Embed Size (px)
Citation preview

14
36
Research ArticleReceived: 7 April 2010 Revised: 8 May 2010 Accepted: 10 May 2010 Published online in Wiley Online Library: 15 June 2010
(wileyonlinelibrary.com) DOI 10.1002/jctb.2447
Electrochemical oxidation of Crystal Violetin the presence of hydrogen peroxideHui Zhang,∗ Jie Wu, Zhongqiong Wang and Daobin Zhang
Abstract
BACKGROUND: The combination of electrochemical oxidation using a Ti/RuO2 –IrO2 anode with hydrogen peroxide has beenused for the degradation of Crystal Violet. The effect of major parameters such as initial pH, hydrogen peroxide concentration,current density, electrolyte concentration and hydroxyl radical scavenger on the decolorisation was investigated.
RESULTS: The decolorisation rate increased with initial pH and hydrogen peroxide concentration, but decreased withelectrolyte and radical scavenger concentration. The decolorisation rate increased with current density, but the increasebecame insignificant after current density exceeded 47.6 mA cm−2. On the other hand, hydrogen peroxide decompositionrate increased with initial pH and current density, but decreased with electrolyte and radical scavenger concentration. Theamount of hydrogen peroxide decomposed during 30 min reaction increased linearly with hydrogen peroxide dosage. The mainintermediates were separated and identified by gas chromatography–mass spectrometry (GC–MS) technique and a plausibledegradation pathway of Crystal Violet was proposed. At neutral pH, the electrochemical process in the presence of hydrogenperoxide was more efficient than that in the presence of Fenton’s reagent (electro-Fenton process).
CONCLUSION: The anodic oxidation process could decolorise Crystal Violet effectively when hydrogen peroxide was present.Almost complete decolorisation was achieved after 30 min reaction under the conditions 2.43 mmol L−1 hydrogen peroxide,47.6 mA cm−2 current density and pH0 7, while 62% COD removal efficiency was obtained when the reaction time was prolongedto 90 min.c© 2010 Society of Chemical Industry
Keywords: Crystal Violet; electrochemical degradation; advanced oxidation process; hydrogen peroxide; decolorisation
INTRODUCTIONCrystal Violet (C.I. Basic Violet 3) is a well-known cationic triph-enylmethane dye, which has been extensively used for textiledying and printing of paper; it also has a number of otheruses such as a biological stain, dermatological agent, veteri-nary medicine, additive to poultry feed to inhibit propagationof mold, intestinal parasites, fungus, etc.1 – 7 The disposal ofwastewater containing Crystal Violet is an environmental concernsince it is a mutagen and mitotic poison.3,4,7 Therefore, variousmethods have been employed to remove it from wastewater,including adsorption,3,4,6 biological processes,8 – 11 TiO2 or ZnO-mediated photocatalytic degradation,1,2,12 – 15 copper (II) aminecomplexes/hydrogen peroxide system,16 UV/hydrogen peroxidetreatment,17 microwave induced catalytic degradation,7 Fentonor Fenton-like processes,17,18 and electrochemical processes.19,20
Among these technologies, electrochemical methods are consid-ered an environment-friendly and effective technology for thetreatment of waste-water containing synthetic organic dyes.21,22
Electrochemical oxidation of dye waste-water can be achievedvia direct oxidation on the anode or indirect oxidation usingelectro-generated strong oxidants such as hydroxyl radicals.23,24
Electrons are needed in both approaches and consequently thedecolorisation of synthetic organic dyes depends on the quantityof charge (charge loading). However, the production of hydroxylradicals electrochemically generated with water is small duringindirect oxidation.25 Therefore, a long reaction time or a high
current density is usually required to achieve satisfactory colourremoval. This would result in high energy consumption when anelectrochemical process is employed alone.26 The introductionof oxidants into the electrolytic system may solve this problem.Recently, the combination of Fenton’s reagent with electrochem-istry, i.e. the electro-Fenton process, has been used to eliminateCrystal Violet.5,27,28 Although the electro-Fenton process is moreefficient than the Fenton and electrochemical processes sepa-rately, the reaction could proceed optimally in the narrow pHrange 3–4.29 – 33 Moreover, the cationic structure of Crystal Violetfavours decolorisation in a basic medium.15 Alternatively, whenhydrogen peroxide instead of Fenton’s reagent is applied to theelectrochemical system, the radicals would be electro-generatedwith hydrogen peroxide,34,35 which would attack the pollutantsin a subsequent step. Consequently, the combination of electro-chemical oxidation with hydrogen peroxide has been employedfor the degradation of organic contaminants such as phenol36 and2,4-dichlorophenoxyacetic acid (2,4-D).37,38 Brillas et al. observedthat in comparison with simple anodic oxidation of 2,4-D, slightlyhigher mineralisation was achieved for anodic oxidation in the
∗ Correspondence to: Hui Zhang, Department of Environmental Engineering,Wuhan University, PO Box C319 Luoyu Road 129# , Wuhan 430079, China.E-mail: [email protected]
Department of Environmental Engineering, Wuhan University, PO Box C319Luoyu Road 129# , Wuhan 430079, China
J Chem Technol Biotechnol 2010; 85: 1436–1444 www.soci.org c© 2010 Society of Chemical Industry

14
37
Electrochemical oxidation of Crystal Violet in the presence of H2O2 www.soci.org
Table 1. The characteristics of Crystal Violet
Name Crystal Violet (Basic Violet 3)
Colour Index (C.I.) 42555
Formula C25H30N3Cl
Structure N+(CH3)2
N(CH3)2
Cl−
(H3C)2N
Formula weight (g mol−1) 408.0
Absorbance (λmax, nm) 584
0 5 10 15 20 25 30
0
20
40
60
80
100
Dec
olor
izat
ion
effic
ienc
y (%
)
Time (min)
Figure 1. Decolorisation of Crystal Violet solution with 0.1 mol L−1 Na2SO4and pH0 7 in different systems: (�) hydrogen peroxide alone ([H2O2]= 2.43 mmol L−1); (◦) anodic oxidation alone (i = 47.6 mA cm−2) (�)Fenton process ([H2O2] = 2.43 mmol L−1, [H2O2]/[Fe2+] = 20) (♦) electro-Fenton (i = 47.6 mA cm−2, [H2O2] = 2.43 mmol L−1, [H2O2]/[Fe2+] =20) (�) anodic oxidation process in presence of hydrogen peroxide(i = 47.6 mA cm−2, [H2O2] = 2.43 mmol L−1).
presence of hydrogen peroxide generated from the two-electronreduction of sparged oxygen on a graphite bar cathode.37 To ourknowledge, there is no report on the electrochemical oxidationof dyes when hydrogen peroxide instead of Fenton’s reagent ispresent. Therefore, in this study, Crystal Violet was degraded inan electrochemical reactor in the presence of hydrogen peroxide.The effect of operating conditions such as initial pH, hydrogenperoxide concentration, current density, electrolyte concentrationand hydroxyl radical scavenger on the decolorisation was inves-tigated. The main intermediate products were identified and thedegradation of Crystal Violet in terms of COD removal was alsoexplored.
EXPERIMENTALCrystal Violet (AR grade, 98%) was obtained from Shenyang No.3 Chemicals Reagent Factory (China) and used without furtherpurification. The structure and relevant data of Crystal Violet areshown in Table 1. All other reagents were of analytical grade.Before each run, a fresh stock solution of Crystal Violet wasprepared in deionised water and the initial concentration (C0) waskept at 200 mg L−1. Sodium sulfate was added as a supporting
0 5 10 15 20 25 30
0
20
40
60
80
100
Dec
olor
izat
ion
effic
ienc
y (%
)
Time (min)
(a)
0 5 10 15 20 25 300.0
0.4
0.8
1.2
1.6
2.0
2.4
H2O
2 co
ncen
trat
ion
(mm
ol L
-1)
Time (min)
(b)
Figure 2. Evolution of decolorisation efficiency (a) and hydrogen peroxideconcentration (b) with reaction time at different pH0 values: (�) pH0 3; (�)pH0 5; (◦) pH0 7; (�) pH0 9. (i = 47.6 mA cm−2, [H2O2] = 2.43 mmol L−1,[Na2SO4] = 0.1 mol L−1).
electrolyte, and sulfuric acid or sodium hydroxide used to adjustthe initial pH (pH0) of the dye solution. The solution pH wasmeasured with an Orion 420Aplus pH-meter (Thermo Electron Co.,US).
Batch experiments were performed in a rectangular electrolyticreactor (plexy glass) containing 200 mL of solution. The reactor wasimmersed in a water bath to keep the temperature constant aroundroom temperature. Electrolysis was conducted under constantcurrent conditions using a direct current (DC) power supply(Model WYK-305) from Yangzhou Jintong Source Co. Ltd. (China).A 5 × 11.9 cm plate anode (Ti/RuO2 –IrO2) and a plate cathode(stainless steel) of the same dimensions were arranged parallel toeach other at a distance of 3.8 cm. The working surface area of theelectrode was 31.5 cm2. A magnetic stirrer (Model 78-1, HangzhouInstrument Motors Factory, China) provided mixing of the solutionin the reactor. When the DC power supply was initiated, ahydrogen peroxide solution was applied to the electrolytic cell.At pre-selected time intervals, samples were withdrawn from theelectrolytic cell. The dye concentration was measured at λmax =584 nm using a Shimadzu UV-1700 spectrophotometer (ShimadzaCo., Japan). The concentration of hydrogen peroxide was analyzedusing a titanium sulfate spectrophotometric method.39 Chemicaloxygen demand (COD) was determined using a closed refluxtitrimetric method based on Standard Methods40 and theinterference of residual hydrogen peroxide in the COD analysis waseliminated. The intermediate products during the reaction weredetected by gas chromatography/mass spectrometry (GC/MS)(Shimadzu GCMS-QP2010 Plus). Samples for GC/MS analysis were
J Chem Technol Biotechnol 2010; 85: 1436–1444 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/jctb

14
38
www.soci.org H. Zhang et al.
0 5 10 15 20 25 30
0
20
40
60
80
100D
ecol
oriz
atio
n ef
ficie
ncy
(%)
Time (min)
0 5 10 15 20 25 300
1
2
3
4
5
H2O
2 C
once
ntra
tion
(mm
ol L
-1)
Time (min)
0 2 50
1
2
3
4
H2O2concentration (mmol L-1)
Dec
ompo
sed
H2O
2 (m
mol
L-1
)
0
20
40
60
80
100
Dec
olor
izat
ion
effic
ienc
y (%
)
1 3 4
(a) (b)
(c)
Figure 3. Evolution of decolorisation efficiency (a) and hydrogen peroxide concentration (b) with reaction time, and hydrogen peroxide decompositionat different hydrogen peroxide concentrations (c): (�) 0.49 mmol L−1; (◦) 0.97 mmol L−1; (�) 2.43 mmol L−1; (♦) 4.85 mmol L−1. (i = 47.6 mA cm−2,[Na2SO4] = 0.1 mol L−1, pH0 7).
prepared as follows: dye solution (200 mL) was extracted withdichloromethane (total volume 200 mL) ten times; the extractswere then concentrated by rotary evaporator at 40 ◦C to about1 mL before being analysed by GC/MS. A HP-5 MS capillarycolumn (30 m length × 0.25 mm ID × 0.25 µm film thickness)was employed for GC separation. The GC equipment was operatedin a temperature programmed mode with an initial temperature of40 ◦C held for 4 min, then ramped first to 80 ◦C with a 4 ◦C min−1
rate and held for 2 min; then ramped to 280 ◦C at 8 ◦C min−1
and held at that temperature for 9 min.18 Helium was used as acarrier gas at a flow-rate of 5.79 mL min−1. Electron impact (EI)mass spectra were scanned from 10–300 m/z. The injector, ionsource and interface temperatures were set at 280, 220 and 280 ◦C,respectively.
RESULTS AND DISCUSSIONDecolorisation of Crystal Violet in different systemsTo investigate the decolorisation using different systems, CrystalViolet was treated for 30 min by hydrogen peroxide alone; anodicoxidation alone; Fenton’s reagent alone; anodic oxidation in thepresence of hydrogen peroxide; and indirect electrochemicaloxidation using Fenton’s reagent (electro-Fenton process). Asshown in Fig. 1, negligible colour removal was observed whenCrystal Violet was treated by hydrogen peroxide alone due to itslimited oxidising power (E0 = 1.763 V), in agreement with the
results of Alshamsi et al.17 The Fenton process terminates at pH0
7 and so Crystal Violet was hardly oxidised by Fenton’s reagent.Anodic oxidation removed 26.1% of the initial Crystal Violet, whichwas accounted for by the generation of hydroxyl radicals at theanode from oxidation of water:
H2O −−−→ •OH + H+ + e− (1)
However, the electro-generated hydroxyl radicals were insufficientto oxidise Crystal Violet completely within the 30 min reaction time.When hydrogen peroxide was applied to the electrochemicalsystem, both hydroxyl radicals and hydroperoxyl radicals wereproduced with hydrogen peroxide at the cathode and anode,respectively:34,35
H2O2 + e −−−→ •OH + OH− (2)
H2O2 −−−→ HO2• + H+ + e− (3)
It is possible that the hydroperoxyl radicals produced couldreact with the dye as follows:17
Dye + HO2• −−−→ Dye• + H2O2 (4)
However the oxidation of Crystal Violet by hydroperoxyl radicalscan be neglected as the hydroperoxyl radical (E0 = 1.65 V) is less
wileyonlinelibrary.com/jctb c© 2010 Society of Chemical Industry J Chem Technol Biotechnol 2010; 85: 1436–1444
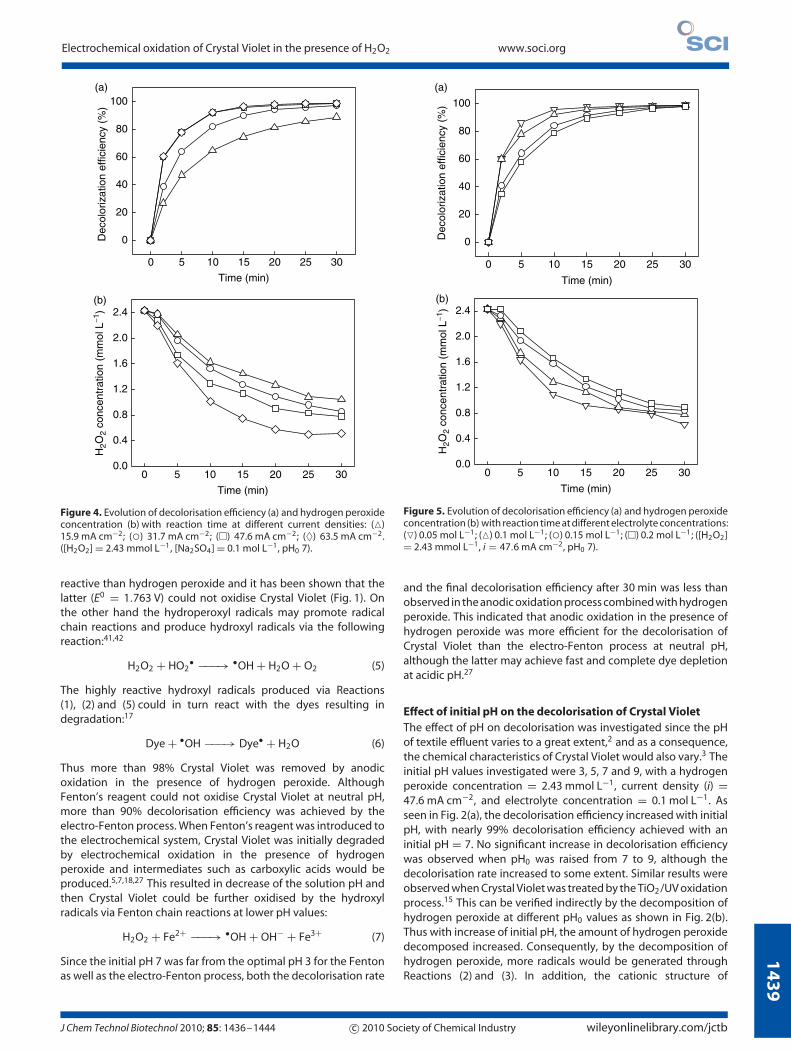
14
39
Electrochemical oxidation of Crystal Violet in the presence of H2O2 www.soci.org
0 5 10 15 20 25 30
0
20
40
60
80
100D
ecol
oriz
atio
n ef
ficie
ncy
(%)
Time (min)
0 5 10 15 20 25 300.0
0.4
0.8
1.2
1.6
2.0
2.4
H2O
2 co
ncen
trat
ion
(mm
ol L
−1)
Time (min)
(a)
(b)
Figure 4. Evolution of decolorisation efficiency (a) and hydrogen peroxideconcentration (b) with reaction time at different current densities: (�)15.9 mA cm−2; (◦) 31.7 mA cm−2; (�) 47.6 mA cm−2; (♦) 63.5 mA cm−2.([H2O2] = 2.43 mmol L−1, [Na2SO4] = 0.1 mol L−1, pH0 7).
reactive than hydrogen peroxide and it has been shown that thelatter (E0 = 1.763 V) could not oxidise Crystal Violet (Fig. 1). Onthe other hand the hydroperoxyl radicals may promote radicalchain reactions and produce hydroxyl radicals via the followingreaction:41,42
H2O2 + HO2• −−−→ •OH + H2O + O2 (5)
The highly reactive hydroxyl radicals produced via Reactions(1), (2) and (5) could in turn react with the dyes resulting indegradation:17
Dye + •OH −−−→ Dye• + H2O (6)
Thus more than 98% Crystal Violet was removed by anodicoxidation in the presence of hydrogen peroxide. AlthoughFenton’s reagent could not oxidise Crystal Violet at neutral pH,more than 90% decolorisation efficiency was achieved by theelectro-Fenton process. When Fenton’s reagent was introduced tothe electrochemical system, Crystal Violet was initially degradedby electrochemical oxidation in the presence of hydrogenperoxide and intermediates such as carboxylic acids would beproduced.5,7,18,27 This resulted in decrease of the solution pH andthen Crystal Violet could be further oxidised by the hydroxylradicals via Fenton chain reactions at lower pH values:
H2O2 + Fe2+ −−−→ •OH + OH− + Fe3+ (7)
Since the initial pH 7 was far from the optimal pH 3 for the Fentonas well as the electro-Fenton process, both the decolorisation rate
0 5 10 15 20 25 30
0
20
40
60
80
100
Dec
olor
izat
ion
effic
ienc
y (%
)
Time (min)
0 5 10 15 20 25 300.0
0.4
0.8
1.2
1.6
2.0
2.4
H2O
2 co
ncen
trat
ion
(mm
ol L
−1)
Time (min)
(a)
(b)
Figure 5. Evolution of decolorisation efficiency (a) and hydrogen peroxideconcentration (b) with reaction time at different electrolyte concentrations:(�) 0.05 mol L−1; (�) 0.1 mol L−1; (◦) 0.15 mol L−1; (�) 0.2 mol L−1; ([H2O2]= 2.43 mmol L−1, i = 47.6 mA cm−2, pH0 7).
and the final decolorisation efficiency after 30 min was less thanobserved in the anodic oxidation process combined with hydrogenperoxide. This indicated that anodic oxidation in the presence ofhydrogen peroxide was more efficient for the decolorisation ofCrystal Violet than the electro-Fenton process at neutral pH,although the latter may achieve fast and complete dye depletionat acidic pH.27
Effect of initial pH on the decolorisation of Crystal VioletThe effect of pH on decolorisation was investigated since the pHof textile effluent varies to a great extent,2 and as a consequence,the chemical characteristics of Crystal Violet would also vary.3 Theinitial pH values investigated were 3, 5, 7 and 9, with a hydrogenperoxide concentration = 2.43 mmol L−1, current density (i) =47.6 mA cm−2, and electrolyte concentration = 0.1 mol L−1. Asseen in Fig. 2(a), the decolorisation efficiency increased with initialpH, with nearly 99% decolorisation efficiency achieved with aninitial pH = 7. No significant increase in decolorisation efficiencywas observed when pH0 was raised from 7 to 9, although thedecolorisation rate increased to some extent. Similar results wereobserved when Crystal Violet was treated by the TiO2 /UV oxidationprocess.15 This can be verified indirectly by the decomposition ofhydrogen peroxide at different pH0 values as shown in Fig. 2(b).Thus with increase of initial pH, the amount of hydrogen peroxidedecomposed increased. Consequently, by the decomposition ofhydrogen peroxide, more radicals would be generated throughReactions (2) and (3). In addition, the cationic structure of
J Chem Technol Biotechnol 2010; 85: 1436–1444 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/jctb

14
40
www.soci.org H. Zhang et al.
0 5 10 15 20 25 30
0
20
40
60
80
100D
ecol
oriz
atio
n ef
ficie
ncy
(%)
Time (min)
0 5 10 15 20 25 300.0
0.4
0.8
1.2
1.6
2.0
2.4
H2O
2 C
once
ntra
tion
(mm
ol L
−1)
Time (min)
(a)
(b)
Figure 6. Evolution of decolorisation efficiency (a) and hydrogen peroxideconcentration (b) with reaction time at different bicarbonate concentra-tions: (�) 0 mol L−1; (�) 0.05 mol L−1; (◦) 0.1 mol L−1; (�) 0.15 mol L−1;(♦) 0.2 mol L−1. ([H2O2] = 2.43 mmol L−1, i = 47.6 mA cm−2, [Na2SO4] =0.1 mol L−1, pH0 9).
Crystal Violet favours decolorisation in basic medium.15 Therefore,increase of initial pH would result in increased colour removal.
Effect of hydrogen peroxide concentration on the decolorisa-tion of Crystal VioletFigure 3 illustrates the decolorisation of Crystal Violet at differenthydrogen peroxide concentrations when the current density= 47.6 mA cm−2, electrolyte concentration = 0.1 mol L−1, andinitial pH value = 7. It can be seen that decolorisation rateincreased with hydrogen peroxide concentration although thedecolorisation efficiency reached a plateau when hydrogenperoxide concentration was increased to 2.43 mmol L−1. Whenhydrogen peroxide concentrations were 2.43 and 4.85 mmol L−1
decolorisation efficiencies were 98.6% and 99.5%, respectively.Hydrogen peroxide is a source of free radicals (Reactions(2) and (3)) and more free radicals would be generated athigher hydrogen peroxide concentrations.17,43,44 However, furtherincrease in hydrogen peroxide concentration would also leadto the quenching reaction of hydroxyl radicals with hydrogenperoxide:17
H2O2 + •OH −−−→ HO2• + H2O (8)
The decomposition of hydrogen peroxide at different hydrogenperoxide concentrations is illustrated in Fig. 3(c). The concentra-tions of decomposed hydrogen peroxide were 0.35, 0.54, 1.65 and3.49 mmol L−1, respectively when hydrogen peroxide dosages
200 300 400 500 600 700
0.0
0.5
1.0
1.5
Abs
orba
nce
Wavelength (nm)
0 min3 min5 min10 min15 min20 min25 min30 min
Figure 7. UV–Vis spectral changes with reaction time ([H2O2] =2.43 mmol L−1, i = 47.6 mA cm−2, [Na2SO4] = 0.1 mol L−1, pH0 7).
were 0.49, 0.97, 2.43 and 4.85 mmol L−1. However, the corre-sponding decolorisation efficiencies were 50.4%, 81.6%, 98.6%and 99.5%, respectively. This indicated that the amount of decom-posed hydrogen peroxide increased almost linearly with hydrogenperoxide dosage (R2 = 0.9977), whereas decolorisation efficiencydid not increase as much as the applied hydrogen peroxide.
Effect of current density on the decolorisationof Crystal VioletThe current densities investigated in this study were 15.9, 31.7,47.6, and 63.5 mA cm−2, with hydrogen peroxide concentration2.43 mmol L−1, electrolyte concentration 0.1 mol L−1, and initialpH value 7. As shown in Fig. 4(a), the decolorisation efficiencyincreased from 88.8 to 97.1% when the current density wasincreased from 15.9 to 31.7 mA cm−2. This indicated that a currentdensity of 31.7 mA cm−2 was sufficient to decolorise Crystal Violetalmost completely with 2.43 mmol L−1 hydrogen peroxide. Furtherincrease in current density resulted in little increase in efficiency.Although the decolorisation rate continued to increase when thecurrent density was increased from 31.7 to 47.6 mA cm−2, it thenlevelled off. Similar results were observed when Crystal Violet wasoxidised by the electro-Fenton process.28 Increase of currentdensity would initially enhance the free radicals generation,but further increase would produce the competitive electrodereactions, such as the discharge of oxygen at the anode and theevolution of hydrogen at the cathode:2,29,31
2H2O −−−→ 4H+ + O2 + 4e (9)
2H+ + 2e −−−→ H2 (10)
These would inhibit the main reactions generating hydroxylradicals to some extent. In addition, the amount of decomposedhydrogen peroxide increased with current density, with the lowestresidual hydrogen peroxide at a current density of 63.5 mA cm−2
after 30 min reaction (Fig. 4(b)). Therefore, the excess hydroxylradicals would react with hydrogen peroxide and so both thedecolorisation efficiency and decolorisation rate were nearly thesame when current densities were 47.6 and 63.5 mA cm−2.
wileyonlinelibrary.com/jctb c© 2010 Society of Chemical Industry J Chem Technol Biotechnol 2010; 85: 1436–1444

14
41
Electrochemical oxidation of Crystal Violet in the presence of H2O2 www.soci.org
5 15 25 35 455x104
1x106
2x106
3x106
4x106
Inte
nsity
(co
unts
)
Retention time (min)
39.63
20.437.55 26.22
24.05
NH3C H
OH
N NH3C
CH3
CH3
CH3
O
OH
NH3C CH3
O
N
OHO
H3CCH3
5 15 25 35 455x104
1x106
2x106
3x106
4x106
Inte
nsity
(co
unts
)
Retention time (min)
24.05
39.58
7.55
26.5925.94
20.98
N NH3C
CH3
CH3
CH3
O
OH
OH
O
NH3C
CH3
O
NH
OHO
H3C
N
OHO
H3CCH3
(a)
(b)
Figure 8. GC–MS chromatograph of reaction products after 5 min (a) and 30 min (b) electrolysis.
Effect of electrolyte concentration on the decolorisationof Crystal VioletFigure 5(a) compares the decolorisation of Crystal Violet underdifferent electrolyte (sodium sulfate) concentrations with hy-drogen peroxide concentration 2.43 mmol L−1, current density47.6 mA cm−2, and initial pH value 7. It can be seen that the de-colorisation rate slows down when the electrolyte concentrationis increased, although the decolorisation efficiency was nearly thesame over the range of electrolyte concentrations investigated.Sihem et al.15 also reported that increase of sulfate concentra-tion diminished the decolorisation rate when Crystal Violet waseliminated by the TiO2/UV oxidation process.
With increase in sulfate concentration, the amount of decom-posed hydrogen peroxide decreased to some extent (Fig. 5(b)). Inaddition, the generated hydroxyl radicals would be quenched bysulfate ion:45,46
•OH + SO42− −−−→ OH− + SO4
•− (11)
Therefore, the rate of decolorisation would be retarded accord-ingly.
Effect of hydroxyl radical scavenger on the decolorisation ofCrystal VioletTo further investigate the effect of a hydroxyl radical scavengeron the decolorisation of Crystal Violet, the experiments werecarried out in the presence of hydroxyl radical scavenger, sodiumbicarbonate. Sodium bicarbonate concentrations investigatedwere 0.05, 0.10, 0.15 and 0.20 mol L−1, with hydrogen peroxide
concentration 2.43 mmol L−1, current density 47.6 mA cm−2,electrolyte concentration 0.1 mol L−1, and initial pH value 9. Asseen in Fig. 6(a), both the decolorisation rate and decolorisationefficiency decreased with bicarbonate concentration, comparedwith retardation of the rate of decolorisation only in the presenceof sulfate. This is because bicarbonate is a stronger hydroxyl radicalscavenger than sulphate:45,47
•OH + HCO3− −−−→ •CO3
− + H2O k = 1.5 × 107 M−1 s−1
(12)The presence of bicarbonate also hindered the decompositionof hydrogen peroxide (Fig. 6(b)). In the absence of bicarbonateafter 30 min reaction 84.4% hydrogen peroxide was decomposed,however, only 45.3% hydrogen peroxide was decomposed whenbicarbonate concentration rose to 0.2 mol L−1. As a consequence,in the absence of bicarbonate the decolorisation efficiency was99.2%, and this was hindered by the presence of bicarbonate,decreasing to 54.5% with bicarbonate concentration 0.2 mol L−1.This indicated that the main reactive species in the anodicoxidation process in the presence of hydrogen peroxide werehydroxyl radicals.
The degradation mechanism of Crystal VioletTo clarify the change in molecular and structural characteristicsof Crystal Violet as a result of the anodic oxidation process in thepresence of hydrogen peroxide, representative UV–visible (vis)spectra changes in the dye solution as a function of reaction timeare shown in Fig. 7. As can be observed from these spectra, be-fore oxidation, the absorption spectrum of Crystal Violet in water
J Chem Technol Biotechnol 2010; 85: 1436–1444 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/jctb

14
42
www.soci.org H. Zhang et al.
N NH3C
CH3
CH3
CH3
NH3C CH3
NH3C CH3
OH
NH3C CH3
OH
NH3C CH3
+ +
N NH3C
CH3
CH3
CH3
O
(A)
N
OHO
H3C CH3
(B)
NH3C CH3
O
(C)
NH3C H
OH
(D)
NH
OHO
H3C
(E)
OH
O
(F)
OH
(G)
Route 1 Route 2
Figure 9. Proposed degradation mechanism of Crystal Violet under anodic oxidation process in the presence of hydrogen peroxide.
exhibits three bands with the most intense located at 584 nm.Following oxidation almost total suppression of the absorbancesignals in the ultraviolet and visible region was observed. Usu-ally, the primary attack on a dye molecule by hydroxyl radicalsbegins with the chromophore with the formation of a colour-less intermediate.5 It was thought that during TiO2 mediatedphotocatalytic degradation of Crystal Violet there were two com-petitive primary processes, i.e. N-demethylation and destructionof the conjugated structure.12 The N-demethylation would resultin hypsochromic shifts.7,12 Chen and co-workers reported the oc-currence of N-demethylation during the degradation of CrystalViolet by TiO2-assisted photodegradation14,15 and in Fenton andFenton-like systems.18 A similar phenomenon was also observedin the microwave induced catalytic degradation process.7 On theother hand, cleavage of the conjugated chromophore structurewould lead to the decay of absorbance. As illustrated in Fig. 7,the hypsochromic shift was insignificant compared with the swiftdisappearance of all three bands.
GC/MS was employed to further identify the intermediate prod-ucts formed during electrolysis, The main intermediate productsafter 5 min and 30 min reaction were detected and are illustratedin Fig. 8(a) and (b), respectively. Based on the results and previousstudies,10,18,24 a plausible degradation pathway is proposed (Fig. 9)with the primary attack of hydroxyl radicals leading to cleavageof the chromophore structure and the generation of 4-(N,N-
dimethylamino)-4′-(N′ ,N′-dimethylamino)benzophenone (A)and4-(N,N-dimethylamino)phenol. N-demethylation of 4-(N,N-di-methylamino)phenol results in the production of 4-(N-methyl-amino)phenol (D), which is further degraded into phe-nol (G). The cleavage of 4-(N,N-dimethylamino)-4′-(N′,N′-dimethylamino)benzophenone (A) involves two different path-ways (Routes 1 and 2). 4-(N,N-dimethylamino)-4′-(N′ ,N′-dime-thylamino)benzophenone (A) on reduction splits into not only 4-dimethylaminobenzoic acid (B) and N,N-dimethylaminobenzene(Route 1), but also N,N-dimethylaminobenzaldehyde (C) and4-(N,N-dimethylamino)phenol (Route 2). 4-(N,N-dimethylamino)phenol, 4-dimethylaminobenzoic acid (B) and N,N-dimethyl-aminobenzaldehyde (C) are subsequently N-demethylated to 4-(N-methylamino)phenol (D), 4-methylaminobenzoic acid (E) and4-hydroxybenzaldehyde (F), respectively. Then, gradual cleavageof the aromatic intermediates would result in the formation ofcarboxylic acids prior to conversion into carbon dioxide,5,27 notshown in Fig. 9.
The change of COD with reaction timeIt is known that complete decolorisation does not mean thatthe dye is completely oxidised,43,44,49 so the degradation ofCrystal Violet in terms of COD removal was investigated. Asseen in Fig. 10, only 46.4% COD was removed after 30 minreaction compared with 98.6% decolorisation efficiency with
wileyonlinelibrary.com/jctb c© 2010 Society of Chemical Industry J Chem Technol Biotechnol 2010; 85: 1436–1444

14
43
Electrochemical oxidation of Crystal Violet in the presence of H2O2 www.soci.org
0 20 40 60 80 100 1200
10
20
30
40
50
60C
OD
rem
oval
effi
cien
cy (
%)
Time (min)
Figure 10. Variation of COD removal efficiency with reaction time ([H2O2]= 2.43 mmol L−1, i = 47.6 mA cm−2, [Na2SO4] = 0.1 mol L−1, pH0 7).
hydrogen peroxide concentration 2.43 mmol L−1, current density47.6 mA cm−2, electrolyte concentration 0.1 mol L−1, and initialpH value 7. After extending the reaction time to 90 min, 62% CODremoval was achieved but further prolongation of reaction timehad little effect on COD removal. The small change in COD removalefficiency during the last stage of the reaction may result from theproduction of intermediates such as short chain carboxylic acidsreleased during cleavage of the previous aryl moieties.5,7,18,27
These carboxylic acids are less reactive towards oxidation byhydroxyl radicals than their parent compounds.
CONCLUSIONSThis study showed that anodic oxidation in the presence of hy-drogen peroxide can decolorise Crystal Violet effectively, and itwas more efficient than the electro-Fenton process at neutral pH.The decolorisation rate increased with initial pH and hydrogenperoxide concentration. Current density favoured increase in therate of decolorisation, but further increase in current density re-sulted in little increase in decolorisation rate when the currentdensity exceeded 47.6 mA cm−2. The presence of sulfate and bi-carbonate hindered the decolorisation reaction. The attack ofhydroxyl radicals led to cleavage of the chromophore structure,and N-demethylation of the primary intermediates occurs subse-quently. Nearly complete decolorisation was achieved after 30 minreaction under the conditions 2.43 mmol L−1 hydrogen peroxide,47.6 mA cm−2 current density and pH0 7, while 62% COD removalefficiency was obtained when the reaction time was prolonged to90 min.
ACKNOWLEDGEMENTSThis study was supported by China Hubei Provincial Science andTechnology Department through ‘The Gongguan Project’ (GrantNo. 2003AA307B01), Wuhan Science and Technology Bureauthrough ‘The Gongguan Project’, China (Grant No. 201060723313)and the National High-Tech R&D Program (863 Program) of China(Grant No. 2008AA06Z332). We appreciate the valuable commentsof the anonymous reviewers.
REFERENCES1 Saquib M and Muneer M, TiO2-mediated photocatalytic degradation
of a triphenylmethane dye (gentian violet) in aqueous suspensions.Dyes Pigment 56:37–49 (2003).
2 Sahoo C, Gupta AK and Pal A, Photocatalytic degradation of CrystalViolet (C.I. Basic Violet 3) on silver ion doped TiO2. Dyes Pigment66:189–196 (2005).
3 Adak A, Bandyopadhyay M and Pal A, Removal of Crystal Violet dyefrom wastewater by surfactant-modified alumina. Sep Purif Technol44:139–144 (2005).
4 Adak A, Bandyopadhyay M and Pal A, Fixed bed column study forthe removal of Crystal Violet (C.I. Basic Violet 3) dye fromaquatic environment by surfactant-modified alumina. Dyes Pigment69:245–251 (2006).
5 Siminiceanu I, Alexandru CI and Brillas E, A kinetic model for the CrystalViolet mineralisation in water by the electro-Fenton process. EnvironEng Manage J 7:9–12 (2008).
6 Monash P and Pugazhenthi G, Adsorption of Crystal Violet dye fromaqueous solution using mesoporous materials synthesized at roomtemperature. Adsorption 15:390–405 (2009).
7 He H, Yang S, Yu K, Ju Y, Sun C and Wang L, Microwave inducedcatalytic degradation of Crystal Violet in nano-nickel dioxidesuspensions. J Hazard Mater 173:393–400 (2010).
8 Manal MA, El-Naggar S, El-Aasar A and Khlood IB, Bioremediation ofCrystal Violet using air bubble bioreactor packed with Pseudomonasaeruginosa. Water Res 39:5045–5054 (2005).
9 Chen CC, Liao HJ, Cheng CY, Yen CY and Chung YC, Biodegradation ofCrystal Violet by Pseudomonas putida. Biotechnol Lett 29:391–396(2007).
10 Chen CH, Chang CF, Ho CH, Tsai TL and Liu SM, Biodegradationof Crystal Violet by a Shewanella sp. NTOU1. Chemosphere72:1712–1720 (2008).
11 Yan K, Wang H and Zhang X, Biodegradation of Crystal Violet bylow molecular mass fraction secreted by fungus. J Biosci Bioeng108:421–424 (2009).
12 Li XZ, Liu GM and Zhao JC, Two competitive primary processes in thephotodegradation of cationic triarylmethane dyes under visibleirradiation in TiO2 dispersions. New J Chem 23:1193–1196 (1999).
13 Couto SR, Dominguez A and Sanroman A, Photocatalytic degradationof dyes in aqueous solution operating in a fluidized bed reactor.Chemosphere 46:83–86 (2002).
14 Chen CC, Mai F, Chen KT, Wu CW and Lu CS, Photocatalyzed N-de-methylation and degradation of Crystal Violet in titania dispersionsunder UV irradiation. Dyes Pigment 75:434–442 (2007).
15 Sihem A, Kamel D, Halima C, Tahar S, Abdlkader B and Azzedine RD,Elimination of a cationic dye (Crystal Violet) in aqueous medium byTiO2/UV oxidation process. Asian J Chem 20:5581–5590 (2008).
16 Salem IA, Activation of H2O2 by Amberlyst-15 resin supportedwith copper(II)-complexes towards oxidation of Crystal Violet.Chemosphere 44:1109–1119 (2001).
17 Alshamsi FA, Albadwawi AS, Alnuaimi MM, Rauf MA and Ashraf SS,Comparative efficiencies of the degradation of Crystal Violetusing UV/hydrogen peroxide and Fenton’s reagent. Dyes Pigment74:283–287 (2007).
18 Fan HJ, Huang ST, Chung WH, Jan JL, Lin WY and Chen CC,Degradation pathways of Crystal Violet by Fenton and Fenton-like systems: condition optimization and intermediate separationand identification. J Hazard Mater 171:1032–1044 (2009).
19 Sanroman MA, Pazos M, Ricart MT and Cameselle C, Electrochemicaldecolourisation of structurally different dyes. Chemosphere57:233–239 (2004).
20 Kobotaeva NS, Sirotkina EE and Mikubaeva EV, Electrochemicaloxidation of tritane dyes. Russ J Electrochem 42:268–271 (2006).
21 Martinez-Huitle CA and Brillas E, Decontamination of wastewaterscontaining synthetic organic dyes by electrochemical methods: ageneral review. Appl Catal B – Environ 87:105–145 (2009).
22 Brillas E, Sires I and Oturan MA, Electro-Fenton process andrelated electrochemical technologies based on Fenton’s reactionchemistry. Chem Rev 109:6570–6631 (2009).
23 Hammami S, Oturan N, Bellakhal N, Dachraoui M and Oturan MA,Oxidative degradation of direct orange 61 by electro-Fentonprocess using a carbon felt electrode: application of theexperimental design methodology. J Electroanal Chem 610:75–84(2007).
24 Lahkimi A, Oturan MA, Oturan N and Chaouch M, Removal of textiledyes from water by the electro-Fenton process. Environ Chem Lett5:35–39 (2007).
25 Boye B, Dieng MM and Brillas E, Anodic oxidation, electro-Fenton andphotoelectro-Fenton treatments of 2,4,5-trichlorophenoxyaceticacid. J Electroanal Chem 557:135–146 (2003).
J Chem Technol Biotechnol 2010; 85: 1436–1444 c© 2010 Society of Chemical Industry wileyonlinelibrary.com/jctb

14
44
www.soci.org H. Zhang et al.
26 Deng Y and Englehardt JD, Electrochemical oxidation for landfillleachate treatment. Waste Manage 27:380–388 (2007).
27 Sires I, Guivarch E, Oturan N and Oturan MA, Efficient removalof triphenylmethane dyes from aqueous medium by in situelectrogenerated Fenton’s reagent at carbon-felt cathode.Chemosphere 72:592–600 (2008).
28 Siminiceanu I, Alexandru CI and Brillas E, Study of the Crystal Violetmineralization in water by the electro-Fenton method. Rev Chim57:1082–1085 (2006).
29 Zhang H, Zhang D and Zhou J, Removal of COD from landfill leachateby electro-Fenton method. J Hazard Mater 135:106–111 (2006).
30 Rao NN, Bose G, Khare P and Kaul SN, Fenton and electro-Fentonmethods for oxidation of H-acid and Reactive Black 5. J EnvironEng – ASCE 132:367–376 (2006).
31 Zhang H, Cheng Z and Zhang D, Treatment of landfill leachate byelectro-Fenton process. Fresen Environ Bull 16:1216–1219 (2007).
32 Brillas E, Banos MA, Skoumal M, Cabot PL, Garrido JA and Rodriguez RM,Degradation of the herbicide 2,4-DP by anodic oxidation, electro-Fenton and photoelectro-Fenton using platinum and boron-dopeddiamond anodes. Chemosphere 68:199–209 (2007).
33 Dhaouadi A, Monser L and Adhoum N, Anodic oxidation and electro-Fenton treatment of rotenone. Electrochim Acta 54:4473–4480(2009).
34 Brillas E, Mur E, Sauleda R, Sanchez L, Peral J, Domenech X et al., Anilinemineralization by AOPs: anodic oxidation, photocatalysis, electro-Fenton and photoelectro-Fenton processes. Appl Catal B – Environ16:31–42 (1998).
35 Socha A, Sochocka E, Podsiadly R and Sokolowska J, Electrochemicaland photoelectrochemical degradation of direct dyes. Color Technol122:207–212 (2006).
36 He YY, Wang XC and Wu ML, Electrochemical advanced oxidativedegradation of phenol in the presence of hydrogen peroxide.J Xi’an Univ Architect Technol 39:345–348 (2007).
37 Brillas E, Calpe JC and Casado J, Mineralization of 2,4-D by advancedelectrochemical oxidation processes. Water Res 34:2253–2262(2000).
38 Brillas E, Boye B, Sires I, Garrido JA, Rodriguez RM, Arias C, et al.,Electrochemical destruction of chlorophenoxy herbicides by anodicoxidation and electro-Fenton using a boron-doped diamondelectrode. Electrochim Acta 49:4487–4496 (2004).
39 Pobiner H, Determination of hydroperoxides in hydrocarbon byconversion to hydrogen peroxide and measurement by titaniumcomplexing. Anal Chem 33:1423–1426 (1961).
40 APHA, AWWA, WPCF, Standard Methods for the Examination of Waterand Wastewater, 20th edn. American Public Health Association,American Water Works Association, Water Pollution ControlFederation, Washington DC, USA (1998).
41 Rathinam A, Nishtar NF, Jonnalagadda RR and Balachandran UN, Wetoxidation of acid brown dye by hydrogen peroxide usingheterogeneous catalyst Mn–salen–Y zeolite: a potential catalyst.J Hazard Mater 138:152–159 (2006).
42 Hameed BH and Lee TW, Degradation of malachite green in aqueoussolution by Fenton process. J Hazard Mater 164:468–472 (2009).
43 Zhang H, Fu H and Zhang D, Degradation of C.I. Acid Orange 7 byultrasound enhanced heterogeneous Fenton-like process. J HazardMater 172:654–660 (2009).
44 Zhang H, Zhang J, Zhang C, Liu F and Zhang D, Degradation of C.I.Acid Orange 7 by the advanced Fenton process in combinationwith ultrasonic irradiation. Ultrason Sonochem 16:325–330 (2009).
45 Lipczynska-Kochany E, Sprah G and Harms S, Influence of somegroundwater and surface waters constituents on the degradationof 4-chlorophenol by the Fenton reaction. Chemosphere 30:9–20(1995).
46 Zhou MH, Yu QH, Lei LC and Barton G, Electro-Fenton method for theremoval of methyl red in an efficient electrochemical system. SepPurif Technol 57:380–387 (2007).
47 Staehelin J and Hoigne J, Decomposition of ozone in water: rate ofinitiation by hydroxide ions and hydrogen peroxide. Environ SciTechnol 16:676–681 (1982).
48 Chen CC and Lu CS, Mechanistic studies of the photocatalyticdegradation of methyl green: an investigation of products ofthe decomposition processes. Environ Sci Technol 41:4389–4396(2007).
49 Ramirez JH, Costa CA and Madeira LM, Experimental design tooptimize the degradation of the synthetic dye Orange II usingFenton’s reagent. Catal Today 107–108:68–76 (2005).
wileyonlinelibrary.com/jctb c© 2010 Society of Chemical Industry J Chem Technol Biotechnol 2010; 85: 1436–1444





























![Electrochemical reduction of hydrogen peroxide on SIMFUEL ......amorphization and decomposition [32]. Although neutral to slightly alkaline conditions (pH 6–9.5) are expected to](https://img.pdfslide.us/doc/110x75/60f691a589db74567b7b1496/electrochemical-reduction-of-hydrogen-peroxide-on-simfuel-amorphization.jpg)