Embed Size (px)
Citation preview

J. clin. Path. (1964), 17, 371
Fluorescence of solutions: A review
R. T. WILLIAMS AND J. W. BRIDGES
From the Department ofBiochemistry, St. Mary's HospitalMedical School, London
The use of fluorescence as an analytical techniquein clinical biochemistry and pathology is in theprocess of developing and it is therefore importantthat those who use it should understand the under-lying principles governing the phenomenon offluorescence and its application, so that this elegantand sensitive technique can be used to its greatestadvantage. Fluorescence has been studied byphysicists and photochemists for over a century butthe results of these studies have penetrated butslowly into biology and medicine, partly due todifficulties of communication and partly becausethe compounds, temperatures, and solvents usedwere of little interest from the biological point ofview.
Present-day research in medicine and biochemistryrequires many highly sensitive analytical techniquesand workers in these fields frequently have todevelop their own physical instruments to meet theneeds of their researches. This type of developmenthas occurred in the field of fluorescence, for thefirst commercial spectrofluorimeter (Aminco-Bow-man spectrophotofluorometer; see section onfluorimeters) was based upon a model devised by amedically qualified scientist, Dr. R. L. Bowman(Bowman, Caulfield, and Udenfriend, 1955). Withthis instrument is it possible to measure not onlyvisible but also ultraviolet fluorescence, and torecord the wavelengths of maximum fluorescenceand of maximum excitation or activation. Somevitamins and drugs have been assayed fluori-metrically for many years (Bowen and Wokes, 1953)but this earlier work was done with filter fluori-meters which are less sensitive and less selectivethan the modern spectrofluorimeters (or fluorescencespectrometers) and usually only measure visiblefluorescence.
Fluorescence is a highly sensitive analytical toolwhich can be used to measure concentrations as lowas 10-8 to 10-10 g./ml. (0-01 to 0-0001 jug./ml.)whereas few substances can be estimated colori-metrically below 10-7 g./ml. (0-1 ,ug./ml.). Since theuse of fluorescence in analysis is in its infancy onecan expect improvements in instrumentation suchas better detectors of fluorescence and better light
sources. There is no doubt that fluorescence analysiswill become even more sensitive and more accuratethan it is at present.
THE MECHANISM OF FLUORESCENCE
In this section is it proposed to give a simplifiedaccount of the origin of fluorescence and to givesome guidance as to what sort of molecules onecould expect to fluoresce. Fluorescence is essentiallyan electronic phenomenon and is primarily con-cerned with light of wavelengths in the region of 200to 800 m,t. Some compounds when illuminated withlight of this region only absorb specific wavelengthsof this light and the wavelengths absorbed arecharacteristic of the particular- compound beingexamined. The extent to which the light of thesewavelengths is absorbed constitutes the absorptionspectrum of the compound. As a consequence of theabsorption of light some of the molecules of the com-pound become excited because certain electrons inthe molecule are raised to a higher energy level. Thisis shown diagrammatically in Figure 1. The energylevels1 are represented in Fig. 1 by horizontal linesand the directions of the energy transitions byvertical or inclined arrows. On absorption of light,the molecule is raised from the ground state G tothe excited state E as indicated by the verticalarrow A. The molecule returns to the ground state,emitting some of its absorbed energy as fluorescence,as indicated by the arrow marked F. An electronictransition due to light absorption is almost instant-aneous (10-15 second), whereas the lifetime of theexcited state is about 10-8 second, and therefore,the whole process of light absorption and fluor-escence emission takes place in about 10-8 second.Now some of the absorbed energy is lost partly bycollisions with other molecules and partly by othermeans, so that less energy is emitted as fluorescencethan was absorbed from the exciting light. Accordingto the quantum theory, light is absorbed in discreteunits called quanta and the energy (E) of a quantumis related to vibrational frequency (v), thus:'To simplify the picture only the lowest vibrational level of eachenergy level is shown.
371
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
E
G
Excited state (singlet)
Triplet state
FIG. 1. Diagram relating energy levels,absorption, fluorescence, and phosphorescence.
Ground state (singlet)
E = hv, where h = Planck's constant. Frequencyis related to wavelength (A) according to the
cexpression, v = A' where c is the velocity of light.
From these two equations, E = hA and, since h and
c are constants, E varies inversely as A. As mentionedabove, the energy emitted as fluorescence is lessthan the light energy absorbed and therefore fromthe last equation the wavelength of fluorescence islonger than that of the absorbed light.The mechanism of phosphorescence is also
illustrated in Figure 1. This phenomenon isdistinguished from fluorescence by the much longerlife time of the excited state (which may be up toseveral seconds). To understand the reason for thisdifference one must consider the spin of the electronsof a molecule. Electrons in most molecules arefound in even numbers and are paired. In each pair,the two electrons spin about their own axes inopposite directions (anti-parallel spins, see below)and, for reasons which need not be considered here,such molecules are said to have a 'singlet' electroniclevel. When the molecule is raised to the excitedstate two things could happen: (1) the electrons mayremain 'singlet' and the molecule can then returnto the ground state with the emission of fluorescence(path F), or, (2) one electron, by some internalenergy transition, may have its spin reversed(parallel spins, see below) and the molecule is thensaid to have a 'triplet' electronic level:
anti-parallel spin parallel spin
In Fig. 1, this change in indicated by the topinclined arrow t and the 'triplet' excited state isindicated by the horizontal line T. Such a moleculeis regarded to be in a metastable state. The life timeof the triplet excited state is longer than that of thesinglet excited state, for the molecule has to return
to the ground singlet state by what is known as a'forbidden transition' which has a low probability.During this return there is emission of phosphor-escence as indicated by the inclined arrow P. In thetransition from the singlet excited state to the tripletstate there is a loss of energy, for the energy level Tis lower than E. Therefore, there is less energyemitted in the transition from T to G, so that thewavelength of phosphorescence is longer than thewavelength of the fluorescence that would have beenproduced by the same excitation. Fluorescence isthus an emission from a singlet excited state (electronspin paired) whereas phosphorescence is emissionfrom a triplet excited state (electrons unpaired).Phosphorescence persists longer than fluorescenceand this persistence is prolonged and the intensityenhanced by low temperatures. The biologicalapplications of phosphorescence are as yet limited.
FLUORESCENT COMPOUNDS An examination of theliterature shows that not all organic compounds arefluorescent. However, those which show fluorescenceare usually aromatic or contain conjugated doublebonds (i.e., alternating single and double bondsbetween atoms). Since fluorescence is an electronicphenomenon, it might be expected that thosemolecules containing electrons which undergoenergy transitions readily would be capable offluorescing. Such electrons are (a) the so-called'IT', 'mobile', or 'delocalized', electrons and (b) the'lone pair' electrons2. The nature ofi'-electrons maybe explained briefly as follows. The application ofquantum mechanics to molecular theory has shownthat substances containing two or more conjugateddouble bonds (i.e., -C=C.C=C-, etc.) have acertain number of electrons possessing greatermobility than the other electrons (i.e., a electrons) ofthe molecule. One of these 'ir-electrons is derived fromeach carbon atom associated with the double bond,and these electrons form a cloud which circulatesthe molecule (Fig. 2). Thus benzene, which has six2The 'lone pair' is a pair of electrons not directly concerned inchemical bonding and they are found particularly associated withN, 0, P, and S atoms in compounds. They may interact with 7r-electrons to increase an electron cloud, but usually on excitation theyyield phosphorescence rather than fluorescence (Kasha, 1960).
372
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solutions: A review
carbon atoms and the equivalent of three conjugateddouble bonds, has six of these electrons whoseorbits form a cloud above and below the molecule.None of these electrons has any special relation toone particular carbon atom, but all six electrons areequally related to all six carbon atoms. The otherelectrons of the molecule, termed a electrons, arelocalized and tend to be located along the linejoining the nuclei of the two participating atoms.The freedom of the IT-electrons, however, can beinfluenced by substituent groups and, in hetero-cyclic systems, by heteroatoms, i.e., atoms otherthan carbon, such as 0, N, and S, so that theIT-electrons become relatively more or less delocalizedunder their influence.
a) -C=C-C=C-
b) :C::C:C::C:
[ 7T<; 7-electron cloudc) :C:C:C:C:
FIG. 2. Diagram showing the 7r-electrons of conjugateddouble bonds.(a) Formal way ofshowing conjugated double bonds(b) The electrons of the bonds(c) Representation of the 7r-electrons; the electronsbetween the C atoms are the a electrons.
If a compound contains 7T-electrons there is agood possibility that it will fluoresce, and if asubstituent, which increases the freedom of theseelectrons, is added to the compound, then thesubstituted compound is likely to be more fluor-escent than the unsubstituted parent compound.On the other hand, if the substituent tends tolocalize the I-electrons, there will be a diminution or
abolition of fluorescence. Let us take the simpleexamples of cyclohexane, benzene, and vitamin A.Cyclohexane contains no conjugated double bondsand is non-fluorescent. Benzene is an aromaticcompound and is weakly fluorescent. Vitamin A isnot aromatic, but contains five conjugated doublebonds and is therefore fluorescent (see below):
H2
OHa Ha
H2( )2Ha
Cyclohexane, non-fluorescent no absorptionpeak
Benzene absorption peak, 254 mM;excitation max. 269 mA (observed);fluorescence max. 291 mA (Bridges andWilliams, 1963)
CH, CH3 CH, CH3CH,OH
CH3
Vitamin A absorption 325 m,u; excitation 327 m,(corrected); fluorescence 510 m j. (Hagins and Jennings,
1959)
It will be noted that the excitation maximum ofbenzene has a longer wavelength than the absorptionpeak, whereas the excitation of vitamin A is fairlyclose to the absorption peak. Theoretically theabsorption and excitation peaks should coincide,and the differences that occur are due to instru-mental errors largely as a result of the variation inenergy output with wavelength of the light sourceused for excitation (see p. 16 and Fig. 11). Theseerrors can be corrected if necessary and whenthese corrections are made the excitation maximumis found to be close to the absorption peak.The effects of substituents upon fluorescence can
be illustrated with benzene, aniline, and nitro-benzene. In dilute solutions in water, aniline is40 to 50 times more fluorescent than benzene,whereas nitrobenzene is non-fluorescent (Bridgesand Williams, 1962).
NH2
0AnilineExc. 290 mmiFlu. 345 mmA
NO2
Nitrobenzenenon-fluorescent
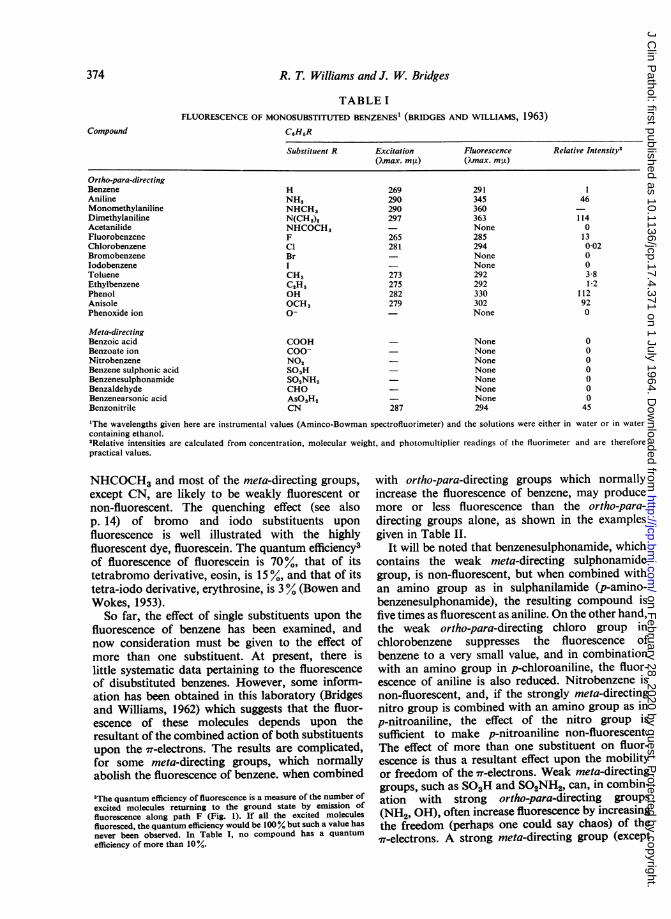
The NH2 group in aniline tends to activate thebenzene ring and thus increases the freedom of the7T-electrons. The NO2 group in nitrobenzene, on theother hand, deactivates the ring by tending towithdraw the 7rT-electrons from the ring thus reducingtheir freedom by increasing their localization. Nowthe NH2 group is one of the classical ortho-para-directing groups and the NO2 group is one of theclassical meta-directing groups, so that one mightconclude that mono-substituted benzenes containingortho-para-directing groups are fluorescent whereasthose containing meta-directing groups are non-fluorescent. This conclusion is partly, but notwholly, true, as can be seen from Table I.The data quoted in Table I were obtained in this
laboratory (Bridges and Williams, 1962; 1963) withthe Aminco-Bowman spectrofluorimeter and it isimportant to indicate how much of the compoundsquoted were needed to produce fluorescence.Benzene, ethylbenzene, and chlorobenzene areweakly fluorescent and with the first two at least20 to 25 ,ug./ml. was needed to detect fluorescence,whilst with chlorobenzene at least 500 ,ug./ml. wasneeded. With aniline and phenol, fluorescencecould be detected down to concentrations of 0-01,ug./ml. The main points about this table are thataromatic compounds containing NH2, OH, F,OCH3, NHCH3, and N(CH3)2 groups are likely tobe fluorescent, whilst those containing Cl, Br, I,
373
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
374
Compound
R. T. Williams and J. W. Bridges
TABLE IFLUORESCENCE OF MONOSUBSTITUTED BENZENES1 (BRIDGES AND WILLIAMS, 1963)
C,H,R
Substituent R
Ortho-para-directingBenzeneAnilineMonomethylanilineDimethylanilineAcetanilideFluorobenzeneChlorobenzeneBromobenzeneIodobenzeneTolueneEthylbenzenePhenolAnisolePhenoxide ion
Meta-directingBenzoic acidBenzoate ionNitrobenzeneBenzene sulphonic acidBenzenesulphonamideBenzaldehydeBenzenearsonic acidBenzonitrile
HNH,NHCH,N(CH3)2NHCOCH,FClBrICH,C,H,OHOCH,0-
COOHCOO-NO,SO,HSO,NH,CHOAsO3H2CN
ExcitationQ,max. m)
269290290297
265281
273275282279
287
Fluorescence(Qmax. m i)
291345360363None285294NoneNone292292330302None
NoneNoneNoneNoneNoneNoneNone294
Relative Intensity'
461140130-02003-8i 2
112920
0000000
45
'The wavelengths given here are instrumental values (Aminco-Bowman spectrofluorimeter) and the solutions were either in water or in watercontaining ethanol.2Relative intensities are calculated from concentration, molecular weight, and photomultiplier readings of the fluorimeter and are thereforepractical values.
NHCOCH3 and most of the meta-directing groups,
except CN, are likely to be weakly fluorescent or
non-fluorescent. The quenching effect (see alsop. 14) of bromo and iodo substituents upon
fluorescence is well illustrated with the highlyfluorescent dye, fluorescein. The quantum efficiency3of fluorescence of fluorescein is 70%, that of itstetrabromo derivative, eosin, is 15 %, and that of itstetra-iodo derivative, erythrosine, is 3 % (Bowen andWokes, 1953).So far, the effect of single substituents upon the
fluorescence of benzene has been examined, andnow consideration must be given to the effect ofmore than one substituent. At present, there islittle systematic data pertaining to the fluorescenceof disubstituted benzenes. However, some inform-ation has been obtained in this laboratory (Bridgesand Williams, 1962) which suggests that the fluor-escence of these molecules depends upon theresultant of the combined action of both substituentsupon the 7r-electrons. The results are complicated,for some meta-directing groups, which normallyabolish the fluorescence of benzene. when combined
'The quantum efficiency of fluorescence is a measure of the number of
excited molecules returning to the ground state by emission of
fluorescence along path F (Fig. 1). If all the excited molecules
fluoresced, the quantum efficiency would be 100% but such a value has
never been observed. In Table I, no compound has a quantumefficiency of more than 10 %.
with ortho-para-directing groups which normallyincrease the fluorescence of benzene, may producemore or less fluorescence than the ortho-para-directing groups alone, as shown in the examplesgiven in Table II.
It will be noted that benzenesulphonamide, whichcontains the weak meta-directing sulphonamidegroup, is non-fluorescent, but when combined withan amino group as in sulphanilamide (p-amino-benzenesulphonamide), the resulting compound isfive times as fluorescent as aniline. On the other hand,the weak ortho-para-directing chloro group inchlorobenzene suppresses the fluorescence ofbenzene to a very small value, and in combinationwith an amino group in p-chloroaniline, the fluor-escence of aniline is also reduced. Nitrobenzene isnon-fluorescent, and, if the strongly meta-directingnitro group is combined with an amino group as inp-nitroaniline, the effect of the nitro group issufficient to make p-nitroaniline non-fluorescent.The effect of more than one substituent on fluor-escence is thus a resultant effect upon the mobilityor freedom of the w-electrons. Weak meta-directinggroups, such as SO3H and SO2NH2, can, in combin-ation with strong ortho-para-directing groups(NH2, OH), often increase fluorescence by increasingthe freedom (perhaps one could say chaos) of theit-electrons. A strong meta-directing group (except
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solhtions: A review
TABLE IIFLUORESCENCE OF SOME DISUBSTITUTED BENZENES COMPARED WITH
RELATED MONOSUBSTITUTED BENZENES (BRIDGES AND WILLIAMS, 1962)
R O R'
R
AnilineBenzenesulphc namideSulphanilamideChlorobenzenep-ChloroanilineNitrobenzenep-Nitroaniline
NH,HNH,HNH,HNH,
HSO,NH,SO,NH,Cl
Cl
NO,NO,
R' Excitation Fluorescence Relative Intensity'
290
275281300
350None350294360NoneNone
460
2200-02
320
0
'Benzene = 1
CN)4 will usually tend to diminish or suppressfluorescence. A good example is the nitro group, formost simple nitro derivatives are non-fluorescent.Two examples are shown below. Diphenyloxazole(PPO) (Ott, Hayes, Hansbury, and Kerr, 1957) isfluorescent but its nitro derivative is not.
2, S-Diphenyloxazole(PPO) fluorescent (flu: 365and 381 mg in toluene)
ON
2-Phenyl-S (p-nitrophenyl)oxazole non-fluorescent
(Diphenyloxazole derivatives are used for scintil-lation counting which depends on the fluorescenceof the scintillator when excited by radioactivity.The scintillator POPOP is 2,2'-p-phenylenebis-(5-phenyloxazole) which has a fluorescence efficiencyapproaching 100%.)
0
2,2'-p-phenylenebis-(5-phenyloxazole);POPOP (flu: 420 and 440 mjs in toluene)
Nevertheless, if there is sufficient conjugation in themolecule to overcome the effect of the nitro group,a molecule containing a nitro group can be fluor-escent, as in the case of 5-dimethylamino-4'-nitro-stilbene below (Lippert, Luder, and Moll, 1959).
CJ\N e CH=CH N02
Fluorescent (A max. ca. 740 m,u in isobutanol)
Again benzoic acid, which contains the m-
directing COOH group, is non-fluorescent, but incombination with the OH group, as in the hydroxy-
4The CN group has a triple bond, i.e., C N, and the electrons ofthis bond can interact with the Tr-electron cloud of the benzene ringto produce the conditions for fluorescence.
benzoic acids or the NH2 group as in the amino-benzoic acids, is does not abolish fluorescence andin some cases, e.g. o-hydroxybenzoic (salicylic) acidand o-aminobenzoic (anthranilic) acid, the fluor-escence is greater than that of either phenol oraniline.
In polycyclic aromatic systems, the number of7r-electrons available is greater than in benzene andtherefore these compounds and their derivatives areusually much more fluorescent than benzene and itsderivatives. Naphthalene, anthracene, and biphenylderivatives, for example, are much more fluorescentthan the corresponding fluorescent benzene deriv-atives.
Naphthalene (flu: ca.323, 340 mA inethanol; Parker andRees, 1962)
Anthracene (flu: ca.372, 400, 425 mg inethanol; Parker andRees, 1962)
Biphenyl (flu: 318mjA in ethanol;Bridges, Creaven,and Williams, 1963)
The fluorescence of heterocyclic systems dependsupon the nature of the hetero atom and uponsubstituents. The hetero atoms or groups usuallyencountered are -N=, >NH, -0-, and -S-. Doublybound nitrogen, i.e., -N=, in a heterocyclic systemtends to deactivate the ring by drawing the 7r-
electrons towards it. Such rings are said to be7T-deficient. Heterocyclic systems containing doublybound nitrogen tend to be non-fluorescent unlessthere are substituents present which counteract theeffect. Thus pyridine is non-fluorescent (Kerr,Hayes, and Ott, 1957), whereas 3-hydroxypyridine isfluorescent (Bridges, Davies, and Williams, 1963), be-causeof the effect of the electron-donating OH group.
N
HOr<
Pyridine non-fluorescent 3-Hydroxypyridine fluorescent(see p. 20)
C onmpound
375
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
When >NH, -0-, and --- occur in the heterocyclicsystem, there is a tendency for them to contributeto the wr-electron system. Such rings are callediT-excessive and the order of contribution is-NH->-O->-S-. There is, therefore, a tendencyfor compounds containing such ring systems to befluorescent. i.e., those containing the pyrrol, furan,and thiophene rings, although the fundamentalring compounds themselves may not be fluorescentor only weakly fluorescent. When the heterocyclic
NiH
Pyrrol Furan Thiophene
system has more than one type of hetero atom,then fluorescence will depend upon the resultant ofthe effect of the two hetero atoms on the 7T-electrons,and on the effect of any substituents. Thus thiazoleand isothiazole derivatives tend to be non-fluor-escent because the effect of the -N= upon thenf-electrons is greater than -S-, but oxazole andisoxazole derivatives could be expected to fluoresce(Ott et al., 1957) because the effect of -O- is greaterthancyclicfluor4374).
CHEMpounpossesimp]comitheirtypeinduc
and, although the nature of these products5 isunknown, their fluorescence can be used for theassay of cortisol in minute quantities (Mattingly,1962). Chemically induced fluorescence can also beused in other ways. Sometimes the 'native' fluor-escence of a compound is too weak for it to be usefulquantitatively, but by a simple chemical reactionthe compound may be converted into a highlyfluorescent product which can be readily assayedfluorimetrically. Thus the tetracycline antibiotics,which probably have a relatively weak nativefluorescence, can be converted by calcium ions and abarbiturate (usually barbital) into highly fluorescentcomplexes (Kohn, 1961), the suggested structure ofone of which is shown below.
HO CH3 N(CH3)2H H
H H CONH2
HO O 0 ° (Exc. 405 m,u; flu;c / 530 mji)Ca
0 NH ' o
C2 N
0
-N=. However, the substituents in hetero- Closely related substances often have native fluor-- systems play a most important role in the escences so similar to each other that it is impossibleescence of such compounds (see pp. 373 and to estimate one of a group of such substances in the
presence of the others. However, it is frequentlypossible to convert one of these compounds intoanother fluorescent species, so that it can be
NUUN estimated in the presence of its congeners. Thuss o adrenaline and noradrenaline show a native fluor-
Thiazole Isothiazole Oxazole Isoxazole escence which is common to most simple catechols,including catechol itself. Both adrenaline and nor-
IICALLY INDUCED FLUORESCENCE Organic com- adrenaline, however, can be oxidized at pH 6 5 toIds which are fluorescent as such are said to 3,5,6-trihydroxyindoles (adrenolutine and nor-.ss 'native' fluorescence. It is often possible by adrenolutine) which are highly fluorescent atle chemical means to convert non-fluorescent alkaline pH values. The estimation of adrenaline)ounds into fluorescent ones and thereby allow in the presence of noradrenaline can be achieved bydetection and estimation by fluorescence. This oxidizing at pH 3*5, for at this pH adrenaline isof fluorescence is referred to as .chemically converted into a trihydroxyindole whereas nor-ced' fluorescence. Thus the adrenal cortical adrenaline is not (Udenfriend, 1962).
steroid, cortisol, is non-fluorescent; in fact, anexamination of its structure shows that it has aninsufficient number of conjugated double bonds forfluorescence. However, if it is dissolved in con-centrated sulphuric acid in the presence of ethanol,it is converted into intensely fluorescent products,
CHHOH
HO H
t0
Cortisolnon-fluorescent
HO CHOH.CHZ.NHCH3 Adrenaline
HOL> Native fluorescence at
330 mt
Adrenolutine formed byHoe l liOH oxidation of adrenaline:
HO %4 } flluorescence 520 miA in'H3 alkalCH,
«It is believed that strong sulphuric acid has the effect of introducingdouble bonds into the cortisol molecule.
376
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
fluorescence of solutions: A review
Changes in the pH of a solution will sometimesallow the differentiation of two substances withsimilar native fluorescence characteristics. Thus bothphenol and anisole fluoresce at 300 to 310 m,u atneutral pH, but at pH 12 phenol is converted intothe non-fluorescent phenoxide ion whereas anisoleremains unchanged (Rosen and Williams, 1961):
04 o
Fluorescent (flu: 310 Non-fluorescentm,) pH 7 (pH 12)
Fluorescent (flu: 300mg) at pH 7 and 12
FACTORS AFFECTING FLUORESCENCEINTENSITY
The intensity of fluorescence of a compound isaffected by a number of factors, the most importantof which are the following: (a) instrumental factors,(b) concentration, (c) solvent, (d)pH, (e) temperature,and (f) stability of the compound in light. Instru-mental factors are discussed in the section on
fluorimeters (see p. 385).
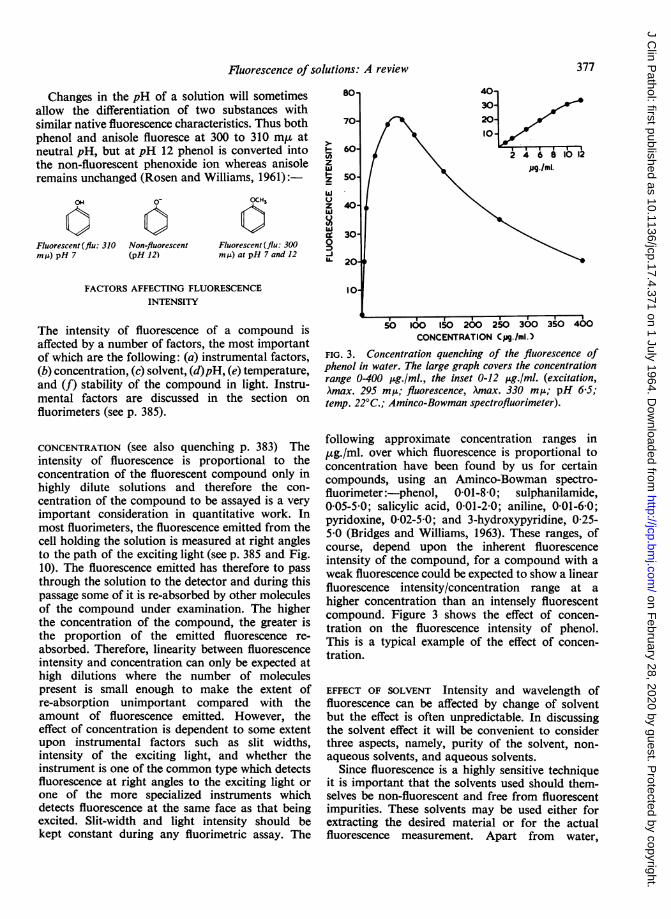
CONCENTRATION (see also quenching p. 383) Theintensity of fluorescence is proportional to theconcentration of the fluorescent compound only inhighly dilute solutions and therefore the con-
centration of the compound to be assayed is a veryimportant consideration in quantitative work. Inmost fluorimeters, the fluorescence emitted from thecell holding the solution is measured at right anglesto the path of the exciting light (see p. 385 and Fig.10). The fluorescence emitted has therefore to passthrough the solution to the detector and during thispassage some of it is re-absorbed by other moleculesof the compound under examination. The higherthe concentration of the compound, the greater isthe proportion of the emitted fluorescence re-absorbed. Therefore, linearity between fluorescenceintensity and concentration can only be expected athigh dilutions where the number of moleculespresent is small enough to make the extent ofre-absorption unimportant compared with theamount of fluorescence emitted. However, theeffect of concentration is dependent to some extentupon instrumental factors such as slit widths,intensity of the exciting light, and whether theinstrument is one of the common type which detectsfluorescence at right angles to the exciting light or
one of the more specialized instruments whichdetects fluorescence at the same face as that beingexcited. Slit-width and light intensity should bekept constant during any fluorimetric assay. The
60 24681012z
50ug./ml.
23I-so
U> 20-
10-
50 100 15O 200 250 300 350 400CONCENTRATION Cpg./mi.)
FIG. 3. Concentration quenching of the fluorescence ofphenol in water. The large graph covers the concentrationrange 0-400 ,ug./ml., the inset 0-12 ,ug./ml. (excitation,Amax. 295 m,u; fluorescence, Amax. 330 mp; pH 6-5;temp. 22°C.; Aminco-Bowman spectrofluorimeter).
following approximate concentration ranges in,ug./ml. over which fluorescence is proportional toconcentration have been found by us for certaincompounds, using an Aminco-Bowman spectro-fluorimeter:-phenol, 0-01-8-0; sulphanilamide,005-50; salicylic acid, 001-2-0; aniline, 001-6-0;pyridoxine, 0-02-5-0; and 3-hydroxypyridine, 0-25-5 0 (Bridges and Williams, 1963). These ranges, ofcourse, depend upon the inherent fluorescenceintensity of the compound, for a compound with aweak fluorescence could be expected to show a linearfluorescence intensity/concentration range at ahigher concentration than an intensely fluorescentcompound. Figure 3 shows the effect of concen-tration on the fluorescence intensity of phenol.This is a typical example of the effect of concen-tration.
EFFECT OF SOLVENT Intensity and wavelength offluorescence can be affected by change of solventbut the effect is often unpredictable. In discussingthe solvent effect it will be convenient to considerthree aspects, namely, purity of the solvent, non-aqueous solvents, and aqueous solvents.
Since fluorescence is a highly sensitive techniqueit is important that the solvents used should them-selves be non-fluorescent and free from fluorescentimpurities. These solvents may be used either forextracting the desired material or for the actualfluorescence measurement. Apart from water,
377
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
solvents used in fluorescence work may include thesimple alcohols from methanol to butanol, ether,benzene, ethylene dichloride, hexane, heptane, etc.All solvents should be checked that they do notcontain any undesirable fluorescence, and otherwiserigorously purified. Details of the purification ofsolvents can be obtained from standard texts(e.g., Udenfriend, 1962). There are other sources offluorescent impurities apart from solvents andparticular attention should be paid to detergentsused to clean glassware. These detergents maythemselves be fluorescent. On the other hand, thereare some cleansing agents which quench fluorescenceand these should be avoided or carefully removed.Chromic acid, for example, absorbs ultraviolet light(see p. 383) and it is preferable to clean cuvettes innitric acid rather than in chromic acid. Somesolvents also absorb specific wavelengths of lighte.g., benzene, and should be avoided for fluorescenceinvolving the regions of their absorption. Apart fromsolvents, the preparation of solutions for fluorescenceassay often involves materials such as adsorbents,e.g., alumina and buffer chemicals, and obviouslythe possibility of the addition of fluorescent impuri-ties or quenching agents from these materials to thesolution to be assayed must be borne in mind.Non-aqueous solvents are not frequently used as
media for fluorescent substances in biological work,but there is no reason why they should not be used.Fluorescence often varies with solvent and severalreasons have been put forward to explain this(Van Duuren, 1963), such as the dielectric constantof the solvent, the association of solvent and soluteby hydrogen bonding, quenching by solventmolecules, and ionization. Indole, for example,shows the same maximum excitation of 285 mp infive solvents, but the wavelength of maximumfluorescence is 297 in cyclohexane, 305 in benzene,310 in dioxan, 330 in ethanol, and 350 m,u in water.The fluorescence wavelength thus increases with thedielectric constant of the solvent, due to an effect onthe 7r-electrons. At 5 ,ug./ml., indole-3-acetic acid isnot fluorescent in cyclohexane or benzene, butfluoresces at 325 in dioxan, 340 in ethanol, and360 m, in water. Chlorophyll shows very littlefluorescence in rigorously dried non-polar solvents,but the addition of a polar solvent (methanol,ethanol, or water) causes a remarkable enhance-ment of fluorescence probably due to hydrogenbonding between chlorophyll and the polar solvent(Livingston, Watson, and McArdle, 1949). Pyrid-oxine fluoresces at 335 m,u in dioxan and 400 m,u inwater and this change is due to ionization. In dioxan,pyridoxine occurs in the uncharged form whilstin water as the dipolar ion (Bridges et al.,1963):
CH2OH CH2.OHCH,OH OH CH1OH CO_
N CH, O H
H
Pyridoxine (molecular). Exc. 296 Pyridoxine (dipolar ion). Exc.mM; flu: 335 mA in dioxan 332 mu; flu: 400 my in water
Another interesting example of ionization is2-hydroxybiphenyl which fluoresces at 348 m,u inethanol and at 415 m,u in water although theexcitation (295 m,t) is the same in both solvents(Bridges, Creaven, Davies, and Williams, 1963).In this case, the difference in fluorescent wave-lengths is due to excited state ionization in waterbut not in ethanol (see p. 380).A good illustration of the effect of solvent upon
fluorescence is shown in Table III, where the intensityof fluorescence of solutions of sulphanilamide(2 ug./ml.) in various solvents is shown. It will benoted that sulphanilamide does not fluoresce in twoketones, in two highly chlorinated methanes, in thenitro compound, nitromethane, and p-xylene. Thefluorescence of sulphanilamide is most intense inthe lower alcohols and least in the aromatic hydro-carbons (Bridges, 1963).
TABLE IIIINTENSITY OF FLUORESCENCE OF SULPHANILAMIDE
IN VARIOUS SOLVENTS1 (BRIDGES, 1963)Solvent Relative
Intensity
WaterMethanolEthanolPropanolIsopropanoln-Butanol
Isobutanoln-PentanolAcetoneButan-2-oneChloroformCarbon tetrachloride
10072107899469
7480000
Solvent RelativeIntensity
1,2-Dichloroethanen-HexaneBenzeneToluenep-XyleneLight petroleum
(b.p. 60-80°)EtherEthyl acetateFormamideDimethylformamideDioxanNitromethane
6942179024
78337044240
'Measured in an Aminco-Bowman spectrofluorimeter. The excitationwavelengths varied slightly with the solvent from 270 m,u in iso-butanol to 305 m,u in toluene. The fluorescence occurred at 350 m«in every case. The fluorescence intensity in water is taken as 100.Concentration of sulphanilamide, 2 ,ug./ml.
RAMAN SPECTRA OF SOLVENTS When very smallamounts of a fluorescent substance in solution haveto be measured, there is the possibility that themeasurement may be interfered with by the so-calledRaman scattering of the solvent. When this occursit is advisable to change the solvent. Ramanscattering is a phenomenon which is commonto gases, liquids, and solids, for when monochro-matic light falls on a substance, some of it isscattered by the molecules of that substance. If thespectrum of the scattered light, which is called a
378
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solutions: A review
Raman spectrum, is examined, it is found to containwavelengths of light which are characteristic of thesubstance illuminated and there is a constantrelationship between the wavelengths of the Ramanlines and the wavelength of the incident light. Infact, Raman spectra are useful in determiningmolecular structure (Cleveland, 1955). However,Raman spectra, unlike fluorescence spectra, have no
absolute excitation and emission wavelengths andcan arise from any wavelength of incident light. If aRaman line of the solvent happens to coincide withthe fluorescence maximum of the compound beingestimated, it could interfere considerably with theestimation and the solvent should be changed.However, Raman scattering is usually weak and mayinterfere only at high dilutions of the fluorescentsubstance. According to Parker (1959), the Ramanspectrum of a solvent can provide a most usefulmeans of checking the day-to-day sensitivity of a
spectrofluorimeter.
BUFFERS In biological work, fluorescence isfrequently measured in aqueous buffer solutions.It is therefore important to know whether theconstituents of the buffer affect the fluorescence.Table IV shows the effect of various buffers uponthe fluorescence of sulphanilamide at 2 ,ug./ml. and itcan be seen that buffers containing 0- IM phosphate,borate, citrate, and phthalate and 0-05M tris-(hydroxymethyl)aminomethane (Tris) have an effecton fluorescence intensity although not a very markedone (Bridges, 1963). It has been reported by Cowgill(1963) that 0-05M Tris buffer (pH 7-5) had noquenching effect upon the fluorescence of indole orphenol. Phosphate, however, quenched both indoleand phenol fluorescence, and in the quenching ofphenol, the di-anion, HPO2-, was more effective
TABLE IVEFFECT OF BUFFER COMPOSITION ON THE
FLUORESCENCE OF SULPHANILAMIDE' (BRIDGES, 1963)Solvent pH of Solution Fluorescence
Intensity'
Water3 4-3 27Water3 8-1 27
[40 25152 26
0OM-Citrate-Na,HPO4 buffer 6-0 247-1 23
L8-0 22O-IM-K,HPO,-KH,PO4 buffer 7-0 21-50-05-M-Tris-HCI buffer 7-0 26-5Burroughs & Wellcome buffers:0 IM-phthalate 4-0 2501 M-phosphate 6-99 240- IM-borate 915 23
12 ,ug./ml.'Paotomultiplier reading on the Aminco-Bowman spectrofluori-meter with excitation at 273 m,u and fluorescence at 350 mnL.3Glass-distilled water to which a drop of dilute HC1 or NaOH wasadded.
than the mono-anion, H2PO , whilst in thequenching of indole the mono-anion was moreeffective than the di-anion. Both acetic acid andsodium acetate are weak quenchers of tyrosinefluorescence but, in the case of the fluorescence oftryptophan, acetic acid but not sodium acetate is aquencher. Commercial buffer solutions containconstituents which are not always disclosed by themanufacturers. These constituents could have afluorescence of their own or they could be quenchingagents. Buffers therefore should always be checkedfor their effects on fluorescence. In the case ofphosphate buffers, an increase of phosphate con-centration frequently leads to a diminution influorescence intensity.
pH EFFECTS Many biologically important com-pounds are capable of undergoing ionization and theionic forms of a compound often have differentfluorescence characteristics from the unionized form.The effect of pH upon the fluorescence of a com-pound is thus of considerable importance, and aknowledge of the changes in fluorescence broughtabout bypH changes in the medium can be valuablefrom several aspects. Thus a compound may befluorescent only over a short range of pH, as in thecase of sulphapyridine, which is practically non-fluorescent abovepH 4 and is maximally fluorescentatpH 0 5 to 1 0 (Bridges, 1963). Again, a compoundmay be fluorescent over a considerable range ofpH,but over a certain section of that range it may bemuch more fluorescent than over the rest. Thussalicylic acid shows some fluorescence from pH0 to 14, but over the rangepH 4 to 12, it is 100 timesas fluorescent as at pH 1 (Rosen and Williams, 1961).pH can also be used to distinguish compounds ofsimilar structure as in the case of tryptamine and5-hydroxytryptamine (5HT). At pH 2 to 10, boththese compounds fluoresce at about 350 to 360 minu,but at pH approximately -05 (about 3N acid)5-hydroxytryptamine fluoresces at 540 m,u, whereastryptamine is non-fluorescent (Bridges and Williams,1963).pH-Fluorescence changes are also the basis of the
so-called fluorescent indicators. In this case thefluorescence is usually visible, and the appearance ofvisible fluorescence is useful as an indicator in thetitration of cloudy or coloured solutions wherenormal indicators, which show pH changes byalteration in colour, are of little use. A good exampleof such an indicator is 2-naphthol which, whenviewed under ultraviolet light, changes fromcolourless to blue at pH 6-8. At low pH values,2-naphthol emits the invisible ultraviolet fluor-escence of unionized naphthol, but above pH 6-8 itemits the blue fluorescence of the naphtholate anion.
379
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
R. T. Williams and J. W. Bridges
pH-Fluorescence changes can also be used for thedetermination of the approximate pKa of acids andbases in minute amounts. These can be determinedfrom pH/intensity curves and sometimes frompH/excitation or fluorescence wavelength diagrams,provided that there is a gradual change in wave-length from unionized to ionized forms.The points mentioned above in this section can
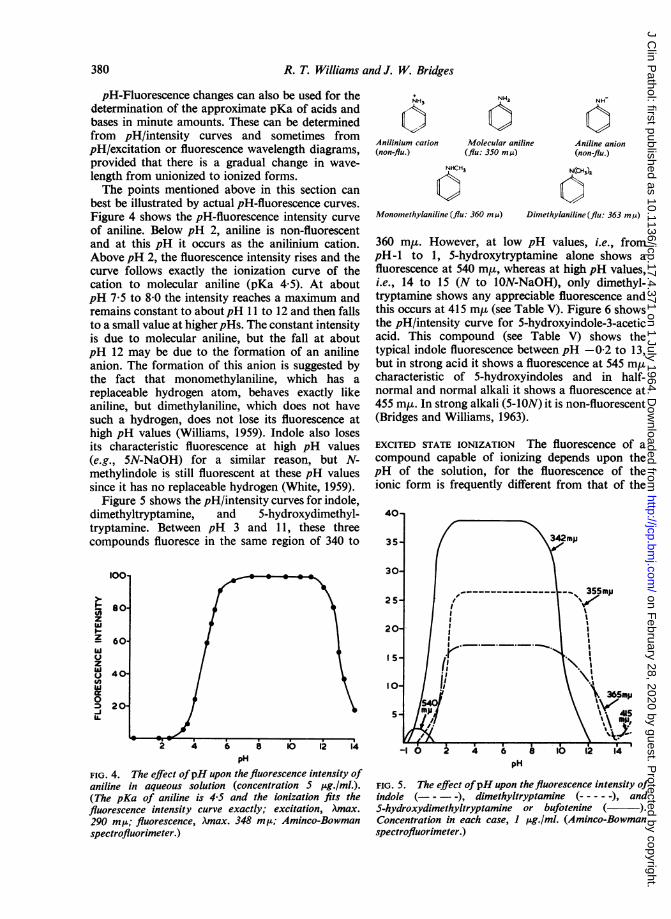
best be illustrated by actual pH-fluorescence curves.Figure 4 shows the pH-fluorescence intensity curveof aniline. Below pH 2, aniline is non-fluorescentand at this pH it occurs as the anilinium cation.Above pH 2, the fluorescence intensity rises and thecurve follows exactly the ionization curve of thecation to molecular aniline (pKa 4 5). At aboutpH 7-5 to 8-0 the intensity reaches a maximum andremains constant to aboutpH 11 to 12 and then fallsto a small value at higher pHs. The constant intensityis due to molecular aniline, but the fall at aboutpH 12 may be due to the formation of an anilineanion. The formation of this anion is suggested bythe fact that monomethylaniline, which has areplaceable hydrogen atom, behaves exactly likeaniline, but dimethylaniline, which does not havesuch a hydrogen, does not lose its fluorescence athigh pH values (Williams, 1959). Indole also losesits characteristic fluorescence at high pH values(e.g., 5N-NaOH) for a similar reason, but N-methylindole is still fluorescent at these pH valuessince it has no replaceable hydrogen (White, 1959).
Figure 5 shows the pH/intensity curves for indole,dimethyltryptamine, and 5-hydroxydimethyl-tryptamine. Between pH 3 and 11, these threecompounds fluoresce in the same region of 340 to
100*
l.
Io-z- 60-
zc40t
020
U.
NH3
Anilinium cation(non-flu.)
NH2
Molecular aniline(flu: 350 m,u)
NH
Aniline anion(non-flu.)
NHCH3 N(CHS
Monomethylaniline (flu: 360 m ,u) Dimethylaniline (flu: 363 m,u)
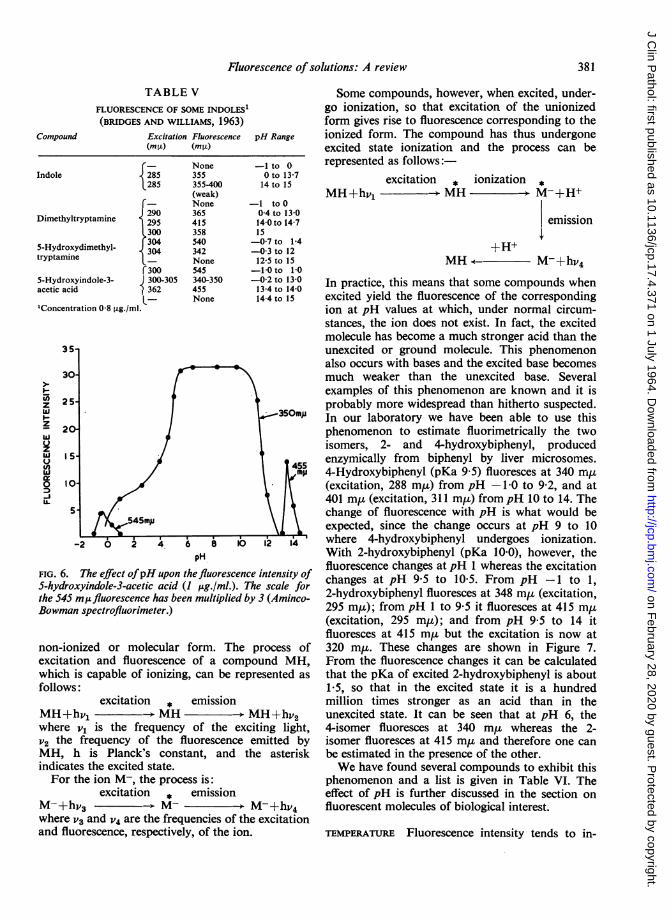
360 miu. However, at low pH values, i.e., frompH-1 to 1, 5-hydroxytryptamine alone shows afluorescence at 540 m,, whereas at high pH values,i.e., 14 to 15 (N to 1ON-NaOH), only dimethyl-tryptamine shows any appreciable fluorescence andthis occurs at 415 m,u (see Table V). Figure 6 showsthe pH/intensity curve for 5-hydroxyindole-3-aceticacid. This compound (see Table V) shows thetypical indole fluorescence between pH -0-2 to 13,but in strong acid it shows a fluorescence at 545 m,ucharacteristic of 5-hydroxyindoles and in half-normal and normal alkali it shows a fluorescence at455 my. In strong alkali (5-ION) it is non-fluorescent(Bridges and Williams, 1963).
EXCITED STATE IONIZATION The fluorescence of acompound capable of ionizing depends upon thepH of the solution, for the fluorescence of theionic form is frequently different from that of the
pH
FIG. 4. The effect ofpH upon the fluorescence intensity ofaniline in aqueous solution (concentration S ILg./ml.).(The pKa of aniline is 4-5 and the ionization fits thefluorescence intensity curve exactly; excitation, Amax.290 mIs; fluorescence, Amax. 348 m,t; Aminco-Bowmanspectrofluorimeter.)
pH
FIG. 5. The effect ofpH upon the fluorescence intensity ofindole (- - - -), dimethyltryptamine (-----), andS-hydroxydimethyltryptamine or bufotenine ( ).Concentration in each case, I ug./ml. (Aminco-Bowmanspectrofluorimeter.)
380
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
Fluorescence of solutions: A review
TABLE VFLUORESCENCE OF SOME INDOLES1(BRIDGES AND WILLIAMS, 1963)
Excitation Fluorescence pH Range
rIndole
Dimethyltryptamine I5-Hydroxydimethyl-tryptamine
r5-Hydroxyindole-3-acetic acid
'Concentration 0-8 ,ug./ml.
(mpO) (mO±)
¢- None285 355285 355-400
(weak)5- None290 365295 415300 358304 540304 342L- Noner300 545C300-305 340-3501362 455L- None
-1 to 00 to 13-714 to 15
-1 toO04 to 13014-0 to 14-715-07 to 1-4-0-3 to 1212-5 to 15-10 to 10-02 to 13-013-4 to 14-014-4 to 15
pH
FIG. 6. The effect ofpH upon the fluorescence intensity ofS-hydroxyindole-3-acetic acid (I pg./ml.). The scale forthe 545 mufluorescence has been multiplied by 3 (Aminco-Bowman spectrofluorimeter.)
non-ionized or molecular form. The process ofexcitation and fluorescence of a compound MH,which is capable of ionizing, can be represented asfollows:
excitation * emissionMH+hvl - MH MH+hV2where v1 is the frequency of the exciting light,V2 the frequency of the fluorescence emitted byMH, h is Planck's constant, and the asteriskindicates the excited state.For the ion M-, the process is:
excitation * emissionM-+hv3 M- M-+hV4where V3 and V4 are the frequencies of the excitationand fluorescence, respectively, of the ion.
Some compounds, however, when excited, under-go ionization, so that excitation of the unionizedform gives rise to fluorescence corresponding to theionized form. The compound has thus undergoneexcited state ionization and the process can berepresented as follows:
excitation * ionization *MH+hvl MH M-+H+
emission
+H+MH < M-+hV4
In practice, this means that some compounds whenexcited yield the fluorescence of the correspondingion at pH values at which, under normal circum-stances, the ion does not exist. In fact, the excitedmolecule has become a much stronger acid than theunexcited or ground molecule. This phenomenonalso occurs with bases and the excited base becomesmuch weaker than the unexcited base. Severalexamples of this phenomenon are known and it isprobably more widespread than hitherto suspected.In our laboratory we have been able to use thisphenomenon to estimate fluorimetrically the twoisomers, 2- and 4-hydroxybiphenyl, producedenzymically from biphenyl by liver microsomes.4-Hydroxybiphenyl (pKa 9 5) fluoresces at 340 mp.t(excitation, 288 m,t) from pH -1 0 to 9-2, and at401 m,u (excitation, 311 m,u) from pH 10 to 14. Thechange of fluorescence with pH is what would beexpected, since the change occurs at pH 9 to 10where 4-hydroxybiphenyl undergoes ionization.With 2-hydroxybiphenyl (pKa 10-0), however, thefluorescence changes at pH 1 whereas the excitationchanges at pH 9 5 to 10-5. From pH -1 to 1,2-hydroxybiphenyl fluoresces at 348 m,u (excitation,295 m,L); from pH 1 to 9-5 it fluoresces at 415 m,u(excitation, 295 m,u); and from pH 9 5 to 14 itfluoresces at 415 mu but the excitation is now at320 m,ut. These changes are shown in Figure 7.From the fluorescence changes it can be calculatedthat the pKa of excited 2-hydroxybiphenyl is about1-5, so that in the excited state it is a hundredmillion times stronger as an acid than in theunexcited state. It can be seen that at pH 6, the4-isomer fluoresces at 340 m,u whereas the 2-isomer fluoresces at 415 m,u and therefore one canbe estimated in the presence of the other.We have found several compounds to exhibit this
phenomenon and a list is given in Table VI. Theeffect of pH is further discussed in the section onfluorescent molecules of biological interest.
TEMPERATURE Fluorescence intensity tends to in-
Compound
viz1-.z
v)U
0-JU.
381
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
R. T. Williams and J. W. Bridges
420-
400
380-
E 360
I
8 340-zw-J
> 320-3
300-
85 -
80-
ziiz
3
I-
zwu
U)0-JUa.
-I 0 2 4 6 a lo 1pH
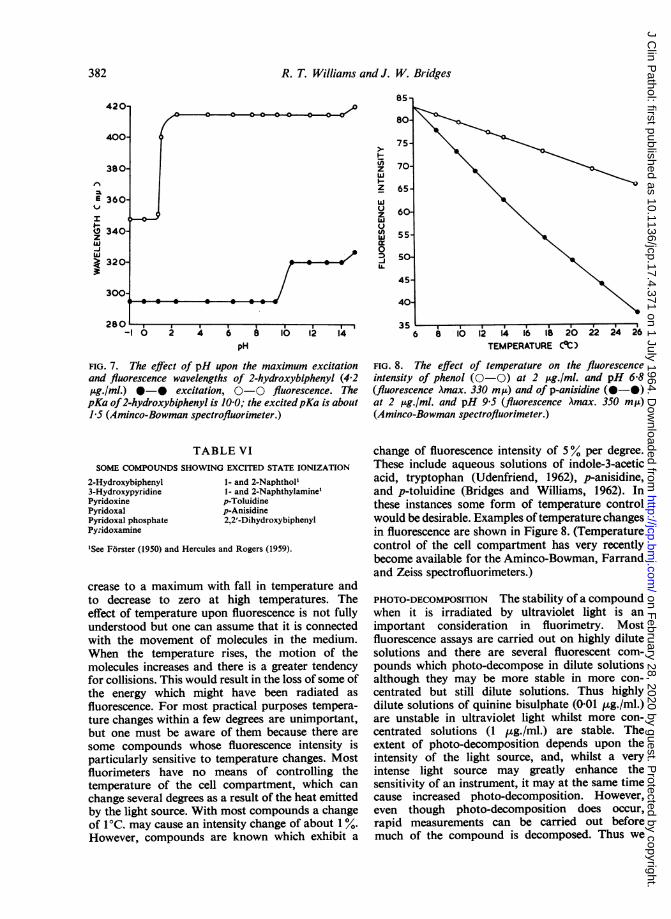
FIG. 7. The effect of pH upon the maximum excitationand fluorescence wavelengths of 2-hydroxybiphenyl (4-2,ug./ml.) *-* excitation, 0-0 fluorescence. ThepKa of2-hydroxybiphenyl is 10 0; the excitedpKa is about1.5 (Aminco-Bowman spectrofluorimeter.)
TABLE VISOME COMPOUNDS SHOWING EXCITED STATE IONIZATION
2-Hydroxybiphenyl3-HydroxypyridinePyridoxinePyridoxalPyridoxal phosphatePy.idoxamine
1- and 2-Naphtholl1- and 2-Naphthylamine'p-Toluidinep-Anisidine2,2'-Dihydroxybiphenyl
'See Forster (1950) and Hercules and Rogers (1959).
crease to a maximum with fall in temperature andto decrease to zero at high temperatures. Theeffect of temperature upon fluorescence is not fullyunderstood but one can assume that it is connectedwith the movement of molecules in the medium.When the temperature rises, the motion of themolecules increases and there is a greater tendencyfor collisions. This would result in the loss of some ofthe energy which might have been radiated as
fluorescence. For most practical purposes tempera-ture changes within a few degrees are unimportant,but one must be aware of them because there are
some compounds whose fluorescence intensity isparticularly sensitive to temperature changes. Mostfluorimeters have no means of controlling thetemperature of the cell compartment, which can
change several degrees as a result of the heat emittedby the light source. With most compounds a changeof 1°C. may cause an intensity change of about 1 %.However, compounds are known which exhibit a
75-
70-
65.-
60.
55-
50-
45-
40-
35 L
6 8 10 12 4 16 lb 20 22 24 26TEMPERATURE C*C)
FIG. 8. The effect of temperature on the fluorescenceintensity of phenol (0-0) at 2 ,ug./ml. and pH 68(fluorescence Amax. 330 mu) and of p-anisidine (0-*)at 2 pLg./ml. and pH 9-5 (fluorescence Amax. 350 m,u)(Aminco-Bowman spectrofluorimeter.)
change of fluorescence intensity of 5% per degree.These include aqueous solutions of indole-3-aceticacid, tryptophan (Udenfriend, 1962), p-anisidine,and p-toluidine (Bridges and Williams, 1962). Inthese instances some form of temperature controlwould be desirable. Examples oftemperature changesin fluorescence are shown in Figure 8. (Temperaturecontrol of the cell compartment has very recentlybecome available for the Aminco-Bowman, Farrandand Zeiss spectrofluorimeters.)
PHOTO-DECOMPOSITION The stability of a compoundwhen it is irradiated by ultraviolet light is animportant consideration in fluorimetry. Mostfluorescence assays are carried out on highly dilutesolutions and there are several fluorescent com-pounds which photo-decompose in dilute solutionsalthough they may be more stable in more con-centrated but still dilute solutions. Thus highlydilute solutions of quinine bisulphate (001 ,ug./ml.)are unstable in ultraviolet light whilst more con-centrated solutions (1 ,ug./ml.) are stable. Theextent of photo-decomposition depends upon theintensity of the light source, and, whilst a veryintense light source may greatly enhance thesensitivity of an instrument, it may at the same timecause increased photo-decomposition. However,even though photo-decomposition does occur,rapid measurements can be carried out beforemuch of the compound is decomposed. Thus we
-~~~~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~~ --_OU I
382
I* 0 0 0 0r
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
Flutorescence of solutions: A review
i-
z
z
Lu
z
w
uJ
lL
0D-j
UA.
100-
90-
80-
0 -0 *----O.
70160-
50-
40-
30-
20-
I 0-
10 20 3o 40 so o 70o 8o 90 16oTIME (mim)
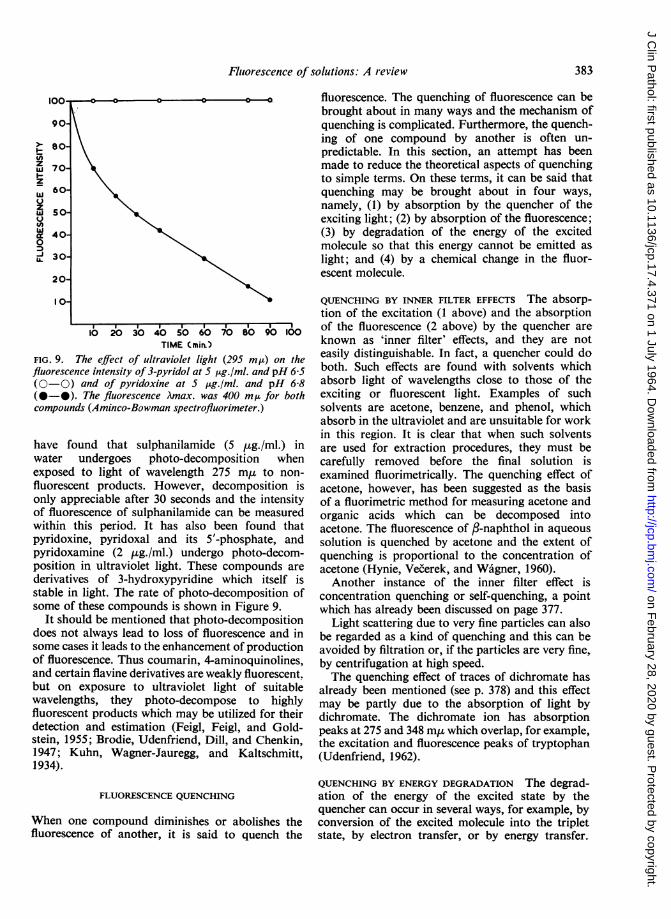
FIG. 9. The effect of ultraviolet light (295 mp) on thefluorescence intensity of 3-pyridol at 5 ttg./ml. and pH 6-5(0-0) and of pyridoxine at 5 pg./ml. and pH 6-8(0-0). The fluorescence Amax. was 400 m,u for bothcompounds (Aminco-Bowman spectrofluorimeter.)
have found that sulphanilamide (5 jug./ml.) inwater undergoes photo-decomposition whenexposed to light of wavelength 275 m,u to non-fluorescent products. However, decomposition isonly appreciable after 30 seconds and the intensityof fluorescence of sulphanilamide can be measuredwithin this period. It has also been found thatpyridoxine, pyridoxal and its 5'-phosphate, andpyridoxamine (2 ,ug./ml.) undergo photo-decom-position in ultraviolet light. These compounds are
derivatives of 3-hydroxypyridine which itself isstable in light. The rate of photo-decomposition ofsome of these compounds is shown in Figure 9.
It should be mentioned that photo-decompositiondoes not always lead to loss of fluorescence and insome cases it leads to the enhancement of productionof fluorescence. Thus coumarin, 4-aminoquinolines,and certain flavine derivatives are weakly fluorescent.but on exposure to ultraviolet light of suitablewavelengths, they photo-decompose to highlyfluorescent products which may be utilized for theirdetection and estimation (Feigl, Feigl, and Gold-stein, 1955; Brodie, Udenfriend, Dill, and Chenkin,1947; Kuhn, Wagner-Jauregg, and Kaltschmitt,
1934).
FLUORESCENCE QUENCHING
When one compound diminishes or abolishes thefluorescence of another, it is said to quench the
fluorescence. The quenching of fluorescence can bebrought about in many ways and the mechanism ofquenching is complicated. Furthermore, the quench-ing of one compound by another is often un-predictable. In this section, an attempt has beenmade to reduce the theoretical aspects of quenchingto simple terms. On these terms, it can be said thatquenching may be brought about in four ways,namely, (1) by absorption by the quencher of theexciting light; (2) by absorption of the fluorescence;(3) by degradation of the energy of the excitedmolecule so that this energy cannot be emitted aslight; and (4) by a chemical change in the fluor-escent molecule.
QUENCHING BY INNER FILTER EFFECTS The absorp-tion of the excitation (1 above) and the absorptionof the fluorescence (2 above) by the quencher areknown as 'inner filter' effects, and they are noteasily distinguishable. In fact, a quencher could doboth. Such effects are found with solvents whichabsorb light of wavelengths close to those of theexciting or fluorescent light. Examples of suchsolvents are acetone, benzene, and phenol, whichabsorb in the ultraviolet and are unsuitable for workin this region. It is clear that when such solventsare used for extraction procedures, they must becarefully removed before the final solution isexamined fluorimetrically. The quenching effect ofacetone, however, has been suggested as the basisof a fluorimetric method for measuring acetone andorganic acids which can be decomposed intoacetone. The fluorescence of f-naphthol in aqueoussolution is quenched by acetone and the extent ofquenching is proportional to the concentration ofacetone (Hynie, Vecerek, and Wagner, 1960).Another instance of the inner filter effect is
concentration quenching or self-quenching, a pointwhich has already been discussed on page 377.
Light scattering due to very fine particles can alsobe regarded as a kind of quenching and this can beavoided by filtration or, if the particles are very fine,by centrifugation at high speed.The quenching effect of traces of dichromate has
already been mentioned (see p. 378) and this effectmay be partly due to the absorption of light bydichromate. The dichromate ion has absorptionpeaks at 275 and 348 m,u which overlap, for example,the excitation and fluorescence peaks of tryptophan(Udenfriend, 1962).
QUENCHING BY ENERGY DEGRADATION The degrad-ation of the energy of the excited state by thequencher can occur in several ways, for example, byconversion of the excited molecule into the tripletstate, by electron transfer, or by energy transfer.
s~~~~~~~I I I I -
383
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
These forms of quenching are often known as 'true'quenching.The quenching molecule may affect the fluor-
escent molecule in such a way as to convert it fromthe singlet excited state to the triplet state so that itno longer emits its energy as fluorescence (see p. 372).The quenching effect of oxygen, certain iodo, bromo,and nitro compounds, and probably pyridine maybe due to this effect. In the case of the halogencompounds the large magnetic fields of the halogenatoms may be responsible for the conversion. In thecase of oxygen, it may be due to the fact that theground state of the oxygen molecule is a triplet,which, when in contact with the singlet excited stateof the molecules of some compounds, exchanges itstriplet state for the singlet state of the other molecule:
Otriplet + Msinglet osinglet + Mtriplet
The quenching effect of oxygen can be avoided bybubbling nitrogen through the solution during themeasurement of fluorescence. However, oxygen isnot a universal quencher of fluorescence, but itdoes quench the fluorescence of several aromaticcompounds. This observation has been used for thedetermination of oxygen, employing the fluorescenceof a borate-benzoin complex which is quenched byoxygen in proportion to the concentration of thelatter (Parker and Barnes, 1957).The degradation of the energy of the excited
molecule can also occur by electron transfer. Theexcited molecule, but not the corresponding groundmolecule, may be able to donate or accept anelectron from the quenching molecule and in thisway the excited state may be destroyed. Thus ferrousions destroy the fluorescence of excited methyleneblue by electron donation, but have no effect on theunexcited dye. Some fluorescent molecules arequenched by electron-donating anions such asI-, Br-, SCN-, and S203, and others by electron-accepting anions such as I03, NO-, and S406, thedirection of electron transfer depending upon theredox potential of the excited molecule which isdifferent from that of the unexcited molecule(Bowen and Wokes, 1953). The fluorescence oftryptophan is quenched by thiosulphate or nitrateions and this effect has been used for identifyingtryptophan in tissue fluids (Duggan and Udenfriend,1956).For quenching to occur by energy transfer, the
energy level of the excited quencher must be justbelow that of the excited fluorescent molecule.Usually the quencher molecule is one which, onexcitation, is converted into the triplet state, so thatwhen the fluorescent molecule is excited, its energyis immediately taken up by the quencher, which thenassumes a non-radiative triplet state and the energy
absorbed by the fluorescent molecule cannot nowbe emitted as fluorescence. In effect, the quencherhas robbed the excited fluorescent molecule of itsenergy because of the proximity of their energylevels. This type of quenching can readily occur insolids where the fluorescent and quenching moleculesare close together, but it is more difficult to producein solution unless high concentrations are used, andthen, of course, energy transfer quenching may bedifficult to distinguish from concentration quenching.An example of energy transfer quenching is shownby phenazine in solid anthracene. Anthracene has ablue fluorescence, but if it contains one part in athousand of phenazine, the fluorescence is quenched.The energy level of the triplet state of excitedphenazine is just below that of the singlet excitedstate of anthracene (Bowen and Wokes, 1953).Energy transfer mechanisms are thought to play
an important role in biological systems, particularlythose involving proteins (Porter and Weber, 1959;Szent-Gyorgyi, 1957). The fluorescence of NADPis thought to involve an energy transfer mechanism(see p. 389).
QUENCHING BY CHEMICAL CHANGE If a fluorescentcompound undergoes a chemical change as a resultof the presence of a second compound it could beconverted into a non-fluorescent product. Althoughthe fluorescence of the compound is quenched bythe addition of the second compound, this form ofquenching is to be distinguished from 'true' quench-ing during which no net chemical change occurs.Quenching by chemical change (cf. photo-de-composition, p. 382) can occur in many ways but theend product is usually a non-fluorescent compound.Furthermore this type of quenching can be veryspecific as the examples given below will show.Quenching can occur as a result ofpH changes in
the solution. A simple case is that of aniline. If acidis added to an aqueous solution of aniline, thefluorescence begins to be quenched at pH 6, and atpH 2 the solution is non-fluorescent. This is due tothe conversion of aniline into the non-fluorescentanilinium ion which is a different chemical speciesfrom aniline:
C6H5NH2 C6H5NH3fluorescent non-fluorescent
anilinium ionThe fluorescence of phenol is quenched when thesolution is made alkaline and in this case phenol isconverted into the non-fluorescent phenoxide ion.An example of specific quenching by an acid is
that of quinine. In O-N-H2SO4, quinine (1 ,ug./ml.)is highly fluorescent but in 01N-HCl, quinine isnon-fluorescent (Bowen and Wokes, 1953). Quinine
384
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence ofsolutions: A review
in the excited state probably forms some kind ofcompound with HCl which is non-fluorescent.An extensive study of the quenching effects of
purines was made by Weil-Malherbe (1946) whofound xanthine, hypoxanthine, caffeine, uric acid,and N-methylxanthines to quench the fluorescenceof several ccrnpounds, probably by formingmolecular compounds or complexes. Thus caffeinein acid solution quenches the fluorescence ofpolycyclic hydrocarbons, whilst in neutral solutionit quenches the fluorescence of riboflavin, andintensifies that of thiochrome, mepacrine, and eosin.In the case of tryptophan its fluorescence is quenchedby ascorbic acid, the nitrate ion, and the thio-sulphate ion, and it is probable that tryptophan isoxidized by these quenchers.
ADSORPTION AND QUENCHING Adsorption of thefluorescent substance on to glassware (includingfluorimeter cells) and precipitates, especially protein,frequently occurs in dilute solutions especially withnon-polar solvents such as hexane. Such adsorptionresults in the loss of fluorescent material andconsequently in low recoveries. There are manyways in which adsorption can be avoided and theseinclude suitable treatment of new glassware,changing the protein precipitants, changing thesolvent, adding a small amount of a polar solventsuch as alcohol, etc., but each case has to be dealtwith on its merits (see Udenfriend, 1962).
FLUORIMETERS
A fluorimeter is an instrument used for the measure-
O light source
Ifprimary filter or monochromator
exciting light
_ - fluorescence
o - 0
4'L
ment of the intensity of fluorescence. If the instru-ment has means of measuring the wavelength of thefluorescence and of the excitation, it is then called aspectrofluorimeter or more accurately, a fluorescencespectrometer. All fluorimeters, simple or complex,contain three essential units, namely, a source ofexciting light, a sample container, usually a cell orcuvette of square section, and a detecting unit formeasuring the fluorescence emitted by the sample.To increase the specificity of both the exciting andfluorescent light, most fluorimeters contain two setsof light filters, and the spectrofluorimeters, twomonochromators, one to select the exciting light andthe other to analyse the fluorescent light. Themonochromators are devices consisting of either adiffraction grating or a prism and the necessaryslits, lenses, and mirrors, which can be movedmechanically to select a desired wavelength or bandof light, which is measured on a wavelength scale ordrum attached to the monochromator. A simplifiedoutline of a fluorimeter is shown in Figure 10.When the exciting light strikes the cuvette
fluorescence is emitted from all its faces, but mostfluorimeters measure only the fluorescence which isemitted from one face at right angles to the directionof the exciting light. The faces in the direction of theincident light emit fluorescence and exciting light,but even at right angles there is some scatteredexciting light to be contended with and this showsup on recording spectrofluorimeters at the wave-length of the exciting light on the fluorescence sideof the instrument. In filter instruments, this scatteredlight can be eliminated to a large degree by suitablesecondary filters (see Fig. 10). Filters can also beused in spectrofluorimeters, if scattered light istroublesome as a result of a slight impurity orturbidity in the sample.
LIGHT SOURCES The source of exciting light is animportant factor in fluorimeters, and the lamps nowin use are the xenon-arc, the high pressure mercury,the tungsten filament, and zinc lamps. Modernspectrofluorimeters utilize the xenon-arc lamp whichemits a continuous spectrum from 200 to 800 m,r.Its brightness in the ultraviolet region is muchgreater than the tungsten filament lamp and it doesnot develop line spectra as does the mercury vapourlamp. The mercury lamp gives a discontinuousspectrum consisting of high intensity lines, the
photo-detector FIG. 10. A simplified outline ofafluorimeter.
secondary filter or monochromator
385
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
R. T. Williams and J. W. Bridges
main ones being at 365, 405, 436, and 546 m,u. If acompound is maximally excited in the region of theselines, the mercury lamp is then very suitable forexciting such a compound, but it is less satisfactoryfor compounds maximally excited at other wave-lengths. The fluorimetric estimation of vitamin B1as thiochrome, of adrenaline as adrenolutine or as aquinoxaline derivative, and of riboflavin areexcellent examples of compounds whose maximumexcitations almost coincide with one of the mercurylines, thus:Mercury Line Maximum Excitation for
366 m,u405 m,u
436 m,u
Thiochrome from vitamin Bl, 370 mgAdrenolutine from adrenaline, 405 m,
rRiboflavin, 440 m,uQuinoxaline derivative
ifrom adrenaline, 435 m,u
Although the xenon-arc gives a continuousspectrum over the ultraviolet and visible regions itsintensity varies with wavelength and tends to
WAVENUMBER (,uCl)32 3-4 3-6 3-8 4-0 4-2 4-4
100 , . . .
90'
80
so-I
z
40
zI
W
WAVELENGTH Cmp)
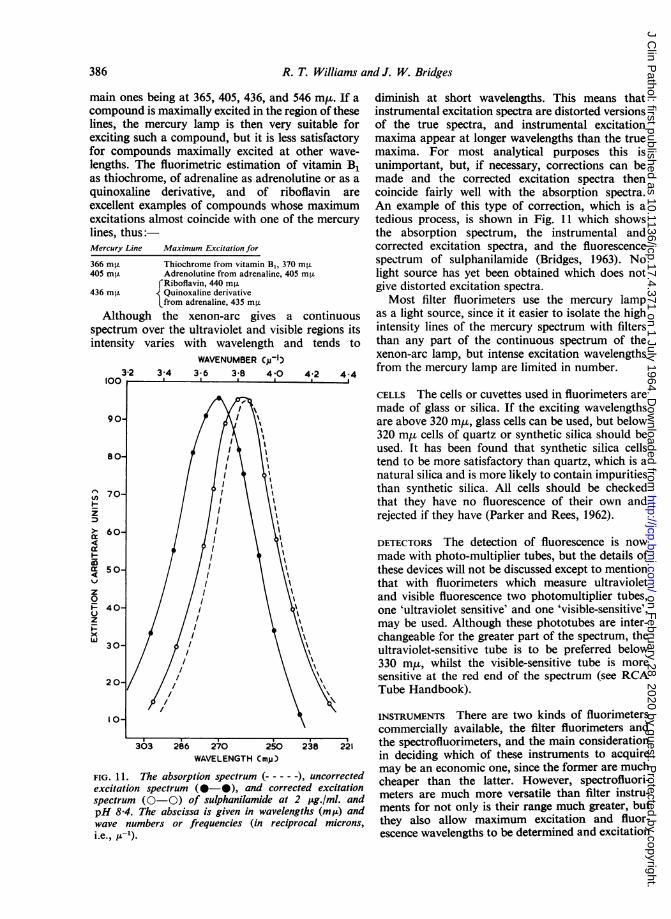
FIG. 11. The absorption spectrum (-), uncorrectedexcitation spectrum (0-0), and corrected excitationspectrum (0-0) of sulphanilamide at 2 pLg./ml. andpH 8-4. The abscissa is given in wavelengths (mua) andwave numbers or frequencies (in reciprocal microns,i.e., p-c).
diminish at short wavelengths. This means thatinstrumental excitation spectra are distorted versionsof the true spectra, and instrumental excitationmaxima appear at longer wavelengths than the truemaxima. For most analytical purposes this isunimportant, but, if necessary, corrections can bemade and the corrected excitation spectra thencoincide fairly well with the absorption spectra.An example of this type of correction, which is atedious process, is shown in Fig. 11 which showsthe absorption spectrum, the instrumental andcorrected excitation spectra, and the fluorescencespectrum of sulphanilamide (Bridges, 1963). Nolight source has yet been obtained which does notgive distorted excitation spectra.Most filter fluorimeters use the mercury lamp
as a light source, since it it easier to isolate the highintensity lines of the mercury spectrum with filtersthan any part of the continuous spectrum of thexenon-arc lamp, but intense excitation wavelengthsfrom the mercury lamp are limited in number.
CELLS The cells or cuvettes used in fluorimeters aremade of glass or silica. If the exciting wavelengthsare above 320 m,t, glass cells can be used, but below320 myt cells of quartz or synthetic silica should beused. It has been found that synthetic silica cellstend to be more satisfactory than quartz, which is anatural silica and is more likely to contain impuritiesthan synthetic silica. All cells should be checkedthat they have no fluorescence of their own andrejected if they have (Parker and Rees, 1962).
DETECTORS The detection of fluorescence is nowmade with photo-multiplier tubes, but the details ofthese devices will not be discussed except to mentionthat with fluorimeters which measure ultravioletand visible fluorescence two photomultiplier tubes,one 'ultraviolet sensitive' and one 'visible-sensitive',may be used. Although these phototubes are inter-changeable for the greater part of the spectrum, theultraviolet-sensitive tube is to be preferred below330 m,L, whilst the visible-sensitive tube is moresensitive at the red end of the spectrum (see RCATube Handbook).
INSTRUMENTS There are two kinds of fluorimeterscommercially available, the filter fluorimeters andthe spectrofluorimeters, and the main considerationin deciding which of these instruments to acquiremay be an economic one, since the former are muchcheaper than the latter. However, spectrofluori-meters are much more versatile than filter instru-ments for not only is their range much greater, butthey also allow maximum excitation and fluor-escence wavelengths to be determined and excitation
386
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solutions: A review
and fluorescence spectra to be drawn. In fact, theygive enough data to the operator to allow analyses tobe carried out under optimal conditions. But formany routine analyses, they offer no great advantagesover good filter instruments. However, if an institu-tion has a number of filter instruments, a spectro-fluorimeter can be very useful in indicating whatfilters ought to be used for a specific analysis in thefilter fluorimeters.
There are several good filter instruments com-mercially available including those made by theBeckman, Coleman, EEL, Farrand, Hilger-Watt,Locarte, Turner, Photovolt, and Zeiss Instrumentcompanies. Their basic design is similar, a mercurylamp being employed as a light source in most cases.(These instruments generally cost between £200 and£500.)The main commercial spectrofluorimeters are the
Aminco-Bowman, Farrand, and Zeiss instruments.The first two employ diffraction gratings as mono-chromators whilst the third, a recently introducedinstrument, uses quartz prisms. The use of prismsresults in a loss of sensitivity, which is, however,compensated by an increased resolving power, andwavelengths can be determined more accurately.The light sources in all three instruments are xenon-arc lamps, the Aminco-Bowman and Farrandinstruments employing a 150 watt lamp and theZeiss instrument a 500 watt lamp whose higher lightintensity is intended to compensate for the loss ofsensitivity due to the prism optical system. (Thebasic cost of these instruments is between £1,250 and£3,000.)
marins, tetracyclines, chlorpromazine derivatives,etc. One can usually tell from the structural formulaof a compound whether it is likely to be fluorescent,by examining its structure for conjugated double
TABLE VIISOME FLUORESCENT NATURAL COMPOUNDS'
Compound Excitation FluorescenceMaximum Maximum(mP.) (mg)
AdenosineAdenylic acidAdrenalinep-Aminobenzoic acidAnthranilic acidATPFolic acidFolinic acidHomogentisic acidHomovanillic acid5-Hydroxyindolacetic acidKynurenineNADP.OestradiolOestronePyridoxalPyridoxine
Riboflavine
Serotonin
TocopherolTryptophanTryptamineTyrosineTyramineVitamin AVitamin Bu,
285285285295300285365370290270300370340285285330340270)370445J295295295285290275375325275
395395325345405395450460340315355490435330325385400
520
340540330365360310310470305
pH orSolvent
17
77777
7EthanolEthanol77
7
20Ethanol11771
Ethanol7
'Taken from Duggan, Bowman, Brodie, and Udenfriend (1957); themaxima quoted are instrumental values.
FLUORESCENT MOLECULES OFBIOLOGICAL INTEREST
It should now be clear what types of chemicalstructures are likely to be fluorescent and whysimple non-aromatic amino-acids, saturated lipids,steroids (except oestrogens), and carbohydrates arenon-fluorescent. There is no reason, however, whyit should not be possible to convert the above typesof non-fluorescent compounds into fluorescentproducts by suitable chemical reactions (Udenfriend,1962). There are a large number of biologicallyimportant compounds which contain the necessaryconjugated double bond systems and are, therefore,potentially or actually fluorescent. These includearomatic amino-acids, certain proteins, carotinoids,porphyrins, purines, indoles, phenols, oestrogens,vitamins (A, K, pyridoxine, thiamine, riboflavine,tocopherol), hormones (adrenaline, serotonin), andco-enzymes. Furthermore, there are a large numberof drugs which are fluorescent e.g., quinine, salicy-lates, barbiturates, quinoline, antimalarials, cou-
Compound
TABLE VIIISOME FLUORESCENT DRUGS1
Excitation FluorescenceMaximum Maximum
AminopterinCinchonineChloroquineChlorpromazineDromoranHarmineLysergic acid diethylamide
(L.S.D.)MenadioneNeocinchophenPhenylephrinePiperoxanePamaquinPodophyllotoxinProcaineProcainamideQuinineReserpineOxytetracyclineYohimbine
(ml±) (mpO)
280, 370320335350275300, 365
325335275, 345270290300, 370280275275250, 350300390270
'Taken from Udenfriend, Duggan, Vasta,maxima quoted are instrumental values.
460420400480320400
465480455305325530325345385450375520360
pH orSolvent
11
111111
7Ethanol
7I7
13111111
11I
387
and Brodie (1957); the
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
bonds and aromatic systems and for the side chainswhich contribute so much to fluorescence intensity;and also noting the presence of deactivating atomsor systems in the molecule and the possibility ofionization if pH changes were to be introduced intothe medium. A list of some fluorescent naturalcompounds is given in Table VII, and of some drugsin Table VIII.
It is not possible, within the limits of the review,to deal with the fluorescent properties of all thecompounds of importance in biochemistry andmedicine, although several such compounds havealready been referred to earlier in this text. However,in what follows some examples have been selectedto illustrate the possibilities of fluorescence.
PYRIMIDINES AND PURINES These compounds are ofconsiderable interest because of their occurrence innucleic acids and nucleotides. If the structure ofpyrimidine and purine is examined (see below), itwill be seen that both molecules contain doublybound nitrogen (-N=), and as stated earlier (seep. 375), such compounds might be expected tobe non-fluorescent. However, as also stated earlier, theeffects of these nitrogens could be overcome by
N O N
N N N
Pyrimidine'
CH,
N
Purine Thymine (flu: 380 m A atpH 11-13 as anion)
suitable substituents and by ionization. Udenfriendand Zaltzman (1962) have examined a number ofpyrimidiness for fluorescence and found onlythymine (2,4-dihydroxy-5-methylpyrimidine) to befluorescent. The fluorescence of thymine was veryweak and only occurred at pH 11 to 13 wherethymine exists as an anion.
Purine, however, is weakly fluorescent as ananion, and presumably the >NH group whenionized is sufficiently electron-donating to overcomethe effect of the doubly bound nitrogens in the ringsystem. Hypoxanthine, xanthine, and uric acid are
NIlI Purine anion(flu: 370 m at
non-fluorescent, but adenine and guanine and thenucleotides derived from them are fluorescent atcertain pH values. Adenine is fluorescent only asthe mono-cation. Adenosine, adenylic acid, adeno-sine diphosphate (ADP), and adenosine tri-phosphate (ATP) are also weakly fluorescent inBorresen (1963) has recently claimed that pyrimidine fluoresces weaklyat high concentrations. This finding is contrary to accepted theoryalthough Borresen has attempted to explain it.
dilute acid solution, and in 5N-H2SO4 they are morefluorescent than adenine itself. Guanine is the mostfluorescent of the natural purines, for its mono-cation and mono-anion are fluorescent. Its nucleo-side and nucleotides, guanosine, guanylic acid,guanosine diphosphate, and guanosine triphosphate,are also fluorescent (Udenfriend and Zaltzman,1962; Borresen, 1963). Guanosine and guanylic acidare fluorescent in acid solution, but they have verylittle fluorescence at pH 11 where guanine isappreciably fluorescent. These observations havebeen used as the basis of a fluorimetric assay ofguanine in nucleic acid hydrolysates (Udenfriendand Zaltzman, 1962).
NH2
N N
OH
N NH
H2NK N
0-
NH
Adenine cation (flu: Guanine cation (flu: Guanine mono-anion380 mI at pH 1) 360 mg at pH 1) (flu: 350 mA at pH 1)
BARBITURATES An examination of the structuralformula of a simple barbiturate (see below) showsthat the molecule does not contain conjugateddouble bonds and is therefore unlikely to befluorescent. In fact, barbiturates are not fluorescentin aqueous solution, but in 01N or N-NaOH someof them show appreciable fluorescence. Pheno-barbitone, pentobarbitone, amylobarbitone, and thethiobarbiturates, thiopental and surital, are fluor-escent at pH 13 to 14 (Udenfriend, Duggan, Vasta,and Brodie, 1957). Barbiturates have two pKa's, one
R R'
HNyNH
0
Barbiturate unionized (eon-fluorescent)
Ft Rl0 0-N N
0i-
R R'0 0
N >yNH
Barbiturate mono-anion(non-fluorescent)
0A-S¶Kf
Barbiturate di-anion (flu: 440 m,u) Thiobarbiturate di-anion(flu: 530 mA)
in the region of 7 to 8 corresponding to the formationof a mono-anion, and the other in the region of12 to 13 corresponding to the formation of thedi-anion (Butler, Ruth, and Tucker, 1955). Since thefluorescence of the barbiturates occurs atpH 13 to 14,the fluorescent species is probably the di-anion.Probably the two ionized OH groups opposing thetwo doubly bound nitrogens are the main contribu-tors in producing the conditions responsible forfluorescence. The thiobarbiturate fluorescence is alsoprobably due to a di-anion (see above). It occurs at a
388
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solutions: A review
longer wavelength (530 m,u) than the barbituratefluorescence (440 m,). N-Substituted barbituratessuch as N-methyl-phenobarbitone are unable toform a di-anion and are probably not fluorescent.
PYRIDINE NUCLEOTIDES Nicotinamide is non-fluorescent and this agrees with the fact that itsmolecule contains doubly bound nitrogen and themeta-directing carbamyl group both of which wouldtend to produce a non-fluorescent molecule. The
H H
CONH,OCO NH
CH3
N C.NFA
CH3
Nicotinamide 4-Hydro-N- Fluorescent form of 4-hydro-(non-fluorescent) methylnicotinamide methylnicotinamide (exc.
360 m p, flu: 468 m )H H 0
C.NH,
CH
&ADR
Fluorescent form ofNADH (exc. 260, 340mis, flu: 457 mu)
oxidized forms of the pyridine nucleotides (NAD+and NADP+) are also non-fluorescent, but thereduced forms (NADH and NADPH) are fluor-escent, a property which has been used for theassay of a number of dehydrogenases, includinglactic, glutamic, and malic dehydrogenases, and tostudy the oxidation-reduction mechanisms of intactcells and subcellular particules (Lowry, Roberts,and Kapphahn, 1957; Lowry, Roberts, and Lewis,1956; Duysens and Amesz, 1957; Duysens andSweep, 1957; Chance and Baltscheffsky, 1958;Chance and Legallais, 1959). The components ofthese coenzymes (NADH and NADPH) areadenine, reduced nicotinamide, ribose, and phos-phate. The reduced form of N-methylnicotinamideis fluorescent (excitation, 360 m,u, and fluorescent,468 m,u) and the fluorescent form of this moleculeas proposed by Weber (1958) is shown above.Adenine is also fluorescent. The corrected excitationsof NADH in aqueous solution are at 260 and340 m,u, and the fluorescence is at 457 m,u (Weber,1957). The 260 m,u excitation is due to the adeninemoiety and the fluorescence resulting from it is dueto energy transfer from the purine portion of themolecule to the substituted dihydronicotinamideportion, the excitation of which is at 340 m,u. Thefluorescent form of NADH proposed by Weberis shown above.The oxidized forms of the pyridine nucleotides
can be induced to fluoresce by heating with alkali(Kaplan, Colowick, and Barnes, 1951; Lowry et al.,
1957), and this procedure can be adapted to measureboth the oxidized and reduced coenzymes fluori-metrically. Fluorimetric methods have also beendevised for N-methylnicotinamide, by condensationwith ketones in the presence of alkali to give highlyfluorescent products (Udenfriend, 1962, for refer-ences).
PORPHYRINS The basic ring system in the porphy-rins is that of porphin, which, as can be seen fromits structure, contains the necessary conjugateddouble bonds which give rise to fluorescence.Porphyrins in acid solution and in organic solventsare, in fact, highly fluorescent, the fluorescence
Porphin
N HN
occurring in the red and infrared regions of thespectrum. However, it is important to note that thebiologically active forms of porphyrins in animals,that is, the metalloporphyrins containing iron andcopper, are non-fluorescent. When these metals areremoved fluorescence appears. Chlorophyll, on theother hand, is a magnesium porphyrin which isfluorescent in water and alcohol (for details ofporphyrin fluorescences, see Vannotti, 1954; andUdenfriend, 1962).
Porphyrins show several fluorescent bands butthe principal bands in pyridine are the following:
mmJProtoporphyrin ....... ...... 634Coproporphyrin ........ ..... 620Uroporphyrin ............. 623Haematoporphyrin ............. 625Mesoporphyrin...... ....... 621Aetioporphyrin............. 621
When haemoglobin undergoes degradation in thebody, one of the products is urobilinogen. In thiscompound, the methine bridges (-CH=) which occurin the original porphin structure have been reducedso that the conjugated double bond system has beendestroyed. Urobilinogen is, therefore, non-fluor-escent. However, if it is oxidized to urobilin, con-jugation is partly restored, and urobilin is fluorescent.
H H H H
Urobilinogen ring system (non-fluorescent)
H H H
Urobilin ring system (diffuse fluorescence, 600-640 my)
389
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
PYRIDOXINE AND RELATED COMPOUNDS Pyridoxine(vitamin B6), pyridoxal, pyridoxal 5-phosphate, andpyridoxamine play an important role in the activityof several enzymes, e.g., transaminases, decarboxy-lases, etc. These compounds are derivatives of3-pyridol and they show very interesting fluor-escence changes at different pHs (Bridges et al.,1963). The fluorescence characteristics of the
()09 3-Pyridol
N
R ,
OH
OCH3
Pyridoxine; RI = Ra = CH20HPyridoxal; R, = CHO; R, = CH20HPyridoxal S-phosphate; RI = CHO; R2 =CH2OPO,H2Pyridoxamine; RI = CH,NH,; R2 =CH,OH
pyridoxine series formulated above are similar tothose of 3-pyridol and it will be simpler to describethe fluorescences of the latter than to describe thedetailed fluorescences of the pyridoxine compounds.3-Pyridol can occur at different pH values in fourdifferent forms each having a characteristic fluor-escence:-
HOQ m [HO Q- Q ]0Q
Cation (flu: Neutral molecule Dipolar ion (54 %) Anion340 mM) (46%)rflu: 37.5 m,4) (flu: 392 m/l) (flu:
365mM)
The cation, neutral molecule, and dipolar ion of3-pyridol are only weakly fluorescent, but the anionis strongly fluorescent, its intensity being 50 timesthat of the cation and 16 times that of the dipolarion. The cation shows excited state ionization, so
that atpH values more than -1 up to 7, it shows thefluorescence of the dipolar ion. The true fluorescenceof the cation, i.e., at 340 mj,, is only shown atpH -1 (1ON-HCl) and less. The unexcited cationionizes to the dipolar ion at pH 4 to 5 (pKa 4-86),so that in the excited state (pKa -0-95) the cationis about a million times weaker as a base than in theunexcited state.The phenomenon of excited state ionization is
also shown by the cations of pyridoxine, pyridoxaland its phosphate, and pyridoxamine. However,all the forms of these compounds are more fluor-escent than the corresponding forms of 3-pyridol.The most fluorescent forms of pyridoxine and ofpyridoxamine are the corresponding dipolar ions(see Table IX) whereas those of pyridoxal and itsphosphate are the anions. In the case of pyridoxalthe most fluorescent form is probably the anion ofthe cyclic hemiacetal of pyridoxal, and in the case of
pyridoxal phosphate it is probably the tri-anion ofthe aldehyde hydrate of pyridoxal phosphate(see Table IX). At highpH values (12 to 14) pyridoxalphosphate shows two fluorescences, one at 370 m,ucorresponding to the tri-anion of the hydrate, andanother, eight times less intense, at 525 m,u whichappears to correspond to the tri-anion of the freealdehyde form of pyridoxal phosphate. 3-Pyridol is
O CHO
O=1.OCH20 Aldehvde.form tri-anion0- CHI (flu: 525 m)
stable in ultraviolet light but pyridoxine and itsthree congeners show photo-decomposition so thatmeasurements of fluorescence have to be madewithin 30 seconds of exposure to the exciting light.The fluorimetric assay of these compounds in
biological materials is carried out by oxidation to thehighly fluorescent pyridoxic acid which is eventuallyconverted into its lactone which, at pH 9, is some25 times more fluorescent than the acid (Huff andPerlzweig, 1944; Reddy, Reynolds, and Price, 1958).
COOH
HO>CHC20H
C9tN
Pyridoxic acid (no distinctfluorescence peak)
CO-0
HO .CHz
CH3,KN
Pyridoxic acid lactone(flu: 450 mM)
PROTEIN FLUORESCENCE AND FLUORESCENCE POLAR-IZATION The study of the fluorescence of proteinsis a difficult but interesting field and up to thepresent it has hardly been exploited. Here it isintended but to mention a few of its simpler aspectsand for further information the reader is referredto Chapter 6 of the book by Udenfriend (1962) andto reviews and papers by Weber and his associates(e.g., Weber, 1953; Weber, 1960 a, b; Teale, 1960;see also Udenfriend, 1962, for other references).
Simple proteins absorb light in the region270-300 m,u and this absorption is due mainly tothe aromatic amino-acids, tyrosine and tryptophan.Proteins containing these amino-acids could there-fore be expected to fluoresce. In addition to theseamino-acids, proteins frequently contain phenyl-alanine which is also fluorescent, but its fluorescenceis much weaker than that of the other two. Thefluorescence maxima and quantum efficiencies(Q.E.) of these acids in neutral aqueous solutionare: tryptophan, Amax. 348 m,u, Q.E. 20%; tyrosine,Amax. 303 m,u, Q.E. 21 %; and phenylalanine,Amax. 282 m,u, Q.E. 4% (Teale and Weber, 1957).A large number of proteins have been found tofluoresce (Shore and Pardee, 1956; Konev, 1957).Those containing phenylalanine and tyrosine but notryptophan show a maximum emission at 303 m,u,
390
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from
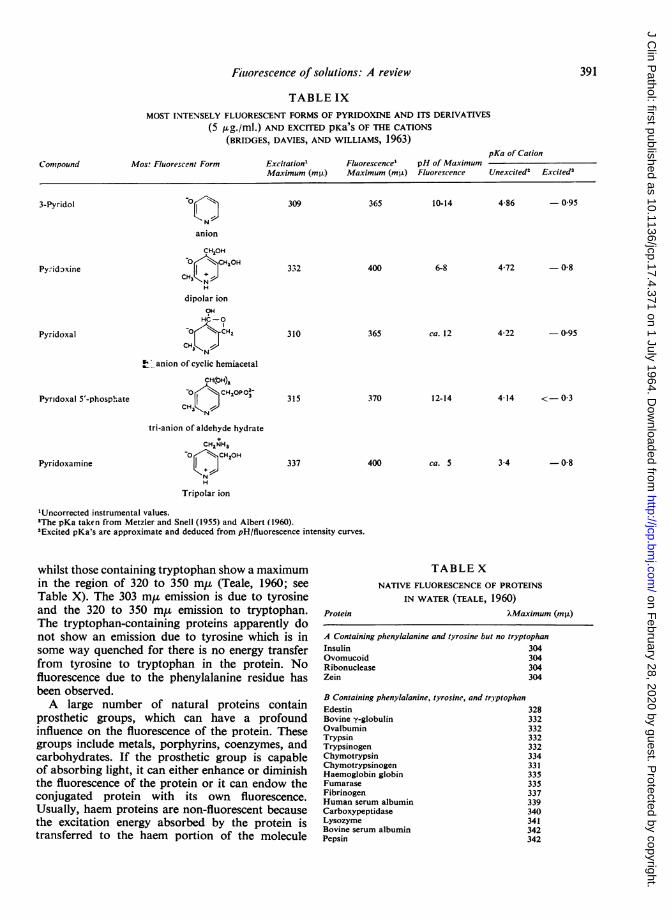
391Fiulorescence of solutions: A review
TABLE IXMOST INTENSELY FLUORESCENT FORMS OF PYRIDOXINE AND ITS DERIVATIVES
(5 /sg./mi.) AND EXCITED pKa'S OF THE CATIONS(BRIDGES, DAVIES, AND WILLIAMS, 1963)
Mos! Fluorescent FormpKa of Cation
Excitation' Fluorescence' pH of MaximumMaximum (mRL) Maximum (mis) Fluorescence Unexcited2 Excited3
3-Pyridol
Py.-idoxine
Pyridoxal
Pyridoxal 5'-phosphate
Pyridoxamine
0
N
anion
CH,OH0 N.H,OH
CH3 NJH
dipolar ionOH
HC-O
o CH,2
CH3c anion of cyclic hemiacetal
CH(OH),
-0 CH1OP0-
CH5
tri-anion of aidehyde hydrate
CHINH5o CHsOH
H
Tripolar ion
309 365 10-14 4-86 - 095
400 6-8 4-72 -0-8332
310
315
337
365 ca. 12
370 12-14
400 ca. 5
4-22 - 095
4-14 <-03
3-4 -0-8
'Uncorrected instrumental values.2The pKa taken from Metzler and Snell (1955) and Albert (1960).3Excited pKa's are approximate and deduced from pH/fluorescence intensity curves.
whilst those containing tryptophan show a maximumin the region of 320 to 350 m,u (Teale, 1960; seeTable X). The 303 m,u emission is due to tyrosineand the 320 to 350 m,u emission to tryptophan.The tryptophan-containing proteins apparently donot show an emission due to tyrosine which is insome way quenched for there is no energy transferfrom tyrosine to tryptophan in the protein. Nofluorescence due to the phenylalanine residue hasbeen observed.A large number of natural proteins contain
prosthetic groups, which can have a profoundinfluence on the fluorescence of the protein. Thesegroups include metals, porphyrins, coenzymes, andcarbohydrates. If the prosthetic group is capableof absorbing light, it can either enhance or diminishthe fluorescence of the protein or it can endow theconjugated protein with its own fluorescence.Usually, haem proteins are non-fluorescent becausethe excitation energy absorbed by the protein istransferred to the haem portion of the molecule
Protein
TABLE XNATIVE FLUORESCENCE OF PROTEINS
IN WATER (TEALE, 1960)
?Maximum (mg)
A Containing phenylalanine and tyrosine but no tryptophanInsulin 304Ovomucoid 304Ribonuclease 304Zein 304
B Containing phenylalanine, tyrosine, and tryptophanEdestin 328Bovine y-globulin 332Ovalbumin 332Trypsin 332Trypsinogen 332Chymotrypsin 334Chymotrypsinogen 331Haemoglobin globin 335Fumarase 335Fibrinogen 337Human serum albumin 339Carboxypeptidase 340Lysozyme 341Bovine serum albumin 342Pepsin 342
Compound
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
where it is degraded (Weber and Teale, 1959)(see p. 384). In the case of proteins conjugated withthe reduced pyridine nucleotides (e.g., alcohol,lactic, glutamic and malic dehydrogenases) theeffect of the prosthetic group is to endow the proteinwith its own fluorescence which is often intensifiedand shifted to shorter wavelengths (Shifrin andKaplan, 1960).
Fluorescent molecules can also be combined withproteins to give highly fluorescent conjugates whichcan be used for many purposes such as the assay ofserum proteins and the distribution of antigens andvarious proteins in tissues (Udenfriend, 1962).One aspect of fluorescence which is of particular
application to the study of proteins is fluorescencepolarization. This phenomenon can be explained asfollows. Light consists of waves whose amplitude inall planes is at right angles to the light path. Lightis plane polarized if its waves are in one plane onlyand this polarization can be achieved with a Nicolprism. If molecules are excited by plane polarizedlight, then those molecules whose axes happen to beorientated in a direction parallel to the light pathare preferentially excited. If these molecules are in arigid medium, the fluorescence they emit will beplane polarized, i.e., the waves are in one plane only.However, if the medium is not rigid, the excitedmolecules may be able to rotate about their axesbefore the fluorescence is emitted. In this case thefluorescence will be partially polarized or non-polarized depending upon the rate at which the
excited molecules rotate, the life time of the excitedstate, and the viscosity of the medium or solvent.The time taken for these excited molecules todisorientate completely is called the relaxation time.Each fluorescent molecule has a characteristicrelaxation time for a given solvent under givenconditions. For small molecules in low viscositysolvents the relaxation time is short compared withthe life time of the excited state (10-8 second) sothat no polarized fluorescence is observed with suchmolecules when they are excited by plane polarizedlight. Large molecules such as proteins, however,may emit polarized fluorescence since their relaxationtimes are long compared with the life time of theexcited state. The degree of polarization can bemeasured with a suitable Nicol prism analyzer andfrom this the relaxation time of the molecule can beestimated.The relaxation time gives useful inform-ation about the physical characteristics, e.g., shapeand size, of a macromolecule. If the macromoleculeis one which has no native fluorescence, its relaxationtime could still be measured provided it can beconjugated with a small fluorescent molecule (seeWeber, 1953; Laurence, 1957).
ADDITIONAL REFERENCES ON COMPOUNDS OF BIO-LOGICAL INTEREST Many references to the applica-tions of fluorescence methods in biology and medi-cine are given in the book by Udenfriend (1962). Inaddition to these, the following list of recentreferences may be quoted.
NATURAL PRODUCTS AND THEIR DERIVATIVES
FIchii, Forchielli, Perloff, and Dorfman (1963)[Rudd, Cowper, and Crawford (1961)
Estrone, -estradiol, cortisone and related compounds Stewart, Albert-Recht, and Osman, 1961)De Moor, Osinski, Deckx, and Steeno (1962)
LMattingly, (1962)17a substituted steroids Steinetz, Beach, Dubnick, Meli, and Fujimoto (1963)Methandrostenolone Tishler, Sheth, Giaimo, and Mader (1962)Bile acids Levin and Johnston (1962)Coproporphyrin Mentz and Grotepass (1960)Phospholipids Harris and Gambal (1963)Glycerol Mendelsohn and Antonis (1961)Acetone Hynie, et al., (1960)Malonaldehyde Sawicki, Stanley, and Johnson (1963)Coumarins Crosby and Berthold (1962)Anthranilic acid Sanders and Parks (1962)Hippuric acid Ellman, Burkhalter, and LaDou (1961)Carbohydrates Towne and Spikner (1963)
rMerrills (1963)Adrenaline, noradrenaline Jacobs, Sobel, and Henry (1961)
LMahler and Humoller (1962)Total catecholamines Small (1963)5-Hydroxy-and 5-methoxy-indoles Quay (1963)Serotonin f Crosti and Luchcelli (1962)
Crawford and Rudd (1962)HNoah and Brand (1961)
Hiatamine Zachariae (1963)Tetrahydrofolate and related compounds Uyeda and Rabinowitz (1963)Pyridoxamine Toepfer, Polansky, and Hewston (1961)NADP Estabrook (1962)Phenylalanine McCaman and Robins (1962)2-Dimethylamino-6-hydroxypurine Udenfriend, Zaltzman-Nirenberg, and Cantoni (1963)Guanine Udenfriend and Zaltzman (1962)
392
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

Fluorescence of solutions: A review
y-GlobulinsPlasma proteinsFibrinolysinLactic dehydrogenaseLipaseA6-3-Ketosteroid isomerase
DrugsDigitoxin and digoxinEmetine hydrochlorideReserpineChlordiazepoxide (librium)BenzoquinolizinesPhenothiazinesTetracyclinesOxytetracyclineAcrifiavineMechlorethamine (mustargen)Isoniazid
Metals in biological materialSelenium
Calcium
Magnesium, calcium, and zincMagnesium
Sokol, Hana, and Albrecht (1961)Hiraoka and Glick (1963)Pappenhagen, Koppel, and Olwin (1962)Laursen (1962)Kramer and Guilbault (1963)Wang, Kawahara, and Talalay (1963)
Jakovljevic (1963)Ichimura (1961)Tishler, Sheth and Giaimo (1963)Koechlin and D'Arconte (1963)Schwartz and Rieder (1961)Mellinger and Keeler (1963)Worsley, McKenna, and Beck (1963)Ibsen, Saunders, and Urist (1963)Bi6an-Fifter (1960)Mellett and Woods (1960)Peters (1960)
Dye, Bretthauer, Seim, and Blincoe (1963)Kepner and Hercules (1963)Wallach and Steck (1963)Hofman (1961)Watanabe, Frantz, and Trottier (1963)Schachter (1961)
REFERENCES
Albert, A. (1960). J. chem. Soc., P. 1020.Bidan-Fister, T. (1960). Acta pharm. jugosl., 10, 161. (Chem. Abstr.
(1961). 55, 14820c.)Borresen, Hans Chr. (1963). Acia chem. scand., 17, 921.Bowen, E. J., and Wokes, F. (1953). Fluorescence of Solutions.
Longmans, Green, LondonBowman, R. L., Caulfield, P. A., and Udenfriend, S. (1955). Science,
122, 32.Bridges, J. W. (1963). Ph.D. Thesis, University of London.
, Creaven, P. J., and Williams, R. T. (1963). Unpublished data.-, Davies, D. S., and Williams, R. T. (1963). Biochem J.88, 65P.
, Davies, D. S., and Williams, R. T. (1963). Unpublished data., and Williams, R. T. (1962). Nature (Lond.), 196, 59.
, (1963). Unpublished data.Brodie, B. B., Udenfriend, S., Dill, W., and Chenkin, T. (1947).
J. biol. Chem., 168, 319.Butler, T. C., Ruth, J. M., and Tucker, G. F. (1955). J. Amer. chem.
Soc., 77, 1486.Chance, B., and Baltscheffsky, H. (1958). J. biol. Chem., 233, 736.-, and Legallais, V. (1959). Rev. sci. Instr., 30, 732.Cleveland, F. F. (1955). Raman Spectra In Determination of Organic
Structures by Physical Methods, edited by E. A. Braude andF. C. Nachod, p. 231. Academic Press, New York.
Cowgill, R. W. (1963). Arch. Biochem., 100, 36.Crawford, N., and Rudd, B. T. (1962). Clin. chim. Acta, 7, 114.Crosby, D. G., and Berthold, R. V. (1962). Analyt. Biochem., 4, 349.Crosti, P. F., and Lucchelli, P. E. (1962). J. clin. Path., 15, 191.De Moor, P., Osinski, P., Deckx, R., and Steeno, 0. (1962). Clin.
chim. Acta, 7, 475.Duggan, D. E., Bowman, R. L., Brodie, B. B., and Udenfriend, S.
(1957). Arch. Biochem., 68, 1.-, and Udenfriend, S. (1956). J. biol. Chem., 223, 313.Duysens, L. N. M., and Amesz, J. (1957). Biochim. biophys. Acta
(Amst.), 24, 19.-, and Sweep, G. (1957). Ibid., 25, 13.Dye, W. B., Bretthauer, E., Seim, H. J., and Blincoe, C. (1963).
Analyt. Chem., 35, 1687.Ellman, G. L., Burkhalter, A., and LaDou, J. (1961). J. Lab. clin.
Med., 57, 813.Estabrook, R. W. (1962). Analyt. Biochem., 4, 231.Feigl, F., Feigl, H. E., and Goldstein, D. (1955). J. Amer. chem. Soc.,
77, 4162Forster, T. (1950). Z. Elektrochem., 54, 531.Hagins, W. A., and Jennings, W. H. (1959). Disc. Faraday Soc., no. 27,
180.Harris, R. A., and Gambal, D. (1963). Analyt. Biochem., 5, 479.
Hercules, D. M., and Rogers, L. B. (1959). Spectrochim. Acta, 14, 393.Hiraoka, T., and Glick, D. (1963). Analyt. Biochem., 5, 497.Hofman, J. (1961). Cas. Lek. ces., 100, 1171.Huff, J. W., and Perlzweig, W. A. (1944). J. biol. Chem., 155, 345.Hynie, I., Vederek, B., and Wagner, J. (1960). Cas. Lek. xes., 99, 88.Ibsen, K. H., Saunders, R. L., and Urist, M. R. (1963). Analyt.
Biochem., 5, 505.Ichii, S., Forchielli, E., Perloff, W. H., and Dorfman, R. I. (1963).
Ibid., 5, 422.Ichimura, Y. (1961). Bunseki Kagaku, 10, 623. (Chem. Abstr. (1962).
56, 1530e.)Jacobs, S. L., Sobel, C., and Henry, R. J. (1961). J. clin. Endocr., 21,
305.Jakovljevic, I. M. (1963). Analyt. Chem., 35, 1513.Kaplan, N. O., Colowick, S. P., and Barnes, C. C. (1951). J. biol.
Chem., 191, 461.Kasha, M. (1960). Radiat. Res., suppl., 2, 243.Kepner, B. L., and Hercules, D. M. (1963). Analyt. Chem., 35, 1238.Kerr, V. N., Hayes, F. N., and Ott, D. G. (1957). Int. J. appl. Radiat.,
1, 284.Koechlin, B. A., and D'Arconte, L. (1963). Analyt. Biochem., 5, 195.Kohn, K. W. (1961). Analyt. Chem., 33, 862.Konev, S. V. (1957). Dok. Akad. Nauk SSSR Otd. Biokh., 116, 594.Kramer, D. N., and Guilbault, G. G. (1963). Analyt. Chem., 35, 588.Kuhn, R., Wagner-Jauregg, T., and Kaltschmitt, H. (1934). Ber.
dtsch. chem. Ges., 67, 1452.Laurence, D. J. R. (1957). In Methods of Enzymology, edited by
S. P. Colowick and N. 0. Kaplan, vol. IV, p. 174. AcademicPress, New York.
Laursen, T. (1962). Scand. J. clin. Lab. Invest., 14, 152.Levin, S. J., and Johnston, C. G. (1962). J. Lab. clin. Med., 59, 681.Lippert, E., Luder, W., and Moll, F. (1959). Spectrochim. Acta, 10, 858Livingston, R., Watson, W. F., and McArdle, J. (1949). J. Amer.
chem. Soc., 71, 1542.Lowry, 0. H., Roberts, N. R., and Kapphahn, J. I. (1957). J. biol.
Chem., 224, 1047.-, , and Lewis, C. (1956). Ibid., 220, 879.Mahler, D. J., and Humoller, F. L. (1962). Clin. Chem., 8, 47.Mattingly, D. (1962). J. clin. Path., 15, 374.McCaman, M. W., and Robins, E. (1962). J. Lab. clin. Med., 59, 885.Mellett, L. B., and Woods, L. A., (1960). Cancer Res., 20, 518.Mellinger, T. J., and Keeler, C. E. (1963). Analyt. Chem., 35, 554.Mendelsohn, D., and Antonis, A. (1961). J. Lipid Res., 2, 45.Mentz, H. E. A., and Grotepass, W. (1960). S. Afr. J. Lab. clin. Med.,
6,43.Merrills, R. J. (1963). Analyt. Biochem., 6, 272.Metzler, D. E., and Snell, E. E. (1955). J. Amer. Chem. Soc., 77, 2431.Noah, J. W., and Brand, A. (1961). J. Allergy, 31, 236.Ott, D. G., Hayes, F. N., Hansbury, E., and Kerr, V. N. (1957).
J. Amer. chem. Soc., 79, 5448.
393
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from

R. T. Williams and J. W. Bridges
Pappenhagen, A. R., Koppel, J. L., and Olwin, J. H. (1962). J. Lab.clin. Med., 59, 1039.
Parker, C. A. (1959). Analyst, 84, 446.and Barnes, W. J. (1957). Ibid., 82, 606.
-, and Rees, W. T. (1962). Ibid., 87, 83.Peters, J. H. (1960). Amer. Rev. resp. Dis., 81, 485.Porter, G., and Weber, G. (1959). Nature (Lond.), 184, 688.Quay, W. B. (1963). Analyt. Biochem., 5, 51.R.C.A. Tube Handbook, HB-3. Radio Corporation of America,
Electron Tube Division, Harrison, New Jersey.Reddy, S. K., Reynolds, M. S., and Price, J. M. (1958). J. biol. Chem.,
233, 691.Rosen, A., and Williams, R. T. (1961). Bull. photoelect. Spectrom.
Grp, no. 13, 339.Rudd, B. T., Cowper, J. M., and Crawford, N. (1961). Clin. chini.
Acta, 6, 686.Sanders, P. P., and Parks, L. W. (1962). Analyt. Biochemii., 3, 354.Sawicki, E., Stanley, T. W., and Johnson, H. (1963). Analyt. Chean.,
35, 199.Schachter, D. (1961). J. Lab. clin. Med., 58, 495.Schwartz, D. E., and Rieder, J. (1961). Clin. chim. Acta, 6, 453.Shifrin, S., and Kaplan, N. 0. (1960). Advanc. Enzymol., 22, 337.Shore, V. G., and Pardee, A. B. (1956). Arch. Biochem., 60, 100.Small, N. A. (1963). Clin. chim. Acta, 8, 803.Sokol, F., Hana, L., and Albrecht, P. (196l). Folia Microbiol. (Praha),
6, 145.Steinetz, B. G., Beach, V. L., Dubnick, B., Meli, A., and Fujimoto, G.
(1963). Steroids, 1, 395.Stewart, C. P., Albert-Recht, F., and Osman, L. M. (161). Clin.
chim. Acta, 6, 696.Szent-Gyorgyi, A. E. (1957). Bioenergetics. Academic Press, New
York.Teale, F. W. J. (1960). Biochem. J., 76, 381.
, and Weber ,G. (1957). Ibid., 65, 476.
Tishler, F., Sheth, P. B., and Giaimo, M. B. (1963). J. Ass. off. agric.Chem., 46, 448.,-, and Mader, W. J. (1962). J. pharm. Sci., 51, 1175.
Toepfer, E. W., Polansky, M. M., and Hewston, E. M. (1961).Analyt. Biochem., 2, 463.
Towne, J. C., and Spikner, J. E. (1963). Analyt. Chem., 35, 211.Udenfriend, S. (1962). Fluorescence Assay in Biology and Medicine.
Academic Press, New York.Duggan, D. E., Vasta, B. M., and Brodie, B. B. (1957). J.Pharmacol. exp. Ther., 120, 26.and Zaltzman, P. (1962). Analyt. Biochem., 3, 49.Zaltzman-Nirenberg, P., and Cantoni, G. L. (1963). Ibid., 5, 258.
Uyeda, K., and Rabinowitz, J. C. (1963). Ibid., 6, 100.Van Duuren, B. L. (1963). Chem. Rev., 63, 325.Vannotti, A. (1954). Porphyrins-Their Biological and Chemical
Importance, translated by C. Rimington, p. 20. Hilger andWatts, London.
Wallach, D. F. H., and Steck, T. L. (1963). Analyt. Biocheml., 6, 176.Wang, S., Kawahara, F. S., and Talalay, P. (1963). J. biol. Chem.,
238, 576.Watanabe, S., Frantz, W., and Trottier, D. (1963). Analyt. Biochenm.,
5, 345.Weber, G. (1953). Advanc. Protein Chenm., 8, 415.
(1957). Nature (Lond.), 180, 1409.
(1958). J. Chim. Phys., 55, 878.-(1960a). Biochem. J., 75, 335.-(1960b). Ibid., 75, 345.
, and Teale, F. W. J. (1959). Disc. Faraday Soc., no. 27, 134.Weil-Malherbe, H. (1946). Biochem. J., 40, 363.White, A. (1959). Ibid., 71, 217.Williams, R. T. (1959). J. roy. Inst. Chem., 83, 611.Worsley, G. H., McKenna, R. D., and Beck, I. T. (1963). Canad. mled.
Ass. J., 88, 1272.Zachariae, H. (1963). Scand. J. clin. Lab. Invest., 15, 173.
394
on February 28, 2020 by guest. P
rotected by copyright.http://jcp.bm
j.com/
J Clin P
athol: first published as 10.1136/jcp.17.4.371 on 1 July 1964. Dow
nloaded from





























