Embed Size (px)
Citation preview

Indian Journal of Chemistry Vol. 55B, November 2016, pp. 1384-1399
Feasibility and diastereoselectivity of acid-mediated three-component aza-Diels-Alder reactions: Preparation of diversely substituted hexahydro-2H-pyrano
[3,2-c]quinolines
Patricia Niño#a, Marta Caba#a, Nuria Aguilarb, Emma Terricabrasb, Fernando Albericioc,d,e & Joan-Carles Fernàndez*a a Almirall-Barcelona Science Park Unit, Barcelona Science Park, Baldiri i Reixac 10-12, 08028-Barcelona, Spain
b Research Center, Almirall, Laureà Miró 408-410, E-08980 Barcelona, Spain c School of Chemistry and Physics, University of Kwazulu-Natal, Durban 4001, South Africa
d CIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona Science Park, Baldiri Reixac 10, 08028 Barcelona, Spain
e Department of Organic Chemistry, University of Barcelona, Martí i Franqués 1-11, 08028 Barcelona, Spain E-mail: [email protected]
Received 1 January 2016; accepted (revised) 13 September 2016
Synthesis of highly functionalized substituted hexahydropyrano[3,2-c]quinolines 1 in high to moderate yields is described herein. The diastereoselectivity and regioselectivity obtained in the Povarov reactions of dihydropyrans is shown to depend on the nature of the catalytic acid and/or effect of the substitution of the aldehyde reactants used in the Povarov reaction. Thus, Lanthanide Lewis acid catalysts give higher endo diastereoselectivity than protic Brönsted acid catalysts such as trifluoroacetic acid (TFA), 4-nitrophatlic acid (4-NO2PhA) and sulfamic acid (HOTf) which give higher exo diastereoselectivity. Furthermore, it is found that ortho and di-ortho substitution of the aldehyde precursor increased the exo diastereoselectivity which is higher when the bulk of the ortho-group increases. In addition, substitution of the protic Brönsted acid (TFA) for TMSCl catalyst improves the yield of the Povarov reaction maintaining the high exo diastereoselectivity. The procedure allows for scale up of the Povarov reaction and the hexahydropyrano[3,2-c]quinolines can be obtained on a multigram scale.
Keywords: Povarov reaction, multicomponent reaction, cycloaddition, imines, aldehydes, alkenes, diastereoselectivity, scandium, heterocycles, tetrahydroquinolines, parallel synthesis
Aza-Diels–Alder is one of the most powerful synthetic routes for constructing nitrogen containing six-membered heterocycles1. Imines derived from anilines can act as hetero-dienes and undergo an imino-Diels–Alder reaction with various dienophiles2, leading to tetrahydroquinolines. Lewis acids (NbCl5, SnCl2, InCl3, TiCl3, TiCl4, BiCl3, VCl3, SbCl3, ZrCl4, BF3·Et2O, LiBF4, etc.)1a,3 are known to catalyze these reactions and advantageously replace Brönsted acids (HCl, TFA, 4-NO2PhA, HOTf, etc.)4. More recently, lanthanide derivatives (Sc(OTf)3, Yb(OTf)3, Sm(OTf)3, SmI2, GdCl3, etc.)5,1a have also been used as catalysts for this reaction. Despite improvements by performing these aza-Diels–Alder reactions in one-pot by coupling with aldehydes, new and milder conditions
are still desirable for these transformations as well as to gain greater diastereoselectivity.
In our previous publication about the preparation of novel tetrahydro-1H-cyclopenta[c]quinolines via the one-pot condensation of aldehydes, cyclopentadiene and anilines in the presence of scandium (III) triflate as the catalyst, we found a high diastereoselectivity to generate the tetrahydroquinoline scaffold (Figure 1)6. The cycloaddition reaction resulted in most cases in the formation of the endo isomer (cis ring-fused as shown in Figure 1) as the major diastereomer. However, the synthesis of analogs with modifications to the central tetrahydroquinoline core changing the dienophile to enolethers such as dihydropyran or dihydrofuran led to the loss of this high endo diastereoselectivity.
In this report, we describe an efficient one-pot aza-Diels-Alder type reaction between the imine (formed in situ from benzaldehyde and amines) and dihydropyran catalyzed by lanthanide derivatives, Lewis acids, or Brönsted acids to afford pyrano[3,2-
# Both authors have contributed equally. List of Abbreviations: ACN = acetonitrile; DCM = dichloromethane; IEDAA = Inverse electron-deman aza Diels-Alder; NBS = N-Bromosuccinimide; NMP = N-methylpyrrolidone; TMEDA = Tetramethyletylenediamine.

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1385
c]quinolines in moderate to high yields, including investigations on the control of exo/endo diastereoselectivity depending on the nature of the catalytic acid and/or effect of the substitution of the aldehyde reactant used in the Povarov reaction.
Results and Discussions Preliminary studies of the Povarov reaction were
carried out in a parallel synthetic approach choosing commercially available substituted anilines and benzaldehydes which contains functional groups (such as esters, carboxylic acids, nitriles) susceptible to be further derivatized or chemically modified to prepare analogues and build small libraries. Moreover, these substitution pattern molecules can hardly be found in the bibliography.
The use of dihydropyran as dienophile and scandium (III) triflate as Lewis acid in acetonitrile led to the hexahydro-2H-pyrano[3,2-c]quinolines in moderate to good yields (Povarov reaction gives better yield using activated electron-rich alkenes)6. However, low diastereoselectivity was found for these compounds obtaining nearly equimolecular mixtures of exo/endo isomers (Table I).
It should be noted that the purification and separation of the exo/endo diastereoisomers was sometimes very difficult either by SiO2 or reverse C18 chromatography. Yields are based in the pure isolated diastereoisomer. 1H NMR analysis of the Povarov adducts 1 was generally sufficient to distinguish between the endo and exo diastereomers. The most useful diagnostic observations for the determination of the relative stereochemistry were the chemical shift of the H-5 proton and the coupling constant between protons H-5 and H-4a. The chemical shift of the H-5 proton was consistently more downfield in the endo series by approximately 0.06 ppm (e.g., H-5; endo = 4.76 ppm, exo = 4.70 ppm for 1d carboxylic acid form). The coupling constant values for JH5-4a were in the range of 0-3 Hz for the endo series and 8-11 Hz in the exo series (e.g., JH5-4a = 0 Hz for endo-1d carboxylic acid form and J H5-4a = 9.2 Hz for exo-1d carboxylic acid form). 13C, DEPT, NOESY 1D and gHSQC NMR experiments were performed to confirm this elucidation (Figure 2).
In general, the formation of endo adducts in often believed to occur via an asynchronous concerted pathway, whereas the corresponding formation of the more thermodynamically stable exo adducts is believed to occur via a stepwise pathway7.
The resultant low diastereselectivity in the preparation of hexahydro-2H-pyrano[3,2-c]quinolones using Sc(OTf)3 as catalyst, encouraged us to study how changes in the Lewis acid or in the reaction conditions could increase the diastereselectivity of the multicomponent reaction. To carry out these
Figure 1 — endo-Tetrahydro-1H-cyclopenta[c]quinolines
Table I — Formation of hexahydroquinolines 1 from the Sc(OTf)3-catalyzed three-component coupling reaction of anilines, aromatic aldehydes and 3,4-dihydro-2H-pyrana
Entry Aniline (R1) Aldehyde (R2=R3) Product drb (exo/endo) Yieldc (exo/endo) 1 Br H 1a 55:45 3% / 4% 2 Me H 1b 40:60 4% / 8% 3 Me Me 1cd 36:64 20% / 52% 4 OMe Me 1d 40:60 22% / 43% 5 Cl Me 1e 50:50 31% / 37%
aAniline (1eq.), Aldehyde (1eq.), 3,4-dihydro-2H-pyran (2eq.). bDetermined by the relative integrations of HPLC chromatogram peaks corresponding to the exo/endoisomers in the crude reaction mixture. c Yield described for the pure exo/endo diastereomers which have been isolated, purified and separated from each other. d The compound exo-1c is described in the experimental section as a carboxylic acid form after saponification of the ester group.

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1386
investigations, methyl 4-formyl-3-methylbenzoate, 4-methoxyaniline and 3,4-dihydro-2H-pyran were submitted to several Lewis and Brönsted acids under different reaction conditions (Table II). 4-Meth-oxyaniline was chosen because it was the aniline that gave lower diastereoselectivity in previous studies6.
From these results we can conclude that when lanthanides were used as Lewis acids no improvements in the diastereoselectivity were found (Table II, entries 1-6). Great exo diastereoselectivity was achieved when TiCl4 was used (Table II, entry 7),
however the chromatogram showed an unknown impurity of 30% with a high mass of [(M+H)+82]u. Acetonitrile (ACN) is the best solvent for high conversion to the hexahydroquinoline final products (Table II, entries 1, 4, 6, 7). When BF3·EtO2 was used as catalyst no reaction was achieved at RT (Table II, entries 9, 10). However, high exo diastereselectivity was found when TFA was used as protic acid, leading to a good conversion to the exo hexahydroquinoline isomer (Table II, entry 11). According to these results, we can conclude that the annulated ring orientation
Figure 2 — 3D Representation of exo/endo-diastereomeric hexahydro-2H-pyrano[3,2-c]quinolines
Table II — Diastereoselective synthesis of hexahydroquinolines 1 from acid catalyzed three-component coupling reaction of anilines, aromatic aldehydes and 3,4-dihydro-2H-pyrana
Entry Catalytic Acid Solvent Reaction conditions drb (exo/endo)
1 Sc(OTf)3 (0.1eq) ACN RT, 16 h 40:60
2 Sc(OTf)3(0.1eq) DCM/Toluene RT, 16 h 30:70c
3 Sc(OTf)3 (0.1eq) DCM/ACN RT, 16 h 30:70 c
4 Yb(OTf)3 (0.1eq) ACN RT, 16 h 36:64
5 Yb(OTf)3 (0.1 q) DCM RT, 16 h 30:70 c
6 InCl3 (0.2eq) ACN RT, 16 h 34:66
7 TiCl4 (0.2eq) ACN RT, 16 h 95:5d
8 FeCl3 (10%mol) DCE 60ºC, 16 h 50:50 c
9 BF3EtO2 (2eq) ACN RT, 16 h No reaction
10 BF3EtO2 (2eq) CHCl3 RT, 16 h No reaction
11 TFA (0.22eq) ACN RT, 16 h 75:25
aAniline (1eq.), Aldehyde (1eq.), 3,4-dihydro-2H-pyran (2eq.). bDetermined by the relative integrations of HPLC chromatogram peaks corresponding to the exo/endoisomers in the crude reaction mixture. c Low conversion was found by HPLC. d Excelent diastereoselectivity, however an unknown impurity (30%) was detected in the HPLC chromatogram.

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1387
(endo-exo selectivity) primarily depends on the catalyst nature.
In order to confirm the obtained favoured exo diastereoselectivity in the Povarov reaction under TFA catalysis, a variety of substituted aldehydes and anilines were submitted to these reaction conditions (Table III).
When the benzaldehyde precursor is ortho-methyl substituted or non-substituted a moderate exo diastereoselectivity was observed (Table III, entries 3, 10 and 4, 11). Surprisingly, ortho-fluorobenzaldehyde gave slightly higher exo diastereoselectivity than the ortho-methyl analogue (Table III, entries 1, 8 and 6, 7). However, ortho,ortho-disubstitution in the benzaldehyde gave a much higher exo diastereoselectivity. Additionally, according to the results in Table III, bulky group substitution increases this exo diastereoselectivity in the following order:
o,o-difluoro<o,o-fluoromethyl<o,o-dichloro<o,o-chloromethyl<o,o-dimethyl (Table III, entries 12, 15, 18, 17, 19). The 4-chloroaniline gave slightly higher exo diastereoselectivity than the other p-substituted anilines although it gave lower reaction yields (Table III, entries 14 vs 13 and 16 vs 17).
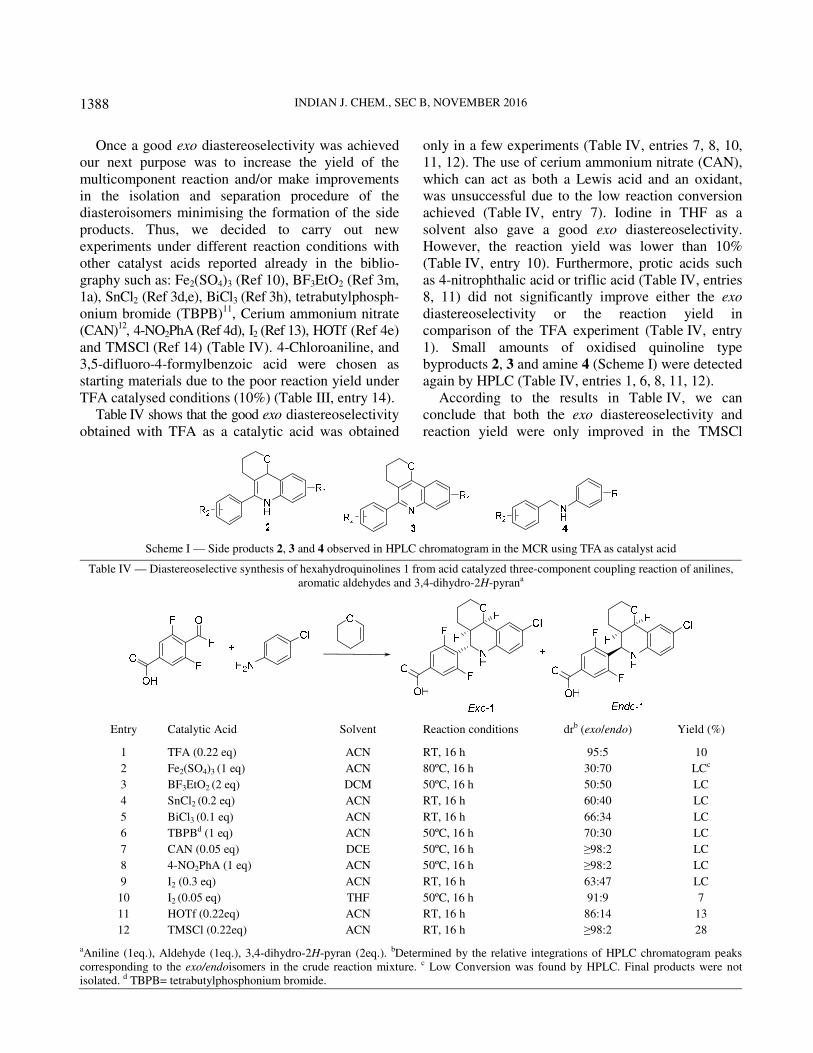
On the other hand, HPLC chromatograms were more complex (dirtier) when TFA was used as Lewis acid in comparison with Sc(OTf)3 catalysis, perhaps due to competing oligomerization reactions8. In addition, small amounts of oxidised quinoline type byproducts 2, 3 and amine 4 were also detected (Scheme I).
The formation of these side products is most likely the result of a hydrogen transfer mechanism9. These facts are the reasons why the purification and separation of the exo/endo diasteroisomers were sometimes very difficult, either by SiO2 or reverse C18 chromatography.
Table III — Preparation of substituted hexahydropyrano[3,2-c]quinolines 1 from the TFA-catalyzed three-component coupling reaction of anilines, aromatic aldehydes and 3,4-dihydro-2H-pyrana
Entry Aniline (R1) Aldehyde (R2) Product Yieldb (%) drcexo/endo
1 Me 2-Me-4-COOMe 1c 50 60:40 2 OMe 2-Me-4-COOMe 1d 45 65:35 3 OCHF2 2-Me-4-COOMe 1f 32 65:35 4 OCF3 2-Me-4-COOMe 1g 32 65:35 5 OCpr 2-Me-4-COOMe 1h 41 75:25 6 Cpr 2-Me-4-COOMe 1i 40 65:35 7 Cpr 2-F-4-COOH 1j 16 70:30 8 Me 2-F-4-COOH 1k 30 85:15 9 Et 2-F-4-COOH 1l 25 77:23
10 OCHF2 4-COOH 1m 25 60:40 11 OCF3 4-COOMe 1nd 28 60:40 12 Me 2,6-F-4-COOH 1o 26 80:20 13 OMe 2,6-F-4-COOH 1p 28 80:20 14 Cl 2,6-F-4-COOH 1q 10 95:5 15 Cl 2-F-6-Me-4-COOH 1r 45 88:12 16 Cl 2-Cl-6-Me-4-COOH (Ref 15) 1s 10 ≥98:2 17 Me 2-Cl-6-Me-4-COOH (Ref 15) 1t 20 90:10 18 Me 2,6-Cl-4-COOMe lu 50 85:15 19 Me 2,6-Me-4-COOMe 1v 27 ≥98:2 20 OMe 2,6-Me-4-COOMe 1w 27 ≥98:2 21 Cl 2,6-Me-4-COOMe 1x 33 ≥98:2 22 OCHF2 2,6-Me-4-COOMe 1y 38 ≥98:2
aAniline (1eq.), Aldehyde (1eq.), 3,4-dihydro-2H-pyran (2eq.). Same results were found using 1 eq. or 0.22 eq. of TFA. b The given yield is only for the exo diastereoisomer after purification. cDetermined by the relative integrations of HPLC chromatogram peaks corresponding to the exo/endoisomers in the crude reaction mixture. d The compound exo-1n is described in the experimental section as a carboxylic acid form after saponification of the ester group.
INDIAN J. CHEM., SEC B, NOVEMBER 2016
1388
Once a good exo diastereoselectivity was achieved our next purpose was to increase the yield of the multicomponent reaction and/or make improvements in the isolation and separation procedure of the diasteroisomers minimising the formation of the side products. Thus, we decided to carry out new experiments under different reaction conditions with other catalyst acids reported already in the biblio-graphy such as: Fe2(SO4)3 (Ref 10), BF3EtO2 (Ref 3m, 1a), SnCl2 (Ref 3d,e), BiCl3 (Ref 3h), tetrabutylphosph-onium bromide (TBPB)11, Cerium ammonium nitrate (CAN)12, 4-NO2PhA (Ref 4d), I2 (Ref 13), HOTf (Ref 4e) and TMSCl (Ref 14) (Table IV). 4-Chloroaniline, and 3,5-difluoro-4-formylbenzoic acid were chosen as starting materials due to the poor reaction yield under TFA catalysed conditions (10%) (Table III, entry 14).
Table IV shows that the good exo diastereoselectivity obtained with TFA as a catalytic acid was obtained
only in a few experiments (Table IV, entries 7, 8, 10, 11, 12). The use of cerium ammonium nitrate (CAN), which can act as both a Lewis acid and an oxidant, was unsuccessful due to the low reaction conversion achieved (Table IV, entry 7). Iodine in THF as a solvent also gave a good exo diastereoselectivity. However, the reaction yield was lower than 10% (Table IV, entry 10). Furthermore, protic acids such as 4-nitrophthalic acid or triflic acid (Table IV, entries 8, 11) did not significantly improve either the exo diastereoselectivity or the reaction yield in comparison of the TFA experiment (Table IV, entry 1). Small amounts of oxidised quinoline type byproducts 2, 3 and amine 4 (Scheme I) were detected again by HPLC (Table IV, entries 1, 6, 8, 11, 12).
According to the results in Table IV, we can conclude that both the exo diastereoselectivity and reaction yield were only improved in the TMSCl
Scheme I — Side products 2, 3 and 4 observed in HPLC chromatogram in the MCR using TFA as catalyst acid
Table IV — Diastereoselective synthesis of hexahydroquinolines 1 from acid catalyzed three-component coupling reaction of anilines, aromatic aldehydes and 3,4-dihydro-2H-pyrana
Entry Catalytic Acid Solvent Reaction conditions drb (exo/endo) Yield (%)
1 TFA (0.22 eq) ACN RT, 16 h 95:5 10
2 Fe2(SO4)3 (1 eq) ACN 80ºC, 16 h 30:70 LCc
3 BF3EtO2 (2 eq) DCM 50ºC, 16 h 50:50 LC
4 SnCl2 (0.2 eq) ACN RT, 16 h 60:40 LC
5 BiCl3 (0.1 eq) ACN RT, 16 h 66:34 LC
6 TBPBd (1 eq) ACN 50ºC, 16 h 70:30 LC
7 CAN (0.05 eq) DCE 50ºC, 16 h ≥98:2 LC
8 4-NO2PhA (1 eq) ACN 50ºC, 16 h ≥98:2 LC
9 I2 (0.3 eq) ACN RT, 16 h 63:47 LC
10 I2 (0.05 eq) THF 50ºC, 16 h 91:9 7
11 HOTf (0.22eq) ACN RT, 16 h 86:14 13
12 TMSCl (0.22eq) ACN RT, 16 h ≥98:2 28
aAniline (1eq.), Aldehyde (1eq.), 3,4-dihydro-2H-pyran (2eq.). bDetermined by the relative integrations of HPLC chromatogram peaks corresponding to the exo/endoisomers in the crude reaction mixture. c Low Conversion was found by HPLC. Final products were not isolated. d TBPB= tetrabutylphosphonium bromide.

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1389
experiment (Table IV, entry 12). In order to confirm these results, application of the optimized catalysis conditions with TMSCl using the above aldehydes and anilines led in better yields the formation of hexahydroquinolines 1 (Table V).
Comparing data from Table III and Table V we can conclude that reaction yields improved when TMSCl as catalyst acid in the Povarov reaction was used keeping a high exo diastereoselectivity (Table V, entries 1, 2, 3 vs Table III, entries 14, 12, 10 respectively). Furthermore, the TMSCl experimental procedure allowed to scale up the formation of hexahydroquinolines due to the easy isolation of the diastereoisomers by filtration from the precipitate formed in the reaction mixture (up to 25 g from starting material for compound 1m, Table V, entry 3). The separation of the exo/endo isomers can be achieved by sequential washings of the filtered solid with ACN because of the higher solubility of the endo isomer in this solvent. Experimental Section
HPLC-UV-MS was performed on a Waters Alliance HT 2795 HPLC with detection performed by Photodiode Array Detector 2996 and Micromass ZQ spectrometer operating in electrospray ionization mode. The HPLC column used is a XBridgeTM C18 (3.5 µm, 4.6 × 50 mm), temperature: 25ºC; flow rate 2.0 mL/min; eluent: A = HCOOH 10 mM, B = ACN:MeOH:HCOOH (1:1:0.05); gradient: 95 to 0% A in 5.5 min, 0% A 3 min, 0 to 95% A 0.5 min. 1H NMR spectra were recorded on a Varian Mercury-400 Fourier Transform Spectrometer operating at 400 MHz. NMR spectra were obtained as CDCl3
solutions (reported in δ, ppm), using chloroform as the reference standard (δ 7.26) or DMSO-d6 (δ 2.50). When peak multiplicities are reported, the following abbreviations are used s (singlet), d (doublet), t (triplet), m (multiplet), br (broadened), dd (doublet of doublets), dt (doublet of triplets), and td (triplet of doublets). Coupling constants, when given are reported in Hertz (Hz).
Compounds were prepared in a parallel approach synthesis, as assessed using the aforementioned standard spectroscopic techniques, in ≥ 95% purity unless otherwise stated. Starting materials were either procured from commercial sources or prepared as described herein. Column chromatography was performed either by flash chromatography (40-65 µm silica gel) or using an automated purification system (SP1 Purification System from Biotage®), which was also used for the reverse C18 chromatography using water (0.1% formic acid) and acetonitrile (0.1% formic acid) as eluents unless otherwise stated. Microwave reactions were performed in a Discovery CEM® system.
4-((4aR,5S,10bR)-9-Bromo-3,4,4a,5,6,10b-hexahydro-
2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid (exo-1a)
and 4-((4aR,5R,10bR)-9-bromo-3,4,4a,5,6,10b-hexahydro-
2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid (endo-1a)
General procedure for Povarov Reaction To a solution of 4-formylbenzoic acid (150 mg, 1
mmol) in anhydrous ACN (10 mL), 4-aminobromo-benzene (1 mmol, 172 mg), 3,4-dihydro-2H-pyran (2 mmol, 0.18 mL) and Sc(OTf)3 (0.05 mmol, 25 mg) were added. The reaction mixture was stirred 16 h at RT. Solvent was evaporated and purification of the
Table V — Preparation of substituted hexahydropyrano[3,2-c]quinolines 1 from the TMSCl-catalyzed three-component coupling reaction of anilines, aromatic aldehydes and 3,4-dihydro-2H-pyrana
Entry Aniline (R1) Aldehyde (R2) Product Yieldb (%) Drcexo/endo
1 4-Cl 2,6-F-4-COOH 1q 28% ≥98:2 2 4-Me 2,6-F-4-COOH 1o 45% 85:15 3 4-OCHF2 4-COOH 1m 39% 75:25d 4 4-Me 4-COOMe 1z 19% 70:30
aAniline (1eq.), Aldehyde (1eq.), 3,4-dihydro-2H-pyran (2eq.). bIsolated as a only exo diastereoisomer. cDetermined by the relative integrations of HPLC chromatogram peaks corresponding to the exo/endoisomers in the crude reaction mixture. d Determined by the relative integrations of H-5 in the 1H-RMN of the crude reaction mixture.

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1390
crude material (55:45 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 6% methanol in ethyl acetate afforded 92 mg (Yield 23%) of a 52:48 mixture of exo/endo diastereomers as a white solid which was purified using reverse C18 chromatography. Only pure fractions were collected to give 11 mg of the title exo-isomer (exo-1a) and 14 mg of the title endo-isomer (endo-1a) both as white solids.
4-((4aR,5S,10bR)-9-Bromo-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)benzoic(exo-1a): 1H NMR (400 MHz, DMSO-d6): δ 7.93 (2H, d, J = 8 Hz), 7.51 (2H, d, J = 8 Hz), 7.17 (1H, d, J = 2.4 Hz), 7.11 (1H, dd, J = 8.8 & 2.4 Hz), 6.55 (1H, d, J = 8.5 Hz), 6.45 (1H, s), 4.56 (1H, d, J = 10 Hz), 4.27 (1H, d, J = 2.4 Hz), 3.85 (1H, d, J = 10.8 Hz), 3.58 (1H, td, J = 10.8 & 2.4 Hz), 1.93-1.91 (1H, m), 1.78-1.69 (1H, m), 1.65-1.57 (1H, m), 1.30-1.26 (1H, m), 1.23-1.18 (1H, m); ESI-MS: m/z 388 [M + H]+.
4-((4aR,5R,10bR)-9-Bromo-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid(endo-1a): 1H NMR (400 MHz, DMSO-d6): δ 7.93 (2H, d, J = 8.4 Hz), 7.51 (2H, d, J = 8 Hz), 7.23 (1H, d, J = 1.6 Hz), 7.12 (1H, td, J = 8.4, 2.4 & 0.4 Hz), 6.65 (1H, d, J = 8.8 Hz), 6.27 (1H, s), 5.22 (1H, d, J = 5.6 Hz), 4.70 (1H, d, J = 2.4 Hz), 3.50 (1H, d, J = 11.2Hz), 3.17-3.28 (1H, m), 2.06-2.04 (1H, m), 1.37-1.31 (2H, m), 1.30-1.20 (1H, m), 1.03-1.00 (1H, m); ESI-MS: m/z 388 [M + H]+.
4-((4aR,5S,10bR)-3,4,4a,5,6,10b-Hexahydro-9-methyl-
2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid (exo-1b) and
4-((4aR,5R,10bR)-3,4,4a,5,6,10b-hexahydro-9-methyl-2H-
pyrano[3,2-c]quinolin-5-yl)benzoic acid (endo-1b)
The following compound was prepared using the same methodology as in compound 1a, using 4-formylbenzoic acid (1.6 mmol, 240 mg) and p-toluidine (1.6 mmol, 178 mg). Solvent was evaporated and purification of the crude material (40:60 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 55% EtAcO in hexanes afforded 170 mg (Yield 32%) of a 35:65 mixture of exo/endo diastereomers as a white solid which was purified using reverse C18 chromatography to give 14 mg of the title exo-isomer (exo-1b) and 50 mg of the title endo-isomer (endo-1b) both as a white solids (Overall yield 12%).
4-((4aR,5S,10bR)-3,4,4a,5,6,10b-Hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid(exo-1b): 1H NMR (400 MHz, DMSO-d6): δ 7.98 (2H, d, J = 8 Hz), 7.58 (2H, d, J = 8.4 Hz), 6.92 (1H,s), 6.86 (1H,
d, J = 8.4 Hz), 6.55 (1H, d, J = 8.4 Hz), 6.04 (1H, s), 4.60 (1H, d, J = 10 Hz), 4.29 (1H, d, J = 2.4 Hz), 3.92 (1H, d, J = 10.8 Hz), 3.61-3.66 (1H, m), 2.19 (3H, s), 1.98-1.94 (1H, m), 1.84-1.74 (1H, m), 1.70-1.62 (1H, m), 1.35-1.26 (2H, m); ESI-MS: m/z 324 [M + H]+.
4-((4aR,5R,10bR)-3,4,4a,5,6,10b-Hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid(endo-1b): 1H NMR (400 MHz, DMSO-d6): δ 7.94 (2H, d, J = 8.4 Hz), 7.54 (2H, d, J = 8.4 Hz), 7.01 (1H,s), 6.81 (1H, d, J = 8 Hz), 6.60 (1H, d, J = 8 Hz), 5.82 (1H, s), 5.21 (1H, d, J = 5.2 Hz), 4.66 (1H, d, J = 1.6 Hz), 3.47 (1H, d, J = 11.2 Hz), 3.33-3.20 (1H, m), 2.18 (3H, s), 2.07-2.02 (1H, m), 1.37-1.32 (3H, m), 1.04-1.00 (1H, m); ESI-MS: m/z 324 [M + H]+.
Methyl 4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6, 10b-
hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)-
3-methylbenzoate (endo-1c) and methyl 4-((4aR, 5S,
10bR)-3, 4, 4a, 5, 6, 10b-hexahydro-9-methyl-2H-
pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (exo-1c)
Method 1: The following compound was prepared using the same methodology as in compound 1a, using methyl 4-formyl-3-methylbenzoate6 (1.97 mmol, 350 mg) p-toluidine (1.97 mmol, 211 mg). Solvent was evaporated and purification of the crude material (36:64 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 7% ethyl acetate in hexanes afforded 136 mg (Yield 20%) of the title exo-isomer (exo-1c) and 363 mg (Yield 52%) of the title endo-isomer (endo-1c).
Method 2: In order to increase the reaction ratio of exo isomer a solution of methyl 4-formyl-3-methylbenzoate6 (5.61 mmol, 1 g) in anhydrous ACN (60 mL), p-toluidine (5.1 mmol, 547 mg), 3,4-dihydro-2H-pyran (10.2 mmol, 0.93 mL) and TFA (1.12 mmol, 0.087mL) were added. The reaction mixture was stirred 16 h at RT. Solvent was evaporated and purification of the crude material (60:40 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 15% ethyl acetate in hexanes afforded 1 g (Yield 50%) of the title exo-isomer (exo-1c) and 290 mg (Yield 14%) of the title endo-isomer (endo-1c).
Methyl 4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6, 10b-
hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (endo-1c): 1H NMR (400 MHz, DMSO-d6): δ 7.83-7.80 (2H, m), 7.67 (1H, d, J = 8 Hz), 7.02 (1H, s), 6.81 (1H, d, J = 8 Hz), 6.60 (1H, d, J = 8 Hz), 5.67 (1H, s), 5.23 (1H, d, J = 5.2 Hz), 4.80 (1H, d, J = 1.6 Hz), 3.85 (3H, s), 3.49-3.46 (1H, m),

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1391
3.26-3.20 (1H, m), 2.38 (3H, s), 2.18 (3H, s), 2.03-2.01 (1H, m), 1.50-1.46 (1H, m), 1.44-1.38 (2H, m), 1.04-1.03 (1H, m); ESI-MS: m/z 352 [M + H]+.
4-((4aR, 5S, 10bR)-3, 4, 4a, 5, 6, 10b-Hexahydro-9-
methyl-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoic
acid (exo-1c carboxylic acidform) To a solution of methyl 4-((4aR, 5S, 10bR)-3, 4, 4a, 5,
6, 10b-hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (exo-1c) (0.39 mmol, 136 mg) in THF (10 mL) was added 1M LiOH (0.78 mmol, 0.78 mL). The reaction mixture was stirred 16 h at RT. Total conversion was not achieved, 1M LiOH (0.8 mmol, 0.8 mL) was added again and was stirred for 24 h at RT. Solvent was evaporated and purification of the crude material by reverse C18 chromatography to give the pure title compound exo-1c carboxylic acid (62 mg, Yield 47%). 1H NMR (400 MHz, DMSO-d6): δ 7.78 (1H, s), 7.74 (1H, d, J = 8Hz), 7.42 (1H, d, J = 8Hz), 6.88 (1H, s), 6.81 (1H, dd, J = 2 & 8Hz), 6.48 (1H, d, J = 8Hz), 5.88 (1H, s), 4.71 (1H, d, J = 9.6Hz), 4.29 (1H, d, J = 2.8Hz), 3.83-3.81 (1H, m), 3.57-3.52 (1H, m), 2.45 (3H, s), 2.15 (3H, s), 2.10-2.07 (1H, m), 1.69-1.59 (2H, m), 1.42-1.36 (1H, m), 1.35-1.24 (1H, m); ESI-MS: m/z 338 [M + H]+.
Methyl 4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6, 10b-
hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-
yl)-3-methylbenzoate (endo-1d) and methyl 4-
((4aR, 5S, 10bR)-3, 4, 4a, 5, 6, 10b-hexahydro-9-
methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-
methylbenzoate (exo-1d)
Method 1: The following compound was prepared using the same methodology as in compound 1a, using methyl 4-formyl-3-methylbenzoate (1.92 mmol, 342 mg) in anhydrous ACN (20 mL), p-anisidine (1.92 mmol, 236 mg), 3,4-dihydro-2H-pyran (3.84 mmol, 0.35 mL) and Sc(OTf)3 (0.096 mmol, 48 mg). The reaction mixture was stirred 16 h at RT. Solvent was evaporated and purification of the crude material (40:60 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 18% ethyl acetate in hexanes afforded 154 mg (Yield 22%) of the title exo-isomer (exo-1d) and 310 mg (Yield 43%) of the title endo-isomer (endo-1d).
Method 2: The following compound was prepared using the same methodology as in compound 1c (method 2), using methyl 4-formyl-3-methylbenzoate (5.61 mmol, 1 g) in anhydrous ACN (60 mL), p-anisidine (5.1 mmol, 629 mg), 3,4-dihydro-2H-pyran (10.2 mmol, 0.93 mL) and TFA (1.12 mmol, 0.087 mL). The reaction mixture was stirred 16 h at RT. Solvent
was evaporated and purification of the crude material (65:35 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 25% ethyl acetate in hexanes afforded 914 mg (Yield 45%) of the title exo-isomer (exo-1d) and 227 mg (Yield 11%) of the title endo-isomer (endo-1d).
Methyl 4-((4aR, 5S, 10bR)-3, 4, 4a, 5, 6, 10b-
hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (exo-1d): 1H NMR (400 MHz, CDCl3): δ 7.87-7.84 (2H, m), 7.49 (1H, d, J = 8 Hz), 6.85 (1H, d, J = 2.8 Hz), 6.75 (1H, dd, J = 8.4 & 2.8 Hz), 6.50 (1H, d, J = 8.8 Hz), 4.91 (1H, d, J = 10 Hz), 4.42 (1H, d, J = 3.2 Hz), 4.03 (1H, m), 3.91 (3H, s), 3.77 (3H, s), 3.72-7.67 (1H, m), 2.50 (3H, s), 2.26-2.22 (1H, m), 1.71-1.69 (2H, m), 1.46-1.39 (2H, m); ESI-MS: m/z 368 [M + H]+.
Methyl 4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6, 10b-
hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-
yl)-3-methylbenzoate (endo-1d): 1H NMR (400 MHz, DMSO-d6): δ 7.81 (1H, d, J = 8 Hz), 7.79 (1H, s), 7.69 (1H, d, J = 8 Hz), 6.79 (1H, s), 6.65 (2H, m), 5.52 (1H, s), 5.24 (1H, 5.6Hz), 4.77 (1H, s), 3.85 (3H, s), 3.66 (3H, s), 3.49 (1H, d, J = 12 Hz), 3.27-3.25 (1H, m), 2.38 (3H, s), 2.02 (1H, m), 1.43-1.39 (3H, m), 1.04 (1H, m); ESI-MS: m/z 368 [M + H]+.
4-((4aR, 5S, 10bR)-3, 4, 4a, 5, 6, 10b-Hexahydro-9-
methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoic
acid (exo-1d carboxylic acid form)
The following compound was prepared using the same methodology as exo-1c carboxylic acid form using methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (exo-1d) (0.42 mmol, 154 mg). Total conversion was not achieved, 1M LiOH (0.84 mmol, 0.8 mL) was added again and was stirred for 24 h at RT. Solvent was evaporated and purification of the crude material by reverse C18 chromatography gave the title benzoic acid. (55 mg. Yield 37%). 1H NMR (400 MHz, DMSO-d6): δ 7.78 (1H, s), 7.75 (1H, d, J = 8Hz), 7.43 (1H, d, J = 8.4Hz), 6.69-6.65 (2H, m), 6.52 (1H, d, J = 8.4Hz), 5.73 (1H, s), 4.70 (1H, d, J = 9.2Hz, H-5), 4.32 (1H, d, J = 2Hz, H-10b), 3.85-3.82 (1H, m), 3.65 (3H, s), 3.60-3.55 (1H, m), 2.46 (3H, s), 2.09-2.07 (1H, m, H-4a), 1.66-1.60 (2H, m), 1.36-1.25 (2H, m); 13C NMR (400 MHz, DMSO-d6): δ 19.3 (CH3), 22.7 (CH2), 23.8 (CH2), 36.8 (CH, C-4a), 50.9 (CH,
C-5), 55.4 (OCH3), 66.5 (CH2), 73.3 (CH, C-10b), 114,4 (CH), 114,8 (CH), 115,9 (CH), 120,0 (C), 126.9 (CH), 127.2 (C), 131.2 (CH), 135.3 (C), 139.8 (C), 143.5 (C), 150.38 (C), 165.7 (COOH); ESI-MS: m/z 354 [M + H]+.

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1392
4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6, 10b-Hexahydro-9-
methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-
methylbenzoic acid (endo-1d carboxylic acid form) To a solution of methyl 4-((4aR,5R,10bR)-
3,4,4a,5,6,10b-hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (endo-1d) (0.84 mmol, 310 mg) in THF (10 mL) was added 1M LiOH (1.68 mmol, 1.7 mL). The reaction mixture was stirred 16 h at RT. Total conversion was not achieved, 1M LiOH (1.68 mmol, 1.7 mL) was added again and was stirred for 48 h at RT. Solvent was evaporated and purification of the crude material by reverse C18 chromatography gave the 4-((4aR, 5R, 10bR)-3, 4, 4a, 5, 6,10b-hexahydro-9-methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylben-zoic acid. (99mg. Yield 33%). 1H NMR (400 MHz, DMSO-d6): δ 7.79-7.76 (2H, m), 7.63 (1H, d, J = 8Hz), 6.79 (1H, s), 6.65 (2H, m), 5.50 (1H, s), 5.23 (1H, d, J = 5.6Hz, H-10b), 4.76 (1H, d, J = 1Hz, H-5), 3.66 (3H, s), 3.49 (1H, d, J = 11.2Hz), 3.28-3.25 (1H, m), 2.36 (3H, s), 2.07-1.99 (1H, m), 1.41-1.39 (3H, m), 1.20-1.15 (1H, m); 13C NMR (400 MHz, DMSO-d6): δ 18.2 (CH2), 18.5 (CH3), 24.9 (CH2), 34.8 (CH, C-4a), 54.8 (CH, C-5), 55.3 (OCH3), 59.9 (CH2), 71.9 (CH, C-10b), 111,1 (CH), 114,4 (CH), 116,0 (CH), 120,1 (C), 126.2 (CH), 127.2 (CH), 129.3 (C), 131.2 (CH), 135.1 (C), 140.2 (C), 144.2 (C), 151.7 (C), 167.4 (COOH); ESI-MS: m/z 354 [M + H]+.
Methyl 4-((4aR, 5R, 10bR)-9-chloro-3, 4, 4a, 5,
6, 10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-
3-methylbenzoate (endo-1e) and methyl 4-((4aR,
5S, 10bR)-9-chloro-3, 4, 4a, 5, 6, 10b-hexahydro-
2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate
(exo-1e)
The following compound was prepared using the same methodology as in compound 1a using 4-chloroaniline (1.97 mmol, 252 mg). Solvent was evaporated and purification of the crude material (50:50 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 6% ethyl acetate in hexanes afforded 226 mg (Yield 31%) of the title exo-isomer (exo-1e) and 277 mg (Yield 37%) of the title endo-isomer (endo-1e).
Methyl 4-((4aR, 5S, 10bR)-9-chloro-3, 4, 4a, 5, 6,
10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3-
methylbenzoate (exo-1e): 1H NMR (400 MHz, CDCl3): δ 7.88-7.85 (2H, m), 7.50-7.44 (1H, m), 7.24 (1H, d, J = 2.4 Hz), 7.04 (1H, dd, J = 8.4 & 2.4 Hz), 6.47 (1H, d, J = 8.8 Hz), 4.93 (1H, d, J = 9.6 Hz), 4.41 (1H, d, J = 3.2 Hz), 4.05-4.01 (1H, m), 3.91 (3H, s), 3.69-3.67 (1H, m), 2.49 (3H, s), 2.25-2.21 (1H, m),
1.72-1.68 (1H, m), 1.58-1.49 (2H, m), 1.47-1.44 (1H, m); ESI-MS: m/z 372 [M + H]+.
Methyl 4-((4aR, 5R, 10bR)-9-chloro-3, 4, 4a, 5, 6,
10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate (endo-1e): 1H NMR (400 MHz, CDCl3): δ 7.90-7.88 (2H, m), 7.64 (1H, d, J = 8 Hz), 7.41 (1H, d, J = 2.4 Hz), 7.04 (1H, dd, J = 8.4 & 2.4 Hz), 6.55 (1H, d, J = 8.8 Hz), 5.26 (1H, d, J = 5.6 Hz), 4.87 (1H, d, J = 2.4 Hz), 3.918 (3H, s), 3.63-3.59 (1H, m), 3.44-3.41 (1H, m), 2.38 (3H, s), 2.18-2.15 (1H, m), 1.89-1.81 (1H, m), 1.60-1.48 (3H, m), 1.28-1.24 (1H, m); ESI-MS: m/z 372 [M + H]+.
Methyl 4-((4aR,5S,10bR)-9-(difluoromethoxy)-
3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-
c]quinolin-5-yl)-3-methylbenzoate (exo-1f)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 4-(difluoromethoxy)aniline (1.40 mmol, 174 mL) and TFA (1eq., 1.40 mmol, 108 mL). Solvent was evaporated and purification of the crude material (65:35 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 22% ethyl acetate in hexanes afforded 181 mg (Yield 32%) of the pure title compound exo-1f. 1H NMR (400 MHz, CDCl3): δ 7.89-7.86 (2H, m), 7.48 (1H, d, J = 8 Hz), 7.08 (1H, d, J = 2.4 Hz), 6.92 (1H, dd, J = 8.8 & 2.4 Hz), 6.51 (1H, d, J = 8.8 Hz), 6.40 (1H, t, J = 74.8 Hz), 4.95 (1H, d, J = 9.6 Hz), 4.43 (1H, d, J = 3.2 Hz), 4.06-4.00 (1H, m), 3.93 (3H, s), 3.74-3.68 (1H, m), 2.51 (3H, s), 2.26-2.20 (1H, m), 1.74-1.70 (2H, m), 1.48-1.42 (2H, m); ESI-MS: m/z 390 [M + H]+.
Methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-(trifluoromethoxy)-2H-pyrano[3,2-c]quinolin-5-yl)-3-
methylbenzoate (exo-1g)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 4-(trifluoromethoxy)aniline (1.40 mmol, 188 mL) and TFA (1eq., 1.40 mmol, 108 mL). Solvent was evaporated and purification of the crude material (65:35 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 14% ethyl acetate in hexanes afforded 192 mg (Yield 32%) of the pure title compound exo-1g. 1H NMR (400 MHz, CDCl3): δ 7.88-7.85 (2H, m), 7.46 (1H, d, J = 7.6 Hz), 7.14 (1H, d, J = 2 Hz), 6.97 (1H, dd, J = 8.4 & 2.4 Hz), 6.50 (1H, d, J = 8.4 Hz), 4.94 (1H, d, J = 9.6 Hz), 4.42 (1H, d, J = 3.2 Hz), 4.04-4.00 (1H, m), 3.92 (3H, s), 3.72-3.66 (1H, m), 2.49 (3H, s), 2.24-2.20 (1H, m), 1.74-1.70 (2H, m), 1.48-1.42 (2H, m); ESI-MS: m/z 408 [M + H]+.

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1393
Methyl 4-((4aR,5S,10bR)-9-cyclopropoxy-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3-methylbenzoate
(exo-1h)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 4-cyclopropoxybenzenamine (3.12 mmol, 600 mg). Solvent was evaporated and purification of the crude material (75:25 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 21% ethyl acetate in hexanes afforded 263 mg (Yield 41%) of the pure title compound exo-1h. 1H NMR (400 MHz, DMSO-d6): δ 7.85-7.76 (2H, m), 7.50-7.46 (2H, m), 6.83 (1H, d, J = 2.8 Hz), 6.72 (1H, dd, J = 9 & 3 Hz), 6.52 (1H, d, J = 8.4 Hz), 5.77 (1H, s), 4.71 (1H, d, J = 9.6 Hz), 4.32 (1H, d, J = 2.8 Hz), 3.84-3.80 (1H, m), 3.84 (3H, s), 3.71-3.67 (1H, m), 3.60-3.54 (1H, m), 2.47 (3H, s), 2.12-2.07 (1H, m), 1.69-1.56 (2H, m), 1.38-1.22 (2H, m), 0.71-0.67 (2H, m), 0.62-0.56 (2H, m); ESI-MS: m/z 394 [M + H]+.
Methyl 4-((4aR,5S,10bR)-9-cyclopropyl-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3-
methylbenzoate (exo-1i) The following compound was prepared using the
same methodology as in compound 1c (method 2) using 4-cyclopropylbenzenamine (3.7 mmol, 490 mg) and TFA (0.22 eq., 0.81 mmol, 0.065 mL). Solvent was evaporated and purification of the crude material (65:35 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 20% ethyl acetate in hexanes afforded 565 mg (Yield 40%) of the pure title compound exo-1i. 1H NMR (400 MHz, CDCl3): δ 7.87-7.83 (2H, m), 7.48 (1H, d, J = 8 Hz), 7.02 (1H, d, J = 2.0 Hz), 6.85 (1H, dd, J = 8.4 & 2.4 Hz), 6.46 (1H, d, J = 8.0 Hz), 4.94 (1H, d, J = 10.4 Hz), 4.41 (1H, d, J = 3.2 Hz), 4.04 (1H, m), 3.91 (3H, s), 3.72-3.65 (1H, m), 2.49 (3H, s), 2.24-2.20 (1H, m), 1.85-1.78 (1H, m), 1.75-1.65 (2H, m), 1.45-1.38 (2H, m), 0.86-0.81 (2H, m), 0.62-0.57 (2H, m); ESI-MS: m/z 364 [M + H]+.
4-((4aR,5S,10bR)-9-Cyclopropyl-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3-fluorobenzoic
acid (exo-1j) The following compound was prepared using the
same methodology as in compound 1c (method 2) using 3-fluoro-4-formylbenzoic acid6 (4.46 mmol, 750 mg), 4-cyclopropylbenzenamine (4.46 mmol, 595 mg) and TFA (1 eq., 4.46 mmol, 0.344 mL). Solvent was evaporated. The crude material (70:30 mixture of exo/endo diastereomers) was precipitated in ACN and the solid thus obtained was purified by reverse C18
chromatography to afford 260 mg (Yield 16%) of the pure title compound (exo-1j). 1H NMR (400 MHz, DMSO-d6): δ 7.79 (1H, dd, J = 7.6 & 1.2 Hz), 7.69-7.62 (2H, m), 6.80 (1H, d, J = 2.4 Hz), 6.76 (1H, dd, J = 8.8 & 2.4 Hz), 6.49 (1H, d, J = 8.4 Hz), 6.03 (1H, s), 4.86 (1H, d, J = 10 Hz), 4.27 (1H, d, J = 2.4 Hz), 3.88-3.84 (1H, m), 3.61-3.55 (1H, m), 2.00-1.97 (1H, m), 1.80-1.75 (1H, m), 1.73-1.66 (2H, m), 1.33-1.24 (2H, m), 0.82-0.77 (2H, m), 0.51-0.48 (2H, m); ESI-MS: m/z 368 [M + H]+.
3-Fluoro-4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid
(exo-1k)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 3-fluoro-4-formylbenzoic6 acid (5.95 mmol, 1 g), p-toluidine (5.95 mmol, 0.64 g) and TFA (1 eq., 5.95 mmol, 0.458 mL). Solvent was evaporated and the crude material (85:15 mixture of exo/endo diastereomers) was purified by reverse C18 chromatography to give the title compound slightly impure, which was triturated in acetonitrile, filtered and dried to obtain 601 mg (Yield 30%) of the pure title compound exo-1k. 1H NMR (400 MHz, DMSO-d6): δ 13.24 (1H, brs), 7.80 (1H, dd, J = 8.2 & 1.4 Hz), 7.70-7.62 (2H, m), 6.90-6.82 (2H, m), 6.49 (1H, d, J = 8 Hz), 6.01 (1H, s), 4.86 (1H, d, J = 10.4 Hz), 4.27 (1H, d, J = 2.4 Hz), 3.88-3.84 (1H, m), 3.62-3.55 (1H, m), 2.15 (3H, s), 2.01-1.97 (1H, m), 1.70-1.64 (2H, m), 1.34-1.22 (2H, m); ESI-MS: m/z 342 [M + H]+.
4-((4aR,5S,10bR)-9-Ethyl-3,4,4a,5,6,10b-hexahydro-2H-
pyrano[3,2-c]quinolin-5-yl)-3-fluorobenzoic acid (exo-1l)
The following compound was prepared using the same methodology as in compound 1c (method 2), using 3-fluoro-4-formylbenzoic acid6 (5.95 mmol, 1 g), 4-ethylbenzenamine (5.95 mmol, 0.74 mL) and TFA (1 eq., 5.95 mmol, 0.458 mL). Solvent was evaporated and the crude material (77:23 mixture of exo/endo diastereomers) was purified by reverse C18 chromatography to give the title compound slightly impure, which was triturated in acetonitrile, filtered and dried to obtain 531 mg (Yield 25%) of the pure title compound exo-1l as a solid. 1H NMR (400 MHz, DMSO-d6): δ 13.24 (1H, brs), 7.80 (1H, dd, J = 8.2 & 1.4 Hz), 7.70-7.63 (2H, m), 6.92-6.85 (2H, m), 6.51 (1H, d, J = 8.4 Hz), 6.03 (1H, s), 4.88 (1H, d, J = 10.8 Hz), 4.29 (1H, d, J = 2.8 Hz), 3.90-3.85 (1H, m), 3.62-3.56 (1H, m), 2.46 (2H, q, J = 7.4 Hz),2.02-1.98 (1H, m), 1.70-1.66 (2H, m), 1.34-1.22 (2H, m), 1.12 (3H, t, J = 7.6 Hz); ESI-MS: m/z 340 [M + H]+.

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1394
4-((4aR,5S,10bR)-9-(Difluoromethoxy)-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid
(exo-1m)
Method 1: The following compound was prepared using the same methodology as in compound 1c
(method 2), using 4-formylbenzoic acid (9.99 mmol, 1.5 g) and 4-(difluoromethoxy)benzenamine (9.99 mmol, 1.2 mL). Solvent was evaporated and purification of the crude material (60:40 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 2% methanol in ethyl acetate afforded 935 mg (Yield 25%) of the pure title compound exo-1m.
Method 2: To a solution of 4-formylbenzoic acid (130.9 mmol, 19.6 g), 4-(difluoromethoxy)benzenamine (157.1 mmol, 25 g) and 3,4-dihydro-2H-pyran (91.12 mmol, 8.31 mL) in anhydrous ACN (91 mL), chloro-trimethylsilane (28.8 mmol, 3.75 mL) was added. The reaction mixture was stirred 16 h at RT. The suspension reaction (75:25 mixture of exo/endo diastereomers) was cooled, filtered and the solid washed with the minimum amount of cold acetonitrile and hexane gave 19.2 g (Yield 39%) of the pure title compound exo-1m. 1H NMR (400 MHz, DMSO-d6): δ 12.90 (1H, brs), 7.95 (2H, d, J = 8 Hz), 7.54 (2H, d, J= 8 Hz), 6.95 (1H, t, J = 75.6 Hz), 6.90 (1H, d, J = 2.8 Hz), 6.87 (1H, dd, J = 8.4 & 2.8 Hz), 6.62 (1H, d, J = 8.8 Hz), 6.32 (1H, s), 4.57 (1H, d, J = 10 Hz), 4.29 (1H, d, J = 2.8 Hz), 3.90-3.86 (1H, m), 3.64-3.56 (1H, m), 1.98-1.92 (1H, m), 1.80-1.60 (2H, m), 1.34-1.20 (2H, m); ESI-MS: m/z 376 [M + H]+.
4-((4aR,5S,10bR)-3,4,4a,5,6,10b-Hexahydro-9-
(trifluoromethoxy)-2H-pyrano[3,2-c]quinolin-5-yl)benzoic
acid (exo-1n carboxylic acid form)
The following compound was prepared using the same methodology as in compound 1c (method 2) using methyl 4-formylbenzoate (6.09 mmol, 1 g), 4-(trifluoro-methoxy)benzenamine (6.09 mmol, 0.82 mL) and TFA (1eq., 6.09 mmol, 0.47 mL). Solvent was evaporated and purification of the crude material (60:40 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 18% ethyl acetate in hexanes afforded a mixture exo/endo diastereomers as a solid. This solid was purified again by flash chromato-graphy over silica gel using an elution of 17% ethyl acetate in hexanes afforded 700 mg (Yield 28%) of methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-9-(trifluoromethoxy)-2H-pyrano[3,2-c] quinolin-5-yl)benzoate
(exo-1n). ESI-MS: m/z 408 [M + H]+. The methyl benzoate exo-1n (1.72 mmol, 700 mg)
in THF (10 mL) was treated with 1M LiOH (6.88
mmol, 6.9 mL) at RT for 16 h. Total conversion was not achieved, 1M LiOH (1.15 mmol, 2 mL) was added again and stirred for additional 24 h. The solvent was evaporated. Water was added, the mixture cooled with an ice/water bath and treated with 1N HCl until a white solid appeared. The solid was separated by filtration, washed with cold water and dried under vacuum at 40°C to give the pure title exo-isomer compound (exo-
1n carboxylic acid form) (572 mg, Yield 84%). 1H NMR (400 MHz, DMSO-d6): δ 7.95 (2H, d, J = 8.4 Hz), 7.54 (2H, d, J = 8 Hz), 7.03-6.99 (2H, m), 6.65 (1H, d, J = 8.4 Hz), 6.53 (1H, s), 4.59 (1H, d, J = 10 Hz), 4.32 (1H, d, J = 2.8 Hz), 3.90-3.86 (1H, m), 3.63-3.58 (1H, m), 1.98-1.94 (1H, m), 1.79-1.75 (1H, m), 1.67-1.63 (1H, m), 1.33-1.29 (1H,m), 1.25-1.21 (1H, m); ESI-MS: m/z 394 [M + H]+.
3,5-Difluoro-4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-9-
methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid (exo-1o) Method 1: The following compound was prepared
using the same methodology as in compound 1c (method 2) using 3,5-difluoro-4-formylbenzoic acid6 (5 mmol, 930 mg) and 4-methylaniline (5.00 mmol, 535 mg). Solvent was evaporated and purification of the crude material (80:20 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 36% ethyl acetate in hexanes afforded the title exo-isomer which was triturated in the minimum amount of DCM. The precipitated solid was filtered, washed with hexane and dried to give 475 mg (Yield 26%) of the title exo compound (exo-1o).
Method 2: The following compound was prepared using the same methodology as in compound 1m (method 2) using 3,5-difluoro-4-formylbenzoic acid6 (32.88 mmol, 6.11 g) 4-methylaniline (27.4 mmol, 2.93 g). The reaction mixture was stirred 16 h at RT. The suspension (85:15 mixture of exo/endo diastereomers) was filtered and the solid washed by the minimum amount of cold acetonitrile and hexane to afford 4.45 g (Yield 45%) of the title exo compound (exo-1o) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 13.56 (1H, brs), 7.57 (2H, d, J = 9.6 Hz), 6.85 (1H, s), 6.80 (1H, dd, J = 8.4 & 2.0 Hz), 6.42 (1H, d, J = 7.6 Hz), 6.05 (1H, s), 5.04 (1H, d, J = 11.2 Hz), 4.29 (1H, d, J = 2.4 Hz), 3.93-3.90 (1H, m), 3.58 (1H, t, J = 10.0 Hz), 2.32-2.28 (1H, m), 2.13 (3H, s), 1.80-1.72 (1H, m), 1.64-1.56 (1H, m), 1.31-1.24 (2H, m); ESI-MS: m/z 360 [M + H]+.
3,5-Difluoro-4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-9-
methoxy-2H-pyrano[3,2-c]quinolin-5-yl)benzoic acid (exo-1p) The following compound was prepared using the
same methodology as in compound 1c (method 2)

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1395
using 3,5-difluoro-4-formylbenzoic acid6 (5 mmol, 930 mg) and 4-methoxyaniline (5.00 mmol, 615 mg). Solvent was evaporated and purification of the crude material (80:20 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 47% ethyl acetate in hexanes afforded 1.3 g (71% purity) of the title exo-isomer which was purified by reverse C18 chromatography to give 530 mg (Yield 28%) of the pure title compound exo-1p. 1H NMR (400 MHz, DMSO-d6): δ 13.56 (1H, brs), 7.57 (2H, d, J = 9.6 Hz), 6.68-6.64 (2H, m), 6.48-6.45 (1H, m), 5.89 (1H, s), 5.02 (1H, d, J = 11.6 Hz), 4.32 (1H, d, J = 2.4 Hz), 3.94-3.91 (1H, m), 3.63 (3H, s), 3.62-3.5 (1H, m), 2.33-2.30 (1H, m), 1.80-1.72 (1H, m), 1.63-1.52 (1H, m), 1.28 (1H, t, J = 13.6 Hz); ESI-MS: m/z 376 [M + H]+.
4-((4aR,5S,10bR)-9-Chloro-3,4,4a,5,6,10b-hexahydro-
2H-pyrano[3,2-c]quinolin-5-yl)-3,5-difluorobenzoic acid
(exo-1q)
Method 1: The following compound was prepared using the same methodology as in compound 1c (method 2) using 3,5-difluoro-4-formylbenzoic acid6 (5.37 mmol, 1 g) and 4-chloroaniline (5.37 mmol, 685 mg). Solvent was evaporated and the crude material was purified (95:5 mixture of exo/endo diastereomers) by flash chromatography over silica gel using ethyl acetate and hexane as eluents afforded the title exo isomer, which was triturated in hexane and drops of DCM or ACN. The precipitated solid was filtered, washed with hexane and dried to give 205 mg (Yield 10%) of the pure title compound (exo-1q).
Method 2: The following compound was prepared using the same methodology as in compound 1m
(method 2) using 3,5-difluoro-4-formylbenzoic acid6 (45.56 mmol, 8.48 g) in anhydrous ACN (91 mL), 4-chlorobenzenamine (45.56 mmol, 5.8 g), 3,4-dihydro-2H-pyran (91.12 mmol, 8.31 mL) and chlorotri-methylsilane (10 mmol, 1.3 mL). The reaction mixture was stirred 16 h at RT. The suspension (≥98:2 mixture of exo/endo diastereomers) was filtered and the solid washed by the minimum amount of cold acetonitrile and hexane to afford 4.9 g (Yield 28%) of the title compound exo-1q as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 13.56 (1H, brs), 7.58 (2H, d, J = 9.6 Hz), 7.07 (1H, d, J= 2.4 Hz), 7.01 (1H, dd, J = 8.8 & 2.4 Hz), 6.51 (1H, d, J = 8.8 Hz), 6.49 (1H, s), 5.05 (1H, d, J = 11.2 Hz), 4.35 (1H, d, J = 2.4 Hz), 3.94-3.91(1H, m), 3.58 (1H, t, J = 10.8 Hz), 2.28 (1H, d, J = 12.8 Hz), 1.81-1.73 (1H, m), 1.65-1.58 (1H, m), 1.28 (2H, d, J = 10.4 Hz); ESI-MS: m/z 380 [M + H]+.
4-((4aR,5S,10bR)-9-Chloro-3,4,4a,5,6,10b-hexahydro-2H-
pyrano[3,2-c]quinolin-5-yl)-3-fluoro-5-methylbenzoic
acid (exo-1r)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 3-fluoro-4-formyl-5-methylbenzoic acid6 (3.07 mmol, 560 mg), 4-chlorobenzenamine (3.07 mmol, 392 mg) and TFA (1 eq, 3.07 mmol, 0.237 mL). Solvent was evaporated and purification of the crude material (88:12 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of MeOH in DCM and then reverse C18 chromatography to give 525 mg (Yield 45%) of the title compound exo-1r. 1H NMR (400 MHz, DMSO-d6): δ 7.66 (1H, s), 7.50 (1H, d, J = 11.2 Hz), 7.08 (1H, d, J = 2.4 Hz), 7.10 (1H, dd, J = 9.2 & 2.8 Hz), 6.55 (1H, d, J = 8.8 Hz), 6.37 (1H, s), 5.05 (1H, d, J = 11.2 Hz), 4.34 (1H, d, J = 2.8 Hz), 3.95-3.93 (1H, m), 3.62-3.56 (1H, m), 2.39-2.36 (1H, m), 1.75-1.61 (2H, m), 1.31-1.24 (2H, m); ESI-MS: m/z 376 [M + H]+.
3-Chloro-4-((4aR,5S,10bR)-9-chloro-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-5-methylbenzoic
acid (exo-1s)
The following compound was prepared using the same methodology as in compound 1c (method 2) using 3-chloro-4-formyl-5-methylbenzoic acid 9 (Scheme II, Ref 15) (0.61 mmol, 120 mg), 4-chlorobenzenamine (0.61 mmol, 78 mg) and TFA (1 eq., 0.61 mmol, 0.047 mL). Solvent was evaporated and the crude material (≥98:2 mixture of exo/endo diastereomers) was purified by reverse C18 chromatography to afford an impure solid. Diethyl ether was added and the solid was separated by filtration, then washed with cold diethyl ether and dried under vacuum at 40ºC to give 26 mg (Yield 10%) of the title compound exo-1s. 1H NMR (400 MHz, DMSO-d6): δ 7.79-7.73 (2H, m), 7.08 (1H, s), 7.02 (1H, d, J = 8 Hz), 6.56 (1H, d, J = 8 Hz), 6.43 (1H, s), 5.42 (1H, brs), 4.33 (1H, d, J = 2 Hz), 3.96-3.94 (1H, m), 3.61-3.56 (1H, m), 2.50-2.48 (1H, m), 1.93-1.67 (2H, m), 1.31-1.17 (2H, m); ESI-MS: m/z 394 [M + H]+.
3-Chloro-4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)-5-methylbenzoic
acid (exo-1t) The following compound was prepared using the
same methodology as in compound 1c (method 2) using 3-chloro-4-formyl-5-methylbenzoic acid 9 (Scheme II, Ref 15) (0.27 mmol, 54 mg), p-toluidine (0.27 mmol, 29 mg) and TFA (1 eq., 0.27 mmol, 0.021 mL). Solvent was evaporated and purification of the crude material (90:10 mixture of exo/endo

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1396
diastereomers) by flash chromatography over silica gel using an elution of 38% ethyl acetate in hexanes and reverse C18 chromatography to give 15 mg (Yield 20%) of the pure title compound exo-1t. 1H NMR (400 MHz, DMSO-d6): δ 7.76-7.69 (2H, m), 6.86 (1H, s), 6.81 (1H, d, J = 7.6 Hz), 6.47 (1H, d, J = 8.4 Hz), 5.97 (1H, s), 5.14 (1H, brs), 4.27 (1H, d, J = 2.4 Hz), 3.95-3.92 (1H, m), 3.60-3.54 (1H, m), 2.50-2.48 (1H, m), 2.14 (3H, s), 1.93-1.61 (2H, m), 1.28-1.17(2H, m); ESI-MS: m/z 372 [M + H]+.
Methyl 3,5-dichloro-4-((4aR,5S,10bR)-3,4,4a,5,6,10b-
hexahydro-9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoate
(exo-1u) The following compound was prepared using the
same methodology as in compound 1c (method 2) using methyl 3,5-dichloro-4-formylbenzoate6 (4.3 mmol, 1 g), p-toluidine (4.3 mmol, 461 mg) and TFA (1 eq., 4.3 mmol, 0.33 mL). Solvent was evaporated and purification of the crude material (85:15 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 10% ethyl acetate in hexanes afforded 874 mg (Yield 50%) of the pure title compound exo-1u. 1H NMR (400 MHz, CDCl3): δ 7.98 (2H, s), 7.04 (1H, s), 6.93 (1H, d, J = 8.4 Hz), 6.49 (1H, d, J = 8 Hz), 5.72 (1H, d, J = 11.6 Hz), 4.37 (1H, s), 4.15-4.12 (1H, m), 3.94 (3H, s), 3.73-3.67 (2H, m), 3.05-3.02 (1H, m), 2.24 (3H, s), 1.80-1.73 (1H, m), 1.39-1.28 (2H, m); ESI-MS: m/z 406 [M + H]+.
Methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)-3,5-dimethylbenzoate
(exo-1v)
The following compound was prepared using the same methodology as in compound 1c (method 2), using p-toluidine(0.70 mmol, 75 mg) and methyl 4-formyl-3,5-dimethylbenzoate6 (0.70 mmol, 135 mg).
Solvent was evaporated and purification of the crude material (≥98:2 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 25% ethyl acetate in hexanes afforded 58 mg (Yield 27%) of the pure title compound exo-1v. 1H NMR (400 MHz, DMSO-d6): δ 7.72 (2H, brs), 7.09 (1H, s), 6.93 (1H, m), 6.49 (1H, d, J = 7.2 Hz), 5.28 (1H, d, J = 12 Hz), 4.38 (1H, d, J = 2.8 Hz), 4.15-4.10 (1H, m), 3.92 (3H, s), 3.74-3.66 (1H, m), 2.74-2.66 (1H, m), 2.57 (3H, brs), 2.25 (3H, brs), 1.74-1.26 (5H, m); ESI-MS: m/z 366 [M + H]+.
Methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methoxy-2H-pyrano[3,2-c]quinolin-5-yl)-3,5-dimethylbenzoate
(exo-1w)
The following compound was prepared using the same methodology as in compound 1c (method 2) using methyl 4-formyl-3,5-dimethylbenzoate6 (0.52 mmol, 100 mg) and TFA (1eq., 0.52 mmol, 40 mL). Solvent was evaporated and purification of the crude material (≥98:2 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 20% ethyl acetate in hexanes afforded 52 mg (Yield 27%) of the pure title compound exo-1w. 1H NMR (400 MHz, DMSO-d6): δ 7.66-7.60 (2H, m), 6.68-6.65 (2H, m), 6.53 (1H, d, J = 9.2 Hz), 5.67 (1H, s), 5.01 (1H, d, J =12 Hz), 4.30 (1H, d, J = 2.4 Hz), 3.98-3.92 (1H, m), 3.83 (3H, s), 3.65 (3H, s), 3.62-3.53 (1H, m), 2.50-2.42 (6H, brs), 1.74-1.62 (1H, m), 1.60-1.48 (1H, m), 1.29-1.14 (3H, m); ESI-MS: m/z 382 [M + H]+.
Methyl 4-((4aR,5S,10bR)-9-chloro-3,4,4a,5,6,10b-
hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)-3,5-dimethylbenzoate
(exo-1x) [
The following compound was prepared using the same methodology as in compound 1c (method 2) using 4-chloroaniline (0.52 mmol, 66 mg), methyl 4-
Scheme II (Ref 15)

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1397
formyl-3,5-dimethylbenzoate6 (0.52 mmol, 100 mg) and TFA (1eq., 0.52 mmol, 40 mL).Solvent was evaporated and purification of the crude material (≥98:2 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 20% ethyl acetate in hexanes afforded 67 mg (Yield 33%) of the pure title compound exo-1x. 1H NMR (400 MHz, DMSO-d6): δ 7.68-7.60 (2H, m), 7.08 (1H, d, J = 2.8 Hz), 7.01 (1H, dd, J = 8.8 & 2.8 Hz), 6.58 (1H, d, J = 8.4 Hz), 6.30 (1H, s), 5.03 (1H, d, J = 11.6 Hz), 4.32 (1H, d, J = 2.8 Hz), 3.98-3.92 (1H, m), 3.83 (3H, s), 3.62-3.55 (1H, m), 3.31 (3H, s), 2.50-2.42 (6H, brs), 1.74-1.54 (2H, m), 1.60-1.48 (1H, m), 1.32-1.14 (3H, m); ESI-MS: m/z 386 [M + H]+.
Methyl 4-((4aR,5S,10bR)-9-(difluoromethoxy)-
3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinolin-
5-yl)-3,5-dimethylbenzoate (exo-1y)
The following compound was prepared using the same methodology as in compound 1c (method 2) using methyl 4-formyl-3,5-dimethylbenzoate6 (3.12 mmol, 600 mg), 4-(difluoromethoxy)aniline (3.12 mmol, 387 mL) and TFA (1eq., 3.12 mmol, 240 mL). Solvent was evaporated and purification of the crude material (≥98:2 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 12% ethyl acetate in hexanes afforded 493 mg (Yield 38%) of the pure title compound exo-1y. 1H NMR (400 MHz, DMSO-d6): δ 7.68-7.60 (2H, m), 6.95 (1H, t, J = 75.2 Hz), 6.91 (1H, d, J = 2.8 Hz), 6.86 (1H, dd, J = 8.4 & 2.8 Hz), 6.59 (1H, d, J = 8.8 Hz), 6.17 (1H, s), 5.04 (1H, d, J = 11.6 Hz), 4.32 (1H, d, J = 2.8 Hz), 3.98-3.92 (1H, m), 3.84 (3H, s), 3.62-3.54 (1H, m), 2.50-2.42 (7H, m), 1.74-1.54 (2H, m), 1.32-1.16 (2H, m); ESI-MS: m/z 418 [M + H]+.
Methyl 4-((4aR,5R,10bR)-3,4,4a,5,6,10b-hexahydro-9-
methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoate (endo-1z)
and methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-9-
methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoate (exo-1z)
The following compounds were prepared using the same methodology as in compound 1m (method 2), using methyl 4-formylbenzoate (3 mmol, 500 mg) and p-toluidine (3 mmol, 322 mg). Solvent was evaporated and purification of the crude material (70:30 mixture of exo/endo diastereomers) by flash chromatography over silica gel using an elution of 16% ethyl acetate in hexanes afforded 110 mg (Yield 11%) of the title endo-isomer (endo-1z) and 190 mg (Yield 19%) of the title exo-isomer (exo-1z) which was precipitated with hexane, filtered and washed to afford the pure title compound (exo-1z) as a solid.
Methyl 4-((4aR,5S,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoate
(exo-1z): 1H NMR (400 MHz, DMSO-d6): δ 7.96 (2H, d, J = 8 Hz), 7.57 (2H, d, J = 8 Hz), 6.86 (1H, s), 6.81 (1H, d, J = 7.6 Hz), 6.51 (1H, d, J = 8 Hz), 6.01 (1H, s), 4.57 (1H, d, J = 10.4 Hz), 4.24 (1H, s), 3.86 (4H, m), 3.61-3.55 (1H, m), 2.15 (3H, s), 1.93-1.90 (1H, m), 1.77-1.73 (1H, m), 1.65-1.61 (1H, m), 1.30-1.19 (2H, m); ESI-MS: m/z 338 [M + H]+.
Methyl 4-((4aR,5R,10bR)-3,4,4a,5,6,10b-hexahydro-
9-methyl-2H-pyrano[3,2-c]quinolin-5-yl)benzoate
(endo-1z): 1H NMR (400 MHz, DMSO-d6): δ 7.96 (2H, d, J = 8.8 Hz), 7.57 (2H, d, J = 8 Hz), 7.01 (1H, s), 6.81 (1H, d, J = 8.4 Hz), 6.61 (1H, d, J = 7.6 Hz), 5.34 (1H, s), 5.21 (1H, d, J = 5.6 Hz), 4.67 (1H, s), 3.86 (3H, s), 3.49-3.45 (1H, m), 3.25-3.23 (1H, m), 2.18 (3H, s), 2.08-2.04 (1H, m), 1.36-1.32 (3H, m), 1.15-1.01 (1H, m); ESI-MS: m/z 338 [M + H]+.
4-Amino-3-chloro-5-methylbenzonitrile, 5 (Scheme II, Ref 15)
To a solution of 4-bromo-2-chloro-6-methylbenzen-amine (4.53 mmol, 1 g) in DMF (10 mL) zinc cyanide (3.17 mmol, 373 mg) and tetrakis(triphenylphosphine)-palladium (0.136 mmol, 157 mg) were added. The reaction mixture was heated at 220°C for 16 h, then cooled to RT and diluted with ethyl acetate and water. Organic layer was then separated, washed with brine and dried over anhydrous sodium sulphate, filtered and concentrated under reduce pressure to afford the desired product which was purified by flash chromatography over silica gel using an elution of 17% ethyl acetate in hexanes to afford 4-amino-3-chloro-5-methylbenzonitrile (390 mg, Yield 51%). 1H NMR (400 MHz, CDCl3): δ 7.39 (1H, s), 7.20 (1H, s), 4.61 (2H, brs), 2.17 (3H, s)
3-Chloro-4-iodo-5-methylbenzonitrile, 6 (Scheme II, Ref 15)
4-Amino-3-chloro-5-methylbenzonitrile 5 (9 mmol, 1.5 g) was gradually added to concentrated sulphuric acid (20 mL). The mixture was then stirred at RT for 20 min and then cooled to 0ºC. To this mixture sodium nitrite (9.9 mmol, 683 mg) was added gradually and stirred at 5ºC over 2.5 h. The mixture thus obtained was then added to ice-water (30 g) and potassium iodide (10.8 mmol, 1.8 g), stirred at 5ºC for 15 min and then at RT for 1 h. The reaction mixture was extracted with diethyl ether. The organic layer was washed with an aqueous solution of sodium thiosulfate, dried over magnesium sulphate, filtered and concentrated. The reaction crude was purified by

INDIAN J. CHEM., SEC B, NOVEMBER 2016
1398
flash chromatography over silica gel using an elution of 8% ethyl acetate in hexanes to afford 3-chloro-4-iodo-5-methylbenzonitrile (1.17g, Yield 46%). 1H NMR (400 MHz, CDCl3): δ 7.54 (1H, s), 7.37 (1H, s), 2.57 (3H, s).
3-Chloro-5-methyl-4-vinylbenzonitrile, 7 (Scheme II, Ref 15)
A solution of 3-chloro-4-iodo-5-methylbenzonitrile 6 (4.48 mmol, 1.17 g), 2-vinyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (8.96 mmol, 1.38 g), tetrakis-(triphenylphosphine) palladium (5% w/w, 59 mg) and 2N aqueous sodium carbonate solution (15.23 mmol, 7.6 mL) in toluene/ethanol (2:1, 22.5 mL). The suspension was degassed by argon and stirred at 90ºC for 16 h, then cooled to RT and concentrated under vacuum. The reaction crude was purified by flash chromatography over silica gel using an elution of 5% ethyl acetate in hexanes to afford 3-chloro-5-methyl-4-vinylbenzonitrile (594 mg, Yield 74%). 1H NMR (400 MHz, CDCl3): δ 7.53 (1H, s), 7.39 (1H, s), 6.68 (1H, dd, J = 18 & 11.6Hz), 5.75 (1H, d, J = 12 Hz), 5.53 (1H, d, J = 18 Hz), 2.39 (3H, s).
3-Chloro-4-formyl-5-methylbenzonitrile, 8 (Scheme II, Ref 15)
3-Chloro-5-methyl-4-vinylbenzonitrile 7 (3.35 mmol, 594 mg) in acetone/water (6:1, 35 mL), OsO4 (2.48 mmol, 630 mg) and NaIO4 (10.5 mmol, 2.15 g) were added. The resulting mixture was kept at RT for 1.5 h and then filtered. The solid was washed with ethyl acetate and the filtrate was concentrated under reduce pressure to afford the desired product which was purified by flash chromatography over silica gel using an elution of 6% ethyl acetate in hexanes to afford the title compound as a solid (410 mg, Yield 68%). 1H NMR (400 MHz, CDCl3): δ 10.62 (1H, s), 7.62 (1H, s), 7.47 (1H, s), 2.61 (3H, s).
3-Chloro-4-formyl-5-methylbenzoic acid, 9 (Scheme II, Ref 15)
A solution of 3-chloro-4-formyl-5-methylbenzonitrile 8 (3.57 mmol, 640 mg) was dissolved in concentrated HCl (15 mL). The reaction mixture was stirred at 90ºC for 16 h and cooled to RT. The crude product was filtered, the solid was washed with water and dried in vacuum to afford the title compound which was used in the next step without further purification (620 mg, Yield 87%). 1H NMR (400 MHz, DMSO-d6): δ 10.41 (1H, s), 7.69 (1H, s), 7.61 (1H, s), 2.59 (3H, s).
Conclusions We have synthesized highly functionalized substituted
hexahydropyrano[3,2-c]quinolones 1 in high to
moderate yields in a parallel synthetic approach. These products are highly functionalized natural product-like tricyclic systems, which may be useful as biologically relevant targets. The diastereoselectivity and region-selectivity obtained in the Povarov reactions of dihydropyrans is shown to depend on the nature of the catalytic acid and/or effect of the substitution of the aldehyde used in the Povarov reaction. Thus, protic Brönsted acid catalysts such as TFA, 4-NO2PhA and HOTf gave higher exo diastereoselectivity. Furthermore, it has been found that ortho and di-ortho substitution of the aldehyde increased the exo diastereoselectivity which was higher when the bulky size of the ortho group increases. This factor is attributed to additional steric interactions that occur in the transition states for the dihydropyrane-based reactions and the competing concerted and stepwise pathways. In addition, we managed to improve the yield of the Povarov reaction keeping the high exo diastereoselectivity by replacing a protic Brönsted acid (TFA) by TMSCl catalyst, a procedure which allowed to scale up the reaction and obtain the hexahydropyrano[3,2-c]quinolines on a multigram scale. Further studies are presently focussed on the enantiomeric resolution of the prepared diasteromeric compounds. Acknowedgements
This work was funded by the CENIT-E program of the Center for Technological and Industrial Development (CDTI) of Spain, under the research project NEOGENIUS PHARMA (ref. CEN 20091003) and by the consortium of companies NEOGENIUS PHARMA AIE established by Almirall S.A., Draconis Pharma S.L., Laboratorios Dr. Esteve S.A. and Proteomika S.L.U.
References 1 (a) Smith C D, Gavrilyuk J I, Lough A J & Batey R A, J Org
Chem, 75 (2010) 702; (b) Xie M, Chen X, Zhu Y, Gao B, Lin L, Liu X & Feng X, Angew Chem Int Ed, 49 (2010) 3799 and references cited therein; (c) Liu H, Dagousset G, Masson G, Retailleau P & Zhu J, J Am Chem Soc, 131 (2009) 4598 and references cited therein; (d) Xing X, Wua J & Dai W-M, Tetrahedron, 62 (2006) 11200 and references cited therein.
2 (a) Diaz J L, Christmann U, Fernandez A, Luengo M, Bordas M, Enrech R, Carro M, Pascual R, Burgueno J & Merlos M, J
Med Chem, 56 (2013) 3656; (b) Liu R H, Yu X, Hu L & Yu N F, Chinese Chem Lett, 23 (2012) 1027; (c) Li M, Sun E, Wen L, Sun J, Li Y & Yang H, J Comb Chem, 9 (2007) 903; (d) Gao K, Li Y, Sun H, Fan R & Wu J, Synth Commun, 37 (2007) 4425; (e) Beifuss U, Ledderhose S & Ondrus V, ARKIVOC (v) (2005) 147; (f) Shi M, Shao L-X & Xu B, Org
Lett, 5,(2003) 579; (g) Shao L-X & Shi M, Adv Synth Catal, 343 (2003) 963; (h) Hadden M & Stevenson P J, Tetrahedron

NIÑO et al.: AZA-DIELS-ALDER REACTIONS
1399
Lett, 40 (1999) 1215; (i) Annunziata R, Benaglia M, Cinquini M & Cozzi F, Eur J Org Chem (2002) 1184; (j) Cheng D, Zhou J, Saiah E & Beaton G, Org Lett, 4 (2002) 4411; (k) Annunziata R, Cinquini M, Cozzi F, Molteni V & Schupp O, Tetrahedron, 53 (1997) 9715.
3 (a) Torquato da Silva B H S, Martins L M & da Silva-Filho L C, Synlett, 23 (2012) 1973; (b) da Silva-Filho L C & Frenhe M, The Electronic Journal of Chemistry, 3 (2011) 1; (c) da Silva-Filho L C, Lacerda V Jr, Constantino M G & Jose da Silva G V, Synthesis, 16 (2008) 2527; (d) Nagarapu L, Bantu R & Puligoundla G, Eur J Org Chem, 2 (2011) 260; (e) Kantevari S, Yempala T, Surineni G, Sridhar B, Yogeeswari P & Sriram D, Eur J Med Chem, 46 (2011) 4827; (f) Ramesh E, Elamparuthi E & Raghunathan R, Lett Org Chem, 5 (2008) 82; (g) Babu G, Nagarajan R, Natarajan R & Perumal P T, Synthesis, 5 (2000) 661; (h) Babu G & Perumal P T, Tetrahedron Lett, 39 (1998) 3225; (i) Mayekar N V, Nayak S K & Chattopadhyay S, Natural Product Communications, 1 (2006) 767; (j) Sundararajan G, Prabagaran N & Varghese B, Org Lett, 3 (2001) 1973; (k) Reddy B V S, Srinivas R, Yadav J S & Ramalingam T, Synth Commun, 31 (2001) 1075; (l) Sabitha G, Reddy M S K, Arundhathi K & Yadav J S, ARKIVOC, 6 (2006) 153; (m) Mahajan D, Ganai B A, Sharma R L & Kapoor K K, Tetrahedron Lett, 47 (2006) 7919; (n) Goudar M A, Jayadevappa H, Sudhakara A & Mahadevan K M, Lett Org Chem, 5 (2008) 628; (o) Mahesh M, Reddy Ch V, Reddy K S, Raju P & Reddy V V N, Synth Commun, 34 (2004) 4089; (p) Merchan Arenas D R, Rojas Ruiz F A & Kouznetsov V V, Tetrahedron Lett, 52 (2011) 1388; (q) Yadav J S, Reddy B V S, Madhuri C R & Sabitha G, Synthesis, 7 (2001) 1065.
4 (a) Liu R H, Yu X, Hu L & Yu N F, Chin Chem Lett, 23 (2012) 1027; (b) Xing X, Wu J, Dai W-M, Tetrahedron, 62 (2006) 11200; (c) Burai R, Ramesh C, Shorty M, Curpan R, Bologa C, Sklar L A, Oprea T, Prossnitzd E R & Arterburn J B, Org Biomol Chem, 8 (2010) 2252; (d) Srinivasa A, Mahadevan K M, Hosamani K M & Hulikal V, Monatsh
Chem, 139 (2008) 141; (e) Nagarajan R, Magesh C J & Perumal P T, Synthesis (2004) 69.
5 (a) Makioka Y, Shindo T, Taniguchi Y, Takaki K & Fujiwara Y, Synthesis, 7 (1995) 801; (b) Narsaiah A V, Reddy A R,
Reddy B V S & Yadav J S, Synth Commun, 40 (2010) 1750; (c) Zhou Z, Xu F, Han X, Zhou J & Qi S, Eur J Org Chem (2007) 5265; (d) Yu Y, Zhou J, Yao Z, Xu F & Shen Q, Heteroatom Chem, 21 (2010) 351; (e) Ma Y, Qian C, Xie M & Sun J, J Org Chem, 64 (1999) 6462.
6 (a) Niño P, Caba M, Aguilar N, Terricabras E, Albericio F & Fernàndez J-C, Indian J Chem (Section B) 2016, submitted for publication; (b) Aguilar N, Fernandez J-C, Terricabras E, Carceller Gonzalez E & Salas Solana J, PCT Int Appl WO 2013149997A1, October 10, 2013; (c) Aguilar N, Fernandez J-C, Terricabras E, Carceller Gonzalez E & Garcia Garcia F-J, Eur Pat 2647638A1, October 9, 2013.
7 (a) Glushkov V A & Tolstikov A G, 77, Russ Chem Rev (2008) 137; (b) Hermitage S, Howard J A K, Jay D, Pritchard R G, Probert M R & Whiting A, Org Biomol Chem, 2 (2004) 2451.
8 Ralbovsky J L & Beckett R P, US Patent 20080214537, September 4, 2008.
9 (a) Shindoh N, Tokuyama H, Takemoto Y & Takasu K, J Org
Chem, 73 (2008) 7451; (b) Itoh T, Nagata K, Kurihara A, Miyazaki M & Ohsawa A, Tetrahedron Lett, 43 (2002) 3105.
10 Khan A T, Das D K & Khan Md M, Tetrahedron Lett, 52 (2011) 4539.
11 Salehi J, Veisi H, Khodaei M M & Khosropour A R, J
Heterocycl Chem, 48 (2011) 484. 12 (a) Sridharan V, Avendano C & Menendez J C, Synthesis
(2008) 1039; (b) Sridharan V, Avendano C & Menendez J C, Tetrahedron, 63 (2007) 673; (c) Ravindranath N, Ramesh C, Reddy M R & Das B, Chem Lett, 32 (2003) 222.
13 (a) Gal E, Cristea C, Silaghi-Dumitrescu L, Lovasz T & Csampai A, Tetrahedron, 66 (2010) 9938; (b) Wang X-S, Zhou J, Yin M-Y, Yang K & Tua S-J, J Comb Chem,12 (2010) 266; (c) Wang X-S, Zhou J, Yin M-Y, Yang K & Tua S-J, J
Heterocycl Chem, 47 (2010) 873; (d) Jin G, Zhao J, Han J, Zhu S & Zhang J, Tetrahedron, 66 (2010) 913; (e) Rai N P, Shashikanth S & Arunachalam P N, Synth Commun, 39 (2009) 2125; (f) Xia M & Lu Y-D, Synlett (2005) 2357.
14 More S V & Sastry M N V, Yao C-F, Synlett (2006) 1399. 15 3-Chloro-4-formyl-5-methylbenzoic acid 9 was prepared from
4-bromo-2-chloro-6-methylbenzenamine as shown in Scheme II.





























