama, R. & other authors (2007). Siamycin
attenuates fsr quorum sensing mediated by a gelatinase biosynthesis-activating
pheromone in Enterococcus faecalis. J Bacteriol 189, 1358-1365.
Nakayama, J., Uemura, Y., Nishiguchi, K., Yoshimura, N., Igarashi, Y. &
Sonomoto, K. (2009). Ambuic acid inhibits the biosynthesis of cyclic peptide
quormones in gram-positive bacteria. Antimicrob Agents Chemother 53, 580-586.
Nakayama, J., Yokohata, R., Sato, M. & other authors (2013). Development of a
peptide antagonist against fsr quorum sensing of Enterococcus faecalis. ACS Chem
Biol 8, 804-811.

Chapter VI
188
Park, S. Y., Kim, K. M., Lee, J. H., Seo, S. J. & Lee, I. H. (2007). Extracellular
gelatinase of Enterococcus faecalis destroys a defense system in insect hemolymph
and human serum. Infect Immun 75, 1861-1869.
Park, S. Y., Shin, Y. P., Kim, C. H., Park, H. J., Seong, Y. S., Kim, B. S., Seo, S. J.
& Lee, I. H. (2008). Immune evasion of Enterococcus faecalis by an extracellular
gelatinase that cleaves C3 and iC3b. J Immunol 181, 6328-6336.
Paulander, W., Nissen Varming, A., Baek, K. T., Haaber, J., Frees, D. & Ingmer,
H. (2013). Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus
aureus. MBio 3, e00459-00412.
Raina, S., De Vizio, D., Odell, M., Clements, M., Vanhulle, S. & Keshavarz, T.
(2009). Microbial quorum sensing: a tool or a target for antimicrobial therapy?
Biotechnology and applied biochemistry 54, 65-84.
Rumbaugh, K. P., Trivedi, U., Watters, C., Burton-Chellew, M. N., Diggle, S. P. &
West, S. A. (2012). Kin selection, quorum sensing and virulence in pathogenic
bacteria. Proc Biol Sci 279, 3584-3588.
Sieradzki, K. & Tomasz, A. (1997). Inhibition of cell wall turnover and autolysis by
vancomycin in a highly vancomycin-resistant mutant of Staphylococcus aureus. J
Bacteriol 179, 2557-2566.
Singh, K. V., Nallapareddy, S. R., Nannini, E. C. & Murray, B. E. (2005). Fsr-
independent production of protease(s) may explain the lack of attenuation of an
Enterococcus faecalis fsr mutant versus a gelE-sprE mutant in induction of
endocarditis. Infect Immun 73, 4888-4894.

GENERAL DISCUSSION
189
Cha
pter
VI
Strickertsson, J. A., Desler, C., Martin-Bertelsen, T., Machado, A. M., Wadstrom,
T., Winther, O., Rasmussen, L. J. & Friis-Hansen, L. (2013). Enterococcus
faecalis infection causes inflammation, intracellular oxphos-independent ROS
production, and DNA damage in human gastric cancer cells. PLoS One 8, e63147.
Tang, H. (2009). Regulation and function of the melanization reaction in Drosophila.
Fly 3, 105-111.
Tao, T., Snavely, M. D., Farr, S. G. & Maguire, M. E. (1995). Magnesium transport
in Salmonella typhimurium: mgtA encodes a P-type ATPase and is regulated by
Mg2+ in a manner similar to that of the mgtB P-type ATPase. J Bacteriol 177, 2654-
2662.
Teixeira, N., Varahan, S., Gorman, M. J. & other authors (2013). Drosophila host
model reveals new enterococcus faecalis quorum-sensing associated virulence
factors. PLoS One 8, e64740.
Thomas, V. C., Thurlow, L. R., Boyle, D. & Hancock, L. E. (2008). Regulation of
autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular
proteases influences biofilm development. J Bacteriol 190, 5690-5698.
Thomas, V. C., Hiromasa, Y., Harms, N., Thurlow, L., Tomich, J. & Hancock, L.
E. (2009). A fratricidal mechanism is responsible for eDNA release and contributes to
biofilm development of Enterococcus faecalis. Mol Microbiol 72, 1022-1036.
Uratani, Y. & Hoshino, T. (1984). Pyocin R1 inhibits active transport in
Pseudomonas aeruginosa and depolarizes membrane potential. J Bacteriol 157,
632-636.

Chapter VI
190
van Delden, C., Kohler, T., Brunner-Ferber, F., Francois, B., Carlet, J. &
Pechere, J. C. (2012). Azithromycin to prevent Pseudomonas aeruginosa ventilator-
associated pneumonia by inhibition of quorum sensing: a randomized controlled trial.
Intensive Care Med 38, 1118-1125.

GENERAL DISCUSSION
191
Cha
pter
VI






![Natural Anti-Quorum Sensing agents against Pseudomonas ... · 2. Quorum Sensing: a Novel Target Vfr Quorum sensing (QS) is a population-dependent event [13]. The capability to sense](https://img.pdfslide.us/doc/110x75/5edbcc02ad6a402d66663060/natural-anti-quorum-sensing-agents-against-pseudomonas-2-quorum-sensing-a.jpg)


![A MATHEMATICAL MODEL OF QUORUM SENSING IN PATCHY … · QUORUM SENSING IN BIOFILMS 269 [11, 52], a comprehensive understanding of quorum sensing systems is highly desirable. 1.3 Modelling](https://img.pdfslide.us/doc/110x75/5fad596118f3d853d1006484/a-mathematical-model-of-quorum-sensing-in-patchy-quorum-sensing-in-biofilms-269.jpg)



![Research Article Broad Spectrum Anti-Quorum Sensing ...downloads.hindawi.com/journals/scientifica/2016/5823013.pdf · isms is called quorum sensing (QS) []. Quorum sensing is a process](https://img.pdfslide.us/doc/110x75/5edbc5d7ad6a402d66662749/research-article-broad-spectrum-anti-quorum-sensing-isms-is-called-quorum-sensing.jpg)















