Embed Size (px)
Citation preview

Flexibility of Protein Structure
• Proteins show varying degree of conformational flexibility
• Due to movements of atoms in molecules • vibration in bond length and angles
• Reflects the existence of populations of alternative conformations
• Severe constraints on the extent of conformational variations
• Integral proteins are less flexible than water- soluble proteins

Hydrogen Exchange
• The best evidence for extensive mobility of protein structure
• Hydrogen atoms bonded covalently to various atoms exchange with solvent at different intrinsic rates, depending on the tendency of that atom to ionize– H atoms on oxygen, nitrogen, or sulfur exchange rapidly– H atoms on carbon change at slow rate
• Intrinsic rate of exchange is temperature dependent– 3 fold increase with 10 C increase– Also, influenced by the environment and by inductive and charge
effects on the amide

Hydrogen Exchange
• Local unfolding by hydrogen exchange in interior group atoms– By diffusion of solvent molecules to various sites
• Rates are correlated with the solvent accessibility of the residue and of its neighbors, not distance
• Rate is decreased by increased pressure• Wide variations in processes depending on the
proteins and conditions

Stability of the Folded Conformation
• The folded conformations of proteins are only marginally stable even under the best conditions
• Can be easily denatured by changes in environmental conditions – Temperature increase– pH changes– Pressure increase– Addition of denaturants

Reversible folding & unfolding
• Anfinsen (1950’s) – showed reversibility of denaturation with urea for RNAse A– amino acid
sequence encodes structure: thermodynamic hypothesis
– Folding is “cooperative”
Denaturation-studiedby various methodsis reversible
S-S bonds in brown

Stability of Proteins
Stability of the folded structure of a globular protein depends on the interplay of three factors.
1. The unfavorable conformational entropy change, which favors random chains instead.
2. The favorable enthalpy contribution arising from intramolecular side group interactions.
3. The favorable entropy change arising from the burying of hydrophobic groups within the molecule.
• Factor 1 works against folding, whereas 2 and 3 help stabilize folding.

Denaturation
• Do need not changes in covalent structure• Usually reversible if it is due to unfolding• Proteins can be unfolded by
– Breaking any disulfide bonds present– Removing any essential cofactors– Mutation in certain crucial residues– Deleting residues from the primary structure

Reversible Unfolding Transition
• No decrease in biological activity• Unfolding is a two-state phenomenon, with only fully
folded (F) and fully folded (U) states• Partially folded structures are unstable relative to U or F• Under conditions that favor unfolding,
– The folded conformation changes very little if at all– Increase in flexibility– Localized conformational alterations– Average structure not changed
• Then protein unfolds completely within a limited range of conditions

Nature of the Unfolded State
• Unfolded states produced under various unfolding conditions have different physical properties
• Unfolded proteins do not contain cooperative folded structure
• But, unfolded and folded proteins are indistinguishable thermodynamically

Molten Globule State• Conformation that are neither fully folded nor
fully unfolded– The overall dimensions of the polypeptide chain are
much less than those of a random coil– Average content of secondary structure is similar to
folded– The side chains are in homogeneous surroundings– Many amide groups exchange hydrogen atoms with
the solvent much rapidly than the folded – The enthalpy is nearly the sale as the folded state– Interconversions of the molten globule state with the
fully folded state are rapid

Physical Basis for Protein Denaturation
• Extreme pH– Non-ionized His and Tyr buried in folded structure can
be ionized– Disruption of salt bridges
• Denaturants: Urea and Guanidinium salts– Break H bonds– Have hydrogen bonding capabilities comparable to
that of water, but different geometries– Increase the solubilities of both polar and nonpolar
molecules

Thermodynamics of Unfolding
• Elevated temperature increase unfolding– Proteins from thermophiles are more heat stable– Unfolded state has a greater heat capacity than the
folded state– Large temperature dependence of the changes in
enthalpy and entrophy– The more hydrophobic the interior of a protein, the
lower its net stability

Forces that hold protein structure together
• Dispersion (van der Waals) forces• Electrostatic interactions• Hydrogen bonds• Hydrophobic interactions
• What pulls it apart?• Conformational entrophy

Stabilizing Forces of Folded Conformations
• Hydrogen bonds– The major stabilizing force for folded conformation– H-bonds in the folded conformation is stronger than those in
unfolded ones– H-bonds with water and a protein are present a fraction of a time
while that within protein present are present all of the time (more negative enthalpy)
• Electrostatic interactions– More important van der Waals interactions
• Van der Waals interactions• Hydrophobic interactions
– Unique in folded structure– Driving force for folding

Contributions to protein stability
type of interaction total contribution*hydrophobic group burial ~200 kcal/molhydrogen bonding small??ion pairs/salt bridges <15 kcal/moldisulfide bonds 4 kcal/mol per link
*For globular protein of 150 residues hydrophobic burial is the chief interaction favoring protein stability, but this is balanced by a huge loss of conformational entropy that opposes folding.
Consequently typical net protein stabilities are 5-20 kcal/mol--> so even minor interactions can make a difference!

Hydrophobicity and the Stability of Folded Conformation
• Hydrophobicity is not the Primary Force to the Stability of Folded Conformation. Why?
• Although the interior of proteins are densely packed, they are not closely packed with all adjacent atoms in Van der Waals contact simultaneously
• Imperfect Van der Waals interactions
• Van der Waals interactions in proteins are in less than optimal conditions and weaker than those occur in liquid

Stability of the Folded conformation
• Folded state has only marginal net stability because of the large conformational entrophy of the unfolded state
• Depends on its primary structure
• Single amino acid replacement can alter the stability of a folded protein quite drastically

Alanine scanning• A way of assessing the importance of amino-acid side chains for
structure/stability etc.• “Remove” each residue one by one by replacement with Ala• Many Ala mutations have no effect on stability--> about half!• A large group also cause significant effects-->several kcal/mol• Occasional a mutant will stabilize the protein--> natural proteins not
maximally stable!

The interior is more important for stability than the exterior
side chains with stability-neutral Ala mutations
side chains with destabilizing Ala mutations

Side-chain packing in the hydrophobic core
• Protein interiors have a “jigsaw puzzle”-like aspect.
• Their packing densities are similar to those of crystals of organic molecules.
• This dense packing are importance for stability and a precise three- dimensional structure
• Over or under filling can be detrimental to stability
• However, not all cores with equal volume are equally stable.

Effect of nondisruptive hydrophobic core mutations
leucinein core
mutation to alanine
difference in water-octanol transferfree energies of leucine and alanineis ~2 kcal/mol. Effects of Leu-->Alamutations are typically larger thanthis. Why?
Not all buried Leu-->Ala mutationsgive the same destabilization. Why?
nondisruptivemeans not causingany steric clashesor uncompensatedburied charges, H-bondsetc

The cost of cavity formation in protein cores
• A Leu-->Ala core mutation leaves a cavity in the hydrophobic core.
• In addition to the ~2 kcal/mol transfer free energy difference between Leu and Ala, there is a penalty of 20 cal-mol/Å2 for forming this cavity.
• This is due to loss of van der Waals interactions with the mutated side chain.
• This increases the cost of a Leu-->Ala mutation from 2 to about 5 kcal/mol!
• Since proteins are only stable toward unfolding to the extent of 5 to 15 kcal/mol, such mutations are potentially devastating
• This suggests that having good packing in terms of not having cavities is important to stability.

Disruptive mutations in hydrophobic cores
Three kinds:steric mutants change in shape, not volumeextreme volume mutants increase core volumepolar mutants put polar/charged residue in core
• Polar/charged core mutants are almost invariably very destabilizing, for obvious reasons.
• Charged groups or groups that can form hydrogen bonds that are isolated within a protein interior are bad for stability
• Energetic effects of increased volume are often hard to predict-- subtle backbone shifts often occur to accommodate the extra volume

Mutations of surface (solvent-exposed) residues
• Although the surface of proteins are very polar overall, individual surface positions can usually be replaced by many other residues including hydrophobics without much effect on stability.
• average effect on stability of surface mutations is small• little stability penalty for change of individual surface polars to
hydrophobics
However:• too many hydrophobics on a protein’s surface will reduce solubility and
promote aggregation• surface polar-to-hydrophobic mutations can reduce structural specificity by
favoring alternative conformations in which the introduced hydrophobic side chain becomes buried.

“Hydrogen bond inventory”
• Definition: Although hydrogen bonds probably do not stabilize proteins per se, it is nonetheless important that all potential hydrogen bond donors and acceptors be hydrogen bonded to something - solvent, protein backbone, or protein side chains.
• Removal of one partner of a hydrogen bonded pair by mutation can be quite destabilizing if the remaining partner is not able to satisfy its hydrogen bond potential by interacting with solvent.
• Also applicable to ion pairing/salt bridge interactions.
• Even though ion pairs don’t contribute much to stability, charged groups which are neither paired with oppositely charged groups nor solvated by water can be very destabilizing!

Role of solvent-exposed salt bridges
• Typical mutations of surface salt bridges are destabilizing by less than 1 kcal/mol, but there are cases where larger effects are observed.
• Surface salt bridges are thus not large contributors to protein stability.
• However, some salt bridges may be important at the level of specifying a particular precise structure, much in the way that hydrophobic packing interactions are.
• Buried salt bridges (and buried polar interactions in general) not important for stability per se, but removal of individual partners can be hugely destabilizing.
• Buried polar interactions serve more to impart specificity to the structure rather than stability, due to the strict requirement for satisfaction of H-bond potential (H-bond inventory) and compensation of charge.

Stability-activity trade-offs?
• Proteins have not evolved to maximize stability.
• They generally evolve to preserve adequate stability.
• However, sometimes stability and activity are selected at the expense of the other.
• Some mutations in the active sites are more stable but less active.
• For instance, the active site of barnase is highly positively charged because it has to bind a negatively charged pentacoordinate phosphate at the transition state.
• When substrate is not bound, the positively charged side chains repel each other, reducing stability.

protein ΔTm ΔΔGf , activity, °C kcal mol-1 (relative)
wild-type 0 0 1E11F 4.3 1.7 <0.0001E11M 4.1 1.6 <0.0001E11A 2.6 1.1 <0.0001E11H 0.1 0.1 <0.0001E11N -0.6 -0.1 <0.0001D20N 3.1 1.3 <0.0001D20T 2.2 0.9 <0.0001D20S 1.6 0.7 <0.0001D20A -0.8 -0.3 0.0005
Mutation of catalytic residues in T4 lysozyme
Glu 11
Asp 20
Replacement with some amino acids increases stability but strongly diminishes activity. This same phenomenon was found to occur for residues involved in substrate binding. Glu11 and Asp20 are examples of what has been referred to as “electrostatic strain” in enzyme active sites. However, not all mutations which remove the charge stabilize the protein, emphasizing that the situation is complex.
active site cleft
Shoichet et al. PNAS 92, 452 (1995)

29
Factors Determining Secondary and Tertiary
Structure

30
The Information for Protein Folding
• The information for determining the 3-dimensional structure of a protein is carried entirely in the amino acid sequence of that protein.
• Native structure is the natural 3-dimensional structure of a protein.
• Denaturation is a process for the lost of natural structure of a protein, along with many of its specific properties.
• In a cell, a newly synthesized polypeptide chain will spontaneously fold into the proper conformation.

Protein Folding• Proteins normally want to fold into their “native,” or
lowest-energy, conformation – the most stable
• Christian B. Anfinsen won the Nobel Prize in 1973 for finding that this folding is based on “directions” from the amino acid sequence and from its hydrophobic/phillic nature
• He started by unfolding proteins into random coils, and found the proteins folded back into their native shape spontaneously, without any other catalysts

32
Thermodynamics of Folding The overall free energy change on folding must be
negative.
• Conformational Entropy– ΔG = ΔH – TΔS
• Charge-Charge Interactions– Salt bridges
• Internal Hydrogen Bonds• van der Waals Interactions• The Hydrophobic Effect

Gibbs Free Energy (ΔG)
• A thermodynamic potential that measures the "useful" or process-initiating work obtainable from an isothermal, isobaric thermodynamic system
• The maximum amount of non-expansion work that can be extracted from a closed system
• Gibbs free energy (ΔG) or available energy is the chemical potential that is minimized when a system reaches equilibrium at constant pressure and temperature.

What features of protein folding produce large, negative ΔHor large positive ΔS changes, to compensate for the conformational entropy loss ?
Contributions to ΔG of folding

35
The Role of Disulfide Bonds
• The formation of disulfide bonds between cysteine residues further stabilize the 3-D structure of proteins.
• Formation of the disulfide bonds is depend on the protein primary structure.
• Disulfide bonds are not essential for correct protein folding, but for contribution to the stability of the structure once it is folded.
• Site-directed mutagenesis, a powerful method for the test of the effect of changing one or more amino acid residues or adding or removing disulfide bonds.


37
Kinetics of Disulfide Bond Formation
• The process of refolding of a protein from a state in which disulfide bonds have been cleaved is complicated and slower.
• Some disulfide bonds that are not in the native structure are formed in the intermediate stages of the folding.
• The protein can utilize a number of alternative pathways to fold but ultimately finds both its proper tertiary structure and the correct set of disulfide bonds.
• It is aided in vivo by enzymatic catalysis of -s-s- bond rearrangement.


When Folding Goes Wrong
• Protein Misfolding: Proteins fold into conformations which are local energy minima, but not the global minimum
• States reaching local, but not global, minima are sometimes called “intermediate states”
• Misfolding is responsible for many diseases due to the accumulation of “plaque” of misfolded proteins – Alzheimers, Parkinsons, Mad Cow Disease, etc.
• “chaperone” proteins are present to help the target protein reach the desired functional (native) conformation

Prions - Protein Folding and Mad Cow Disease
• A class of diseases that is transmitted by a protein.• Bovine spongiform encephalopathy or mad cow disease is
the best known of these diseases.• The infectious agent is called prion, and the protein is
called prion-related protein, or PrP.• PrP protein is normally present in many animals including
human in a nonpathological form of PrPc (prion-related protein cellular).
• Under certain circumstances (not yet known), PrPc, can change the conformation to a different structure called PrPsc
(or prion-related protein scrapie).• The N-terminal portion of PrPsc is partially folded into a β-
sheet which wreaks havoc with the nervous system.• It can induce conversion of PrPc in the recipient to PrPsc.

The Protein Folding Problem: Levinthal’s paradox
How many conformations could a protein chain adopt?
• there are about 3-5 combinations of φ/ψ for each amino acid that would be acceptable to its neighbour.
• so its 3-5n conformations that a protein can adopt (n = number of amino acids)
• 100 amino acids = 4100 i.e; about 1 x 1060 conformations

Levinthal paradox
• If a protein can sample 1014 conformations per second (average frequency of bond rotation is about 10-14s, 0.01 ps)
• Then it would take our 100 residue protein 1 x 1045
seconds, or 5 x 1038 years to sample all its options.
• Folding cannot simply occur by the protein trying every conformation until it finds the most stable one. There isn't enough time.
• HOW DO PROTEINS EVER FOLD????

• cytochrome b562: 5 μs• lambda repressor: 0.67 ms• rat IFABP: 33 ms• CRABP 1: 24.5 sec• tryptophan synthase β2-subunit: 992 sec (396 aa)
Time-scales for folding

Thermodynamic vs. Kinetic control?
• Folding is driven by thermodynamic considerations.
• The native state must be thermodynamically stable.
• But the kinetics of folding is also important.
• Folding must occur rapidly.
• Do folded structures represent true global energy minimum, or just “kinetically accessible” local minima?
• What causes slow folding: a high transition-state barrier, or just a large space to search?

Kinetics of Protein Folding
• Folding takes place through a series of intermediate states, unfolded protein U, nucleation protein, partially folded II , nearly folded IN and the final rearrangement to become native protein F.
• The funneling model for nucleation of proteins proposes that there is not just one but many possible paths from the denatured state to the folded state, and each path leads downhill in energy.
• The molten globule, a compact structure in which much of the secondary and tertiary folding has occurred, but the internalized hydrophobic residues have not yet formed.
• There is also off-path state in which some key element is incorrectly folded, and the final folding time will be delayed.

Lowest energy state?
• The forces of macromolecular interaction:– Covalent bonds– Electrostatics – Hydrogen bonds – van der Waals – Hydrophobic effect
– If not optimal then each of these forces contributes an energy cost to the system.
– If optimal then the forces above are satisfied and the energy is minimal

Balls roll downhill, and Levinthal is satisfied
• Levinthal’s problem is removed if the folding pathway is biased towards the correct structure.
• i.e. once you start moving in the right direction, you’ll get there
correctly folded state is the lowest energy state

Folding theory: Energy landscapes
• formation of local structure stabilizes
• An intermediate called the “transition state ensemble” is thought to exist – Metastable– Key contacts– Same topology as fully folded
structure
• enables collapse to native fold
energy

Energy Landscapes in 2-D
• In the process of folding from a random coil to the native state (at the global minimum), proteins “pass through” intermediate states (at local minima)
• Sometimes the proteins can get “stuck” in these intermediate states and cannot get over the “energy hump” needed (at transition states) to get to the final native conformation
• The study of protein folding from the denatured to the native state of vice versa, over time, is kinetics
Local minima
Global minimum
ETransition states

N
TS
D
∆GTS
-N
xu
xf
∆∆GTS-N
∆∆GD-N
When you add FORCE (F):Relative to the native state, N,
the barrier to unfolding (∆GTS-N ) is lowered by: Fxu
and the free energy of unfolding (∆GD-N ) is lowered by: F(xu + xf )
The protein is less stable and unfolds more rapidly - the unfolding rate (ku ) is a measure of the height of the barrier between N and TS
How does force modify the unfolding landscape?

The Funnel (3-D) Landscape Model
• Protein Folding actually proceeds (as down a funnel) through multiple kinetic pathways
• Unstable (High Energy) unfolded conformations at rim can follow many energy gradients to get to the single global minimum (Lowest Energy)
• The steepest path is the fastest folding trajectory, but other slower trajectories to the minimum exist
• Note the diverse multitude of unfolded conformations, but only one conformation for the native state
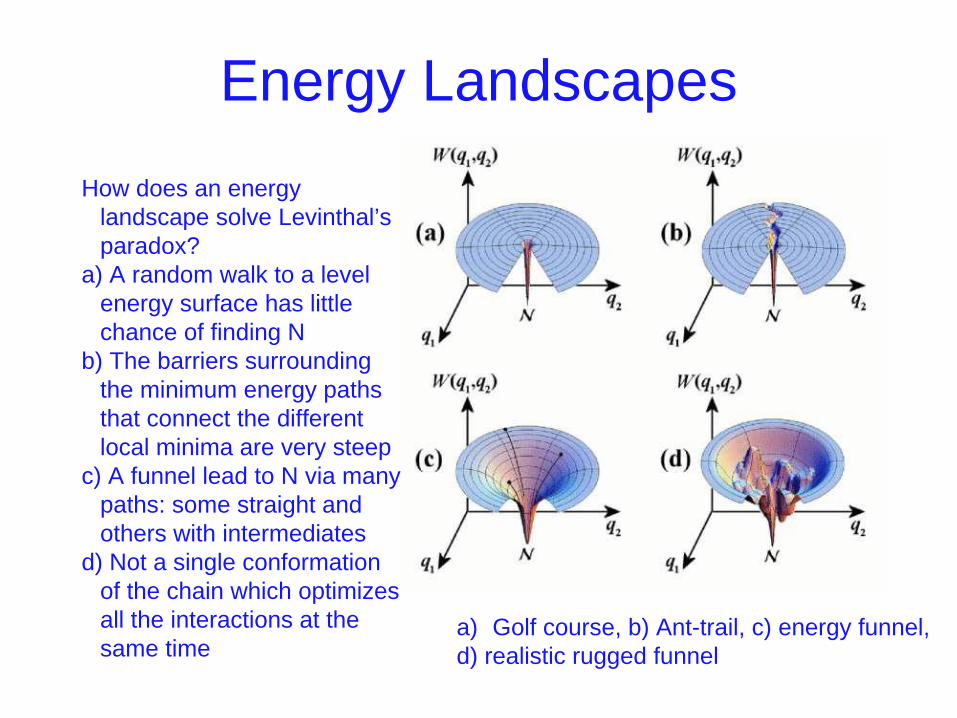
Energy Landscapes
a) Golf course, b) Ant-trail, c) energy funnel, d) realistic rugged funnel
How does an energy landscape solve Levinthal’s paradox?
a) A random walk to a level energy surface has little chance of finding N
b) The barriers surrounding the minimum energy paths that connect the different local minima are very steep
c) A funnel lead to N via many paths: some straight and others with intermediates
d) Not a single conformation of the chain which optimizes all the interactions at the same time

Molten Globule
• An intermediate state in the folding of protein pathway of a protein that has some secondary and tertiary structure, but lacks the well packed amino acid side chains that characterize the native state of a protein.
• Observed for many protein under both equilibrium and non-equilibrium conditions.
• By contrast, for fast folding proteins without intermediates, the search for a core or nucleus is likely to be the rate-determine step; once the core is formed, folding to the native state is fast

• A class of proteins which "chaperone" a protein to help keep it properly folded and non-aggregated.
• Aggregation is a problem for unfolded proteins
• The hydrophobic residues, which normally are deep inside of a protein, may be exposed when the protein is released from the ribosome.
• If they are exposed to hydrophobic residues in other strands, the two strands may associate with each other hydrophobically (to aggregate) instead of folding properly.
• It provides a central cavity (shelter) in which new protein chains can be "incubated" until they have folded properl
• ATP is required to drive the process in one direction. 24
Chaperonins

Chaperonins

Enzymes that Speed Folding
• Protein disulphide isomerase Facilitates formation of correct disulphide bridges
• Peptidyl proline isomerase Catalyses cis- trans isomerisation of peptide bonds involving proline
• Molecular chaperones Help folding, especially of large proteins, by preventing interaction with other proteins

Prediction of Protein Structure
• Can we predict the protein structure?
• Yes, if we could obtain all the necessary information, including the amino acid and understanding the rules of folding.
• Secondary structure can be predicted fairly well to-date, but not for the tertiary structure.

Problem of protein structure prediction
• For over 30 years, there has been an ardent search for methods to the predict three- dimensional (3D) structure from the sequence
• Many methods were found which looked initially very promising - but always the hope has been dashed

Techniques of Structure Prediction
• Computer simulation based on energy calculation
– Based on physio-chemical principles
– Thermodynamic equilibrium with a minimum free energy
– Global minimum free energy of protein surface
• Knowledge Based approaches
– Homology Based Approach
– Threading Protein Sequence
– Hierarchical Methods