Embed Size (px)
Citation preview

Excited-State Kinetics of Azoalkanes Some Further Studies with Azoethane,
Azo-n -propane, and Azoisopropane
G. 0. PRITCHARD and P. E. MARCHANT Department of Chemistry, University of California, Santa Barbara, California 9.7106
C. STEEL Department of Chemistry, Rrandeis liniuersity, Waltham, Massachuwtts 021.54
Abstract
The photochemistry of azo-n-propane is investigated a t 366 nm up to 1 atm pressure, and over a range of temperature from 50 to 190OC. Some additional experiments with azoethane a t room temperature and azoisopropane a t 180 and 190°C are also reported. From a con- sideration of the pressure dependence of the quantum yields for photodissociation a gener- alized mechanism is proposed which accounts for the known experimental observations in acyclic azoalkane photochemistry. These observations include the extensive photoisomer- ization data which were previously obtained for azoisopropane. In the mechanistic scheme dissociation a t low pressures is believed to occur mainly from Sy and Ty, the vibrationally excited and randomized first excited singlet and triplet states. A t high pressures and low temperatures (5 100°C) the major dissociation channel is probably a nonrandom SI state. In direct or singlet sensitized photolysis isomerization occurs predominantly at high pressure and is postulated to occur by internal conversion from S:, the thermalized singlet, to the ground state. During the process partitioning to the cis or trans isomer is equally probable. In triplet sensitized photolysis isomerization occurs via intersystem crossing from TI to the ground state. A t elevated temperatures (>150°C) dissociation from Sy, which has a significant ac- tivation energy, can compete with return to the ground state.
Introduction
There has been much recent interest in the photochemistry of acyclic azoalkanes, R-N=N-R, in particular in trying to determine the state(s) from which photoreaction (mainly decomposition and isomerization) occurs [l-71. Although there has been discussion on this subject for many years, the picture is still far from clear. A major reason for this is undoubtedly the lack of observable fluorescence or phosphorescence, which seriously hampers meaningful photophysical measurements [8,9]. Chemical studies have included gas-phase measurements of decomposition and isomerization yields over a range of pressures using both direct and sensitized excitation.
International Journal of Chemical Kinetics, Vol. XI, 951-967 (1979) (C' 1979 John Wiley & Sons, Inc. 0538-8066/79/0O11-0951$01 .OO

952 PRITCHARD, MARCHANT, A N D STEEL
Comparable solution-phase studies have also been reported [ 1,3,7]. The results have been described in terms of three rather different models. (a) Fogel and Steel (FS) proposed [7] that the major dissociative channels in the gas phase are the vibrationally excited ground states (Sz(cis) and Si(trans)) formed by internal conversion (IC) or intersystem crossing (ISC) from the electronically excited states. (b) Chervinsky and Oref (CO) identify [6] the source of dissociation with the vibrationally excited triplet (T;O and a “fast” state (F) which is most likely a random or nonrandom form of the first singlet. The F state is responsible for the dissociation occurring at very high pressures in the gas phase or in solution. (c) Pritchard and co-workers (PSM) on the other hand explain their data in terms of disso- ciation from the vibrationally excited singlet Sy, from G, and from the thermalized triplet TY [4,11,12].
In this paper we shall be mainly concerned with azoethane (AE), azo- n-propane (ANP), azoisopropane (AIP), and hexafluoroazomethane (HFAM). When these compounds are studied in the gas phase, an im- portant source of information as to the mechanism is the plot of the re- ciprocal of the quantum yield of decomposition versus pressure. Because of their kinetic similarity to Stern-Volmer (SV) quenching plots of elec- tronically excited states, such figures are also sometimes referred to as Stern-Volmer [ll). In this case the terminology has simply been extended to include possible vibrational as well as electronic relaxation of the dis- sociative state(s). Curvature in the plot provides evidence that decom- position is originating from at least two states with different lifetimes. For all acylic azoalkanes to date where measurements have been carried out over a substantial pressure range there has been evidence for curvature. Below we report on such studies for trans-ANP. The photochemistry of this molecule has been much less investigated than trans-AE [4,5,13] or trans-AIP [7,12], for example. It has been used as a photolytic source of n-propyl radicals [14-161, but it is only in the work of Terry and Futrell [ 16,171 that low-pressure quantum yield data are reported (2-16 torr a t room temperature), and they fit a linear SV curve. I t is not possible to detect curvature in the quenching plot over such a limited pressure range [4-7,181, so we were interested in extending the latter to see if curvature could be detected for ANP as for the other azoalkanes. Also our previous data [4] on trans-AE showed significant scatter, and we were only able to infer that there could be curvature in the quenching plots a t low pressures, if the limiting yield as P - 0 ( O ~ N ~ ) was in fact unity. In an effort to con- firm the predicted low-pressure curvature we have therefore reinvestigated the photolysis of trans-AE.
Finally in an attempt to distinguish between the various mechanisms which have been proposed to explain acyclic azoalkane photochemistry we have extended dissociation yield measurements on trans-AIP to 190°C, since this is the azoalkane for which there are probably most data over a

KINETICS OF AZOALKANES 953
20
( 5 -
% I O -
5 -
0 -
wide range of conditions [7,12]. As will be seen, the proposed models predict different results, so comparisons with experiment can be made.
1 I I I -
0 15OoC -
0
0 -
0 0 -
0 . .
0 0
170°C 0
0 18OOC
8 : rn W 190°C
0 -
1 : . 1 I I I
Experimental and Results
Most of the experimental procedures and details are described elsewhere [4,12]. Dissociation yields for trans-ANP as a function of concentration are shown in Figures 1 and 2 where [MI = (l/RT) (PANP + P b u h n e ) . Below 1.1 X M trans-ANP was the only component, and concentration was varied by changing the ANP pressure. To obtain higher pressures, ANP
[ M I x lo3 Figure 1. centration [MI a t 50 and 100°C and 366 nm.
Plot for ANP of reciprocal NP quantum yield versus total molar con-

954 PRITCHARD, MARCHANT, AND STEEL
was kept constant a t 1.1 x 10-3M and n-butane was added as a vibrational relaxer. A constant fraction (approximately 25%) of the incident intensity a t 366 nm was absorbed in each run. The curvature in the low-temperature quenching plots can clearly be seen so that ANP fits in with other acyclic azoalkanes in that more than one dissociative state must be involved. The increasing dissociation a t higher temperatures (Fig. 2) is also quite similar to the data previously obtained with AIP [12]. The dark reaction correction due to pyrolysis of trans-ANP typically amounted to less than 5% of the total N2 formed a t the highest temperature (190°C) when the vessel was properly "seasoned" (see later). These corrections are included in the data shown. However, by analogy with AIP [7], cis-ANP is undoubtedly being formed a t the higher pressures and should decompose preferentially a t the more elevated temperatures.
This was shown directly by the following sequence of experiments. Trans- ANP-butane mixtures a t three different total pressures were pho- tolyzed at 50°C. Under these conditions decomposition is quite small, @N~ < 0.03 (see Fig. 1). On terminating the photolysis and warming the pho- tolysis mixtures a t 180"C, N2 was evolved amounting to 30% of that formed in the direct photolysis a t 180°C. It is important to note that the quantum yields shown in Figures 1 and 2 have not been corrected for this cis pyrol- ysis, hut reflect the measurement of all dissociative events occurring sub- sequent to the absorption of a photon by an azo molecule. In some ex- periments a t 180°C the incident intensity was reduced by approximately a factor of 10 with no change in the measured + N ~ yields.
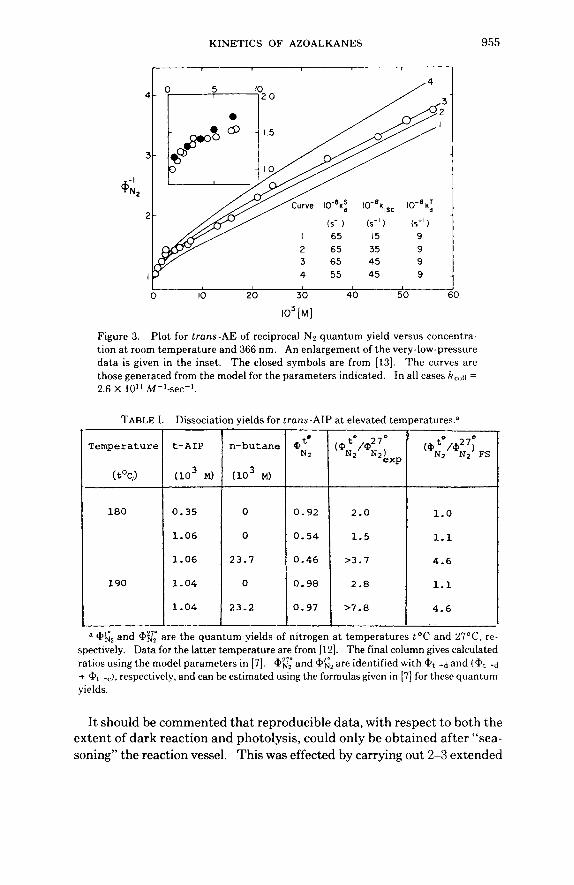
The data from the reexamination of the AE system a t room temperature are shown in Figure 3. The inset includes the data in [13], which led to the original conclusion that there was an intercept that was greater than unity. In our experiments n-butane was added as a vibrational quencher above 30 torr (1.6 X 10-:3M). Under these circumstances the absorbed light in- tensity was always about 25% of the incident intensity a t 366 nm. Below 30 torr the absorbed intensity was both measured directly (it amounted to 10% a t 10 torr) and calculated from Beer's law. Both methods led to coincident 4+~~ values. It is now possible to discern curvature a t very low pressures for the A E data in Figure 3, and to draw a smoothed curve into an intercept of unity.
The photolysis of trans-AIP was studied a t two temperatures, 180 and 19O"C, carried out both in the presence and in the absence of n-butane. Typical quantum yield data are shown in column 4 of Table I. A t both temperatures there was a significant correction for direct thermal decom- position (-50% a t 190°C); this has been allowed for in the quantum yields. Because the dark correction was found to increase very rapidly a t higher temperatures, data were limited to 19O"C, but i t is apparent that even a t high pressures the quantum yields of dissociation are approaching unity. This is in contrast to the small yields a t lower temperatures.
KINETICS OF AZOALKANES
23.2
955
4.6 0.97 >?.a
( 5 " ) ( 5 . ' ) (5")
65 45 I
0 10 20 30 40 50 60
lo3 [MI Figure 3. Plot for trans-AE of reciprocal N:! quantum yield versus concentra- tion a t room temperature and 366 nm. An enlargement of the very-low-pressure data is given in the inset. The closed symbols are from [13]. The curves are those generated from the model for the parameters indicated. In all cases hcc)~i = 2.6 X 10" M-'.sec-'.
TABLE I. Dissociation yields for trans-AIP a t elevated temperatures.=
Temperature
180
190
t - A I P
(lo3 MI
0.35
1.06
1.06
1.04
1.04
0 10.98 I 2.8 1 1.1
a as2 and are the quantum yields of nitrogen a t temperatures t"C and 27"C, re- spectively. Data for the latter temperature are from 1121. The final column gives calculated ratios using the model parameters in [7]. a"'," and as2 are identified with at -d and (aL .d
+ @L .cj, respectively, and can be estimated using the formulas given in (71 for these quantum yields.
It should be commented that reproducible data, with respect t,o both the extent of dark reaction and photolysis, could only be obtained after "sea- soning" the reaction vessel. This was effected by carrying out 2-3 extended

956 PRITCHARD, MARCHANT, AND STEEL
dark runs. The original data on + N ~ for AIP a t 180°C gave values of -0.7 when t -AIP = 1.06 X 10-3M and n-butane = 8.5 X 10-3M. The differences can be attributed to the smaller dark corrections that were used in the latter work [19].
Discussion
General Implications of Curvature
To be satisfactory a model should be able to rationalize the totality of experimental results, both in the gas phase and in solution. It is therefore worthwhile to sketch some of the main results to date and to indicate the major themes of the current models.
As mentioned in the Introduction, one of the features of the gas-phase photochemistry of the azoalkanes considered (namely, AE, ANP, AIP, and HFAM) is the curvature in the Stern-Volmer plots for dissociation. This indicates that at least two states must be involved in the decomposition, a simple mechanism being
(2) A* + A t
(343) A * ( $ ) --* N2 + 2R
(4a,b) A*($) + A (or M) - A + A (or M)
It is only necessary to postulate that the A* state has a longer lifetime than A* to account for curvature in the quenching plot. Photon absorption produces an 5'; state through an n + a* transition and the double arrow in eq. (1) serves to signify that A* need not in principle be identified with this initial state. Wu and Rice [18] originally proposed that A* represents a configuration in which randomization of the excess vibrational energy has not occurred while At could refer to molecules in which the excitation energy has had ample time to be statistically distributed among the normal modes.
CO Mechanism
CO identify the fast state (F), that is, the state with the shorter lifetime, with one of the following: random 5'1, nonrandom S1, nonrandom T I (all of which require a large value for the intersystem crossing rate constant), and the slow state with the vibrationally excited randomized TI state (Table 11). They conclude that it is dissociation from the F state which leads to

KINETICS OF AZOALKANES 957
uthors
SM L41
's C71
:o [6]
TABLE 11. Models in acyclic azoalkane photochemistry. States Involved
In Dissociation
S: (cis), SX (trans)
F (=Sl (random) or S1 (non-random) or
Ti (non-rarndorn) , T:
Synopsis of Model
Dissociation from S': and TY at room
temperature. Explains curvature in
v. P plots in 0-1 atrn. region. At
high pressures dissociation is from
TY which occurs with an activation energy.
Dissociation predominantly from S x (cis)
and S;(trans). Explains curvature in
v. P plots in 0-1 atm. region and
observed isomerization yields.
Dissociation from F and TY-
curvature in high-pressure region. Dis-
sociation at high pressures from F.
Explains
the residual quantum yields of nitrogen formation at very high pressures or in solution. Pressures up to 140 atm of He and 27 atm of COZ were used. Under these conditions the yields are comparable to but still somewhat larger than solution yields [1,3]. I t should also be noted that in solution dissociation yields can be quite solvent dependent, generally dropping markedly in polar, especially hydrogen-bonding, solvents.
The F state cannot be used as one of the states to explain the curvature observed by both PSM and FS in the 0-1-atm region since it has too short a lifetime to be intercepted in this pressure range.
PSM Mechanism
To explain their curvature PSM [4] suggested that the two states were Sy and 7?; (both energy randomized), and that dissociation competes with vibrational quenching in both manifolds. This differs from the CO pos- tulate in that collisional deactivation is competitive with dissociation and ISC for the Sy state. In the mechanism vibrationally relaxed S1 molecules (5'7) also undergo ISC, and all collisional processes eventually populate the vibrationally equilibrated 2'1 state (c). At sufficiently high temperatures thermal decomposition can occur from the latter, and this may compete effectively with ultimate return to the ground state.

958 PRITCHARD, MARCHANT, AND STEEL
FS Mechanism
Both PSM and CO were mainly concerned with explaining their disso- ciation yields, and isomerization is not discussed in detail. However, there is the implicit assumption that isomerization must arise from intersystem crossing from T I onto the ground-state SO surface. FS [7] on the other hand were concerned with rationalizing both dissociation and isomerization which they observed to occur concomitantly in the gas phase. A s was the case with P S M , their gas-phase studies were mainly limited to the O-l-atm region. FS noted decreasing quantum yields of decomposition (ad = +NJ and increasing isomerization yields as pressure was increased (that is, increasing +t-t and +t-c if trans-AIP was the starting compound and increasing @c-t and +c-c if cis-AIP was the starting compound). In ad- dition to the curved SV plots for the decomposition, FS further observed whichever isomer they started with, the buildup of trans always occurred first as pressure was increased. In order to explain this nonequality of +t+t
and (or aC-+ and +c-c) in the O-l-atm region as well as the curved SV plots for decomposition, FS suggested that decomposition originated from Sl;(trans) and Sg(cis). These states were believed to be formed rapidly by IC from S1. In presenting their mechanism FS were concerned with the major channels, that is, dissociation at low pressures and isomer- ization a t high pressures or in solution. They did not consider the source of the small amount of dissociation occurring in solution. However, if the latter occurs from an F state, provided the latter is S1 random or nonran- dom, it is perfectly compatible with their mechanism. It is interesting to note that recent CIDNP studies [20] indicate that solution-phase decom- position does indeed originate from the singlet. A major reason why FS did not favor triplet involvement in direct photolysis was the observation that in solution the triplet sensitized yields for decomposition and isom- erization were quite different from the corresponding nonsensitized yields. However CO [6] have rebutted this viewpoint; they pointed out that if isomerization occurs while the azoalkane is part of the donor-acceptor complex, then the isomerization yield could be different from that obtained from “isolated” triplets formed in the direct excitation followed by inter- system crossing. However, although steric hindrance to the rate of energy transfer from donor to acceptor is well established [21], this does not mean that the partitioning ratio of the excited acceptor to cis and trans isomers should be significantly affected, and it is not clear why the presence of the donor should favor the trans isomer (T+c-t > T+c-c) [7]. Not only is the partitioning about equal in direct photolysis (ac+ = +c-c ) , but also in singlet sensitized photolysis the yields are comparable to the direct yields [9,22], so in singlet sensitization the donor apparently plays no role in di- recting the stereochemistry. In our opinion therefore the problem of the difference in the sensitized and direct isomerization yields in solution still remains and we shall return to i t later in the discussion.

KINETICS OF AZOALKANES 959
Nonequality of Isornerization Yields
Despite the rather attractive aspects of the FS model, it will be seen that it fails to predict correctly the outcome of one rather simple and direct experiment. This is described in detail in the next section. It is therefore necessary to see if there are other ways in which the nonequality of the gas-phase yields +t-t and +t-c, and +c-t and +c,c can be rationalized.
(a) In terms of a simple PSM mechanism the following may be suggested. Collisionally deactivated triplet molecules are formed by sequences such as SY - TY+'MTy and S;('--MS(I - T? [4]. These are then followed by
S:(cis)
ST (trans )
Si(cis) - Si(trans) S:(cis) + M - S,(cis) + M
S:(trans) + M - SJtrans) + M
Notice that in contrast to the FS model dissociation does not originate from Sg(cis) and SE;(trans) since these states have been formed from the bottom of the triplet manifold, as distinct from S;( as postulated by FS, so that the energy is too small for dissociation to be effective in the experimental pressure range. In order to accomodate the experimental fact that a t very high pressues (= solution) +,,, = +t-.c = +c-t = +c,c, it follows that k s Z= k6. Also if the reasonable assumption ks = kg = kcoll is made and if it is assumed that the triplet is common to both isomers [23,24], then
The thermal reaction analogous to eq. (7) has been studied for AIP and is known to occur with facility [25]. The inverse reaction, Sg(trans) - Sz(cis) can be ignored because it will occur too slowly. It will be noticed that qualitatively eq. (10) is of the correct form since +,-, 2 +t--c. Quantita- tively the situation is less satisfactory. Data given in [7] are plotted in the form of eq. (10) in Figure 4, but they do not give a good straight line. However, taking the initial slope and assuming kcC,ll = 3.1 X 10" M-'.sec-' one obtains k7 = sec-1. Unfortunately this value is much too high. The triplet energy ET with respect to the stable trans isomer is usually set a t -55 kcal/mol [2,3,10,21,26], although there has been some speculation that it might be even lower [23]. Also AHf(cis-AIP) - AHf(trans-AIP) - 8 kcal/mol[7], thus Sg(cis) - 47 kcal/mol. Calculations of the value of k7 as a function of energy have been carried out by the method of Yau and Pritchard [27] using the thermal parameters for the isomerization of cis-AIP and a plausible vibrational analysis. The value obtained [28] is k7 = 103*1.6 sec-' a t E = 47 kcal/mol, the uncertainty in being associated with a

960 PRITCHARD, MARCHANT, AND STEEL
r, I00 200 300 400 [MI-' (Iifer/mol)
Figure 4. Ratio of isomerization yields as a function of effective total molar con- centration [MI for t-AIP. [MI = (l/KZW'azo + 0.34 Pco,). Data from (71. Pazo constant at 0.25 torr; Xi,, = 334.2 nm; 7' = 25OC.
corresponding uncertainty in the thermal A factor. Similar values are obtained using a regular RRKM calculation. It is unlikely that both ap- proaches are in error by a factor of lo6, so the suggested mechanism cannot account quantitatively for the data.
(b) The PSM mechanism for the gas-phase photochemistry may be modified as shown in Figure 5. The main change is that the major reaction at low temperature from the bottom of ST is now IC rather than ISC. A rationale for this is given later in this section. I t will also be noted that as far as dissociation is concerned, the mechanism is formally similar to the
N, +R*
Trans Cis Trans Cis Figure 5. Modification of PSM model.

KINETICS OF AZOALKANES 961
scheme (eqs. (1144) of the Discussion) suggested 10 years ago by Wu and Rice [18] with ksa = k;, kz = k I s c , kgb = ki. A t elevated temperature thermal dissociation from S: can occur a t an appreciable rate ( k d ) . Also the scheme includes the small amount of dissociation from the nonrandom F state to accommodate solution-phase dissociation in direct photolysis at low temperature.
Neglecting dissociation from the F state and assuming that the tem- perature is low enough so that dissociation from S? is negligible, the model yields under steady-state conditions
(11) *t--d = k ; T s + PISCk; fTT
(12)
(13)
at-t = a k c o I I [ M ] T s + P P I S C h c o l l [ M ] T T
*t-c = (1 - a ) k c o l l [ M ] T s + (1 - P ) P I S C k c o l l [ M ] ~ T
where 1/rS = (k; + k ~ s c + kco1,[M]), ~ / T T = (k: + hco11[M]), and PIS(; = k ISCTS.
In the case of AIP the solution-phase value for triplet sensitized isom- erization T*t-c lies between 0.04 and 0.08 [7], so that P = 0.94. For direct irradiation in solution = 0.5, so a must also be = 0.5. A diameter of 7 A yields kcoll = 3.1 X 10" A4-l - sec-' at 25°C. Model curves for @t-.d
using various sets of k;, kIsc and k i are shown in Figure 6. The slope at high concentrations is largely governed by k;, while the height of the curve
I ' I
I 14 18 2 0
2 10 I8 2 0
3 I0 18 1 7
4 10 25 2 0 I
0 I 2 3 4 5 lo3 [M I
Figure 6. Reciprocal of decomposition yield for t-AIP at 25°C as a function of effective total molar concentration for Xirr = 365.5 nm. [MI = ( l /RT) (Pa,, + 0.34Pc0,). 0-Azo alone; 0-COz as added gas, Pa,,, = 0.25 torr. Data from (341. The curves are generated from the model with the rate constants given in the inset. In all cases kcall = 3.1 X 10" M-'.sec-'.

962 PRITCHARD, MARCHANT, AND STEEL
is controlled by krsc. Both the latter and kx influence the effective cur- vature in the low concentration region (see Fig. 9). Figure 7 shows the plot for all three quantum yields. The best values of k3, i z ~ s c , and k ; f are sum- marized in Table 111, and we shall return later to a more detailed analysis of the data given there.
It is necessary to explain that if ISC is important for Sp, why has it been neglected in comparison to IC for S?. A possible explanation is to be seen in Figure 8. Although the exact positions of the So, S1, and T1 manifolds on the energy scale are open to some question, there seems to be little doubt as to the general shapes of the potential energy surfaces [23,24]. The SO surface approaches the S1 surface close to the minimum of the latter. In this region internal conversion should become quite effective. On the other hand a t higher energies, crossing from S; to TY should be relatively more
10
00
04
0 2
0
Figure 7. Quantum yield for t-AIP as a function of effective total molar concen- tration (MI for A,,, = 365.5 nm a t T = 25°C. 0 , A, .-Azo alone; 0, A, O-coz as added gas. Data from [XI. The curves are generated from the model using the same parameters as set 3, Figure 6, with a = 0.57 and p = 0.94.
TABLE 111. Rate constants for the photodissociation of azoalkanes a t 366 n m a
Compound
t - A I P
a Values in columns 1-3 from this work, except for HFAM [18]. k R is from [6]. For estimating E see text.

KINETICS OF AZOALKANES 963
Ll Trans
Figure 8. Schematic potential energy surfaces for acyclic azoalkanes to show why internal conversion (IC) may be favored for low energies on the SI surface while a t higher energies intersystem crossing (ISC) becomes the dominant photo- physical channel.
favorable because of the large energy mismatch between S1 and SO. As has been pointed out previously [23], such surfaces also rationalize the observed lack of fluorescence or phosphorescence from acyclic azoalkanes. From Figure 5 it is seen that a t high pressures most molecules populate the thermalized singlet Sy and return from there by IC to the ground state if the temperature is low enough so that there is no thermal dissociation from 5'7. Under these conditions the only decomposition is from the F state. At elevated temperatures dissociation from Sy, which has a significant acti- vation energy, can compete with IC. Because of this the net quantum yield of dissociation tends toward unity as temperature is increased (Fig. 2).
A n Experimental Distinction Between the P S M and FS Mechanism
The above discussion has shown that a modified PSM mechanism is capable of explaining the data of FS, in particular the nonequality of and +t.-c in the gas phase and the nonequality of the isomerization yields for direct and sensitized photolysis in solution. Is it then possible to dis- tinguish experimentally between a modified PSM and a FS mechanism? I t is an experimental fact that in the gas phase +t--d increases with tem- perature; see, for example, Figure 2 and [12]. In terms of the FS mechanism this would be attributed to decomposition of cis formed in the primary photolysis. This means that for AIP in the limit of high pressures or con- centrations and elevated temperatures at+ - 0.5 since (+t--d + +t +J - 0.5 a t high pressures and a t room temperature where the cis isomer is stable [7]. However, the data in Table I show that in the high-concentra- tion region +t-d - 1.0 as temperature is raised. Even in the intermediate concentration region (51 X 10-3M) the FS mechanism fails to give quan- titative agreement. Columns 5 of Table I gives the ratios of the experi- mental quantum yields a t the elevated temperature (180 or 190OC) to those at 27"C, the data for the latter temperature being taken from [12]. These

964 PRITCHARD, MARCHANT, AND STEEL
ratios are a measure of the increased quantum yields of dissociation a t el- evated temperature. The final column gives the values predicted using the FS mechanism and the data in "71. The quantitative agreement in the intermediate concentration region is seen to be poor, with the FS mecha- nism predicting smaller increases than are actually observed. Although data for AE and HFAM cannot be analyzed in terms of the FS mechanism, since isomerization yields have not been determined for these compounds, there seems every reason to believe that the FS mechanism would also not account for the relatively large increases in quantum yield with increasing temperature in the intermediate concentration region obtained for these compounds [4], since in this region the FS mechanism predicts little cis formation a t low temperature and hence little increase in decomposition yield with increasing temperature.
There is the possibility that a t elevated temperatures secondary N2 sources may be present which could tend to invalidate the above conclu- sions. A possible reaction sequence is
However, these reactions would be minimized in the presence of excess n-butane which will compete with AIP for the C3H7 radicals. The fact that a t 190°C + N ~ is the same in the presence and absence of added butane (Table I) indicates that in fact the above reactions do not contribute sig- nificantly to the rate of N2 formation. Of course in the HFAM system such reactions cannot even be postulated as a possible source of N2 forma- tion.
Extension of Model to Other Azoalkanes
The above analysis indicates that the modified PSM mechanism accounts well for the experimental data for AIP, with respect to both isomerization and decomposition in the 0-1-atm range. In addition if it is assumed that there is some dissociation from a nonranclom Sy state (the fast state) competing with randomization to Sy (random), it can also account for the small amount of dissociation remaining in solution a t low temperatures for the small azoalkanes AM, AE, ANP, AIP, and HFAM.' m + ~ 2 = 0.15 for AM, 0.023 for AE, 0.007 for ANP, and 0.025 for AIP [3]. HFAM does photolyze in solution, but quantum yields are not reported [29]. These low quantum yields suggest the relative thermal stability of the cis isomers of these compounds [22,30,31]. In contrast the large dissociative yield obtained at room temperature for bulky azoalkanes like ATB ( m + ~ 2 = 0.46) is known to be due to the thermal dissociation of the unstable cis isomer [3].
In the original PSM mechanism dissociation in solution occurred from c, but this is in- consistent with CIDNP and other recent studies 120,351.

KINETICS OF AZOALKANES 965
Moreover the above mechanism justifies that in solution the triplet-sen- sitized yields for isomerization are different from the direct yields or sin- glet-sensitized yields, without having to assume that the presence of the sensitizer has a steric influence favoring trans formation. Because of these considerations we thought it worthwhile to extend the analysis to other azoalkanes. Such model curves are shown in Figures 3 and 9, while in Table I11 the rate constants determined in this fashion have been assembled to- gether with Wu and Rice's data for HFAM [18]. Several points may be noted. First, k i l k x - constant, as might be expected for a series of com- pounds which in all probability have rather similar potential energy sur- faces. Also ka and k x decrease with increasing molecular complexity. For example, in terms of RRK theory [32] HFAM and AE have about the same number of effective oscillators [33] , while ANP and AIP have significantly more, but also are of comparable complexity. On the other hand KISC shows no such systematic trend. Again this is not surprising in that these rate constants should depend not so much on molecular complexity as on the rate of curve crossing from S1 to 2'1. In column 6 the rate constants for the random long-lived state obtained in [6] are tabulated. Because of the large pressure range used in this study the curvature arising from multistate dissociation in the 0-1-atm region was not considered. This means that these rate constants should in fact represent an average given approxi- mately by
ISC + k z X ksd - k = k i x
( k i + hsc) ( h 3 + h s c )
5 0 . I
40-
30.
4 25 0.5 4 50 0.7 4 80 0 .5
lo3 [MI Figure 9. Reciprocal of decomposition yield for t-ANP a t 5OoC as a function of total molar concentration (see Figure l), @.t-d 3 @N? Curves are generated from the model with the rate constants given in the inset. In all cases kcoll = 3.35 X 10" M-*.sec-'.

966 PRITCHARD, MARCHANT, AND STEEL
The values of k calculated in this way are shown in the final column. I t will be noted that the correlation with k R is good.
Conclusion and Dedication
In this paper we have tried to present a unified mechanism that is con- sistent with the known experimental observations in acyclic azoalkane photochemistry. Dissociation a t low pressures is believed to occur mainly from Sy and TY, the vibrationally excited and randomized first excited singlet and triplet states. A t high pressures a t low temperatures ( r l00”C) the bulk of dissociation occurs from the F state, which is probably a non- random S1 state. In direct or singlet sensitized photolysis isomerization occurs predominantly a t high pressure and is believed to originate by IC from S:, the thermalized singlet, to the ground state. The partitioning to cis and trans would appear to occur with more or less equal probability. In triplet-sensitized photolysis isomerization occurs by ISC from T1 to the ground state. Much of the pioneering work in this field was carried out by the late 0. K. Rice. As mentioned above, the underlying basis of the general mechanism which we have proposed is to be found in his research and we dedicate this paper to his memory.
Acknowledgment
We thank A. W. Yau and H. 0. Pritchard for carrying out the calculation of k7. We also thank I. Oref for some helpful comments. This work was supported by the National Science Foundation under Grant MPS-75- 17732.
Bibliography
[I] R. J. Drewer, in “The Chemistry of Hydrazo, Azo and Azoxy Groups,” pt. 2, S. Patai, Ed.,
[2] H. Durr and B. Ruge, in “Topics in Current Chemistry,” Vol. 66, Springer, New York,
(31 P. S. Engel and C. Steel, Acct. Chem. Res., 6,275 (1973). [4] G. 0. Pritchard, F. M. Servedio, and P. E. Marchant, Int. J . Chem. Kinet., 8, 959
[5) S. Chervinsky and I. Oref, J . Phys. Chem.. 79, 1050 (1975). [6] S. Chervinsky and I. Oref, J . Phys. Chem., 81,1967 (1977). 17) L. D. Fogel and C . Steel, J . Am. Chem. Soc., 98,4859 (1976). [8] S. S. Collier, D. H. Slater, and J. G . Calvert, Photochem. Photobiol., 7,737 (1968). [9] I. I. Abram, G . S. Milne, B. S. Solomon, and C. Steel, J . Am. Chem. Soc., 91, 1220
Wiley-Interscience, New York, 1975, Chap. 20.
1976, p. 53.
(1976).
( 1969). [lo] H. Rau, Angew Chem., Int. Ed. (Engl . ) , 12,224 (1973). Ill] G . 0. Pritchard, W. A. Mattinen, and J. R. Dacey, Int. J . Chem. Kinet., 2. 191 (1970). [12] G . 0. Pritchard and F. M. Servedio, Int. J . Chem. Kinet . , 7,99, 195 (1975). [13] H. Cerfontain and K. 0. Kutschke, Can. J . Chem., 36,344 (1958). [14] J. A. Kerrand J. G. Calvert, J . Am. Chem. Soc., 83,3391 (1961).

KINETICS OF AZOALKANES 967
R. E. Berkeley, G. N. C. Woodall, 0. P. Strausz, and H. E. Gunning, Can. J . Chem., 47, 3305 (1969). cJ. 0. Terry and J. H. Futrell, Can. J . Chem., 45, 2327 (1967). J. 0. Terry, M.S. thesis, Air Force Institute of Technology, Wright-Patterson Air Force Base, 1964. E.-C. Wu and 0. K. Rice, J . Phys. Chem., 46,2021 (1967). F. M. Servedio, Ph.D. dissertation, University of California, Santa Barbara, 1973. cJ. A. den Hollander, J . Chem. Soc.. Chem. Commun., 403 (1976). C. C. Wamser, R. 1’. Medary, I. E. Kochevar, N. J. Turro, and P. L. Chang,J. Am. Chem. Soc.. 97,4864 (1975). M. N. Ackermann, N. C. Craig, R. R. Iseberg, D. M. Lanter, R. A. MacPhail, and W. G. Young, J . Am. Chem. SOC., 99,1661 (1977). R. N. Camp, I. R. Epstein, and C. Steel, J . Am. Chem. SOC., 99,2453 (1977). N. C. Baird and J. R. Swenson, Can. J . Chem., 51,3097 (1973). I,. D. Fogel, A. M. Rennert, and C. Steel, J . Chem. Soc.. Chem. Commun., 537 (1975). 0. A. Mosher, M. S. Foster, W. M. Flicker, J. L. Beauchamp, and A. Kupperman,J. Chem. Phys., 62,3424 (1975). A. W. Yau and H. 0. Pritchard, Can. J . Chem., 56, 1389 (1978). A. W. Yau and H. 0. Pritchard, private communication. 0. Dobis, J. M. Pearson, and M. Szwarc, J . Am. Chem. SOC., 90,278 (1968). R. F. Hutton and C. Steel, J . Am. Chem. Soc., 86,745 (1964). V. I. Pergushov, 0. N. Bormot’ko, and V. S. Gurman, Chem. Phys. Lett., 51, 269 (1977). P. J. Robinson and K. A. Holbrook, “Unimolecular Reactions,” Wiley, New York, 1972. E. Leventhal, C. R. Simonds, and C. Steel, Can. J . Chem., 40,930 (1962). L. D. Fogel, Ph.D. dissertation, Brandeis University, Waltham, MA., 1974. P. S. Engel, D. J. Bishop, and M. A. Page, J . Am. Chem. SOC., 100,7009 (1978).
Received January 10,1978 Revised April 4, 1979 Accepted April 12,1979





























