Embed Size (px)
DESCRIPTION
Local Flexibility Aids Protein Multiple Structure Alignment. Matt Menke Bonnie Berger Lenore Cowen. The Protein Multiple Structure Alignment Problem. Input: The 3D coordinates of the atomic structures of k proteins - PowerPoint PPT Presentation
Citation preview

Local Flexibility Aids Protein Multiple Structure Alignment
Matt Menke Bonnie Berger
Lenore Cowen

The Protein Multiple Structure Alignment Problem
Input: The 3D coordinates of the atomic structures of k proteins
Output: A multiple sequence alignment, together with a set of rigid body transformations that superimpose the structures

What Makes a Good Alignment?
Geometric criteria:
Good multiple structure alignments MAXIMIZE number of residues places in alignment while MINIMIZING distances between aligned residues.

What Makes a Good Alignment?
Geometric criteria:
BICRITERIA OPTIMIZATION PROBLEM:
Place everything in the core, and residue distances are bad.
Place a single residue in the core, all distances are great!

What Makes a Good Alignment?
Biological criteria:
Good multiple structure alignments align structures (and portions within structures) that are supposed to align.

History of the Protein Structure Alignment Problem
• Studied as long as the better-known multiple sequence alignment problem
• Pairwise and multiple structure versions
• Wikipedia has links to over 50 different methods (programs/server/papers)
• NP-hard for ever simple variants (reference)
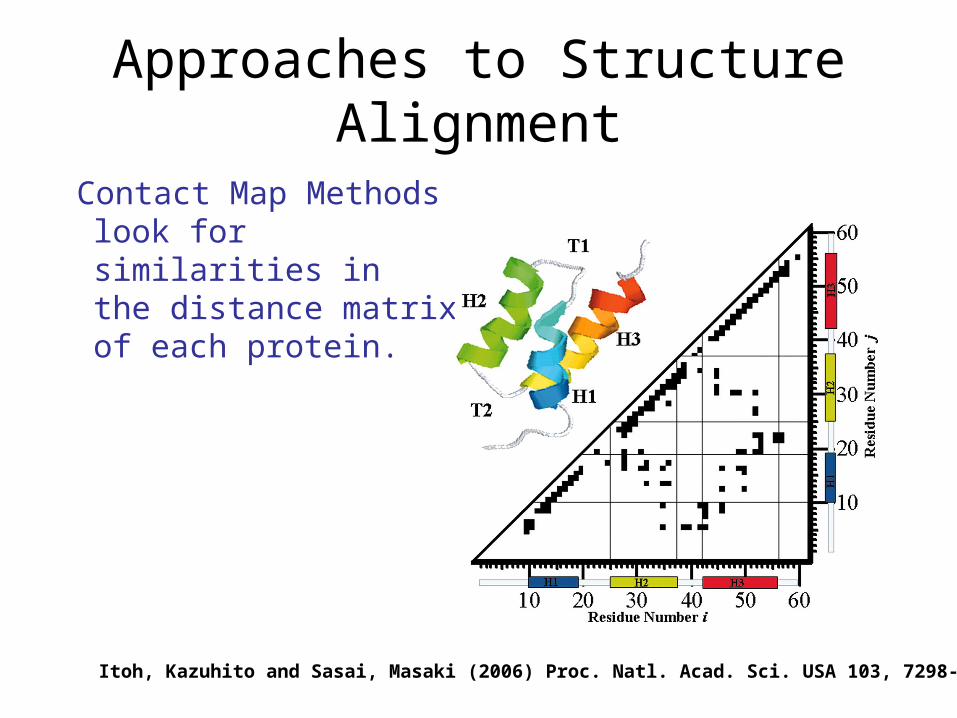
Approaches to Structure Alignment
Contact Map Methods look for similarities in the distance matrix of each protein.
Itoh, Kazuhito and Sasai, Masaki (2006) Proc. Natl. Acad. Sci. USA 103, 7298-7303

Approaches to Structure Alignment
• AFP chaining methods align all short pieces and chain together using dynamic programming
• Geometric hashing, secondary structure elements, etc.

The Benchmark Datasets
• Globins
• Homstrad– 1028 alignments – Each alignment contains 2-41 structures– 399 sets with > 2 structures

Why Another Structure Aligner?

The Benchmark Datasets
Sabmark – more distant homologySuperfamily set: – 3645 domains in 426 subsetsTwilight zone set: – 1740 domains in 209 subsetsBoth sets contain: – Between 3 and 25 structures– Decoy structures (sequence matches that
reside in different SCOP domains)

Matt: Multiple Alignment with Translation and Twists
• Matt is an AFP chaining method that additionally adds flexibility in the form of geometrically impossible bends and breaks.

Other work modeling flexibility
• In structure alignment: – Flexprot [Shatsky et al., 2002]– Fatcat/POSA [Ye&Godzik, 2004, 2005]
• For other reasons: – Molecular docking [Echols et al,03; Bonvin,06]– Ligand binding [Lemmen et al, 2006]– Decoy construction [Singh&Berger, 2006]

Matt: Pairwise alignment algorithm
1. Align all-against-all 5-9 residue fragments
2. Assemble fragment pairs with dynamic programming, allowing “impossible” local rotations & translations (bent alignment)
3. Keeping residue correspondences, find best rigid body superimposition (unbent alignment)


Outline of the Matt Algorithm

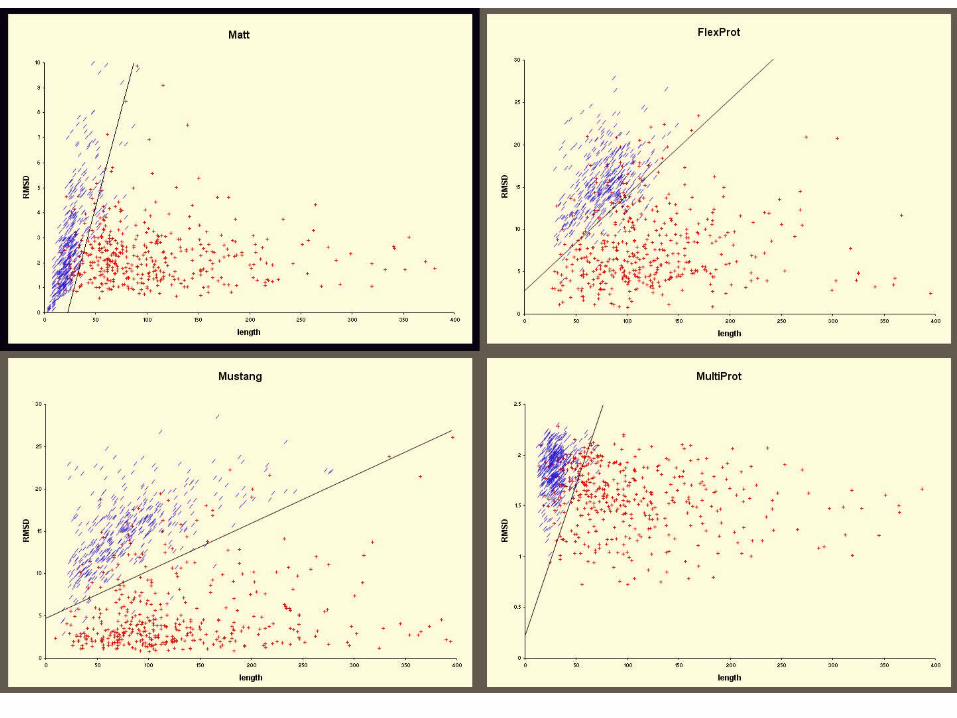
Results on Sabmark (Superfamily)
Program Name Avg. Core Size Avg. RMSD
Multiprot 68.701 1.498
Mustang 104.162 4.146
Matt 104.692 2.639

Results on Sabmark (Twilight Zone)
Program Name Avg. Core Size Avg. RMSD
Multiprot 36.54 1.536
Mustang 66.833 5.035
Matt 66.967 2.916



Sabmark Decoy Set
• For each SCOP superfamily, positive examples of the fold, and negative examples that are – Random examples from a different
superfamily– Examples from a different superfamily that are
nonetheless good BLAST hits

On the Web
• Matt source code and Windows binaries can be downloaded from: http://matt.cs.tufts.edu or http://groups.csail.mit.edu/cb/matt/
• Licensed under GPL 2.0; talk to us for commercial resale licensing.
• Accepts PDB files; outputs bent and unbent alignments in FASTA, PDB and RASMOL format.
• Matt paper: “M. Menke, B. Berger, L. Cowen, "Matt: Local Flexibility Aids Protein Multiple Structure Alignment", PLOS Computational Biology, Vol. 4, No 1., 2008.

Acknowledgements
• National Science Foundation
• National Institutes of Health