Embed Size (px)
Citation preview

of April 4, 2019.This information is current as
Lymphoproliferative SyndromeMechanism in the Human Autoimmune FAS Haploinsufficiency Is a Common Disease
Rao, Joie Davis, Thomas A. Fleisher and João B. OliveiraHye Sun Kuehn, Iusta Caminha, Julie E. Niemela, V. Koneti
ol.1100021http://www.jimmunol.org/content/early/2011/04/12/jimmun
published online 13 April 2011J Immunol
MaterialSupplementary
1.DC1http://www.jimmunol.org/content/suppl/2011/04/12/jimmunol.110002
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
FAS Haploinsufficiency Is a Common Disease Mechanism inthe Human Autoimmune Lymphoproliferative Syndrome
Hye Sun Kuehn,* Iusta Caminha,* Julie E. Niemela,* V. Koneti Rao,† Joie Davis,†
Thomas A. Fleisher,* and Joao B. Oliveira*
The autoimmune lymphoproliferative syndrome (ALPS) is characterized by early-onset lymphadenopathy, splenomegaly, immune
cytopenias, and an increased risk for B cell lymphomas. Most ALPS patients harbor mutations in the FAS gene, which
regulates lymphocyte apoptosis. These are commonly missense mutations affecting the intracellular region of the protein and
have a dominant-negative effect on the signaling pathway. However, analysis of a large cohort of ALPS patients revealed that
∼30% have mutations affecting the extracellular region of FAS, and among these, 70% are nonsense, splice site, or insertions/
deletions with frameshift for which no dominant-negative effect would be expected. We evaluated the latter patients to understand
the mechanism(s) by which these mutations disrupted the FAS pathway and resulted in clinical disease. We demonstrated that
most extracellular-region FAS mutations induce low FAS expression due to nonsense-mediated RNA decay or protein instability,
resulting in defective death-inducing signaling complex formation and impaired apoptosis, although to a lesser extent as compared
with intracellular mutations. The apoptosis defect could be corrected by FAS overexpression in vitro. Our findings define
haploinsufficiency as a common disease mechanism in ALPS patients with extracellular FAS mutations. The Journal of Immu-
nology, 2011, 186: 000–000.
The autoimmune lymphoproliferative syndrome (ALPS)is characterized by early-onset development of benignlymphadenopathy and splenomegaly, multilineage cyto-
penias due to autoimmune peripheral destruction and splenic se-questration of blood cells, and increased risk for B cell lympho-mas (1–3). Patients typically accumulate a hallmark population ofmature TCRab+ T cells that are CD4 and CD8 negative (4, 5).ALPS is caused by defects in proteins involved in the FAS pathwayof lymphocyte apoptosis. Most (∼65%) patients have mutations inthe FAS (TNFRSF6/APO1/CD95) gene, whereas a minority hasmutations in the gene encoding FAS ligand (FASL) or caspase-10(6–12). Germline mutations in CASP8 or somatic mutations inNRAS or KRAS cause ALPS-related syndromes currently classi-fied separately (12–15).The FAS gene contains nine exons spanning 26 kb on chro-
mosome 10q24.1 (16). The first five exons encode the extracellularportion of the protein containing three cysteine-rich domains thatare involved in receptor trimerization and FASL binding requiredfor triggering of the apoptotic signal. Exon 6 codes for thetransmembrane domain, and the intracellular portion is encoded
by exons 7–9. The FAS death domain (DD), an 85-aa-longstructure encoded by exon 9, is required for FAS-induced apo-ptosis of lymphocytes under physiological conditions (17).The majority of FAS defects associated with ALPS are het-
erozygous missense mutations that affect the intracellular DD,allowing for the expression of a defective protein with a dominant-negative effect on the signaling pathway (7, 18). These mutationsdemonstrate high penetrance for clinical symptoms including re-fractory cytopenias and an increased risk for lymphoma (2, 18).However, evaluation of a large cohort of ALPS patients at ourcenter has revealed a significant subpopulation harboring muta-tions affecting the extracellular region of the protein, and theseare commonly nonsense, or insertions, deletions, and splice sitemutations with frameshift (Fig. 1). These mutations are predictedto abolish protein expression and hence are not expected to resultin dominant-negative interference, and the mechanism by whichthis affects FAS apoptosis signaling is not clearly defined. FAShaploinsufficiency is one possible disease mechanism in ALPSassociated with extracellular mutations, but currently there areonly very limited data supporting this mechanism (19, 20). In fact,FAS haploinsufficiency was clearly demonstrated in only oneALPS patient to date, such that the prevalence, long-term clinicalimpact, and biochemical consequences are unknown (19).To understand the mechanism by which extracellular region
FAS mutations disrupt the apoptotic machinery, we analyzed the
functional impact of 29 representative mutations across all regions
of FAS using patient-derived B cell lines. We demonstrate that
most (8 of the 11) mutations affecting the extracellular regions
of the FAS receptor we studied are associated with haploinsuffi-
ciency. These mutations induced less severe disruption of the
FAS-mediated apoptosis-signaling platform when compared with
missense intracellular mutations, and the apoptotic defect could
be rescued in vitro by FAS overexpression. These findings define
haploinsufficiency as an ALPS-causing disease mechanism, in
addition to the well-documented dominant-negative interference
seen with intracellular mutations.
*Department of Laboratory Medicine, Clinical Center, National Institutes of Health,Bethesda, MD 20814; and †ALPS Unit, Laboratory of Clinical Infectious Diseases,National Institute of Allergy and Infectious Diseases, National Institutes of Health,Bethesda, MD 20814
Received for publication January 5, 2011. Accepted for publication March 17, 2011.
This work was supported by the National Institutes of Health Intramural Program.
H.S.K. performed research, analyzed data, made the figures, and wrote the paper; I.C.performed analysis of the biomarkers; J.E.N. performed genomic DNA sequencinganalysis; V.K.R. and J.D. were responsible for patient care; T.A.F. supervised re-search; and J.B.O. designed and supervised the project and wrote the paper.
Address correspondence and reprint requests to Dr. Joao Bosco Oliveira, Departmentof Laboratory Medicine, Clinical Center, National Institutes of Health, 10 CenterDrive, Bethesda, MD 20814. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ALPS, autoimmune lymphoproliferative syn-drome; DD, death domain; DiOC6, 3,39-dihexyloxacarbocyanine iodide; DISC,death-inducing signaling complex; EC, extracellular; FADD, Fas-associated deathdomain protein; FASL, FAS ligand; NMD, nonsense-mediated mRNA decay.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100021
Published April 13, 2011, doi:10.4049/jimmunol.1100021 by guest on A
pril 4, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
Materials and MethodsPatient samples, cell culture, and gene sequencing
All patients were studied at the National Institutes of Health under In-stitutional Review Board-approved protocols (93-I-0063 and 95-I-0066).Genomic DNA samples were PCR-amplified and sequenced as pre-viously described (21). For mRNA sequencing, cDNA was prepared fromEBV-transformed B cell lines using the RNeasy plus mini kit (Qiagen). Torule out the presence of contaminating DNA, RNA samples were subjectedto PCR amplification using intron-specific primers; the absence of am-plified product (DNA) was confirmed by denaturing agarose gel electro-phoresis (data not shown). cDNA was PCR amplified (forward: 59-GTG-AGGGAAGCGGTTTACGAGTGA-39; and reverse: 59-AGTGGGGTTAG-CCTGTGGATAGAC-39), and the products were subjected to sequencing(primer sequences available upon request). To determine the ratio of mu-tant to wild-type FAS transcripts, cDNAwas amplified and cloned into thepcDNA 3.0-HAvector (modified from pcDNA3.0 [Invitrogen]) with EcoRIand XhoI enzyme sites. After transformation, we picked 10–20 coloniesand performed sequencing. Three samples with DD mutations (D260N,Q276X, L294Dfs) were used as controls. For functional studies, EBV-transformed B cell lines from the patients were cultured in RPMI 1640media with 10% FBS, 100 units/ml penicillin, 100 mg/ml streptomycin,and 2 mM L-glutamine (Life Technologies, Invitrogen).
Apoptosis measurement
EBV-transformed B cells (200,000/well) were aliquoted in triplicate into96-well plates and cultured with or without APO-1-3 (1 mg/ml) (ENZO LifeSciences) in the presence of protein A (1 mg/ml). After 24 h, cell loss wasdetermined by measuring the loss of the mitochondrial transmembranepotential using 3,39-dihexyloxacarbocyanine iodide (DiOC6) (Calbiochem,EMC Biosciences) staining. Briefly, cells were incubated with 40 nMDiOC6 for 15 min at 37˚C, and live cells (DiOC6 high) were counted byflow cytometry (BD FACSCanto II; BD Biosciences) by constant timeacquisition. The percentage of cell loss was calculated according to thefollowing formula: (number of live cells without APO-1-3 treatment 2number of live cells with APO-1-3 treatment/number of live cells withoutAPO-1-3 treatment) 3 100.
FAS cell surface expression
To determine FAS (CD95) expression, EBV-transformed B cells (0.53 106
cells/sample) were washed with PBS and incubated with 10 mg/ml PE-CD95 (BD Biosciences) or IgG control for 30 min at 4˚C (dark) in 100 ml5% FCS in PBS. After washing with PBS two times, 10,000 live cells wereanalyzed by flow cytometry. Absolute number of FAS molecules on thecell surface was established by developing a mean equivalent solublefluorochrome standard curve using QuantiBRITE PE (BD Biosciences)
beads run in parallel for each experiment, according to the manufacturer’sprotocol.
Immunoprecipitation
EBV-transformed B cells (3 3 106 cells/sample) were cultured with orwithout APO-1-3 (0.5 mg/ml) in the presence of protein A (1 mg/ml) for 20min at 37˚C. Immunoprecipitation was performed according to the man-ufacturer’s instructions (Pierce Classic IP Kit; Thermo Scientific). Briefly,after exposure, cells were washed with cold PBS, lysed, and spun down(13,000 rpm, 4˚C, 20 min). Supernatants were incubated with anti-FAS Ab(A-20; Santa Cruz Biotechnology) and protein A/G agarose for 2 h, afterwhich the immune complexes were washed, and proteins were separatedby electrophoresis on 4–12% NuPAGE Bis–Tris gels (Invitrogen), trans-ferred to nitrocellulose membranes, and probed using the anti–Fas-associated DD protein (FADD) (BD Biosciences) or anti–caspase-8 (CellSignaling Technology) Abs. To quantitate changes in protein level, theECL films were scanned using a Quantity One scanner (Bio-Rad).
FAS transfection
EBV-transformed B cells (3 3 106 cells /sample) were transfected with5 mg GFP (pmaxGFP) or 5 mg YFP-FAS (pEYFP-N1-FAS) using theAmaxa Human B-cell Nucleofector kit (Program U-015; Amaxa). Twenty-four hours after transfection, medium was replaced with fresh completemedium. After 48 h of transfection, expression of FAS on the cell surfacewas determined by flow cytometry as described above, gating on live GFP-or YFP-transfected cells. To evaluate apoptosis, the transfected cell lineswere treated with vehicle or APO-1-3 in the presence of protein A (asabove) and live GFP+ or YFP+ cells counted as described above.
Biomarkers
The quantification of IL-10, soluble Fas ligand, vitamin B12, IL-18, TNF-a,and TCRab+CD42CD82 T cells on patient samples was performed aspreviously described (22).
Statistical analysis
Data are represented as the mean 6 SEM, except where noted. The sta-tistical analyses were performed by unpaired Student’s t test or Mann–Whitney rank-sum test. Differences were considered significant when p ,0.05.
ResultsFAS cell-surface expression in ALPS-associated FAS mutations
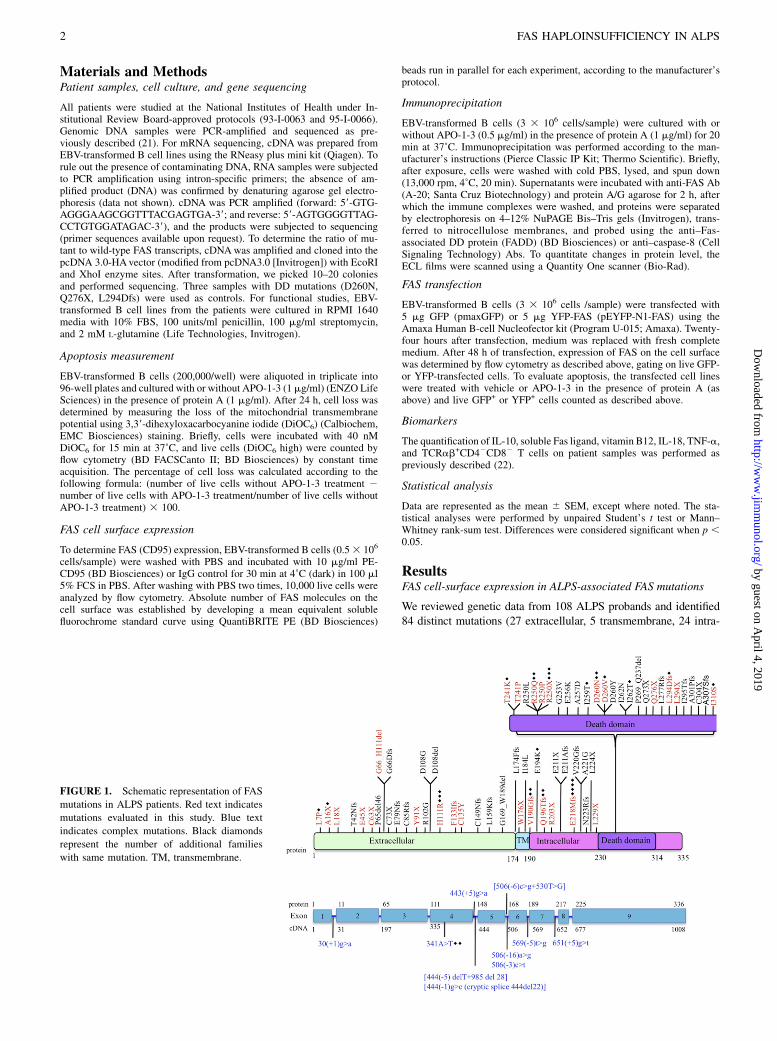
We reviewed genetic data from 108 ALPS probands and identified84 distinct mutations (27 extracellular, 5 transmembrane, 24 intra-
FIGURE 1. Schematic representation of FAS
mutations in ALPS patients. Red text indicates
mutations evaluated in this study. Blue text
indicates complex mutations. Black diamonds
represent the number of additional families
with same mutation. TM, transmembrane.
2 FAS HAPLOINSUFFICIENCY IN ALPS
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

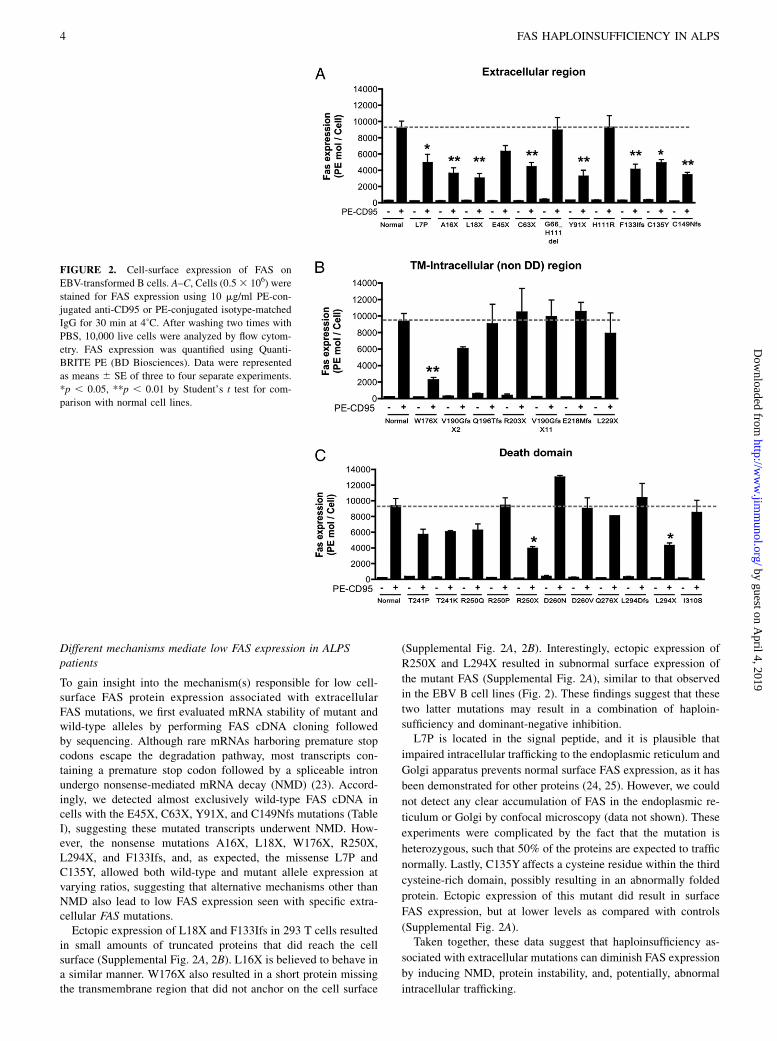
cellular non-DD, and 28 intracellular DD mutations) (Fig. 1). Themajority of the non-DD mutations were either nonsense (11 of 57)or splice site or insertions/deletions with predicted frameshifts (36of 57). More importantly, most (19 of 27) of the mutations affect-ing the extracellular regions of the protein were also nonsense,insertions/deletions, or splice site with frameshifts. We selected 29mutations across all regions to determine their functional effect(Fig. 1, Table I). To assess the impact of the different FAS mutationson the expression of FAS, we determined the number of FASmolecules on the cell surface of patient-derived EBV-transformedB cell lines using a flow cytometry-based assay. The majority of themutations affecting the extracellular region (8 out of 11) demon-strated significantly reduced FAS expression (Fig. 2A, SupplementalFig. 1). The mutation W176X, which abolishes the entire trans-membrane region, also resulted in low FAS expression (Fig. 2B).The mutation H111R and the inframe deletion G66_H111 affectthe FASL-binding domain and were not expected to result in lowerprotein expression. As predicted, most mutations affecting in-tracellular regions of the FAS protein did not significantly changecell-surface FAS expression (Fig. 2B, 2C, Supplemental Fig. 1).Exceptions included the intracellular nonsense mutations R250Xand L294X, which significantly reduced FAS surface expression(Fig. 2B, 2C, Supplemental Fig. 1). Taken together, these datasuggested haploinsufficiency as a possible disease mechanism inALPS caused by FAS extracellular region mutations.
Low FAS expression is associated with apoptosis dysfunctiondue to impaired death-inducing signaling complex formation
We next evaluated the functional impact of the 29 selectedmutations in the FAS pathway by the treatment of EBV-transformed B cell lines with the agonistic anti-FAS Ab APO-1-3
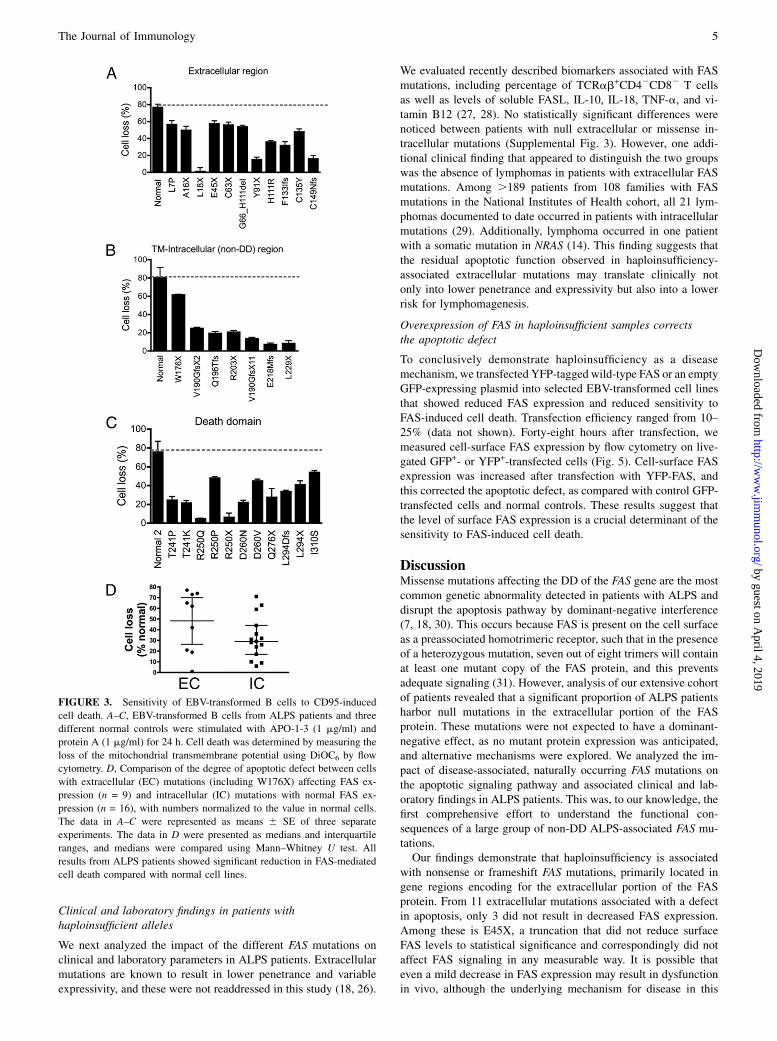
followed by cross-linking. All cell lines with extracellular, trans-membrane, or intracellular mutations showed significant resis-tance to FAS-mediated cell death when compared with controlcell lines (Fig. 3A–C). The apoptotic defect could also be observedon primary cultured T cells (Supplemental Table I). Mutations inthe extracellular domains resulted in milder apoptotic defectswhen compared with intracellular mutations, with median celllosses of 62 and 29%, respectively (Fig. 3D). However, this dif-ference did not reach statistical significance (p = 0.17), likely dueto the large data spread in the extracellular group and the limitedsample number in each group.To further dissect the impact of FAS mutations on the down-
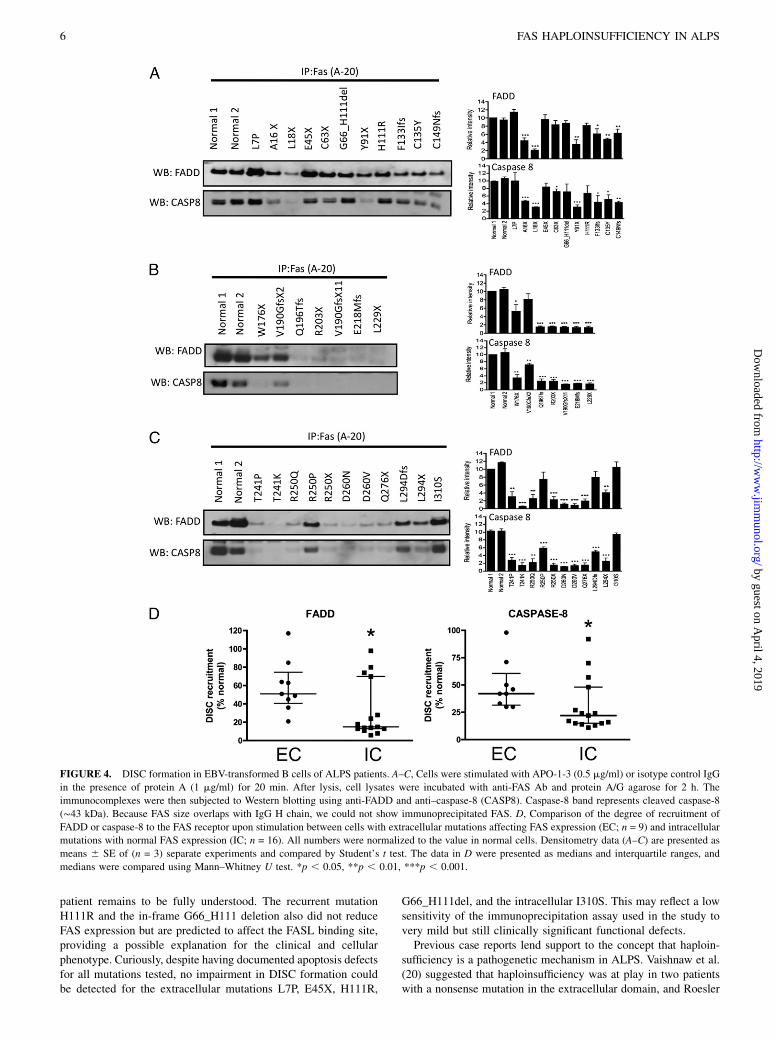
stream signaling pathways, we evaluated death-inducing signalingcomplex (DISC) formation in response to FAS activation. Thiscomplex includes FADD together with caspase-8/10 and is formedwithin seconds following FAS stimulation, resulting in the pro-cessing and activation of caspase-8/10 with propagation of theapoptotic signal. Following FAS activation with APO-1-3 andprotein A, immunoprecipitated protein complexes were examinedbyWestern blotting to measure the levels of coimmunoprecipitatedFADD and caspase-8 bound to the intracellular portion of the FASreceptor. Compared to mutations affecting the intracellular region(Fig. 4B, 4C), the extracellular mutations (Fig. 4A) showed con-sistent but more limited impairment in FADD (p = 0.032) andcaspase-8 (p = 0.02) recruitment to the signaling complex, basedon multiple experiments (Fig. 4D). These data demonstrate thatextracellular mutations with low FAS expression attenuate down-stream signaling and impair apoptosis, although to a lesser extentwhen compared with the intracellular mutations, and confirm in-terference of apoptosis by haploinsufficiency at the molecularlevel.
Table I. Summary of FAS mutations and their effects
IdentificationNo. cDNA Protein Location
Surface Fas(% Normal)
Cell Loss(% Normal)
FADDRecruitment(% Normal)
Caspase-8Recruitment(% Normal)
RNA Stability(% MutantClones)a
127 c.20T . C p.L7P Exon 1 54 74 117 98 Stable (44.5)220 c.46_47delGC p.A16X Exon 2 39 65 45 46 Stable (15)74 c.53T . G p.L18X Exon 2 33 1 21 30 Stable (31)153 c.133G . T p.E45X Exon 2 69 75 99 82 Unstable (0)50 c.189T . A p.C63X Exon 2 48 73 85 71 Unstable (0)111 c.197(-1)g . a p.G66_H111del Intron 2 98 70 89 70 Stable89 c.273C . A p.Y91X Exon 3 36 19 36 30 Unstable (8)180 c.332A . G p.H111R Exon 3 101 47 83 66 Stable77 c.397_398delTTinsA p.F133IfsX54 Exon 4 45 42 63 42 Stable (15)205 c.404G . A p.C135Y Exon 4 53 62 49 50 Stable (22)34 c.444(-1)g . c p.C149NfsX32 Intron 4 37 21 64 42 Unstable (0)175 c.528G . A p.W176X Exon 6 24 77 51 33 Stable (6)4 c.569(-2)a . c p.V190GfsX2 Intron 6 64 31 80 70 Stable45 c.585_595del11 p.Q196TfsX12 Exon 7 97 24 14 24 Stable72 c.607A . T p.R203X Exon 7 112 26 15 24 Stable149 c.651(+2)t . a p.V190GfsX11 Intron 7 106 17 14 15 Stable62 c.676(+2)t . c p.E218MfsX4 Intron 8 109 9 13 17 Stable98 c.686delT+690_694del5 p.L229X Exon 9 84 10 13 16 Stable3 c.721A . C p.T241P Exon 9 63 32 28 27 Stable110 c.722C-.A p.T241K Exon 9 67 28 6 14 Stable29 c.749G . A p.R250Q Exon 9 69 6 24 22 Stable31 c.749G . C p.R250P Exon 9 104 63 70 57 Stable55 c.748C . T p.R250X Exon 9 44 8 21 15 Stable (37.5)197 c.778G . A p.D260N Exon 9 145 29 11 11 Stable (28.6)137 c.779A . G p.D260V Exon 9 100 59 8 13 Stable33 c.826C . T p.Q276X Exon 9 90 36 18 15 Stable (60)121 c.879_880delAT p.L294DfsX2 Exon 9 115 44 74 48 Stable (60)30 c.880delT p.L294X Exon 9 48 54 38 25 Stable (28)17 c.929T . G p.I310S Exon 9 94 71 98 92 Stable
aStable: both wild-type and mutant forms were detected; Unstable: .90% of the clones were wild-type.
The Journal of Immunology 3
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
Different mechanisms mediate low FAS expression in ALPSpatients
To gain insight into the mechanism(s) responsible for low cell-surface FAS protein expression associated with extracellularFAS mutations, we first evaluated mRNA stability of mutant andwild-type alleles by performing FAS cDNA cloning followedby sequencing. Although rare mRNAs harboring premature stopcodons escape the degradation pathway, most transcripts con-taining a premature stop codon followed by a spliceable intronundergo nonsense-mediated mRNA decay (NMD) (23). Accord-ingly, we detected almost exclusively wild-type FAS cDNA incells with the E45X, C63X, Y91X, and C149Nfs mutations (TableI), suggesting these mutated transcripts underwent NMD. How-ever, the nonsense mutations A16X, L18X, W176X, R250X,L294X, and F133Ifs, and, as expected, the missense L7P andC135Y, allowed both wild-type and mutant allele expression atvarying ratios, suggesting that alternative mechanisms other thanNMD also lead to low FAS expression seen with specific extra-cellular FAS mutations.Ectopic expression of L18X and F133Ifs in 293 T cells resulted
in small amounts of truncated proteins that did reach the cellsurface (Supplemental Fig. 2A, 2B). L16X is believed to behave ina similar manner. W176X also resulted in a short protein missingthe transmembrane region that did not anchor on the cell surface
(Supplemental Fig. 2A, 2B). Interestingly, ectopic expression of
R250X and L294X resulted in subnormal surface expression of
the mutant FAS (Supplemental Fig. 2A), similar to that observed
in the EBV B cell lines (Fig. 2). These findings suggest that these
two latter mutations may result in a combination of haploin-
sufficiency and dominant-negative inhibition.L7P is located in the signal peptide, and it is plausible that
impaired intracellular trafficking to the endoplasmic reticulum and
Golgi apparatus prevents normal surface FAS expression, as it has
been demonstrated for other proteins (24, 25). However, we could
not detect any clear accumulation of FAS in the endoplasmic re-
ticulum or Golgi by confocal microscopy (data not shown). These
experiments were complicated by the fact that the mutation is
heterozygous, such that 50% of the proteins are expected to traffic
normally. Lastly, C135Y affects a cysteine residue within the third
cysteine-rich domain, possibly resulting in an abnormally folded
protein. Ectopic expression of this mutant did result in surface
FAS expression, but at lower levels as compared with controls
(Supplemental Fig. 2A).Taken together, these data suggest that haploinsufficiency as-
sociated with extracellular mutations can diminish FAS expression
by inducing NMD, protein instability, and, potentially, abnormal
intracellular trafficking.
FIGURE 2. Cell-surface expression of FAS on
EBV-transformed B cells. A–C, Cells (0.53 106) were
stained for FAS expression using 10 mg/ml PE-con-
jugated anti-CD95 or PE-conjugated isotype-matched
IgG for 30 min at 4˚C. After washing two times with
PBS, 10,000 live cells were analyzed by flow cytom-
etry. FAS expression was quantified using Quanti-
BRITE PE (BD Biosciences). Data were represented
as means 6 SE of three to four separate experiments.
*p , 0.05, **p , 0.01 by Student’s t test for com-
parison with normal cell lines.
4 FAS HAPLOINSUFFICIENCY IN ALPS
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
Clinical and laboratory findings in patients withhaploinsufficient alleles
We next analyzed the impact of the different FAS mutations onclinical and laboratory parameters in ALPS patients. Extracellularmutations are known to result in lower penetrance and variableexpressivity, and these were not readdressed in this study (18, 26).
We evaluated recently described biomarkers associated with FASmutations, including percentage of TCRab+CD42CD82 T cellsas well as levels of soluble FASL, IL-10, IL-18, TNF-a, and vi-tamin B12 (27, 28). No statistically significant differences werenoticed between patients with null extracellular or missense in-tracellular mutations (Supplemental Fig. 3). However, one addi-tional clinical finding that appeared to distinguish the two groupswas the absence of lymphomas in patients with extracellular FASmutations. Among .189 patients from 108 families with FASmutations in the National Institutes of Health cohort, all 21 lym-phomas documented to date occurred in patients with intracellularmutations (29). Additionally, lymphoma occurred in one patientwith a somatic mutation in NRAS (14). This finding suggests thatthe residual apoptotic function observed in haploinsufficiency-associated extracellular mutations may translate clinically notonly into lower penetrance and expressivity but also into a lowerrisk for lymphomagenesis.
Overexpression of FAS in haploinsufficient samples correctsthe apoptotic defect
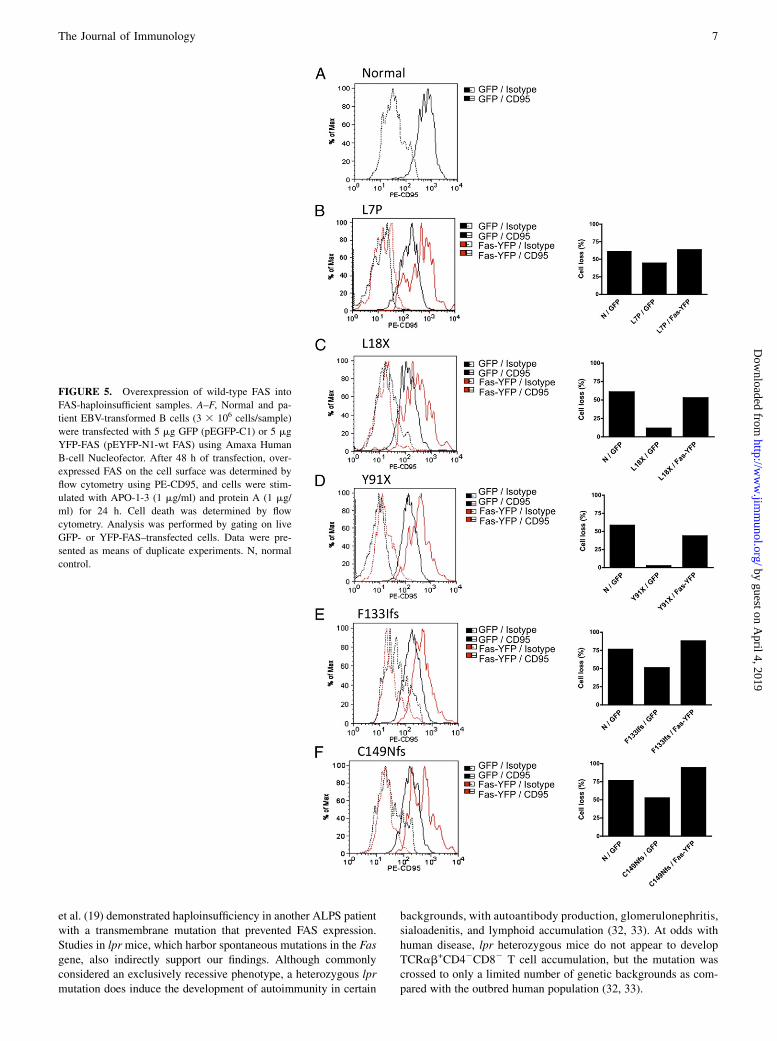
To conclusively demonstrate haploinsufficiency as a diseasemechanism, we transfected YFP-tagged wild-type FAS or an emptyGFP-expressing plasmid into selected EBV-transformed cell linesthat showed reduced FAS expression and reduced sensitivity toFAS-induced cell death. Transfection efficiency ranged from 10–25% (data not shown). Forty-eight hours after transfection, wemeasured cell-surface FAS expression by flow cytometry on live-gated GFP+- or YFP+-transfected cells (Fig. 5). Cell-surface FASexpression was increased after transfection with YFP-FAS, andthis corrected the apoptotic defect, as compared with control GFP-transfected cells and normal controls. These results suggest thatthe level of surface FAS expression is a crucial determinant of thesensitivity to FAS-induced cell death.
DiscussionMissense mutations affecting the DD of the FAS gene are the mostcommon genetic abnormality detected in patients with ALPS anddisrupt the apoptosis pathway by dominant-negative interference(7, 18, 30). This occurs because FAS is present on the cell surfaceas a preassociated homotrimeric receptor, such that in the presenceof a heterozygous mutation, seven out of eight trimers will containat least one mutant copy of the FAS protein, and this preventsadequate signaling (31). However, analysis of our extensive cohortof patients revealed that a significant proportion of ALPS patientsharbor null mutations in the extracellular portion of the FASprotein. These mutations were not expected to have a dominant-negative effect, as no mutant protein expression was anticipated,and alternative mechanisms were explored. We analyzed the im-pact of disease-associated, naturally occurring FAS mutations onthe apoptotic signaling pathway and associated clinical and lab-oratory findings in ALPS patients. This was, to our knowledge, thefirst comprehensive effort to understand the functional con-sequences of a large group of non-DD ALPS-associated FAS mu-tations.Our findings demonstrate that haploinsufficiency is associated
with nonsense or frameshift FAS mutations, primarily located ingene regions encoding for the extracellular portion of the FASprotein. From 11 extracellular mutations associated with a defectin apoptosis, only 3 did not result in decreased FAS expression.Among these is E45X, a truncation that did not reduce surfaceFAS levels to statistical significance and correspondingly did notaffect FAS signaling in any measurable way. It is possible thateven a mild decrease in FAS expression may result in dysfunctionin vivo, although the underlying mechanism for disease in this
FIGURE 3. Sensitivity of EBV-transformed B cells to CD95-induced
cell death. A–C, EBV-transformed B cells from ALPS patients and three
different normal controls were stimulated with APO-1-3 (1 mg/ml) and
protein A (1 mg/ml) for 24 h. Cell death was determined by measuring the
loss of the mitochondrial transmembrane potential using DiOC6 by flow
cytometry. D, Comparison of the degree of apoptotic defect between cells
with extracellular (EC) mutations (including W176X) affecting FAS ex-
pression (n = 9) and intracellular (IC) mutations with normal FAS ex-
pression (n = 16), with numbers normalized to the value in normal cells.
The data in A–C were represented as means 6 SE of three separate
experiments. The data in D were presented as medians and interquartile
ranges, and medians were compared using Mann–Whitney U test. All
results from ALPS patients showed significant reduction in FAS-mediated
cell death compared with normal cell lines.
The Journal of Immunology 5
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
patient remains to be fully understood. The recurrent mutationH111R and the in-frame G66_H111 deletion also did not reduceFAS expression but are predicted to affect the FASL binding site,providing a possible explanation for the clinical and cellularphenotype. Curiously, despite having documented apoptosis defectsfor all mutations tested, no impairment in DISC formation couldbe detected for the extracellular mutations L7P, E45X, H111R,
G66_H111del, and the intracellular I310S. This may reflect a lowsensitivity of the immunoprecipitation assay used in the study tovery mild but still clinically significant functional defects.Previous case reports lend support to the concept that haploin-
sufficiency is a pathogenetic mechanism in ALPS. Vaishnaw et al.(20) suggested that haploinsufficiency was at play in two patientswith a nonsense mutation in the extracellular domain, and Roesler
FIGURE 4. DISC formation in EBV-transformed B cells of ALPS patients. A–C, Cells were stimulated with APO-1-3 (0.5 mg/ml) or isotype control IgG
in the presence of protein A (1 mg/ml) for 20 min. After lysis, cell lysates were incubated with anti-FAS Ab and protein A/G agarose for 2 h. The
immunocomplexes were then subjected to Western blotting using anti-FADD and anti–caspase-8 (CASP8). Caspase-8 band represents cleaved caspase-8
(∼43 kDa). Because FAS size overlaps with IgG H chain, we could not show immunoprecipitated FAS. D, Comparison of the degree of recruitment of
FADD or caspase-8 to the FAS receptor upon stimulation between cells with extracellular mutations affecting FAS expression (EC; n = 9) and intracellular
mutations with normal FAS expression (IC; n = 16). All numbers were normalized to the value in normal cells. Densitometry data (A–C) are presented as
means 6 SE of (n = 3) separate experiments and compared by Student’s t test. The data in D were presented as medians and interquartile ranges, and
medians were compared using Mann–Whitney U test. *p , 0.05, **p , 0.01, ***p , 0.001.
6 FAS HAPLOINSUFFICIENCY IN ALPS
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from
et al. (19) demonstrated haploinsufficiency in another ALPS patientwith a transmembrane mutation that prevented FAS expression.Studies in lprmice, which harbor spontaneous mutations in the Fasgene, also indirectly support our findings. Although commonlyconsidered an exclusively recessive phenotype, a heterozygous lprmutation does induce the development of autoimmunity in certain
backgrounds, with autoantibody production, glomerulonephritis,sialoadenitis, and lymphoid accumulation (32, 33). At odds withhuman disease, lpr heterozygous mice do not appear to developTCRab+CD42CD82 T cell accumulation, but the mutation wascrossed to only a limited number of genetic backgrounds as com-pared with the outbred human population (32, 33).
FIGURE 5. Overexpression of wild-type FAS into
FAS-haploinsufficient samples. A–F, Normal and pa-
tient EBV-transformed B cells (3 3 106 cells/sample)
were transfected with 5 mg GFP (pEGFP-C1) or 5 mg
YFP-FAS (pEYFP-N1-wt FAS) using Amaxa Human
B-cell Nucleofector. After 48 h of transfection, over-
expressed FAS on the cell surface was determined by
flow cytometry using PE-CD95, and cells were stim-
ulated with APO-1-3 (1 mg/ml) and protein A (1 mg/
ml) for 24 h. Cell death was determined by flow
cytometry. Analysis was performed by gating on live
GFP- or YFP-FAS–transfected cells. Data were pre-
sented as means of duplicate experiments. N, normal
control.
The Journal of Immunology 7
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

Haploinsufficiency has been commonly reported for genes in-volved in nonlinear signaling processes, such as DNA transcriptionand assembly of macromolecular complexes (34). Accordingly,productive FAS signaling requires the assembly of large signalingplatforms on the cell surface (35). This is thought to be required toconcentrate procaspase-8 in close proximity allowing for self-cleavage, activation, and propagation of the apoptotic signal (36,37). Thus, lower amounts of surface FAS may prevent the for-mation of a critical threshold of these complexes resulting ina disrupted apoptotic signal. Indeed, we showed that DISC for-mation is adversely affected in most of these patients, although toa lesser extent than in patients with intracellular mutations.The milder nature of the apoptotic defect seen in haploinsuf-
ficiency-associated FAS mutations can be molecularly explainedby the fact that the intact allele will allow expression of normal FASproteins on the cell surface, which will presumably preassociatethrough their preligand assembly domain and form functionaltrimers, albeit at levels ∼50% of that seen in healthy controls. Incontrast, missense intracellular mutations with dominant-negativeeffect will result in the incorporation of mutant proteins in sevenout of eight FAS trimers on the cells surface, rendering themnonfunctional.The residual FAS function seen in ALPS patients with haplo-
insufficiency-associated extracellular mutations can potentiallyexplain the incomplete clinical penetrance and variable expressivity,typical also of other diseases associated with haploinsufficientalleles (18, 20, 30, 34). More strikingly, the absence of lymphomacases to date in patients with FAS extracellular (EC) mutationssuggests that this level of residual FAS function may be sufficientfor tumor suppression, placing these patients at a lower cancer risk(3, 29). However, further long-term follow-up of ALPS patients atour and other centers will be necessary to fully substantiate thisinitial observation.It is plausible that modifying factors, including genetic, epi-
genetic, or environmental, may make a larger contribution to dis-ease phenotype in patients with EC mutations as compared withmore severe dominant-negative mutations. Along these lines,Magerus-Chatinet et al. (38) demonstrated in recent work that agroup of ALPS patients with low-penetrance EC mutations pres-ent with a somatic event in the second FAS allele in double-negative T cells, and this was associated with the presence ofclinical symptoms.Lastly, one can also speculate that the definition of haplo-
insufficiency as a common disease mechanism in ALPS makes genetherapy a future possibility for this group of patients based on thefinding that increasing FAS cell-surface expression can re-establishnormal apoptosis. In contrast, the current approach of viral vectorgene insertion would likely not be effective in correcting the apoptoticdefect associated with dominant-negative FAS mutations.
AcknowledgmentsWe thank the patients and their families for contributions to the study. We
also thank Richard Siegel for the FAS-YFP plasmid construct.
DisclosuresThe authors have no financial conflicts of interest.
References1. Sneller, M. C., J. Wang, J. K. Dale, W. Strober, L. A. Middelton, Y. Choi,
T. A. Fleisher, M. S. Lim, E. S. Jaffe, J. M. Puck, et al. 1997. Clincal, immu-nologic, and genetic features of an autoimmune lymphoproliferative syndromeassociated with abnormal lymphocyte apoptosis. Blood 89: 1341–1348.
2. Le Deist, F., J. F. Emile, F. Rieux-Laucat, M. Benkerrou, I. Roberts, N. Brousse,and A. Fischer. 1996. Clinical, immunological, and pathological consequences ofFas-deficient conditions. Lancet 348: 719–723.
3. Straus, S. E., E. S. Jaffe, J. M. Puck, J. K. Dale, K. B. Elkon, A. Rosen-Wolff,A. M. Peters, M. C. Sneller, C. W. Hallahan, J. Wang, et al. 2001. The de-velopment of lymphomas in families with autoimmune lymphoproliferativesyndrome with germline Fas mutations and defective lymphocyte apoptosis.Blood 98: 194–200.
4. Sneller, M. C., S. E. Straus, E. S. Jaffe, J. S. Jaffe, T. A. Fleisher, M. Stetler-Stevenson, and W. Strober. 1992. A novel lymphoproliferative/autoimmunesyndrome resembling murine lpr/gld disease. J. Clin. Invest. 90: 334–341.
5. Bleesing, J. J., M. R. Brown, S. E. Straus, J. K. Dale, R. M. Siegel, M. Johnson,M. J. Lenardo, J. M. Puck, and T. A. Fleisher. 2001. Immunophenotypic profilesin families with autoimmune lymphoproliferative syndrome. Blood 98: 2466–2473.
6. Rieux-Laucat, F., F. Le Deist, C. Hivroz, I. A. Roberts, K. M. Debatin,A. Fischer, and J. P. de Villartay. 1995. Mutations in Fas associated with humanlymphoproliferative syndrome and autoimmunity. Science 268: 1347–1349.
7. Fisher, G. H., F. J. Rosenberg, S. E. Straus, J. K. Dale, L. A. Middleton,A. Y. Lin, W. Strober, M. J. Lenardo, and J. M. Puck. 1995. Dominant interferingFas gene mutations impair apoptosis in a human autoimmune lymphoprolifer-ative syndrome. Cell 81: 935–946.
8. Wang, J., L. Zheng, A. Lobito, F. K. Chan, J. Dale, M. Sneller, X. Yao,J. M. Puck, S. E. Straus, and M. J. Lenardo. 1999. Inherited human Caspase 10mutations underlie defective lymphocyte and dendritic cell apoptosis in auto-immune lymphoproliferative syndrome type II. Cell 98: 47–58.
9. Wu, J., J. Wilson, J. He, L. Xiang, P. H. Schur, and J. D. Mountz. 1996. Fasligand mutation in a patient with systemic lupus erythematosus and lympho-proliferative disease. J. Clin. Invest. 98: 1107–1113.
10. Del-Rey, M., J. Ruiz-Contreras, A. Bosque, S. Calleja, J. Gomez-Rial,E. Roldan, P. Morales, A. Serrano, A. Anel, E. Paz-Artal, and L. M. Allende.2006. A homozygous Fas ligand gene mutation in a patient causes a new type ofautoimmune lymphoproliferative syndrome. Blood 108: 1306–1312.
11. Bi, L. L., G. Pan, T. P. Atkinson, L. Zheng, J. K. Dale, C. Makris, V. Reddy,J. M. McDonald, R. M. Siegel, J. M. Puck, et al. 2007. Dominant inhibition of Fasligand-mediated apoptosis due to a heterozygous mutation associated with auto-immune lymphoproliferative syndrome (ALPS) Type Ib. BMC Med. Genet. 8: 41.
12. Oliveira, J. B., J. J. Bleesing, U. Dianzani, T. A. Fleisher, E. S. Jaffe,M. J. Lenardo, F. Rieux-Laucat, R. M. Siegel, H. C. Su, D. T. Teachey, andV. K. Rao. 2010. Revised diagnostic criteria and classification for the autoim-mune lymphoproliferative syndrome (ALPS): report from the 2009 NIH In-ternational Workshop. Blood 116: e35–e40.
13. Chun, H. J., L. Zheng, M. Ahmad, J. Wang, C. K. Speirs, R. M. Siegel,J. K. Dale, J. Puck, J. Davis, C. G. Hall, et al. 2002. Pleiotropic defects inlymphocyte activation caused by caspase-8 mutations lead to human immuno-deficiency. Nature 419: 395–399.
14. Oliveira, J. B., N. Bidere, J. E. Niemela, L. Zheng, K. Sakai, C. P. Nix,R. L. Danner, J. Barb, P. J. Munson, J. M. Puck, et al. 2007. NRAS mutationcauses a human autoimmune lymphoproliferative syndrome. Proc. Natl. Acad.Sci. USA 104: 8953–8958.
15. Niemela, J. E., L. Lu, T. A. Fleisher, J. Davis, I. Caminha, M. Natter, L. A. Beer,K. C. Dowdell, S. Pittaluga, M. Raffeld, V. K. Rao, and J. B. Oliveira. 2011.Somatic KRAS mutations associated with a human non-malignant syndrome ofautoimmunity and abnormal leukocyte homeostasis. Blood 117: 2883–2886.
16. Behrmann, I., H. Walczak, and P. H. Krammer. 1994. Structure of the humanAPO-1 gene. Eur. J. Immunol. 24: 3057–3062.
17. Chinnaiyan, A. M., K. O’Rourke, M. Tewari, and V. M. Dixit. 1995. FADD,a novel death domain-containing protein, interacts with the death domain of Fasand initiates apoptosis. Cell 81: 505–512.
18. Jackson, C. E., R. E. Fischer, A. P. Hsu, S. M. Anderson, Y. Choi, J. Wang,J. K. Dale, T. A. Fleisher, L. A. Middelton, M. C. Sneller, et al. 1999. Auto-immune lymphoproliferative syndrome with defective Fas: genotype influencespenetrance. Am. J. Hum. Genet. 64: 1002–1014.
19. Roesler, J., J. M. Izquierdo, M. Ryser, A. Rosen-Wolff, M. Gahr, J. Valcarcel,M. J. Lenardo, and L. Zheng. 2005. Haploinsufficiency, rather than the effect ofan excessive production of soluble CD95 (CD95DeltaTM), is the basis for ALPSIa in a family with duplicated 39 splice site AG in CD95 intron 5 on one allele.Blood 106: 1652–1659.
20. Vaishnaw, A. K., J. R. Orlinick, J. L. Chu, P. H. Krammer, M. V. Chao, andK. B. Elkon. 1999. The molecular basis for apoptotic defects in patients withCD95 (Fas/Apo-1) mutations. J. Clin. Invest. 103: 355–363.
21. Niemela, J. E., A. P. Hsu, T. A. Fleisher, and J. M. Puck. 2006. Single nucleotidepolymorphisms in the apoptosis receptor gene TNFRSF6. Mol. Cell. Probes 20:21–26.
22. Caminha, I., T. A. Fleisher, R. L. Hornung, J. K. Dale, J. E. Niemela, S. Price,J. Davis, K. Perkins, K. C. Dowdell, M. R. Brown, V. K. Rao, and J. B. Oliveira.Using biomarkers to predict the presence of FAS mutations in patients withfeatures of the autoimmune lymphoproliferative syndrome. J. Allergy Clin.Immunol. 125: 946–949.
23. Chang, Y. F., J. S. Imam, and M. F. Wilkinson. 2007. The nonsense-mediateddecay RNA surveillance pathway. Annu. Rev. Biochem. 76: 51–74.
24. Siggaard, C., S. Rittig, T. J. Corydon, P. H. Andreasen, T. G. Jensen,B. S. Andresen, G. L. Robertson, N. Gregersen, L. Bolund, and E. B. Pedersen.1999. Clinical and molecular evidence of abnormal processing and trafficking ofthe vasopressin preprohormone in a large kindred with familial neurohypophy-seal diabetes insipidus due to a signal peptide mutation. J. Clin. Endocrinol.Metab. 84: 2933–2941.
25. Birney, E., J. A. Stamatoyannopoulos, A. Dutta, R. Guigo, T. R. Gingeras,E. H. Margulies, Z. Weng, M. Snyder, E. T. Dermitzakis, R. E. Thurman, et al;ENCODE Project Consortium; NISC Comparative Sequencing Program; Baylor
8 FAS HAPLOINSUFFICIENCY IN ALPS
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

College of Medicine Human Genome Sequencing Center; Washington Univer-sity Genome Sequencing Center; Broad Institute; Children’s Hospital OaklandResearch Institute. 2007. Identification and analysis of functional elements in 1%of the human genome by the ENCODE pilot project. Nature 447: 799–816.
26. Rieux-Laucat, F., S. Blachere, S. Danielan, J. P. De Villartay, M. Oleastro,E. Solary, B. Bader-Meunier, P. Arkwright, C. Pondare, F. Bernaudin, et al. 1999.Lymphoproliferative syndrome with autoimmunity: A possible genetic basis fordominant expression of the clinical manifestations. Blood 94: 2575–2582.
27. Caminha, I., T. A. Fleisher, R. L. Hornung, J. K. Dale, J. E. Niemela, S. Price,J. Davis, K. Perkins, K. C. Dowdell, M. R. Brown, V. K. Rao, and J. B. Oliveira.2010. Using biomarkers to predict the presence of FAS mutations in patientswith features of the autoimmune lymphoproliferative syndrome. J. Allergy Clin.Immunol. 125: 946–949.
28. Magerus-Chatinet, A., M. C. Stolzenberg, M. S. Loffredo, B. Neven,C. Schaffner, N. Ducrot, P. D. Arkwright, B. Bader-Meunier, J. Barbot,S. Blanche, et al. 2009. FAS-L, IL-10, and double-negative CD4-CD8-TCRalpha/beta+ T cells are reliable markers of ALPS associated with FAS loss offunction. Blood 113: 3027–3030.
29. Rao, V. K., S. Price, J. Davis, K. Perkins, F. Gill, S. Pittaluga, T. Fleisher, andE. Jaffe. 2010. Development of lymphomas in families with autoimmune lym-phoproliferative syndrome (ALPS). Pediatr. Blood Cancer 54: 813 (Abstr. 171).
30. de Villartay, J. P., F. Rieux-Laucat, A. Fischer, and F. Le Deist. 1998. Clinicaleffects of mutations to CD95 (Fas): relevance to autoimmunity? Springer Semin.Immunopathol. 19: 301–310.
31. Siegel, R. M., J. K. Frederiksen, D. A. Zacharias, F. K. Chan, M. Johnson,D. Lynch, R. Y. Tsien, and M. J. Lenardo. 2000. Fas preassociation required for
apoptosis signaling and dominant inhibition by pathogenic mutations. Science288: 2354–2357.
32. Carlsten, H., A. Tarkowski, R. Jonsson, and L. A. Nilsson. 1990. Expression ofheterozygous lpr gene in MRL mice. II. Acceleration of glomerulonephritis,sialadenitis, and autoantibody production. Scand. J. Immunol. 32: 21–28.
33. Ogata, Y., M. Kimura, K. Shimada, T. Wakabayashi, H. Onoda, T. Katagiri, andA. Matsuzawa. 1993. Distinctive expression of lprcg in the heterozygous state ondifferent genetic backgrounds. Cell. Immunol. 148: 91–102.
34. Veitia, R. A., and J. A. Birchler. 2010. Dominance and gene dosage balance inhealth and disease: why levels matter! J. Pathol. 220: 174–185.
35. Siegel, R. M., J. R. Muppidi, M. Sarker, A. Lobito, M. Jen, D. Martin,S. E. Straus, and M. J. Lenardo. 2004. SPOTS: signaling protein oligomerictransduction structures are early mediators of death receptor-induced apoptosisat the plasma membrane. J. Cell Biol. 167: 735–744.
36. Muzio, M., A. M. Chinnaiyan, F. C. Kischkel, K. O’Rourke, A. Shevchenko,J. Ni, C. Scaffidi, J. D. Bretz, M. Zhang, R. Gentz, et al. 1996. FLICE, a novelFADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death—inducing signaling complex. Cell 85: 817–827.
37. Muzio, M., B. R. Stockwell, H. R. Stennicke, G. S. Salvesen, and V. M. Dixit.1998. An induced proximity model for caspase-8 activation. J. Biol. Chem. 273:2926–2930.
38. Magerus-Chatinet, A., B. Neven, M. C. Stolzenberg, C. Daussy, P. D. Arkwright,N. Lanzarotti, C. Schaffner, S. Cluet-Dennetiere, F. Haerynck, G. Michel, et al.2011. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans asa consequence of genetic defect accumulation. J. Clin. Invest. 121: 106–112.
The Journal of Immunology 9
by guest on April 4, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from









![1 Pensions (FAS 87); Post Retirement Benefits (FAS 106); Post Employment Benefits (FAS 112); Disclosure about Pensions, etc. (FAS 132 [R]) – amendment](https://img.pdfslide.us/doc/110x75/56649d1f5503460f949f3b1c/1-pensions-fas-87-post-retirement-benefits-fas-106-post-employment-benefits.jpg)



















