Embed Size (px)
Citation preview

ENERGETICS, THERMAL AND STRUCTURAL
PROPERTIES OF HAFNIUM CLUSTERS VIA
MOLECULAR DYNAMICS SIMULATION
by
NG WEI CHUN
Thesis submitted in fulfillment of the requirements
for the degree of
Master of Science
September 2016

ii
ACKNOWLEDGEMENT
First of all, I wish to express my gratitude to my supervisor, Dr. Yoon Tiem
Leong, and my co-supervisor, Dr. Lim Thong Leng, for their professional guidance
and suggestions throughout the whole period of my project and thesis writing. Their
motivation and valuable ideas, as well as the tireless commitment in this research, are
utmost helpful, especially in leading my learning path as a researcher. For their help
and concerns in my studies, I am greatly indebted to both of them.
I would like to thank the Ministry of Higher Education for financial support in
term of Fundamental Research Grant Scheme (FRGS) (Project number:
203/PFIZIK/6711348) as well as MyMaster scholarship in covering the tuition fees.
For the immediate colleagues from the theoretical and computational group, I
would like to thank them for helping me in my research and pleasantly accommodate
my presence. We shared many fruitful discussions that involved a lot of general
knowledge, essentially a wide coverage on the latest world news that brings insights
to each of us. Special thanks to Mr. Min Tjun Kit, my senior who has offered all kinds
of operational and technical support in LAMMPS. Next in line are juniors Ms. Soon
Yee Yeen and Ms. Ong Yee Pin for their help in organizing various meetings and
sharing of paperwork.
I would like to acknowledge the collaborating group from Taiwan National
Central University, Prof. Lai San Kiong and his fellow students, especially Peter Yen
for their academic support. Their ideas and comments are the most valuable in helping
the competing of this thesis.

iii
Last but not least, I am grateful for my family who supports me in all aspects.
They understand and respect my decisions during the completion of my project, and
thesis. The hard works and sacrifices they have made encourage me even more to
succeed both in life and in academic.

iv
TABLE OF CONTENTS
Acknowledgement ii
Table of Contents iv
List of Tables viii
List of Figures ix
List of Abbreviations xii
List of Symbols xiv
Abstrak xviii
Abstract xx
CHAPTER 1:
INTRODUCTION
1
1.1 Computational Simulation of Atomic Cluster 2
1.2 Objectives of Study 4
1.3 Organization of Thesis 5
CHAPTER 2:
REVIEWS ON RELATED TOPICS
7
2.1 All about Nanoclusters 8

v
2.2 Melting in Bulk and Cluster 12
2.3 Chemical Similarity and Shape Recognition 22
2.4 Empirical Interatomic Potential 30
2.5 The Method of Basin Hopping 36
CHAPTER 3:
METHODOLOGY
41
3.1 PTMBHGA 42
3.2 Molecular Dynamics Simulation of Hafnium Clusters 47
3.2.1 Simulated Annealing Process 47
3.2.2 COMB Potential 52
3.2.3 Cluster Structures Generation 56
3.2.4 Chemical Similarity Comparison 60
3.2.5 Flying Ice Cube Problem 63
3.3 Post-Processing 68
3.3.1 Global Similarity Index 71

vi
CHAPTER 4: DEPENDABILITY OF COMB POTENTIAL 83
4.1 Geometrical Re-Optimization of Hafnium Clusters 83
4.2 Structural Confirmation of Hafnium Clusters 90
CHAPTER 5:
SIMULATED ANNEALING OF THE HAFNIUM
CLUSTERS
94
5.1 The Melting Point of Hafnium Clusters 94
5.2 Melting Temperature and Cluster Sizes 102
5.3 Similarity Index and Cluster Melting 106
5.3.1 Hf30 107
5.3.2 Hf50 109
5.3.3 Hf99 111
CHAPTER 6:
CONCLUSIONS AND FUTURE STUDIES
114
6.1 Conclusions 114
6.2 Future Studies 115

vii
REFERENCES
118
APPENDIX A
Functionality Form of COMB Potential
125

viii
LIST OF TABLES
Page
Table 2.1 The example parameters of LJ potential for the noble gases. 14
Table 3.1 The potential parameters of Hf for the COMB potential. 53
Table 4.1 Comparing the clusters obtained via COMB potential after 1 K
relaxation and those via DFT geometrical re-optimization with
B3LYP basis set (the plmp intra-line comparison).
84
Table 4.2 Comparing the structures of clusters obtained via plmp with
COMB (left) and those upon DFT geometrical re-optimization
(right) side by side. The size of the cluster is labeled just below
the respective pairs of clusters in comparison.
88
Table 4.3 The hafnium clusters of size Hf4 to Hf8 in stage 2 of the pg3
process line, along with their DFT total energy value in hartree.
(*) indicates structures with lowest energy, while (**) indicates
similar structures that are also obtained in the plmp process line.
91
Table 5.1 The melting and pre-melting temperatures obtained from three
different approaches. The first four columns are obtained from
caloric curves and 𝑐𝑣 curves
103

ix
LIST OF FIGURES
Page
Figure 2.1 Schematic diagram used by Ihsan Boustani (1997) to illustrate
the growth of boron cluster from the basic unit of hexagonal
pyramid B7. By adding the repetitive geometrical motif, the
cluster eventually forms the infinite quasi-planar surfaces or
nanotubes.
11
Figure 2.2 Sample of SCOP2 graph viewer result given by Andreeva et
al. (2013), showing the Cro types protein sequence and
structure.
23
Figure 2.3 Commonly in use interatomic potential in increasing
computational cost Ng et al. (2015a).
35
Figure 2.4 A schematic sketch to illustrate the effect of BH
transformation to the PES of a one-dimensional example.
37
Figure 2.5 A schematic sketch indicating the strategy to obtain true
global minimum by the way of sampling LLS at a coarse level
search with BH method without structural optimization (stage
1), and subsequently undergo a refined geometrical re-
optimization of these LLS using DFT method (stage 2). The
doted profile in stage 1 represent the simplified staircase
topology of the PES.
40
Figure 3.1 Flow chart of the general layout of methodology 41
Figure 3.2 Flow chart for the algorithm in the hybrid PTMBHGA +
(LAMMPS / G03) package.
45
Figure 3.3 The melting point and pre-melting point of Hf50 with various
heating rate. The circle region marks the convergence of
melting point lower than certain heating rate.
50
Figure 3.4 The plots of temperature, 𝑇 against the simulation time step,
∆𝑡 a) without and b) with time averaging in each 300∆𝑡 time
interval.
51

x
Figure 3.5 A schemetic flow chart of the two parallel process lines. The
plmp process line in the left and the pg3 process line in the
right. Quantum refinement (geometrical re-optimization)
steps are carried out with G03 using the same basis sets and
settings in both process lines.
57
Figure 3.6 A schemetic flow chart of intra-line comparison within the
plmp process line.
62
Figure 3.7 A schemetic flow chart of inter-line comparison between the
plmp and pg3 process lines.
63
Figure 3.8 The condition of Hf13 cluster during the heating procedure
which encountered flying ice cube artifact, generating
excessive kinetic energy. a). The cluster begin to spin in a
clockwise manner along the red arrows direction shown, at
the beginning of heating procedure. b). The Hf13 cluster
around 1800K~1900K where the whole cluster start to drift
across the simulation box, in addition to the rotation motion,
while remain closely bonded like an ‘ice’ body. The dynamic
bonding shown in the figure is kept below 3.2Å, slightly
longer than the actual bond length in bulk hafnium.
64
Figure 3.9 The condition of Hf13 cluster, showing the bond breaking and
bond formation at a) ~850 K, b) ~900 K, c) ~1100 K and d)
~2050 K. Along the simulation time, the cluster did not rotate
nor drift across the simulation box, each atom vibrate relative
to one another, carry the kinetic energy in them.
68
Figure 3.10 The LLS of Hf7 cluster. a) The ground state structure. b) The
second lowest energy isomer. c) A slight modification was
made based on the ground state structure where the bipyramid
top was moved closer to the pentagonal base. The green cross
indicates center of mass.
73
Figure 4.1 Plot of graphs comparing a) the average bond length and b)
the global similarity indexd between COMB structures and
that after DFT re-optimization.
85
Figure 5.1 a) The caloric curve and b) 𝑐𝑣 curve of Hf20 obtained via
prolonged annealing process (TNA = total number of atoms
in the cluster). The green arrow indicates the pre-melting
temperature at 𝑇𝑝𝑟𝑒 = 1400 K, and the red arrow indicates the
melting point at 𝑇𝑚 = 1850 K.
96

xi
Figure 5.2 a) The caloric curve and b) 𝑐𝑣 curve of Hf10 obtained via
direct heating process. The green arrow indicates the pre-
melting temperature at 𝑇𝑝𝑟𝑒 = 1350 K , and the red arrow
indicates the melting point at 𝑇𝑚 = 2200 K.
99
Figure 5.3 a) The similarity index 𝜉𝑖 and b) fluctuation of similarity
index 𝑆𝜉𝑖 of Hf13 obtain via a direct heating process. The
green arrow indicates the pre-melting temperature at 𝑇𝑝𝑟𝑒 =
1600 K, and the red arrow indicates the melting point at 𝑇𝑚 =
2050 K.
101
Figure 5.4 Plotting together the estimated pre-melting temperature, 𝑇𝑝𝑟𝑒
and the exact melting point, 𝑇𝑚 of Hf clusters of various size
𝑛 for a) prolonged simulated annealing, b) direct heating
process, and c) the global similarity index.
104
Figure 5.5 The estimated melting point of the hafnium cluster against the
cluster size 𝑛, based on three different approaches.
105
Figure 5.6 a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity
index 𝑆𝜉𝑖 of Hf30. The screenshots show the configuration of
the cluster Hf30 during that particular temperature.
108
Figure 5.7 a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity
index 𝑆𝜉𝑖 of Hf50. The screenshots shows the configurations
of the cluster Hf50 during that particular temperature.
109
Figure 5.8 The artifact of single atom drifting away observed in the case
of a) Hf18 and b) Hf26.
111
Figure 5.9 a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity
index 𝑆𝜉𝑖 of Hf99. The screenshots shows the configurations
of the cluster Hf99 during that particular temperature
112
Figure 5.10 Hf99 upon the equilibration at 𝑇 = 3000K. 113

xii
LIST OF ABBREVIATIONS
AIREBO Adaptive Intermolecular Reactive Empirical Bond Order Potential
BCC Body-Centered Cubic
BH Basin Hopping
BOP Bond Order Potential
B3LYP Becke Three Parameter Hybrid Functionals with Correlation functional
of Lee, Yang, and Parr
CM Center of Mass
CNT Carbon Nanotubes
COMB Charged-Optimized Many-Body Potential
COR Center of Reference (Generalized Center of Mass)
DFT Density Functional Theory
eFF Electron Force Field
FCC Face-Centered Cubic
GA Genetic Algorithm
G03 Gaussian 03 Program
LAMMPS Large-scale Atomic/Molecular Massively Parallel Simulator
.lammpstrj LAMMPS Output Trajectory File

xiii
LanL2DZ Los Alamos ECP Plus DZ Pseudopotential for Hafnium
LJ Lennard-Jones Potential
LLS Low-Lying Structures
.log LAMMPS Output Log File
MD Molecular Dynamics
PES Potential Energy Surface
pg3 PTMBHGA + G03 Hybrid Package
plmp PTMBHGA + LAMMPS Hybrid Package
PTMBHGA Parallel Tempering Multi-Canonical Basin Hopping and Genetic
Algorithm
Qeq Charge Equilibration
ReaxFF Reactive Force Field
REBO Reactive Empirical Bond Order Potential
SCF Self-Consistent Field Procedure
SW Stillinger-Weber
TEA Tersoff-Erhart-Albe Potential
USR Ultrafast Shape Recognition
VMD Visual Molecular Dynamics Software

xiv
LIST OF SYMBOLS
Ca Center of mass of cluster a
𝑐𝑣 Constant temperature specific heat capacity
𝐷𝑎𝑣𝑒𝜒
Average bond length
𝑑𝑚𝜒
Distance between atoms in a cluster 𝜒, by sorting sequence of 𝑚
𝑑𝑁(𝐴, 𝐵) Rogan similarity measure for cluster 𝐴 and 𝐵; 𝐷𝑁(𝐴, 𝐵) the
normalized form
𝑑𝑆(𝐴, 𝐵) Springborg similarity measure for cluster 𝐴 and 𝐵; 𝐷𝑆(𝐴, 𝐵) the
normalized form
𝑑𝑠,𝑖 Distance of atom 𝑠 from the center of mass of 𝑖th cluster
�̃�(𝑋) Transformed energy topology
𝐸𝑡 or 𝐸𝑇 Total energy
𝐹 Force
𝑓𝑖 Fitness value of candidate cluster 𝑖
𝑘𝐵 Boltzmann constant
𝑘𝑠,𝑖 Difference between the distances of atom 𝑠 from 𝑖th cluster and 0th
cluster
𝑚𝑖 Mass of atom 𝑖

xv
𝑀𝑙 Moments of shape descriptors
𝑀𝑝(𝑥1, … , 𝑥𝑛) Generalized Mean of variables 𝑥
𝑛 Cluster size, or number of atoms
𝑃 Pressure
𝑝 Power of generalized mean, a non-zero real number
𝑝𝑖 Gaussian weight
𝑞𝑖 Charge of atom 𝑖
𝑟 Distance or position
𝑟𝑏 Dynamic bond length imposed in visualization
𝑆𝐴𝐵 Normalized similarity index, Tanimoto similarity index
𝑆𝑞𝑖 USR similarity index
𝑆𝜉𝑖 Fluctuation of global similarity index 𝜉𝑖
𝑇 Temperature
𝑇𝑐 Critical temperature in a phase diagram
𝑇𝑚 Melting Temperature
𝑇𝑚𝑏𝑢𝑙𝑘 Bulk melting point
𝑇𝑝𝑟𝑒 Pre-melting temperature

xvi
𝑉𝐴𝐵 Overlapping volume of structure 𝐴 and 𝐵
𝑉𝑒𝑓𝑓(1, … , 𝑛) Effective interatomic potential for 𝑛 interacting particles
𝑉𝑖 Potential energy of the cluster 𝑖
𝑉𝑖𝑗 Interaction potential between atom 𝑖 and 𝑗
𝑣𝑖𝑔
Volume of an atom 𝑖; 𝑣𝑖𝑗𝑔
is the intersection volume of the pair of atoms
𝑖 and 𝑗
𝑉𝐿𝐽 Lennard-Jones potential
𝑉𝑛 𝑛-body Gupta potential
𝑉𝑝𝑎𝑖𝑟 Pair-wise potential
𝑤𝑖 Weightage factor
𝛿 Lindemann index
Δ𝑡 MD simulation timestep
휀 Depth of the potential well
𝜉𝑖 Global similarity index
𝜌 Density
𝜌𝑖𝑔(𝒓𝒊) Spherical Gaussian as a function of vector position 𝒓𝒊 of atom 𝑖
𝜌𝜒𝑔
Gaussian densities
𝜎 Interatomic separation at equilibrium

xvii
𝜎𝑖 ‘Radius’ of an atom
⟨𝜎𝑖(𝑡)⟩𝑠𝑡𝑎 Short-time average distance
𝜒 Structures label, such as 𝜒 = 𝐴 or 𝐵
𝜒𝑖 Chemical potential of atom 𝑖

xviii
CIRI-CIRI BERTENAGA, HABA, DAN STRUKTUR BAGI GUGUSAN
HAFNIUM MELALUI SIMULASI DINAMIK MOLEKUL
ABSTRAK
Kelakuan keleburan gugusan hafnium (saiz 2 < 𝑛 < 99 ) dikaji melalui
simulasi dinamik molekul (MD). Interaksi antara atom hafnium diperihalkan dengan
keupayaan Charged-Optimized Many-Body (COMB). Keupayaan COMB yang sama
digunakan bersama dengan algoritma pengoptimuman global yang dikenali
PTMBHGA untuk menjanakan struktur input pada keadaan asas untuk proses MD.
Struktur keadaan asas yang diandai telah disahkan apabila berbanding dengan rujukan
dan pengiraan prinsip pertama. Selanjutnya, mengesahkan pergantungan potensi
COMB dalam proses MD. Biasanya, parameter tenaga digunakan untuk menilai sifat-
sifat gugusan. Tesis ini telah menggunakan geometri gugusan selain daripada profil
kalori untuk mengaji dinamik semasa keleburan gugusan. Untuk mencapai matlamat
ini, algoritma indeks keserupaan global telah direka untuk mengukur tahap persamaan
antara dua gugusan. Ia diperoleh berasalkan keserupaan kimia bagi molekul dan
mematuhi prinsip sifat serupa. Proses pemanasan MD dijalankan sama ada
menggunakan pemanasan langsung atau penyepuhlindapan simulasi berpanjangan.
Takat lebur dikenalpasti dengan menggunakan lengkungan kalori, keluk isipadu malar
muatan haba dan indeks keserupaan global. Takat lebur gugusan hafnium berubah
dengan saiz gugusan, 𝑛. Di samping itu, peralihan takat lebur berlaku di pelbagai suhu,
bermula dengan peringkat pra-lebur pada suhu 𝑇𝑝𝑟𝑒 sampai peringkat terlebur pada
suhu 𝑇𝑚 yang lebih tinggi. Ketiga-tiga kaedah bersetuju dengan satu sama lain untuk
julat suhu lebur untuk gugusan hafnium. Walau bagaimanapun, didapati bahawa

xix
indeks keserupaan global lebih unggul, kerana ia juga dapat mengesan mekanisme
lebur gugusan hafnium.

xx
ENERGETICS, THERMAL AND STRUCTURAL PROPERTIES OF
HAFNIUM CLUSTERS VIA MOLECULAR DYNAMICS SIMULATION
ABSTRACT
The melting behavior of hafnium clusters (of sizes 2 < 𝑛 < 99) are studied via
molecular dynamics (MD) simulation. The interaction between the hafnium atoms is
described by Charged-Optimized Many-Body (COMB) potential. The same COMB
potential is used with a global optimization algorithm called PTMBHGA to generate
the input ground state structures for MD processes. These assumed ground state
structures are verified as compared to the literature and first-principles calculation,
which further confirm the dependability of COMB potential within the MD processes.
Conventionally, the energy parameters are used to evaluate the properties of a cluster.
This thesis implements the use of geometry of the clusters in additional to the caloric
profile to evaluate the dynamics during cluster melting. Global similarity index, a
purpose-designed algorithm to quantify the degree of similarity between two clusters
is formulated to achieve this objective. It is derived based on the chemical similarity
of molecule and fulfil similar property principle. The heating MD process is carried
out either using direct heating or prolonged simulated annealing. Melting point is
identified by caloric curve, heat capacity curve and global similarity index. The
melting point of hafnium cluster changes with the size of the cluster, 𝑛. In addition to
that, the melting transition happens across a range of temperature, starting with a pre-
melting stage at temperature 𝑇𝑝𝑟𝑒 to total melting at a higher temperature 𝑇𝑚. All the
three methods agree with each other for the range of melting temperature for hafnium

xxi
clusters. However, it is found that global similarity index is much more superior, as it
also traces the melting mechanism of hafnium clusters.

1
CHAPTER 1
INTRODUCTION
We have already entered into an age of uncertainty about Moore’s Law.
(The key conclusion of a presentation by some of the leading technologists at the
Intel Corporation during a press conference dated 4th May 2011.)
The world has lived through the digital revolution, and it is still progressing
rapidly. The shrinking of the silicon microchips is expected to meet its end in these
few years. This is one of the major topics of interest discussed during the latest 2015
International Solid-State Circuits Conference (ISSCC 2015) (Antoniadis, 2015). To
date, one of the latest models is the 14 nm 6th generation core processor
microarchitecture with the codename Skylake by Intel. Beyond the sub-10 nm,
Moore’s Law poses many challenges to microchip manufacturer, such as a more
demanding device geometry design, higher packing density of transistors, and better
performance per cost of manufacturing (Kim, 2015). To resolve the 10 nm
technological bottleneck in the near future, researchers are hoping for a new material
as a replacement for silicon. Schlom et al. (2008) reported the existing problems within
the silicon oxides transistors and a possible replacement by a hafnium-based dielectric.
Some other possible candidates do exist, such as the rare-earth LaLuO3 which has a
higher dielectric constant. However, the high melting temperature of the proposed
alternative substances increases the cost of fabricating transistors made of these
substances.

2
The understanding of the properties of hafnium is essential in order to fully
utilize this element in microchip manufacturing. In particular, the properties and
thermal behavior of nanoscale hafnium allotropes have not been well studied so far.
Studying the properties of hafnium at the nanoscale is experimentally challenging.
Theoretical modeling and computational simulation hence provide a convenient and
viable approach to complement experimental investigation of nanoscale hafnium.
This work studied the element hafnium in the form of nanoclusters with ranges
from 2 to 99 atoms. The stable ground state structures of hafnium clusters are sought,
and their thermal properties, including their melting behavior, are numerically studied
using molecular dynamics (MD) simulation.
1.1 Computational Simulation of Atomic Cluster
The recent progress in nanotechnology has caused a surge in the interest of
searching for a new generation of nanomaterials with exotic or desirable functionalities.
Some of these newly established materials are nanoclusters and nanoalloys.
Nanoclusters are comprised of fixed number of atoms or molecules that are closely
bonded to each other by atomic forces. The number of atoms or molecules that makes
up a cluster, 𝑛, is normally referred as the size of the nanocluster. Nanoalloys are
clusters which composed of more than one element. In such form, an element is no
longer behaving like an individual atom, molecule or bulk solid. On top of its varying
properties, the structural and energetic behavior of the cluster may also change with
size 𝑛 (Taherkhani and Rezania, 2012).

3
Besides attempts to understand the behavior of the cluster which is dependent
on the size 𝑛 , attention is also focused on addressing the issue of engineering
applications in nanotechnology. In fact, the purpose of studying these nanoclusters is
to obtain a better theoretical understanding at atomic level, and to better control the
production and their application (Baletto and Ferrando, 2005). One trait of nanocluster
is that the properties of the cluster vary as 𝑛 changes. This enables effective tuning of
cluster properties by controlling the size, 𝑛. In some cases, certain properties of the
cluster could be strongly amplified when the size takes on some specific ‘magic
number’. The size-dependent properties and existence of magic number provide a
handy way for nanomaterial design. The second trait of nanocluster is the exhibition
of unique properties that do not occur in their elemental form.
Nanoclusters find their applications in catalysis, magnets design and medical
uses (Ferrando et al., 2006). The catalytic effect of nanoclusters is strongly related to
their geometry, such as the core-shell structures which are commonly found in
bimetallic nanoalloy. For example, Son et al. (2004) demonstrated the example of
Ni/Pd core-shell nanoparticles in catalyzing the Sonogashira coupling reactions in a
more economical way. The magnetic behavior of some bulk metals sometimes displays
a useful nature when they are in the form of a cluster. For example, Park and Choen
(2001) managed to synthesize a magnetic nanoalloy of cobalt-platinum via
experiments. They claimed that these nanoclusters could be used in nanodevice
applications. In biomedical applications, Sun et al. (2006) reported a theoretical study
of the effects of gold coating on the magnetic and structural properties of iron clusters
of various sizes. In particularly gold metal clusters are of interest in the medical field
due to their enhanced optical properties and inert nature of chemical reactions
(Giasuddin et al. 2012).

4
Experiments on a free-standing atomic cluster are rarely reported. For that
reason, the understanding of various properties of the nanoclusters requires
complementary input through computational simulations and theoretical modeling.
The validity of computational simulations founded on the theories of condensed matter.
MD, for instance, required the microscopic variables of the ensemble to rescale
correctly and the interactions between the particles to be appropriately described by an
interatomic potential. Computational simulations of condensed matter systems only
become an intensive and active field of research in recent years due to improving CPU
capability.
The main aim of the studies mentioned above, among others, include
understanding and predicting the properties of material systems at the nanoscale. There
are also studies aimed to improve the technique of simulation. This thesis is an
endeavor to contribute to the research field of computational nanomaterials by
targeting a specific system, the hafnium clusters. Specifically, this thesis attempts an
unbiased search algorithm that is able to locate the global minimum of a free-standing
cluster in a vacuum and performs MD simulations on the cluster systems at elevated
temperatures. The detailed dynamics of the system are analyzed by a novel quantifying
method that detects the chemical similarity of the candidate structures.
1.2 Objectives of Study
This work predicts the melting point and analyze the melting behavior of a
hafnium nanocluster via MD simulations. The interaction is described by an
interatomic potential developed recently, the Charged-Optimized Many-Body (COMB)
potential. The dependability of COMB potential in generating the ground state

5
structures and later on in MD simulations is verified by chemical similarity properties
of clusters. In this thesis, the detailed melting behavior of a nanocluster can be
visualized in a frame by frame video mode by putting together the coordinates of the
atoms in each time step. This approach is successfully being represented by a similarity
index analysis created in this study for the visualized trajectory of clusters’ geometry.
Some of the commonly recognized properties of cluster such as the repetitive geometry
motif and size-dependent melting point are being considered in this thesis as well. The
simulation is capable of yielding quantitative information and providing convenient
qualitative visualization of the atomistic behavior of the cluster during heating process
and melting transition.
1.3 Organization of Thesis
Chapter 1 briefly laid out the recent progress of microchips architect as well as
some background for the computational simulation of nanoclusters. Moreover, the
objectives of study are described in this chapter. The thermal characteristics of a
nanocluster, especially those relevant to the melting transition, are discussed in
Chapter 2. This chapter also introduced the concept of shape recognition, the role of
interatomic potential in molecular dynamics simulation and the method of basin-
hopping as a global optimization method. Chapter 3 covered the computational
methodology used to define the compatibility of COMB potential as well as the MD
simulated annealing procedures for hafnium cluster. The dynamics of heating and
melting transition of hafnium clusters are simulated by using the LAMMPS package.
This chapter also illustrated the steps taken to overcome the problems arose during the
simulation. In Chapter 4, the appropriateness of the choice of COMB potential is

6
discussed by using the obtained ground state hafnium clusters. The results from MD
simulations are then discussed in Chapter 5 by using different post-processing
approaches. Lastly, the conclusions and suggestions are given in Chapter 6.

7
CHAPTER 2
REVIEWS ON RELATED TOPICS
Silicon has played an important part in our lives. However, the jamming of a
circuit will soon become one of the obstacles to bring the world another steps forward.
According to Moore’s Law, the observed number of transistors in an integrated circuit
doubles every two years. Researchers are looking for a replacement for silicon as a
possible way out to overcome the die shrinkage limit of the silicon transistors. Hafnium
was expected as one of the possible element that fulfills all the preliminary tests
according to Schlom et al. (2008). Different allotropes of hafnium might provide a
possible candidate as a replacement for the silicon. To date, the search for silicon
substitution relentlessly continues. This thesis was an effort to investigate the
properties of one of the possible substitutes, hafnium, in the form of nanoclusters. The
result of this study shall contribute a better understanding of hafnium from the
atomistic point of view.
In the first two sections in this chapter, some of the past studies on nanoclusters
and their thermal properties are discussed in general. Section 2.3 gives a brief
introduction of chemical similarity and some of its latest progress. Furthermore, this
thesis also proposes a novel method of similarity index which was derived from the
shape recognition method of chemical similarity. This tool is used to study the detailed
melting mechanism during the phase change in addition to locating the melting point.
Furthermore, the interatomic potential, which is an essential aspect in every MD

8
simulation, is also discussed in the next section. Finally, the last section of this chapter
covers the method of Basin Hopping (BH) which was implemented in a global
optimization algorithm known as PTMBHGA (were further discussed in Section 3.1)
to generate the ground state structures of hafnium clusters.
2.1 All about Nanoclusters
The keyword atomic nanoclusters refer to a group of atoms with the number of
atom, 𝑛, larger than two but smaller than the bulk sized thermodynamic limit. The
main interest of studying nanoclusters was to find the link that relates the properties of
material between the molecular and bulk level. Despite much advancement in the
research front, there were still limited experimental data available on cluster. Even if
there was, most of the studies on atomic nanoclusters were theoretical and simulations.
As a matter of fact, using computational simulation to investigate microscopic system
at the atomic level was consensually accepted as an efficacious tool, as the numerical
modelling used were parameterized based on the experimental data or first-principles
calculations. Commonly accepted methods for first-principles calculation are the
density functional theory (DFT) and the Hartree-Fock method.
Predicting the correct ground state structure of a cluster was a non-trivial task.
From the point of view of computational simulation, the ground state structure
obtained had to be in high geometrical resemblance with the predictions by the first-
principles calculations (Soulé et al., 2004). The set of parameters in particular
interatomic potential that produces the correct structure can later be used to predict
some other physical properties of the nanoclusters. This statement applied generally

9
for all range of empirical interatomic potential and even the high precision quantum
mechanical tight binding approaches.
The small value of 𝑛 in a cluster gave rise to certain unique properties not
present in the bulk solid. The unique symmetry arrangement in the cluster directly
influenced the way electrons were arranged, specifically the valence electrons. For
instance, the electrical conductivity of carbon nanotubes (CNT) can be modulated by
varying the structural orientation of the carbon atoms (Ebbesen et al., 1996). In a
metallic solid, the electrons were scattered on the surface of the atoms as a sea of
electrons. The mobility of electrons on the surface gave rise to the electronic behavior,
such as electrical and heat conductivity. However, the electrons in the nanoclusters
were arranged into ‘shell’ and ‘core’ sites that were induced from the small value of
𝑛. Gould et al. (2015) named this as ‘onion’ like shells of arrangement. The non-
uniform distribution of valence electrons in atomic nanoclusters is a phenomenon
known as the electronic charge transfer, which in turn gives rise to certain electronic
properties that vary independently from their bulk counterpart.
From computational point of view, the limit of the number of atoms, 𝑛 in a
single cluster was limited by the computing power. However, this was not the case in
experiment. Martin et al. (1993) have attempted experimentally to determine the
relationship between melting temperature of sodium clusters and the size up to 𝑛~104.
In fact, 𝑛~104 was not large enough to be considered as a bulk, as the melting
temperature was still lower than the bulk melting temperature, 𝑇𝑚𝑏𝑢𝑙𝑘. Nonetheless, a
group of atoms as large as this was difficult to be processed with computational
simulation.

10
The study of clusters through molecular dynamics simulations was mostly
focused on thermodynamical investigation (Calvo and Spiegelmann, 1999). The
methodology to do this was well established and diverse, but obtaining the lowest
energy structures had always been the primary objective.
The most apparent difference between a cluster and a bulk is the relative
binding energy of the structures. An atomic cluster has a higher surface-atoms-to-
body-atoms ratio as compared to the bulk. Hence, the surface effect in a cluster is
relatively stronger. This causes the atoms in a cluster to be less bonded to each other.
Often the atoms are arranged in core-shell order, and some are stable with only the
shell, without the core atoms. For examples, nanotubes and fullerenes which were
discovered as early as in year 1985. Kroto et al. (1985) showed that C60, which has a
unique geometry, is more stable than all other allotropes of carbon.
Some chemists would describe the arrangement of electrons into a new set of
orbitals. These orbitals are attributed to the entire group of atoms which act as a single
entity. The clusters sometime work as a new chemical species referred to as
superatoms, such as the case of Al13 which acted like a super chlorine discovered by
Bergeron et al. (2004). This cluster was known to be a magic cluster, with magic
number 𝑛 = 13. Magic clusters are clusters that are chemically more stable than other
non-magic clusters.
In order to obtain the resultant orbitals, the construction of a cluster often
involves the imposition of a-priori symmetry constraints. On the other hand, physicists
rely on unbiased search algorithm to obtain not a single structure but sometimes
multiple lowest energy structures. Chuang et al. (2006) reported a few highly
symmetry candidates for the case of Al13, and extended the magic number to 𝑛 = 7,

11
13, 20, and 22. They also showed that the motif of geometry in single elemental
clusters always shows a repetitive unit.
Beside Chuang et al. (2006), Kiran et al. (2005) also confirmed the notion of
the geometrical motif in clusters. They showed that boron nanotubes were formed with
B20 as the cradle motif. B20 was found to have a double ring structure. They showed a
strong connection between the ring and double ring structures for being the ‘embryo’
of single-walled nanotubes. In fact, pure boron clusters have been studied earlier by
Boustani (1997) and Boustani et al. (1999) via a systematic ab-initio method. It was
reported that the boron clusters can be constructed with either hexagonal or pentagonal
pyramids, as shown in Figure 2.1. The nanotubular and the quasi-planar structures
were shown to be relatively stable and acted as basic building motif of larger boron
clusters. Another similar evidence was the case of C20, where the repetitive geometry
motif was observed in the ring-to-fullerene transition (Taylor et al., 1994).
Figure 2.1: Schematic diagram used by Boustani (1997) to illustrate the growth of
boron cluster from the basic unit of hexagonal pyramid B7. By adding the repetitive
geometrical motif, the cluster eventually forms the infinite quasi-planar surfaces or
nanotubes.

12
Besides single element nanocluster, dual element cluster was also interested in
this field of research. Clusters with multiple elements showed larger varieties of
geometrical motifs. Ions in these clusters may arranged themselves into core-shell,
pancake (top-bottom or left-right) or even completely randomized arrangements
(Rossi et al., 2005). Hsu and Lai (2006) arrived at the same groups of geometries after
an extensive search. They included the mixing energy of the clusters to ensure that the
potential energy surface (PES) is thoroughly searched for CunAu38-n (0 ≤ 𝑛 ≤ 38).
The magic number also work on bimetallic clusters such as 𝑛 = 15 and 𝑛 = 38. A
more recent study by Wu et al. (2011) has laid out the exact combination of atom
numbers for Ag-Pd clusters, where the interactions of atoms were modelled with Gupta
potential. In that study, Wu et al. prepared two different sets of parameters for the
Gupta potential. The first one was fitted using experimental data while the second set
was obtained from DFT fitting. They compared the cluster structures for both sets of
parameters and found that silver atoms have the tendency to stay at the surface.
2.2 Melting in Bulk and Cluster
The term bulk solid refers to a collection of sufficiently large number of atoms,
𝑛. On the other hand, the number of atom of a cluster is less as compared to a bulk.
When 𝑛 grows to a certain large number, transition from a cluster to a bulk will occur.
Thus, at this thermodynamic limit of 𝑛, the cluster will eventually behave like a bulk
solid. Crystals and the amorphous solid are two general types of bulk solid. Bulk
metallic solid refers to atoms or ions that occupy the Bravais lattice and possess
periodical symmetry of translation in all the axes. For a perfect crystal, all ions have
the same arrangement and orientation along every direction. Their arrangements are

13
classified into distinctive space group. However, in reality, it is implausible to arrange
a group of 𝑛 atoms in such a uniformity over a wide range of displacement. There are
always imperfections and dislocations within a crystal. Following that, an amorphous
solid does not possess any systematic order in the arrangement of the atoms. The
random arrangement of the atoms of an amorphous solid gives rise to a totally different
macroscopic behavior as compared to the crystalline solid. Some of the common
examples showing such distinctly different behaviors are the existence of high thermal
conductivity, electrical conductivity and magnetism in metallic crystals but not in
amorphous solid.
Atomic nanoclusters share some characteristics of a bulk solid. On top of this,
atomic nanoclusters also resemble the characteristics of a molecule (Menard et al.,
2006). Although nanoclusters do not have finite periodicity in the arrangement of
atoms, they carry a system of their own symmetry similar to that of the molecular
symmetry, which is also known as the point group symmetry. Perfect symmetry only
happens to certain magic number clusters. Aside from these magic numbers, some
clusters are arranged obviously more random than the others (Ahlrichs and Elliott,
1999). This might result in a behavior integrating both crystalline and the amorphous
state.
The crystallization of liquid is a rather complex process, despite the existence
of well-tabulated experimental data. Earlier simulations showed that noble gas (e.g.
helium gas) forms a face-centered cubic structure with the Lennard-Jones (LJ)
potential when the system was cooled. The simulation result agreed with known
experimental observation. However, Barron and Domb (1955) predicted the possibility
of a different form of liquid crystallization at a lower temperature. This complicates

14
the crystallization process of liquid. In addition, as for simple pair potential such as
the LJ potential, the simulation result would change significantly with any slight
modification of the parameters. The functional form of the LJ potential is given in
Equation (2.1)
𝑉𝐿𝐽(𝑟) = 4휀 [(𝜎
𝑟)
12
− (𝜎
𝑟)
6
] (2.1)
where 휀 represents the depth of the potential well, 𝜎 is the interatomic separation at
equilibrium and 𝑟 is the distance between the particles. The fixed parameters here refer
to the numerical value of 휀 and 𝜎 uniquely for different element in different chemical
environment. Any change to these two parameters will completely alter the potential
to model a different element (or no element at all). Table 2.1 presents some examples
of parameters for interaction of noble gases (from He to Xe), some as reported by
Whalley and Schneider (1955).
Table 2.1: The example parameters of LJ potential for the noble gases.
Interactions pair 휀/𝑘𝐵 (K) 𝜎 (Å)
He-He 10.80 2.57
Ne-Ne 36.38 2.79
Ar-Ar 119.49 3.38
Kr-Kr 166.67 3.60
Xe-Xe 225.30 4.10
The multiple transitions of a crystalline form, or commonly known as allotropy
is observed in metal as well. For instance, metal iron (Fe) which is a body-centered
cubic (BCC) at room temperature, becomes face-centered cubic (FCC) at 1183 K, but
back to BCC at 1667 K and finally melts when it goes beyond 1808 K.
Oxtoby (1990) laid out some of the most common problems in the liquid-solid
transition in a bulk crystal. The notion of equilibrium that underlies phase change,

15
namely the coexistence of phases at critical temperature, 𝑇𝑐 is widely accepted. During
the latent heat of fusion (melting), the thermodynamic properties are easily
documented, but the microscopic changes to the structures between the coexisting
phases are not quite understood. There are still some unsolved phenomena such as the
local nucleation within a bulk or the dislocation of impurities and surfaces. The
dynamical studies of non-equilibrium growth in the microscopic level are very
different from the macroscopic observables.
In fact, some of the known premonitory effects close to the melting transition
are observed in bulk crystal. For example, substantial changes in volume,
compressibility, heat capacity and electric conductivity are observed in bulk crystal
long before the bulk melting point 𝑇𝑚𝑏𝑢𝑙𝑘 (Dash, 2002). These changes are more
apparent in clusters, and are known as pre-melting effect. It happens at a temperature
𝑇𝑝𝑟𝑒 before the actual melting point 𝑇𝑚. Breaux et al. (2005) studied the pre-melting
effect in aluminium clusters, where the surface melting is observed to occur at a
temperature much lower than 𝑇𝑚.
Surface melting occurs as a major event in clusters. In fact, surface melting is
an important observation that leads to a complete theory of bulk melting. Faraday
(1859) pointed out that the surface melting occurs naturally in any bulk solid. For
instance, the melting of water on the surface of ice causes it to be slippery. In fact, this
finding can be explained indirectly as the wetting of a solid surface. During this
process, any liquid remaining on the solid surface is actually melted from its own
surface. There is a model describing the surface energy in term of contact angle
(Subedi, 2011). Thus, the macroscopic measurable contact angle becomes a direct
measure of the microscopic free energy of the surface liquid layer. Through this

16
approach, we are able to predict more properties of surface melting and the existence
of the metastable state.
Naturally, the interest of study is to find out the asymptotic behavior of atom
to bulk. The gradual change of thermal properties with the of size of the clusters, 𝑛, is
not fully understood. Nevertheless, melting temperature of a cluster being size-
dependent is widely accepted, due to most findings supporting the statement (Duan et
al., 2007; Liu et al., 2013; Neyts and Bogaerts, 2009; Zhao et al., 2001). However, as
reported by Martin et al. (1993), even at 𝑛~104, the melting temperature of sodium
clusters is still lower than the bulk value. It was stated otherwise by Calvo and
Spiegelmann (1999) where they suggested the pre-melting, 𝑇𝑝𝑟𝑒 effect to be taken into
consideration. From their findings, the melting transition in sodium clusters is said to
be at 𝑛 > 93. The core-shell structures of the clusters contribute a significant effect to
the surface melting and thus the pre-melting phenomena. Experiments have led to a
homogeneous melting model (Effremov et al., 2000) that relates the reduced melting
point of a cluster, 𝑇𝑚 to that of the bulk, 𝑇𝑚𝑏𝑢𝑙𝑘 using
𝑇𝑚 = 𝑇𝑚𝑏𝑢𝑙𝑘 −
𝛼
𝑟 (2.2)
where 𝛼 is a positive quantity with the dimension of [L][θ] which can be determined
experimentally, and 𝑟 is the radius of the particle or cluster. This relationship was
derived by Buffat and Borel (1976) from Gibbs-Duhem equation. In the following year,
Couchman and Jesser (1977) outlined a direct theoretical study on Sn, In and Au
clusters to predict their surface melting. The thermodynamic theory has successfully
quantified three features of cluster melting:
1. Melting is initiated at the surface.

17
2. The existence of an upper and lower limit of the range of melting
temperature, 𝑇𝑚.
3. Characteristics of surface nucleation and liquid layer growth are
qualitatively captured.
The thermal properties of bulk solid can be obtained through some
macroscopically measurable quantities, specifically thermodynamics quantities such
as pressure, 𝑃, density, 𝜌, and temperature, 𝑇. The thermal properties of a cluster, on
the other hand, needs to be obtained by indirect means. Schmidt and Haberland (2002)
revised an approach to measure the melting temperature, latent heat, and entropy of
some bigger sodium clusters of size ranging from 𝑛 = 55 to 𝑛 = 357 , which are
derived indirectly from the caloric curve.
Many experimental studies (Schmidt and Haberland, 2002; Martin et al. 1993)
have established the following fact regarding the melting of free cluster in comparison
to bulk counterpart, namely:
1. The melting temperature, 𝑇𝑚 of a cluster is generally lower than the
bulk value, 𝑇𝑚𝑏𝑢𝑙𝑘.
2. The latent heat of transformation is smaller than the bulk.
3. The melting stage does not occur at a fixed temperature, but begin with
pre-melting, 𝑇𝑝𝑟𝑒 over a finite range of temperature.
4. The heat capacity of the finite-sized system can sometimes take a
negative value.
In fact, statement one and two are analogous, since the melting temperature,
𝑇𝑚 and the latent heat of a cluster show similar fluctuations, while pre-melting is

18
widely observed though experiments. The first three statements were discussed earlier,
but the fourth seems unnatural. A negative heat capacity implies that energy is
absorbed with decreasing in the cluster temperature. Heat absorption during melting is
commonly understood in terms of latent heat of transformation, where the mean kinetic
energy tends to remain constant. In their paper, Schmidt and Haberland (2002) have
explained that a finite sized system tends to convert some of the kinetic energy into
potential energy in order to avoid partially molten states. Phenomenological
observations related to the thermal behavior of cluster will be further discussed in the
next chapter from the view point of computational simulation.
In computational simulation, the concept of melting as applicable in the bulk
can be similarly applied in microscopic systems. This is a benefit to molecular dynamic
simulation whereby the same set of equations of motion is used to solve the interatomic
interaction for any kind of system, be it bulk or microscopic.
In a bulk system which is typified by a size of ~1023 atoms, MD simulation is
performed by imposing periodic boundary condition to mimic an extensive body
which is formed by a periodic repetition of supercells. However, for finite system such
as a cluster, it is possible to simulate the movement of every single atom. The
microscopic properties, such as binding energy, temperature, entropy and density of
the cluster could be easily computed by sampling the trajectory of every atom for every
time step. As far as molecular dynamics are concerned, a free-standing cluster in the
vacuum which is practically difficult to set up in experiment can be computationally
simulated by choosing a proper empirical interatomic potential. In many occasions,
the computational simulation can become relatively cheap to perform. With the ease

19
of acquiring simulation data, the focus of research effort can hence be devoted to the
extraction of physical information from the cluster system.
A cluster behaves differently from both crystals and amorphous solid. The
commonly accepted Lindemann criterion of melting is not completely accurate to
predict the phase change in cluster. One particular reason for this discrepancy is that
the thermal instability of a free-standing cluster creates some errors within the effective
range of the interatomic potential in the simulation. This arises due to the existence of
multiple basins along the PES. Thus, the initial structure for the simulation has to be
ensured such that it lies within the basin of the global minimum (Leary 2000).
However, for an expensive interatomic potential, the time required to globally
optimize the initial structure is tremendously long and computationally expensive. The
numerous approximations and functional form of empirical interatomic potential
become a limiting factor as to how accurate a simulation can resemble the real system.
This also gives rise to error while solving the equation of motions.
MD simulations allow us to study various characteristics, such as the surface
and the core-shells models. Duan et al. (2007) in their molecular simulation of pure Fe
cluster showed that for large clusters such as Fe300, the surface can exist as molten
phase while the core as solid phase at the same time. The temperature range of melting
is, however, narrow and precise. This coexistence of different phases on the surface
and in the core proves that the melting of different shells at a different temperature is
probable even for pure element clusters. Logically, one would expect that only a core-
shell clusters of different elements to undergo this kind of melting pattern. Duan et al.
(2007) also made a conclusion that the coexistence is over time for small clusters;
whereas the coexistence is over space for big clusters.

20
When the cluster size is relatively small, the values of melting point, 𝑇𝑚 ,
instead of decreasing with √𝑛3
, could show oscillation. The peaks in the oscillation can
be explained by the existence of magic number in finite sized clusters. Such
observation was made by Schmidt et al. (1998) in their experiment where the melting
point of Na147 is higher than Na130 by ~60 K. The experiment by Schmidt et al. (1998)
was considered very sophisticated at that time. It can be said that when the cluster size
is very small, the experimental studies become extremely difficult. On the other hand,
computational simulation enables easy manipulation of cluster species which will be
a complement to the experimental limit.
Another benefit of computational simulation is that the composition and
geometrical constraint can be controlled to create a large variety of structures. Kuntová
et al. (2008) performed extensive studies on Ag-Ni and Ag-Co bimetallic nanoalloys.
They picked only the highly symmetric magic clusters as their candidates, namely
Ag72Ni55, Ag72Co55, Ag32Ni13, and Ag32Co13. According to the simulation result Ag
atoms tend to be sitting in the shell while both the Ni and Co atoms were in the core.
Even though the structures of the nanoalloys are similar, the melting behaviors are
different. Melting of clusters is greatly affected by the nature of the potential energy
surface (PES) that causes a large behavioral change.
The melting data obtained via simulation can be used to study the dynamics of
phase change. Comparison between commonly used post-processing methods were
discussed by Lu et al. (2009). The most commonly used post-processing method is
caloric curves, whereby the binding energy (or binding energy per atom) is plotted
against temperature. Another method is the constant temperature specific heat capacity,

21
𝑐𝑣, as a function of temperature, which is actually the fluctuations of the caloric curve,
given by the equation
𝑐𝑣 =⟨𝐸𝑡
2⟩𝑇 − ⟨𝐸𝑡⟩𝑇2
2𝑛𝑘𝐵𝑇2
(2.3)
where 𝐸𝑡 is the total energy of cluster, 𝑘𝐵 the Boltzmann constant, 𝑛 the total number
of atoms in the cluster and ⟨ ⟩𝑇 represent the thermal average at temperature 𝑇. A
typical 𝑐𝑣 curve appears in the form of a sharp peak at the melting temperature, 𝑇𝑚.
Nevertheless, the two methods mentioned above are not the only methods for
quantifying the melting behavior of clusters. Some characteristics from simulated
annealing process have to be obtained with special treatments. Lu et al. (2009) showed
that there is a mismatched in the melting temperature of Co13 and Co14 clusters, if
Lindemann index, 𝛿, was used as a mean to gauge the melting process, as compared
to 𝑐𝑣 curve. Lindemann index is given by the equations
𝛿 =1
𝑛∑ 𝛿𝑖
𝑖
(2.4)
𝛿𝑖 =1
𝑛 − 1∑
(⟨𝑟𝑖𝑗2⟩𝑇 − ⟨𝑟𝑖𝑗⟩𝑇
2 )12
⟨𝑟𝑖𝑗⟩𝑇𝑗≠𝑖
(2.5)
where 𝑟𝑖𝑗 is the distance between the 𝑖th and 𝑗th atoms. Lastly, the dynamics during
the cluster melting is studied by short-time averaged distance ⟨𝜎𝑖(𝑡)⟩𝑠𝑡𝑎, given by the
equation
⟨𝜎𝑖(𝑡)⟩𝑠𝑡𝑎 = ∑|𝑟𝑖(𝑡) − 𝑟𝑗(𝑡)|
𝑗
(2.6)

22
where 𝑟𝑖(𝑡) represents the position of the 𝑖th atom at time 𝑡, while ⟨ ⟩𝑠𝑡𝑎 denotes that
the average is taken for a short interval of time steps and then plotted against the time.
Similar method had been used earlier by Aguado et al. (2001) on Na cluster.
Computing the time-average value is troublesome but somewhat could be a solution
to the thermal instability in the simulated annealing procedure. The problem of thermal
stability is a big obstacle in simulation of cluster melting. Details on molecular
dynamics methods used in this thesis were discussed further in Chapter 3.
2.3 Chemical Similarity and Shape Recognition
The method of molecular shape comparison is used in comparing the geometry
or spatial configuration of two or more molecular structures to identify the chemical
similarity between them (Grant et al., 1996). Molecular shape comparison is an
important field of research and application. It is a method with a wide range of
applications in the field of informatics, cheminformatics and bioinformatics, such as
drug discovery, screening in pharmaceutical studies, nucleic acid sequencing of
biological data, protein classifications and identification. The development of
structural classification of proteins remain important today, and it is continually
improving. Andreeva et al. (2013) recently improved their prototype of structural
classification of proteins to the second generation SCOP2 (http://scop2.mrc-
lmb.cam.ac.uk/ 1st March 2016). A sample screenshot of using SCOP2 graph viewer
is attached in Figure 2.2, showing the classification of Cro regulator proteins based on
structural properties and relationships.
The usefulness of molecular shape comparison lies in its ability to transform
data into information which in turn leads to a better decision making in drug lead

23
identification and optimization (Brown, 2005). Shape recognition is by far a method
most suitably applied on static molecules. One practical example of dynamical system
is the sequential changes of biological molecules across generations. However, it lacks
the flexibility to make pattern prediction in dynamical systems which involve constant
change in their configuration throughout their historical evolution.
Figure 2.2: Sample of SCOP2 graph viewer result given by Andreeva et al. (2013),
showing the Cro types protein sequence and structure.

24
Virtual screening is a computational procedure to search for chemical
similarity to identify and compare the structures of molecules or coupounds from a
standard database library, such as the protein data bank. There are different approaches
to virtual screening, one of which is similarity-based virtual screening. The formalism
of similarity based virtual screening is based on the similar property by Johnson and
Maggiora (1990), which stated that the coupounds with higher structural similarity
tend to have similar chemical and biological activities. With a suitable approach, one
can assign a probability to the activity of the structure under study with reference to
the known sample from the database of compounds. However, in the field of
informatics, different organizations or companies provide their own unique ways to
test for chemical similarity. The degree of similarity between the compared structures
predicted by different approach can be differed from one another.
The selected structures for shape identification are normally represented by a
binary molecular fingerprint of descriptors, also known as structural keys that containt
various visualisable information. For example, the size of the molecule, the number of
bonds or type of bondings involved, the active functional groups and the pattern of
target structure or substructure (http://www.daylight.com, 1st March 2016). Some
descriptors might carry a certain portion of information that outweight others and
become less universal for a certain group of compounds. Thus, certain descriptors can
work better and faster when the information density is lower, which does not always
necessarily so. The descriptors are generally calssified into three types based on the
dimensionality of the descriptors. The two-dimensional descriptors such as MACCS,
MDL keys, and Daylight are said to perform better than the three-dimensional ones
(Oprea, 2002). These two-dimensional descriptors are mostly patented under their own
company signature. MDL two dimensional descriptors have been designed to be used

25
for the saearch of substructures. Durant et al. (2002) managed to re-optimize the
existing 166 bit and 960-bit keysets of the time to increase the number of success
measurements. The newly designed descriptor was found to have equal performance
although the keysets are composed differently without overlapping. It seems that the
construction of the keysets is bound to some known constraints, thus prompting a
possibility of further studies to enable the construction of keyset that is data size
independent (Zhu et al., 2016).
Three-dimensional descriptors were difficult to compute until Ballester and
Richards (2007) proposed the ultrafast shape recognition (USR) method based on four
sets of distance distribution of the atoms in a molecule defined at different points of
reference. The geometries of the atomic configurations are described by a total of three
statistical moments, namely the mean, variance and skewness. It turns out that these
USR descriptors are orientation independent, so that molecules being screened do not
need to be aligned. USR is said to perform at least three order of magnitude faster than
other descriptors. The similarity index is denoted as 𝑆𝑞𝑖 ∈ (0,1) . A value of 1
represents “totally identical” while 0 means “vastly differed”. 𝑆𝑞𝑖 is given by the
equation
𝑆𝑞𝑖 = (1 +1
12∑|𝑀𝑙
𝑞 − 𝑀𝑙𝑖|
12
𝑙=1
)
−1
(2.7)
where the moments of shape descriptors 𝑀𝑞 and 𝑀𝑖 represent the query and the 𝑖th
molecule.
Later in the same year, Cannon et al. (2008) attempted to combine the binary
166 bit MACCS keys and the USR with an additional four extra moments based on

26
kurtosis of the distributions. The hybrid descriptors were shown to yield a better result
as compared to binary 166 bit MACCS keys or USR. The performance of the above
hybrid descriptors was assessed by considering the accuracy and effectiveness of the
algorithm. The performance is tabulated according to different measures, such as the
percentage of actives recalled in the top 1% and top 5% of the ranked validation sets,
precision of predicted positives, area under the Receiver Operating Characteristic
curve (AUC), the F-measure and Matthew Correlation Coefficient (MCC). All these
methods are statistical set-up to measures the performance of the algorithm.
To get a glimpse of how robust the work of Ballester and Cannon is, other
works with similar task were referred. The details of the formulation will not be fully
discussed here; only the concept of matching is explained, along with the commons
and differences as compared to other similarity indices. The choice of notation may be
modified from the references for easy comparison. Grant et al. (1996) were among the
earlier successes, where the process of matching two molecules was worked out by
aligning two structures A and B in order to obtain a maximum intersection volume.
The alignment problem was solved by optimizing the rotation and translation of the
comparing structures with respect to one another. Then, the normalized similarity
index, 𝑆𝐴𝐵 can be obtained via the equation
𝑆𝐴𝐵 =2 ∫ 𝑑𝒓𝜌𝐴
𝑔𝜌𝐵
𝑔
∫ 𝑑𝒓(𝜌𝐴2 + 𝜌𝐵
2)≡
2𝑉𝐴𝐵𝑔
𝑉𝐴2 + 𝑉𝐵
2 (2.8)
where the volume of intersection, 𝑉𝐴𝐵𝑔
is the numerator part of the 𝑆𝐴𝐵, which is given
by

27
𝑉𝐴𝐵𝑔
= ∫ 𝑑𝒓𝜌𝐴𝑔
𝜌𝐵𝑔
(2.9)
The term 𝜌𝜒𝑔
, 𝜒 = 𝐴 or 𝐵 are the Gaussian densities and can be represented in terms
of the spherical Gaussian, 𝜌𝑖𝑔(𝑟𝑖) as the product formula
𝜌𝜒𝑔
= 1 − ∏(1 − 𝜌𝑖𝑔
)
𝑖∈𝜒
, 𝜒 = 𝐴 𝑜𝑟 𝐵
(2.10)
𝜌𝑖𝑔(𝒓𝒊) = 𝑝𝑖𝑒
−(3𝑝𝑖𝜋
12⁄
4𝜎𝑖3 )
23⁄
(𝑟−𝒓𝒊)2
(2.11)
where 𝜎𝑖 is the radius of the atom and the Gaussian weight is assumed to be 𝑝𝑖 = 2.70.
The difficulties in shape matching of dissimilar molecules are solved by the idea of
shape multipoles, where each atom is described by merely two parameters, 𝜎𝑖 and 𝑝𝑖.
The shape multipoles are the product of radius and the Gaussian density.
Another recent work by Yan et al. (2013) saw the implementation of the
weighted Gaussian function instead of the common Gaussian approximation with the
use of Tanimoto similarity index. Tanimoto similarity index (Bajusz et al., 2015) is
said to be the most widely used measure of chemical similarity, given by the equation
𝑆𝐴𝐵 =𝑉𝐴𝐵
𝑉𝐴𝐴 + 𝑉𝐵𝐵 − 𝑉𝐴𝐵
(2.12)
In this study, the overlapping volume of the two molecules are given by
𝑉𝐴𝐵𝑔
= ∑ 𝑤𝑖𝑤𝑗𝑣𝑖𝑗𝑔
𝑖∈𝐴,𝑗∈𝐵
(2.13)
The difference from previous works lies in the existence of the weighting factor

28
𝑤𝑖 =𝑣𝑖
𝑔
𝑣𝑖𝑔
+ 𝑘 ∑ 𝑣𝑖𝑗𝑔
𝑗≠𝑖
(2.14)
where 𝑘 is a constant fitted to hard-sphere volume, while the volume terms 𝑣𝑖𝑔
for
atom 𝑖 and 𝑣𝑖𝑗𝑔
for the volume of atom pair intersection are given by equations
𝑣𝑖𝑔
= ∫ 𝑑𝒓𝒊𝜌𝑖𝑔(𝒓𝒊)
(2.15)
𝑣𝑖𝑗𝑔
= ∫ 𝑑𝒓 𝜌𝑖𝑔(𝒓)𝜌𝑗
𝑔(𝒓) (2.16)
Lastly, the spherical Gaussian 𝜌𝑖𝑔(𝒓𝒊) is the same as introduced by Grant et al. (1996),
but the Gaussian weight 𝑝 = 2√2 is multiplied with the same weightage factor, 𝑤𝑖.
Apparently, both the Grant et al. (1996) and Yan et al. (2013) version of 𝑆𝐴𝐵 satisfy
the condition 0 ≤ 𝑆𝐴𝐵 ≤ 1 with same representations of 𝑆𝑞𝑖 by Ballester and Richards
(2007).
The three-dimensional descriptors introduced can be separated into two types.
The first is the direct alignment and explicit shape comparison. This method is said to
be less efficient but fulfills the similar property principle (Fang et al., 2009). The
second method is indirect comparison using the shape descriptor such as the USR.
Although it could be computed rather easily and fast, the representation of the shape
is incomplete. The fast computing USR method has led Hsu (2014) to adopt it in the
study of melting of finite size clusters. The simplicity of USR method is required in
order to handle up to 108 frames of coordinate profile. Another advantage of using
shape recognition method in the study of melting transition is that it can handle any
type of structural sampling. This is due to the fact that this approach concerns solely

29
the geometry of the structures without the need to consider of the nature of the atoms.
The fundamental characteristics of the atoms such as the atomic radius and atomic
mass do not affect how the method is applied to the task. It could reveal the interaction
behavior that is difficult to be detected by other methods as its job only involve
observing the evolution of the trajectories. The effect of shape recognition method in
tracking the trajectory can be viewed as though each frame of coordinates is being
traced like a movie.
Besides the direct shape comparison method, Rogan et al. (2013) revised an
approach for cluster conformation which is based on the distances between atoms in
the cluster. The similarity measure is actually an idea originally proposed by Grigoryan
and Springborg (2003), where the original definitions are given by the equations
𝑑𝑆(𝐴, 𝐵) = [2
𝑛(𝑛 − 1)∑ (𝑑𝑚
𝐴 − 𝑑𝑚𝐵 )2
𝑛(𝑛−1) 2⁄
𝑚=1
]
12⁄
(2.17)
𝑆𝐴𝐵 =1
1 + 𝑑𝑆(𝐴, 𝐵)
(2.18)
where 𝑑𝑚𝜒
is the distance between the atoms in the cluster 𝜒, 𝜒 is the label for cluster
in comparison: 𝐴 𝑜𝑟 𝐵, while 𝑚 labels the distances in ascending order within the
cluster 𝜒. The suffix 𝑆 stands for Springborg. The modified version by Rogan et al.
(2013) is given by the equation
𝑑𝑁(𝐴, 𝐵) = [2
𝑛(𝑛 − 1)∑ (
𝑑𝑚𝐴
𝐷𝑎𝑣𝑒𝐴
−𝑑𝑚
𝐵
𝐷𝑎𝑣𝑒𝐵
)
2𝑛(𝑛−1) 2⁄
𝑚=1
]
12⁄
(2.19)

30
where 𝐷𝑎𝑣𝑒𝜒
represents the average bond length for clusters 𝜒 = 𝐴 𝑜𝑟 𝐵 . Note that
𝑑𝑆(𝐴, 𝐵) has a dimension of length [L] but 𝑑𝑁(𝐴, 𝐵) is dimensionless, and it also
includes a scaling effect by including the sum of the ratio in its definition. Rogan et al.
(2013) tried to plot the comparison of clusters with a color map instead of taking the
same approach of calculating the similarity index 𝑆𝐴𝐵 . Hence, the normalization is
done the other way round with 0 and 1 for maximum and minimum similarity
respectively. The normalization is given by the equations
𝐷𝑆(𝐴, 𝐵) =𝑑𝑆(𝐴, 𝐵) − 𝑑𝑆,𝑚𝑖𝑛
𝑑𝑆,𝑚𝑎𝑥 − 𝑑𝑆,𝑚𝑖𝑛
(2.20)
𝐷𝑁(𝐴, 𝐵) =𝑑𝑁(𝐴, 𝐵) − 𝑑𝑁,𝑚𝑖𝑛
𝑑𝑁,𝑚𝑎𝑥 − 𝑑𝑁,𝑚𝑖𝑛
(2.21)
2.4 Empirical Interatomic Potential
It has become a common practice in the research community whereby
computer simulations are performed to complement experimental investigation. When
a system is approaching microscopic level, molecular dynamics (MD) simulations can
play a crucial role in their theoretical study as experiments became relatively difficult
to be conducted. Over the years, theoretical approaches are based on mathematical
methods developed during earlier time with lots of assumptions and approximations.
On the other hand, MD simulations have attempted to solve the root problem as it is.
Many MD applications are still facing challenges to completely represent the quantum
mechanical problems. MD practitioners try to overcome this problem by working
backward from the existing experimental data. This approach is widely known as the
fitting of parameters empirically or reversed engineering. The confidence in a MD

31
simulation result relies on the choice of a good theoretical model coupled with a
sensible methodology.
In MD simulations involving interatomic interactions, the functional form of
the equations of motion depend on the choice of potential energy term (a.k.a. force
field) that appear in the Hamiltonian of the system. A simulation is bound by a set of
approximations. The approximation methods can be classified into three main types
according to their formulations, namely the first-principle or ab initio, semi-empirical
and empirical (classical). The most widely used first-principles methods are either
density functional theory (DFT) or Hartree-Fock (HF) methods. Although the results
from first-principle calculations agree well with the experiment, it is computationally
expensive and time-consuming. The semi-empirical method uses a certain amount of
experimental data as input parameters for the calculation. The added approximations
and constraints speed up the computational time, but sometimes reduce the accuracy
of the modeling itself. Within this modeling framework, the parameters are
parameterized such that the results agree best with either experimental data or ab-initio
results. Empirical approach presumes systems of balls and springs connected to each
other. The interatomic interaction can be either operating between a pair of atoms, or
include a third entry (angles). Some more advance interaction is many-body in nature
(dihedral).
One of the most commonly used pair potentials is the LJ potential, 𝑉𝐿𝐽(𝑟),
which has been introduced by Equation (2.1) in Section 2.2. Although being less
accurate, the simplicity of calculation has led to its extensive use in many simulations.
The term 𝑟−12 is the short range Pauli repulsion while the term 𝑟−6 is the long range
attraction such as Van der Waals’ interaction and the London dispersion force. Some

32
studies require the LJ potential to work with other effective short range potential to
model a large system with long range interaction. Due to its simple interaction that
resembles the inert system, one of the early motivations of LJ potential in clusters was
to calculate the gas-liquid nucleation of noble gases (Zeng and Oxtoby, 1990). In fact,
the MD study of gas-liquid nucleation of LJ fluid has never stop but continue to gain
more interest in search for better nucleation theories (Laasonen, 2000). Another
notable pair potential is the Morse potential, sometimes replacing the LJ potential for
a better description of long range interaction.
Gupta (1981) has succeeded in improving the classical interaction to account
for the surface separation correction of face-centered-cubic metals. The electronic
charge transfer at the surface was corrected earlier by Finnis and Heine (1974). The 𝑛-
body Gupta potential is fast to compute and converge, according to the equation
𝑉𝑛 = ∑ { ∑ 𝐴𝑖𝑗𝑒𝑥𝑝 (−𝑝𝑖𝑗 (𝑟𝑖𝑗
𝑟𝑖𝑗(0)
− 1))
𝑛
𝑗=1(𝑗≠𝑖)
𝑛
𝑖=1
− [ ∑ 𝜉𝑖𝑗2 𝑒𝑥𝑝 (−2𝑞𝑖𝑗 (
𝑟𝑖𝑗
𝑟𝑖𝑗(0)
− 1))
𝑛
𝑗=1(𝑗≠𝑖)
]
1 2⁄
}
(2.22)
where 𝐴𝑖𝑗, 𝜉𝑖𝑗, 𝑝𝑖𝑗, 𝑞𝑖𝑗 and 𝑟𝑖𝑗(0)
are the parameters to be fitted with bulk values. The
fitting enables a certain degree of correction to the classical potential. As can be seen
in the form of equation, Gupta potential remains intact as pair-wise potential with the
following general form of equation
𝑉𝑝𝑎𝑖𝑟 = ∑(𝑉𝑟𝑒𝑝𝑢𝑙𝑠𝑒 + 𝑉𝑎𝑡𝑡𝑟𝑎𝑐𝑡) (2.23)

33
A recent study by Rogan et al. (2013) demonstrated that the choice of pair
potential is almost subtle when quantum refinement is introduced. The comparison
was done on the Gupta, Sutton-Chen, and the LJ potentials, where these three
potentials basically yield almost similar structures for small clusters of Ni and Cu after
a further re-optimization with DFT. The most concerned objective of MD simulation
is the accuracy of an empirical interatomic potential being able to predict the dynamics
of its respective system. The outcome of the prediction need to be tally with the DFT
(at zero Kelvin) or the experimental result as well. In this thesis, this procedure is used
to ensure the appropriateness of the choice of interatomic potential.
One can generalize the interaction for 𝑛 interacting particles beyond the pair
potential as the sum of contributions from one-body, two-body, three-body terms, etc.
as the serie,
𝑉𝑒𝑓𝑓(1, … , 𝑛) = ∑ 𝑣1(𝑖)
𝑖
+ ∑ 𝑣2(𝑖, 𝑗)
𝑖,𝑗>𝑖
+ ∑ 𝑣3(𝑖, 𝑗, 𝑘)
𝑖,𝑗>𝑖,𝑘>𝑗
+ ⋯
+ 𝑣𝑛(1, … , 𝑛) (2.24)
For an effective representation, 𝑣𝑛 should converge to zero as 𝑛 increases. The
first term corresponds to the external forces and is normally not included in the
Hamiltonian. Many recent popular interatomic potentials are derived based on this
form of equation with their own significant successes. To name a few, there are
Stillinger-Weber (SW) potential (Stillinger and Weber, 1985), Tersoff potential
(Tersoff, 1988), the improved version of both Reactive Empirical Bond Order (REBO)
potential (Brenner et al., 2002) and the Adaptive Intermolecular Reactive Empirical
Bond Order (AIREBO) potential (Stuart et al., 2000). The term ‘generation’ is usually
adopted to differentiate the version of their development, such as the first generation

34
Brenner potential for the 1990 REBO potential (Brenner, 1990) for hydrocarbons and
second generation Brenner potential in 2002. Nevertheless, for Brenner case, the first
generation REBO potential underwent a drastic improvement in both the analytical
equation and the extension in the fitting database.
The practicality of many-body potentials has been routinely demonstrated
whereby numerous structures are successfully predicted. One of the latest examples is
the growth of graphene on a silicon carbide substrate by the simulated annealing
process (Yoon et al., 2013). In the study, Tersoff potential and a modified version of
Tersoff-Erhart-Albe (TEA) (Erhard and Albe, 2005) potential were compared with the
formation of the graphene layers. Even though graphene was not discovered during
1988, Tersoff was able to predict every possible combinations and permutations of
carbon-carbon interaction. The capabilities of the potentials to predict structure rely
on the parameters during fitting. These type of potentials are known as bond order
potentials (BOP). In MD, BOP are best suited to describe the bonding states of the
atoms to includes various bonding states between a pair of atoms. The fitting of the
parameters includes the consideration from the number of bonds, angles, and bond
length.
Pair potential is not suitable to describe directional interaction where a third
particle is involved. The shortcoming can be solved by incorporating a term, 𝑣3, which
works to include more properties of the structures by taking into account the
contribution from more experimental data. Consequently, the additional terms stabilize
the structure. Thus, most MD simulations are carried out with many body potentials.
Ng et al. (2015a) discussed some of the widely used semi-empirical interatomic

35
potential in MD simulations in Figure 2.3, in the direction of increasing computational
cost.
Figure 2.3: Commonly in use interatomic potential in increasing computational cost
Ng et al. (2015a).
Perhaps the most widely studied material in computational simulation is silicon.
Biswas and Hamann (1987) stated that three-body potential was insufficient to fully
represent the silicon clusters. The structures are more realistic when the potential
involves four- or five-body terms, but it would have too many parameters to be fitted.
The idea digresses from the of n-body form, focusing on building a potential that
directly contain the physics required to describe the structure of the element. Tersoff
(1988) was among the earliest to propose the role of geometry in the form of potential
function. The functional form of analytical function was formulated to include the
bond order parameters, fitted based on the coordination of the candidates.
Material scientists are trying to include more properties into the formulation of
classical potential. The quantum mechanical behaviors are often taken into
consideration as much as possible when constructing such potential. Electron Force
Field (eFF), which is still at developing stage at Caltech (Jaramillo-Botero et al., 2011),
is an example of such type of potential. They demonstrated an effective dynamics
modeling with the first-principles based eFF, with parallel computing. In eFF, both
nuclear and electron are mobile and contained a set of equation for their motion. The
time-dependent Schrodinger equation is used to obtain the semi-empirical relation for
SW Ab initio
EAM Tersoff
REBO
SPC/E
CHARMM
MEAM
BOP
AIREBO
ReaxFF
eFF
COMB
SNAP DFT
Cost (sec/atom, time-step)

36
their independent motion. This consideration enables eFF to be applied in any
calculations that involve electron excitation.
Rather than relying on a computationally expensive interatomic potential, an
improved MD method will help to boost up the performance of simulations as well.
Rick et al. (1994) revised an additional step in MD by enabling point charges to be
considered in the interaction via electronegativity equalization (Qeq) method. This
additional charge equilibration step has been included in the formulation of Charge-
Optimized Many-Body (COMB) potential by Yu et al. (2007) for the system of Si and
SiO2. It is effective in predicting the correct properties of the elements. The COMB
potential is modified by the Tersoff potential. The fitting procedure of the parameters
is similar to that of Tersoff potential.
This thesis uses the second generation COMB potential (Shan et al., 2010) in
predicting the structure of clusters. The development of COMB potential and the key
characteristics were discussed in details in Chapter 3 along with the methodology
involved.
2.5 The Method of Basin Hopping
Basin-Hopping (BH) is an unbiased global optimization method introduced by
Wales and Doye (1997). The complexity of the PES determines the complication in
obtaining the global minimum structure, which corresponds to the structure with the
lowest energy. The objective of BH is to scan and later transform the PES into a
simplified staircase topology. Hence, it is very efficient to locate the minima. The
transformed energy �̃�(𝑋) is defined by

37
�̃�(𝑋) = 𝑚𝑖𝑛{𝐸(𝑋)} (2.25)
where 𝐸(𝑋) represents a certain point on the PES with 𝑋 being the 3𝑛-dimensional
position coordinates of the atoms, {𝑟1, 𝑟2, … , 𝑟𝑛} . {𝑟𝑖} carries the set of Cartesian
coordinate of atom 𝑖 in the form of {𝑥𝑖, 𝑦𝑖 , 𝑧𝑖}.
Figure 2.4: A schematic sketch to illustrate the effect of BH transformation to the
PES of a one-dimensional example.
Wales and Doye (1997) presented a schematic example to illustrate the effect
of the BH method in a one-dimensional PES as shown in Figure 2.4. The solid line is
the original energy profile while the dashed line is the staircase topology after the
transformation of Equation (2.25). Based on the sketch, the effect of scanning through
the PES should yield the same minima as the staircase topology. Essentially, the BH
method works in combining the deterministic and stochastic methods in a Monte Carlo
approach.
The BH method was adopted as part of a global search algorithm by Hsu and
Lai (2006), in their full-length algorithm called parallel tempering multi-canonical
basin hopping plus genetic algorithm, PTMBHGA. The effect of transforming the PES
simplifies the dynamics of geometrical search, even with much more complicated

38
potential such as Gupta potential instead of the simplified LJ potential. The tendency
of trapping in saddle point was also solved by working the multi-canonical BH (MBH)
together with Genetic Algorithm (GA) and parallel tempering Monte-Carlo (PT)
method, hence the name PTMBHGA.
Many who study cluster structures have come to realize that the global
minimum of an interatomic potential does not always correspond to the result obtained
from DFT calculations. Oganov and Valle (2009) proposed that there are probabilities
that the low-lying structures (LLS) might include the global minimum. Sometimes, the
quantum refined global minimum happens to coincide with one of the higher energy
local minimum instead of the global minimum of the empirical potential. Hence, the
method of BH became even more powerful because the purpose of searching the
minimum was aimed at locating a set of minima along the PES. This approach is
adopted in this thesis to locate the unique ground state structures of hafnium cluster.
During the search for the global minimum structure of a cluster, the effect of
electronic charge transfer is an important factor affecting the outcome of the structural
geometry (Mollenhauer and Gaston, 2016). However, the effect of electronic charge
transfer is not taken in account in most of the less computationally intensive potentials.
Hence, the global minimum obtained from empirical interatomic potentials may not
always be the one correspond to the real case.
The use of LLS was introduced by Hartke (1996, 1998) in an attempt to
improve the global minimum search based on ab initio methods such as DFT. The
example of using LLS in the global minimum search can be explained in a schematic
sketch as shown in Figure 2.5. Bear in mind that the final aim is to obtain the true
global minimum of given cluster size at DFT level. In practice, the true global

39
minimum is formidably difficult to identify directly with finite computational resource.
The overall strategy is to arrive at this true global minimum via an indirect route. In
stage 1 of Figure 2.5, BH method is used to transform the PES into a simplified
staircase topology which contains a collection of LLS in different basins. These LLS
are indicated as green triangles in Figure 2.5. The LLS obtained in this stage will not
be minimized into the local minimum of each basin as only a minimal default setting
for energy calculation will be used (where no powerful minimization algorithm will
be called) so that the PES can be scanned through at a higher efficiency (at a fixed
computational resource constraint). Stage 1 is a slightly faster process as compared to
stage 2. In stage 2, the LLS from stage 1 are geometrically re-optimized (opt; indicated
as down arrows in Figure 2.5) into their corresponding local minima using an
expensive computational setting. These geometrically re-optimized structures are
labelled by red diamonds in Figure 2.5 which are seen sitting at the local minimum of
each basin. These red diamond are originally the LLS at stage 2 level. Geometrical re-
optimization process in this strategy provides a mechanism to transform the LLS in
stage 1 into the local minimum in each basin. Now, each basin contains a cluster
structure with unique geometry. The lowest energy structure among these LLS is
considered as the true global minimum structure.
The same strategy as described above was employed to generate free-standing
hafnium cluster for geometrical comparison in this thesis, which was discussed in
detail in Chapter 4.

40
Figure 2.5: A schematic sketch indicating the strategy to obtain true global
minimum by the way of sampling LLS at a coarse level search with BH method
without structural optimization (stage 1), and subsequently undergo a refined
geometrical re-optimization of these LLS using DFT method (stage 2). The doted
profile in stage 1 represent the simplified staircase topology of the PES.
Computationally expensive
process
Relatively fast process
Stage 1
Stage 2
PES
Basin containing the global minimum.
True global minimum
LLS of BH scanning at stage 1 Local minimum of each basin
upon geometrical optimization Opt

41
CHAPTER 3
METHODOLOGY
The simulation in this thesis can be conceptually separated into three
distinctive stages as shown in Figure 3.1. The first stage is to locate the ground state
structures of hafnium clusters with COMB potential to minimize the preliminary error
that could propagate into the second stage, which is the simulated annealing process.
Although the two stages are carried out separately, the choice of the interatomic
potential has to be consistent, as if a single set of laws is governing the whole process.
The resultant output from stage two is then brought to a series of post-processing
algorithm to understand the dynamic mechanism of the heating process.
Figure 3.1: Flow chart of the general layout of methodology.
Generating the ground
state structures
• generate using PTMBHGA
• tunning of the global minimisation
• validate the appropriateness of interatomic potential
Simulated annealing procedure
• using the same interatomic potential from the previous stage
• optimized the setting for simulations, such as heating method, heating rate, thermostat, etc.
• fixing errors arosed from the simulation
• 1 K relaxation
• heating to a target temperature and equilibrated for an extended period of time
Post-processing
• tabulating of data
• plot caloric and specific heat capacity curves
• produce the global similarity index
Stage 1
Stage 2
Stage 3

42
3.1 PTMBHGA
As mentioned in Section 2.2, the initial structure (input structure) of MD
processes should lie within the basin which contains the global minimum. In cluster
science, the geometry of the cluster should agree well from those obtained from DFT.
This means that the input structure is expected to yield the global minimum after the
quantum refinement, an issue which was discussed earlier in Section 2.5. The success
of a global optimization search algorithm depends on its ability to align the PES of the
empirical potential in use against a reliable DFT reference. MD simulation is
computationally cheaper than DFT. The former has another advantage over the latter
for being able to measure the temperature-dependent dynamics at a much lower
computational cost. The reliability of an interatomic potential depends on its ability to
obtain an accurate ground state structure which should agree well with the DFT result.
Locating a correct ground state structure is the prerequisites in any MD simulation of
a cluster. In fact, this is the first step as indicated in Figure 3.1.
To accurately locate the global minimum of a cluster, this thesis adopts part of
the global searching algorithm developed by Lai et al. (2002) called parallel tempering
multi-canonical basin hopping plus genetic algorithm (PTMBHGA). The algorithm
contains two global optimizers that complement each other. The basin hoping (BH)
scans through the PES and generates 20 parent candidates which are scattered over the
minima of the PES. Genetic algorithm (GA) would then discard 5 and regenerate new
candidates to explore the PES in a guided way so that a new breed of next-generation
atomic clusters is formed. The process is then repeated, i.e. for every generation, 5
‘genetically’ unfit candidates are discarded and 5 new candidates are generated
through GA, making the collection of atomic clusters always fixed at 20. GA also helps

43
BH to escape the trap in saddle point by introducing new offspring which diverges into
the different basin. This BH-GA process will cease once the number of similar
structure, all having local minimum energy value, achieved a predefined value, 7
structures to be exact. Overall, the fitness value 𝑓𝑖 of the candidates is evaluated during
all stages as a normalized control in deciding which candidates are to be discarded
based on the equations
𝑓𝑖 =𝐹𝑖
∑ 𝐹𝑗20𝑗=1
(3.1)
𝐹𝑖 =𝑉𝑚𝑎𝑥 − 𝑉𝑖
𝑉𝑚𝑎𝑥 − 𝑉𝑚𝑖𝑛
(3.2)
where 𝑉𝑖 is the potential energy of the cluster 𝑖, to be calculated using COMB potential
in this thesis.
It can be seen that from the equation for 𝑓𝑖, the fitness value is defined for a
particular cluster 𝑖 by comparing it to the group of currently available candidates at
that particular generation. Basically, the single structure might have different fitness
value when some candidates were being discarded and replaced. Hence, the same
fitness value can also act as a tolerance control in the loop, as to when the algorithm is
expected to yield the global minimum. The loop control of this current work requests
more than 6 lowest candidates with identical fitness to simultaneously exist.
The PTMBHGA code is comprised of two independent parts, the global-search
algorithm part (comprised of GA and BH search algorithm) and an ‘energy calculator’
part which calculates the energy of any given cluster configuration generated by the
global search algorithm. The original energy calculator in the PTMBHGA code was a
built-in Gupta potential. In the current study, the code was extended and modified in

44
order to integrate with the MD package LAMMPS (Plimpton 1995) and the DFT
package, Gaussian 03 (dubbed G03). LAMMPS is an efficient MD software with
numerous preloaded interatomic potentials. This ‘plug and play’ package enables a
versatile energy calculator selection to the already powerful global-search algorithm.
This modified version of PTMBHGA code will use LAMMPS and G03 as the ‘energy
calculator’. The COMB potential with charge optimization from the energy calculator
LAMMPS is called when evaluating the energies of the cluster generated by the GA-
BH algorithm in the PTMBHGA. The PTMBHGA + LAMMPS hybrid package is
dubbed ‘plmp’ in this thesis. In the case where G03 is used as energy calculator, the
PTMBHGA + G03 hybrid package is dubbed ‘pg3’.
Another technical detail to mention here is the option ‘switching off’ which is
available in the GA part of PTMBHGA. It controls how a new generation of
configurations are generated and discarded. When the option ‘switching off’ is evoked,
the discard and regeneration of configurations were done by stochastic means of
Monte-Carlo method. This option speeds up the global minimum search when the
potential function becomes relatively complicated. The reduced cycle cuts down
unnecessary iterations when computing the potential energy to achieve the objective
function.
A schematic flow chart of the modified versions of PTMBHGA used in this
thesis are shown in Figure 3.2, adapted from Lai et al. (2002). This flow chart laid out
the important steps in the algorithm.

45
Figure 3.2: Flow chart for the algorithm in the hybrid PTMBHGA + (LAMMPS /
G03) package.
After they are obtained from plmp (using the COMB potential) or pg3, the
clusters are geometrically re-optimized with G03. The geometry re-optimization
implementation is a powerful function offered by G03 based on the Berny algorithm
(Li and Frisch, 2006). This quantum refinement algorithm enables a local
START
Initialize stochastically a population of 20 candidates. The
distances between the atoms in
cluster are within the cutoff distance
imposed by the COMB potential.
Calculate the individual
potential energy Vi
for each candidates i by
COMB potential or G03 and locally
minimize the candidates by
applying the BH method.
Calculate the fitness value fi for each candidates.
Discard 5 candidates with the lowest value of fi.
5 new candidates are generated
again via stochastic means. The value of Vi are
calculated for these new candidates.
Calculate the fitness value, fi
again for every candidates.
Check if 6 or more candidates have the same lowest fitness value fi.
Output global minimum energy
and the coordinates of the
lowest energy candidate cluster.
STOP
YES
NO

46
minimization of the structure at DFT level by adjusting the relative position of every
atom in the cluster. For the case of hafnium cluster, the method and basis set used in
the re-optimization are B3LYP/LanL2DZ pseudopotential (Hay and Wadt, 1985) as
suggested by (Sun et al., 2010).
As discussed earlier in Section 2.2, Kuntová et al. (2008) mentioned that the
similarity in geometry is the key to relate whether the structures of different PES lie in
the same basin. Comparing the chemical similarity of the structures lying in an
empirical potential PES and that in a DFT PES is a convenient way to computationally
access the correspondence between two PES, specifically, whether both PES share a
common basin. As a matter of principle, the structures obtained from plmp are the
global minima in the COMB PES (COMB structures) only. In theory they are not to
be taken as presenting the global minima in the DFrT PES. In this work, it is
hypothesizing that the COMB structures lies within the basin containing the global
minima of the DFT PES. Additional tests were then carried out to justify the hypothesis
by systematically comparing the structures obtained from the quantum refinement of
COMB structures and the global minimum structures obtained from DFT approach.
Basically the comparison involves the global similarity index, symmetry point
group, as well as the average bond length. Global similarity index is a novel quantity
specifically developed in this thesis based on the principle of molecular shape
comparison discussed in Section 2.3. It is a very effective measure to quantify the
similarity in the structural geometry between two clusters with the same number of
atoms (see details in Section 3.3.1). A high degree of similarity between the COMB
structures and the global minimum structures obtain from DFT approach indicates that
the structures lying at certain point in the COMB PES are close to some local minimum

47
of that DFT basin. The overall details were laid out in Section 3.2.4. By justifying the
scheme as mentioned above, COMB can then be used for simulated annealing process
later. The scheme also provides a convenient and cheap method to generate global
cluster structures with DFT accuracy using empirical potential in place of the
computationally expensive full DFT approach.
In this thesis, the minimum structures obtained via pg3 were taken as
representing the true global minimum, which were made as the reference structures.
To date, experimental data on small hafnium cluster is yet to be available. Therefore,
the minimal structures obtained via pg3 are predictions to be experimentally justified.
3.2 Molecular Dynamics Simulation of Hafnium Clusters
Ab initio method such as DFT gives detailed result for the properties of a
system. This method is bounded to zero Kelvin temperature and applicable up to only
hundreds of atom sizes because it is computationally expensive and time consuming
(Charles, 2008). Thus, classical MD is a more practical approach to simulate the
melting of hafnium cluster. In addition to the capability to handle large system, MD
also allows the user to keep track of the thermal behavior sensitively.
3.2.1 Simulated Annealing Process
Simulated annealing process refers to heating and quenching procedures in MD.
In this thesis, it involved the heating of candidate structures from 1 K to the desired
target temperature, and then equilibrating the system at that temperature for an

48
extended period of time during which the simulated data is sampled for statistical
analysis. The overall process described in this section is the second stage shown in the
flow chart of Figure 3.1.
Simulated annealing process was performed using LAMMPS package with the
COMB potential to describe the interactions among the hafnium atoms. The hafnium
clusters were positioned at the center of a simulation box with fixed boundary
condition to resemble a vacuum condition. The vacuum condition is essential to
simulate a free-standing cluster model. Each hafnium cluster is essentially the ground
state structure obtained from the procedure as described in Section 3.1 with plmp.
The simulation box is fixed with reflective walls at 100Å from the origin in
each direction. The simulation timestep, Δ𝑡 is 0.5 fs . The temperature control is
carried out with canonical (NVT) Nose-Hoover thermostat, by constantly rescale the
velocities of every atom in the sample.
The output of MD simulation was continuously monitored at various stages to
assure that the dynamics of the cluster during simulated annealing process closely
resemble the real situation. Proper preparation of ground state structures prior to the
annealing simulation has been mentioned in Section 2.1. However, to give the hafnium
cluster some initial configuration of microstates, the whole simulation need to undergo
relaxation at 0~1 K before the actual heating process commence. Next, the heating
rate is repeatedly revised to ensure the ergodicity of the system during melting. The
size of the cluster was taken into consideration as well when deciding the heating rate.
The following criteria were used to decide the choice of heating rate:

49
1. Beyond the optimum heating rate, any slower rate of heating gives a 𝑐𝑣
curve that clearly shows a consistent range of melting point for clusters of all
sizes (applied to both 𝑇𝑚 and 𝑇𝑝𝑟𝑒).
2. The computational time is not too demanding in order to heat up the
system to the desired target temperature, and equilibrate it at that temperature
for an extended period of time.
3. The pre-melting behavior should be visibly observed in every samples.
4. The melting point is within a reasonable range for the cluster of
neighboring sizes. The difference should be below 15% deviation.
Figure 3.3 shows the resultant plot of melting and pre-melting point for Hf50 against
the heating rate. Apparently, the first three statement converge once the heating rate
come close to 7 × 1012 Ks−1 (the melting points become constant value, as shown in
the marked area of Figure 3.3). It is found that, for smaller cluster size 𝑛, the difference
in melting point tends to be larger as compared to the neighboring size. But the forth
statement is met even for higher heating rate, up to > 10 × 1012 Ks−1. The heating
rate is set at slightly lower than the converge point as indicates by the red arrow in
Figure 3.3, which is 5 × 1012 Ks−1 (≡ 400 Δ𝑡K−1).

50
Figure 3.3: The melting point and pre-melting point of Hf50 with various heating
rate. The circle region marks the convergence of melting point lower than certain
heating rate.
The temperatures in the simulated annealing of a small cluster display noises
with large fluctuation over a broad range of temperature. As shown in Figure 3.4a, the
spread in the value of the temperature over the course of simulation is very large. The
reason of the temperature fluctuations is due to the thermostat effect on a very small
sized cluster. The plot in Figure 3.4a represents the temperature readings that are
recorded once every 300Δ𝑡. The large fluctuation in simulated temperature can be
artificially reduced to that shown in Figure 3.4b by performing time averaging over
the course of annealing procedure (Berry and Smirnov, 2004). To be consistent, the
average value of temperature is evaluated over a time window of 300Δ𝑡. Thus, instead
of just taking the reading once every 300Δ𝑡, calculating time averages enable the
temperature data to be recorded every timesteps. During the simulation, time averaging
procedure do not take away the fluctuations in temperature, but it acts as a tool to
dissolve the fluctuations. As seen in Figure 3.4b, the noise becomes relatively smaller.
Taking time averages are also useful to avoid information lost during the logging of

51
data while saving up the disk space. In addition to the temperature readings, time
averaging was applied to every measurement in the simulation, include energy value,
pressure, forces and coordinates of each atom in the cluster. At the meantime, the
atomic configurations (coordinates of every particle) of the clusters are recorded in a
separated trajectory file every 300Δ𝑡 for post-processing purposes.
a)
b)
Figure 3.4: The plots of temperature, 𝑇 against the simulation time step, ∆𝑡 a)
without and b) with time averaging in each 300∆𝑡 time interval.

52
As a rule of thumb, the melting temperature, 𝑇𝑚, is expected to increase with
the number of atoms in the clusters, 𝑛. The surface-to-body-atom ratio will decrease
with cluster size, 𝑛 . Due to the reduction of surface-to-body-atom ratio, 𝑇𝑚 will
converge to bulk melting temperature, 𝑇𝑚𝑏𝑢𝑙𝑘 beyond certain size, whereby thermal
behavior starts to resemble those of bulk value. It is beyond the scope of this thesis to
go as big as 104 in size, but the size effect is being studied up to 𝑛 = 50. An additional
cluster of size 𝑛 = 99 is designed to compare the results.
In addition to the temperature profile, LAMMPS has enabled more information,
such as the energy profile and atomic positions of each time step. To maintain
consistency, all the calculated data (to be used for post-processing) are recorded every
300∆𝑡.
3.2.2 COMB Potential
The COMB2 potential adopted in LAMMPS is based on the work of Shan et
al. (2010). Its parameters are as indicated in Table 3.1. The numbers are fit into the
total potential energy in the following equations
𝐸𝑇 = ∑ [𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖) +
1
2∑ 𝑉𝑖𝑗(𝑟𝑖𝑗, 𝑞𝑖, 𝑞𝑗)
𝑗≠𝑖
+ 𝐸𝑖𝐵𝐵]
𝑖
(3.3)
𝑉𝑖𝑗(𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗) = 𝑈𝑖𝑗𝑅(𝑟𝑖𝑗) + 𝑈𝑖𝑗
𝐴(𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗) + 𝑈𝑖𝑗𝐼 (𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗) + 𝑈𝑖𝑗
𝑉(𝑟𝑖𝑗) (3.4)
where 𝐸𝑖𝑠𝑒𝑙𝑓
is the self-energy of atom 𝑖, 𝐸𝑖𝐵𝐵 is the bond-bending energy of atom 𝑖,
𝑉𝑖𝑗 is the interaction potential between the 𝑖 th and 𝑗th atom, 𝑟𝑖𝑗 is the interatomic

53
distance and 𝑞𝑖 and 𝑞𝑗 are the charges of the atom 𝑖 and atom 𝑗. The 𝑉𝑖𝑗 comprises of
four parts: short-range repulsion, 𝑈𝑖𝑗𝑅 , short-range attraction, 𝑈𝑖𝑗
𝐴 , long-range
Coulombic, 𝑈𝑖𝑗𝐼 , and long range van der Waals force, 𝑈𝑖𝑗
𝑉 . Full description of the
functional form of COMB potential can be found in Appendix A.
Table 3.1: The potential parameters of Hf for the COMB potential.
𝐴(eV) 707.5303 𝑅𝑠 3.40 𝜒 0 𝐾𝐿𝑃6 0.008
𝐵(eV) 55.94216 𝑆𝑠 4.20 𝐽 3.13952 𝑅Ω 0.14
𝜆(Å-1) 2.069563 𝑄𝐿 -4.0 𝐾 0 𝐸0 0.16
𝛼(Å-1) 0.959614 𝑄𝑈 4.0 𝐿 0.00941 𝛾 0.10
𝛽 0.046511 𝐷𝐿 0.26152 𝜉 0.679131
𝑛 1.011011 𝐷𝑈 -0.25918 𝜌1 -3.928750
𝑚 1 𝑛𝐵 10 𝜌2 4.839580
𝑐 0 𝐶𝑉𝐷𝑊 0
𝑑 1
ℎ 0
Yu et al. (2007) were the first to develop the COMB potential based on the
Tersoff potential for Si and SiO2. Tersoff (1998a) is a bond order potential and is able
to correctly predict the bond breaking and bond formation of the Si systems. The
original Tersoff (1988b) was able to predict many properties of Si and Si base system,
but the charge distribution was not included in the Tersoff formalism. Yusakawa (1996)
then proposed an extension of the Tersoff model by including charge determination
and electrostatic terms which work similarly like the Rappé and Goddard (1991)
version of charge equilibration (QEq). COMB is effective such that the charge transfer
scheme was a separate procedure in MD. As a comparison, other charge optimized
potentials such as ReaxFF (Duin et al., 2003) proposed an extra constraint in the fitting
of the parameters. The functional form of ReaxFF comprised of a total of nine
separable functions. Duin et al. (2003) optimized the parameters by using the essential
training sets to meet all the parameter search requirements. The target of the

54
optimization was aimed to reproduce the heats of formation, the bond lengths and bond
angles based on the experimental values.
Yusakawa’s version of Tersoff potential includes charge transfer properties to
the existing short-range covalent bond, which is contained in the 𝐸𝑖𝑠𝑒𝑙𝑓
term
(Yusakawa, 1996). The early form of the potential was
𝐸𝑇 = ∑ [𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖) +
1
2∑ 𝑉𝑖𝑗(𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗)
𝑗≠𝑖
]
𝑖
(3.5)
The self-energy term 𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖) as suggested by Rappé and Goddard (1991) is a second
order function of 𝑞𝑖. The charges are treated classically in the self-consistent equations
of motion with Euler-Lagrange equations in the form
𝑚𝑖�̈�𝑖 = −𝜕
𝜕𝑟𝑖𝑇(𝑟𝑖, 𝑞𝑖)
(3.6)
𝑠𝑖�̈�𝑖 = −𝜕
𝜕𝑞𝑖𝑇(𝑟𝑖, 𝑞𝑖)
(3.7)
where 𝑇 is the kinetic energy with an additional term contributed by the charge,
𝑇 =1
2∑ 𝑚𝑖�̇�𝑖
2
𝑖
+1
2∑ 𝑠𝑖�̇�𝑖
2
𝑖
(3.8)
The variable 𝑠𝑖 holds the same significance as 𝑚𝑖, and it is assumed as the mass of the
charge particle, 𝑚𝑞 . QEq requires the derivatives 𝜕𝑇
𝜕𝑞𝑖 which is the microscopic
chemical potential, 𝜒𝑖, to remain constant over the course of the simulation, such that

55
𝜒1 = 𝜒2 = ⋯ = 𝜒𝑛. The equality of 𝜒𝑖 can also be seen as the mean value of chemical
potential, namely
𝜒𝑖 = �̅� =1
𝑛∑ 𝜒𝑖
𝑖
(3.9)
At equilibrium, the force acting on the charge transfer should be zero, thus, the
condition of the charge transfer can be written as
𝑚𝑞�̈�𝑖 = 𝜒𝑖 − �̅� (3.10)
along with the constraint imposed for the charge to be always a constant, 𝐶 (𝐶 = 0 in
this case),
∑ 𝑞𝑖
𝑛
𝑖
= 𝐶 (3.11)
The extension by Yu et al. (2007) was made to account for the additional
properties concerning the energy state, geometrical stability, bond length and the bond
angles of the Si and SiO2 polymorphs. Thus, the first generation COMB potential
(COMB1) has the same form of equation as COMB2. This enables the COMB2
potential to be applicable equally across generations to some of the elements
previously fitted with COMB1. COMB2 and COMB1 are distinguished from each
other, where COMB2 has been improved by including the amorphous properties of
silica (Shan et al., 2010b).
COMB potential is an empirical potential with parameters fitting to
experimental data. The dependability of COMB potential to finally lead to the correct
ground state structures, which provides justification for its further use in subsequent

56
MD simulations, is an important factor that determines the accuracy of this study. As
far as generation of correct ground states is concerned, one practical approach to check
for the dependability of an empirical potential is by determining how close its PES
resembles that of an ab initio method. If these two PES have close resemblance, the
cluster configurations closed to the global minima of the PES of the empirical potential
can be mapped to the global minima in the PES of an ab initio method upon a local
optimization procedure of these LLS.
In this thesis, the confirmation of the dependability of COMB potential in
generating the hafnium clusters were performed at two independent levels. Before
these two independent levels of confirmation are discussed, the procedure of
generating the data of cluster structures have to be described first.
3.2.3 Cluster Structures Generation
Ground state structures of hafnium clusters were generated in two parallel
processes, referred as the plmp and pg3 process lines. Figure 3.5 depicts the flow chart
of the two parallel process lines.

57
Figure 3.5: A schemetic flow chart of the two parallel process lines. The plmp
process line in the left and the pg3 process line in the right. Quantum refinement
(geometrical re-optimization) steps are carried out with G03 using the same basis
sets and settings in both process lines.
The plmp process line: The PTMBHGA + LAMMPS process line has already
been mentioned briefly in Section 3.1. Candidate trial structures were generated by the
PTMBHGA algorithm with the GA part switched off, while the total energy of these
structures were calculated using the COMB potential implemented with the LAMMPS
package. COMB potential has been discussed previously (Section 3.2.2). All the trial
structures generated by PTMBHGA were allowed to undergo a short interval of
relaxation using LAMMPS package at 𝑇 = 1 K. Each hafnium atom was allowed to
equilibrate with a minimal vibration. The total energy of these clusters (which solely
depend on the coordinates of the atoms in a cluster) were determined as it is without
any sophisticated geometrical optimization. LAMMPS in this case played the role of
so-called ‘energy calculator’. Due to the built-in global search algorithm of
PTMBHGA + LAMMPS (plmp)
COMB structures
DFT optimized structures
PTMBHGA + G03 (pg3)
LLS of fast DFT procedure
DFT optimized structures
Stage 1 Stage 1
Stage 2 Stage 2
Geometrical re-
optimization with
G03
Geometrical re-
optimization with
G03

58
PTMBHGA, a collection of low energy structures was generated upon a sufficiently
long period of time subjected to a controllable pre-set stopping criteria in PTMBHGA.
The resultant structure was the global minimum of the COMB PES (this structure is
referred as COMB structure). This was the first stage of cluster generation in the plmp
process line. The computational bottle neck of the first stage of the plmp process line
lies in the PTMBHGA generation of candidate configurations, not in the energy
calculator. In the second stage, the COMB structures from the first stage was
geometrically re-optimized using the DFT software package G03 by activating the
option ‘UltraFine’ in the integral grid size. This switched on the algorithm to
implement two-electron integral computation in the package for the purpose of high
accuracy calculation. The quadratically convergent self-consistent field (SCF)
procedure (Bacskay, 1981) was activated in this stage as well. The basis set
B3LYP/LanL2DZ was used in stage 2.
The output from stage 1 in the plmp process line was a collection of LLS for
COMB PES. However, in practice, it was found that, the output structures were often
unique in most cases. Hence, only one COMB structure (which was also the global
minimum of the COMB PES) was fed into G03 for geometrical re-optimization,
yielding the sole lowest energy structure as the final output for the plmp process line.
The pg3 process line: As for the plmp process line, the pg3 process line was
separated into two stages, a relative less computationally intensive PES scan in stage
1, followed by a more computationally intensive geometrical re-optimization in the
second stage. The common basis set B3LYP/LanL2Dz was used in both stages (recall
that the same basis set was also used in the second stage in the plmp process line).

59
In the PES scan in the first stage, candidate clusters generated via the
PTMBHGA algorithm were fed into the G03 package (which acts as the energy
calculator) so that the total energy of these clusters were determined as it is without
any geometrical optimization. This was a relatively fast but coarse PES scan procedure,
as each round of total energy determination does not involve the operation of local
optimization. As such, it is possible to sample (despite at a relatively coarse degree)
the DFT structures as much as practically possible. This has the practical significance
that, given limited available computing resource, the first stage in pg3 line can perform
a reasonably good mapping of the PES to provide better quality candidate structures
for geometrical optimization in the next stage. As an illustration, for 𝑛 = 7 hafnium
cluster, the pg3 stage 1 process run for ~100 hours using eight computing cores
(Intel® Xeon® Processor E5-1620 v2, 10Mb cache, 3.70 GHz) in parallel mode. During
this period, all stochastically generated configurations by PTMBHGA were scanned
through by the energy calculator, of which only 999 converged structures with the
lowest energies are kept for record. These were the LLS generated in stage 1. In
contrast to the stage 1 in the plmp process line, the computational bottle neck of the
stage 1 of the pg3 process line was lied in the energy calculator but not in the
generation of candidate configurations by PTMBHGA.
The second stage in pg3 was similar to the second stage in the plmp process
line, except that the input candidate LLS for geometrical re-optimization were
generated in stage 1 in which G03 was used as energy calculator. Geometrical re-
optimization in the second stage yield geometrically optimized structures at the DFT
level at the end of stage 2.

60
The pg3 process to generate true ground state structures has been proven
workable in an early study reported in Ng et al. (2015b) for the boron clusters. It is
part of the work of this thesis at the early stage to verify the workability of the pg3
process line. However, this process line is only for small-size cluster due to the
expensive computational cost.
3.2.4 Chemical Similarity Comparison
While visual inspection provides a convenient and qualitative way to compare
the geometrical configurations between two clusters, an unambiguous quantitative
algorithm to quantify the degree of similarity is a necessity. Some past studies on
chemical similarity have been laid out in Section 2.3. The quantitative resemblance
between two clusters with the same atom number 𝑛 is referred as chemical similarity.
In this thesis, a purpose-specific algorithm that could capture certain global features of
the geometrical configuration between two clusters in comparison was proposed. It
was referred as the ‘global similarity index’, denoted as 𝜉𝑖 . The proposed global
similarity index is just one possible form of chemical similarity. More technical details
of the global similarity index were deferred to Section 3.3.1.
In the context of this thesis, chemical similarity comparison is to be carried out
at two independent levels, which were referred as the inter-line comparison and intra-
line comparison. Each of these two comparisons has its own physical interpretation.
The intra-line chemical similarity comparison was performed to determine
whether the COMB structures generated by the COMB potential lie within the basin
of the global minimum (or one of the lowest energy local minimum) at the DFT level.

61
If the comparison returns a high similarity measure, the previous statement is deemed
positive. Whereas, the inter-line comparison is a process to determine whether the
ground state structures obtained at the end of the stage 2 of the two independent process
lines agree with each other. In this way, the dependability of COMB potential in
generating true ground state structures of hafnium clusters can be quantitatively and
objectively accessed.
The process of performing chemical similarity comparison was discussed in
more details in the following paragraphs.
Intra-line comparison within the plmp process: Figure 3.6 depicts the flow
chart of the intra-line comparison. Comparison was made for the hafnium clusters
before and after re-optimization (i.e., comparing the structures from stage 1 against
that from stage 2). In the intra-line comparison, the following three measures were
numerically monitored, namely (i) the global similarity index, (ii) the symmetry point
group, and (iii) the differences in relative average bond lengths. These comparisons
stated the degree of chemical similarity of a specific cluster with fixed atom number 𝑛
before and after geometrical re-optimization. The results from the chemical similarity
comparison indicated the dependability of COMB potential as an empirical potential
to obtain structures close to ab-initio method, at least for the hafnium clusters.

62
Figure 3.6: A schemetic flow chart of intra-line comparison within the plmp
process line.
Inter-line comparison between the plmp and pg3 process lines: Figure 3.7
depicts the flow chart of the inter-line comparison. The final structures obtained after
the quantum refinement from the plmp process line were compared directly with those
obtained from the pg3 process line. This were a direct comparison between the
structures in stage 2 from both process lines which use a common G03 local
optimization. The chemical similarity between the clusters in stage 2 in both process
lines indicated the dependability of COMB potential in generating the ground state
structures as well as in MD simulation.
PTMBHGA + LAMMPS (plmp)
COMB structures
DFT optimized structures
Stage 1
Stage 2
Geometrical re-
optimization with
G03
Chemical
similarity
comparison

63
Figure 3.7: A schemetic flow chart of inter-line comparison between the plmp and
pg3 process lines.
3.2.5 Flying Ice Cube Problem
In this thesis, the flying ice cube problem was encountered during the simulated
annealing procedure. Solving the flying ice cube problem was one of the major part of
the second stage as shown in Figure 3.1. Figure 3.8 shows the condition of a sample
Hf13 cluster during the occurrence of flying ice cube problem.
PTMBHGA + LAMMPS (plmp)
COMB structures
DFT optimized structures
PTMBHGA + G03 (pg3)
LLS of fast DFT procedure
DFT optimized structures
Stage 1 Stage 1
Stage 2 Stage 2
Geometrical re-
optimization with
G03
Geometrical re-
optimization with
G03
Chemical
similarity
comparison

64
a)
b)
Figure 3.8: The condition of Hf13 cluster during the heating procedure which
encountered flying ice cube artifact, generating excessive kinetic energy. a). The
cluster begin to spin in a clockwise manner along the red arrows direction shown,
at the beginning of heating procedure. b). The Hf13 cluster around 1800K~1900K
where the whole cluster start to drift across the simulation box, in addition to the
rotation motion, while remain closely bonded like an ‘ice’ body. The dynamic
bonding shown in the figure is kept below 3.2Å, slightly longer than the actual
bond length in bulk hafnium.
In MD simulation, flying ice cube problem refers to the situation in which the
system under simulation as a whole is rotating and drifting in the vacuum, while
remaining closely bounded to each other (Harvey et al., 1998). The name has its origin

65
due to the visualization as if a rigid iced body flying across the simulation box.
According to the theory of equipartition, when the average energy is not equally
distributed among various form (for kinetic energy, the contributions aroused from the
vibration, translation and rotation of the atoms, etc. …), the system is not in thermal
equilibrium. Flying ice cube problem happens when the individual velocity of each
particle are drained into the momentum of the center of mass. This process happens
due to repeated temperature (velocity) rescaling by imposing a thermostat, especially
during the annealing stages.
The excessive momentum is observed visually for both linear and angular
momentum of the center of mass. To describe how such problems are causing errors
to the outcome of the calculations, certain simulated examples are used to demonstrate
them. First, by imposing the command ‘fix recenter’ in the LAMMPS code, the cluster
as a whole will constantly reposition the center of mass to its original spot in every
time step. This will not affect the outcome since the cluster is in vacuum and does not
interact with anything else. Should flying ice cube artifact happen, each microscopic
vibration of all particles would diminish even though the temperature of the system
continues to rise. The excessive energy is carried in the form linear velocity of the
center of mass, causing the cluster to remain bonded together. At some point, the
system might even start to rotate on its own, converting a portion of the energy into
rotatory motion. As a result, the temperature of the simulation box rose, but not the
cluster.
There are two approaches to correct the errors caused by the flying ice cube
problem. One is by using the Langevin dynamics (thermostat) and another one is by

66
removing the excessive momentum along the calculation via the ‘fix momentum’
command in LAMMPS package.
Langevin dynamics are able to capture the random stochastic fluctuation,
normally arises from interaction with the background implicit solvent. For the cluster
during the occurrence of flying ice cube phenomenon, this approach controls the
perturbation of the omitted degree of freedom. Based on the LAMMPS documentation,
the force on each atom has the form of
𝐹 = 𝐹𝑐 + 𝐹𝑓 + 𝐹𝑟 (3.12)
where 𝐹𝑐 is the conservative force computed via the usual interatomic interactions and
𝐹𝑓 is the frictional drag or viscous damping term proportional to the particles’ velocity.
The proportionality constant for each atom is computed as 𝑚
𝑑𝑎𝑚𝑝, with 𝑚 being the
mass of particle and 𝑑𝑎𝑚𝑝 is the damping factor, and thus
𝐹𝑓 = − (𝑚
𝑑𝑎𝑚𝑝) 𝑣
(3.13)
𝐹𝑟 is the force due to solvent atom at temperature 𝑇 randomly bumping into another
particle. 𝐹𝑟 is proportional to
𝐹𝑟 ∝ √𝑘𝐵𝑇
𝑑𝑡∙
𝑚
𝑑𝑎𝑚𝑝
(3.14)
where 𝑘𝐵 is the Boltzmann constant.
At the same time while solving the flying ice cube problem, the Langevin
dynamics also approximate the canonical ensemble that act as a thermostat. The

67
approach affects the system in such a way that it modifies the forces to effect
thermostatting, which is different from Nose-Hoover thermostat and its time
integration.
In this thesis, the second method was used to retain the thermostat in use. Thus,
the ‘fix momentum’ command was used instead of Langevin dynamics. On the other
hand, by constantly zeroing both the linear and angular momentum of the system, the
errors in controlling the excessive (removing the residual) velocities could be kept to
a minimum. When the momentum was zero, the force was adjusted accordingly for
every time step, thus, the modification will not affect the canonical ensemble. Thus,
the immediate state of the system and temperature were preserved.
The command ‘fix momentum’ will zero the momentum (linear or angular or
both) of the clusters every time step by adjusting the velocities of every single atom
relative to the velocity of the center of mass. After imposing ‘fix momentum’
command, the cluster was not rotating and drifting anymore at low temperature, but
every atom will vibrate with the increasing temperature. The bond breaking and bond
formation were displayed accordingly, as shown in Figure 3.9 for Hf13 cluster.
In order to avoid further complication from flying ice cube problem, the
zeroing of ‘fix momentum’ option is applied thoroughly for every single simulated
annealing process in this thesis. Overall, every MD process was checked to be
consistent to each samples and runs.

68
a) ~850 K c) ~1100 K
b) ~900 K d) ~2050 K
Figure 3.9: The condition of Hf13 cluster, showing the bond breaking and bond
formation at a) ~850 K, b) ~900 K, c) ~1100 K and d) ~2050 K. Along the
simulation time, the cluster neither rotate nor drift across the simulation box, each
atom vibrate relative to one another, carry the kinetic energy in them.
3.3 Post-Processing
The widely accepted method to study the thermal properties of a heating
process is by plotting the caloric curve and the 𝑐𝑣 vs. temperature curve (𝑐𝑣 curve). In
this thesis, both curves were plotted to obtain the melting temperature of hafnium
clusters. The melting temperature is compared to that obtained via the global similarity
index, which was introduced in the next subsection. Together, both method set up the

69
third and final part of the thesis as shown in Figure 3.1. The caloric curve of a hafnium
cluster under thermal annealing can be obtained by plotting its potential energy as a
function of temperature. This information is readily extracted from the log file (.log)
from LAMMPS package. Next to the caloric curve, the 𝑐𝑣 value can be calculated
using Equation (2.3) in Section 2.2, which is restated below,
𝑐𝑣 =⟨𝐸𝑡
2⟩𝑇 − ⟨𝐸𝑡⟩𝑇2
2𝑛𝑘𝐵𝑇2
(3.15)
The caloric profile can be obtained from ‘direct heating’ and ‘prolonged
annealing’. In the direct heating procedure, the cluster is heated directly to a high
temperature with a slow heating rate. To ensure the melting dynamics is well
represented by caloric profile, the heating rate is pre-defined as discussed in Section
3.2.1, which is 5 × 1012 Ks−1 . A slow hearing rate is able to minimize the error
accumulated due to rapid velocity rescaling at every time step. The value of 𝑇∆𝑡 and
𝐸∆𝑡 are taken directly from the readings of a single log file. The subscript ∆𝑡 refer to
the values each taken at the same timestep.
In the second approach, each point on the caloric curve was obtained from a
single annealing process. In this procedure, the candidate structure was heated to the
desired target temperature and allowed to equilibrate for a very long time to ensure the
ergodicity of the microstate, specifically the temperature and the potential energy value.
For example, to obtain a caloric profile from 1000 K to 3000 K, with a temperature
windows of 50 K, a total of 41 simulated annealing processes need to be performed.
The first simulation correspond to the heating to the target temperature of 1000 K, and
equilibrate at 1000 K for an extended period of time and yield the average value of

70
⟨𝐸1000 K⟩ for 𝑇 = 1000 K. The next will be ⟨𝐸1050 K⟩ for 𝑇 = 1050 K, and so on, till
the 41st simulation of ⟨𝐸3000 K⟩ for 𝑇 = 3000 K.
Comparing both heating procedures, although the prolonged annealing process
is very lengthy to simulate, the direct heating process is comparatively trickier to
handle. However, both methods were used in this thesis to compare both approaches.
The outcome of the comparison was presented in Chapter 5.
The other output of LAMMPS package is the trajectory file (.lammpstrj) that
records all the screenshots of the structures over the course of the simulation with fixed
time interval. When the simulation is done accordingly, the trajectories and the log file
should tally with each other. Thus, the thermal behavior can be observed through the
trajectory file while melting temperature is pinpointed using the log file. One of the
benefits in simulated annealing is that one can visualize the simulation by using
visualization program. The screenshots of the trajectory when physically significant
variation occurred during the simulation were presented in Chapter 4.
In this thesis, the dynamical information of the simulation was traced by
comparing the similarity index of each frame along the heating process. The
comparisons are made with respect to the initial configuration at 0 K (after relaxation)
prior to the heating process. The trajectories were recorded every 300Δ𝑡 , which
corresponds to 0.15 ps of simulation time. With the heating rate of 5 × 1012 Ks−1, the
simulated data were expected to cover at least one record per unit temperature.

71
3.3.1 Global Similarity Index
The global similarity index, 𝜉𝑖 proposed in this thesis is a novel approach and
derived based on generic chemical similarity idea to detect the changes along the
trajectory of the heating process. This is a new measure deserving an
acknowledgement of novelty, an original idea first proposed in this thesis. The global
similarity index proposed in this thesis is used to identify the chemical similarity
between the clusters of the same size. It is used as a measure to determine the
dependability of COMB potential as well as to detect changes in the structures during
the heating procedure. As compared to all the existing method, global similarity index
is able to predict the detailed melting mechanism which was discussed later in this
section.
The functional form of 𝜉𝑖 is proposed to take the form of
𝜉𝑖 =1
𝑛∑(𝑘𝑠,𝑖 + 1)
−1𝑛
𝑠=1
(3.16)
𝑘𝑠,𝑖 = |√𝑑𝑠,𝑖 − √𝑑𝑠,0| (3.17)
where 𝑑𝑠,𝑖 and 𝑑𝑠,0 represent the sorted distance of atoms relative to the average
positions (center of mass) of all the atoms in the cluster for the 𝑖th (denoted as the
subscript 𝑖) and the 0th frame (for the simulated annealing process, this is the input
structure), while 𝑛 corresponds to the number of atoms, which is an integer equals to
the number of pairs of 𝑘𝑠,𝑖. The value of 𝜉𝑖 = 1 corresponds to totally identicalness
and 𝜉𝑖 → 0 for vast difference.

72
The application of 𝜉𝑖 was demonstrated in the example using the LLS of Hf7.
Figure 3.10 shows the visualization of Hf7 clusters by using Visual Molecular
Dynamics (VMD) (Humphrey et al., 1996) software. In this example, the similarity
index is applied to the structures in Figure 3.10b and 3.10c relative to 3.10a. In this
illustration, the structure of Figure 3.10a play the role as the 0th frame. The Hf7 in
Figure 3.10a is the ground state structure as obtained in this thesis, while 3.10b is the
second lowest energy isomer. Figure 3.10c was obtained via modification to the
ground state Hf7 where both the top and bottom atoms of the pentagonal bipyramid is
moved closer to the pentagonal base. Minor movement of atoms such as the case of
Figure 3.10c from 3.10a were expected during the heating process of the cluster. In
these figures, a visualisation setting known as dynamic bonds is enabled. In this
visualisation setting, bonds will be automatically shown as a connection between two
atoms that are separated at a distance of equal or less than a specified value 𝑟𝑏. In this
illustration, a value of 𝑟𝑏 = 3.2Å was to distinguish visually the differences between
the structures (notice the connection appeared for the top-bottom atoms in Figure
3.10c). Figure 3.10c was artificially prepared for the purpose to demonstrate the
characteristics of 𝜉𝑖.

73
a) c)
b)
Figure 3.10: The LLS of Hf7 cluster. a) The ground state structure. b) The second
lowest energy isomer. c) A slight modification was made based on the ground state
structure where the bipyramid top was moved closer to the pentagonal base. The
green cross indicates center of mass.
The average position of each structures in Figure 3.10 was marked as the center
of mass (CM) with a green cross. For 3.10a, which was the ground state structure, the
center of mass was located right at the center of the bipyramid frame, say at Ca (0, 0,
0). In order to apply Equation (3.17) to Figure 3.10a, the distance of each atom from
Ca is calculated and sorted in ascending order. The sorted distances in this example are
{𝑑𝑠,0}={1.62966, 1.62967, 2.4062, 2.40638, 2.40712, 2.40729, 2.40807}, where 𝑠 =
1, 2, … , 7. The same calculations when applied to the other two structures, give {𝑑𝑠,1}
(Cb as the center of mass, Figure 3.10b) and {𝑑𝑠,2} (Cc as the center of mass, Figure
3.10c). Once 𝑑𝑠,0 , 𝑑𝑠,1 and 𝑑𝑠,2 have been calculated, 𝑘𝑠,𝑖 can be evaluated as
Equation (3.17) for 𝑖 = 1, 2 in this illustration. By applying Equation (3.16), the value

74
of 𝜉𝑖 for the structure of Figure 3.10b as compared to Figure 3.10a is 𝜉1 =0.988913
while for Figure 3.10c is 𝜉2 =0.979697. In actual case, 𝑖 = 1, 2, … until a very large
number of frames.
The pair of structures in Figure 3.10a and 3.10c clearly appear more alike than
the pair of structures in Figure 3.10a and 3.10b. However, the similarity measure as
defined by Equation (3.16) reveals the otherwise. This means the definition of 𝜉𝑖 as
stated in Equation (3.16) did not correctly reproduce the similarity measure. Some
improvement was deemed necessary so that 𝜉𝑖 could sensitively accommodate the
changes such as that occurred in Figure 3.10c. To achieve this purpose, the functional
form of 𝜉𝑖 was modified according to the prescription below.
Instead of a single, conventionally defined center of mass, this thesis used three
special cases of generalized mean positions to act as the center of mass of a cluster,
namely the arithmetic mean, the harmonic mean, and the quadratic mean (a.k.a. root
mean square). Each of mean is viewed as a center of reference (COR) for the
immediate state of clusters. The idea of COR works like the center of mass, where the
whole cluster is represented by a single point mass. With this generalization, there are
now three COR for each frame of cluster. The generalized mean, also known as the
power mean, can be expressed in the following form
𝑀𝑝(𝑥1, … , 𝑥𝑛) = (1
𝑛∑ 𝑥𝑖
𝑝
𝑛
𝑖=1
)
1𝑝
(3.18)
where 𝑝 must be a non-zero real number, and 𝑥1, … , 𝑥𝑛 are variables. The special cases
of Equation (3.18) when 𝑝 takes on the value 𝑝 = 1, −1, 2 are arithmetic mean,

75
harmonic mean, and quadratic mean respectively. These are just the common
definition of means which we are all familiar with, i.e.,
𝑀1(𝑥1, … , 𝑥𝑛) =𝑥1 + ⋯ + 𝑥𝑛
𝑛
(3.19)
𝑀−1(𝑥1, … , 𝑥𝑛) =𝑛
1𝑥1
+ ⋯ +1
𝑥𝑛
(3.20)
𝑀2(𝑥1, … , 𝑥𝑛) = √𝑥1
2 + ⋯ + 𝑥𝑛2
𝑛
(3.21)
The cluster was quantified by three different sets of 𝑘𝑠,𝑖 , each based on a
different COR. The definitions of Equation (3.16, 3.17) ware hence generalized to
include an COR index to indicate which COR is being referred to when evaluating a
𝜉𝑖,
𝜉𝑖𝐶𝑂𝑅 =
1
𝑛∑(𝑘𝑠,𝑖
𝐶𝑂𝑅 + 1)−1
𝑛
𝑠=1
(3.22)
𝑘𝑠,𝑖𝐶𝑂𝑅 = |√𝑑𝑠,𝑖
𝐶𝑂𝑅 − √𝑑𝑠,0𝐶𝑂𝑅|
(3.23)
In each COR, there are 𝑛 sorted atomic distances, giving a total of 3𝑛 pairs of
𝑘𝑠,𝑖. The distances of atoms from the mean position are sorted in ascending order from
the shortest to the longest. The difference in Equation (3.17, 3.23) are measured with
respect to the distance of atoms in the 0 K frame of the same sorting sequence. After
the value of 𝜉𝑖 is calculated for each COR, averaging of 𝜉𝑖 over these COR was then
obtained, via

76
𝜉�̅� =1
𝑁𝐶𝑂𝑅∑ 𝜉𝑖
𝐶𝑂𝑅
𝐶𝑂𝑅
=1
3∑
1
𝑛∑(𝑘𝑠,𝑖
𝐶𝑂𝑅 + 1)−1
𝑛
𝑠=1𝐶𝑂𝑅
=1
3𝑛∑ ∑(𝑘𝑠,𝑖
𝐶𝑂𝑅 + 1)−1
𝑛
𝑠=1𝐶𝑂𝑅
(3.24)
where 𝑁𝐶𝑂𝑅 = 3 , the number of COR used in this thesis. With the inclusion of
additional COR, the quadratic mean of the structure in Figure 3.10a was calculated to
be (1.43865, 1.43828, 0.871093). The sorted distances in ascending order of every
atom from this COR were {0.945749, 2.1706, 2.76504, 2.77303, 3.22416, 4.31185,
4.31648}. The harmonic mean and the corresponding sorted distances for Figure 3.10a
can also be similarly calculated (figures were not provided for the sake of brevity).
The same calculation was also applied to the structures 3.10b and 3.10c. Finally, by
applying Equation (3.24), the 𝜉�̅� measured the for structure in Figure 3.10c as
compared to 3.10a yields a value of 𝜉2̅ = 0.93887 while for Figure 3.10b yields 𝜉1̅ =
0.911066. Apparently, this generalization improves the sensibility of 𝜉𝑖 in accessing
the similarity of the clusters. In the subsequent discussion, the bar in the 𝜉�̅� symbol
shall be dropped, with the understanding that whenever global similarity index is
mentioned, it is referring to the COR-averaged version of 𝜉�̅�, unless it is explicitly
stated otherwise. As demonstrated in Chapter 5 by using COR-averaged Equation
(3.24), the pre-melting phase can be distinctively identified as a sharp transition in 𝜉𝑖.
It was found that taking the square of Equation (3.24) improves the sensitivity
of 𝜉𝑖 in detecting similarity in the clusters. Since a 𝜉𝑖 ranges from 0 → 1, squaring
Equation (3.24) does not change the normalization property of 𝜉𝑖. In fact, by squaring
Equation (3.24), the variation in 𝜉𝑖2
becomes numerically more pronounced and

77
sensitive due to the parabolic behavior of squaring a number within the range of (0,1).
For the structure in Figure 3.10c, the value of 𝜉𝑖2 is 𝜉2
2 = 0.938872 = 0.881477;
while for the structure in Figure 3.10b, 𝜉12 = 0.9110662 = 0.830042. In practice, the
value of 𝜉𝑖 tends to vary only very slightly especially when comparing the structures
before and after geometrical re-optimization with G03. Hence, squaring the 𝜉𝑖 is a
convenient way to enhance the visibility of the variation effect. The squared version
of similarity index, 𝜉𝑖2 was adopted in the subsequent chapters.
It is instructive to compare Equation (3.24) to Equation (2.7), which is the USR
defined by Ballester and Richards (2007)
𝑆𝑞𝑖 = (1 +1
12∑|𝑀𝑙
𝑞 − 𝑀𝑙𝑖|
12
𝑙=1
)
−1
𝑆𝑞𝑖 as defined in the definition of USR has a very different working principle from that
of the global similarity index. 𝑆𝑞𝑖 has the general form of
𝑆𝑞𝑖~1
1 +1
12∑ (⋯ )𝑙
12𝑙=1
, (⋯ )𝑙 ≡ |𝑀𝑙𝑞 − 𝑀𝑙
𝑖|
(3.25)
whereas 𝜉𝑖 has the general form
𝜉𝑖~1
3𝑛∑ ∑
1
1 + (⋯ )𝑠
𝑛
𝑠=1𝐶𝑂𝑅
, (⋯ )𝑠 ≡ |√𝑑𝑠,𝑖𝐶𝑂𝑅 − √𝑑𝑠,0
𝐶𝑂𝑅| (3.26)
The fraction of 1
12 in 𝑆𝑞𝑖 is sitting inside the reciprocal, whereas the fraction
1
3𝑛 in 𝜉𝑖 is
placed outside the reciprocal. This will ensure that 𝑘𝑠,𝑖𝐶𝑂𝑅 is not averaged before the
index 𝑠 is summed over. As it was originally proposed by Ballester and Richards

78
(2007), the 𝑀𝑙 terms as appear in 𝑆𝑞𝑖 have to undergo a prescribed normalization
procedure so that they give values with the same order of magnitude for all 𝑙 (𝑀𝑙 will
give values with different order of magnitude for different 𝑙 if not normalized). On the
other hand, normalization procedure is not required for 𝜉𝑖 as each 𝑘𝑠,𝑖𝐶𝑂𝑅 , being the
differences in the distances of atoms from the COR, has the same order of magnitude.
The main idea of the global similarity index is to compare the configuration of
one image of the molecule (cluster) to another. Imagine two images of identical cluster
with one of them is obtained by performing a linear transformation (including
translation, rotation, reflection and rescaling) on the other. The global similarity index
must be able to discern the fact that these two images, despite having different
coordinates, are in fact the same cluster, and returns a unique value of 𝜉𝑖 = 1. In
general, when two images are being compared, the transformational relation between
them have to be factored in while evaluating the global similarity index.
Now, consider two images (atomic configurations of two clusters) which are
related to each other via a linear transformation. The transformation is said to be
congruent if it undergoes mapping operation of translation, rotation, reflection, or the
combination of more than one of these. If the image also undergoes rescaling, it is
similar but not congruent. Conceptually, congruence is a subset to similarity. The
primary aim of the 𝜉𝑖 measure as defined in this thesis was to recognize whether the
transformation falls into any group of equality, and numerically quantify the degree of
congruence or similarity. If one wishes to identify two congruent images, the definition
of their 𝑘𝑠,𝑖𝐶𝑂𝑅 have to fulfill the following requirements: 𝑘𝑠,𝑖
𝐶𝑂𝑅 should be

79
1) invariant under translational, rotational or reflection transformations,
i.e., 𝑘𝑠,𝑖𝐶𝑂𝑅 = 𝑘𝑠,𝑖′
𝐶𝑂𝑅, where 𝑖, 𝑖′ refer to two images related by a
congruent transformation;
2) of the same order of magnitude;
3) able to capture any perturbation to the position of any atomic points in
an image.
For 2) to be true would require |𝑑𝑠,𝑖𝐶𝑂𝑅|~|𝑑𝑠,𝑖′
𝐶𝑂𝑅|, where 𝑠 refers to the same points
(atoms) in image 𝑖 and 𝑖’ . Requirement 3) can be achieved by including various
independent COR into the definition of 𝜉𝑖 . There is a possibility that certain
perturbation may be insensitive to a particular COR but sensitive to other. By including
various COR, perturbation missed by one particular COR could be picked up by the
other COR which is more susceptible to this perturbation. The example as discussed
for Figure 3.10 was designed to illustrate the improved discerning ability of 𝜉𝑖 with
the inclusion of various COR into its definition. In fact, the three theoretical criterion
discussed above form the basic working principle on which the global similarity index
was originally proposed.
If two images are similar, in addition to the three requirements as stated above,
𝑘𝑠,𝑖𝐶𝑂𝑅 also needs to be invariant under rescaling transformation. The effect of rescaling
operator shall also be considered in the definition of 𝑘𝑠,𝑖𝐶𝑂𝑅, such that it returns a value
of close to 0 (≡ 𝜉𝑖 → 1). However, it would be in general not possible for requirement
2), i.e., |𝑑𝑠,𝑖𝐶𝑂𝑅|~|𝑑𝑠,𝑖′
𝐶𝑂𝑅|, to be fulfilled due to rescaling transformation. For example, if
𝑖′ corresponds to an enlarged frame of a cluster 𝑖 , |𝑑𝑠,𝑖′𝐶𝑂𝑅| will be way larger than
|𝑑𝑠,𝑖𝐶𝑂𝑅|. Hence 𝜉𝑖 will not return a value close to 1 even though they are in fact similar.

80
Hence, the definition of 𝑘𝑠,𝑖𝐶𝑂𝑅 as defined in Equation (3.23) has to be supplanted by a
new form, 𝑘′𝑠,𝑖𝐶𝑂𝑅
, when calculating global similarity index for rescaled images:
𝑘′𝑠,𝑖𝐶𝑂𝑅 = |√
𝑑𝑠,𝑖𝐶𝑂𝑅
𝐷𝑖𝐶𝑂𝑅 − √
𝑑𝑠,0𝐶𝑂𝑅
𝐷0𝐶𝑂𝑅|
(3.27)
𝐷𝑖𝐶𝑂𝑅 is defined as a characteristic length of the 𝑖th frame, such that when the image 𝑖
is blown up by an arbitrary scale, 𝑑𝑠,𝑖
𝐶𝑂𝑅
𝐷𝑖𝐶𝑂𝑅 would remain unchanged. The choice of 𝐷𝑖
𝐶𝑂𝑅
that could provide such an invariant effect can be conveniently taken to be, without
loss of generality, 𝐷𝑖𝐶𝑂𝑅 = |𝑑𝑚
𝐶𝑂𝑅 − 𝑑𝑚−1𝐶𝑂𝑅 | for any 𝑚 ∈ [2, 𝑛].
As it turns out, in the MD heating simulation to be carried out on the hafnium
clusters, the clusters in general do not undergo rescaling transformation up to the point
before they become completely melted. Their sizes are maintained throughout the MD
heating process up to the melting point. In a MD heating simulation, rescaling of
cluster structures is a natural outcome. Hence, in order for the global similarity index
to correctly quantify the characteristics of the cluster evolution, it must be able to
capture the rescaling effect. In other words, if a cluster’s geometry remains the same
but its overall size gets blow up by some factor, the global similarity index must show
a variation in its numerical value, signifying the occurrence of rescaling in the cluster.
On the other hand, in the case of intra-line comparison, COMB structures tend to be
rescaled by geometrical re-optimization by G03. However, these structures actually
remain in the same basin of the DFT PES. Thus, in this case, global similarity index
used to compare the clusters before and after the re-optimization operation should be
treated as similar.

81
Global similarity index should be able to recognize the congruence of the
cluster frames in the MD heating process, while in the plmp intra-line comparison, it
must be able to recognize both congruence and similarity between the structures. In
practical terms, this means when rescaling effect is considered as a change in chemical
similarity e.g., in a heating process, the definition for 𝑘𝑠,𝑖𝐶𝑂𝑅 in Equation (3.23) is used.
Otherwise, e.g., in the case of geometrical re-optimization of a cluster by G03,
Equation (3.27) is used instead. As a note, the inter-line comparison is a direct yes-or-
no problem; the resultant structures from both process lines are either the same or
otherwise. The answer does not require the application of global similarity index.
The measurement protocol of the similarity index 𝜉𝑖 can be further studied by
computing the fluctuation of 𝜉𝑖 over the course of simulation. In general, 𝜉𝑖, which is
related to the microscopic property, is not directly measurable via experiment.
However, its fluctuation could be related to a macroscopic observable. As in the case
of 𝑐𝑣, it is a measure of fluctuation in the binding energy. The fluctuation of 𝜉𝑖 remains
a prospect for future study. The fluctuation equation is considered to be proportional
to the variance of 𝜉𝑖 as
𝑆𝜉𝑖∝ 𝜎2(𝜉𝑖) (3.28)
The 𝑆𝜉𝑖 curve shows a sudden change in the similarity index 𝜉𝑖 in the form of sharp
peaks at the particular moment when major transition occurs in the configuration.
In this thesis, the global similarity index 𝜉𝑖 was used to study the dynamics of
hafnium clusters during the simulated annealing processes. The value of 𝜉𝑖 in each
instance 𝑖 is a measure of similarity of the cluster structure in that instance as compared
to the ground state structure (the structure during the initial 1 K equilibration) in the

82
0th frame. During the early part of the heating-up process, only little variation in 𝜉𝑖 is
detected, indicating that only minor change to the geometry is happening in the cluster.
Up to this point, 𝜉𝑖 tends to measure an almost constant value close to 1. The abrupt
change to the global similarity index will only happen during (or close to) the melting
temperature. Visually, the melting transition happens with a total distortion in the
structure spanning the whole cluster. Following the sequential evolution of 𝜉𝑖 allows
one to locate the sudden change in the structure of the clusters during a heating process.
After the melting point, the whole cluster is expected to be fluid-like and each atom
tends to roam across the simulation box randomly. Thus, the global similarity index is
expected to be constantly close to 0. Note that, 𝜉𝑖 stays almost constant during most of
the simulated annealing process, except during the range of temperature in which the
melting process occurs. Thus, the fluctuation of similarity index 𝑆𝜉𝑖 becomes useful to
locate the temperature where these transition happened.
In addition to the melting transition, patterns of distortion to the clusters during
the simulated annealing process are also detectable by the global similarity index based
on the fluctuation curve 𝑆𝜉𝑖. This was discussed in Chapter 5.

83
CHAPTER 4
DEPENDABILITY OF COMB POTENTIAL
4.1 Geometrical Re-Optimization of Hafnium Clusters
The structures obtained at the end of the stage 1 of the plmp process line (the
COMB structures) were geometrically re-optimized with G03 package using B3LYP
and basis set LanL2DZ. This is the intra-line comparison mentioned in Section 3.2.4.
The comparison procedure has to be carried out in order to evaluate the chemical
similarity of the hafnium clusters before subjecting them for MD simulation.
Specifically, the average bond length, symmetry point group and global similarity
index were compared. Intra-line comparison accessed the dependability of the COMB
potential in MD simulations to be carried subsequently.
The outcome of intra-line comparison was numerically tabulated in Table 4.1
for the clusters with the size of 𝑛 = 2 to 𝑛 = 20. The plots as shown in Figure 4.1
were graphical representation of Table 4.1 for the average bond length and global
similarity index.
The average bond lengths were calculated by considering only the nearest
neighbor atoms in the clusters. The calculated bond lengths were similar for this
sample of hafnium clusters. As shown in Figure 4.1a, the difference in the bond lengths
for the clusters of neighboring sizes remains minimal as 𝑛 increases. However, COMB
structures ded not show gradual increment in term of 𝑛 as the structures of DFT
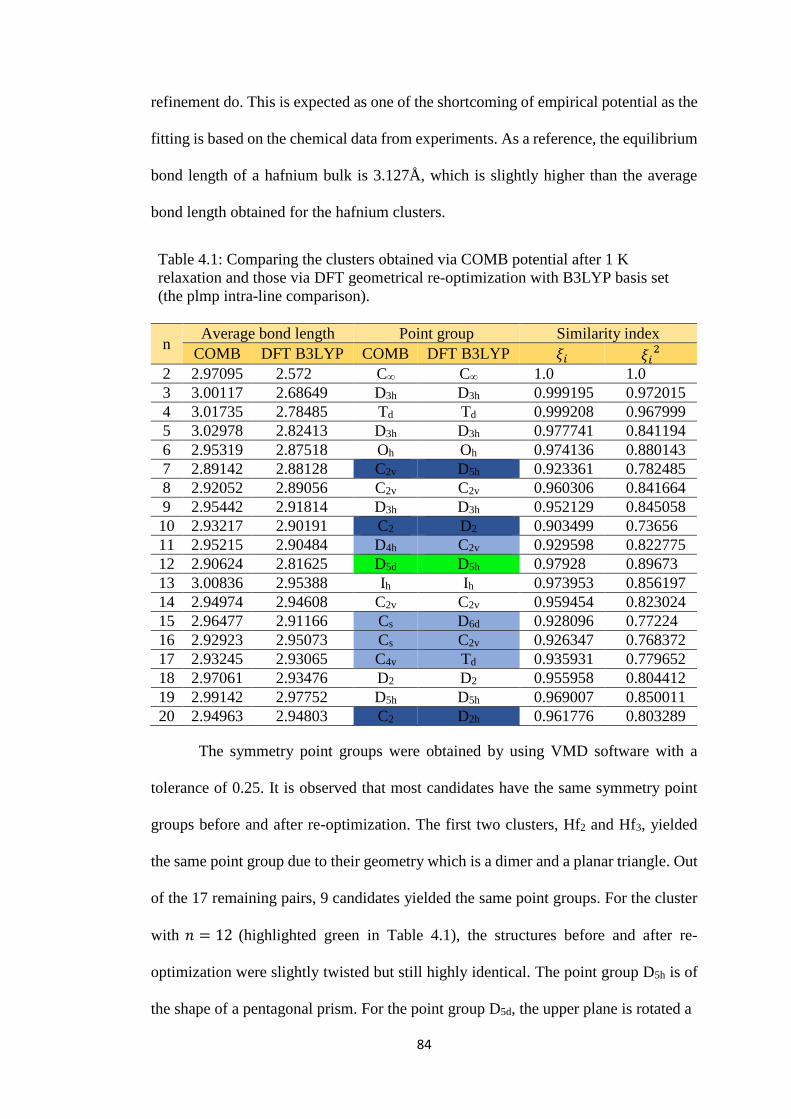
84
refinement do. This is expected as one of the shortcoming of empirical potential as the
fitting is based on the chemical data from experiments. As a reference, the equilibrium
bond length of a hafnium bulk is 3.127Å, which is slightly higher than the average
bond length obtained for the hafnium clusters.
Table 4.1: Comparing the clusters obtained via COMB potential after 1 K
relaxation and those via DFT geometrical re-optimization with B3LYP basis set
(the plmp intra-line comparison).
n Average bond length Point group Similarity index
COMB DFT B3LYP COMB DFT B3LYP 𝜉𝑖 𝜉𝑖2
2 2.97095 2.572 C∞ C∞ 1.0 1.0
3 3.00117 2.68649 D3h D3h 0.999195 0.972015
4 3.01735 2.78485 Td Td 0.999208 0.967999
5 3.02978 2.82413 D3h D3h 0.977741 0.841194
6 2.95319 2.87518 Oh Oh 0.974136 0.880143
7 2.89142 2.88128 C2v D5h 0.923361 0.782485
8 2.92052 2.89056 C2v C2v 0.960306 0.841664
9 2.95442 2.91814 D3h D3h 0.952129 0.845058
10 2.93217 2.90191 C2 D2 0.903499 0.73656
11 2.95215 2.90484 D4h C2v 0.929598 0.822775
12 2.90624 2.81625 D5d D5h 0.97928 0.89673
13 3.00836 2.95388 Ih Ih 0.973953 0.856197
14 2.94974 2.94608 C2v C2v 0.959454 0.823024
15 2.96477 2.91166 Cs D6d 0.928096 0.77224
16 2.92923 2.95073 Cs C2v 0.926347 0.768372
17 2.93245 2.93065 C4v Td 0.935931 0.779652
18 2.97061 2.93476 D2 D2 0.955958 0.804412
19 2.99142 2.97752 D5h D5h 0.969007 0.850011
20 2.94963 2.94803 C2 D2h 0.961776 0.803289
The symmetry point groups were obtained by using VMD software with a
tolerance of 0.25. It is observed that most candidates have the same symmetry point
groups before and after re-optimization. The first two clusters, Hf2 and Hf3, yielded
the same point group due to their geometry which is a dimer and a planar triangle. Out
of the 17 remaining pairs, 9 candidates yielded the same point groups. For the cluster
with 𝑛 = 12 (highlighted green in Table 4.1), the structures before and after re-
optimization were slightly twisted but still highly identical. The point group D5h is of
the shape of a pentagonal prism. For the point group D5d, the upper plane is rotated a
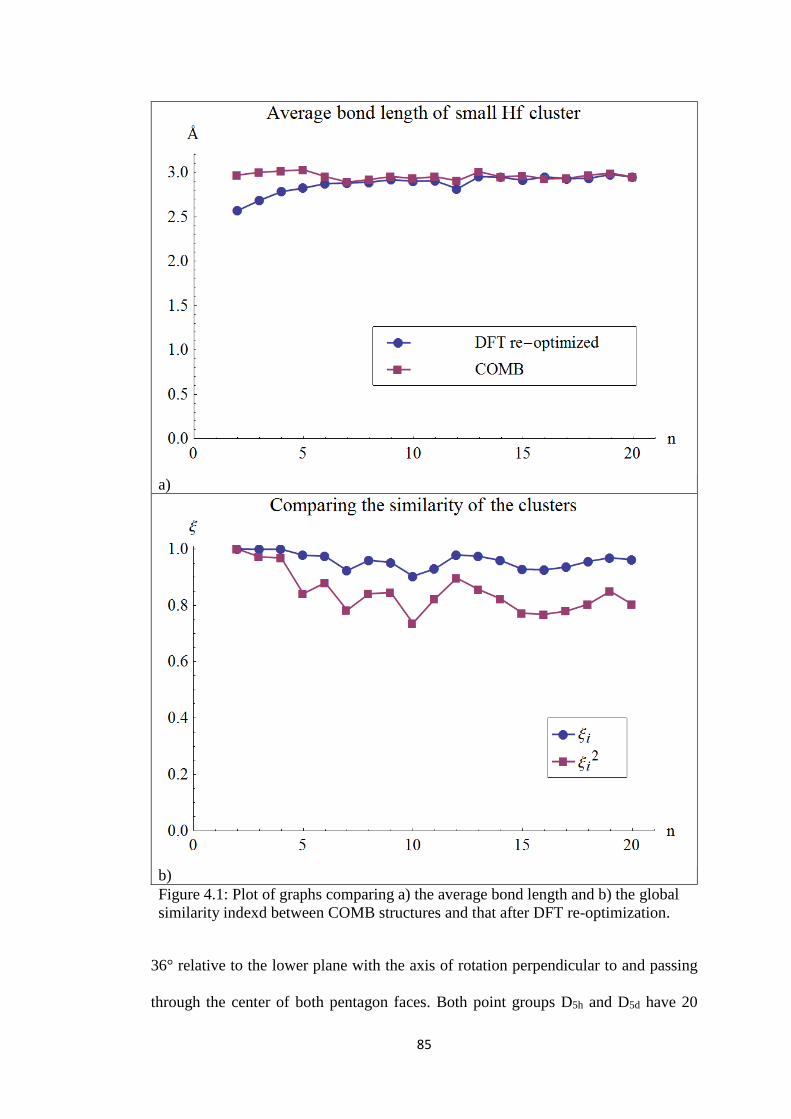
85
36° relative to the lower plane with the axis of rotation perpendicular to and passing
through the center of both pentagon faces. Both point groups D5h and D5d have 20
a)
b)
Figure 4.1: Plot of graphs comparing a) the average bond length and b) the global
similarity indexd between COMB structures and that after DFT re-optimization.

86
symmetry elements. Hf12 cluster was identical before and after the geometrical re-
optimization. Four other structures are slightly deviated (𝑛 = 11, 15, 16, 17; shaded
light blue in Table 4.1), and only three are heavily re-orientated (𝑛 = 7, 10, 20; shaded
dark blue). The order of dissimilarity in the point group symmetries were highlighted
with the increasing color intensity in Table 4.1 according to their order of differences.
Symmetry point group can only give a qualitative estimation to the structural similarity
and is somewhat lacking of quantitative preciseness. Nonetheless, this method still
predicts a good agreement to the other two methods, which were discussed in the
following paragraphs. Another way to access structural likeliness is by calculating the
global similarity index for the structures before and after re-optimization. In this
context, both the congruent and similar identifiers were applied. The role and
characteristic of both identifiers have been explained in Section 3.3.1.
Geometrical optimization in the G03 package has the tendency to rescale the
size of the cluster, and this effect has been taken into consideration in the global
similarity index by using the definition of 𝑘′𝑠,𝑖𝐶𝑂𝑅 as described in Equation (3.27). The
rescaling effect is seen, e.g., in the Hf2 to Hf6 clusters. As explained in Section 3.3.1,
since the variation in these structures before and after the geometrical re-optimization
is generally very minimum (very similar to each other), the function of the global
similarity index 𝜉𝑖 is to be squared to provide a more sensitive and visible reading
while monitoring the comparison process. In this case, Equation (3.24) can be written
as
𝜉𝑖2 =
1
9𝑛2∑ ∑(𝑘′𝑠,𝑖
𝐶𝑂𝑅 + 1)−2
𝑛
𝑠=1𝐶𝑂𝑅
(4.1)

87
As an illustration, Figure 4.1b shows the scores of global similarity index with
and without squaring. It can be seen that 𝜉𝑖2
displays a more visually distinctive
variation in its spectrum than 𝜉𝑖 . Despite both 𝜉𝑖2 and 𝜉𝑖 contains identical amount of
information, it was visually more convenient to monitor 𝜉𝑖2
, instead of 𝜉𝑖 , for
identifying abrupt changes in the evolution of the cluster structures. Hence, the choice
of 𝜉𝑖2 was merely due to visual convenience.
Table 4.2 displays the snapshots of the 19 structures obtained from plmp with
COMB potential and their corresponding DFT re-optimized counterparts. In Table 4.2,
two clusters of the same size are displayed side by side for the ease of direct
comparison. The left structures are those obtained from plmp, while the right ones are
the corresponding structures which have underwent DFT geometrical re-optimization
process. Observing the frames of structures tabulated in Table 4.2, it is visually
obvious that cluster with 𝑛 = 7,10,15,16,17 will yield the lowest similarity score.
This observation tallies well with the results tabulated in Table 4.1 and Figure 4.1b
(𝜉𝑖 = 1 is for total identity while 𝜉𝑖 → 0 for full structural dissimilarity).
Repetitive motif of pure elemental cluster can be observed in Hf7, Hf13 and
Hf19, which showed pentagonal bi-pyramidal like structure. It is shown in the next
section that this repetitive motif of Hf7 is in fact the lowest energy isomers for hafnium
cluster.
To sum up the plmp intra-line comparison, COMB potential produced COMB
structures that are geometrically close to the structures after G03 re-optimization. This
can be seen from the high degree of chemical similarity between them. This provided
the first part for the confirmation of the dependability of COMB potential in terms of
producing true global minimum of hafnium clusters. The full verification for COMB

88
dependability involves not only the intra-line comparison result but also from the inter-
line comparison, which was discussed in the next section.
Table 4.2: Comparing the structures of clusters obtained via plmp with COMB
(left) and those upon DFT geometrical re-optimization (right) side by side. The
size of the cluster is labeled just below the respective pairs of clusters in
comparison.
COMB DFT COMB DFT
Hf4 Hf5
Hf6 Hf7
Hf8 Hf9
Hf10 Hf11

89
COMB DFT COMB DFT
Hf12 Hf13
Hf14 Hf15
Hf16 Hf17
Hf18 Hf19
Hf20

90
4.2 Structural Confirmation of Hafnium Clusters
The previous section concluded that the reliability of COMB potential in
obtaining structures of hafnium clusters close to the local minimum in the DFT PES.
The objective of this section, on the other hand, is to determine, by way of inter-line
comparison, whether these local minimum are the global minimum or one of the lowest
energy isomers for the hafnium clusters in the DFT PES. As shown in schematic sketch
of Figure 3.5, both the process lines underwent geometrical re-optimization with the
same method and same basis set to ensure the consistency during comparison.
The hafnium clusters generated via pg3 (stage 1 in the pg3 process line) are
shown in Table 4.3. Due to the expensive computational cost, only small clusters of
Hf4, Hf5, Hf6, Hf7, and Hf8 are generated for the pg3 process line. As it is known a
priori, Hf2 and Hf3 will surely yield dimer and planar triangle structures. Each column
in Table 4.3 was filled with the screenshot of the geometry of the hafnium clusters, the
SCF energy in atomic units (hartree) rounded down to four decimal places, and the
cluster size. It was found that for smaller cluster sizes (Hf4, Hf5 and Hf6), only one
unique structure is resulted upon re-optimization by G03 (stage 2 in the pg3 process
line). In other words, the generally different structures (of a fixed 𝑛-sized cluster, with
𝑛 = 4, 5, 6) in stage 1 converged to the same geometry upon re-optimization in stage
2. On the other hand, Hf7 yields two while Hf8 yields four distinctive yet energetically
close-by structures in stage 2. The resultant clusters (with size 𝑛 = 4, 5, 6, 7, 8 )
obtained at the end of stage 2 in the pg3 process line are tabulated in Table 4.3.

91
Table 4.3: The hafnium clusters of size Hf4 to Hf8 in stage 2 of the pg3 process
line, along with their DFT total energy value in hartree. (*) indicates structures
with lowest energy, while (**) indicates similar structures that are also obtained in
the plmp process line.
-195.3351 -244.2367
Hf4 (*, **) Hf5 (*, **)
-293.1347 -341.9995
Hf6 (*, **) Hf7
-342.0305 -390.8634
Hf7 (*, **) Hf8
-390.8866 -390.8891
Hf8 (**) Hf8
-390.9112
Hf8 (*)

92
A single asterisk (*) in Table 4.3 indicates structures with lowest energy, while
double asterisk (**) indicates coincidental structures that are also obtained in stage 2
of the plmp process line. In general, the structural geometry of hafnium clusters
obtained via the pg3 process line agreed well with the findings by Charles (2008).
Besides Hf6, the remaining hafnium clusters generated in this thesis have the same
lowest energy structures as presented by Charles (2008). The overall agreement of the
resultant global structures of the hafnium clusters with the findings from the literature
provides a justification, albeit only for the limited range of cluster size, to the pg3
process line as a reliable method to generate true global minimum structures, if not for
the expansive computing cost.
The hafnium clusters marked with (**) are clear evidence that these ground
state structures produced via the COMB potential coincide with that generated via the
pg3 approach. The only exception is Hf8, where the structure predicted by the plmp
process line is in fact one of the low lying structures given by the pg3 approach.
Although the accuracy of COMB potential in predicting ground state structure is not
100%, but it is acceptable to a reliable extend. Four out of five ground state structures
obtained via the plmp process line are highly similar or coincidental with that produced
via the DFT approach, the pg3 process line, whereas the only exception happened to
be coincidental with one of LLS in the pg3 prcoess line. The largest cluster size which
COMB can reproduce the correct global structures can only be verified up to a size of
𝑛 = 8, within the affordability of the computational resource in this work. Due to the
expensive computational cost, ground state structures of larger cluster size from pg3
were not generated to provide comparison (and justification) to that generated via the
COMB potential. On the other hand, the structures obtained from COMB are also
supported by previous findings in the literature (Charles, 2008), further supported the

93
COMB potential for hafnium is in fact suitable to be used in finding the global
minimum structure of a cluster. As stated in the discussion of COMB potential, the
empirical potential used to generate the input structures was subsequently used for the
MD simulation.
To sum up, the intra-line and inter-line comparisons as discussed in this
Chapter have provided combine verifications to the dependability of the COMB
potential. Taken together, COMB potential is suitable to describe inter-atomic
interactions of hafnium atom in cluster environment, which agree with that obtained
with ab initio methods (pg3). After verifying the dependability of the COMB potential,
it was further used in MD simulation in the next Chapter.

94
CHAPTER 5
SIMULATED ANNEALING OF THE HAFNIUM CLUSTERS
5.1 The Melting Point of Hafnium Clusters
In previous chapter, the dependability of COMB potential has been verified. In
this chapter, the melting behavior of the hafnium clusters upon simulated annealing at
elevated temperatures using COMB potential was investigated. The converged COMB
structures (the global minimum in the stage 1 of the plmp process line) were used as
input in the subsequent annealing MD simulation. The output of MD heating
simulation by using COMB as the input potential was examined in detail in the
following discussions.
The results of simulated annealing presented in this chapter are based on the
few post-processing methods mentioned in Section 3.3. The COMB structures were
used as input for the simulated annealing to ensure that the same set of laws governing
the geometry of the structures were also used to govern the dynamic interactions
between the hafnium atoms during the simulated annealing procedure. There is a subtle
weak connection between the initial input structure and the end resultant melting
configuration. In fact, Lu et al. (2013) recently proved that the choice of input structure
essentially affects the outcome of simulated annealing procedure.
To begin, the MD heating simulation of Hf20 as an illustrative example was
discussed. The caloric and 𝑐𝑣 curves of Hf20 generated by prolonged annealing process

95
were presented in Figure 5.1. Each point on the caloric curve has a corresponding
counterpart on the 𝑐𝑣 curve at the same temperature. It is easy to understand the
connection between the two graphs based on thermodynamic interpretation for the
constant volume specific heat capacity 𝑐𝑣, which is given by the gradient of the caloric
curve according to the following equation
𝑐𝑣 = (𝜕𝐸
𝜕𝑇)
𝑣
(5.1)
where 𝐸 are the potential energy value, 𝑇 the temperature, and the subscript 𝑣 denotes
the derivatives taken under constant volume thermosetting. However, the method used
to obtain the 𝑐𝑣 curve in this thesis is by calculating the fluctuations of the caloric
curve with Equation 2.3.
Melting transition is expected to happen when the potential energy of the
cluster changed drastically during the melting point, 𝑇𝑚. This temperature corresponds
to the latent heat of fusion. Based on the definition of Equation 5.1, this drastic change
of 𝐸 value is expecting to return a relatively larger value of 𝑐𝑣 in the vicinity of 𝑇𝑚.
Thus, melting transition corresponds to a large peak in 𝑐𝑣 curve. Apparently, from
Figure 5.1, the caloric curve was less effective in giving the exact temperature for both
the melting point and pre-melting point as compared to the 𝑐𝑣 curve. There were
many random peaks in the 𝑐𝑣 curve (Figure 5.1b), the first apparent peak is the pre-
melting, while the highest peak is the melting point. The melting point corresponds to
the most obvious transition in the caloric curve (Figure 5.1a). The green arrow
indicates the pre-melting temperature 𝑇𝑝𝑟𝑒 = 1400 K while the red arrow indicates the
melting temperature 𝑇𝑚 = 1850 K in both graphs.

96
a)
b)
Figure 5.1: a) The caloric curve and b) 𝑐𝑣 curve of Hf20 obtained via prolonged
annealing process (TNA = total number of atoms in the cluster). The green arrow
indicates the pre-melting temperature at 𝑇𝑝𝑟𝑒 = 1400 K, and the red arrow
indicates the melting point at 𝑇𝑚 = 1850 K.
In addition to the prolonged annealing method, an independent method to
obtain melting behaviour of the clusters was also attempted, as discussed in Section
3.3, which is the direct heating method. The overall idea of direct heating process was
described in Section 3.3. The purposes for conducting this procedure on top of the

97
prolonged method are two-fold: (1) it was used as an independent check to the results
obtained via the prolonged method, and (2) to generate the evolutional history of global
similarity index of the clusters, which carries the dynamical information throughout
the evolutionary history of a MD simulation. (2) can be obtained via post-processing
the LAMMPS trajectory file (.lammpstrj). The trajectory file contained the frames of
the atoms that reflect the heating process as recorded in the corresponding log (.log)
file. However, in the prolonged annealing simulation, in which the target temperatures
were generally set to a very high value, e.g. 𝑇 = 3000 K, a large fluctuation in the
temperature is usually present throughout the equilibration steps (see the example in
Figure 3.4). Due to the large fluctuation in the temperature, the trajectory of the atoms
also fluctuates rigorously. Over an extended equilibration time step, the trajectory of
each individual hafnium atom in the cluster will move all over the simulation box and
tends to be ergodic. The ergodicity is not desired for the coordinates of the cluster
system as the average position of each atom will overlap in the confined space of
simulation box. The coordinates as recorded in the trajectory file throughout the
equilibration stage do not corresponds to the target temperature at which the system is
in. Therefore, global similarity index cannot be used in the prolonged heating
procedure to quantify the heating process.
In contrast, the direct heating method, due to the imposition of the 300∆𝑡 time
averaging protocol, the fluctuations of all recorded data in both the LAMMPS output
files (.log and .lammpstrj) were confined within the 300∆𝑡 time window. As a result,
the original thick slope in Figure 3.4a was now transformed into a much refined and
narrower slope in Figure 3.4b. The coordinates as recorded in the trajectory file in the
300∆𝑡 time window do correspond to the temperature at which the system is currently

98
in. This allowed the information of heating mechanism to be extracted by using global
similarity index in the direct heating procedure.
In addition, the direct heating method is also able to predict the melting
temperature 𝑇𝑚 and pre-melting temperature 𝑇𝑝𝑟𝑒 by plotting the caloric curve and the
𝑐𝑣 curve. In this respect, the caloric and 𝑐𝑣 curve are known as ‘indicators’, meaning
MD quantities from which thermodynamically interesting transitions can be monitored.
In fact, the caloric profile is readily to be extracted from the log file of LAMMPS
output, since both 𝐸 and 𝑇 are recorded in separated columns of the log file. Figure
5.2 shows the caloric curve and 𝑐𝑣 curve of Hf10 via direct heating method. The 𝑐𝑣
curve shows a lot of peaks (Figure 5.2b). The first apparent peak is the pre-melting
and the tallest peak is the melting point. The melting point corresponds to the largest
transition in caloric curve (Figure 5.2a). By using the same color code as for Figure
5.1, green arrow indicates the pre-melting temperature 𝑇𝑝𝑟𝑒 = 1350 K, and the red
arrow indicates the melting point 𝑇𝑚 = 2200 K. Both curves provide complementary
information to help locating the transition temperatures. Only one unique transition
temperature was reported based on these two indicators after comparing and
contrasting both curves to estimate the most precise value for the transition
temperatures. Due to its higher sensitivity, the transition temperatures reported in this
thesis were in all cases read off from the 𝑐𝑣 curve. As it turns out, indication of
transition in the caloric curve is relatively less distinctive (hence higher in uncertainty),
hence it was used only as a supplimentary confirmation to the transition value reported
from the 𝑐𝑣 curve.

99
a)
b)
Figure 5.2: a) The caloric curve and b) 𝑐𝑣 curve of Hf10 obtained via direct heating
process. The green arrow indicates the pre-melting temperature at 𝑇𝑝𝑟𝑒 = 1350 K,
and the red arrow indicates the melting point at 𝑇𝑚 = 2200 K.
The identification of transition temperatures can also be made based on another
independent indicator, which is the global similarity index. In this case, 𝜉𝑖 shall
consider only the congruent identifier (rescaling is expected to be observed during
heating procedure) as described by Equation (3.24), where 𝑘𝑠,𝑖𝐶𝑂𝑅 takes the form of
𝑐𝑣(eV/K)

100
Equation 3.23. It also reveals the structural changes occurring in the clusters during
the heating procedure. The trajectory file used to obtain the similarity index profile has
based on the direct heating method. The dynamical evolution of global similarity index
as a function of temperature is shown in Figure 5.3 for Hf13 as a sample candidate.
Figure 5.3a shows the similarity index 𝜉𝑖 profile for each frames during the heating
process as compared to the ground state structure (the 0th frame) by using Equation
3.22. Figure 5.3b is the fluctuation (variance) of similarity index 𝑆𝜉𝑖 corresponds to the
same trajectory calculated by using Equation 3.28.
The features which are thermodynamical interest in the example of Figure 5.3
is now interpreted. Melting and pre-melting features can be identified from both the
𝑆𝜉𝑖 vs 𝑇 and 𝜉𝑖 vs 𝑇 curves, albeit with different sensitivity. The pre-melting transition
is not obvious in the 𝑆𝜉𝑖 curve due to the suppression of the overall scale in 𝑆𝜉𝑖
by the
high peak at 𝑇 ≈ 2050 K. The pre-melting transition can merely become visible by
zooming in to the 𝑆𝜉𝑖 curve at around 𝑇 ≈ 1050 K, as displayed by the inset of Figure
5.3b. As is observed throughout the MD calculations in this thesis, it was found that
usually the signal of pre-melting or melting in the 𝑆𝜉𝑖 curve waas moderate unless the
transitions in the clusters happened in a really abrupt manner. In practice, both the 𝑆𝜉𝑖
and 𝜉𝑖 curves have to be simultaneously deployed so that the complementary features
in both curves can pinpoint a transition with a more reliable precision. As in the case
of Figure 5.3, the pre-melting point correspond to the first obvious drop in the
similarity index (indicated by the green arrow) at 𝑇𝑝𝑟𝑒 = 1600 K. On the other hand,
the melting point was visibly apparent in both graphs (indicated by the red arrow) at
𝑇𝑚 = 2050 K.
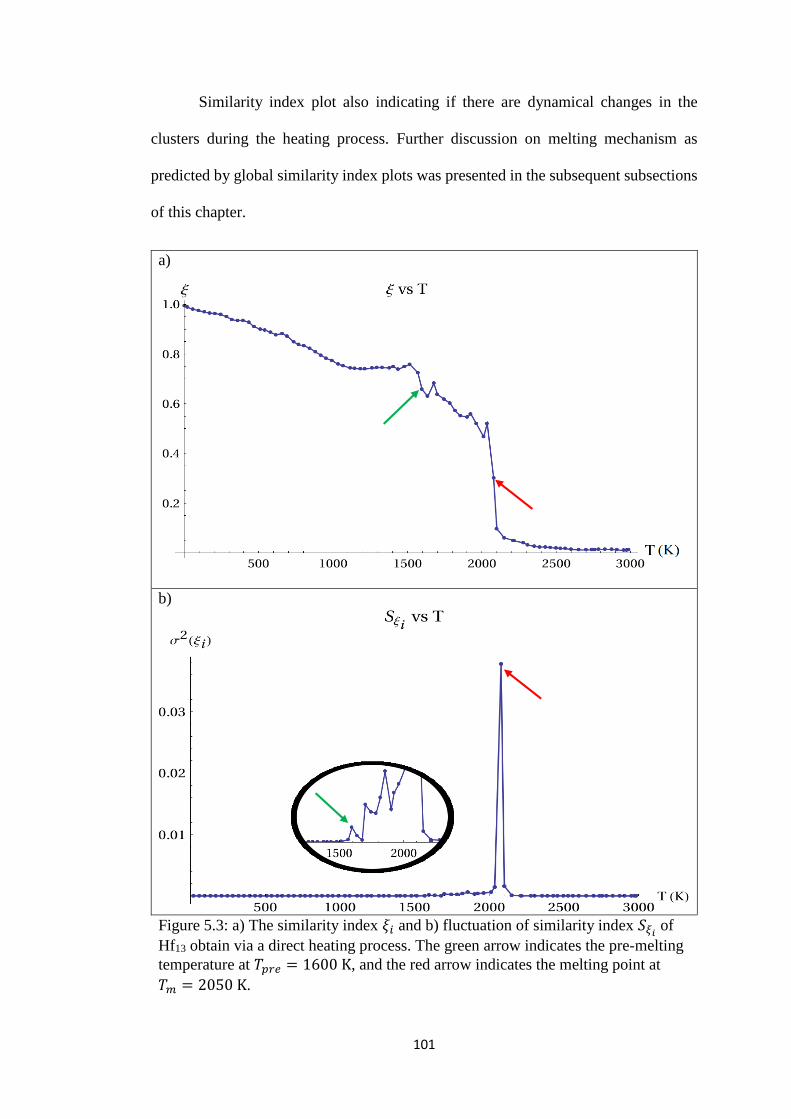
101
Similarity index plot also indicating if there are dynamical changes in the
clusters during the heating process. Further discussion on melting mechanism as
predicted by global similarity index plots was presented in the subsequent subsections
of this chapter.
a)
b)
Figure 5.3: a) The similarity index 𝜉𝑖 and b) fluctuation of similarity index 𝑆𝜉𝑖
of
Hf13 obtain via a direct heating process. The green arrow indicates the pre-melting
temperature at 𝑇𝑝𝑟𝑒 = 1600 K, and the red arrow indicates the melting point at
𝑇𝑚 = 2050 K.

102
5.2 Melting Temperature and Cluster Sizes
Three different indicators were used in this thesis to identify melting transitions
in two independent heating procedures. In the prolonged heating method, only two
indicators were used to identify melting transitions, namely the caloric and 𝑐𝑣 curves.
In the direct heating method, the caloric, the 𝑐𝑣 curves and global similarity index were
used as indicators. In this section, the transition temperatures as a function of cluster
size 𝑛 were reported.
Table 5.1 shows the results of melting and pre-melting temperatures in both
heating procedures. The first two columns were the melting temperatures obtained
from prolonged annealing methods. The third and fourth columns were directly
extracted from a direct heating simulation elevated from 0 K to 3000 K at the
optimum rate of 5 × 1012 Ks−1. These transition temperatures were determined from
the 𝑐𝑣 curve but not the caloric curve (for the reason already explained in the previous
subsection). Meanwhile the last two are transition temperatures deduced from the
graph of fluctuations in the global similarity index, 𝑆𝜉𝑖. Recall that global similarity
index is only applicable in the direct heating process. The distinctions between the
prolonged annealing and the direct heating process have been mentioned in Section
3.3. In fact, the prolonged simulated annealing method involves compilation of many
heating processes equilibrated at different target temperatures. Every single data point
in the caloric curve was the result of statistically sampling a cluster’s energy over a
lengthy period of equilibration at the fixed temperature. The pre-melting temperature,
𝑇𝑝𝑟𝑒 , and the exact melting temperature, 𝑇𝑚, were plotted in the same graph in Figure
5.4 for each method. The melting temperature 𝑇𝑚 vs 𝑛 obtained from the three

103
different approaches listed in Table 5.1 were also compiled into a single graph as
shown in Figure 5.5 for a better comparison.
Table 5.1: The melting and pre-melting temperatures obtained from three different
approaches. The first four columns are obtained from caloric curves and 𝑐𝑣 curves.
𝑛
Prolonged simulated
annealing
Direct heating
process
Fluctuation of the
similarity index, 𝑆𝜉𝑖
curves
𝑇𝑝𝑟𝑒 (K) 𝑇𝑚 (K) 𝑇𝑝𝑟𝑒 (K) 𝑇𝑚 (K) 𝑇𝑝𝑟𝑒 (K) 𝑇𝑚 (K)
6 1250 1750 1250 2000 1250 1950
10 1500 1750 1350 2200 1850 2250
14 1550 1800 1200 1900 1700 2200
17 1200 1900 1250 2250 1700 2100
18 1450 1700 1450 2200 1850 2100
20 1400 1850 1500 2150 1750 2150
22 1600 1850 1500 2150 1750 2150
24 1350 1800 1700 2050 1800 2100
26 1500 1850 1950 2300 2050 2250
30 1650 1750 1650 2250 1750 1850
34 1500 1800 1950 2300 1850 2100
38 1550 1850 1650 2500 1850 2000
39 1600 1900 1700 2350 1600 2000
40 1350 1900 1400 2100 1800 2150
42 1650 1950 1800 2100 1800 1950
46 1600 1850 1800 2250 1800 2200
50 1450 1900 1600 2250 1800 2150
99 1650 2050 2150 2500 1900 2300
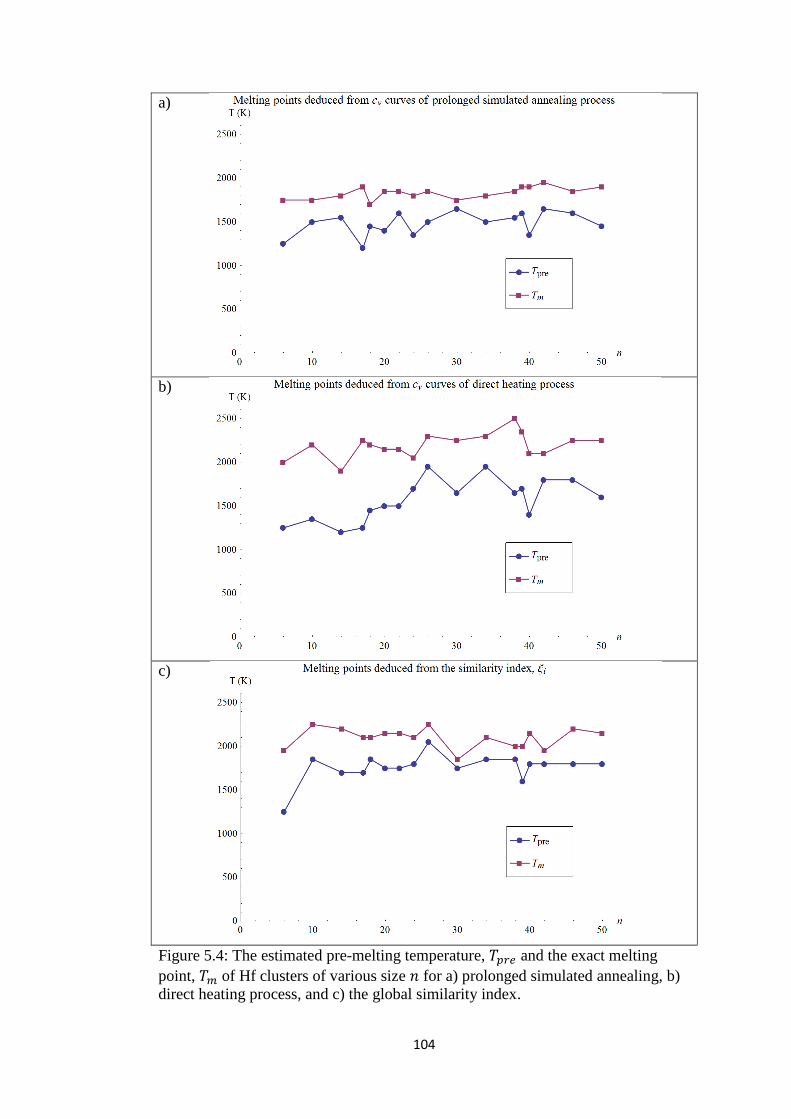
104
a)
b)
c)
Figure 5.4: The estimated pre-melting temperature, 𝑇𝑝𝑟𝑒 and the exact melting
point, 𝑇𝑚 of Hf clusters of various size 𝑛 for a) prolonged simulated annealing, b)
direct heating process, and c) the global similarity index.

105
Figure 5.5: The estimated melting point of the hafnium cluster against the cluster
size 𝑛, based on three different approaches.
As mentioned in Section 3.3, direct heating of the structure to a high
temperature allows one to yield detailed dynamics of the clusters. It can be seen from
Figure 5.5 that this approach overall predicts a higher melting point than that from the
prolonged annealing method, but the prediction still agreed well with expectation as
compared to the bulk melting of hafnium, which is 𝑇𝑚𝑏𝑢𝑙𝑘(Hf)~2504 K according to
the periodic table. Nevertheless, both simulation approaches agreed that the melting
temperature of hafnium cluster increases with the size 𝑛.
One of the most important results obtained from this thesis is that global
similarity index, 𝜉𝑖, has the capacity to predict the dynamics of the heating process
from the MD trajectory of clusters. A few samples of 𝜉 plotted against the temperature
𝑇 were shown in next section, along with their respective fluctuations, 𝑆𝜉𝑖. Snapshots
of the coordinates of the clusters were presented directly from VMD visualization.
Based on the 𝜉𝑖 graph (Figure 5.3 for example), every obvious drop in the average
value corresponds to a significant change in trajectory, starting with changes from

106
surface and slowly to the core of the whole cluster. In general, the pre-melting effect
take place at the first significant peak of 𝑆𝜉𝑖, whereby the exterior of the cluster is
usually heavily distorted, but the particle is still bounded relatively close to each other.
As the heating continues, the cluster would be a de-fragmented and start drifting in
random direction, entering the fluidic phase. Section 5.3 presented some of the
screenshots of the clusters over the course of simulation, which corresponds to the
points represented in the similarity index curves for the case of Hf30, Hf50, and Hf99. In
order to portray the distance between atoms in the clusters, the dynamic bond length
was set to be around 3.127Å.
5.3 Similarity Index and Cluster Melting
In the selected cases of the following subsections, the plots of similarity index
𝜉𝑖 were compared to its fluctuation plot 𝑆𝜉𝑖 as a function of temperature. Every major
transitions in 𝜉𝑖 correspond to a significant change in cluster geometry which were
shown by VMD screenshots, add-ons to each graph signified the happening of certain
event during heating process. These transitions were sometimes less apparent in 𝜉𝑖
graphs, but are indicated clearly in the fluctuation plot, 𝑆𝜉𝑖.
As mentioned earlier in the first section of this chapter, the direct heating
procedure enables LAMMPS package to record the dynamic changes of candidate
structures in both the .log and .lammpstrj files. The global similarity index method
used in this thesis enabled the coordinates of the system to be recorded in the trajectory
file to be portrayed graphically such as that shown in the example of Figure 5.3. In

107
fact, the original purpose of the trajectory file was to allow the visualization of the
dynamics of the system.
The following subsections of Section 5.3 focused only to a few specific sizes
of hafnium clusters. The figures shown in each subsection were discussed in details in
the following paragraphs. The choice of the hafnium clusters in the discussions
included all the events that were detected by both the similarity index 𝜉𝑖 plot and the
fluctuation of the similarity index 𝑆𝜉𝑖 plot.
5.3.1 Hf30
From Figure 5.6, the first significant change of Hf30 cluster in the heating
process occurs around 𝑇 = 1000 K to 1300 K, indicated by the sudden drop of 𝜉𝑖. The
outermost layer atoms become further apart from each other but remain intact with the
‘core’ atoms. The pre-melting took place at the first peak of the 𝑆𝜉𝑖, at around 𝑇𝑝𝑟𝑒 =
1750 K with the ‘shell’ atoms begin to drift apart from the cluster. The highest peak
of 𝑆𝜉𝑖, corresponds to a temperature around 𝑇𝑚 = 1850 K, showing a total breakdown
of the cluster. The screenshot at 𝑇𝑚 = 1850 K has not included a drifted hafnium atom
far towards the edge of the simulation box. A final screenshot at 𝑇 = 2150 K showed
the remnant cluster ‘core’ after many surface atoms have drifted away.

108
a)
b)
Figure 5.6: a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity index
𝑆𝜉𝑖 of Hf30. The screenshots show the configuration of the cluster Hf30 during that
particular temperature.

109
5.3.2 Hf50
a)
b)
Figure 5.7: a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity index
𝑆𝜉𝑖 of Hf50. The screenshots shows the configurations of the cluster Hf50 during
that particular temperature.
For the case of Hf50 as shown in Figure 5.7, the 𝜉𝑖 curve displays some unique
features, which were the sudden droped at around 𝑇 = 1850 K and a sudden rose at
around 𝑇 = 1900 K. This behavior was observed to be the effect of a single hafnium
atom leaving the cluster drifting to the wall of the simulation box and bouncing back
to the cluster. This behavioral change was the result of kinetic energy accumulated

110
within a single atom that causes it to escape from the cluster. In order to ensure that
this artifact is not a random error in the simulation, the overall simulated annealing
procedure was re-run with slightly different initial condition (different random initial
velocity). The same feature was showed up when different initial condition is imposed.
The sharp change was also detected by the fluctuation plot against temperature
showing two sharp peaks at about the same temperature. The left one corresponded to
a sudden drop in 𝜉𝑖 while the right one corresponds to a sudden rise of 𝜉𝑖 value. The
true melting is at around 𝑇𝑚 = 2150 K when the cluster broke down completely and
drifted apart. In fact, the same scenario was also observed in the case of Hf18 and Hf26.
Along with their respective graph of similarity index, 𝜉𝑖 curve, Figure 5.8 shows the
particular snapshot where the drifting of a single atom begins to take place. For all
these cases, the drifting of single atom happened close to the melting point, 𝑇𝑚. Other
than atoms leaving the cluster one at a time, the more common observation is that the
clusters were being tore down into few groups and slowly dissolve into a mist of atoms
cloud in liquid-gaseous like phase.

111
a)
b)
Figure 5.8: The artifact of single atom drifting away observed in the case of a) Hf18
and b) Hf26.
5.3.3 Hf99
Cluster size Hf99 was used to describe the complete trace of dynamics during
the single annealing process from 𝑇 = 0 K to 3000 K, as shown in Figure 5.9. The
atomic vibration slowly increased in magnitude upon velocity rescaling. As observed
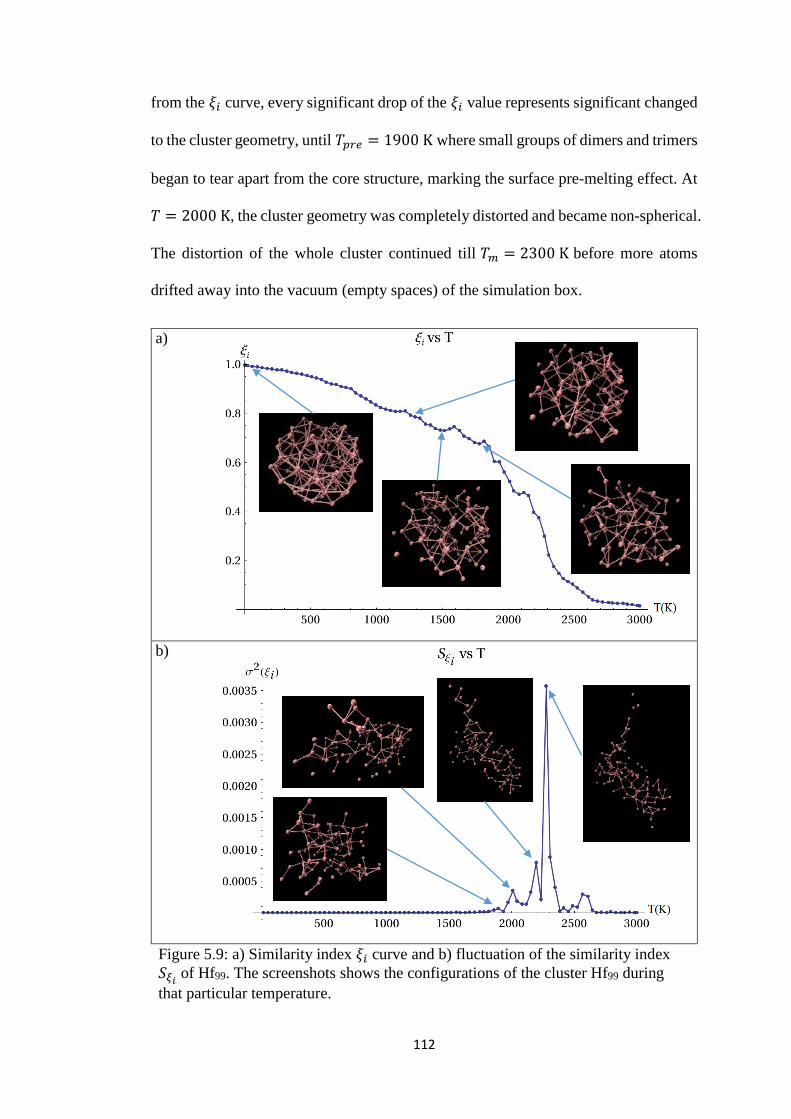
112
from the 𝜉𝑖 curve, every significant drop of the 𝜉𝑖 value represents significant changed
to the cluster geometry, until 𝑇𝑝𝑟𝑒 = 1900 K where small groups of dimers and trimers
began to tear apart from the core structure, marking the surface pre-melting effect. At
𝑇 = 2000 K, the cluster geometry was completely distorted and became non-spherical.
The distortion of the whole cluster continued till 𝑇𝑚 = 2300 K before more atoms
drifted away into the vacuum (empty spaces) of the simulation box.
a)
b)
Figure 5.9: a) Similarity index 𝜉𝑖 curve and b) fluctuation of the similarity index
𝑆𝜉𝑖 of Hf99. The screenshots shows the configurations of the cluster Hf99 during
that particular temperature.

113
Figure 5.10 shows the state of cluster Hf99 upon some long equilibration at 𝑇 =
3000 K. In order to visualize the appearance of the whole cluster Hf99 after it is
completely melted and stayed in fluidic phase at the end of 𝑇 = 3000 K equilibration,
the snapshot is enlarged four times as compared to the previous examples. The frame
of Figure 5.10 was set roughly to the size of the simulation box.
Figure 5.10: Hf99 upon the equilibration at 𝑇 = 3000K.

114
CHAPTER 6
CONCLUSIONS AND FUTURE STUDIES
6.1 Conclusions
In this thesis, the MD simulations have been carried out to yield the detailed
dynamics of the hafnium cluster systems in heating processes. Each part of the
simulations was verified with careful examinations. Chapter 4 laid out the verification
to show that COMB potential is an appropriate interatomic potential in describing
hafnium clusters. The hafnium clusters obtained via the plmp algorithm with COMB
potential for the size 𝑛 = 2 to 𝑛 = 8 were in a good agreement to that reported in the
literatures. COMB potential also produced structures close to the ground state of DFT
method, justified by the principle of chemical similarity. Clusters Hf7, Hf13, and Hf19
showed repetitive geometry motif of pentagonal bipyramid. The plmp algorithm
proved to be very efficient in searching the global minimum structures of the given
interatomic potential. The BH-GA part of the algorithm was capable to handle various
class of empirical potential with well-established convergence. In principle, the
process lines comparison algorithm as described in Chapter 4 can also be adopted as a
practically implementable protocol to verify or falsify the dependability of any generic
empirical potentials for producing true ground state structures at the DFT accuracy.
Chapter 5 reported the melting transition of hafnium clusters for the size of
clusters up to 𝑛 = 99. The melting point of hafnium cluster showed gradual increment
with the increasing of number of atoms, 𝑛, but remained lower than the bulk value of
𝑇𝑚𝑏𝑢𝑙𝑘(𝐻𝑓)~2504 K. This showed the size dependent melting behavior of a cluster. In
addition, hafnium clusters experienced surface atom pre-melting effect at some

115
temperature 𝑇𝑝𝑟𝑒 lower than that of its melting point 𝑇𝑚. Other than the surface pre-
melting, some cluster sizes such as Hf18, Hf26 and Hf50 displayed a featured pre-melting
stage in which a single hafnium atom was observed to escape from the cluster in a brief
moment before the clusters melted thoroughly.
6.2 Future Studies
During the generation of candidate structures for hafnium clusters, both plmp
and pg3 process line come in handy. The process lines comparison algorithm to search
and confirm the global minimum of given empirical potential can be used as a good
addition to the technique currently available in the field of cluster science. If an
empirical potential is found to pass the test of the process line comparisons, then
ground state structures of large cluster size at DFT accuracy could be generated via the
computationally economical plmp procedure. Extended work to gather simulation data
on clusters formed by different types of element, for a variety of interatomic potential
is verified by the protocol, and large size ground state structures at DFT accuracy are
correctly generated via the plmp process. The robustness of the process lines algorithm,
if established with empirical evidences, shall be a very powerful tool for large size,
DFT-accurate, ground state cluster search.
The MD procedures used in this thesis were successful. These include the
solution to some of the fundamental problems such as the flying ice cube problem and
the broad fluctuations in thermodynamic variables for the heating of small size systems.
The MD procedures used in this thesis can be used to study any cluster system of
interest provided a good interatomic potential to describe the system is available. In
fact, MD method is very powerful and is able to perform more than just heating and

116
quenching, bearing a good complement to the experiments. In many instances, MD is
able to mimic ‘experiment’ on small sizes system, such as the work of this thesis.
In this thesis, the method of global similarity index succeeded in studying the
dynamic of the system during heating and melting. The visualization was simplified
into graphs that trace the trajectories of the coordinates of each hafnium atom during
the MD process. The general idea of global similarity index was based upon the
chemical similarity principle. It was rather simplified in term of functional form as
well as working principle aiming to detect any characteristic transition throughout the
evolutional history of a cluster in a MD simulation. The idea and application of global
similarity index has the potential to be further developed into a powerful fingerprint
descriptor that could unambiguously and uniquely recognize the identity of a three
dimensional molecule, such as protein molecule or other large, biochemically
interested molecules, from a numerical database containing some arbitrary,
unidentified molecular structures. The proposed descriptor could find its application
in computational drug design. For example, the design of a drug targeting a particular
protein. Firstly, catalogue all the fingerprint of (the improved version of) the global
similarity index of a list of candidate drugs. Meanwhile, an algorithm can be developed
to computationally scan the entire 3D morphology of a target protein for local sites
containing any signature of complementary fingerprint of the drugs in the catalogue.
If candidate local sites on the protein with complementary similarity to any of the drugs
in the catalogue was reported, then a full scale biomolecular MD can be followed up,
where these drugs can be placed close to the local site to monitor the resultant
interactions between them.

117
The global similarity index can also be expanded independently to take into
consideration of additional physical properties other than the atomic position that has]
been used in this thesis.

118
REFERENCES
Aguado A., López J.M., Alonso J.A. and Stott M.J. (2001). Melting in large sodium
clusters: An orbital-free molecular dynamics study, J. Phys. Chem. B 105: 2386.
Ahlrichs R. and Elliott S.D. (1999). Clusters of aluminium, a density functional study,
Phys. Chem. Chem. Phys. 1: 13.
Andreeva A., Howorth D., Chothia C., Kulesha E. and Muezin A.G. (2013). SCOP2
prototype: a new approach to protein structure mining, Nucl. Acid Res. 42: D310.
Antoniadis D. (2015). EP1: Moore’s Law Challenges Below 10nm: Technology,
Design and Economic Implications, ISSCC 2015, Evening Session, EP1.
Bacskay G.B. (1981). A quadratically convergent Hartree-Fock (QC-SCF) method.
Application to closed shell systems, Chem. Phys. 61: 385.
Bajusz D., Rácz A. and Héberger K. (2015). Why is tanimoto index an appropriate
choice for fingerprint-based similarity calculations? J. Cheminf. 7: 20.
Ballester P.J. and Richards W.G. (2007). Ultrafast shape recognition for similarity
search in molecular databases, Proc. R. Soc. A 463: 1307.
Balleto F. and Ferrando R. (2005). Structural properties of nanoclusters: Energetic,
thermodynamic, and kinetic effects, Rev. Mod. Phys. 77: 371.
Barron T.H.K. and Domb C. (1955). On the cubic and hexagonal close-packed lattices,
Proc. Roy. Soc. A227: 447.
Berry R.S. and Smirnov B.M. (2004). Heat capacity of isolated clusters, J. Exp. Theor.
Phys. 98: 366.
Bergeron D.E., Castleman A.W. Jr, Marisato T. and Khanna S.N. (2004). Formation
of Al13I-: evidence for the superhalogen character of Al13, Science 304: 84.
Biswas R. and Hamann D.R. (1987). New classical models for silicon structural
energies, Phys. Rev. B 36: 6434.
Boustani I. (1997). Systematic ab initio investigation of bare boron clusters:
Determination of the geometry and electronic structures of Bn (n=2-14), Phys. Rev.
B 55: 16426.
Boustani I., Quandt A., Hernández E. and Rubio A. (1999). New boron based
nanostructured materials, J. Chem. Phys. 110: 3176.
Breaux G.A., Neal C.M., Cao B. and Jarrold M.F. (2005). Melting, premelting and
structural transitions in size-selected aluminium clusters with around 55 atoms,
Phys. Rev. Lett. 94: 173401.

119
Brenner D.W. (1990). Empirical potential for hydrocarbons for use in simulating the
chemical vapor deposition of diamond films, Phys. Rev. B 42: 9458.
Brenner D.W., Shenderova O.A., Harrison J.A., Stuart S.J., Ni B. and Sinnott S.B.
(2002). A second-generation reactive empirical bond order (REBO) potential
energy expression for hydrocarbons, J. Phys.: Condens. Matter 14: 783.
Brown F. (2005). Editorial opinion: chemoinformatics – a ten year update, Curr. Opin.
Drug Discov. Dev. 8: 298.
Buffat Ph. and Borel J-P. (1975). Size effect on the melting temperature of gold
particles, Phys. Rev. A 13: 2287.
Calvo F. and Spiegelmann F. (1999). Mechanism of phase transitions in sodium
clusters: From molecular to bulk behavior, J. Chem. Phys. 112: 2888.
Cannon E.O., Nigsch F. and Mitchell J.BO (2008). A novel hybrid ultrafast shape
descriptor method for use in virtual screening, Chem. Cent. J. 2:3.
Charles W.B. Jr. (2008). Clusters of hafnium, Hfn 𝑛 = 2 − 8, Chem. Phys. Lett. 462:
183.
Chuang F.C., Wang C.Z. and Ho K.H. (2006). Structure of neutral aluminium clusters
Aln (2≤n≤23): Genetic algorithm tight-binding calculations, Phys. Rev. B 73:
125431.
Couchman O.R. and Jesser W.A. (1977). Thermodynamic theory of size dependence
of melting temperature in metals, Nature 269: 481.
Dash J.G. (2002). Melting from one to two to three dimensions, Contemp. Phys. 43:
427.
Duan H., Ding F., Rosén A., Harutyunyan A.R., Curtarolo S. and Bolton K. (2007).
Size dependent melting mechanism of iron nanoclusters, Chem. Phys. 333: 57.
Duin A.C.T., Strachan A., Stewman S., Zhang Q., Xu X. and Goddard W.A. III (2003).
ReaxFFsio reactive force field for silicon and silicon oxide systems, J. Phys. Chem.
A 107: 3803.
Durant J.L., Leland B.A., Henry D.R. and Nourse J.G. (2002). Reoptimization of MDL
keys for use in drug discovery, J. Chem. Inf. Comput. Sci. 42: 1273.
Ebbesen T.W., Lezec H.J., Hiura H., Bennett J.W., Ghaemi H.F. and Thio T. (1996).
Electrical conductivity of individual carbon nanotubes, Nature 382: 54.
Efremov M.Y., Schiettekatte F., Zhang M., Olson E.A., Kwan A.T., Berry R.S. and
Allen L.H. (2000). Discrete periodic melting point observations for nanostructure
ensembles, Phys. Rev. Lett. 85: 3560.
Ellis W.Y, Notsch J.D. and McGreevy P.B. (2003). Chemical information systems,
U.S. Patent No. 6,654,736.

120
Erhart P. and Albe K. (2005). Analytical potential for atomistic simulations of silicon,
carbon and silicon carbide, Phys. Rev. B 71: 035211.
Fang Y., Liu Y-S. and Ramani K. (2009). Three dimensional shape comparison of
flexible proteins using the local-diameter descriptor, BMC Struct. Bio. 9: 29.
Faraday M. (1859). Note on regelation, Proc. Roy. Soc. 10: 440.
Ferrando R., Jellinek J. and Johnston R.L. (2008). Nanoalloys: From theory to
applications of alloy clusters and nanoparticles, Chem. Rev. 108: 845.
Finnis M.W. and Heine V. (1974). Theory of lattice contraction at aluminium surface,
J. Phys. F 4: L37.
Giasuddin ASM, Jhuma K.A. and Mujibul Haq A.M. (2012). Use of gold nanoparticles
in diagnostics, surgery and medicine: a review, Bangladesh J. Med. Biochem. 5(2):
56.
Gould A.L., Logsdail A.J. and Catlow C.R.A. (2015). Influence of composition and
chemical arrangement on the kinetic stability of 147-atom Au-Ag bimetallic
nanoclusters, J. Phys. Chem. C 119: 23685.
Grant J.A., Gallardo M.A. and Pickup B.T. (1996). Comparison: A simple application
of a gaussian description of molecular shape, J. Comput. Chem. 17: 1653.
Grigoryan V.G. and Springborg M. (2003). Structure and energetics of Ni clusters with
up to 150 atoms, Chem. Phys. Lett. 375: 219.
Gupta R.P. (1981). Lattice relaxation at a metal surface, Phys. Rev. B 23: 6265.
Hartke B. (1996). Global geometry optimization of clusters guided by N-dependent
model potentials, Chem. Phys. Lett. 258: 144.
Hartke B. (1998). Global geometry optimization of small silicon clusters at the level
of density functional theory, Theor. Chem. Acc. 99: 241.
Harvey S.C., Tan K.Z.R. and Cheatham T.E. III. (1998). The flying ice cube: velocity
rescaling in molecular dynamics leads to violation of energy equipartition, J.
Comput. Chem. 19: 726.
Hay P.J. and Wadt W.R. (1985). Ab initio effective core potentials for molecular
calculations. Potentials for the transition metal atom Sc to Hg, J. Chem. Phys. 82:
270.
Hsu P.J. and Lai S.K. (2006). Structures of bimetallic clusters, J. Chem. Phys. 124:
044711.
Hsu P.J. (2014). A new perspective of shape recognition to discover the phase
transition of finite size clurters, J. Comput. Chem. 35: 1082.
Humphrey W., Dalke A. and Schulten K. (1996). VMD – visual molecular dynamics,
J. Mol. Graphics 14: 33.

121
Jaramillo-Botero A., Su J., Qi A. and WGoddard A. 3rd (2011). Large-scale, long-
term non-adiabatic electron molecular dynamics for describing material properties
and phenomena in extreme environments, J. Comp. Chem. 32: 497.
Johnson A.M. and Maggiora G.M. (1990). Concepts and Applications of Molecular
Similarity, New York: John Willey & Sons.
Khan A, Rahman K, Kim D.S. and Choi K.H. (2012). Direct printing of copper
conductive micro-tracks by multi-nozzle electrohydrodynamic inkjet printing
process, J. Mat. Process. Technol. 212: 700.
Kim D.S., Rahman K., Khan A. and Choi K.H. (2012). Direct Fabrication of Copper
Nanoparticle Patterns through Electrohydrodynamic Printing in Cone-Jet Mode,
Mater. Manuf. Process. 27: 1295.
Kim K. (2015). Silicon Technologies and Solutions for the Data-Driven World, ISSCC
2015, Session 1, Plenary 1.1.
Kiran B., Bulusu S., Zhai H.J., Yoo S., Zeng X.C. and Wang L.S. (2005). Planar-to-
tubular structural transition in boron clusters: B20 as the embryo of single-walled
boron nanotubes, Proc. Natl. Acad. Sci. U.S.A. 102: 961.
Kroto H.W., Heath J.R., O’Brien S.C., Curl R.F. and Smalley R.E. (1985). C60:
Buckminsterfullerene, Nature 318: 162.
Kuntová Z., Rossi G. and Ferrando R. (2008). Melting of core-shell Ag-Ni and Ag-Co
nanoclusters studied via molecular dynamics simulations, Phys. Rev. B 77: 205431.
Laasonen K., Wonczak S., Strey R. and Laaksonen A. (2000). Molecular dynamics
simulations of gas-liquid nucleation of Lennard-Jones fluid, J. Chem. Phys. 113:
9471.
Lai S.K., Hsu P.J., Wu K.L., Liu W.K. and Iwamatsu M. (2002). Structures of metallic
clusters: Mono- and polyvalent metals, J. Chem. Phys. 117: 10715.
Leary R.H. (2000). Global optimization of funneling landscapes, J. Global. Optim. 18:
367.
Li X. and Frisch M.J. (2006). Energy-represented DIIS within a hybrid geometry
optimization method, J. Chem. Theory and Comput. 2: 835.
Liu C-M., Xu C., Cheng Y., Chen X-R. and Cai L-C. (2013). Size-dependent melting
and coalescence of tungsten nanoclusters via molecular dynamics simulation, Phys.
Chem. Chem. Phys. 15: 14069.
Lu S., Zhang J. and Duan H. (2009). Melting behaviors of CoN (N=13, 14, 38, 55, 56)
clusters, Chem. Phys. 363: 7.
Lv M.B., Cheng Yan and Fu Min. (2013). A new global search for the ground state
structure of small cluster: application to S6, J. At. Mol. Sci. 4: 357.

122
Martin T.P., Näher U., Schaber H., and Zimmermann (1993). Evidence for a size-
dependent melting of sodium clusters, J. Chem. Phys. 100: 2322.
Menard L.D., Gao S.P., Xu H., Twesten R.D., Harper A.S., Song Y., Wang G.,
Douglas A.D., Yang J.C., Frenkel A.I., Nuzzo R.G. and Murray R.W. (2006). Sub-
nanometer Au monolayer-protected clusters exhibiting molecule-like electronic
behavior: quantitative high-angle annular dark-field scanning transmission electron
microscopy and electrochemical characterization of clusters with precise atomic
stoichiometry, J. Phys. Chem. B 110: 12874.
Mollenhauer D. and Gaston N., (2016). Phosphine passivated gold clusters: how
charge transfer affects electronic structure and stability, Phys. Chem. Chem. Phys.
2016, advance article.
Neyts E.C. and Bogaerts A. (2009). Numerical study of the size-dependent melting
mechanisms of nickel nanoclusters, J. Phys. Chem. C 113(7): 2771.
Ng W.C., Lim T.L. and Yoon T.L. (2015a). Ground-state structures of hafnium
clusters, AIP Conf. Proc. 1657: 070003.
Ng W.C., Yoon T.L. and Lim T.L. (2015b). Guided basin-hopping search of small
boron clusters with density functional theory, AIP Conf. Proc. 1657: 070004.
Oganov A.R. and Valle M. (2009). How to quantify energy landscapes of solids, J.
Chem. Phys 130: 104504.
Oprea T.I. (2002). On the information content of 2D and 3D descriptors for QSAR, J.
Braz. Chem. Soc. 13: 811.
Oxtoby D.W. (1990). New perspectives on freezing and melting, Nature 347: 725.
Park J. and Cheon J. (2001). Synthesis of “solid solution” and “core-shell” type cobalt-
platinum magnetic nanoparticles via transmetalation reactions, J. Am. Chem. Soc.
123: 5743.
Plimpton S. (1995). Fast parallel algorithms for short-range molecular dynamics, J.
Comp. Phys. 117: 1.
Rappé A.K. and Goddard W.A. III. (1991). Charge equilibration of molecular
dynamics simulations, J. Phys. Chem. 95: 3358.
Rick S.W., Stuart S.J. and Berne B.J. (1994). Dynamical fluctuating charge force fields:
Application to liquid water, J. Chem. Phys. 101: 6141.
Rogan J., Ramírez M., Varas A. and Kiwi M. (2013). How relevant is the choice of
classical potentials in finding minimal energy cluser conformations? Comp. Theor.
Chem. 1021: 155.
Rossi G., Ferrando R., Rapallo A., Fortunelli A., Curley B.C., Lloyd L.D. and Johnston
R.L. (2005). Global optimization of bimetallic cluster structures. II. Size-matched
Ag-Pd, Ag-Au, and Pd-Pt systems, J. Chem. Phys. 122: 194309.

123
Schlom D.G., Guha S. and Datta S. (2008). Gate Oxides beyond SiO2, MRS Bulletin
33: 1017.
Schmidt M., Kusche R., Issendorff B. and Haberland H. (1998). Irregular variations in
the melting point of size-selected atomic clusters, Nature 393: 238.
Schmidt M. and Haberland H. (2002). Phase transition in clusters, C. R. Physique 3:
327.
Shan T.R., Devine B.D., Kemper T.W., Sinnott S.B. and Phillpot S.R. (2010a).
Charge-optimized many-body potential for the hafnium/hafnium oxide system,
Phys. Rev. B 81: 125328.
Shan T.R., Devine B.D., Hawkins J.M., Asthagiri A. Phillpot S.R. and Sinnott S.B.
(2010b). Second-generation charge-optimized many body potential for Si/SiO2 and
amorphous silica, Phys. Rev. B 82: 235302.
Son S.U., Jang Y., Park J., Na H.B., Park H.M., Yun H.J., Lee J. and Hyeon T. (2004).
Designed synthesis of atom-economical Pd/Ni bimetallic nanoparticle-based
catalysts for sonogashira coupling reactions, J. Am. Chem. Soc. 126: 5026.
Soulé B. de B., Ford M.J. and Cortie M.B. (2004). Low energy structures of gold
nanoclusters in the size range 3-38 atoms, J. Mol. Struct. Theochem 686: 193.
Stillinger F.H. and Weber T.A. (1984). Computer simulation of local order in
condensed phases of silicon, Phys. Rev. B 31: 5262.
Stuart S.J., Tutein A.B. and Harrison J.A. (2000). A reactive potential for
hydrocarbons with intermolecular interactions, J. Chem. Phys. 112: 6472.
Subedi D.P. (2011). Contact angle measurement for the surface characterization of
solids, The Himalayan Physics Vol. II: 1.
Sun Q., Kandalam A.K., Wang Q., Jena P., Kawazoe Y. and Marquez M. (2006).
Effect of Au coating on the magnetic and structural properties of Fe nanoclusters
for use in biomedical applications: A density-functional theory study, Phys. Rev. B
73: 134409.
Sun X., Du J., Zhang P. and Jiang G. (2010). A systemic DFT study on several 5d-
electron element dimers: Hf2, Ta2, Re2, W2, and Hg2, J. Clust. Sci. 21: 619.
Taylor P.R., Bylaska E., Weare J.H. and Kawai R. (1995). C20: fullerene, bowl or ring?
New results from coupled-cluster calculations, Chem. Phys. Lett. 235: 558.
Taherkhani F. and Rezania H. (2012). Temperature and size dependency of thermal
conductivity of aluminium nanocluster, J. Nanopart. Res. 14: 1222.
Tersoff J. (1988a). New empirical approach for the structure and energy of covalent
systems, Phys. Rev. B 37: 6991.

124
Tersoff J. (1988b). Empirical interatomic potential for silicon with improved elastic
properties, Phys. Rev. B 38: 9902.
Wales D.J. and Doye J.P.K. (1997). Global optimization by basin-hopping and the
lowest energy structures of Lennard-Jones clusters containing up to 110 atoms, J.
Phys. Chem. A 101: 5111.
Walters W.P., Stahl M.T. and Murcko M.A. (1998). Virtual screening - an overview,
Drug Discov. Today 3: 160.
Whalley E. and Schneider W.G. (1955). Intermolecular potentials of Argon, Krypton
and Xenon, J. Chem. Phys. 23: 1644.
Wu X., Wu Y., Kai X., Wu G. and Chen Y. (2011). Structural optimization of Ag-Pd
clusters based on different potential parameterizations, Chem. Phys. 390: 36.
Yan X., Li J., Liu Z., Zheng M., Ge H. and Xu J. (2013). Enhancing molecular shape
comparison by weighted gaussian functions, J. Chem. Inf. Model. 53: 1967.
Yoon T.L., Lim T.L., Min T.K., Hung S.H., Jakse N. and Lai S.K. (2013). Epitaxial
growth of graphene on 6H-silicon carbide substrate by simulated annealing method,
J. Chem. Phys. 139: 204702.
Yu J.G., Sinnott S.B. and Phillpot S.R. (2007). Charge optimized many-body potential
for the Si/SiO2 system, Phys. Rev. B 75: 085311.
Yusakawa A. (1996). Using an extended Tersoff interatomic potential to analyze the
static-fatigue strength of SiO2 under atmospheric influence, JSME Int. J., Ser. A 39:
313.
Zeng X.C. and Oxtoby D.W. (1990). Gas-liquid nucleation in Lennard-Jones fluids, J.
Chem. Phys. 94: 4472.
Zhao S., Wang S. and Ye H. (2001). Size-dependent melting properties of free silver
nanoclusters, J. Phys. Soc. Jpn. 70: 2953.
Zhou X.W., Zimmerman J.A., Wong B.M. and Hoyt J.J. (2007). An embedded-atom
method interatomic potential for Pd-H alloys, J. Mater. Res. 23: 704.
Zhu L., Amsler M., Fuhrer T., Schaefer B., Faraji S., Rostami S., Ghasemi S.A.,
Sadeghi A., Grauzinyte M., Wolverton C. and Goedecker S. (2016). A fingerprint
based metric for measureing similarities in crystalline structures, J. Chem. Phys.
144: 034203.

125
APPENDIX A
FUNCTIONALITY FORM OF COMB POTENTIAL
The functional form of COMB2 potential is discussed briefly in Section 3.2.2,
given by equations (3.3) and (3.4). The details of each term will be discussed further
in this section according to Shan et al. (2010). The general form of the potential is as
follow,
𝐸𝑇 = ∑ [𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖) +
1
2∑ 𝑉𝑖𝑗(𝑟𝑖𝑗, 𝑞𝑖, 𝑞𝑗)
𝑗≠𝑖
+ 𝐸𝑖𝐵𝐵]
𝑖
(A1)
where 𝐸𝑇 is the total energy of the system, 𝐸𝑖𝑠𝑒𝑙𝑓
is the self-energy of atom 𝑖, 𝐸𝑖𝐵𝐵 is
the bond-bending energy of atom 𝑖, 𝑉𝑖𝑗 is the interaction potential between the 𝑖th and
𝑗th atoms, 𝑟𝑖𝑗 is the interatomic distance and 𝑞𝑖 and 𝑞𝑗 are the charges of the atom 𝑖
and atom 𝑗.
𝑉𝑖𝑗(𝑟𝑖𝑗, 𝑞𝑖, 𝑞𝑗) = 𝑈𝑖𝑗𝑅(𝑟𝑖𝑗) + 𝑈𝑖𝑗
𝐴(𝑟𝑖𝑗, 𝑞𝑖, 𝑞𝑗) + 𝑈𝑖𝑗𝐼 (𝑟𝑖𝑗 , 𝑞𝑖, 𝑞𝑗) + 𝑈𝑖𝑗
𝑉(𝑟𝑖𝑗) (A2)
The 𝑉𝑖𝑗 comprises of four parts: short-range repulsion 𝑈𝑖𝑗𝑅 , short-range
attraction 𝑈𝑖𝑗𝐴, long-range Coulombic 𝑈𝑖𝑗
𝐼 , and long range van der Waals force 𝑈𝑖𝑗𝑉 ,
which are defined as,
𝑈𝑖𝑗𝑅(𝑟𝑖𝑗) = 𝑓𝑠𝑖𝑗
𝐴𝑖𝑗𝑒(−𝜆𝑖𝑗𝑟𝑖𝑗) (A3)
𝑈𝑖𝑗𝐴(𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗) = −𝑓𝑠𝑖𝑗
𝑏𝑖𝑗𝐵𝑖𝑗𝑒(−𝛼𝑖𝑗𝑟𝑖𝑗) (A4)

126
𝑈𝑖𝑗𝐼 (𝑟𝑖𝑗, 𝑞𝑖 , 𝑞𝑗) = 𝐽𝑖𝑗(𝑟𝑖𝑗)𝑞𝑖𝑞𝑗 (A5)
𝑈𝑖𝑗𝑉(𝑟𝑖𝑗) =
𝑓𝐿𝑖𝑗(𝐶𝑉𝐷𝑊𝐶𝑉𝐷𝑊)1 2⁄
𝑟𝑖𝑗6
(A6)
Firstly, we will lay out the definitions of the parameters governing the short-
range attractions given in equations (A3) and (A4). The bond-order term 𝑏𝑖𝑗 is given
by the equations in the following,
𝑏𝑖𝑗 = {1 + [𝛽𝑖 ∑ 𝜉𝑖𝑗𝑘𝑔(𝜃𝑗𝑖𝑘)
𝑘≠𝑖,𝑗
]
𝑛𝑖
}
−1 (2𝑛𝑖)⁄
(A7)
𝜉𝑖𝑗𝑘 = 𝑓𝑠𝑖𝑘𝑒
[𝛼𝑖𝑗
𝑚𝑖(𝑟𝑖𝑗−𝑟𝑖𝑘)𝑚𝑖]
(A8)
𝑔(𝜃𝑗𝑖𝑘) = 1 +𝑐𝑖
2
𝑑𝑖2 −
𝑐𝑖2
[𝑑𝑖2 + (ℎ𝑖 − cos 𝜃𝑗𝑖𝑘)
2]
(A9)
where 𝜉𝑖𝑗𝑘 is the symmetry function and 𝑔(𝜃𝑗𝑖𝑘) is the angular function. The 𝜃𝑗𝑖𝑘 is
the angle between bonds 𝑖𝑗 and 𝑖𝑘. Next, the inverse decay lengths 𝜆𝑖𝑗 and 𝛼𝑖𝑗 as well
as the leading coefficients 𝐴𝑖𝑗 and 𝐵𝑖𝑗 are based on the Lorentz-Berthelot mixing rules
(Allen and Tildesley 1989). They are given by the following set of equations,
𝜆𝑖𝑗 =1
2(𝜆𝑖 + 𝜆𝑗) (A10)
𝛼𝑖𝑗 =1
2(𝛼𝑖 + 𝛼𝑗) (A11)
𝐴𝑖𝑗 = √𝐴𝑆𝑖𝐴𝑆𝑗
(A12)

127
𝐵𝑖𝑗 = √𝐵𝑆𝑖𝐵𝑆𝑗
(A13)
The terms 𝐴𝑆𝑖 and 𝐵𝑆𝑖
are charge dependence,
𝐴𝑆𝑖= 𝐴𝑖𝑒(𝜆𝑖𝐷𝑖) (A14)
𝐵𝑆𝑖= 𝐵𝑖𝑒
(𝛼𝑖𝐷𝑖)[𝛼𝐵𝑖− |𝑏𝐵𝑖
(𝑞𝑖 − 𝑄𝑂𝑖)|
𝑛𝐵𝑖] (A15)
𝐷𝑖 = 𝐷𝑈𝑖+ |𝑏𝐷𝑖
(𝑄𝑈𝑖− 𝑞𝑖)|
𝑛𝐷𝑖 (A16)
𝑏𝐷𝑖=
(𝐷𝐿𝑖− 𝐷𝑈𝑖
)1 𝑛𝐷𝑖
⁄
(𝑄𝑈𝑖− 𝑄𝐿𝑖
)
(A17)
𝑛𝐷𝑖=
ln⌊𝐷𝑈𝑖𝐷𝑈𝑖
− 𝐷𝐿𝑖⁄ ⌋
ln⌊𝑄𝑈𝑖𝑄𝑈𝑖
− 𝑄𝐿𝑖⁄ ⌋
(A18)
𝑏𝐵𝑖=
|𝑎𝐵𝑖|
1 𝑛𝐵𝑖⁄
Δ𝑄𝑖
(A19)
𝑎𝐵𝑖= (1 − |
𝑄𝑂𝑖
Δ𝑄𝑖|
𝑛𝐵𝑖
)
−1
(A20)
Δ𝑄𝑖 =𝑄𝑈𝑖
− 𝑄𝐿𝑖
2 (A21)
𝑄𝑂𝑖=
𝑄𝑈𝑖+ 𝑄𝐿𝑖
2 (A22)
The cutoff function 𝑓𝑠𝑖𝑗 exists to truncate the potential in term of radii 𝑟 = 𝑅 and 𝑟 =
𝑆, according to the piecewise function,

128
𝑓𝑠𝑖𝑗= 𝑓𝑐 ⌊𝑟𝑖𝑗, (𝑅𝑆𝑖
𝑅𝑆𝑗)
1 2⁄
, (𝑆𝑆𝑖𝑆𝑆𝑗
)1 2⁄
⌋ (A23)
𝑓𝑐(𝑟, 𝑅, 𝑆) = {1
{1 2⁄ + 1 2⁄ cos[𝜋(𝑟 − 𝑅) (𝑆 − 𝑅)⁄ ]}0
} ,𝑟 ≤ 𝑅
𝑅 < 𝑟 < 𝑆𝑟 ≥ 𝑆
(A24)
The long-range Coulombic interaction is described by the charge coupling
factor 𝐽𝑖𝑗(𝑟𝑖𝑗) with the following Coulomb integral,
𝐽𝑖𝑗(𝑟𝑖𝑗) = ∫ 𝑑3𝑟𝑖 ∫ 𝑑3𝑟𝑗
𝜌𝑖(𝑟𝑖, 𝑞𝑖)𝜌𝑗(𝑟𝑗 , 𝑞𝑗)
𝑟𝑖𝑗
(A25)
𝜌𝑖(𝑟𝑖, 𝑞𝑖) = 𝑞𝑖
𝜉𝑖3
𝜋𝑒(−2𝜉𝑖|𝑟𝑖𝑗−𝑟𝑖|)
(A26)
where 𝜉𝑖 is an orbital exponent that controls the radial decay of the density.
The energy of charge formation is described by the self-energy term 𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖)
in the following form,
𝐸𝑖𝑠𝑒𝑙𝑓(𝑞𝑖) = 𝜒𝑖𝑞𝑖 + 𝐽𝑖𝑞𝑖
2 + 𝐾𝑖𝑞𝑖3 + 𝐿𝑖𝑞𝑖
4 (A27)
where the coefficients 𝜒𝑖 , 𝐽𝑖 , 𝐾𝑖 and 𝐿𝑖 are fitted to ionization energies and electron
affinities of elements hafnium and oxygen.
Lastly, the bond-bending term 𝐸𝑖𝐵𝐵 of Hf-Hf-Hf bonds is defined as,
𝐸𝐻𝑓−𝐻𝑓−𝐻𝑓 = ∑ ∑ ∑ 𝑓𝑠𝑖𝑗𝑓𝑠𝑖𝑘
[𝐾𝐿𝑃𝑃6(cos 𝜃𝐻𝑓−𝐻𝑓−𝐻𝑓)]
𝑘≠𝑖,𝑗𝑗≠𝑖𝑖
(A28)

129
where 𝑃6 is the third order Legendre polynomial function of the Hf-Hf-Hf bond angle
and 𝐾𝐿𝑃 is the coefficient fitted to the difference in cohesive energies of Hf in different
phases.





























