Embed Size (px)
Citation preview

Eur Respir J 1990, 3, 441-446
Familial elevation of serum angiotensin converting enzyme activity
M. Luisetti*, M. Martinetti**, M. Cuccia***, J.M. Dugoujon+, V. De Rose++, V. Peona*, E. Pozzi++, C. Grassi*
Familial elevation of serum angiotensin converting enzyme activity. M. Luisetti, M. Martinetti, M. Cuccia, J.M. Dugoujon, V. De Rose, V. Peona, E. Pozzi, C. Grassi.
* lstituto di Tisiologia e Malattie dell' Apparato Respiratorio, IRCCS Policlinico S. Matteo, Universita di Pavia, Italy.
ABSTRACT: A clustering of high levels of serum angiotensin converting enzyme ( S-ACE) was found in an Italian family. The elevation affected five subjects, two of whom were completely healthy and free from known causes of S-ACE Increase. The values of S-ACE In hyperACEmic subjects exceeded the values found in normal relatives severalfold. HyperACEmla seemed to be Inherited as an autosomal dominant trait. Immunogenetic studies were performed, but we did not find a genetic marker for this condition. The S-ACE activity was Inhibited in vitro by edetlc acid (EDT A) and SQ 14,225 (captopril). The S-ACE activity was also determined after 1:8 dilution and dialysis against saline of sera. From these experiments we deduced that Lieberman's intrinsic ACE Inhibitor was lacking in the hyperACEmic sera. In the presence of remarkable S-ACE increase, a congenital elevation of S-ACE should be considered and it would be useful to perform a familial investigation.
*"' Laboratorio HLA, Centro Trasfusionale A VIS, Pavia, Italy.
**"' Dipartimento di Genetica e Microbiologia, Univemta di Pavia, Italy.
+ Centre de Recherches sur le Polymorphisme Gen&ique des Populations Humaines, CRPG-CNRS, Toulouse, France.
++ Cattedra di Fisiopatologia Respiratoria, Universita di Torino, Italy.
Correspondence: M. Luisetti, lstituto di Tisiologia e Mal. App. Resp., IRCCS San Matteo, via Taramelli 5, 27100 Pavia, Italy.
Keywords : Alpha -antitrypsin typing; Gm, Km immunoglobulin ahotypes; HLA polymoTJ?hisms; interstitial lung disease: intrinsic serum angtotensin converting enzyme (S-ACE) inhibitor.
Eur Respir J., 1990, 3, 441--446.
Elevation of serum angiotensin converting enzyme (S-ACE) activity is associated with active sarcoidosis [1], and with a number of pulmonary and extra-pulmonary diseases, such as pneumoconiosis [2], Gaucher's disease [3], diabetes mellitus [4], liver cirrhosis [5] and leprosy [6]. In sarcoidosis in particular, serialS-ACE determinations are helpful in the diagnosis and assessment of disease activity [7], so that S-ACE assay is currently required on the diagnostic procedures of interstitial lung disease (ILD). However, in sarcoidosis and in other known causes of S-ACE elevation, S-ACE activity rarely exceeds twofold the mean value of healthy controls [7].
In normal subjects there is no difference in S-ACE activity between sexes and races, whilst it seems to be slightly elevated in children up to 15 yrs with respect to adults [8]. When a large series of healthy controls are considered, scattered increases in S-ACE values are sometimes observed [9].
In 1985, FunsAWA et al. [10] first reported extremely elevated S-ACE levels in four members of a Japanese family not affected by known causes of S-ACE elevation (hyperACEmia). We report a clustering of hyperACEmia in an Italian family, including immunogenetic and in vitro S-ACE inhibition studies.
There is evidence from recent studies [11] of a strong correlation between immunoglobulin A (IgA) serum
Received: July, 1989; accepted after revision, December 18, 1989.
deficiency, immunoglobulin serum levels [12] and particular histocompatibility locus antigen (HLA) supratypes. Moreover, the presence of immune suppression gene(s) closely linked to the HLA-D subregion [13] has recently been postulated. For these reasons it seemed interesting to investigate the epistatic interaction of major histocompatibility complex (MHC) genes in the control of serum levels of ACE.
Furthermore, a correlation has recently been found between congenital elevation of S-ACE levels and a rare variant (Z) of alpha1-antitrypsin [9]. Genes coding for alpha
1-antitrypsin allotypes have been mapped on the
lowest end of chromosome 14, close to the immunoglobulin heavy chain gene cluster. It would not be a coincidence if an unusual Gm haplotype occurred in the relatives with hyperACEmia. For this reason we extended our genetic study of this rare condition to the characterization of the Gm polymorphism and the alpha
1-
antitrypsin phenotype of the whole family under study.
It has previously been reported that human serum may contain an intrinsic ACE inhibitor [14]. The presence of the inhibitor is particularly frequent in sera with elevated ACE levels and its effect can be eliminated by serum dilution [15]. For Lhis reason, a study of intrinsic S-ACE inhibitor is included in this report.
442 M. LUISETII ET AL.
Subjects and methods
S-ACE determination
S-ACE activity was determined by calorimetric assay (ACE Calor, Fujirebio Inc., Japan), using the substrate p-hydroxyhippuryl-1-histidyl-1-leucine [16]. Since it has previously been described that elevated S-ACE levels obtained with one method are not necessarily confirmed when other methods are used [17], the calorimetric S-ACE values were checked by radiometric assay (substrate p-[3H]-benzoylglycylglycylglycine) [18].
Proband history
The proband, a 61 yr old female, was first admitted to our department in October, 1986. Since 1984 she had complained of cough and exertional dyspnoea, with progressive worsening of symptoms. The clinical characteristics of the proband are summarized in table 1. Scalene lymph node and liver biopsies were negative and the patient resolutely refused other biopsy procedures. The S-ACE activity was extremely high (103.95 U·ml-1), pathological increases over 45 U·ml-1
being rare in our laboratory (normal values: mean±so 15.53±5.03).
Table 1. - Clinical characteristics of the proband
Initials Sex Age Chest X-ray Other clinical yrs pattern signs
R.P. ~ 61 lLD (reticula- Liver steatosis nodular pattern) (hepatic biopsy) with hilar lymph node enlargement
Family history
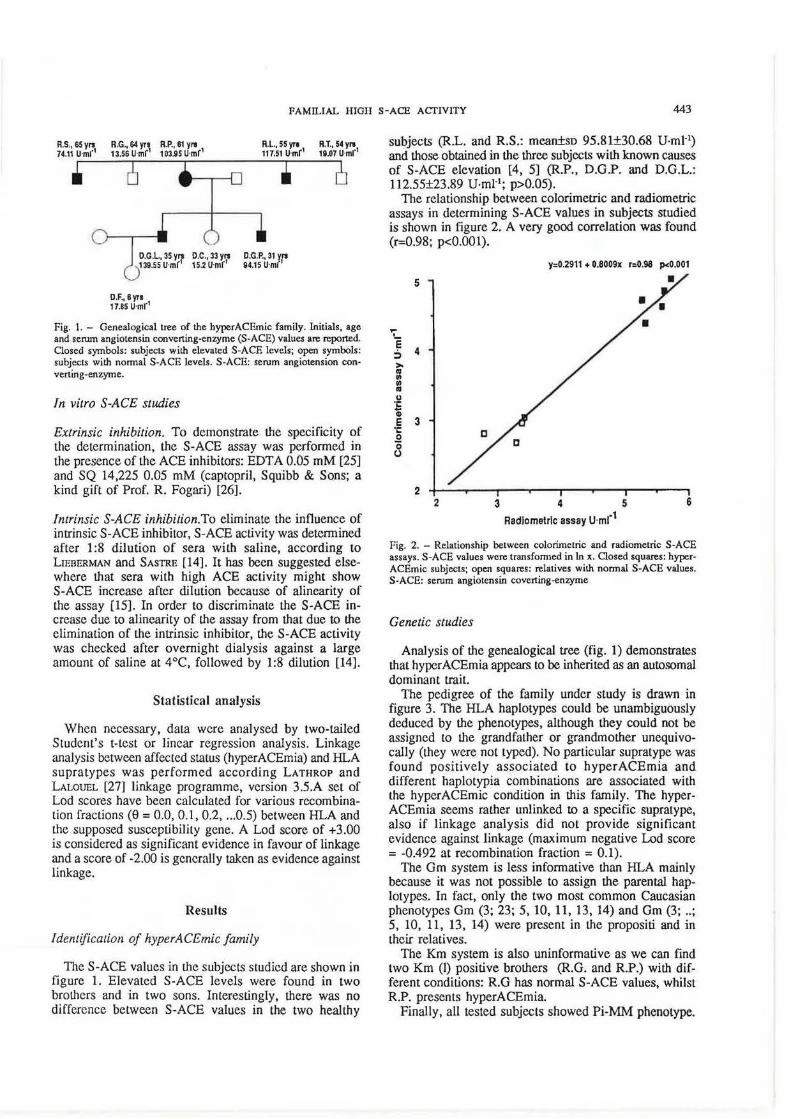
The genealogical tree is shown in figure L The proband family consists of four siblings (four brothers, none of whom had offspring), three offspring (2 sons and 1 daughter) and one grand-daughter (daughter of a son). The four brothers were not affected by known causes of S-ACE elevation: their blood pressure was normal, except for R.S. who showed a mild hypertension. The daughter of the proband (D.C.) was healthy. A son (D.G.P.) was affected by chronic aggressive hepatitis and it was subsequently found that he was a human immunodeficiency virus (HIV)-infected drug addict: therefore, he was no longer studied. D.G.L., the second son, was affected by diabetes mellitus; his daughter (D.F.) was healthy.
Genetic studies
Alpha1-antitrypsin phenotyping (Pi-typing) was deter
mined by isoelectric focusing of sera [19]. HLA class I and 11 polymorphisms were identified by a standard microlymphocytotoxicity test on T and B enriched lymphocyte suspensions from peripheral blood samples [20]. Specificities were assigned according to the lOth International Histocompatibility Workshop definitions [21]. Complement typing was performed: six subjects were analysed for Bf, C4A and C4B polymorphisms;
Respiratory function
tests
FVC 60* TLC 56 FEV
1 48
DLCO 45 Pao2 10.2** Paco
2 5.4**
Ga67-scan
positive
BAL fmdings
150x1Q3-ml·11
M080#-Ly 18 Neu 2
S-ACE value U·ml-1
103.95
*: %predicted; 1: total count·ml-1 recovered fluid; #- : differential count(%); **: kPa; M0: macrophages; Ly: lymphocytes; Neu: neutrophils: BAL: bronchoalveolar lavage: S-ACE: serum angiotensin converting enzyme; lLD: interstitial lung disease; FVC: forced vital capacity; TLC: total lung capacity; FEV
1: forced expiratory volume in one second; DLCo: single breath diffusing
capacity for carbon monoxide; Pao2
and Paco2
: arterial oxygen and carbon dioxide tension, respectively.
The patient was suspected of having sarcoidosis and started receiving prednisone 40 mg daily, orally, in November, 1986. The prednisone was continued until August, 1987, with progressive dose tapering.
At the moment of prednisone withdrawal, the chest Xray was unchanged and the respiratory function tests only slightly improved. Surprisingly, S-ACE activity did not decrease during the prednisone administration. The behaviour of S-ACE in the proband aroused the suspicion of a lack of connection with the lLD and/or the hepatic disease and, therefore, we decided to study the other members of the family.
one individual was only defined for Bf. Bf alleles were determined by immunofixation electrophoresis using the standard method proposed by ALPER et al. [22]. C4 allotypes were defined by immunofixation electrophoresis of neuramidase treated serum samples according to the method of AwnEH and ALPER [23]. Gm and Km typing was performed: all subjects were typed for G 1m (1, 2, 3, 17), G2m (23), G3m (5, 10, 11, 13, 14, 21, 28) and Km (1) allotypes, respectively located on IgGl, IgG2, IgG3 subclasses and on K light chain. The standard haemagglutination inhibition assay was used with reagents described elsewhere [24].
FAMILIAL HIGH S-ACE ACTIVITY 443
R.G., 64 yra R.P., 81 yrt 13.56 U·mr' 103.95 U·mf1
O.F., 6yr1 17.85 U·mf1
D.C., 33 yrt 15.2U·mr'
R.L., 55 yre R.T., 54 yre 117.51 U·mr' 1U7 U·mf1
O.G.P., 31 yrt 94.15 U·mf1
Fig. 1. - Genealogical tree of the hyperACEmic family. Initials, age and serum angiotensin converting-enzyme (S-ACE) values are reported. Closed symbols: subjects with elevated S-ACE levels; open symbols: subjects with normal S -ACE levels. S -ACE: serum angiotension converting-enzyme.
In vitro S-AGE studies
Extrinsic inhibition. To demonstrate the specificity of the determination, the S-ACE assay was performed in the presence of the ACE inhibitors: EDT A 0.05 mM [25] and SQ 14,225 0.05 mM (captopril, Sqtiibb & Sons; a kind gift of Prof. R. Fogari) [26].
Intrinsic S-ACE inhibition.To eliminate the influence of intrinsic S-ACE inhibitor, S-ACE activity was determined after 1:8 dilution of sera with saline, according to LIEBERMAN and SASTRE (14). It has been Suggested elsewhere that sera with high ACE activity might show S-ACE increase after dilution because of alinearity of the assay (15]. In order to discriminate the S-ACE increase due to alinearity of the assay from that due to the elimination of the intrinsic inhibitor, the S-ACE activity was checked after overnight dialysis against a large amount of saline at 4°C, followed by 1:8 dilution [14].
Statistical analysis
When necessary, data were analysed by two-tailed Student's Hest or linear regression analysis. Linkage analysis between affected status (hyperACEmia) and HLA supratypes was performed according LATHROP and LALOUEL [27] linkage programme, version 3.5.A set of Lod scores have been calculated for various recombination fractions (e = 0.0, 0.1, 0.2, ... 0.5) between HLA and the supposed susceptibility gene. A Lod score of +3.00 is considered as significant evidence in favour of linkage and a score of -2.00 is generally taken as evidence against linkage.
Results
Identification of hyperACEmic family
The S-ACE values in the subjects studied are shown in figure 1. Elevated S-ACE levels were found in two brothers and in two sons. Interestingly, there was no difference between S-ACE values in the two healthy
subjects (R.L. and R.S.: mean±so 95.81±30.68 U·mJ·1)
and those obtained in the three subjects with known causes of S-ACE elevation [4, 5] (R.P., D.G.P. and D.G.L.: 112.55±23.89 U.mJ-1; p>0.05).
The relationship between calorimetric and radiometric assays in determining S-ACE values in subjects studied is shown in figure 2. A very good correlation was found (r=0.98; p<0.001).
y:0.2911 + 0.8009x r:0.98 pc0.001
5
';' e 4 :,
>-
= Ill Ill
E Cl)
E 3 '1: 0 0 0
2 2 3 4 5 6
Radiometric assay U·mr1
Fig. 2. - Relationship between colorimetric and radiometric S -ACE assays. S·ACE values were transformed in ln x. Closed squares: hyperACEmic subjects; open squares: relatives with normal S-ACE values. S-ACE: serum angiotensin coverting-enzyme
Genetic studies
Analysis of the genealogical tree (fig. 1) demonstrates that hyper ACEmia appears to be inherited as an autosomal dominant trait.
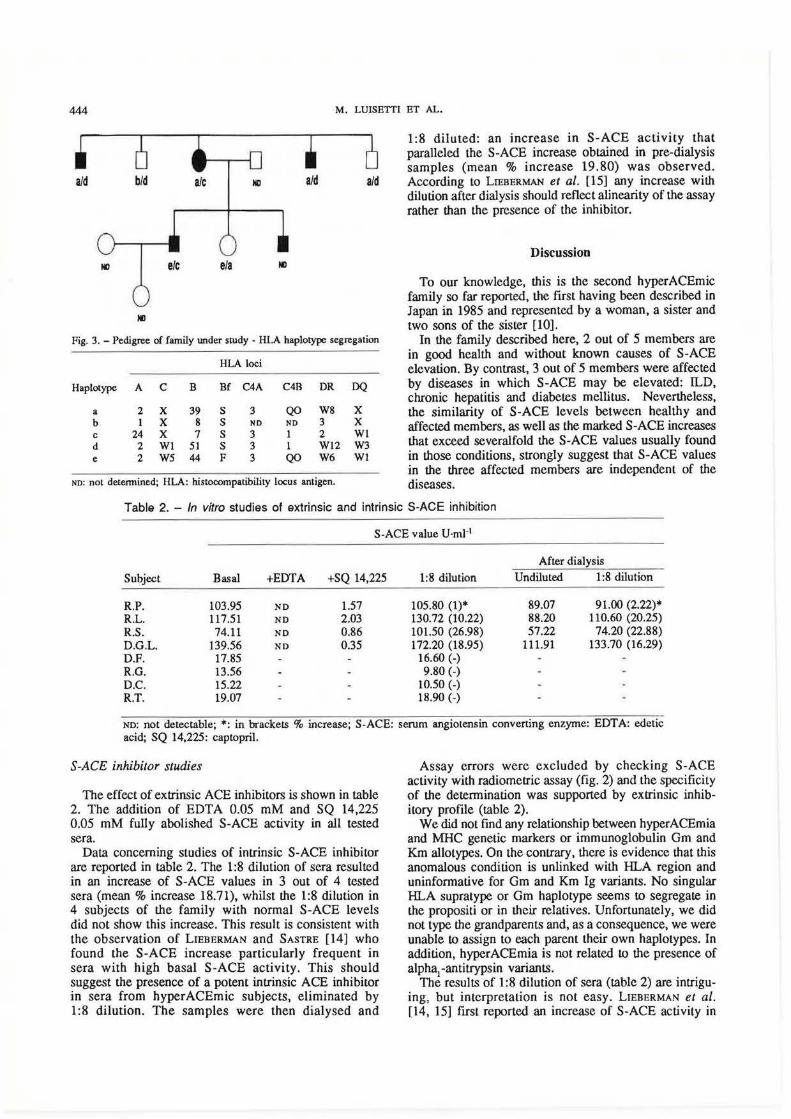
The pedigree of the family under study is drawn in figure 3. The HLA haplotypes could be unambiguously deduced by the phenotypes, although they could not be assigned to the grandfather or grandmother unequivocally (they were not typed). No particular supratype was found positively associated to hyperACEmia and different haplotypia combinations are associated with the hyperACEmic condition in this family. The hyperACEmia seems rather unlinked to a specific supratype, also if linkage analysis did not provide significant evidence against linkage (maximum negative Lod score = -0.492 at recombination fraction = 0.1).
The Gm system is less informative than HLA mainly because it was not possible to assign the parental haplotypes. In fact, only the two most common Caucasian phenotypes Gm (3; 23; 5, 10, 11, 13, 14) and Gm (3; .. ; 5, 10, 11, 13, 14) were present in the propositi and in their relatives.
The Km system is also uninformative as we can find two Km (I) positive brothers (R.G. and R.P.) with different conditions: R.G has normal S-ACE values, whilst R.P. presents hyperACEmia.
Finally, all tested subjects showed Pi-MM phenotype.
444 M. LUISETII ET AL.
aid b/d ale ND aid aid
NO ere eta
NO
Fig. 3. -Pedigree of family under study - HLA haplotype segregation
HLA loci
Haplotype A c B Bf C4A C4B DR DQ
a 2 X 39 s 3 QO W8 X b 1 X 8 s NO NO 3 X c 24 X 7 s 3 1 2 Wl d 2 Wl 51 s 3 1 Wl2 W3 e 2 W5 44 F 3 QO W6 Wl
NO: nol determined; HLA: histocompatibility locus antigen.
1:8 diluted: an increase in S-ACE acttvtty that paralleled the S-ACE increase obtained in pre-dialysis samples (mean % increase 19.80) was observed. According to LIEBERMAN et al. [15] any increase with dilution after dialysis should reflect alinearity of the assay rather than the presence of the inhibitor.
Discussion
To our knowledge, this is the second hyperACEmic family so far reported, the first having been described in Japan in 1985 and represented by a woman, a sister and two sons of the sister [10].
In the family described here, 2 out of 5 members are in good health and without known causes of S-ACE elevation. By contrast, 3 out of 5 members were affected by diseases in which S-ACE may be elevated: ILD, chronic hepatitis and diabetes mellitus. Nevertheless, the similarity of S-ACE levels between healthy and affected members, as well as the marked S-ACE increases that exceed severalfold the S-ACE values usually found in those conditions, strongly suggest that S-ACE values in the three affected members are independent of the diseases.
Table 2. - In vitro studies of extrinsic and intrinsic S-ACE inhibition
S-ACE value U·ml·1
After dialysis
Subject Basal +EDTA +SQ 14,225 1:8 dilution Undiluted 1:8 dilution
R.P. 103.95 NO 1.57 105.80 (1)* 89.07 91.00 (2.22)* R.L. 117.51 NO 2.03 130.72 (10.22) 88.20 110.60 (20.25) R.S. 74.11 NO 0.86 101.50 (26.98) 57.22 74.20 (22.88) D.G.L. 139.56 NO 0.35 172.20 (18.95) 111.91 133.70 (16.29) D.F. 17.85 16.60 (-) R.G. 13.56 9.80 (-) D.C. 15.22 10.50 (-) R.T. 19.07 18.90 (-)
No: not detectable; •: in brackets % increase; S-ACE: serum angiotensin convening enzyme: EDT A: edetic acid; SQ 14,225: captopril.
S-ACE inhibitor studies
The effect of extrinsic ACE inhibitors is shown in table 2. The addition of EDTA 0.05 mM and SQ 14,225 0.05 mM fully abolished S-ACE activity in all tested sera.
Data concerning studies of intrinsic S-ACE inhibitor are reported in table 2. The 1:8 dilution of sera resulted in an increase of S-ACE values in 3 out of 4 tested sera (mean % increase 18.71), whilst the 1:8 dilution in 4 subjects of the family with normal S-ACE levels did not show this increase. This result is consistent with the observation of LIEBERMAN and SASTRE [14) who found the S-ACE increase particularly frequent in sera with high basal S-ACE activity. This should suggest the presence of a potent intrinsic ACE inhibitor in sera from hyperACEmic subjects, eliminated by 1:8 dilution. The samples were then dialysed and
Assay errors were excluded by checking S-ACE activity with radiometric assay (fig. 2) and the specificity of the determination was supported by extrinsic inhibitory profile (table 2).
We did not find any relationship between hyperACEmia and MHC genetic markers or immunoglobulin Gm and Km allotypes. On the contrary, there is evidence that this anomalous condition is unlinked with HLA region and uninformative for Gm and Km Ig variants. No singular HLA supratype or Gm haplotype seems to segregate in the propositi or in their relatives. Unfortunately, we did not type the grandparents and, as a consequence, we were unable to assign to each parent their own haplotypes. In addition, hyperACEmia is not related to the presence of alpha
1-antitrypsin variants.
The results of 1:8 dilution of sera (table 2) are intriguing, but interpretation is not easy. LIEBERMAN et al. [14, 15] first reported an increase of S-ACE activity in

FAMILIAL HIGH S-ACE ACfiVITY 445
25.4% of 280 sera tested after 1:8 dilution of samples. This increase was particularly frequent (47%) in sera with higher S-ACE values (> 35 U.ml·1, spectrophotometric assay). The method of LIEBERMAN [1] requires a 1:1 ratio of serum:substrate solution.
According to LlEBERMAN and SASTRE [14] when sera with a significant elevation after 1:8 dilution by the spectrophotometric assay are investigated by the colorimetric assay (that requires a I :6 ratio of serum:substrate solution), the further 1:8 dilution resulted in minimal increase of S-ACE activity (1 %). We deduce that the increase that was found in 3 out of 4 hyperACEmic sera is noteworthy, particularly considering that an overall 1: 14 dilution was performed.
LIEBERMAN and SASTRE (14] ascribed the post-dilutional increase to a circulating ACE inhibitor, eliminated by 1:8 dilution. The nature and role of the inhibitor are uncertain, but it seems to possess a molecular weight (MW) over 50 kDa and to be labile at 60°C. A peculiar feature of Lieberman 's inhibitor is the irreversible inhibition after dialysis [14]: the further 1:8 dilution of the sample does not produce any increase inS-ACE activity, if the pre-dialysis increase was really due to the inhibitor's effect. The authors' explanation of this phenomenon is a continued irreversible inhibition, rather than a loss of an inhibitor by dialysis [14]. In our samples (table 2), the undiluted and 1:8 diluted post-dialysis S-ACE values are notably lower than pre-dialysis values, but the post-dialysis 1:8 dilution percentage increase was unchanged when compared with pre-dialysis increase. Thus, saline dialysis seems to be helpful in discriminating the presence of the inhibitor from the S-ACE increase due to alinearity of the assay, that is not affected by dialysis.
According to LIEBERMAN et al. [15] it is, therefore, likely that in hyperACEmic sera an intrinsic inhibitor is lacking, the after dilution increase of S-ACE activity being better ascribed to alinearity of the assay; that is understandable in view of the extremely high S-ACE values. Allernatively, it is possible to hypothesize the presence of an inhibitor other than that described by Lieberman and colleagues, not affected by dialysis. There is growing interest in intrinsic S-ACE inhibitors [28, 29] and the characterization of such agents will soon provide the complete profile of circulating and tissue ACE inhibitors. Another question concerns the source of S-ACE. It is known that in normal conditions S-ACE is mainly supplied by endothelial cells [30] whilst in granulomatous disorders the S-ACE increase is due to monocyte/macrophage hypcrfunction (31, 32]. In nongranulomatous disorders it is likely that pathological increase of S-ACE could be due to endothelial cells [9], although direct evidence is so far lacking.
An interesting finding coming from hyperACEmic subjects is the resistance of hyperACEmia to a high dose corticosteroid regimen, whilst the S-ACE decrease occurring in treated sarcoidosis patients is related to suppression of inflammatory process [33). It is likely that molecular biological techniques [34, 35] will soon provide information about site and regulation of S-ACE production in normal and pathological conditions.
In conclusion, this report would suggest that in the presence of a markedly increased S-ACE activity. (severalfold exceeding the normal values), even in patients with known causes of S-ACE increase, a possible familial elevation must be considered.
Acknowledgements: The authors are deeply indebted to E. Guitard and C. Pizzochero for their excellent technical assistance, to G. Merlini for Pi-typing and to L. Bacche.lla for radiometric S·ACE assay.
References
1. Lieberman J. - Elevation of serum angiotensinconverting enzyme (ACE) in sarcoidosis. Am J Med, 1975, 59, 365-372. Erratum: 1976, 60, A23. 2. Gronhagen-Riska C, Kurppa K, Fyhrquist F, Serloos 0. -Angiotensin-converting enzyme and lysozyme in silicosis and asbestosis. Scand J Respir Dis, 1978, 59, 228-231. 3. Lieberman J, Beutler E.- Elevation of serum angiotensinconverting enzyme in Gaucher's disease. N Engl J Med, 1976, 294, 1442- 1444. 4. Lieberman J, Sastre A. - Serum angiotensin-<:onvening enzyme: elevations in diabetes mellitus. Ann lnJern Med, 1980, 93, 825- 826. 5. Borowsky SA, Lieberman J, Strome S, Sastre A. -Elevation of serum angiotensin-converting enzyme levels. Occurrence in alcoholic liver disease. Arch lnlern Med, 1982, 142, 893-895. 6. Lieberman J, Rea TH. - Serum angiotensin-converting enzyme in leprosy and coccidioidomycosis. Ann Intern Med, 1977, 87, 422-425. 7. Rohatgi PK. - Serum angiotensin converting enzyme in pulmonary disease. Lung, 1982, 160, 287-301. 8. Rodriguez GE, Shin BC, Alermathy RS, Kending EL. -Serum angiotensin-<:onverting enzyme activity in normal children and in those with sarcoidosis. J Pediatr, 1981, 99, 68- 72. 9. Lieberman J, Sastre A. - Serum angiotensin converting enzyme levels in patients with alpha1-antitrypsin variants. Am J Med, 1986, 81, 821-824. 10. Fujisawa N, Okabe T, Yotsumoto H, Takaku F, Lanzillo JJ, Fanburg BL. - Familial clustering of elevated serwn angiotensin converting enzyme (abslr.). Sarcoidosis, 1985, 2, 72. 11. Cuccia Belvedere M, Monafo V, Martinetti M, Plebani A, De Paoli F, Burgio GR. - Recurrent extended HLA haplotypes in children with selective IgA deficiency. Tissue Amigens, 1989, 34, 127-132. 12. Salvaneschi L, Martinctti M, Pizzochero C, Folati L, Mazzucco U, Finco 0 , Dugoujon JM, Cuccia Belvedere M. -Marqueurs gcnctiques (HLA, Gm, Km, group sanguin) et niveaux seriques des immunoglobulines dans une population saine de 1 'Italie du Nord (Lombardie ). XIV Congres de la Societe Nationale de Transfusion Sanguine. Rermes 22-24 June, 1988 (abstr.ll.8), 80. 13. Sasazuki T. - HLA-linked immune suppression genes. European Histocompatibility Conference. Strasbourg 6-8 June, 1988, 47. 14. Lieberman J, Sastre A. - An angiotensin-converting enzyme (ACE) inhibitor in human serwn. Increased sensitivity of the serwn ACE assay for detecting active sarcoidosis. Chest, 1986, 90, 867-875. 15. Liebcrman J, Sastre A, Zakria F. - Comparison of an intrinsic serum ACE inhibitor to captopril and enalapril in man. In: Sarcoidosis and other granulomatous disorders. C. Grassi, G. Rizzato and E. Pozzi eds, Elsevier, Amsterdam, 1988, pp. 221-226.

446 M. LUISETTI ET AL.
16. Kasahara Y, Ashihara Y. - Colorimetry of angiotensinI-converting enzyme activity in serum. C/in Chem, 1981, 27, 1922-1925. 17. Thomas AV, Ansari A, Khurona M, Niden AA. -Elevated serum angiotensin converting enzyme in miliary tuberculosis (abstr.). Am Rev Respir Dis, 1979, 119, A83. 18. Rohatgi PK, Ryan JW.- Simple radioassay for measuring serum activity of angiotensin-converting enzyme in sarcoidosis. Chest, 1980, 78, 69-76. 19. Klasen EC, Rigutti A. - Isoelectric focusing of alpha1-
antitrypsin (Pi) using pH-range carrier ampholytes in combination with a highly cross-linked gel and a separator. Electrophoresis, 1982, 3, 168-171. 20. Terasaki PI, McClelland ID. - Microdroplet assay for human serum cytotoxins. Nature, 1964, 204, 998-1000. 21. Bodmer WF, Albert E, Bodmer JG, Dupont B, Mach B, Mayr W, Sasazuki T, Schreuder GMT, Svejgaard A, Terasaki PI. - Nomenclature reports for factors of the HLA system. Tissue Antigens, 1989, 32, 177-187. 22. Alper CA, Boenish T, Watson L.- Genetic polymorphism in human glycine rich beta-glycoprotein. J Exp Med, 1972, 135, 68-80. 23. Awdeh ZL, Alper CA.- Inherited structural polymorphism of the fourth component of human complement. Proc Natl Acad Sci USA, 1980, 77, 3576-3580. 24. Martinetti M, Tafi A, Dugoujon JM, Mazzacane D, Blanc M, Cuccia Belvedere M.- Possible interaction between HLA and Ig light chain markers in susceptibility to uveitis. Disease Markers, 1988, 6, 257-262. 25. Soffer RL. - Angiotensin-converting enzyme and the regulation of vasoactive peptides. Ann Rev Biochem, 1976, 45, 73-94. 26. Ondetti MA, Rubin B, Clishman DW. - Design of specific inhibitors on angiotensin converting enzyme: new class of orally active antihypertensive agents. Science, 1977, 196, 441-444. 27. Lathrop GM, Lalouel JM. - Easy calculations of Lod scores and genetic risks on small computers. Am J Hum Genet, 1984, 36, 460-465. 28. Lieberman J. - Angiotensin-converting enzyme (ACE) and serum lysozyme in sarcoidosis. In: Sarcoidosis. J. Lieberman ed., Grune and Stratton, Orlando, 1985, pp. 145-159. 29. Kohama Y, Maisumoto S, Oka H, Teramoto T, Okabe M, Mimura T. - Isolation of angiotensin-converting enzyme inhibitor from tuna muscle. Biochem Biophys Res Commun, 1988, 155, 332-337. 30. Caldwell PRB, Seegal BC, Hsu KC. - Angiotensin converting enzyme: vascular endothelial localization. Science, 1976, 191. 105~1051. 31. Silverstein E, Pertschuk LP, Friedland J. - Immunofluorescent localization of angiotensin-converting enzyme in epithelioid cells and giant cells of sarcoidosis granulomas. Proc Natl Acad Sci USA, 1979, 76, 6646-6648. 32. Stanislas-Leguem G, Mordclct-Dambrine M, Dusser D, Huesca M, Chretien J, Huchon GJ. - In vitro synthesis
of angiotensin-converting enzyme by alveolar macrophages is increased in disseminated sarcoidosis. Lung, 1986, 164, 269-277. 33. Deremee RA, Rohrbach MS. - Serum angiotensinconverting enzyme activity in evaluating the clinical course of sarcoidosis. Ann Intern Med, 1980, 92, 361-368. 34. Bemstein KE, Martin BM, Bernstein EA, Lintan J, Striker L, Striker J. - The isolation of angiotensin converting enzyme cDNA. J Bioi Chem, 1988, 263, 11021-11024. 35. Roy SN, Kusari J, Soffer RL, Lai CY, Sen GC.- Isolation of cDNA clones from rabbit angiotensin-converting enzyme: identification of two distinct mRNAs for the pulmonary and the testicular isoenzymes. Biochem Biophys Res Commun, 1988, 155, 678-684.
Elevation familiale de l'activite serique de /'enzyme de conversion de l'angiotensine. M. Luisetli, M. Martinetti, M. Cuccia, J.M. Dugoujon, V. De Rose, V. Peona, E. Pozzi, C. Grassi. RESUME: Une agglomeration de hauts niveaux d'enzyme de conversion de l'angiotensine serique (S-ACE) a ete mise en evidence dans une famille italienne. Cette elevation concemait une femme, deux de ses quatre freres, et deux de ses trois descendants. Deux d'entre eux (les deux freres) etaient completement normaux et ne portaient aucune des causes connues d'elevation du taux de S-ACE. Les valeurs de S-ACE (mesure colorimetrique) chez les 5 sujets en question (X±so 105.85±24.60) depassaient plusieurs fois les valeurs trouvees chez leurs familiers normaux (16.45±2.49). Cette elevation du taux d'angiotensine semble etre hereditaire, sous forme d'un trait dominant autosomique. Des etudes immuno-genetiques (HLA, classes I, II et Ill, et le typage Gm, Km) ont ete realisees, mais !'augmentation du taux d'angiotensine n'est apparue liee, ni au phenotype alpha 1 antitrypsine, ni a un supratype particulier HLA, ni a un phenotype d'immunoglobuline. L'activite S-ACE etait inhibee in vitro par l'EDTA 0.05 mM et par SQ 14.225 (Captopril) 0.05 mM. L'activite S-ACE a egalement ete determinee apres dilution des sera a 1:8, afin de determiner l'inhibiteur intrinseque de ACE, qui est elimine par la dilution de l'echantillon; dans 3 de ces 4 sera, cette procedure a montre une augmentation remarquable des valeurs de S-ACE (augmentation x: 18.71 %), suggerant la presence de l'inhibiteur. Afin de distinguer entre celle-ci et !'absence de linearite de l'essai, qui peut se produire dans des sera a activite elevee en S-ACE, nous avons dialyse pendant une nuit les sera contre de la solution saline. Nous avons trouve, malgre la dialyse, une augmentation significative des valeurs de S-ACE dans les memes sera hyperACE dilues au 1:8 (19.80%), et nous en avons deduit que l'inhibiteur intrins~ue de l'ACE faisait defaut dans les sera hyperACEmiques. Nous suggerons qu'en presence d'une augmentation remarquable de S-ACE, une elevation congenitale de S-ACE devrait etre prise en consideration, et qu'une investigation familiale meriterait d'etre realisee. Eur Respir J., 1990, 3, 441-446.


























![[Med ITA] Microbiologia - Terreni Di Coltura e Procedure](https://img.pdfslide.us/doc/110x75/5571fc2d497959916996ac28/med-ita-microbiologia-terreni-di-coltura-e-procedure.jpg)

