Embed Size (px)
Citation preview

Universidad Autónoma de Madrid. Facultad de Ciencias. Departamento de Biología Molecular
El sistema toxina-antitoxina,
,como inhibidor de la
proliferación celular e inductor de
tolerancia
Vrginia S. Lioy Marzo, 2010


Memoria presentada por Virginia Lioy para optar al grado de
Doctor en Ciencias por la Universidad Autónoma de Madrid.
El trabajo ha sido realizado en el Centro Nacional de
Biotecnología (CNB-CSIC) bajo la dirección del Prof. Juan
Carlos Alonso Navarro y la tutela de la Dra. Sylvia Ayora Hirsch.


Abstract
The emergence and spread of pathogenic bacteria that have become resistant to multiple
antibiotics through lateral gene transfer have created the need of novel antimicrobials.
Toxin-antitoxin (TA) modules, which have been implicated in plasmid maintenance and
stress management, are ubiquitous among plasmids from vancomycin, methicillin or
erythromycin resistant bacteria. In the pSM19035-encoded TA loci, the labile antitoxin
binds to free toxin and neutralizes it phosphotranferase activity. When the toxin is freed
from the antitoxin, it induces a reversible state of growth arrest in distantly related
bacteria as Bacillus subtilis and Escherichia coli. Growth arrest is mainly due to the
inhibition of the synthesis of the nucleotide pool, especially GTP and UTP. This inhibition,
which is obtained by an unknown mechanism, leads to a drastic reduction on the rate of
replication, transcription and translation. However, upon prolonged toxin action, the cells
cannot longer be rescued from their stasis state, and the cells die by lysis. It was found that
upon induction, the repression of several essential genes involved in lipid metabolism
could be responsible for cellular lysis.
Bacterial populations contain a large fraction of cells susceptible to antibiotics and a
small fraction in stasis that are refractory to them, called persisters cells. In this work, we
have shown that toxin induces a small subpopulation that is refractory to the toxin effect.
This small subpopulation could be considered as “persisters cells”. In the presence of one
antibiotic and the toxin it was observed an increased multidrug sensitivity, suggesting that
toxin induces a different type of persisters. Furthermore, action is independent of the
culture phase. We have shown that expression of the antitoxin only reversed the
refractory cells selected by the action of toxin. Our results suggested that expression of
toxin should reversibly induce a phenomenon that mimics persistence. However, stasis or
dormancy, triggered by the expression of the toxin, per se is not sufficient for multidrug
tolerance. Indeed, expression increases multidrug sensitivity.
All these results pointed towards system as an ideal candidate for developing new
antimicrobials. If we could find a compound that disrupts the ∙ interaction, it can be
considered as an attractive antimicrobial agent. Using molecular dynamics we were able to
predict that Asp18 and in minor extent Glu22 residues might be relevant for ∙ interaction.
In order to validate the in silico predictions a mutagenesis analysis on the system was
carried out. In addition, a Bioluminescence resonance energy transfer (BRET) assay was
developed for high-throughput screening (HTS). For the BRET assay, luc gene was fused
to the gene (Luc-) and to the gfp gene (-GFP). Luc- efficiently transfers the excited
energy to the fluorescent acceptor molecule (-GFP or K46A-GFP) and rendered high
bioluminescence BRET signals. The mutagenesis was performed in a variant with a
mutation in the active center (K46A-GFP) but that still formed a complex with the Luc-
antitoxin. The exchange of Asp18 to Ala from K46A-GFP (D18A) affects Luc-·D18A
K46A-GFP interaction, but exchange of Glu22 to Ala (E22A) does not. With these results,
we have validated the hypothesis that it is possible to disrupt a TA module, offering novel
and unexploited targets to fight against antibiotic-resistant strains.


Abreviaturas
A: adenina
Aa: aminoácidos
AB: antibiótico
ADN:ácido desoxirribonucleico
ADNcd: ADN cadena doble
ADP: Adenosina-5´-difosfato
ARN: ácido ribonucleico
ARNm: ácido ribonucleico mensajero
ARNt: ácido ribonucleico de transferencia
ARNtm: ácido ribonucleico de tranferencia y mensajero
ATP: Adenosina-5´-trifosfato
C: citosina
DAPI: 4',6-diamidino-2-fenilindol
DNasa I: Desoxiribonucleasa I
dNTPs: 2´-desoxirribonucleósidos-5´-trifosfato
DO: densidad óptica
dPAGE: electroforesis en gel de poliacrilamida desnaturalizante
EDF: factor de muerte extracellular, del inglés-Extracellular Death Factor
EF: factor de elongación, del inglés- Elongation factor
G: guanina
G+: Gram positiva
G-: Gram negativa
GDP: guanosina-5´-difosfato
GFP: proteína verde fluorescente, del inglés-Green Fluorescence Protein
GTP: guanosina-5´-trifosfato
His: histidina
HTH: hélice giro hélice, del inglés-Helix-Turn-Helix
HTS: del inglés-High Through Put Screening
IPTG: isopropyl -D-thiogalactósido
Kb: kilo pares de bases
LB: Medio de cultivo Luria Bertani
Luc: Renilla luciferasa
MMM9: medio mínimo M9
MMS7: medio mínimo S7
MRSA: Estafilococos aureus reisitentes a meticilina, del inglés-Methilin Resistant
Staphilococcus aureus
N: asparagina
ndPAGE: electroforesis en gel de poliacrilamida no desnaturalizante
Ni-NTA: matriz de níquel
NMP: nucleosido monofosfato
nt: nucleótido
(p)ppGpp: guanosina penta o tetrafosfato
pb: pares de bases
PCR: reacción en cadena de la polimerasa (del inglés-polymerase chain reaction)
PEI: polietilénimina

Pir: pirimidinas
RF: factor de libreación, del inglés-Release Factor
RHH: lamina-hélice-hélice, del inglés-Ribbon-Helix-Helix
RMN: resonancia magnética nuclear
ROS: especies reactivas de oxigeno, del inglés-Reactive Oxiygen Species
rpm: revoluciones por minuto
SA: sulfato amónico
SDS: dodecil sulfato sódico
SDS-PAGE: electroforesis en gel de poliacrilamida en presencia de SDS
TA: toxina-antitoxina
TAE: Tris Acetato EDTA
UFC: unidades formadoras de colonias
UTP: uridina-5´- trifosfato
VBNC: del inglés-Viables But Non Culturables
VRE: Enterococos resistentes a Vancomicina, del inglés-Vancomycin Resistant
Enterococcus
W: triptófano
Xil: xilosa

Índice


INTRDUCCIÓN
1. EL PLÁSMIDO PSM19035 1
2. CLASIFICACIÓN DE LOS SISTEMAS DE TOXINA ANTITOXINA 3
2.1 Sistemas tipo I 3
2.2 Sistemas TA tipo II 4
3. SISTEMAS TA QUE INHIBEN LA REPLICACIÓN 4
4. SISTEMAS TA QUE INHIBEN LA TRADUCCIÓN 5
4.1 Sistema RelBE 6
4.3 Sistemas MazEF y Kid/Kis 7
4.3.1 Sistema MazEF 8
4.3.2 Sistema Kid/Kis. 9
4.4 Sistema HipBA 10
5. SISTEMAS CON MECANISMOS DE ACCIÓN DESCONOCIDOS 12
5.1 Sistemas y PezAT 12
6. REGULACIÓN DE LA EXPRESIÓN DE LOS SISTEMAS TA 13
6.1 Regulación de la expresión en sistemas de 2 componentes 13
6.2 Regulación de la expresión del sistema : función y características de la proteína 2 14
7. ESTRUCTURAS CRISTALINAS DE LOS SISTEMAS TA: ANÁLISIS Y DISEÑO DE NUEVOS
ANTIMICROBIANOS 16
7.1 Estructura cristalina del complejo CcdAB 16
7.2 Estructura cristalina del complejo RelBE 17
7.3 Estructura cristalina del complejo Kid-Kis y MazEF 17
7.4 Estructura Cristalina de HipBA 18
7.5 Estructura cristalina del complejo y PezAT 19
OBJETIVOS 21

1. MATERIALES 23
1.1 Cepas 23
1.2. Reactivos y materiales 23
2. MÉTODOS 24
2.1. Manipulación de células 24
2.1.1. Obtención de células competentes. 24
2.1.2 Transformación Bacteriana. 24
2.2. Manipulación del ADN 25
2.2.1. Purificación y cuantificación del ADN 25
2.2.2. Plásmidos 25
2.2.3. Marcaje radiactivo de fragmentos de ADN. 26
2.2.3.1. Marcaje de fragmentos con extremos 5´ protuberantes 26
2.2.3.2. Obtención y purificación de los fragmentos de ADN utilizados para los ensayos de
interacción proteína-ADN. 26
2.2.4. Mutagénesis dirigida. 26
2.3. Obtención y estudio de proteínas 27
2.3.1. Sobreproducción de proteínas. 27
2.3.2. Purificación de proteínas. 27
2.3.2.1. Purificación de las proteínas 27
2.3.2.2. Purificación de -His6)2 y (D60A-His6)2. 28
2.4. Ensayos bioquímicos 28
2.4.1. Medida de interacciones proteína-ADN 28
2.4.1.1. Ensayos de retraso en gel. 28
2.4.1.2. Ensayos de protección a DNasa I. 29
2.5 Modelado Molecular 29
2.6. Ensayos in vivo 29
2.6.1 Medio de Cultivo: 29
2.6.2. Microscopías 30
2.6.2.1 Microscopía de fluorescencia: Visualización de nucleoides y estado de las membranas
30
2.6.2.1 Microscopía electrónica: Visualización del efecto de la sobre-expresión de en células
de E. coli 30
2.6.3 Estudios de viabilidad en B. subtilis y E. coli 30
2.6.3.1 Estudio del efecto de la expresión de Y83C y en la viabilidad de células de B. subtilis
30
2.6.3.2 Identificación de especies reactivas de oxigeno mediadas por en B. subtilis 30
2.6.3.3 Estudio del efecto de la expresión de en células de E. coli 31
2.6.3.4 Estudio de la toxicidad de :GFP en células de E. coli 31
2.6.3.5 Estudio del efecto de la expresión deY83C y en la generación de células persistentes
en B. subtilis 31

2.6.4 Estudio de la inhibición de la síntesis de ARN, ADN y proteínas luego de la inducción de
en B. subtilis 32
2.6.5 Geles en 2D, análisis de imágenes y identificación de proteínas 32
2.6.6 Cuantificación del pool de purinas in vivo. 32
2.6.7 Análisis del transcriptoma de B. subtilis en presencia de Y83C 33
2.6.8 Ensayo de BRET en células de E. coli 33
RESULTADOS
PARTE I: REGULACIÓN DEL SISTEMA DE TA PAPEL DE LA PROTEÍNA EN LA
MODULACIÓN DEL REPRESOR TRANSCRIPCIONAL 35
1.1 La proteína 2 forma complejos específicos y 2 y 2D60A incrementan la formación del
complejo 2·DNA 35
1.3 El aumento de la afinidad de 2 por la región promotora es independiente de la unión de 2 a
ATP y a ADN 36
PARTE II. SISTEMA TA: EL PAPEL DE EN EL SISTEMA 38
2.1. Sistemas Toxina Antitoxina: Antecedentes del tema 38
2.2 Caracterización del efecto tóxico de la proteína en B. subtilis 38
2.2.1 La expresión de la toxina Y83C inhibe la proliferación celular 38
2.2.2 La expresión de la toxina Y83C induce un efecto bacteriostático y la muerte de una
fracción de la población bacteriana 39
2.2.3 La expresión de no induce especies reactivas de oxigeno 40
2.2.4 La expresión de inhibe la replicación, transcripción y traducción in vivo. 41
2.2.5 La toxina inhibe la síntesis de purinas in vivo 42
2.2.6 Efecto de la toxina Y83C en la expresión génica de B. subtilis 44
2.2.6.1 Genes cuya expresión fue reprimida por Y83C 45
2.2.6.2 Genes cuya expresión fue inducida por Y83C 47
PARTE III. SISTEMA TA: EL PAPEL DE 2 EN EL SISTEMA 47
3.1 Según los niveles de expresión de el efecto tóxico producido en las células puede revertirse
por la expresión de la antitoxina 2 47
PARTE IV. LA TOXINA INDUCE UN AUMENTO EN LA SENSIBILIDAD DE CÉLULAS
TOLERANTES A ANTIBIÓTICOS. 50
4.1 La expresión de Y83C induce un aumento de la sensibilidad a antibióticos. 50
4.2 La expresión de no produce un aumento de células tolerantes en presencia de antibióticos.
51
PARTE V. EL SISTEMA COMO DIANA ANTIMICROBIANA. 56

5.1 Identificación de los aminoácidos esenciales para la interacción del complejo ∙ 56
5.2 Estudio in vivo de los mutantes en los residuos esenciales en la interacción ∙ 57
DISCUSIÓN
1. LA SEGREGACIÓN DEL PLÁSMIDO PSM19035 ES MUY EFICIENTE 61
2. CARACTERIZACIÓN FISIOLÓGICA DEL EFECTO TÓXICO DE 61
3. LA ANTITOXINA 2ES CAPAZ DE RECUPERAR LA ACTIVIDAD METABÓLICA DE LAS
CÉLULAS QUE ESTABAN BAJO EL EFECTO DE . 65
4. LA TOXINA PODRÍA MODIFICAR EL POOL DE NUCLEÓTIDOS INDUCIENDO UN
ESTADO BACTERIOSTÁTICO. 65
5. LA TOXINA INDUCE UN ESTADO BACTERIOSTÁTICO Y PODRIA DISPARAR LA
MUERTE DE UNA PEQUEÑA FRACCIÓN DE LA POBLACIÓN, QUE ASEGURARÁ LA
SUPERVIVENCIA DEL RESTO, PERO NO PARECE INDUCIR SENESCENCIA 66
6. LA TOXINA COMO DIANA ANTIMICROBIANA 66
6.1 La toxina aumenta la eficacia de los antibióticos sin producir células persistentes. 66
6.2 Diseño de nuevas dianas antimicrobianas. 68
CONCLUSIONES 69
BIBLIOGRAFÍA 71
ANEXO I 77
1. Genes Afectados por la expresión de Y83C 77
ANEXO II 83

Introducción


P á g i n a | 1
Los microorganismos son capaces de desarrollar resistencia a los agentes
antimicrobianos más utilizados, haciendo que los tratamientos de las infecciones
bacterianas sean cada día más difícil. Los mecanismos de adquisición de resistencias
pueden variar desde la generación de mutaciones espontaneas, que modifican la diana del
antibiótico, hasta la expresión de bombas que se encarguen de expulsar al antibiótico
fuera de la célula. Se cree que la forma más probable de adquirir resistencia a antibióticos
es a través de la transferencia de elementos genéticos móviles entre bacterias. Estos
elementos móviles incluyen a los plásmidos, que son elementos genéticos
extracromosomales que se transfieren horizontalmente dentro del mismo género o entre
géneros distintos por conjugación, transducción o transformación. Los plásmidos pueden
conferir resistencias a múltiples antibióticos, por ej.: -lactámicos, aminoglicósidos,
tetraciclinas, macrólidos y glicopeptidos. Se ha demostrado que la resistencia a
vancomicina en especies de Enterococcus o VRE (del inglés-vancomicin resistant
enterococcus) se encuentra codificada en plásmidos, y se cree que las cepas de
Staphylococcus aureus resistenes a meticilina o MRSA (del inglés-methicilin resistant
Sthapylococcus aureus) que también se hicieron resistentes a vancomicina, adquirieron la
resistencia a vancomicina a través de la transferencia de plásmidos de las especies de
enterococos resistentes (DeNap & Hergenrother, 2005, Williams & Hergenrother, 2008).
Debido a esto, el diseño de nuevas dianas antimicrobianas que permitan eliminar a los
plásmidos que portan las bacterias patógenas abre una esperanza en la lucha contra las
bacterias resistentes. Se han propuesto varias estrategias para combatir la resistencia a
antibióticos, y éstas van desde inhibir la transferencia horizontal hasta la inhibición de la
replicación de los mismos. Pocos intentos se han realizado explotando los sistemas
toxina–antitoxina (TA), que están involucrados en el mantenimiento estable del plásmido
en la célula (Williams & Hergenrother, 2008). Análisis estadísticos revelan que plásmidos
aislados de VRE tenían más de 2 sistemas TA. Entre ellos el sistema TA , identificado
por primera vez en el plásmido pSM19035, estaba presente en el 44% de los plásmidos
analizados (Moritz & Hergenrother, 2007). Este sistema se encuentra ampliamente
propagado en estreptococos, y en especial en Streptoccocus pyogenes. Por esto, el
presente trabajo se centrará en el estudio y la caracterización del sistema y su posible
uso como nueva diana antimicrobiana.
1. El plásmido pSM19035
El plásmido pSM19035 (Figura 1) tiene un tamaño de 28,9 Kb y pertenece a la familia de
plásmidos inc18, junto con los plásmidos pAM1 y pIP501. Este plásmido se aisló de
células de Streptococcus pyogenes y a pesar de sus repeticiones internas (el 80 % de su
genoma está repetido e invertido) es capaz de propagarse de manera muy estable en un
gran número (al menos 18 de las 21 especies) de especies del Filo Firmicutes sin perder
información genética (Ceglowski et al., 1993a, Brantl et al., 1990, Ceglowski et al., 1993b).
La replicación de pSM19035 está controlada por 3 mecanismos independientes: En el
primero, el producto del gen repS, que es esencial para la iniciación de la replicación, es
reprimido por el producto del gen copS (Figura 2). En el segundo, se síntetiza un ARN
antisentido que bloquea la utilización del ARNm repS (Brantl & Wagner, 1997, Heidrich &
Brantl, 2007) (Figura 2). Por último, la utilización de promotor de copS (Pcop) e
indirectamente la síntesis de CopS, está regulada por el producto del gen que está
localizado fuera del replicón mínimo.
Se vio que este plásmido era 10.000 veces más estable de lo esperado si el mecanismo
de segregación fuera al azar. Se identificaron 2 regiones discretas, SegA y SegB
responsables de la estabilidad segregacional del pSM19035, funcionando ambas

P á g i n a | 2
actividades independientemente del replicón (Ceglowski et al., 1993a, Brantl et al., 1990,
Ceglowski et al., 1993b). La región SegA codifica las proteínas y . Esta región está
implicada en la inversión de la mitad de su genoma durante la replicación y la resolución
de los multímeros que se forman tras la replicación del plásmido, optimizando la
replicación y maximizando la segregación al azar. La proteína es una recombinasa
perteneciente a la familia de invertasas-resolvasas que cataliza recombinaciones
específicas de sitio, y se une a una región de 85 pares de bases (denominada sitio six)
dividida en dos sitios adyacentes (sitios I y II) (Canosa et al., 2003, Canosa et al., 1998,
Canosa et al., 1996, Rojo & Alonso, 1994, Alonso et al., 1996). Además de incrementar el
número de moléculas monoméricas, la proteína junto con la proteína (topoisomerasa
III) ejercen control también sobre la replicación del plásmido, mediando el proceso de
inversión del ADN necesario para evitar la colisión entre las dos horquillas de replicación,
ya que el pSM19035 tiene dos orígenes de replicación enfrentados, uno en cada repetición
invertida y la replicación es unidireccional (Figura 1). La función de la proteína es
desconocida, y no presenta homología con ninguna proteína de las bases de datos.
La región SegB contiene 4 marcos abiertos de lectura (y) (Figura 1 y 2) que están
ordenados en dos unidades transcripcionales, la primera codifica la proteína y la
segunda unidad es un operón formado por los genes y (Figura 2) (Ceglowski et al.,
1993a). La proteína (o ParA), que es un dímero en solución (2, 68 kDa) pertenece a la
familia de ATPasas involucradas en partición y presenta alta homología estructural con la
proteína Soj de B. subtilis. La proteína (o ParB), que es también un dímero en solución
(2, 16 kDa), es el regulador global del plásmido, ya que además de mantener los niveles
de expresión del sistema TA, regula el número de copia (de la Hoz et al., 2000) y funciona
junto a estabilizando el plásmido. Sin embargo, la función principal de estabilización del
plásmido se debe a , sistema de TA. Los sistemas TA se clasifican en dos tipos según la
naturaleza de la antitoxina, a continuación se realizará una breve descripción de ellos.
Figura 1. Mapa físico del plásmido pSM19035. Las secuencias duplicadas están indicadas por una línea gruesa y la cabeza de flecha indica su orientación. Las zonas no repetidas (NR1 y NR2) están indicadas por una línea fina. Se muestran las regiones requeridas para la replicación (Rep) y la segregación (SegA y SegB). Las flechas en color fuera del círculo muestran los marcos abiertos de lectura más relevantes y las puntas de flecha
que sobresalen indican los subsitios de unión de , en las regiones promotoras de los genes copS, y . La
línea naranja indica los sitios six, la amarilla los oriS y la roja los sitios de unión de . Las flechas negras en los oriS indican la dirección de la replicación. Como el plásmido se encuentra duplicado, solo se ha coloreado una de las repeticiones.

P á g i n a | 3
2. Clasificación de los sistemas de toxina antitoxina
La principal característica de los sistemas de TA es que están formados por dos
componentes. Uno de ellos es una toxina de larga vida media y el otro una antitoxina, con
una vida media mucho más corta, capaz de antagonizar el efecto tóxico de la toxina. Estos
sistemas se clasifican según la naturaleza de la antitoxina. En los sistemas de tipo I la
antitoxina es un pequeño ARN antisentido, que es complementario al ARN mensajero de
la toxina. De esta forma el pequeño ARN antisentido se une al mensajero de la toxina
evitando la traducción de la misma. En los sistemas de tipo II la antitoxina es una proteína
que inhibe la acción tóxica de la toxina formando un complejo estable. Como se mencionó
anteriormente, la antitoxina tiene una vida media mucho más corta que la toxina, lo que
permite a ésta ultima quedar libre si no hay síntesis de novo y ejecutar su efecto tóxico.
Existen varias familias de TA, y se pueden agrupar según la similitud de sus toxinas. Las
toxinas de los sistemas tipo I suelen ser pequeñas e hidrofóbicas, interaccionando con la
membrana y generando una pérdida de su potencial. Las toxinas del tipo II tienen como
dianas procesos primordiales para la bacteria, como pueden ser la traducción o la
replicación (Gerdes et al., 2005, Van Melderen & Saavedra De Bast, 2009). Los sistemas
TA se pueden encontrar tanto en plásmidos de bajo número de copia como en el
cromosoma de una gran diversidad de microorganismos. Generalmente, los genes de la
toxina y la antitoxina forman un operón, en donde el gen de la antitoxina se encuentra
aguas abajo del de la toxina. Excepto para el sistema (Camacho et al., 2002, Meinhart
et al., 2003), la regulación de la expresión del operón está dada por la antitoxina y/ó por el
complejo TA. En el caso del operón la regulación se debe exclusivamente al producto
del gen . El sistema forma parte de una familia menos conocida, la cual no presenta
homología con otras descritas hasta el momento. Este sistema, se encontró entre otros,
tanto en plásmidos aislados de cepas de enterococos resistentes a vancomicina (Moritz &
Hergenrother, 2007) como en el cromosoma de bacterias patógenas como S. pneumoniae
y E. coli B7A (Khoo et al., 2007, Van Melderen & Saavedra De Bast, 2009). Su actividad
es desconocida hasta el momento, y por eso es objeto de estudio en este trabajo.
2.1 Sistemas tipo I
El sistema tipo I mejor estudiado es el sistema hok/sok del plásmido R1 de Escherichia
coli. En este sistema TA, sok es un pequeño ARN antisentido que inhibe la traducción del
mensajero de la toxina, hok. Cuando sok se encuentra unido a hok, RNasa III rápidamente
Figura 2. Regiones Rep, SegA y SegB del plásmido pSM19035. Detalle de las regiones involucradas en
replicación y segregación estable del plásmido. La proteína 2, en gris, reprime la expresión del operón formado
por y del gen y del gen copS uniéndose a una serie de repeticiones (mostradas como flechas). A su
vez, la proteína CopS, reprime la expresión de repS, cuyo producto RepS es esencial para el inicio de la
replicación del plásmido. Desde el PIII,, se sintetiza un ARN anti-sentido (ARN III) que impide la transcripción de
repS.

P á g i n a | 4
degrada el complejo sok-hok, evitando así que el ARN mensajero de la toxina se traduzca.
El ARN antisentido sok tiene una vida media muy corta (30 s) y cuando el plásmido se
pierde, hok, con vida media más larga (20 min) queda libre y puede ser traducido (Gerdes
& Wagner, 2007). Como en el caso de hok, todas las toxinas de los sistemas tipo I
descritas hasta el momento son pequeñas e hidrofóbicas y generalmente causan daños en
la membranas bacterianas. Se han descrito sistemas tipo I no solo en plásmidos de bajo
número de copia sino también en el cromosoma de E. coli, dos de ellos inducibles por el
sistema SOS. Entre ellos el más importante de destacar es el sistema SymER. La
transcripción de la toxina SymE esta reprimida por LexA, y la traducción de los mensajeros
residuales de SymE están reprimidos por la unión a los mismos de SymR, un ARN
antisentido. La toxina SymE exhibe homología con MazF, pero a diferencia de ella, co-
purifica con ribosomas lo que sugiere que su mecanismo de acción es diferente al descrito
para MazF (Gerdes & Wagner, 2007)
2.2 Sistemas TA tipo II
Los sistemas de TA tipo II se encuentran ampliamente distribuidos en plásmidos y en los
cromosomas de una gran diversidad de bacterias y arqueas. Es importante destacar, que
muchas bacterias presentan múltiples copias de los sistemas TA en sus cromosomas, ya
sea de sistemas TA homólogos o con distintas dianas celulares. Por ej., Nitrosomas
europea tiene al menos 45 sistemas TA y Mycobacterium tuberculosis 38. Sin embargo,
hasta el momento no se han descrito sistemas TA en organismos intracelulares obligados,
por ej., en el patógeno humano Mycobacterium leprae no se han encontrado sistemas TA
completos. También es importante destacar que los organismos intracelulares obligados
carecen de genes relA/spoT intactos. Estos genes, y los de TA, no serían necesarios ya
que estos microorganismos se multiplican en un ambiente constante, por lo tanto no se
beneficiarían al tener sistemas de respuesta a estrés (Gerdes et al., 2005)
Los sistemas de TA tipo II se clasifican en 9 familias, teniendo en cuenta las
características estructurales de las toxinas que conforman el sistema, ya que la naturaleza
de las antitoxinas varía aún en sistemas homólogos. Estas familias contienen tanto
sistemas TA que se encontraron en plásmidos como en cromosomas. En la tabla 1 se
detallan brevemente las características más importantes de los representantes de cada
familia. Hasta el momento, los sistemas TA se pueden dividir en dos grupos, aquellos
cuyas toxinas inhiben la replicación y aquellos cuyas toxinas inhiben la traducción. Es
sorprendente que toxinas que inhiben el mismo proceso celular (traducción) lo pueden
hacen a través de mecanismos muy distintos. Por esto, a continuación se detallarán los
sistemas TA más relevantes para este trabajo, agrupándolos según su diana molecular.
3. Sistemas TA que inhiben la replicación
De los sistemas que inhiben la replicación, CcdAB del plásmido F fue el primero en
describirse como un sistema de muerte post-segregacional, aunque luego se lo encontró
en el cromosoma de E. coli (Wilbaux et al., 2007). Más tarde se caracterizó el sistema
ParDE de RK2 (Jiang et al., 2002). Estos dos sistemas son los únicos descritos hasta el
momento que inhiben la replicación del ADN. Sorprendentemente, sistemas TA con
toxinas que presentan homología estructural a CcdB (Kid y MazF), tienen como diana
celular la degradación de ARN (Kamphuis et al., 2007a).

P á g i n a | 5
Toxina Diana Proceso celular afectado
CcdB ADN girasa Replicación
RelE ARNm unido al ribosoma
Traducción
MazF/Kis ARNs Traducción
ParE ADN girasa Replicación
VapC ARNs Traducción
Doc Ribosoma Traducción
HipA EF-Tu Traducción
HigB ARNm unido al ribosoma
Traducción
Desconocida Desconocido
3.1 Sistema CcdAB del plásmido F
El primer sistema de TA que se identificó en plásmidos fue el sistema CcdAB del
plásmido F de E. coli. Se describierón dos proteínas, CcdB (101 aa) y CcdA (72 aa), en el
locus ccd (del inglés – couple cell division) como las encargadas de mantener el plásmido
en las células (Ogura & Hiraga, 1983). De esta forma, se demostró que CcdB codificaba
una toxina de vida media larga, y CcdA una antitoxina de vida media corta, que era
degrada por la proteasa LonA (Van Melderen et al., 1994). Así, si la célula se dividía y sus
hijas no recibían al menos una copia del plásmido, CcdA era degrada y CcdB, de vida
media larga, podía inducir la muerte post-segregacional de las células sin plásmido. Esta
inhibición post-segregacional del crecimiento estaba acompañada por la inducción del
sistema SOS, la reducción de la síntesis de ADN, la filamentación celular y la formación de
células anucleadas. Gracias a que se pudieron aislar mutantes resistentes a la actividad
tóxica de CcdB, se encontró que la subunidad A de la ADN girasa era la diana molecular
de la toxina. De hecho se aislaron dos mutantes (GyrA64, en donde la arginina 462 fue
cambiada por una cisteína, R462C, y tldC15, donde la glicina 214 cambió a glutamato,
G214Q) en dos laboratorios independientes que confirmaban que CcdB interaccionaba
con esta subunidad de la ADN girasa (Bernard & Couturier, 1992, Miki et al., 1992). Se
propuso que el mecanismo molecular de la actividad de la toxina es el de estabilizar el
complejo de cortado, en donde el ADN está covalentemente unido a la subunidad A de la
girasa, causando la rotura del ADN y, a diferencia de las quinolonas, este proceso es
dependiente de ATP. La interacción CcdB-GirA, pudo ser revertida in vitro si se agregaba
al complejo la antitoxina CcdA (Bahassi et al., 1999, Bernard et al., 1993, Gerdes, 2000,
Maki et al., 1992). Esta reversión, demostró que era probable que la muerte inducida por
este sistema TA sea un evento extremo debido a la sobreproducción de CcdB, sugiriendo
que en condiciones fisiológicas, CcdB pueda inhibir la replicación del ADN pero sin inducir
muerte post-segregacional (Gerdes, 2000).
4. Sistemas TA que inhiben la traducción
Las mayorías de las familias de sistemas TA inhiben la traducción a través de
mecanismos diferentes. Entre ellas se pueden destacar los sistemas TA: RelB, MazE, Kid,
Tabla 1. Familias de toxinas

P á g i n a | 6
YoeB, HipA, HigA y Doc. De estos sistemas, el sistema MazEF y RelBE son los mejores
caracterizados, y su papel en la célula es motivo de grandes debates. De todos estos
sistemas, solo se describirán en detalle los más relevantes para este trabajo.
4.1 Sistema RelBE
El sistema RelBE se encontró en el cromosoma de E. coli y fue el primero que permitió
incluir a los sistemas de TA como moduladores del crecimiento. Este sistema también se
encuentra ampliamente extendido en Gram positivas (G+), Gram negativas (G-) y
Arqueas, tanto en plásmidos como en sus cromosomas (Gerdes et al., 2005). RelE
codifica una toxina de vida media larga y RelB una antitoxina de vida media más corta. Se
demostró que este sistema cromosomal también es capaz de estabilizar plásmidos
(Gotfredsen & Gerdes, 1998). El sistema RelBE forma parte de un operón junto con RelF,
una toxina del tipo I (Hok). La inducción de RelF produce una rápida inhibición de la
proliferación, arresto de la respiración y un colapso del potencial de membrana. Sin
embargo no se conoce el papel fisiológico de RelF, ya que parece que su síntesis se
encuentra reprimida (Gerdes et al., 1986). El operón relBEF se descubrió como resultado
de la observación de que ciertas mutaciones puntuales en el gen relB resultaban en una
respuesta retrasada relajada (del inglés- delayed relax response). Cuando células
silvestres se sometían a una falta de aminoácidos, la síntesis de ARN caía rápidamente.
Esta caída estaba mediada por la producción de (p)ppGpp (producto de RelA). Sin
embargo se vio que en mutantes en el gen relA, la síntesis de ARN era estable durante
todo el tiempo que durase la situación de estrés nutricional. Por esto, este fenotipo se
conoció como respuesta relajada. Células de E. coli con mutaciones puntuales en el gen
relB, tardaban 10 min en tener una síntesis de ARN estable luego de inducir la falta de
aminoácidos, de ahí que la respuesta se llamó “retrasada”. Otra característica de estos
mutantes, es que una vez que se acababa el estrés nutricional, es decir, se volvían a
incorporar aminoácidos al medio, los mutantes en el gen relB reiniciaban el crecimiento de
manera muy lenta, sugiriendo que existía la acumulación de algún compuesto tóxico
durante ese periodo de carencia (Gotfredsen & Gerdes, 1998). Más tarde, se demostró
que la degradación de RelB estaba mediada por la proteasa LonA y se inducía en
respuesta al estrés nutricional (Christensen et al., 2001). De esta forma RelE quedaba
libre, inhibiendo el crecimiento celular y reduciendo el número de unidades formadoras de
colonias (UFC). Este efecto se producía por medio de la inhibición de la síntesis de
proteínas, cuando la toxina degradaba al ARN mensajero unido al sitio A del ribosoma. La
degradación del ARN es dependiente de secuencia, siendo la secuencia UAG y UAA
reconocidas con más eficiencia (Pedersen et al., 2003). Cuando al ARN mensajero es
cortado en el sitio A del ribosoma, se produce una parada de la traducción, “envenenando
al ribosoma”. Este ribosoma “envenenado” es luego rescatado por un ARN que es tanto
ARN de transferencia como ARN mensajero, el ARNtm. El ARNtm permite que la
traducción continúe, colocando una secuencia específica que será reconocida por las
proteasas celulares cuando el ribosoma encuentre el codón de parada y se libere la
proteína (Figura 3). De esta forma, las proteínas cuyos ARNm fueron cortados por RelE
serán degradadas por las proteasas celulares, generando así una fuente de aminoácidos
que serán usados para la biosíntesis de proteínas esenciales para la bacteria (Pedersen et
al., 2003)

P á g i n a | 7
Se demostró que la expresión de RelE no inducía muerte celular, sino un estado
bacteriostático que podía ser revertido por la síntesis de novo de la antitoxina RelB, y que
esto era aplicable a otros sistemas de TA tales como MazEF, generando una gran
controversia (Pedersen et al., 2002, Amitai et al., 2004). De esta manera, la síntesis de
RelB era capaz de revertir la inhibición de la traducción mediada por RelE tanto in vivo
como in vitro. Gracias a estos resultados, surge una nueva teoría, en donde los sistemas
de TA se activarían en respuesta a estrés, como por ejemplo por la carencia de
aminoácidos, e inducirían un estado bacteriostático permitiendo a las células sobrevivir
hasta que esta situación de estrés se revierta.
Nieto y colaboradores fueron los primeros en caracterizar un sistema de TA en el
cromosoma de una bacteria G+, Streptococus pnemuniae. Se encontraron dos locus
cuyos genes presentaban homología con el sistema RelBE de E. coli, pero solamente uno
de ellos, RelBE2Spn resultó ser tóxica en E. coli y en S. pnemuniae (Nieto, Pellicer et al.
2006). Este sistema se encontraba en el cromosoma de las dos cepas de S. pneumoniae
secuenciadas hasta ese momento; en la cepa no patogénica R6 y en la patogénica Tigr4,
abriendo una puerta en la generación de nuevos antimicrobianos. El efecto de la
sobreproducción de la toxina RelB2Spn en S. pneumoniae era más suave si se realizaba
en la cepa silvestre, sin embargo, si la toxina era sobreexpresada en el mutante
relBE2Spn, se observó un efecto mucho más fuerte en la disminución de las UFC (1000
veces) y este efecto se pudo revertir por la expresión de la antitoxina RelE2Spn (Nieto et
al., 2006). También se demostró que la expresión de la toxina RelBE2Spn en E. coli era
tan eficaz como en S. pneumoniae, demostrando que la actividad de RelE2Spn tiene un
amplio espectro de huésped (Nieto, et al. 2006).
4.3 Sistemas MazEF y Kid/Kis
Los sistemas MazEF y Kid/Kis presentan gran homología entre sí, considerándose
sistemas TA de la misma familia. Sin embargo, el sistema Kid/Kis se encontró por primera
vez en el locus parD del plasmido R1 de E. coli (Ruiz-Echevarria et al., 1991a, Ruiz-
Echevarria et al., 1991b). El sistema MazEF se describió por primera vez en el cromosoma
de E. coli, aunque también se encuentra en plásmidos de bajo número de copia. El
sistema CcdAB también posee homología estructural con estos Kid/Kis y MazEF, pero
como la toxina CcdB tiene una diana distinta se lo considera otra familia de TA.
Figura 3. Organización genética y componentes regulatorios del sistema RelBE. Se muestra el operón
RelBE, el sitio de corte en el ARNm mediado por la toxina RelE y el rescate del ribosoma mediado por el ARNtm,
luego de que el ARNm fue cortado. Adapatado de (Pedersen et al., 2002)

P á g i n a | 8
4.3.1 Sistema MazEF
El sistema MazEF, también conocido como ChpAIK, cuando se describió por primera vez
como un sistema TA se demostró que se inducia en la respuesta estricta (del inglés -
stringent response) cuando las células crecían en ausencia de algún aminoácido o fuente
de carbono (Aizenman et al., 1996). Aizenman y colaboradores demostraron que el
producto de la proteína RelA (ppGpp), reprimía la expresión del operón MazEF, activando
a la toxina MazF, e induciendo la muerte celular programada de la población bacteriana.
En el año 2006, Gross y colaboradores encontraron que MazG era un modulador del
sistema de muerte celular programada MazEF y que el gen mazG formaba parte del
operón mazEFG. MazG resultó ser la enzima encargada de degradar el ppGpp sintetizado
por RelA en respuesta a la falta de aminoácidos. Se demostró que el complejo MazEF era
capaz interaccionar con MazG, pero cuando la célula se encontraba en una condición de
estrés nutricional, RelA sintetizaba altas cantidades de ppGpp, inhibiendo la expresión del
promotor de mazEFG (Aizenman et al., 1996). Así, MazE era degrada por ClpPA; MazG y
MazF que tenían una vida media más larga quedaban libres y cada una realizaba su
función: MazF inhibía la síntesis de proteínas y MazG degradaba ppGpp y otros
nucleótidos (Gross et al., 2006). La inducción de MazF también se producía por otros tipos
de estrés celular como pueden ser la inhibición de la transcripción, traducción o
replicación, por el daño en el ADN producido por la falta de timina, así como también por el
tratamiento de células de E. coli con radiación UV y por estrés oxidativo (H2O2) (Hazan &
Engelberg-Kulka, 2004, Hazan et al., 2001, Sat et al., 2001, Sat et al., 2003). Como se
mencionó en el apartado anterior (sistema RelBE), el grupo de Ken Gerdes cuestionó el
modelo establecido para MazEF, y demostró que la inducción de MazF generaba un
estado bacteriostático que podía ser revertido por la expresión de MazE. Sin embargo, el
grupo de Hanna Engelberg-Kulka demostró unos años después que el efecto
bacteriostático inducido por la expresión de MazF solo duraba un determinado tiempo,
luego del cual las células entraban un “punto de no retorno”, induciendo la muerte celular
de las bacterias (Pedersen et al., 2002, Amitai et al., 2004, Engelberg-Kulka et al., 2006)
MazF inhibe la síntesis de proteínas degradando el ARN mensajero en la secuencia 5´-
NCA-3´, donde N puede ser A o U, ya sea ARN de cadena simple o doble y de manera
independiente del ribosoma (Munoz-Gomez et al., 2004, Zhang et al., 2003). Este es el
único sistema TA en el que se encontró que la muerte celular esta mediada por una
molécula señal, un péptido lineal de cinco aminoácidos con la secuencia NNWNN,
conocido como “factor de muerte extracelular” o EDF (del inglés- Extracellular Death
Factor). La producción de este péptido depende de la densidad de la población bacteriana,
de la respuesta al estrés y de dos proteínas: Zwf (glucosa-6-fosfato deshidrogenasa) y
ClpXP. Si este péptido no se produce, el sistema MazEF no es capaz de inducir la muerte
celular programada en E. coli (Kolodkin-Gal & Engelberg-Kulka, 2008, Kolodkin-Gal et al.,
2007). De hecho la adición del EDF en cultivos MazEF+ tratados con rifampicina, producía
un cambio en el efecto antibacteriano del antibiótico, que pasaba de ser bacteriostático a
ser bactericida. De este modo, se propuso que los antibióticos pueden producir muerte por
dos vías distintas, una dependiente de MazEF y la otra independiente de ella (ver Figura
4) (Kohanski et al., 2007, Kolodkin-Gal et al., 2008).
Recientemente se ha demostrado que la inducción de MazF en células de E. coli inhibe
la síntesis de una gran parte de las proteínas, pero que también produce un incremento en
la síntesis de algunas proteínas de bajo peso molecular. El aislamiento y posterior análisis
de las proteínas que se sintetizaban en presencia de MazF desveló que dependían del tipo
de estrés que se utilizaba para inducir el mecanismo de muerte celular. Si este mecanismo

P á g i n a | 9
era independiente de especies reactivas de oxigeno o ROS (del inglés - Reactive Oxygen
Species), se sintetizaban solo proteínas de muerte. Sin embargo, si el mecanismo de
muerte era dependiente de ROS, las proteínas que se seguían sintetizando eran tanto
proteínas de muerte como de supervivencia (Figura 4). Se cree, que la síntesis selectiva
de estas proteínas cuando se induce el sistema MazEF permite la supervivencia de una
pequeña fracción de la población, que será la que luego retomará el crecimiento cuando la
situación de estrés desaparezca. A partir de estos resultados, se propone a MazF como un
regulador de la muerte celular programada, que activa distintos mecanismos de muerte
dependiendo del estrés en el que las células se encuentren. En este modelo, el sistema
MazEF se induciría y la gran mayoría de las células entrarían en muerte celular
programada, mientras una pequeña población sobreviviría y se alimentaria de los restos
de las bacterias muertas. Cuando la situación de estrés revierta, esta pequeña fracción
permitirá la supervivencia de la población (Amitai et al., 2009)
4.3.2 Sistema Kid/Kis.
El sistema Kid/Kis se encuentra codificado en los genes del locus parD del plásmido R1 de
E. coli. También se encuentra en el plásmido R100, pero con el nombre pem, dando lugar
a la toxina pemK y a la antitoxina pemI. Al igual que MazF, la toxina Kid es una
endoribonucleasa que degrada ARN mensajero independiente del ribosoma, inhibiendo la
traducción. Existe cierta controversia sobre el sitio de corte en el ARN mensajero mediado
por Kid. Algunos autores aseguran que PemK, corta la secuencia 5´-UA(C/A/U)-
3´preferentemente entre la U y la A y en cadena sencilla (Zhang et al., 2004). Otros
experimentos demostraron que Kid cortaba en el extremo 5´ de la adenosina en la
secuencia 5´-UA(C/A)-3´ también en cadena simple aunque se observó corte en cadena
doble y en el extremo 3´ de la base adenosina (Munoz-Gomez et al., 2005, Diago-Navarro
et al., 2009b), por último se describió que in vivo Kid cortaba la secuencia 5´-UUACU-3´
con alta afinidad, entre la U y la A en el mensajero policistrónico copB-repA del plásmido
R1, disminuyendo los niveles del represor CopB. Esta disminución en los niveles de CopB,
genera un aumento en la tasa RepA/CopB y en el número de copia de R1 (Pimentel et al.,
Figura 4. Mecanismo de muerte inducido
por antibióticos. El mecanismo puede ser
MazEF dependiente cuando se encuentra
como intermediario la molécula señal EDF
(vía de color), o independiente de ella si no
se expresa (vía color gris) . En el caso de que
el mecanismo de muerte esté mediado por
MazEF éste puede ser dependiente de ROS
(muerte celular de toda la población, flechas
color azul) o independiente de ROS (muerte
de una fracción de la población, flechas color
rojo). Adapatado de (Kolodkin-Gal et al.,
2008, Amitai et al., 2009)

P á g i n a | 10
2005). El mecanismo molecular del corte del ARN mensajero mediado por Kid es el mejor
estudiado hasta el momento. Se sabe que Kid al igual que la RNasa T1 es una RNasa que
cicla el producto de degradación. En el caso de Kid, se encontró que usando el substrato
mínimo 5´-AUACA- 3´ y el di-nucleótido UpA, se producía un intermediario de fosfato
cíclico 2´:3´ a un lado y un grupo 5´-OH libre por el otro. A partir de estos estudios, y
utilizando un fragmento de ARN no degradable, se pudo por primera vez crear un modelo
en donde se identificaron los aminoácido esenciales para el corte e interacción de Kid con
el ARN mensajero (Kamphuis et al., 2006)
Recientemente, se aislaron una serie de mutantes en el gen prfA de E. coli, que
mostraban una mayor sensibilidad a la acción de la toxina RelE. El gen prfA codifica el
factor de liberación de clase I o RFI (del inglés- Release Factor I), el cual permite la
liberación del ARN mensajero del ribosoma cuando se llega a un codón de parada. En
bacterias existen dos factores de liberación el RFI y el RFII. Como se mencionó
anteriormente, la toxina RelE interacciona con el ARN mensajero en el sitio A del ribosoma
y competiría con el RFI por el mismo sitio. Un mutante de RFI que es menos eficiente en la
terminación de los ARN mensajeros explica de manera lógica la mayor sensibilidad a la
toxina, ya que en condiciones silvestres RFI podría competir con RelE por el sitio A del
ribosoma. Sin embargo, este mutante también era más sensible frente a Kid. Este
resultado no era esperado ya que Kid degrada ARN de manera independiente del
ribosoma y su estructura terciaria es distinta a la de RelE, no pudiendo interaccionar con el
sitio A del ribosoma. Una posible explicación al fenotipo observado podía ser que una
actividad reducida de RFI genera una pausa del ARN mensajero que se está traduciendo
en el ribosoma cuando se llega al codón de parada. Esta pausa produce un posterior corte
en el ARN mensajero, por lo tanto un aumento en la toxicidad de Kid puede ser debido
simplemente al aumento en el corte de ARN causado por la pausa del ribosoma en
ausencia de un RFI funcional. Estos mecanismos aún no están claros ya que el papel de
RFI en la toxicidad mediada por Kid sigue siendo objeto de estudio (Diago-Navarro et al.,
2009b).
4.4 Sistema HipBA
Las células persistentes se describieron por primera vez en 1944 por Joseph Bigger,
quien notó que existía una población de células de Staphyloccocus que sobrevivían al
tratamiento con penicilina. Estas células, a diferencia de las resistentes, son variantes
fenotípicas de la cepa silvestre que cuando son re inoculadas producen el mismo número
de células persistentes. El número de células persistentes, varía según la fase de
crecimiento, pero puede llegar a alcanzar el 1% de la población en fase estacionaria o en
biofilms (Lewis, 2007). En los años 80s Harris Moyed y colaboradores buscaban
mutaciones de genes en E. coli que produjeran un aumento en el número de células
persistentes a ampicilina. De esta forma, encontraron que células que tenían un alelo
mutante aumentaban hasta 10.000 veces la frecuencia de células sobrevivientes, y por
esto lo llamaron alelo de híper persistencia o hipA7 (del inglés- high persistance). El
número de células persistentes en la cepa hipA7 también se veía aumentado cuando las
células se trataban con agentes que inhibían la replicación del ADN como ácido nalidíxico
o quinolonas y también como respuesta a la falta de timina. (Moyed & Bertrand, 1983,
Moyed & Broderick, 1986, Scherrer & Moyed, 1988). Intentos fallidos por clonar el gen
hipA llevaron a la conclusión de que su producto era tóxico y que solo se podía clonar en
presencia de una secuencia que se encontraba aguas arriba, el gen hipB. Así, se
descubrió que hipA formaba un operón con hipB y que eran parte de un genuino sistema
de TA, en donde HipB, una proteína de unión a ADN del tipo Cro/CI, contrarrestaba los
efectos tóxicos de HipA. Los primeros ensayos realizados en células de E. coli para

P á g i n a | 11
determinar cuál era la diana de HipA demostraron que ésta inhibía la síntesis del ARN y
proteínas, y como efecto secundario, la replicación del ADN. Esta inhibición en la síntesis
de macromoléculas inducía un estado bacteriostático sin producir muerte celular, que
podía ser revertido por la antitoxina HipB. También se demostró que la toxina silvestre era
capaz de aumentar el número de células persistentes (Korch & Hill, 2006, Black et al.,
1994, Black et al., 1991). En cuanto al fenotipo observado con el alelo hipA7, se encontró
que hipA tenía dos mutaciones puntuales en su secuencia: G22S y D291A. Estudios sobre
estos mutantes demostraron que una variante de HipA conteniendo solamente la
mutación G22S generaba una proteína no tóxica sin fenotipo híper persistente. Sin
embargo, una variante conteniendo solamente el cambio D291A rendía una proteína
tóxica, capaz de producir células persistentes. Además, la generación de células
persistentes de E. coli con la mutación hipA7 sólo era posible en presencia de los genes
relA y spoT, indicando que eran necesarios altos niveles de (p)ppGpp para producir dicho
estado (Korch et al., 2003)
Estudios posteriores en células de E. coli, explicaron que las células persistentes eran
células en un estado fisiológico distinto a los descritos hasta el momento, cuyo patrón de
expresión génica podía llegar a indicar que se trataba de células “durmientes”. Análisis de
“microarrys” mostraron que estas células tenían inducidos varios de los sistemas TA de E.
coli (RelBE, MazEF, dinJ/yafQ, etc). Además se demostró que la sobreexpresión de
toxinas producían un aumento en la tolerancia a antibióticos en E. coli (Keren et al., 2004,
Shah et al., 2006). Gracias a estos resultados se propuso que el papel de las toxinas en la
formación de células persistentes sería el de inactivar las dianas de los antibióticos, de
forma tal que estos ya no puedan corromperla (Figura 5). Sin embargo, los sistemas TA no
son los únicos involucrados en la tolerancia a antibióticos, ya que recientemente se ha
descrito que el producto de los genes glpD y plsB, así como también los niveles de
glicerol-3-fosfato estarían involucrados en la formación de células persistentes (Spoering
et al., 2006).
A partir del análisis de la secuencia primaria de HipA se encontró que esta toxina, a
diferencia de las que estaban descritas hasta el momento, presentaba homología con
proteínas quinasas, conteniendo todos los residuos catalíticos de estas, especialmente el
residuo Aspartato 309 (D309). Además, se demostró que HipA era capaz de
autofosforilarse in vitro en presencia de ATP, en el residuo serina 150 (S150). Si el
residuo D309 era remplazado por glutamina (Q), la autofosforilación del residuo S150 no
se producía y se perdía la capacidad de producir células tolerantes, implicando que la
actividad kinasa era esencial para la generación de este fenotipo (Correia et al., 2006).
Recientemente, se publicó que HipA es capaz de interaccionar con el factor de elongación
Figura 5. Modelo de formación de células persistentes. Variaciones estocásticas inducen la expresión de
la toxina, que se une a su diana y no deja que al antibiótico inhiba la proliferación celular. Adaptado de (Keren
et al., 2004)

P á g i n a | 12
Tu o EF-Tu (del inglés- Elongation Factor Tu). EF-Tu es una de las proteínas más
abundantes de E. coli, pertenece a la superfamilia de guanosina trisfosfatasa y juega un
papel relevante en la traducción catalizando la unión del ARNt-aminoacil al ribosoma
(Schumacher et al., 2009). Ensayos in vitro demostraron que HipA era capaz de fosforilar
a EF-Tu, y que ésta fosforilación era estimulada por GDP, bloqueando de esta forma la
unión de EF-Tu al ARNt-aminoacil. Este resultado podría explicar la toxicidad de HipA
aunque no se descartan otras dianas dado que HipA afecta no sólo la traducción, sino
también la transcripción (Schumacher et al., 2009)
5. Sistemas con mecanismos de acción desconocidos
5.1 Sistemas y PezAT
Como se mencionó en la sección 1, la principal función de estabilidad del plásmido
pSM19035 se debe al sistema . Este sistema pertenece a una familia que no presenta
homología (ni de secuencia ni estructural) con ningún otro sistema TA. A partir de la
caracterización bioquímica de los componentes del sistema se sabe que el gen codifica
una antitoxina de 10 KDa (90 aminoácidos) que forma un dímero en solución, mientras
que el de , codifica una toxina de 32 kDa (287 aminoácidos).Las proteínas2 y forman
un heterotetrámero muy estable en solución, evitando así la actividad tóxica de (Figura
12A). Estudios in vivo realizados en el laboratorio revelaron que cuando la replicación,
traducción o transcripción se detienen en células de B. subtilis que contienen un plásmido
con el sistema , se observa una disminución en las unidades formadoras de colonias
(UFC). Esta caída en las UFC coincide con la degradación de 2, con una vida media de
≈18 min, y con la persistencia de cuya vida media supera los 60 min (Camacho et al.,
2002). Ensayos realizados por otro laboratorio en el mismo sistema indicaron que el efecto
de la toxina en bacterias G+ como B. subtilis era bacteriolítico, y que en células G- (E.
coli) este efecto era bacteriostático, produciendo filamentación celular sin inducción del
sistema SOS (Zielenkiewicz & Ceglowski, 2005). Aunque hasta el momento el mecanismo
por el cual la toxina induce la inhibición de la proliferación es desconocido, postulamos
que éste debe estar relacionado con la unión a nucleótido ya que presenta un dominio
de unión a ATP/GTP y en ese sitio se encuentra plegada como algunas quinasas y
fosfotransferasas (Meinhart et al., 2003). El sistema se encuentra ampliamente
distribuido en plásmidos que otorgan resistencia a vancomicina en enterococos o en las
cepas de S. aureus resistentes a metilicina y en otros plásmidos de G+ (Moritz &
Hergenrother, 2007, Sletvold et al., 2008). Sin embargo, también se ha identificado este
sistema en el cromosoma de otras bacterias, entre ellas de S. pneumoniae y E. coli (Khoo
et al., 2007, Van Melderen & Saavedra De Bast, 2009). El sistema cromosomal mejor
caracterizado es el de S. pneumoniae, y se lo conoce como PezAT. A diferencia del
sistema plasmídico, este sistema no está formado por 3 componentes, ya que la
regulación del operón la realiza la antitoxina PezA. Como se ha mencionado
anteriormente, es muy común que la naturaleza de la antitoxina varíe entre sistemas con
toxinas homólogas y el sistema PezAT es el mejor ejemplo. En este caso, la antitoxina
PezA es más grande que (158 aa, ver Figura 6), posee dominios de unión al ADN del
tipo HTH para regular la expresión del operón (ver próxima sección), y solo presenta una
débil homología con en el extremo carboxilo terminal (Khoo et al., 2007). Estudios in
vivo permitieron determinar que al igual que , PezT era capaz de inhibir la proliferación
celular en E. coli, y que esta inhibición era revertida por la presencia de la antitoxina PezA.
P á g i n a | 13
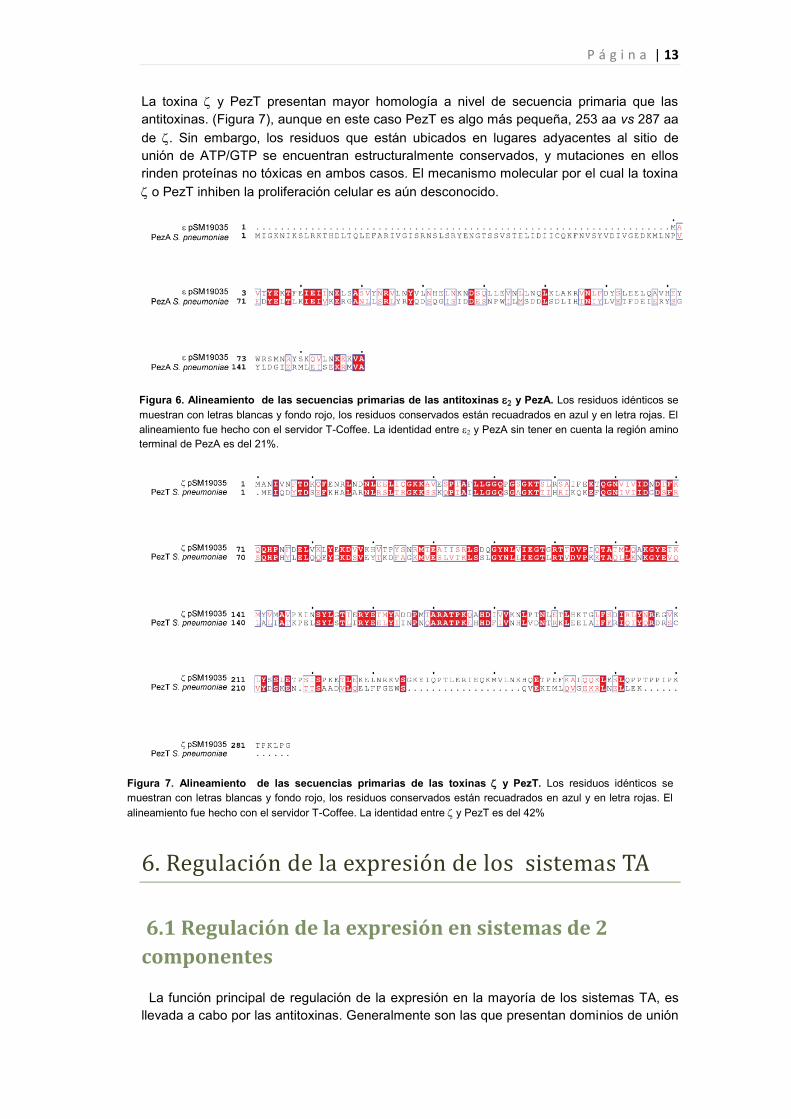
La toxina y PezT presentan mayor homología a nivel de secuencia primaria que las
antitoxinas. (Figura 7), aunque en este caso PezT es algo más pequeña, 253 aa vs 287 aa
de . Sin embargo, los residuos que están ubicados en lugares adyacentes al sitio de
unión de ATP/GTP se encuentran estructuralmente conservados, y mutaciones en ellos
rinden proteínas no tóxicas en ambos casos. El mecanismo molecular por el cual la toxina
o PezT inhiben la proliferación celular es aún desconocido.
6. Regulación de la expresión de los sistemas TA
6.1 Regulación de la expresión en sistemas de 2
componentes
La función principal de regulación de la expresión en la mayoría de los sistemas TA, es
llevada a cabo por las antitoxinas. Generalmente son las que presentan dominios de unión
Figura 6. Alineamiento de las secuencias primarias de las antitoxinas y PezA. Los residuos idénticos se
muestran con letras blancas y fondo rojo, los residuos conservados están recuadrados en azul y en letra rojas. El
alineamiento fue hecho con el servidor T-Coffee. La identidad entre y PezA sin tener en cuenta la región amino
terminal de PezA es del 21%.
Figura 7. Alineamiento de las secuencias primarias de las toxinas y PezT. Los residuos idénticos se
muestran con letras blancas y fondo rojo, los residuos conservados están recuadrados en azul y en letra rojas. El
alineamiento fue hecho con el servidor T-Coffee. La identidad entre y PezT es del 42%

P á g i n a | 14
a ADN, ya sean del tipo HTH ó RHH. Sin embargo, la toxina también actúa como co-
represor transcripcional, de forma tal que cuando se consigue un nivel determinado de
toxina y antitoxina, el complejo inocuo es capaz de auto reprimir su expresión. Por ej., en
el sistema CcdAB, la regulación está mediada por la unión de CcdA2 a través de los
dominios RHH, que se encuentran en el extremo amino terminal, a una secuencia de ADN
palindrómica de 6 pb en la región promotora del operón ccd. La antitoxina CcdA une ADN
sóla ó cuando se encuentra acomplejada con CcdB, pero se demostró que in vivo es
necesaria la presencia del complejo TA para regular la expresión del operón (Tam & Kline,
1989). Esta represión, como también sucede con Kid/Kis, depende de la concentración de
toxina (Monti et al., 2007). El complejo (CcdB2CcdA2)n con una relación CcdB:CcdA=1, se
une al ADN y reprime la transcripción. Sin embargo, si la concentración de CcdB aumenta
y sobrepasa los niveles de CcdA, no se observa unión a la región palindrómica (Madl et
al., 2006).
El sistema RelBE actúa de manera similar a CcdAB, ya que la regulación de la
transcripción del operón está dada por la unión a la secuencia palindrómica de RelB y del
complejo RelBE. En este caso, la antitoxina RelB también se une a la región promotora a
través de su extremo amino terminal, gracias a un dominio RHH y RelE actúa como un co-
represor. (Gotfredsen & Gerdes, 1998, Overgaard et al., 2009). La antitoxina HipB es
capaz de unir cooperativamente las secuencias operadoras del promotor del operón hipBA
y de esta forma regular la expresión del sistema TA (Black, Irwin et al. 1994). Como en los
sistemas TA anteriormente nombrados, el complejo HipA-HipB une las regiones
operadoras con mayor afinidad. Gracias a que la estructura cristalina del complejo HipA-
HipB se obtuvo acomplejada a una secuencia operadora, se pudo ver que HipB forma un
dímero, que interacciona específicamente a través del surco mayor de ADN a través del
motivo HTH, induciendo una curvatura de 70º en el ADN (Schumacher et al., 2009)
PezA es un represor transcripcional de la familia Xre y Cro/CI que reconoce
específicamente una secuencia palindrómica imperfecta de 56 pb que se encuentra aguas
arriba del gen pezA, llamada PS. En presencia de PezT la represión mediada por PezA es
aún mayor, lo que demuestra que al igual que en otros sistemas TA, PezT actúa como un
co-represor transcripcional (Khoo et al., 2007, de la Hoz et al., 2000). A diferencia del
sistema PezAT, no se ha demostrado hasta el momento que el sistema esté
involucrado en la regulación de la expresión, siendo la proteína 2 la encargada de dicha
tarea (Khoo et al., 2007, de la Hoz et al., 2000). La función de la proteína 2 se describirá
a continuación.
6.2 Regulación de la expresión del sistema : función
y características de la proteína 2
La proteína (7,9 KDa), que forma un dímero en solución (2), pertenece a la familia de
proteínas que presentan dominio de unión a ADN del tipo RHH. Al igual que otros
miembros de esta familia, se une al ADN como un dímero a través de dos hojas . La
estructura cristalina de una variante de 2 a la que le faltan los primeros 19 aminoácidos
(219) unida a dos repeticiones en la dirección →→ o en la dirección →← permitió
entender mejor el mecanismo de unión de esta proteína a las secuencias promotoras. A
diferencia de otras proteínas de la misma familia (Arc2, CopG2, y MetJ2), 2 no produce
curvatura en el ADN cuando se une a éste. Además en los surcos mayores del ADN, se
forman interacciones específicas con los aminoácidos treonina 29 y arginina 31
localizados en la hoja Estas uniones permiten un reconocimiento específico y de alta
afinidad del ADN consenso (Weihofen et al., 2006). Las secuencias repetidas que la
proteína 2 reconoce específicamente se encuentran en las regiones promotoras de los

P á g i n a | 15
genes involucrados en el control del número de copia del plásmido (Pcop), en partición (P)
y en el sistema TA (P) (Figura 8A). La unión de 2 a estas secuencias promotoras
reprime la transcripción de los genes a través de un mecanismo desconocido pero que no
es mero impedimento estérico. La proteína 2 reconoce un bloque compuesto por dos
repeticiones directas y una inversa
de 7 pares de bases (5´- A/T ATCAC
A/T- 3´), representado como →→←, que se repite dos
(P) o tres veces (Pcop) más una repetición inversa adicional aguas abajo del último bloque
en el P o una directa aguas abajo del último bloque de Pcop. En cambio, en P hay siete
repeticiones directas y dos invertidas(de la Hoz et al., 2000, de la Hoz et al., 2004) (Figura
8B). Estudios in vivo realizados en B. subtilis utilizando el gen de la -galactosidasa como
reportero de la actividad promotora del P y P demostraron que la proteína 2 era
responsable de la represión transcripcional de todo el operón, mientras que el P es un
promotor muy débil cuya regulación no se ve afectada ni por la presencia del gen ni por
(de la Hoz et al., 2000).
Como se mencionó anteriormente, la proteína 2 forma junto con 2 un sistema de
partición activa. En estos sistemas, la proteína ParB (en este caso 2) reconoce las
regiones centroméricas o parS del ADN y junto con la proteína ParA (2), que actúa como
proteína motora, median la segregación del plásmido (Pratto et al., 2008). En el
pSM19035, se definieron 6 regiones centroméricas, y dos de ellas son el P. Por este
motivo y tratando de encontrar un link entre los mecanismos de estabilidad presentes en el
pSM19035, se estudiará el papel de la proteína 2 en la regulación de la expresión del
sistema TA a través de 2.
Figura 8. Regiones promotoras reconocidas por la proteína 2. En la parte (A) se muestran gráficamente las
posiciones de 3 de las 6 regiones promotoras reconocidas por la proteína 2. En la parte (B) se puede observa en
detalle la las Secuencias promotoras de los genes regulados por 2 en el plásmido pSM19035. Las cajas indican
las regiones -35 y-10 de Pcop, P y P. +1 indica el sitio donde se inicia la transcripción. Las repeticiones de 7 pb y sus orientaciones relativas se muestran con flechas debajo de las secuencias. La denominación de parS1, parS2 y
parS3 hacen referencia a la utilización de estos sitios de unión de 2 como sitios centroméricos del sistema de partición activa del plásmido pSM19035

P á g i n a | 16
7. Estructuras cristalinas de los sistemas TA: análisis y diseño de nuevos antimicrobianos
Como se mencionó anteriormente los sistemas TA se encuentran presentes en plásmidos
aislados de bacterias resistentes a antibióticos. Estos plásmidos presentan varios de los
sistemas TA más representativos. Por esta razón, se podrían buscar inhibidores de la
interacción toxina-antitoxina de forma tal que la toxina quede libre y pueda detener el
crecimiento microbiano. Esto permitirá desarrollar nuevos antimicrobianos que inhibirán el
crecimiento de las bacterias resistentes, y aunque la frecuencia de mutación de la diana o
de la toxina sería muy baja, puede ayudar a la desestabilización del plásmido (ya que se
perderá la presión de selección) (Williams & Hergenrother, 2008). Por este motivo, es
importante estudiar las características estructurales de los complejos TA, de manera que
se pueda conocer el mecanismo de interacción de la antitoxina con la toxina, y se puedan
identificar los aminoácidos esenciales para la estabilidad del complejo. Así se podría
diseñar una molécula capaz de interaccionar con la antitoxina de forma tal que deje libre a
la toxina para que pueda ejecutar su acción. Por esto, a continuación se describirán las
estructuras de los sistemas TA más relevantes para este trabajo
7.1 Estructura cristalina del complejo CcdAB
La estructura cristalina de CcdB fue la primera de todos los sistemas TA en conocerse.
En la Figura 9 se puede ver que tiene una gran homología con Kid y MazF a pesar de que
las dianas son completamente distintas. Al igual que Kid y MazF, CcdB está compuesta en
el extremo amino terminal por 5 láminas antiparalelas y una hélice de 13 residuos en el
extremo carboxilo terminal que están unidas entre sí por 3 hojas anti paralelas. Se cree
que el sitio de unión de CcdA está entre las hojas 3 y 5. La estructura de CcdA se
determinó por medio de resonancia magnética nuclear y carece de similitud con las
antitoxinas Kis y MazE. La antitoxina CcdA pertenece a la familia de proteínas de unión a
ADN por medio de dominios RHH. Sin embargo, al igual que en Kis y MazE, es el
carboxilo terminal de la antitoxina el que interacciona con la toxina, formando un
heterohexámero (CcdB2CcdA2CcdB2) Según los estudios de RMN, el extremo carboxilo
terminal de CcdA carece de estructura cuando no se encuentra acomplejada con CcdB,
explicando su degradación por medio de la proteasa LonA.
La estructura cristalina de CcdB unida a un fragmento de la subunidad A de la ADN
girasa (GirA14) desveló el mecanismo de acción de la toxina. Gracias a esta estructura, se
pudo comprender por qué el mutante R462 era capaz de resistir al efecto tóxico de CcdB.
Este residuo interacciona con los residuos W99, N95 y N92 de CcdB y se encuentra en el
centro de la unión del complejo CcdB-GirA14. A partir del análisis de la estructura de
GirA14 unida a CcdB y GirA14 libre se pudo determinar que cuando CcdB interacciona
con GirA en ausencia de ADN, ésta queda congelada en un estado abierto e incapaz de
unir ADN. Sin embargo, si CcdB interacciona con GirA luego de que se forme el complejo
de cortado (cuando el segmento G del ADN ya ha sido cortado y está unido
covalentemente al complejo GirA-GirB) el segmento T ya no puede pasar a través de la
puerta G debido a que CcdB congela a GirA en la conformación abierta y su presencia
“bloquea” la salida. Así, se forma un complejo ternario Girasa:ADN:CcdB, que genera una
barrera para la replicación del ADN y la transcripción. Este complejo solo puede
desbloquearse por la presencia de CcdA, quien al interaccionar con CcdB, libera a la
girasa para que pueda seguir funcionando (Dao-Thi et al., 2005)

P á g i n a | 17
7.2 Estructura cristalina del complejo RelBE
La primer estructura cristalina de RelBE se obtuvo del sistema RelBE de la arquea
Pyrococcus horikoshii (Figura 10). Se demostró que el sistema RelBE de P. horikoshii
(RelBE) es funcional en E. coli. La estructura de RelBE es diferente a la de MazEF y
Kid/Kis, lo que podría explicar porque la toxina RelE corta el ARN de manera dependiente
del ribosoma, mientras que MazF y Kid lo hacen de manera independiente. La antitoxina
RelB presenta una estructura definida cuando está unida a RelE, pero no parece tener
estructura terciaria cuando está libre, lo que explicaría por qué es rápidamente degrada
por la proteasa LonA. Una vez acomplejada, RelB se une alrededor de la molécula de
RelE. De esta forma, RelB y RelE interaccionan formando un heterotetrámero (RelB-
RelE)2. Se identificó el residuo R85 de RelE como el más importante para la actividad
ribonucleasa de la toxina, así como también se demostró que mutaciones en los residuos
R40, L48, R58 y R65 disminuían la toxicidad de RelE. RelB no interacciona directamente
con los aminoácidos involucrados en la actividad nucleasa de RelE, lo que supone que el
mecanismo de inhibición de la actividad de la toxina mediado por RelB, esta dado por un
cambio en la conformación de la proteína RelE cuando se acompleja con RelB, de forma
tal que no pueda acceder al sitio A del ribosoma para degradar el ARN mensajero.
7.3 Estructura cristalina del complejo Kid-Kis y MazEF
Como se mencionó anteriormente los sistemas Kid/Kis y MazEF presentan una alta
homología estructural. De hecho se demostró que la MazE era capaz de inhibir hasta
cierto punto la toxicidad de Kid (Kamphuis et al., 2007b). La estructura cristalina de MazEF
fue la primera en determinarse, en donde se demostró que el complejo formado por la
toxina y la antitoxina era un heterohexámero, en donde un dímero de la antitoxina
interacciona a través del carboxilo terminal con un dímero de toxina (Figura 9).
Figura 9. Estructuras cristalinas de las toxinas CcdB, Kid y del complejo inactivo MazEF. Se muestran las estructuras de un dímero de CcdB y un dímero de Kid. En el complejo MazEF, los dímeros de toxina MazF están representados de color azul y celeste, mientras que los dímeros de la antitoxina MazE esta representados de color amarillo oscuro y claro.

P á g i n a | 18
El análisis de la estructura cristalina de Kid y de MazF acomplejada con MazE, demostró
que las antitoxinas producen un cambio en la conformación de la toxina cuando se unen a
ellas. Además, el extremo carboxilo de la antitoxina estaría desordenado en ausencia de la
toxina, siendo susceptible de degradación por la proteasa especifica (Kamada et al., 2003,
Kamphuis et al., 2007a). Tiempo después, por medio de RMN se estudió la estructura
terciaria del complejo Kid/Kis, y se observo que los sitios de interacción entre Kis y Kid
eran similares a los de MazF con MazE, confirmando que existía gran similitud estructural
entre estos complejos. Las proteínas Kid y Kis forman distintos complejos dependiendo de
la relación molar en la que se encuentren. Por ej, para relaciones Kid:Kis iguales o
superiores a 1:1, tal y como se encuentra in vivo, se forman varios complejos; desde un
tetrámero Kis2-Kid2 hasta un decámero Kis2-Kid2-Kis2-Kid2-Kis2. Sin embargo, cuando Kid
se encuentra en exceso, como cuando se pierde el plásmido en la célula, la especie más
abundante es el heterohexámero Kid2-Kis2-Kid2 (Kamphuis et al., 2007a). La forma
dimérica de Kid es la activa, ya que necesita residuos que se encuentran en los dos
monómeros para unir una molécula de ARN mensajero. Esto explica por qué el residuo
R85 era importante para la toxicidad, ya que es el encargado de formar un puente salino
con E18 conectando los dos monómeros de Kid y generando el sitio activo de la toxina. El
dímero de Kid posee dos sitios de unión a ARN, aunque sólo es capaz de unir una
molécula de ARN por dímero. Cuando Kis se une a Kid, uno de los sitios de unión de Kid
deja de estar accesible al ARN, y es ocupado por el extremo carboxilo terminal de Kis.
(Kamphuis et al., 2006). Estudios de modelado de Kid unido a un pequeño substrato de
ARN y de mutagénesis dirigida han confirmado que los residuos D75, R73 y H17 son los
residuos catalíticos de Kid, siendo responsables del corte del ARN. También se ha
determinado que el residuo H17 estabiliza el complejo Kid2-ARN, y el corte específico de
secuencia tiene lugar debido a las interacciones de los residuos T46 (altamente
conservado entre otras toxinas del mismo tipo), S47, A55, F57, T69, V71 y R73 con los
nucleótidos UA(A/C). Sin embargo a pesar de la gran homología con MazF, sólo el residuo
catalítico D75 está conservado (en MazF es D76). De manera contraria, y explicando la
diferencia en las dianas celulares, la toxina CcdB carece de muchos de los residuos
involucrados en el corte del ARN. (Kamphuis et al., 2006, Diago-Navarro et al., 2009a).
7.4 Estructura Cristalina de HipBA
Figura 10. Estructura cristalina del complejo
RelBE. En azul y verde se muestran dos
monómeros de RelE, mientras que en naranja y
verde oscuro 2 monómeros de RelB. Adapatado de
(Takagi et al., 2005)

P á g i n a | 19
Recientemente se ha obtenido la estructura cristalina de HipA-ATP y de su forma Apo
(Schumacher et al., 2009), a partir de la cristalización de un mutante de HipA que puede
ser sobre-expresado en ausencia de HipB, el mutante HipAD309Q. También se logró
obtener la estructura cristalina del complejo HipA-HipB unido a la secuencia operadora
que reconoce HipB. La estructura de HipA demuestra que tiene un plegamiento globular,
con 15 láminas y 15 hélices. La proteína HipA se divide en un dominio amino terminal
compuesto por láminas y hélices mientras que el carboxilo terminal sólo tiene
hélices. La zona entre los residuos 185-195 no se encuentra en el cristal, y corresponde
al lazo o loop de activación de las quinasas. La comparación de la estructura HipA con
otras estructuras conocidas demostró que su plegamiento es similar al de la ciclina A
quinasa CDK2, especialmente en el carboxilo terminal donde residen los aminoácidos
catalíticos de la quinasa CDK2. De la comparación de HipA-ATP y apoHipA se pudo ver
que la unión al nucleótido genera una pequeña rotación de 4º tanto del extremo amino
como del extremo carboxilo terminal, pero por homología con otras kinasas, se cree que
una vez unida a su diana se producirá un cambio mayor en la conformación. Dos
moléculas de HipA interaccionan a cada lado del dímero de HipB, a modo de sándwich
(Figura 11). A diferencia de otros sistemas TA como RelBE o Kid/Kis, el extremo amino
terminal de HipA interacciona con un monómero de HipB, mientras que el extremo
carboxilo terminal de HipA interacciona con el otro monómero (Figura 11). De esta forma,
HipB no ocluye el sitio activo de HipA, pero esta interacción la congela en una
conformación abierta, que es inactiva. Si se comparan las estructuras de HipA-ATP y
HipA-HipB, no se visualizan grandes cambios en la conformación de la toxina indicando
que HipA sería capaz de unir ATP aunque esté acomplejada a HipB. (Schumacher et al.,
2009)
7.5 Estructura cristalina del complejo y PezAT
La estructura cristalina del complejo fue la primera estructura que se conoció de un
sistema de TA perteneciente a G+. A partir de ella, se determino que y interaccionan
formando un heterotetrámero, en donde un dímero de se encuentra entre dos moléculas
de (2, Figura 12A). El análisis de la secuencia primaria de indicó que presentaba un
dominio del tipo Walker A, que es típico de proteínas que unen ATP/GTP, sugiriendo que
este dominio podría estar involucrado en la actividad de . La estructura cristalina de en
el complejo 2 demostró que el extremo amino terminal de 2 interaccionaba con ,
ocupando el sitio de unión del nucleótido, y evitando así la entrada del mismo. La
comparación de estructuras terciarias indicó que en el sitio activo la toxina adquiría el
mismo plegamiento que las quinasas de nucleósido monofosfato (quinasas NMP) y que la
Figura 11. Estructura cristalina
del complejo HipBA
interaccionando con ADN. En
amarillo se muestran los
monómeros de HipA, con la
molecula de ATP modelada y en
celeste el dimero de HipB
interaccionando con el ADN (en
rojo). Adaptado de (Schumacher et
al., 2009)

P á g i n a | 20
cloranfenicol fosfotranserasa. Además, mutaciones realizadas en los residuos que forman
parte del dominio de unión a ATP/GTP (K46A, R158A, D68T y R171S) generaron
variantes no tóxicas de sugiriendo que la actividad fosfotransferasa era la responsable
de la toxicidad de (Meinhart et al., 2003).
Años después se obtuvo la estructura cristalina del complejo PezAT, y se confirmó la
gran homología entre este sistema y . El complejo PezAT también es un heterotetrámero
donde un dímero de PezA se encuentra entre dos moléculas de PezT (PezTPezA2PezT,
Figura 12B). A pesar de que PezA2 y 2presentan una homología a nivel de secuencia
solo en el extremo carboxilo terminal, la forma de interaccionar con la toxina se encuentra
conservada. La interacción de PezA2 con PezT es similar a la de 2 con , en donde la
hélice 1 de PezA interacciona con la zona de unión a nucleótido de PezT, al igual que lo
hace 2 con , evitando que el nucleótido entre en el sitio de unión. Sin embargo, es
importante destacar que los aminoácidos involucrados en la interacción toxina-antitoxina
no están conservados, excepto aquellos que juagan un rol importante en la inactivación de
PezT ó . En cuanto a las regiones amino terminales de PezA, no se ha podido determinar
la estructura exacta, pero la densidad de electrones indicó que los dominios de unión a
ADN parecen ser flexibles y no interaccionan ni con PezT ni entre ellas (Khoo et al., 2007).
Figura 12. Comparación de la estructura cristalina de los complejos y PezAT. En (A) se muestra el heterotetrámero
compuesto por dos moléculas de (en rojo y verde) y un dimero de (amarillo). En (B) se muestra el heterotetrámero
compuesto por dos molecluas de PezT (beige y rojo) y un dímero de PezA (celeste)

Objetivos


O b j e t i v o s | 21
Objetivos
El propósito de esta tesis doctoral, fue el estudio del sistema toxina antitoxina () del
plásmido pSM19035 y el análisis detallado de sus componentes, las proteínas 2, y . Para
comprender mejor los mecanismos de estabilidad del plásmido y la función de la toxina se
propusieron los siguientes objetivos:
1. Caracterización de la interrelación entre los sistemas de partición y de muerte post-
segregacional. Se determinará el papel de la proteína 2 en la regulación de la transcripción del
operón .
2. Estudio del efecto tóxico de in vivo
3. Análisis del rol de sobre la actividad de .
4. Caracterización del proceso por el cual la toxina inhibe la proliferación celular
5. Comparación de la inducción de bacterioestasis con la de persistencia a antibióticos
6. Estudio de la tolerancia a la toxina y de la persistencia a antibióticos
7. Caracterización del sistema como una nueva diana antimicrobiana.

O b j e t i v o s | 22

Materiales y
Métodos


P á g i n a | 23
1. Materiales
1.1 Cepas
Las cepas utilizadas y construidas en este trabajo se detallan en la siguiente tabla:
CEPA GENOTIPO UTILIZACIÓN
E. coli
XL1 Blue
recA1 endA1 gyrA96 thi-1
hsdR17 supE44 relA1 lac [F' proAB
lacIqZ∆M15 Tn10 (Tet)] (Stratagene)
Células para construcción y
mantenimiento de plásmidos.
E. coli
CC118 (ara leu) araD lacX74 galE galK phoA thi-1 rpsE spoB argE recA1
Ensayos in vivo para estudiar la
toxicidad de E. coli
BL21(DE3)
pLysS B F- dcm ompT hsdS (rB- mB-)
gal (DE3) [pLysS Cat] (Stratagene)
Sobreproducción de las proteínas:
2, , 219,
-His6, 2D60A-His6
B. subtilis
BG214
trpC2, metB5, amyE, sigB37, xre1,
attSP,
attICEBs1
Cepa silvestre
B. subtilis
BG687
amyE:PxylA trpC2, metB5, sigB37, xre1,
attSP,
attICEBs1
Ensayos de proteomica para
estudiar la toxicidad de
Y83C(cepa control)
B. subtilis
BG689
amyE:PxylA Y83C, trpC2, metB5,
sigB37, xre1, attSP ,
attICEBs1
Ensayos in vivo para estudiar la
toxicidad de Y83C
B. subtilis
BG873
amyE:PxylA, trpC2, sigB37, xre1, attSP,
attICEBs1
Ensayos de proteomica para
estudiar la toxicidad de Y83C
B. subtilis
BG871
amyE:PxylA Y83C, trpC2, sigB37, xre1,
attSP,
attICEBs1
Ensayos in vivo para estudiar la
toxicidad de Y83C(cepa
control)
B. subtilis
BG1125
amyE:PHyperspank trpC2, metB5,
sigB37, xre1, attSP,
attICEBs1
Ensayos in vivo para estudiar la
toxicidad de
B. subtilis
BG1127
amyE:PHyperspank , trpC2, metB5,
sigB37, xre1, attSP,
attICEBs1
Ensayos in vivo para estudiar la
toxicidad de (cepa control)
Tabla 2. Cepas
1.2. Reactivos y materiales
En la tabla 3 se muestran los reactivos y materiales utilizados:
PRODUCTO CASA COMERCIAL
32
P- dATP Amersham Bioscience
Sepharosa G-50, Amersham Pharmacia Biotech
Columna de cromatografía Micro Bio-Spin Bio Rad
DNasa I Boheringer Mannheim
IPTG, rifampicina Calbiochem
SDS, Sulfato amónico ICN
Ascorbato Sódico. Aldrich

P á g i n a | 24
Metanol, Hidrolizado de caseína Fluka
Enzimas de restricción, fragmento klenow de ADN polimerasa I MBI Fermentas
Acetato sódico trihidrato, Ac. bórico, Ac. Clorhídrico, Ac. Fórmico,
Ac. Tricloracético, Alcohol Isoamílico, Azul de Coomassie,
Cloroformo, Cloruro magnésico hexahidratado, Etanol Absoluto,
Imidazol, Isopropanol, Hidróxido de Sodio, Hierro (II) amonio-
sulfato hexahidratado, Dimetilsulfóxido, Isopropanol, L-metionina,
Parafina, Titriplex (EDTA), Tritón-X 100, Urea, 2 nirofenil--
galactopiranósido, PEI
Merck
Filtros de 0.05 µm (tipo VM), 0.45 µm (tipo HAWP), 0.22 µm (tipo
Millex-GS)
Millipore
Glicerol MP Biomedicals
Enzimas de restricción, polinucleótido kinasa (PNK), T4 ligasa New England Biolabs
Acido Acético Glacial, Vaselina filante Panreac
Agarosa, Agar bacteriológico, extracto de levadura Pronadisa
Ni-NTA agarosa Qiagen
FastStart Taq DNA polymerase, RNasa A pancreática, DAPI Roche
Acrilamida, bisacrilamida Serva
Antibioticos, Bromuro de Etidio, Cloranfenicol, Glutaraldehído,
Lisozima, Xylene cianol, Thiourea, DSS,
Sigma
Fosfatasa alcalina (SAP), LB, Tris Ultrapuro UBS
Fosfocelulosa. Whatman
Tabla 3. Materiales y reactivos utilizados
2. Métodos
2.1. Manipulación de células 2.1.1. Obtención de células competentes.
Las células competentes en E. coli se obtuvieron creciéndolas en LB a 37ºC, y cuando
llegan a fase logarímica (DO560=0,4), se centrifugan y se tratan sucesivamente con CaCl2
0,05M, el CaCl2 hace permeable las paredes de las células, de modo que el ADN presente
en el medio, puede entrar en dichas células. Las bacterias competentes se pueden
conservar a –80ºC con glicerol al 15%. Todo el proceso debe desarrollarse a 4ºC. La
eficiencia obtenida suele ser de 106
células transformadas/g de ADN plasmídico
(Hanahan, 1983).
B. subtilis tiene competencia natural bajo determinadas condiciones de desarrollo y
nutricionales. Para obtener células competentes lo que se hace es inocular una colonia en
el medio GM1, se deja crecer durante toda la noche a 30ºC, sin agitación, a la mañana
siguiente se hace un dilución de forma que el cultivo quede a DO560=0,04, se pone a
crecer a 37ºC en agitación y una vez hayan pasado 90 min desde que el cultivo entró en
fase estacionaria, se añade el 15% de glicerol y se congelan rápidamente a -80ºC (Bott &
Wilson, 1968).
Medio GM1: Sbase 1X, glucosa 0,5%, extracto de levadura 0,1%, caseina hidroxilada
0,02%, MgSO4 0,8 mM, D/L triptofano 0.025% y L-Metionina 0,02%.
2.1.2 Transformación Bacteriana.

P á g i n a | 25
La transformación de E. coli se desarrolló siguiendo el método de choque térmico
(Hanahan, 1983). Se mezclan 200 µl de células competentes con 10-100 ng del plásmido
y se incuba a 4 ºC durante 30 min. Después se da un choque térmico a 42 ºC durante 1
min para que los poros de la membrana se abran y el ADN sea capaz de entrar al citosol.
Seguidamente se mantienen las células en hielo durante 2 min para que la membrana se
restablezca. Se plaquea en LB-Agar con el antibiótico seleccionador.
Para transformar B. subtilis se hace una dilución 1:10 de las células competentes en el
medio GM2, se dejan 1h a 37ºC en agitación, en este momento las células se encuentran
en el pico de competencia, se mezclan 200 l de las células con 100-200 ng del ADN y se
dejan 30 min a 37ºC, pasado este tiempo son tranferidas a médio LB-agar con el
antibiótico correspondiente (Bott & Wilson, 1968).
Medio GM2: GM1 más MgSO4 3,3 mM y CaCl2 0,5 mM
2.2. Manipulación del ADN 2.2.1. Purificación y cuantificación del ADN
El ADNcd plasmídico fue purificado por lisis alcalina (Birnboim & Doly, 1979) o bien
utilizando el kit para purificación de ADN de Qiagen.
La concentración del ADN fue cuantificada por absorbancia a 260 nm y su pureza
relacionando la absorbancia a 260 nm y 280 nm.
2.2.2. Plásmidos
En la tabla 4 se detallan los plásmidos utilizados y/o construidos en este trabajo.
NOMBRE DERIVADO DE DESCRIPCIÓN
pT712 Posee el promotor de 10 del fago T7 (GIBCO-
BRL)
pET21b Posee el promotor de 10 del fago T7 e introduce
una secuencia para 6 histidinas en el extremo 3´
del gen (GIBCO-BRL)
pDR111 Vector integrativo para B. subtilis, posee el
promotor Hyperspank controlado por IPTG
(Laboratorio de David Rudner)
pHP13 Vector lanzadera para E. coli y B. subtilis
pLex Posee el promotor Lac controlado por IPTG y un
origen de bajo número de copia en E. coli
(Invitrogen)
pCB30 pUC19 Secuencia promotora del gen orf, en orientación
HindIII-EcoRI. (de la Hoz y col. 2000)
pT712- pT712 Plásmido para sobre producir la proteína (de la
Hoz y col. 2000)
pT712- pT712 Plásmido para sobre producir la proteína
(Pratto et al., 2008)
pCB746 pET21b Plásmido para sobre producir la proteína is6
(Pratto et al., 2008)
pCB755 pET21b Plásmido para sobreproducir la proteína D60Ahis6
(Pratto et al., 2008)

P á g i n a | 26
pCB297 pLEX15A Lleva el gen bajo un promotor inducible por IPTG
(Lioy et al., 2006)
pCB298 pHP13 Lleva el promotor del gen , el gen y el gen
(Lioy et al., 2006)
pBT346 pHP13 Lleva los genes: orf, orf, orf, orf
pCB539 pHP13 Lleva el gen y el gen gfp, bajo el control del
promotor de cloramfenicol (Lioy et al., 2006)
pCB613 pHP13 Lleva el promotor del gen , el gen :Luc y el gen
z:gfp (Lioy et al., 2009)
pCB617 pHP13 Lleva el promotor del gen cloranfenicol y el gen
:gfp (Lioy et al., 2009)
pCB638 pHP13 Lleva el promotor del gen , el gen :Luc y el gen
D18AK46A:gfp (Lioy et al., 2009)
pCB635 pFUS2 Lleva el promotor de arabinosa y el gen
pCB639 pHP13 Lleva el promotor del gen , el gen :Luc y el gen
E22AK46A:gfp (Lioy et al., 2009)
pCB799 pHP13 Lleva el gen bajo el promotor de xilosa (este
trabajo)
Tabla 4. Plásmidos
2.2.3. Marcaje radiactivo de fragmentos de ADN. 2.2.3.1. Marcaje de fragmentos con extremos 5´ protuberantes
Los fragmentos de ADN, con uno de sus extremos 5´ protuberantes, fueron marcados
mediante la incubación del ADN con el fragmento Klenow de la ADN polimerasa I de E.
coli en presencia de 250de dCTP, dGTP, dTTP y [P]dATP, durante 15 min a
temperatura ambiente. El resto de nucleótidos no incorporados se eliminaron de la
muestra filtrándola a través de una columna de Sephadex G-50.
2.2.3.2. Obtención y purificación de los fragmentos de ADN utilizados
para los ensayos de interacción proteína-ADN.
Se digiere el plásmido pCB30 con las enzimas de restricción HindIII y KpnI para
obtener el fragmento de ADN con la región promotora del gen con el extremo 5´
protuberante. La digestión se carga, en un gel de agarosa al 0,8% y la banda de ADN, que
tiene un tamaño de 423 pb, se purifica y posteriormente se marca radiactivamente con
[P]dATP como se describbió en el apartado anterior.
2.2.4. Mutagénesis dirigida.
Para realizar las mutaciones puntuales (D18A y E22A) se utilizó un sistema de doble
amplificación por PCR:
- en la primera PCR se utilizó como molde la secuencia del gen en el que se quería
introducir la mutación puntual. Se hicieron dos PCR, una utilizando como cebadores un
oligonucleótido externo que anilla aguas arriba del gen y otro interno que contiene la
mutación que se desea. Para la segunda PCR se utiliza como cebadores el

P á g i n a | 27
oligonucleótido interno complementario al que tenía la mutación y otro externo que anilla
aguas abajo del gen.
- en la segunda PCR se utiliza como molde los fragmentos de ADN obtenidos con la
primera PCR y como cebadores a los oligonucleótidos externos. De esta manera se
obtuvo un gen que llevaba la mutación puntual.
2.3. Obtención y estudio de proteínas
2.3.1. Sobreproducción de proteínas.
Los genes que codifican las proteínas -His6)2 y (D60A-His6)2 se clonaron
en pT712 o pET21b (GIBCO-BRL) bajo la expresión del promotor 10 del fago T7 para su
sobreproducción. Este promotor es únicamente reconocido por la ARN polimerasa del
fago. De esta manera se pueden sobre-expresar genes y por lo tanto sobre producir las
proteínas codificadas de manera muy efectiva. (Studier y col, 1990).
El hospedador empleado para la transformación de estos plásmidos fue la estirpe E.
coli BL21(DE3)[pLysS]. El bacteriófago DE3, que es un derivado del fago que posee el
gen lacI, el promotor lacUV5, y el gen de la ARN polimerasa de T7 está integrado en el
genoma de BL21. La cepa BL21 es un derivado de E. coli que carece de la proteasa
LonA. En la cepa BL21(DE3) el gen de la ARN polimerasa se expresa a partir del promotor
lacUV5, que es inducible por IPTG (1 mM), que se añade al cultivo cuando este se
encuentra a una DO560=0,6 . Así, mediante la adición de IPTG, que se deja induciendo la
síntesis de ARN polimerasa de T7 durante 20 min y la posterior adición e incubación
durante 2 hr con Rifampicina (200 g/ml), se inhibe la transcripción de la ARN polimerasa
celular, pero no la de T7, de esta manera pueden expresarse de forma eficiente los genes
que se hayan clonado en pT712 o pET21b.
Aquellos genes cuyos productos son tóxicos para la célula no pueden ser expresados
en BL21(DE3), porque el nivel basal de la actividad de la ARN polimerasa puede promover
la transcripción en la célula no inducida. Un modo de reducir este nivel basal es
introduciendo un plásmido codificante del gen 10 de T7 (pLysS). El producto del gen 10 de
T7 secuestra selectivamente a la ARN polimerasa de T7. El plásmido pLysS lleva el gen de
resistencia a cloranfenicol (Studier y col, 1990).
2.3.2. Purificación de proteínas.
2.3.2.1. Purificación de las proteínas
Una vez obtenida masa celular en la cual las distintas proteínas habían sido sobre
producidas según el sistema, ya descrito, del promotor 10 del fago T7, las células fueron
centrifugadas durante 15 min a 9.000 rpm y posteriormente resuspendidas en el tampón A
(fosfato sódico pH 7,5 50 mM, glicerol 10%) con 100 mM de NaCl, en una proporción de 5
ml/g de biomasa húmeda. Una vez obtenida una suspensión homogénea se lisan con la
prensa de French a 1500 psi. Las células lisadas se centrifugaron, a 18000 rpm y a 4º C,
en rotor Sorvall GSA. Todas las variantes de son solubles.
Se midió la absorbancia a 260 nm del sobrenadante de la lisis y a continuación se
añadió PEI hasta una concentración final de 0,25%, cuando DO260=120, y se centrifugó a
13000 rpm, a 40
C, durante 15 min en rotor Sorvall SS-34; las proteínas de interés
permanecen en el sobrenadante.

P á g i n a | 28
Se hizo una precipitación al 50% de sulfato amónico (SA) donde precipitan las
proteínas. Se diluyó el precipitado de SA para obtener una solución con fuerza iónica
correspondiente a 50 mM NaCl. Las proteínas se cargaron en una matriz de fosfocelulosa
previamente activada y equilibrada con tampón A conteniendo 50 mM de NaCl. Las
proteínas y quedaron retenidas en la matriz, que posteriormente se lavó con
concentraciones crecientes de NaCl, en todos los casos las proteínas eluyeron a 200 mM
de NaCl. Una vez eluidas se bajó la concentración de sal hasta 100 mM para cargarlas en
una columna conteniendo la matriz SP-Sepharosa equilibrada a 100 mM de NaCl. Las
proteínas quedan retenidas y se lavó la matriz con concentraciones crecientes de sal. Las
proteínas comenzaron a eluir a 175 mM de NaCl, libres de contaminantes pero muy
diluidas por lo que hubo que pegarlas de nuevo a una matriz de fosfocelulosa, de muy
poco volumen, de nuevo a 50 mM NaCl, para concentrarlas. Una vez cargada toda la
proteína en la segunda columna de fosfocelulosa se eluyó mediante un golpe de sal a 300
mM de NaCl, se añadió glicerol al 50% y se almacenó a -20ºC.
La concentración de cada proteína fue determinada por absorbancia mediante los
coeficientes de absorción de A1%,1cm = 3,63 y 4,8 para y.
(http://www.expasy.ch/tools/protparam.html)
Los diferentes pasos de purificación fueron seguidos por electroforesis en geles de
SDS-PAGE al 17.5%.
2.3.2.2. Purificación de -His6)2 y (D60A-His6)2.
Las proteínas y D60Afueron sobre-expresadas en E. coli BL21(DE3) y para facilitar
su purificación, se decidió añadir a las mismas una cola de 6 histidinas (-His6 y (D60A-
His6). Las células se lisaron en tampón A. Los restos celulares fueron eliminados por
centrifugación. El sobrenadante de lisis se cargó en una columna de Ni-NTA, se lavó a
concentraciones bajas de imidazol y finalmente la proteína fue eluida a imidazol 50 mM. El
eluido de la resina de Ni de se cargó en una columna Q-Sepharosa equilibrada con
tampón A y NaCl 50 mM. Una vez lavada la columna se eluyó la proteína a 200 mM de
NaCl. La proteína pura se dializó frente a fosfato sódico pH 7,5 50 mM, 50% glicerol, NaCl
100 mM y se conservó –20 ºC.
La concentración de cada proteína fue determinada por absorbancia mediante los
coeficientes de absorción de A1%,1 cm= 0,857 y 0,858 para (-His6)2 y (D-His6)2 .
2.4. Ensayos bioquímicos 2.4.1. Medida de interacciones proteína-ADN 2.4.1.1. Ensayos de retraso en gel.
La interacción proteína-ADN fue medida por la variación de la movilidad de un ADN
marcado cuando está libre o cuando forma un complejo con una o varias proteínas. Los
fragmentos de ADN marcados radiactivamente fueron incubados con distintas
concentraciones de las proteínas en tampón B (Tris pH7,0 50mM, MgCl2 10mM, NaCl
50mM) durante 15 min a 37º en 20 µl de volumen final. En el caso de los ensayos con
se añadió ATP o ADP 1 mM.
Las mezclas de reacción con tampón de carga fueron cargadas en geles nd-PAGE del
del 4%. Los geles se corrieron en 1X TAE para los ensayos con , a 4°C, 200 V durante 2
horas y fueron secados y autoradiografiados.

P á g i n a | 29
Para obtener los valores de kd,app de los geles de retrasos y de los ensayos de
protección a DNasa I, las concentraciones del ADN libre y de los complejos ADN-proteína
fueron determinados por densitometría de las autoradigrafías. La concentración de
proteína que transfiere el 50% del [32
P]-ADN en complejos o que protege el 50% del [32
P]-
ADN de la digestión por DNasa I es aproximadamente la constante de disociación (kd,app).
2.4.1.2. Ensayos de protección a DNasa I.
En este caso la interacción ADN-proteína se mide por la capacidad de la proteína que
se una al ADN de proteger la región a la cual se une del ataque de la enzima DNasa I. La
protección se visualiza como una “huella” en el ADN que marca la zona a la que se unió la
proteína de interés.
Las condiciones de reacción para el ensayo de protección a DNasa I fueron las mismas
que para los ensayos de retraso. La concentración de ADN utilizada es de entre 0,5 y 1
nM. Luego de la incubación se añade DNasa I, previamente calibrada. Se considera como
óptima la cantidad suficiente como para que cada molécula de ADN sea cortada por
término medio una vez. Se dejó actuar 5 min a 37ºC. La reacción se paró añadiendo EDTA
hasta una concentración final de 25 mM. El ADN fue precipitado con acetato sódico 0,3 M
y etanol frío. Las muestras fueron resuspendidas en tampón de carga desnaturalizante
[80% (v/v) formamida, 0,1 % (v/v) azul de bromofenol y 0,1% (v/v) xilencianol], separadas
en un gel desnaturalizante de poliacrilamida del 6 o del 15% (dPAGE) y luego
autoradiografiados. Como marcador de tamaño, se obtuvieron patrones utilizando la
reacción química de secuenciación (G+A) con los mismos fragmentos de ADN que se
utilizaron en el experimento.
2.5 Modelado Molecular
Las simulaciones se realizaron, a temperatura y volumen constante. Los datos para la
simulación se tomaron de la estructura cristalina de 22 (PDB:1GVN). Ambas proteínas
fueron preparadas usando el XLEAP modulo para agregar átomos de hidrogeno y cargas
atómicas parciales. Se agregó una caja periódica de moléculas de agua (TIP3P) rodeando
el complejo a una distancia de 10 A de la superficie receptora. El tiempo total de las
simulaciones fue de 1 nanosegundo (ns). Se mantuvo un gradiente de temperatura
constante desde 300K a 400K durante los primeros 250 picosegundos, y luego se
mantuvo la temperatura constante a 400K. Para más detalles consultar (Lioy et al., 2009)
2.6. Ensayos in vivo 2.6.1 Medio de Cultivo:
En todos los casos las células de B. subtilis se crecieron en medio mínimo S7 (MMS7),
que está compuesto por: MOPS 50 mM, (NH4)2SO4 10 mM, Fosfato potásico 5 mM, MgCl2
2 mM, CaCl2 0,7 mM, ZnCl2 1 M, MnCl2 50 M, Fructosa 1%, glutamato 0,1%, D/L
triptofano 0.04 % y L-Metionina 0,04 %. A menos que se indique lo contrario, todos los
ensayos se realizaron a 37ºC.
Las células de E. coli CC118 se crecieron en medio mínimo M9 (MMM9), con glicerol
como fuente de carbono en lugar de glucosa. El MMM9 se preparó con los siguientes
componentes (para 0,5L): Na2HPO4∙7H2O 12,8gr, KH2PO4 3gr, NaCl 0,5gr, NH4Cl 1,0gr.
Luego agregar SO4Mg 2mM, 1% glicerol, CaCl2 0,1mM.

P á g i n a | 30
2.6.2. Microscopías 2.6.2.1 Microscopía de fluorescencia: Visualización de nucleoides y
estado de las membranas
Se utilizaron células de B. subtilis BG689 y BG687 en fase exponencial inoculando cultivos
de toda la noche en MMS7 fresco y creciéndolas luego a 37 ºC hasta una OD560 de 0,2. A
tiempo cero, se agregó xilosa 0,5%, y se tomaron muestras a los tiempos indicados. En
este caso 1 ml de células se fijaron con paraformaldehído al 2%, luego de 10 min a
temperatura ambiente, las células se centrifugaron, lavaron y resuspendieron en tampón
fosfato. Se incubaron 5 min en oscuridad con 4´,6’-diamino-2-fenilindol (DAPI, 0.2g/ml).
Para analizar la integridad de las membranas las células sin fijar fueron teñidas con Ioduro
de Propidio (IP) y SYTO9. SYTO9 es un colorante verde hidrofóbico que penetra en todas
las células, presenten o no defectos en las membranas. IP, es un colorante rojo hidrofílico
que sólo tiñe aquellas células con defectos en las membranas.
2.6.2.1 Microscopía electrónica: Visualización del efecto de la sobre-
expresión de en células de E. coli
En colaboración con el laboratorio del Dr. Rudi Lurz se estudió el efecto de en la
fisiología de E. coli. Para esto se crecieron células conteniendo el sistema en trans II
(pCB635 y pCB297) hasta 1 x 107 células/ml en MMM9 suplementado con 2% glicerol,
0,2% glucosa y 0,05mM IPTG y con los antibióticos correspondientes. Luego el cultivos se
dividió en dos partes iguales y a una de ellas se le agregó arabinosa 0,2% para inducir la
expresión de Se tomaron muestras a los 0, 30 y 60 min post inducción. Para preparar
las muestras de microscopía electrónica las células se fijaron en glutaraldehído, se
embebieron en un medio Spurr de baja viscosidad y se trataron con tetróxido de osmio.
(Lioy et al., 2006)
2.6.3 Estudios de viabilidad en B. subtilis y E. coli
2.6.3.1 Estudio del efecto de la expresión de Y83C y en la viabilidad de
células de B. subtilis
Células de B. subtilis BG687, BG689, BG1125 y BG1127 se crecieron hasta 5x107
células/ml en MMS7, suplementado con los antibióticos correspondientes, y se indujeron
con xilosa 0,5% o IPTG 2 mM según corresponda. Se tomaron muestras a distintos
tiempos y se plaquearon en LB agar para determinar el número de unidades formadoras
de colonias.
En los ensayos para determinar si el efecto de es reversible (células BG1125), se
agregó xilosa a los 30, 60 y 120 min luego de inducir la expresión de y a los tiempos
indicados se plaquearon en placas de LB suplementadas con xilosa 0,5%, mientras que
aquellas células que no habían sido expuestas a la antitoxina se plaquearon en placas de
LB sin suplementar.
2.6.3.2 Identificación de especies reactivas de oxigeno mediadas por en
B. subtilis

P á g i n a | 31
Células de B. subtilis BG1125 se crecieron hasta 5x107 células/ml en medio mínimo S7,
momento en el cual el cultivo se dividió en 4 partes iguales, a una parte se le agregó IPTG
2 mM (inducción de ), a la otra 2,2´-dipiridil 500 mM (DP), a la tercera IPTG 2mM y
500mM DP, y la ultima fracción se dejó como control. A los 120 y 240 min se tomaron
muestras y se plaquearon en placas de LB agar para determinar el número de UFC/ml.
2.6.3.3 Estudio del efecto de la expresión de en células de E. coli
Para estudiar si la toxina afectaba la proliferación células de E. coli, y ver si este efecto
era dependiente de la concentración in vivo de , se utilizaron dos estrategias. En la
primera se utilizaron células de E. coli CC118 conteniendo los plásmidos pCB298 (+) y
pCB297 (+) (sistema en trans I) y en la segunda células de E. coli CC118 conteniendo los
plásmidos pCB635 (+) y pCB297 (+) (sistema en trans II). En el caso de las células
conteniendo el sistema en trans I, se crecieron en MMM9 con los antibióticos
correspondiente, hasta llegar a 5x106 células/ml y en presencia de IPTG 1 mM (para
inducir la expresión de ). En ese momento, los cultivos se centrifugaron y se
resuspendieron en MMM9 precalentado. Luego, estos cultivos se dividieron en dos partes
iguales y a una de ellas se le agregó IPTG 1mM para inducir la expresión de la antitoxina,
mientras que a la otra no, ya que se encuentra bajo un promotor constitutivo. A los 120,
180, 240, 300 y 360 min se tomaron muestras y se agregó IPTG para inducir la expresión
de la antitoxina. Luego las células se plaquearon en LB agar sin suplementar y se
determinaron las UFC/ml. Para estudiar la viabilidad de las células conteniendo el sistema
en trans II, las células fueron crecidas hasta 1 x 107 células/ml en MMM9 suplementado
con 2% glicerol, 0,2% glucosa y 0,05mM IPTG y con los antibióticos correspondientes.
Para inducir la expresión de , se agregó arabinosa al 0,2%. A los 30, 60, 90, 120 y 180
min se agregó 1mM IPTG para inducir la expresión de . Se tomaron muestras a distintos
tiempos y se plaquearon en LB agar con 0,2% glucosa para determinar el número de
unidades formadoras de colonias.
2.6.3.4 Estudio de la toxicidad de :GFP en células de E. coli
Para determinar si la fusión del gen a el gen de la proteína verde fluorescente (gfp) era
funcional, se realizaron una serie de ensayos en E. coli, en donde se comparo la actividad
de la toxina silvestre con la de la toxina fusionada a gfp. Para esto, se crecieron células
de E. coli conteniendo los plásmidos pCB297 (+), pCB298 (+) (sistema en trans I) y
células de E. coli conteniendo el plásmidos pCB617 (:gfp) y pCB297(+) hasta llegar a
fase exponencial en medio LB, en presencia de IPTG 1 mM (para inducir la expresión de
). En ese momento, los cultivos se centrifugaron y se resuspendieron en medio LB
precalentado. Luego, estos cultivos se dividieron en dos partes iguales y a una de ellas se
le agregó IPTG 1mM para inducir la expresión de la antitoxina, mientras que la otra se
siguió creciendo, ya que se encuentra bajo un promotor constitutivo. A los 120 min se
tomaron muestras y se plaquearon en LB agar sin suplementar y se determinaron las
UFC/ml
2.6.3.5 Estudio del efecto de la expresión deY83C y en la generación de
células persistentes en B. subtilis

P á g i n a | 32
Para los ensayos realizados con células de B. subtilis BG687, BG689, BG1125 en fase
exponencial (5x107 células/ml), las células se crecieron en MMS7. Cuando se obtuvo la
DO deseada, los cultivos se dividieron en varias partes iguales, y se indujeron con xilosa
0,5% o IPTG 2 mM según corresponda. Al mismo tiempo, se agregaron los siguientes
antibióticos: Amp (3g/ml), Cip (4g/ml), Eri (20g/ml, solo BG689 y BG687) y Tri (3
g/ml). A los 120 y 240 min se tomaron muestras y se plaquearon en LB agar o en LB
agar suplentado con xilosa 0,5% (solo con la cepa BG1125 y, para permitir la expresión de
) y se determinaron el número de unidades formadoras de colonias luego de incubar las
placas durante 16 hs a 37ºC.
Para los ensayos realizados en fase estacionaria (1x109 células/ml), las células se
crecieron en MMS7 a 37ºC y cuando llegaron a la DO deseada las células se centrifugaron
y resuspendieron en el mismo volumen de MMS7 fresco pre-calentado a 37ºC. Luego, se
procedió de la misma manera que se describió en el párrafo anterior.
2.6.4 Estudio de la inhibición de la síntesis de ARN, ADN y
proteínas luego de la inducción de en B. subtilis
Células de B. subtilis BG689 y BG687 fueron crecidas a 37 ºC en MMS7 hasta una DO
de 0,2 (1x107 células/ml). Se agregó xilosa 0,5% para inducir la expresión de . Se
tomaron muestras de 1 ml en los puntos de tiempo indicados a las cuales se les agregó
2,5 Ci de [6-3H] timidina (síntesis de ADN), 2,5 Ci L-[4,5-
3H] leucina (síntesis de
proteínas) y 2,5 Ci [5-3H] uridina (síntesis de ARN). Luego de 1 min de incorporación, las
muestras fueron seguidas por 2-3 min con 10 mg/ml de timidina, uridina o leucina fría. Las
muestras fueron incubadas 2 min con 1mg/ml de lisozima y luego se agregó TCA con una
concentración final de 20% y se incubaron en hielo por 60 min. Tras la incubación, las
muestras se centrifugaron a 20.000 g por 30 min a 4 ºC. Los precipitados fueron lavados
con 200 l de TCA 20 frío y con 200 l de etanol 96%. Finalmente los precipitados fueron
transferidos a un vial y se cuantificó la radiactividad en un contador de líquido de centelleo.
2.6.5 Geles en 2D, análisis de imágenes y identificación de
proteínas
Las cepas BG873 y BG871 se crecieron medio BM (Stulke et al., 1993) hasta 1x108
células/ml. Las proteínas se marcaron con 10 Ci/ml de L-[35
S] metionina durante 5 min
antes (control) y a distintos tiempos (10, 30 y 60 min) luego de agregar xilosa 0,5%. La
incorporación de L-[35
S] metionina se detuvo con la adición de 1mg/ml de cloranfenicol y
un exceso de L-metionina fría (10 mM). Las células se lisaron y se separó la fracción
soluble de proteínas de los restos celulares a través de centrifugación. La metionina
incorporada se cuantifico por la precipitación de las muestras con 10% TCA en filtros de
papel como se describió previamente en (Bernhardt et al., 1999). Se cuantificó la
concentración total de proteínas por Bradford y 80 g de proteínas marcadas con L-[35
S]
metionina se cargaron en un gel y se separaron por electroforesis en gel de poliacrilamida
en 2 dimensiones (2D-PAGE), en las condiciones que se describe en (Lioy et al., 2006).
Para identificar las proteínas, se realizó el mismo ensayo pero sin incorporar radiactividad.
Los spots se cortaron del gel y se identificaron por espectroscopia de masas.
2.6.6 Cuantificación del pool de purinas in vivo.

P á g i n a | 33
Para estudiar el efecto de Y83C sobre el pool de purinas in vivo, se crecieron células de
BG687 y BG689 en MMS7 pero con 1mM de tampón Fosfato Potásico, y en presencia de
50Ci de 32
Pi. Las células se crecieron hasta DO 0,2, momento en el cual los cultivos se
dividieron en dos partes iguales y a una de ellas se le agregó xilosa 0,5% para inducir la
expresión de Y83C. A los tiempos indicados se tomaron 0,2 ml de células y se incubaron
por 30 min en hielo con 0,04 ml de ácido fórmico 2M, según (Wang et al., 2007). Luego
las muestras se centrifugaron durante 15 min a 4ºC y se cargaron en una cromatografía en
capa delgada (CCD) para separar ATP y GTP. La CCD se corrió en K2HPO4 0,85M. Luego
las placas se expusieron con una pantalla Phosphoimager y la radioactividad incorporada
se cuantificó por medio del software Quantity One de la casa comercial Bio-Rad
2.6.7 Análisis del transcriptoma de B. subtilis en presencia de
Y83C.
En colaboración con el laboratorio del Dr. J. Eduardo González-Pastor, se realizó el
análisis del transcriptoma de B. subtilis en presencia de Y83C. Para esto a cultivos en
fase exponencial de BG687 y BG689 se les agregó xilosa 0,5% para inducir la expresión
de la toxina y se tomaron muestras de 50ml a los tiempos 0, 5 y 15 min. Las muestras se
centrifugaron y se procedió a la extracción de ARN total según protocolos establecidos. El
ARN se transformó en ADNc con la superScrict III transcriptasa en reverso, usando
hexámeros random como cebadores, incluyendo amino-acil y amino-hexil nucleótidos
modificados en la muestra. Después de purificar el ADNc, los fluoróforos Cy3 y Cy5
(Amersahm Biosciences) se acoplaron únicamente a la banda de ADNc modificada en el
amino terminal. La eficiencia de marcaje se determinó utilizando un espectrofotómetro
NanoDrop ND1000 (NanoDrop Technologies. Wilmintong, DE. EE.UU.). Anteriormente a la
hibridación, los cristales del microarray (Eurogentec. Lieja, Bélgica) se bloquearon durante
45 minutos a 42ºC por inmersión en SSC 5x, SDS 0.1% (p/v), 1% (p/v) albúmina sérica
bovina. Los cristales fueron lavados brevemente en agua a temperatura ambiente, seguido
de un nuevo lavado en isopropanol, antes de que se secaran. Se mezclaron cantidades
iguales de cDNAs marcados con Cy3 o Cy5 (alrededor de 300 pM de cada uno) y se
secaron en Speed-Vac. Las muestras se disolvieron en 35 ml de formamida desionizada
50% (v/v), solución Denhardt’s 5x, SSC 6x, SDS 0.5% (p/v), dextransulfato 5% (p/v), pre-
filtrado y precalentado a 42ºC. Después de 2 minutos a 90ºC para desnaturalizar el cDNA,
se aplicó la solución a los cristales y se les cubrió con un cristal cobertor de 24 x 60 mm.
Los cristales se introdujeron en una cámara de hibridación y se incubaron a 42ºC durante
18 horas, evitando su exposición a la luz. Luego, tras quitar el cobertor, los cristales se
realizaron dos lavados de 5 minutos con agitación suave en SSPE 0.5x SDS 0.5% (p/v)
pre-calentado a 37ºC (SSPE 1x se compone 150 mM NaCl, 1 mM EDTA, 11.5 mM
NaH2PO4, PH 7.4). Finalmente se lavó con 0.1x SSPE a temperatura ambiente. Se dejaron
secar los cristales y se escanearon en un escáner de microarrays con láseres de 543 y
633 nm. Se tomaron imágenes a una resolución de 10 mm. La intensidad de los spots se
determinó con el programa informático Genepix Pro 5.0 (Axon. Palo Alto, CA., EE.UU.).
Los datos de intensidades se trataron estadísticamente mediante el programa LIMMA.
Para visualizar los resultados obtenidos por LIMMA se utilizó el programa FIESTA viewer.
Los ensayos para determinar qué genes eran modificados por la toxina se realizaron por
triplicado (replicas biológicas) y se consideraron aquellos genes cuyo valor P era ≤ 0,2.
2.6.8 Ensayo de BRET en células de E. coli

P á g i n a | 34
El ensayo de BRET se realizó in vivo en células de E. coli transformadas con los
distintos plásmidos descritos en la tabla 4. Las células transformadas se crecieron hasta
alcanzar la fase exponencial, a 37ºC y en LB, con los antibióticos correspondientes. La
señal de BRET se midió 10 min luego de adicionar el substrato (tiempo necesario para que
el substrato entre en las células). Los filtros de emisión utilizados para medir la señal
fueron 410 y 515 nm. La intensidad de ambas longitudes de onda se grabaron
automáticamente y la razón 515/410 (señal de BRET) se calculó como medida de BRET
(Lioy et al., 2009).

Resultados


P á g i n a | 35
Parte I: Regulación del sistema de TA papel de la proteína en la modulación del represor transcripcional
El sistema de es el único sistema TA que está formado por un tercer componente, la
proteína 2. La proteína 2 es un represor transcripcional que se une a una región aguas arriba
del promotor (P) del operón . La diana de 2 en este promotor está compuesta por 7
copias de la repetición 5´-A/TATCAC
A/T-3´ (iterones), en orientación directa o inversa. Los 7
iterones están compuestos de 2 repeticiones en orientación directa y una en orientación inversa
y ese motivo está presente dos veces, más un iterón en orientación inversa en el promotor P
(Figura 8).
Como se detalló en la sección 6.2 de introducción, las proteínas 2 o no parecen estar
involucradas en la modulación de la utilización de P, sino que es la proteína 2 la encargada
de dicha tarea. La proteína 2 además de encargarse de regular la expresión del operón, actúa
junto con la proteína 2 otorgando estabilidad al plásmido, ya que juntas componen un sistema
de partición activa. Se demostró que la proteína 2 podía modular la actividad ATPasa de 2,
actividad necesaria para la correcta partición del plásmido (Pratto et al., 2008). Por este motivo,
nos preguntamos si hay una interrelación entre los sistemas de partición y los de muerte post-
segregacional y si 2, el motor molecular del sistema de partición, era capaz de modular la
actividad represora de 2, de forma tal que aumente o disminuya la represión del sistema TA.
De esta manera, para estudiar cuál es el efecto de la proteína en la regulación
transcripcional del operón formado por los genes , y , se purificaron las proteínas y ,
así como también las variantes D60A y 19, y se analizó in vitro el efecto de y su
variante en el cargado de y 19 en la región promotora.
1.1 La proteína 2 forma complejos específicos y 2 y
2D60A incrementan la formación del complejo 2·DNA
Para estudiar el efecto de la proteína 2 en la regulación del operón se realizaron
ensayos de retraso en gel de las proteínas 2, 2 y sus variantes, con el ADN conteniendo la
región P (Figura 13). Como se muestra en la Figura 13A la proteína 2 unida a ATP
(·ATP·Mg2+
)2 se unió al ADN de manera inespecífica y con baja afinidad (Kdap 140 nM, calles
5-7), pero cuando se introduce una mutación en el dominio Walker A’ (D60A·ATP·Mg2+
)2,
dando lugar a una proteína que une ATP, pero no lo hidroliza, se incrementó la afinidad
aparente 4 veces (Kdapp 40 nM calles 8-10). Este aparente incremento de afinidad, se debe a
que se aumenta el tiempo de des-ensamblado del complejo proteína·DNA (off rate), es decir, la
hidrólisis de ATP promueve el desensamblado de 2 del ADN y al no haber hidrólisis, el
desensamblado es más lento ((Pratto et al., 2008) y resultados sin publicar de Soberón N.)
Por el contrario, 2 se une específicamente y con alta afinidad (Figura 13A, 9 nM calles 2-4 y
C1) a la región de ADN que contiene los iterones y la secuencia promotora. A concentraciones
limitantes de 2 (3 nM) y en presencia de concentraciones crecientes de las proteínas
(·ATP·Mg2+
)2 o (D60A·ATP·Mg2+
)2, se observó un aumento de la afinidad de 2 de más de 3
veces por la región promotora. También se formaron complejos de alto peso molecular (C2 y
C3). Tanto (·ATP·Mg2+
)2 como (D60A·ATP·Mg2+
)2 estimularon el cargado de 2 en su diana
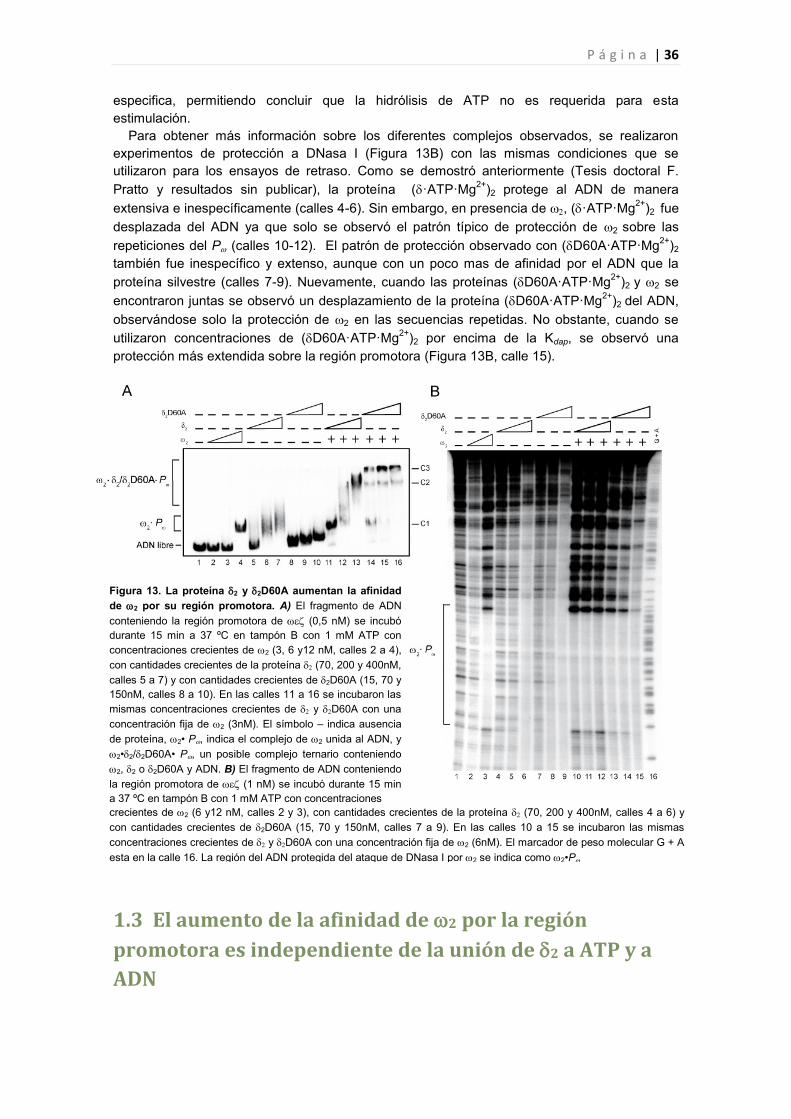
P á g i n a | 36
especifica, permitiendo concluir que la hidrólisis de ATP no es requerida para esta
estimulación.
Para obtener más información sobre los diferentes complejos observados, se realizaron
experimentos de protección a DNasa I (Figura 13B) con las mismas condiciones que se
utilizaron para los ensayos de retraso. Como se demostró anteriormente (Tesis doctoral F.
Pratto y resultados sin publicar), la proteína (·ATP·Mg2+
)2 protege al ADN de manera
extensiva e inespecíficamente (calles 4-6). Sin embargo, en presencia de , (·ATP·Mg2+
)2 fue
desplazada del ADN ya que solo se observó el patrón típico de protección de 2 sobre las
repeticiones del P (calles 10-12). El patrón de protección observado con (D60A·ATP·Mg2+
)2
también fue inespecífico y extenso, aunque con un poco mas de afinidad por el ADN que la
proteína silvestre (calles 7-9). Nuevamente, cuando las proteínas (D60A·ATP·Mg2+
)2 y 2 se
encontraron juntas se observó un desplazamiento de la proteína (D60A·ATP·Mg2+
)2 del ADN,
observándose solo la protección de 2 en las secuencias repetidas. No obstante, cuando se
utilizaron concentraciones de (D60A·ATP·Mg2+
)2 por encima de la Kdap, se observó una
protección más extendida sobre la región promotora (Figura 13B, calle 15).
1.3 El aumento de la afinidad de 2 por la región
promotora es independiente de la unión de 2 a ATP y a
ADN
crecientes de 2 (6 y12 nM, calles 2 y 3), con cantidades crecientes de la proteína (70, 200 y 400nM, calles 4 a 6) y
con cantidades crecientes de 2D60A (15, 70 y 150nM, calles 7 a 9). En las calles 10 a 15 se incubaron las mismas
concentraciones crecientes de y D60A con una concentración fija de 2 (6nM). El marcador de peso molecular G + A
esta en la calle 16. La región del ADN protegida del ataque de DNasa I por 2 se indica como 2•P
Figura 13. La proteína 2 y 2D60A aumentan la afinidad
de 2 por su región promotora. A) El fragmento de ADN
conteniendo la región promotora de (0,5 nM) se incubó
durante 15 min a 37 ºC en tampón B con 1 mM ATP con
concentraciones crecientes de 2 (3, 6 y12 nM, calles 2 a 4),
con cantidades crecientes de la proteína (70, 200 y 400nM,
calles 5 a 7) y con cantidades crecientes de 2D60A (15, 70 y
150nM, calles 8 a 10). En las calles 11 a 16 se incubaron las
mismas concentraciones crecientes de y D60A con una
concentración fija de 2 (3nM). El símbolo – indica ausencia
de proteína, 2• P, indica el complejo de 2 unida al ADN, y
2•2/2D60A• P, un posible complejo ternario conteniendo
2, 2 o 2D60A y ADN. B) El fragmento de ADN conteniendo
la región promotora de (1 nM) se incubó durante 15 min
a 37 ºC en tampón B con 1 mM ATP con concentraciones

P á g i n a | 37
Para que el sistema 2∙2 de partición activa sea funcional, es esencial que la proteína 2
pueda unir e hidrolizar ATP. Por este motivo, se estudió si el aumento producido por 2 en la
afinidad de 2 por la región promotora del operón era también dependiente de la unión a
ATP, y para esto se realizó un ensayo de retraso en gel en ausencia de nucleótido. Como
muestra la Figura 14A (calles 6-8) en estas condiciones 2 no fue capaz de formar un complejo
que migre distinto al ADN libre, así como tampoco pudo hacerlo la proteína 2D60A (calles 9-
11). Sin embargo, cuando concentraciones de 2 que no eran capaces de formar un complejo
estable con el ADN (1,5 nM; calles 11-17) fueron incubadas en presencia de 2 o su variante
2D60A, se observó un aumento de más de 4 veces de afinidad por el ADN conteniendo la
región P. El aumento de afinidad de 2 por su región promotora, podría estar dado por una
interacción proteína-proteína en solución. Para confirmar esta interacción, se realizó el mismo
ensayo pero con una variante de 2 que carece de los primeros 19 residuos (219). El
extremo amino terminal de 2 es necesario para la interacción con 2, pero no lo es para ejercer
su actividad como represor de la transcripción del operón (Pratto et al., 2008, Welfle et al.,
2005). Como muestra la figura 14B, la afinidad de 219 por el ADN específico fue la misma
que su variante silvestre (Figura 14B calles 2-5). Sin embargo, no se observó un aumento de la
afinidad de la proteína 219 por la región P en presencia de 2 o 2D60A (calles 6 y 7).
Estos resultados demostraron que tanto 2 como su variante 2D60A son capaces de
aumentar la afinidad de 2 por la región promotora (independientemente de la unión a ADN y
ATP de 2), haciendo que 2 reprima la expresión de y . De esta forma, los niveles de toxina
y antitoxina se encontrarían perfectamente regulados, y esta regulación sería modulada por la
proteína ParA del sistema de partición activa. El sistema de partición activa no solo minimizaría
el coste de tener un sistema de TA (Brendler et al., 2004), sino que también aseguraría la
correcta regulación de la expresión del mismo.
Figura 14. La proteína 2 aumenta la afinidad de 2 por la región promotora independientemente de su unión a
ATP. A) El fragmento de ADN conteniendo la región promotora de (0,5 nM) se incubó durante 15 min a 37 ºC en
tampón B con concentraciones crecientes de 2 (1.5, 3, 6 y12 nM, calles 2 a 5), con cantidades crecientes de la proteína
(25, 50 y 100nM, calles 6 a 8) y con cantidades crecientes de 2D60A (25, 50 y 100nM, calles 9 a 11). En las calles 12
a 16 se incubaron las mismas concentraciones crecientes de y D60A con una concentración fija de 2 (1.5 nM). B) El
fragmento de ADN conteniendo la región promotora de (0,5 nM) se incubó durante 15 min a 37 ºC en tampón B con
concentraciones crecientes de 219 (1.5, 3, 6 y12 nM, calles 2 a 5). En las calles 6 y 7 se incubaron las proteínas
(100nM) y D60A (100nM) con 1,5 nM de 219.
El símbolo – indica ausencia de proteína, 2• P y 219• P indican el complejo de 2 o y 219 unida al ADN.

P á g i n a | 38
Parte II. Sistema TA: El papel de en el sistema
2.1. Sistemas Toxina Antitoxina: Antecedentes del tema
Actualmente la función de los sistemas de TA no es clara. Se han descrito una gran variedad
de funciones en la que estos sistemas juegan un papel relevante. Sin embargo, la hipótesis
más aceptada hasta el momento es que son elementos de respuesta a estrés capaces de
inducir en las bacterias algunos de los siguientes fenómenos:
1- Muerte celular programada
2- Bacterioestásis o células persistentes.
3- Senescencia celular
Nuestro objetivo fue analizar esas hipótesis y realizar experimentos para determinar cuál es
el rol más probable del sistema en B. subtilis.
2.2 Caracterización del efecto tóxico de la proteína en B.
subtilis
2.2.1 La expresión de la toxina Y83C inhibe la proliferación
celular
En ausencia de la antitoxina , la toxina no puede ser clonada de manera estable dando
lugar a reordenamientos genéticos o mutaciones. Una de las mutaciones aisladas de esta
manera, en donde la tirosina 83 fue reemplazada por una cisteína (Y83C), dió lugar a una
variante que todavía mantenía su actividad tóxica (Lioy et al., 2006), y por ello fue analizada en
más detalle. El gen Y83C fue clonado bajo un promotor débil e integrado como copia única en
el locus amyE del cromosoma de B. subtilis dando como resultado la cepa BG689. Como
control de todos los experimentos se construyó la cepa BG687, en donde una copia vacía del
casete de expresión bajo el promotor de xilosa fue integrada en el mismo sitio por
recombinación homóloga.
Para determinar la cinética de acción de Y83C se crecieron células de BG689 y BG687
hasta DO ≈ 0,2 a las cuales se les agregó xilosa 0.5% para inducir la expresión de la toxina.
Como se puede ver en la Figura 15 la expresión de Y83C fue capaz de reducir más de
10,000 veces la formación de colonias. Esta inhibición es constante en el tiempo, desde los 15
min de inducción hasta los 120 min. Sin embargo, si las diluciones se realizan en medio
conteniendo glucosa 2%, que actúa como represor del promotor de xilosa, se observa durante
los primeros 5 min una disminución de 50 veces en lugar de 100 en el efecto tóxico de Y83C
aunque a tiempos más tardíos ese efecto no es notable (línea punteada de la Figura 15)(Lioy et
al., 2006). Como muestra la Figura 15, en los cultivos control (BG687) inducidos con xilosa no
se produjo ningún efecto sobre las unidades formadoras de colonias, demostrando que la
inhibición del crecimiento está dada por el efecto de la toxina Y83C.
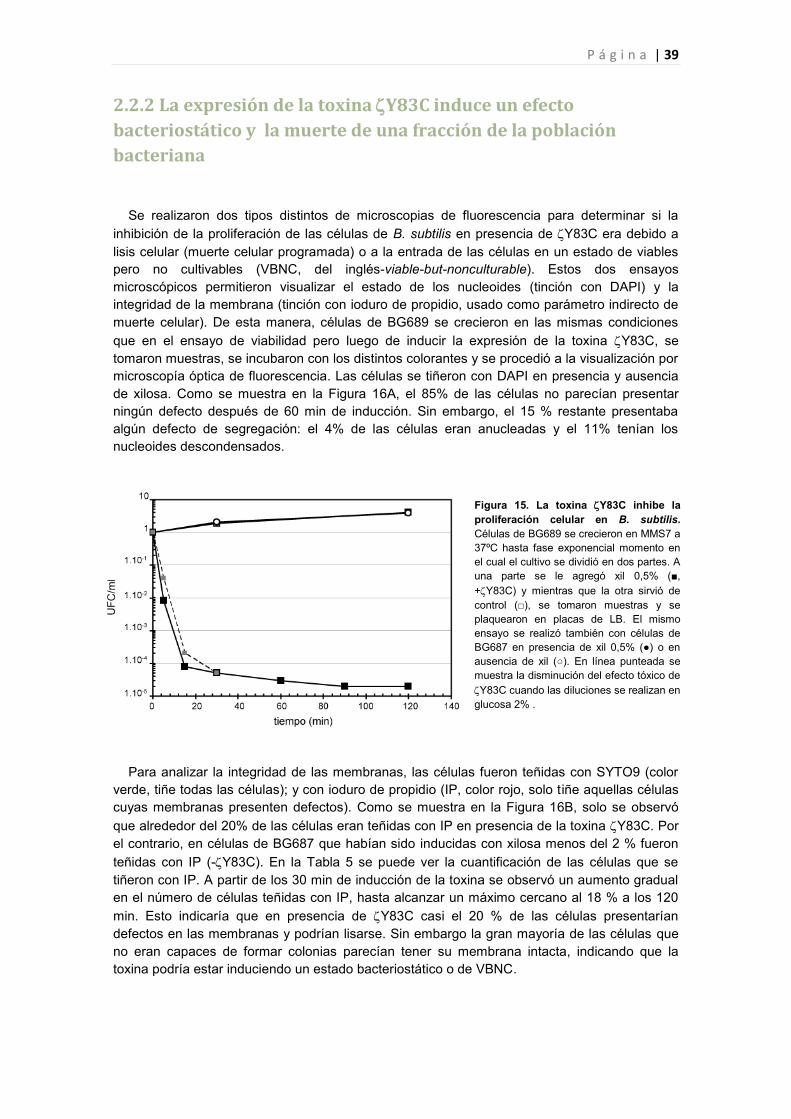
P á g i n a | 39
2.2.2 La expresión de la toxina Y83C induce un efecto
bacteriostático y la muerte de una fracción de la población
bacteriana
Se realizaron dos tipos distintos de microscopias de fluorescencia para determinar si la
inhibición de la proliferación de las células de B. subtilis en presencia de Y83C era debido a
lisis celular (muerte celular programada) o a la entrada de las células en un estado de viables
pero no cultivables (VBNC, del inglés-viable-but-nonculturable). Estos dos ensayos
microscópicos permitieron visualizar el estado de los nucleoides (tinción con DAPI) y la
integridad de la membrana (tinción con ioduro de propidio, usado como parámetro indirecto de
muerte celular). De esta manera, células de BG689 se crecieron en las mismas condiciones
que en el ensayo de viabilidad pero luego de inducir la expresión de la toxina Y83C, se
tomaron muestras, se incubaron con los distintos colorantes y se procedió a la visualización por
microscopía óptica de fluorescencia. Las células se tiñeron con DAPI en presencia y ausencia
de xilosa. Como se muestra en la Figura 16A, el 85% de las células no parecían presentar
ningún defecto después de 60 min de inducción. Sin embargo, el 15 % restante presentaba
algún defecto de segregación: el 4% de las células eran anucleadas y el 11% tenían los
nucleoides descondensados.
Para analizar la integridad de las membranas, las células fueron teñidas con SYTO9 (color
verde, tiñe todas las células); y con ioduro de propidio (IP, color rojo, solo tiñe aquellas células
cuyas membranas presenten defectos). Como se muestra en la Figura 16B, solo se observó
que alrededor del 20% de las células eran teñidas con IP en presencia de la toxina Y83C. Por
el contrario, en células de BG687 que habían sido inducidas con xilosa menos del 2 % fueron
teñidas con IP (-Y83C). En la Tabla 5 se puede ver la cuantificación de las células que se
tiñeron con IP. A partir de los 30 min de inducción de la toxina se observó un aumento gradual
en el número de células teñidas con IP, hasta alcanzar un máximo cercano al 18 % a los 120
min. Esto indicaría que en presencia de Y83C casi el 20 % de las células presentarían
defectos en las membranas y podrían lisarse. Sin embargo la gran mayoría de las células que
no eran capaces de formar colonias parecían tener su membrana intacta, indicando que la
toxina podría estar induciendo un estado bacteriostático o de VBNC.
Figura 15. La toxina Y83C inhibe la
proliferación celular en B. subtilis.
Células de BG689 se crecieron en MMS7 a
37ºC hasta fase exponencial momento en
el cual el cultivo se dividió en dos partes. A
una parte se le agregó xil 0,5% (■,
+Y83C) y mientras que la otra sirvió de
control (□), se tomaron muestras y se
plaquearon en placas de LB. El mismo
ensayo se realizó también con células de
BG687 en presencia de xil 0,5% (●) o en
ausencia de xil (○). En línea punteada se
muestra la disminución del efecto tóxico de
Y83C cuando las diluciones se realizan en
glucosa 2% .

P á g i n a | 40
Tiempo (min)
Porcentaje de células teñidas con IP
+ Y83C - Y83C
0 1,20 1,00
30 7,85 ND 60 16,50 1,40 90 19,00 ND
120 17,75 1,10
2.2.3 La expresión de no induce especies reactivas de oxigeno
La generación de especies reactivas de oxigeno (ROS) produce daños en el ADN y
proteínas (proceso conocido como senescencia), de forma tal que deterioran al
microorganismo dejándolo en un estado de arresto o llevándolo a la lisis celular (Dwyer et al.,
2007, Nystrom, 2003). Recientemente se ha implicado a los sistemas de TA en la producción
de ROS (Kolodkin-Gal et al., 2008, Amitai et al., 2009). Para determinar si la inhibición de la
proliferación causada por era debido a la producción de ROS se llevó a cabo un experimento
en presencia de un agente secuestrador del hierro, el 2,2´- dipiridil (DP). El DP secuestraria al
hierro intracelular y de esta forma se inhibiría la reacción de Fenton (Figura 17) y su
subsecuente producción de radicales hidroxilo que dañarían el ADN y las proteínas (Kohanski
et al., 2007). Para corroborar esta hipótesis, se utilizó una cepa de B subtilis en donde se clonó
la toxina silvestre bajo el control de un promotor inducible por IPTG y se la integró como copia
única, por recombinación homologa, en el locus amyE (BG1125). Como se describió
anteriormente, la toxina silvestre no es capaz de clonarse en ausencia de su antídoto, por lo
que también se clonó el gen de la antitoxina desde un promotor regulable por xilosa, en un
plásmido de bajo número de copia (pCB799). En este sistema la toxina silvestre inhibe la
proliferación celular de la misma manera que lo hace la variante Y83C (Figura 18 y 23). Esta
Figura 16. Efecto de Y83C en la fisiología de B. subtilis. Células de BG689 se crecieron en MMS7 a 37ºC hasta fase
exponencial, momento en el cual el cultivo se dividió en dos partes y a una de ellas se le agregó xil 0,5%. A tiempo 0 min
y 60 min se tomaron muestras y se tiñeron con DAPI, para estudiar la apariencia de los nucleoides (las flechas muestran
células con defectos) (A) o con IP y Syto 9 para analizar el estado de la membrana (B)
Tabla 5. Porcentaje de células teñidas con IP. Los
porcentajes se obtuvieron por citometría de flujo.(ND: no
determinado)
Fe2+
+ H2O2 → Fe3+
+ OH· + OH−
Fe3+
+ H2O2 → Fe2+
+ OOH· + H+
Figura17. Reacción de Fenton. En esta reacción el ión de hierro media la reducción del peróxido de hidrogeno dando
como productos especies reactivas de oxigeno (OH∙ y OOH∙)
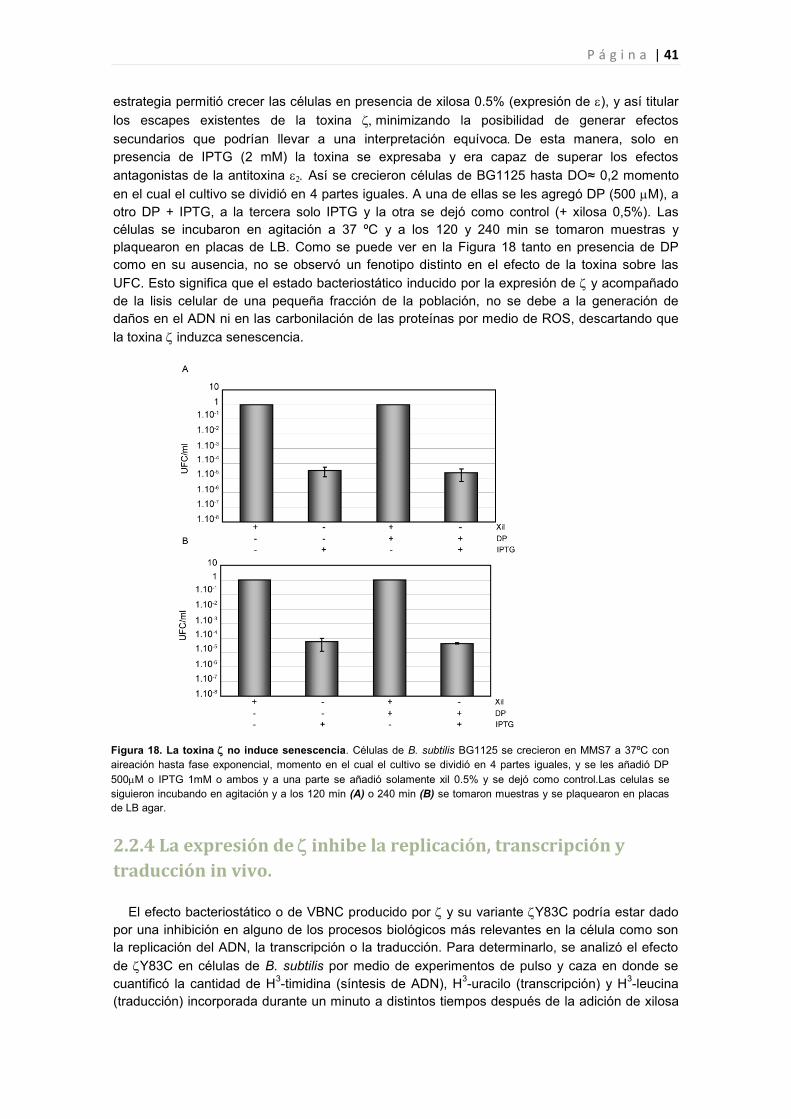
P á g i n a | 41
estrategia permitió crecer las células en presencia de xilosa 0.5% (expresión de ), y así titular
los escapes existentes de la toxina minimizando la posibilidad de generar efectos
secundarios que podrían llevar a una interpretación equívocaDe esta manera, solo en
presencia de IPTG (2 mM) la toxina se expresaba y era capaz de superar los efectos
antagonistas de la antitoxina Así se crecieron células de BG1125 hasta DO≈ 0,2 momento
en el cual el cultivo se dividió en 4 partes iguales. A una de ellas se les agregó DP (500 M), a
otro DP + IPTG, a la tercera solo IPTG y la otra se dejó como control (+ xilosa 0,5%). Las
células se incubaron en agitación a 37 ºC y a los 120 y 240 min se tomaron muestras y
plaquearon en placas de LB. Como se puede ver en la Figura 18 tanto en presencia de DP
como en su ausencia, no se observó un fenotipo distinto en el efecto de la toxina sobre las
UFC. Esto significa que el estado bacteriostático inducido por la expresión de y acompañado
de la lisis celular de una pequeña fracción de la población, no se debe a la generación de
daños en el ADN ni en las carbonilación de las proteínas por medio de ROS, descartando que
la toxina induzca senescencia.
2.2.4 La expresión de inhibe la replicación, transcripción y
traducción in vivo.
El efecto bacteriostático o de VBNC producido por y su variante Y83C podría estar dado
por una inhibición en alguno de los procesos biológicos más relevantes en la célula como son
la replicación del ADN, la transcripción o la traducción. Para determinarlo, se analizó el efecto
de Y83C en células de B. subtilis por medio de experimentos de pulso y caza en donde se
cuantificó la cantidad de H3-timidina (síntesis de ADN), H
3-uracilo (transcripción) y H
3-leucina
(traducción) incorporada durante un minuto a distintos tiempos después de la adición de xilosa
Figura 18. La toxina no induce senescencia. Células de B. subtilis BG1125 se crecieron en MMS7 a 37ºC con
aireación hasta fase exponencial, momento en el cual el cultivo se dividió en 4 partes iguales, y se les añadió DP
500M o IPTG 1mM o ambos y a una parte se añadió solamente xil 0.5% y se dejó como control.Las celulas se
siguieron incubando en agitación y a los 120 min (A) o 240 min (B) se tomaron muestras y se plaquearon en placas
de LB agar.
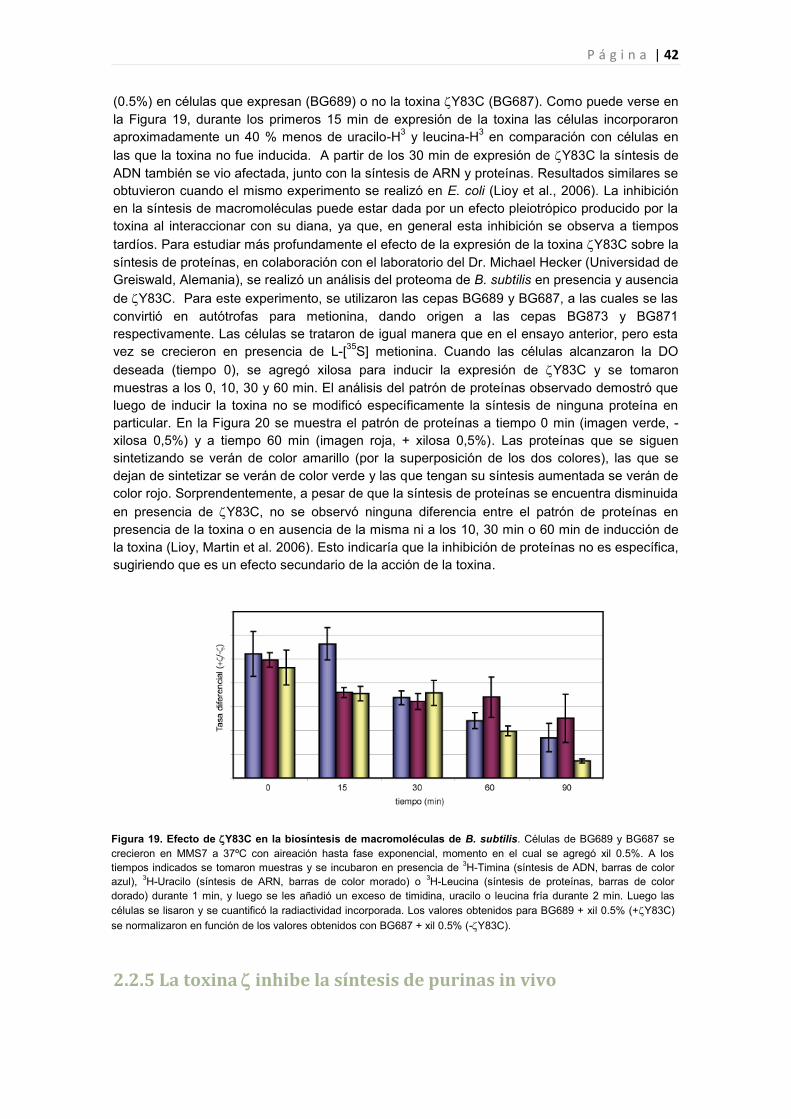
P á g i n a | 42
(0.5%) en células que expresan (BG689) o no la toxina Y83C (BG687). Como puede verse en
la Figura 19, durante los primeros 15 min de expresión de la toxina las células incorporaron
aproximadamente un 40 % menos de uracilo-H3 y leucina-H
3 en comparación con células en
las que la toxina no fue inducida. A partir de los 30 min de expresión de Y83C la síntesis de
ADN también se vio afectada, junto con la síntesis de ARN y proteínas. Resultados similares se
obtuvieron cuando el mismo experimento se realizó en E. coli (Lioy et al., 2006). La inhibición
en la síntesis de macromoléculas puede estar dada por un efecto pleiotrópico producido por la
toxina al interaccionar con su diana, ya que, en general esta inhibición se observa a tiempos
tardíos. Para estudiar más profundamente el efecto de la expresión de la toxina Y83C sobre la
síntesis de proteínas, en colaboración con el laboratorio del Dr. Michael Hecker (Universidad de
Greiswald, Alemania), se realizó un análisis del proteoma de B. subtilis en presencia y ausencia
de Y83C. Para este experimento, se utilizaron las cepas BG689 y BG687, a las cuales se las
convirtió en autótrofas para metionina, dando origen a las cepas BG873 y BG871
respectivamente. Las células se trataron de igual manera que en el ensayo anterior, pero esta
vez se crecieron en presencia de L-[35
S] metionina. Cuando las células alcanzaron la DO
deseada (tiempo 0), se agregó xilosa para inducir la expresión de Y83C y se tomaron
muestras a los 0, 10, 30 y 60 min. El análisis del patrón de proteínas observado demostró que
luego de inducir la toxina no se modificó específicamente la síntesis de ninguna proteína en
particular. En la Figura 20 se muestra el patrón de proteínas a tiempo 0 min (imagen verde, -
xilosa 0,5%) y a tiempo 60 min (imagen roja, + xilosa 0,5%). Las proteínas que se siguen
sintetizando se verán de color amarillo (por la superposición de los dos colores), las que se
dejan de sintetizar se verán de color verde y las que tengan su síntesis aumentada se verán de
color rojo. Sorprendentemente, a pesar de que la síntesis de proteínas se encuentra disminuida
en presencia de Y83C, no se observó ninguna diferencia entre el patrón de proteínas en
presencia de la toxina o en ausencia de la misma ni a los 10, 30 min o 60 min de inducción de
la toxina (Lioy, Martin et al. 2006). Esto indicaría que la inhibición de proteínas no es específica,
sugiriendo que es un efecto secundario de la acción de la toxina.
2.2.5 La toxina inhibe la síntesis de purinas in vivo
Figura 19. Efecto de Y83C en la biosíntesis de macromoléculas de B. subtilis. Células de BG689 y BG687 se
crecieron en MMS7 a 37ºC con aireación hasta fase exponencial, momento en el cual se agregó xil 0.5%. A los
tiempos indicados se tomaron muestras y se incubaron en presencia de 3H-Timina (síntesis de ADN, barras de color
azul), 3H-Uracilo (síntesis de ARN, barras de color morado) o
3H-Leucina (síntesis de proteínas, barras de color
dorado) durante 1 min, y luego se les añadió un exceso de timidina, uracilo o leucina fría durante 2 min. Luego las
células se lisaron y se cuantificó la radiactividad incorporada. Los valores obtenidos para BG689 + xil 0.5% (+Y83C)
se normalizaron en función de los valores obtenidos con BG687 + xil 0.5% (-Y83C).
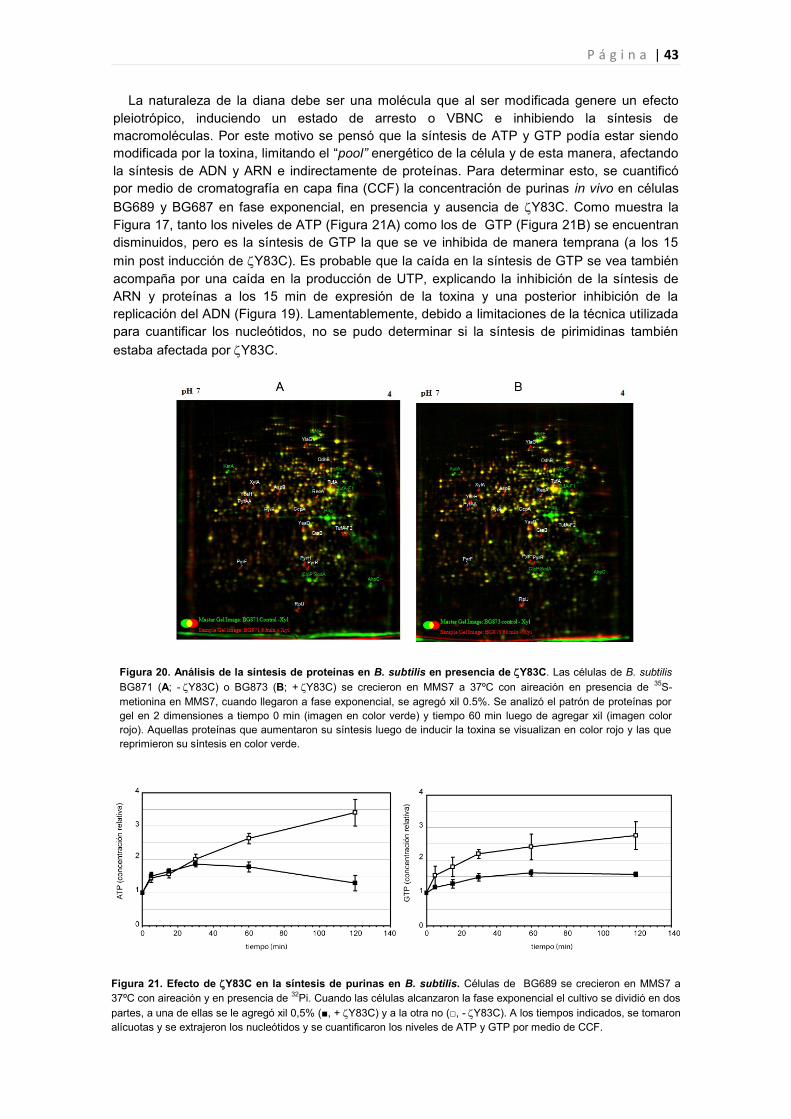
P á g i n a | 43
La naturaleza de la diana debe ser una molécula que al ser modificada genere un efecto
pleiotrópico, induciendo un estado de arresto o VBNC e inhibiendo la síntesis de
macromoléculas. Por este motivo se pensó que la síntesis de ATP y GTP podía estar siendo
modificada por la toxina, limitando el “pool” energético de la célula y de esta manera, afectando
la síntesis de ADN y ARN e indirectamente de proteínas. Para determinar esto, se cuantificó
por medio de cromatografía en capa fina (CCF) la concentración de purinas in vivo en células
BG689 y BG687 en fase exponencial, en presencia y ausencia de Y83C. Como muestra la
Figura 17, tanto los niveles de ATP (Figura 21A) como los de GTP (Figura 21B) se encuentran
disminuidos, pero es la síntesis de GTP la que se ve inhibida de manera temprana (a los 15
min post inducción de Y83C). Es probable que la caída en la síntesis de GTP se vea también
acompaña por una caída en la producción de UTP, explicando la inhibición de la síntesis de
ARN y proteínas a los 15 min de expresión de la toxina y una posterior inhibición de la
replicación del ADN (Figura 19). Lamentablemente, debido a limitaciones de la técnica utilizada
para cuantificar los nucleótidos, no se pudo determinar si la síntesis de pirimidinas también
estaba afectada por Y83C.
Figura 20. Análisis de la síntesis de proteínas en B. subtilis en presencia de Y83C. Las células de B. subtilis
BG871 (A; -Y83C) o BG873 (B; +Y83C) se crecieron en MMS7 a 37ºC con aireación en presencia de 35
S-
metionina en MMS7, cuando llegaron a fase exponencial, se agregó xil 0.5%. Se analizó el patrón de proteínas por
gel en 2 dimensiones a tiempo 0 min (imagen en color verde) y tiempo 60 min luego de agregar xil (imagen color
rojo). Aquellas proteínas que aumentaron su síntesis luego de inducir la toxina se visualizan en color rojo y las que
reprimieron su síntesis en color verde.
Figura 21. Efecto de Y83C en la síntesis de purinas en B. subtilis. Células de BG689 se crecieron en MMS7 a
37ºC con aireación y en presencia de 32
Pi. Cuando las células alcanzaron la fase exponencial el cultivo se dividió en dos
partes, a una de ellas se le agregó xil 0,5% (■, + Y83C) y a la otra no (□, -Y83C). A los tiempos indicados, se tomaron
alícuotas y se extrajeron los nucleótidos y se cuantificaron los niveles de ATP y GTP por medio de CCF.

P á g i n a | 44
2.2.6 Efecto de la toxina Y83C en la expresión génica de B.
subtilis
Para conocer más acerca del mecanismo de acción de la toxina , se analizó en
colaboración con el laboratorio del Dr. Eduardo González-Pastor (Centro de Astrobiología,
Madrid) el patrón de genes afectados por la expresión de la misma. Para esto, células de
BG689 y BG687 se crecieron hasta DO ≈ 0,2 momento en el cual se agregó xilosa 0.5% para
inducir la expresión de Y83C en BG689 y como control en BG687. A tiempos 0, 5 y 15 minutos
post-inducción se tomaron muestras y se procesaron para extraer ARN usando protocolos ya
establecidos. Los tiempos que se seleccionaron para analizar el transcriptoma de B. subtilis en
presencia de la toxina fueron tales que evitasen cualquier efecto secundario en la expresión
génica, pero suficientes para ver un efecto tóxico. De esta forma, se comparó el transcriptoma
de células de B. subtilis expresando Y83C (BG689 + xil 0,5%) con aquellas que no lo
expresaban (BG687 + xil 0.5%). Sorprendentemente, se obtuvo un número reducido de genes
afectados por la toxina (107 genes) y no se alteró la expresión de ningún otro sistema TA (por
ejemplo EndoA/YdcD, SpoIISAB) Resultados similares fueron observados cuando se analizó el
efecto de la toxina en células de E. coli (Lioy et al., 2006). Por el contrario, ensayos de
microarrays para el sistema de TA RelBE dieron como resultados más de 700 genes afectados,
datos consistentes con la actividad de RNasa de RelE (Lioy et al., 2006). Por lo tanto, este
resultado indicaría que la toxina no tiene como diana la degradación de ARN.
Los genes cuya expresión fue inducida o reprimida por la toxina Y83C se detallan en la
Tabla 1 del Anexo I, junto con una breve descripción de la función de la proteína que codifican,
la categoría a la que pertenece el gen, el nivel de modificación o “fold change“ y el valor P
(probabilidad de significancia).
De los 107 genes cuya expresión fue afectada por la expresión de la toxina, un total de 58
fueron reprimidos y 49 inducidos. De los 58 genes reprimidos, 14 de ellos se reprimieron
durante los primeros 5 min de expresión de Y83C, mientras que 46 lo hicieron a las 15 min. Es
destacable que a tiempo 15 min, existen dos genes cuya expresión se encuentra reprimida
desde tiempo 5 min (yqeC, ywcE, ver Figura 22). De los genes inducidos, 20 lo hicieron durante
los primeros 5 min y otros 32 durante los últimos 15 min de inducción de la toxina, de los cuales
3 genes mantuvieron su expresión elevada desde los 5 min hasta el final del experimento
(pyrAB, oppC y pyrP, Figura 22).
Los genes cuya expresión estaba modulada por la toxina fueron clasificados en categorías
según (Kunst et al., 1997). Como puede verse en la Tabla 1 del anexo I, existen genes que
pertenecen a diversas funciones celulares, muchas de ellas relevantes como: síntesis de
carbohidratos y moléculas relacionadas, síntesis de aminoácidos, síntesis de nucleótidos y
ácidos nucleicos o la síntesis de lípidos. La gran mayoría de los genes reprimidos a tiempo 15
min codifican proteínas que forman parte de procesos tan relevantes como la síntesis de
carbohidratos y lípidos. Sin embargo, los únicos genes esenciales afectados por la expresión
de Y83C se encuentran a los 15 min, y están reprimidos. Estos genes esenciales, son genes
que forman parte de la síntesis de lípidos de membrana (accB, accC, acpA, fabD, fabF, fabG,
yhdO y plsX) y de la pared celular (glmS).

P á g i n a | 45
Del análisis de los genes cuya expresión fue modulada por la toxina , se seleccionaron
aquellos que fueron más relevantes para explicar el fenotipo observado. Desafortunadamente,
el 18.7% de los genes afectados son desconocidos o su función lo es y no se pudieron
clasificar en ninguna categoría (Tabla 6 y Tabla 1 del Anexo I). A continuación se describirán
los genes que fueron seleccionados como relevantes.
2.2.6.1 Genes cuya expresión fue reprimida por Y83C
Tras 5 min bajo la acción de la toxina en la célula se reprimió la transcripción de 14 genes,
sin embargo sólo el producto de dos de ellos resultaron poder tener alguna relación con el
efecto tóxico de Y83C:
1. El gen yvdC: Cuando se buscó el gen yvdC en el genoma de B. subtilis 168, no se
encontró la descripción de ningún producto conocido, sin embargo, la búsqueda por BLAST de
la secuencia proteica que codifica si dio resultado positivo. El producto del gen yvdC
presentaba dominios que pertenecían a la familia MazG. La proteína YvdC (106 aa) es más
pequeña que MazG (263 aa) de E. coli (EcMazG), pero contiene los 2 dominos NTPasa que
son necesarios para degradar ppGpp o NTPs. (Lee et al., 2008). La represión del gen yvdC
podría sugerir un efecto indirecto de la toxicidad de Y83C, en donde la célula reprime la
expresión de un gen cuyo producto podría consumir nucleótidos que son necesarios para la
replicación del ADN, y de esta manera priorizar el uso de esos nucleótidos en procesos más
relevantes para el mantenimiento celular. Esta represión podría ir acompañada de un aumento
en los niveles de ppGpp ya que el producto del gen relA se ve aumentado a los 15 min de
expresión de la toxina.
2. El gen yqeC: Este gen potencialmente codifica la 6-fosfogluconato deshidrogenasa,
enzima que forma parte de la vía de las pentosas fosfato y participa en la producción de
ribulosa -5- fosfato a partir de 6- glucofosfonato. La ribulosa-5- fosfato se convierte luego en
ribosa-5-fosfato (Rib-5-P). La Rib-5-P es una de las moléculas que regula la síntesis de
nucleótidos en B. subtilis (Tozzi et al., 2006), lo que podría indicar que la síntesis de
nucleótidos este afectada y que la represión de este gen esté relacionada con esta inhibición.
La localización de esta proteína podría ser en la membrana (según:http://subtiwiki.uni-
goettingen.de/wiki/index.php/Lists_of_genes,_proteins_and_RNAs)
Cuando las células de BG689 fueron sometidas al efecto tóxico de Y83C durante 15 min,
los genes cuya expresión se vieron disminuidas fueron 46. De estos, se seleccionaron los
siguientes genes:
Figura 22. Diagrama de Venny indicando el número de genes cuya expresión se vio afectada por Y83C
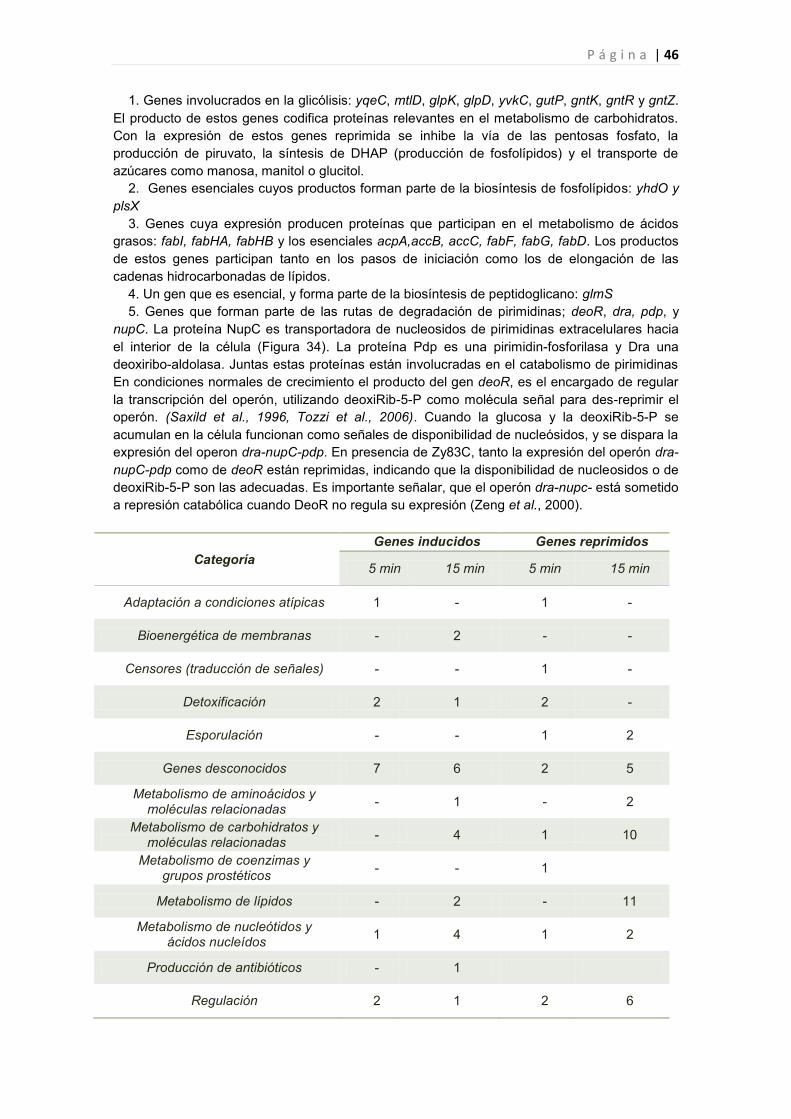
P á g i n a | 46
1. Genes involucrados en la glicólisis: yqeC, mtlD, glpK, glpD, yvkC, gutP, gntK, gntR y gntZ.
El producto de estos genes codifica proteínas relevantes en el metabolismo de carbohidratos.
Con la expresión de estos genes reprimida se inhibe la vía de las pentosas fosfato, la
producción de piruvato, la síntesis de DHAP (producción de fosfolípidos) y el transporte de
azúcares como manosa, manitol o glucitol.
2. Genes esenciales cuyos productos forman parte de la biosíntesis de fosfolípidos: yhdO y
plsX
3. Genes cuya expresión producen proteínas que participan en el metabolismo de ácidos
grasos: fabI, fabHA, fabHB y los esenciales acpA,accB, accC, fabF, fabG, fabD. Los productos
de estos genes participan tanto en los pasos de iniciación como los de elongación de las
cadenas hidrocarbonadas de lípidos.
4. Un gen que es esencial, y forma parte de la biosíntesis de peptidoglicano: glmS
5. Genes que forman parte de las rutas de degradación de pirimidinas; deoR, dra, pdp, y
nupC. La proteína NupC es transportadora de nucleosidos de pirimidinas extracelulares hacia
el interior de la célula (Figura 34). La proteína Pdp es una pirimidin-fosforilasa y Dra una
deoxiribo-aldolasa. Juntas estas proteínas están involucradas en el catabolismo de pirimidinas
En condiciones normales de crecimiento el producto del gen deoR, es el encargado de regular
la transcripción del operón, utilizando deoxiRib-5-P como molécula señal para des-reprimir el
operón. (Saxild et al., 1996, Tozzi et al., 2006). Cuando la glucosa y la deoxiRib-5-P se
acumulan en la célula funcionan como señales de disponibilidad de nucleósidos, y se dispara la
expresión del operon dra-nupC-pdp. En presencia de Zy83C, tanto la expresión del operón dra-
nupC-pdp como de deoR están reprimidas, indicando que la disponibilidad de nucleosidos o de
deoxiRib-5-P son las adecuadas. Es importante señalar, que el operón dra-nupc- está sometido
a represión catabólica cuando DeoR no regula su expresión (Zeng et al., 2000).
Categoría
Genes inducidos Genes reprimidos
5 min 15 min 5 min 15 min
Adaptación a condiciones atípicas 1 - 1 -
Bioenergética de membranas - 2 - -
Censores (traducción de señales) - - 1 -
Detoxificación 2 1 2 -
Esporulación - - 1 2
Genes desconocidos 7 6 2 5
Metabolismo de aminoácidos y moléculas relacionadas
- 1 - 2
Metabolismo de carbohidratos y moléculas relacionadas
- 4 1 10
Metabolismo de coenzimas y grupos prostéticos
- - 1
Metabolismo de lípidos - 2 - 11
Metabolismo de nucleótidos y ácidos nucleídos
1 4 1 2
Producción de antibióticos - 1
Regulación 2 1 2 6

P á g i n a | 47
Síntesis de ARN - - 1 -
Transformación/competencia 2 3 - -
Transporte/proteínas de unión 5 7 1 8
Total 20 32 14 46
2.2.6.2 Genes cuya expresión fue inducida por Y83C
Durante los primeros 5 min de expresión de la toxina, solo dos genes fueron considerados
relevantes para explicar el fenotipo observado y ambos están relacionados aunque
pertenezcan a categorías distintas:
1. Los genes pyrAB y pyrP: Estos genes forman parte del operón pir, el cual está regulado
de diversas maneras. La primera es indirecta es a través de la Rib-5-P (cuya producción está
reprimida por Y83C), molécula que al unirse a DeoR, desreprime el operón dra-pdp-nupC. La
segunda etapa en la regulación del operón pir se da a través de PyrR, que deja de reprimir
transcripcionalmente al operón si los niveles de UTP y GTP son bajos en la célula. En la
sección 2.2.5 se demostró que los niveles de GTP estaban disminuidos en presencia de Y83C
Tras 15 min de expresión de la toxina, de los genes inducidos caben destacar los siguientes:
gamA, gamP, pyrB, pyrE, pyrAB, pyrR, pyRP, relA, comGF, comGD y comGC
1. Los genes gamA y gamP están involucrados en la producción de D-fructosa 6- fosfato
(Fru-6- P), que podría acoplarse a la vía de degradación de la glucosa, minimizando de esta
manera el gasto energético que se produce en otras vías.
2. Los genes pyr y relA forman parte del metabolismo de ácidos nucleídos y nucleótidos. La
inducción de los genes del operón pir podría sugerir una disminución de pirimidinas intracelular.
Sumado a esto, la inducción del gen relA da como resultado la disminución de los niveles de
GTP, acompañado de un aumento en los niveles de ppGpp. El aumento de la expresión del
producto del gen relA y su respectivo efecto en los niveles de GTP pueden explicar el fenotipo
observado en la Figura 21, donde se ve una drástica disminución de la concentración de GTP
in vivo a los 15 min de expresar la toxina Y83C.
3. Los productos de los genes comGF, comGD y comGC forman parte de un pseudopili que
permite que el ADN exógeno entre en contacto con ComEA. La inducción de estos genes se
encuentra relacionada con bajos niveles de GTP y altos niveles de RelA. Podría ser una
estrategia de B. subtlis para intentar captar ADN externo, que pueda tener una copia del
antídoto () y de esta manera revertir el efecto toxico causado por .
3.1 Según los niveles de expresión de el efecto tóxico
producido en las células puede revertirse por la
expresión de la antitoxina 2
Como se mostró en la sección anterior la gran disminución en las unidades formadoras de
colonias mediada por la toxina estaría dada por la entrada de las células en un estado
bacteriostático. Si esto es así, si se inicia la síntesis de novo de la antitoxina 2, se debería ver
Tabla 6. Número de genes afectados por la expresión de Y83C.
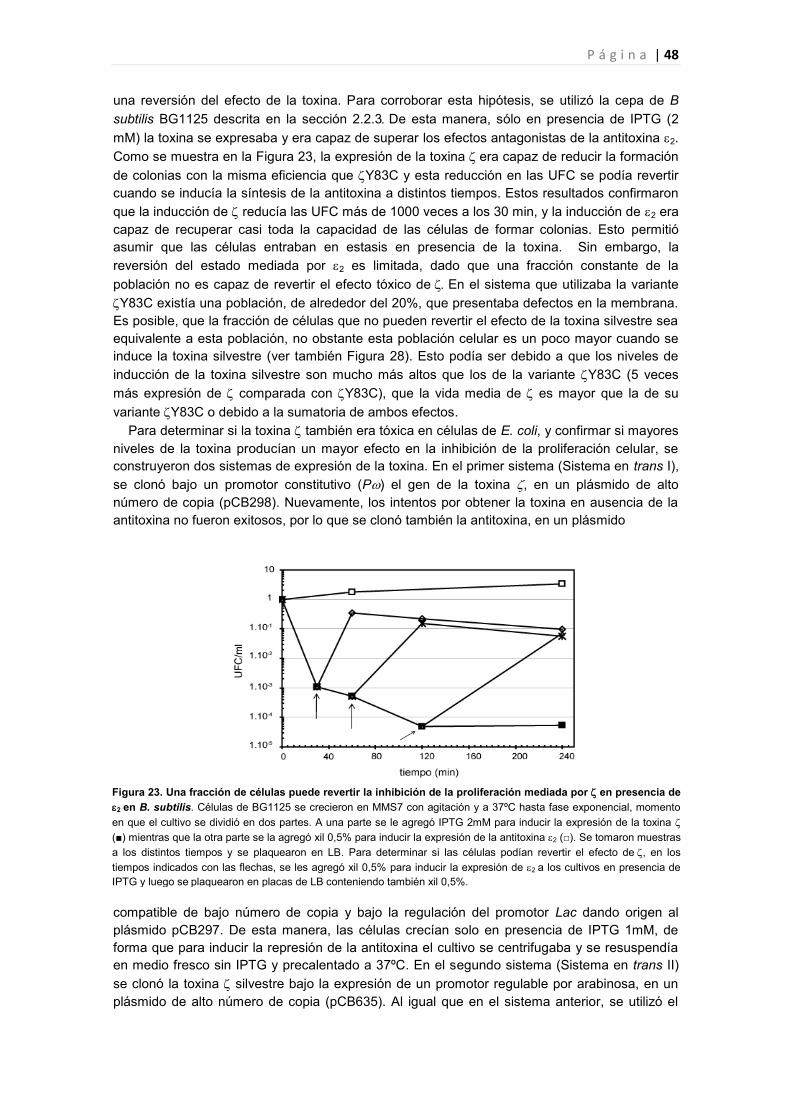
P á g i n a | 48
una reversión del efecto de la toxina. Para corroborar esta hipótesis, se utilizó la cepa de B
subtilis BG1125 descrita en la sección 2.2.3De esta manera, sólo en presencia de IPTG (2
mM) la toxina se expresaba y era capaz de superar los efectos antagonistas de la antitoxina 2.
Como se muestra en la Figura 23, la expresión de la toxina era capaz de reducir la formación
de colonias con la misma eficiencia que Y83C y esta reducción en las UFC se podía revertir
cuando se inducía la síntesis de la antitoxina a distintos tiempos. Estos resultados confirmaron
que la inducción de reducía las UFC más de 1000 veces a los 30 min, y la inducción de 2 era
capaz de recuperar casi toda la capacidad de las células de formar colonias. Esto permitió
asumir que las células entraban en estasis en presencia de la toxina. Sin embargo, la
reversión del estado mediada por 2 es limitada, dado que una fracción constante de la
población no es capaz de revertir el efecto tóxico deEn el sistema que utilizaba la variante
Y83C existía una población, de alrededor del 20%, que presentaba defectos en la membrana.
Es posible, que la fracción de células que no pueden revertir el efecto de la toxina silvestre sea
equivalente a esta población, no obstante esta población celular es un poco mayor cuando se
induce la toxina silvestre (ver también Figura 28). Esto podía ser debido a que los niveles de
inducción de la toxina silvestre son mucho más altos que los de la variante Y83C (5 veces
más expresión de comparada con Y83C), que la vida media de es mayor que la de su
variante Y83C o debido a la sumatoria de ambos efectos.
Para determinar si la toxina también era tóxica en células de E. coli, y confirmar si mayores
niveles de la toxina producían un mayor efecto en la inhibición de la proliferación celular, se
construyeron dos sistemas de expresión de la toxina. En el primer sistema (Sistema en trans I),
se clonó bajo un promotor constitutivo (P) el gen de la toxina , en un plásmido de alto
número de copia (pCB298). Nuevamente, los intentos por obtener la toxina en ausencia de la
antitoxina no fueron exitosos, por lo que se clonó también la antitoxina, en un plásmido
compatible de bajo número de copia y bajo la regulación del promotor Lac dando origen al
plásmido pCB297. De esta manera, las células crecían solo en presencia de IPTG 1mM, de
forma que para inducir la represión de la antitoxina el cultivo se centrifugaba y se resuspendía
en medio fresco sin IPTG y precalentado a 37ºC. En el segundo sistema (Sistema en trans II)
se clonó la toxina silvestre bajo la expresión de un promotor regulable por arabinosa, en un
plásmido de alto número de copia (pCB635). Al igual que en el sistema anterior, se utilizó el
Figura 23. Una fracción de células puede revertir la inhibición de la proliferación mediada por en presencia de
2en B. subtilis. Células de BG1125 se crecieron en MMS7 con agitación y a 37ºC hasta fase exponencial, momento
en que el cultivo se dividió en dos partes. A una parte se le agregó IPTG 2mM para inducir la expresión de la toxina
(■) mientras que la otra parte se la agregó xil 0,5% para inducir la expresión de la antitoxina 2 (□). Se tomaron muestras
a los distintos tiempos y se plaquearon en LB. Para determinar si las células podían revertir el efecto de, en los
tiempos indicados con las flechas, se les agregó xil 0,5% para inducir la expresión de 2 a los cultivos en presencia de
IPTG y luego se plaquearon en placas de LB conteniendo también xil 0,5%.
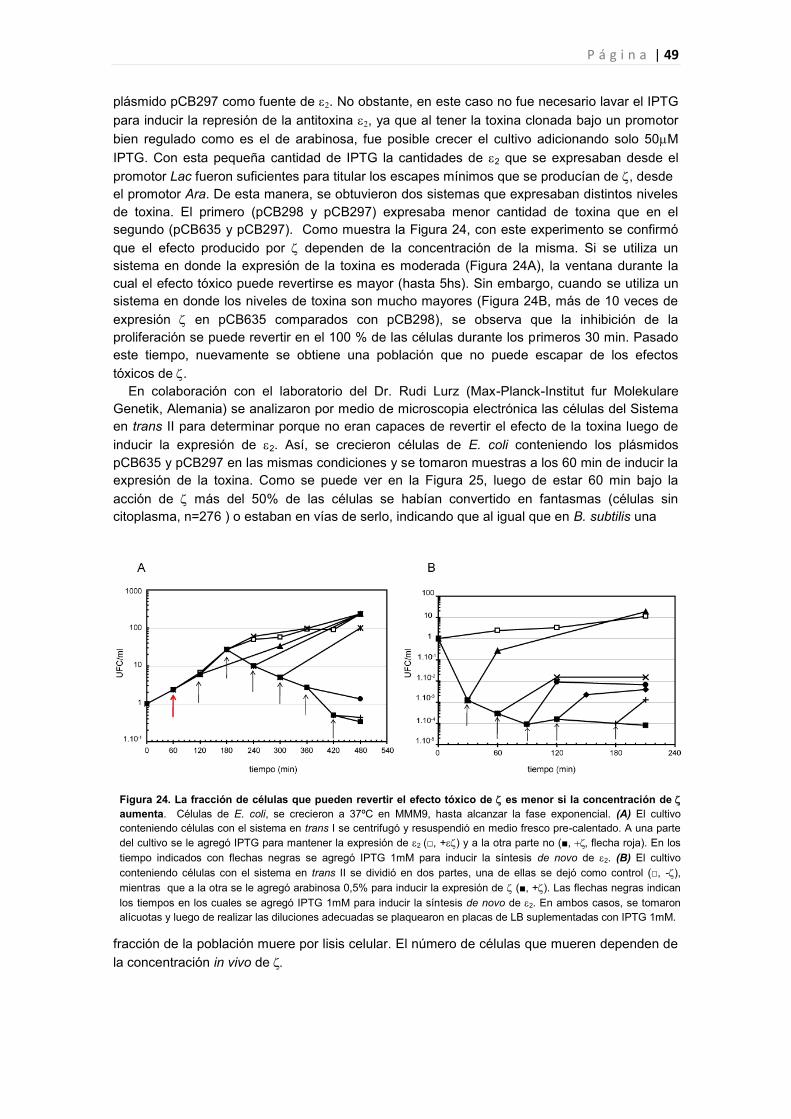
P á g i n a | 49
plásmido pCB297 como fuente de . No obstante, en este caso no fue necesario lavar el IPTG
para inducir la represión de la antitoxina , ya que al tener la toxina clonada bajo un promotor
bien regulado como es el de arabinosa, fue posible crecer el cultivo adicionando solo 50M
IPTG. Con esta pequeña cantidad de IPTG la cantidades de 2 que se expresaban desde el
promotor Lac fueron suficientes para titular los escapes mínimos que se producían de , desde
el promotor Ara. De esta manera, se obtuvieron dos sistemas que expresaban distintos niveles
de toxina. El primero (pCB298 y pCB297) expresaba menor cantidad de toxina que en el
segundo (pCB635 y pCB297). Como muestra la Figura 24, con este experimento se confirmó
que el efecto producido por dependen de la concentración de la misma. Si se utiliza un
sistema en donde la expresión de la toxina es moderada (Figura 24A), la ventana durante la
cual el efecto tóxico puede revertirse es mayor (hasta 5hs). Sin embargo, cuando se utiliza un
sistema en donde los niveles de toxina son mucho mayores (Figura 24B, más de 10 veces de
expresión en pCB635 comparados con pCB298), se observa que la inhibición de la
proliferación se puede revertir en el 100 % de las células durante los primeros 30 min. Pasado
este tiempo, nuevamente se obtiene una población que no puede escapar de los efectos
tóxicos de .
En colaboración con el laboratorio del Dr. Rudi Lurz (Max-Planck-Institut fur Molekulare
Genetik, Alemania) se analizaron por medio de microscopia electrónica las células del Sistema
en trans II para determinar porque no eran capaces de revertir el efecto de la toxina luego de
inducir la expresión de 2. Así, se crecieron células de E. coli conteniendo los plásmidos
pCB635 y pCB297 en las mismas condiciones y se tomaron muestras a los 60 min de inducir la
expresión de la toxina. Como se puede ver en la Figura 25, luego de estar 60 min bajo la
acción de más del 50% de las células se habían convertido en fantasmas (células sin
citoplasma, n=276 ) o estaban en vías de serlo, indicando que al igual que en B. subtilis una
fracción de la población muere por lisis celular. El número de células que mueren dependen de
la concentración in vivo de
Figura 24. La fracción de células que pueden revertir el efecto tóxico de es menor si la concentración de
aumenta. Células de E. coli, se crecieron a 37ºC en MMM9, hasta alcanzar la fase exponencial. (A) El cultivo
conteniendo células con el sistema en trans I se centrifugó y resuspendió en medio fresco pre-calentado. A una parte
del cultivo se le agregó IPTG para mantener la expresión de 2(□, +) y a la otra parte no (■, flecha roja). En los
tiempo indicados con flechas negras se agregó IPTG 1mM para inducir la síntesis de novo de 2. (B) El cultivo
conteniendo células con el sistema en trans II se dividió en dos partes, una de ellas se dejó como control (□, -),
mientras que a la otra se le agregó arabinosa 0,5% para inducir la expresión de (■, +). Las flechas negras indican
los tiempos en los cuales se agregó IPTG 1mM para inducir la síntesis de novo de 2. En ambos casos, se tomaron
alícuotas y luego de realizar las diluciones adecuadas se plaquearon en placas de LB suplementadas con IPTG 1mM.

P á g i n a | 50
Parte IV. La toxina induce un aumento en la sensibilidad de células tolerantes a antibióticos.
4.1 La expresión de Y83C induce un aumento de la
sensibilidad a antibióticos.
Los resultados expuestos hasta el momento han demostrado que cuando una población de
células es expuesta a la toxina , siempre existe una pequeña fracción de células que son
capaces de tolerar el efecto tóxico. Sin embargo, si estas células son diluidas en medio fresco y
puestas nuevamente en contacto con la toxina, se obtiene el mismo número de células que son
insensibles a , mostrando un fenotipo similar a las células persistentes que se describieron en
la introducción en la sección 4.4.
Para determinar si la exposición de las células a la toxina estaba acompañada de la
producción de células persistentes (o tolerantes), como ya se postuló para otros sistemas TA
como HipAB o RelBE, se llevaron a cabo una serie de experimentos en donde se evaluó la
actividad inhibidora del crecimiento producida por la toxina Y83C en presencia y ausencia de
antibióticos con distintas dianas celulares: ampicilina (inhibe la síntesis de la pared celular),
eritromicina (inhibe la traducción), ciprofloxacina (inhibe la replicación) y triclosan (inhibe la
síntesis de lípidos).
Se crecieron células de BG689 hasta OD cercana a 0,2. Se adicionó el antibiótico
correspondiente en concentraciones próximas a 3 veces la concentración mínima inhibitoria
(MIC), y se indujo la expresión de la toxina (+ xil). Como controles se realizaron cultivos a los
que o bien solo se les agregó el antibiótico o solo se les adicionó el inductor de Y83C. Como
se puede ver en la Figura 26 se tomaron muestras a los 120 (A) y 240 min (B) luego de inducir
la expresión de la toxina, de agregar el antibiótico o ambos. En todos los casos, el efecto
producido por Y83C en presencia del antibiótico (cualquiera sea su diana) es mayor que el del
Figura 25. Microscopias electrónicas de células de E. coli sobre-expresando Células de E. coli conteniendo los
plásmidos pCB635 y pCB297 se crecieron hasta fase exponencial en MMM9 a 37ºC, momento en el cual el cultivo se dividió
en partes iguales y a una parte se le agregó arabinosa 0,5% para inducir la expresión de . A los 0 (A y B) y 60 (C y D) min
se tomaron muestras y se prepararon las células para su visualización como se detalla en materiales y métodos.

P á g i n a | 51
antibiótico solo o la toxina sola. El número de células tolerantes en fase exponencial siempre
es menor que en fase estacionaria, donde llegan a formar el 1% de la población total. Por este
motivo, también se realizaron los mismos ensayos pero en células de BG689 en fase
estacionaria (Figura 27). Nuevamente, el número de células que sobrevivieron al efecto de la
toxina junto con el del antibiótico es menor que cualquiera de ellos solos. Incluso, aunque el
efecto del antibiótico sobre las UFC sea más leve en fase estacionaria, el efecto conjunto
toxina-antibiótico sobre la viabilidad es siempre mayor que el efecto sólo antibiótico o sólo
toxina.
4.2 La expresión de no produce un aumento de
células tolerantes en presencia de antibióticos.
Era posible que parte de la actividad sinérgica producida por la toxina en presencia de los
antibióticos pudiera estar dada por el tipo de sistema de expresión que se estaba empleado, en
donde la expresión de la toxina Y83C no podía ser revertida (efecto crónico). Para demostrar
que el efecto producido por la toxina no estaba enmascarando la formación de células
tolerantes, se realizó el mismo ensayo pero utilizando esta vez células BG1125 (ver sección
2.2.3). De este modo, se crecieron células hasta fase exponencial (Figura 28) o fase
estacionaria (Figura 29) (sin xilosa, para minimizar la cantidad de antitoxina en la célula)
momento en el cual el cultivo se dividió en 8 partes iguales. A cada nuevo cultivo se le
adicionó IPTG (+), xilosa (+), el antibiótico correspondiente (Amp, Cip o Tri), o según
corresponda: IPTG + antibiótico (para estudiar la expresión de en conjunto con el antibiótico)
o xilosa + Antibiótico (para estudiar solo el efecto del antibiótico) como lo indican las Figuras 28
y 29. Nuevamente, en presencia de los antibióticos junto con la toxina silvestre, se observó
mayor muerte que con cualquiera de ellas solas. Sólo una pequeña fracción de células eran
capaces de sobrevivir (< 1.10-7
células/ml). De hecho el efecto conjunto toxina-antibiótico fue
mayor de lo esperado debido a la gran expresión de toxina desde el promotor Lac. Para
determinar si la disminución en las UFC en presencia de y el antibiótico generaba células
tolerantes al mismo, una alícuota de células que habían estado en contacto con la toxina y el
antimicrobiano se plaqueó en presencia de xilosa, el inductor de la antitoxina . Como se puede
ver en las Figuras 28 y 29, tanto en fase exponencial como en fase estacionaria, las células
lograron revertir el efecto de la toxina pero no del antimicrobiano. Esto indica que tanto la
exposición prolongada, como la temporal a la toxina no produce tolerancia a los antibióticos,
sino por el contrario, aumenta la eficacia de los mismos.
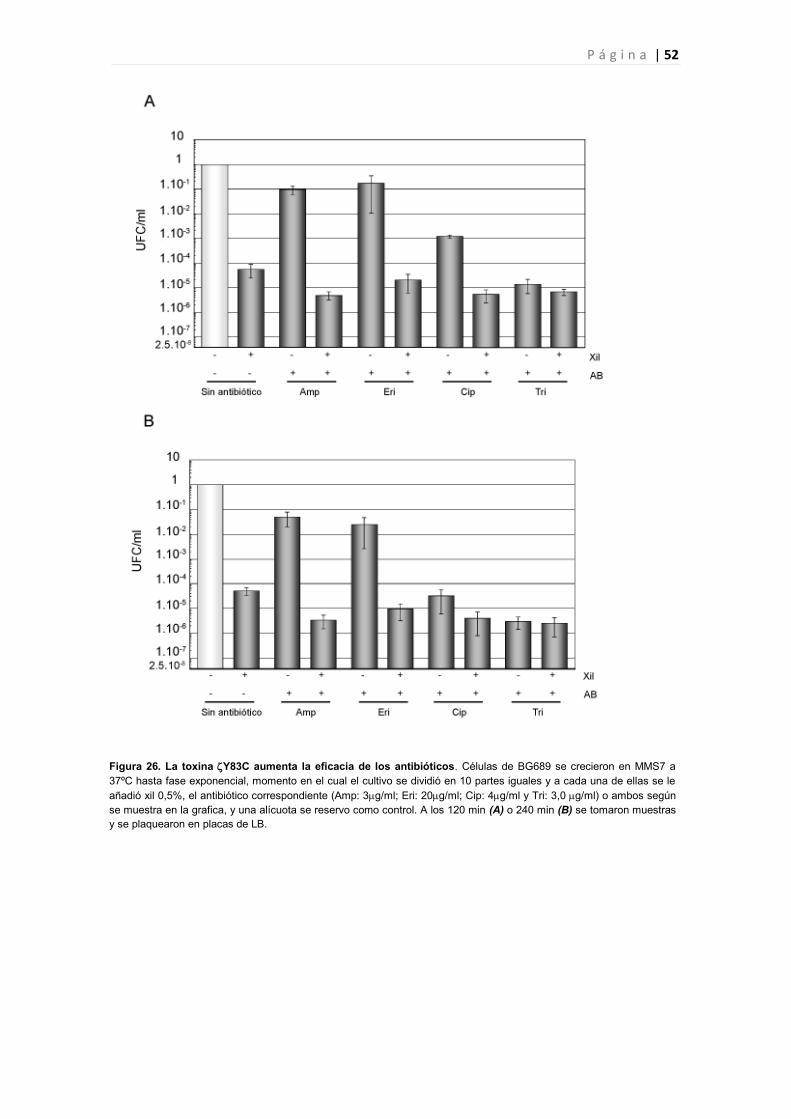
P á g i n a | 52
Figura 26. La toxina Y83C aumenta la eficacia de los antibióticos. Células de BG689 se crecieron en MMS7 a
37ºC hasta fase exponencial, momento en el cual el cultivo se dividió en 10 partes iguales y a cada una de ellas se le
añadió xil 0,5%, el antibiótico correspondiente (Amp: 3g/ml; Eri: 20g/ml; Cip: 4g/ml y Tri: 3,0 g/ml) o ambos según
se muestra en la grafica, y una alícuota se reservo como control. A los 120 min (A) o 240 min (B) se tomaron muestras
y se plaquearon en placas de LB.
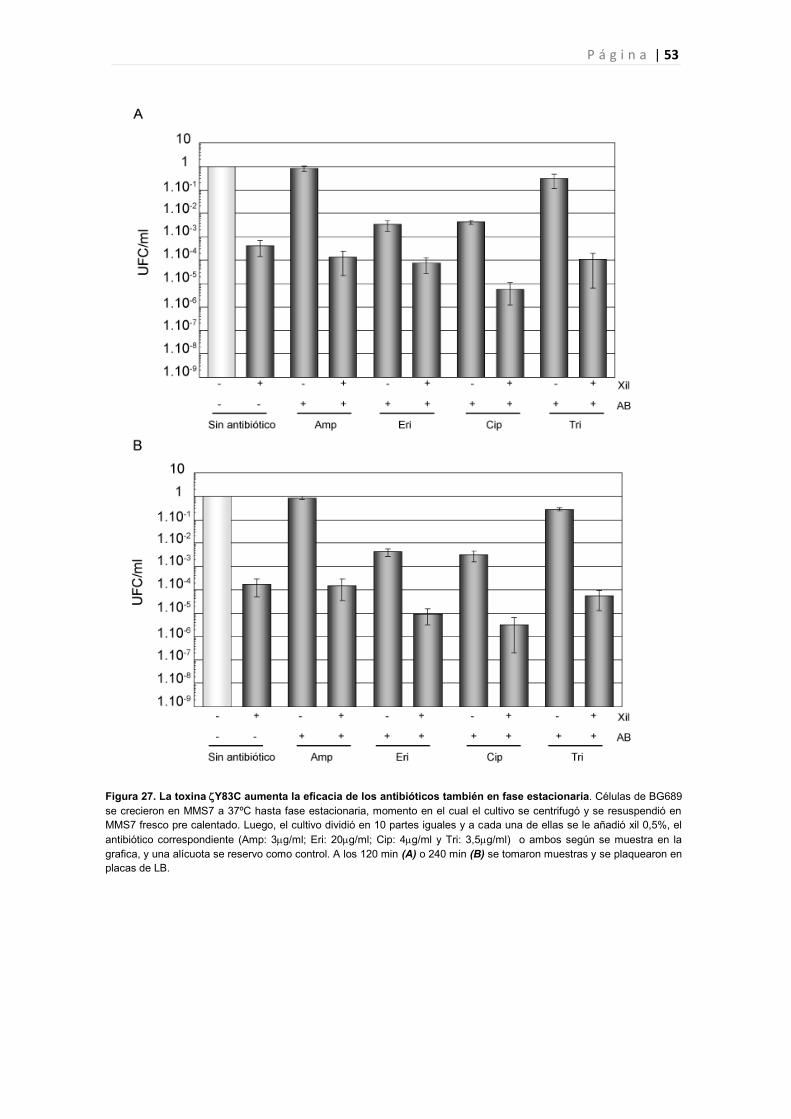
P á g i n a | 53
Figura 27. La toxina Y83C aumenta la eficacia de los antibióticos también en fase estacionaria. Células de BG689
se crecieron en MMS7 a 37ºC hasta fase estacionaria, momento en el cual el cultivo se centrifugó y se resuspendió en
MMS7 fresco pre calentado. Luego, el cultivo dividió en 10 partes iguales y a cada una de ellas se le añadió xil 0,5%, el
antibiótico correspondiente (Amp: 3g/ml; Eri: 20g/ml; Cip: 4g/ml y Tri: 3,5g/ml) o ambos según se muestra en la
grafica, y una alícuota se reservo como control. A los 120 min (A) o 240 min (B) se tomaron muestras y se plaquearon en
placas de LB.
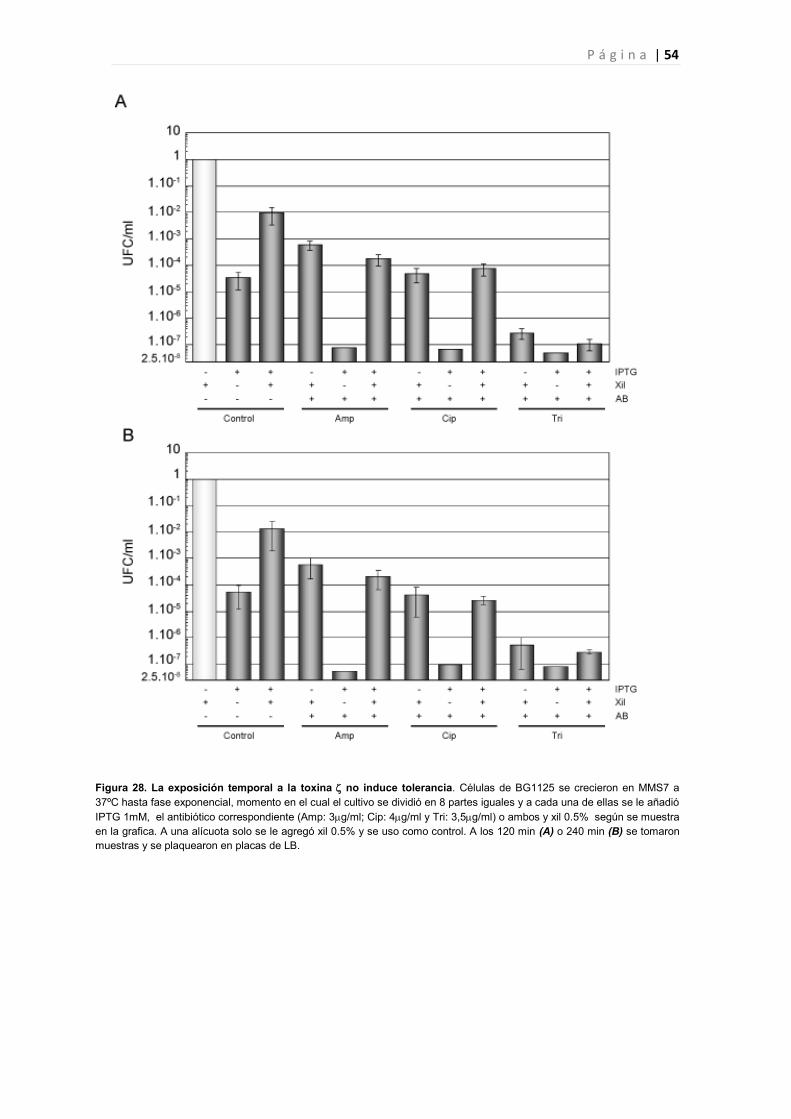
P á g i n a | 54
Figura 28. La exposición temporal a la toxina no induce tolerancia. Células de BG1125 se crecieron en MMS7 a
37ºC hasta fase exponencial, momento en el cual el cultivo se dividió en 8 partes iguales y a cada una de ellas se le añadió
IPTG 1mM, el antibiótico correspondiente (Amp: 3g/ml; Cip: 4g/ml y Tri: 3,5g/ml) o ambos y xil 0.5% según se muestra
en la grafica. A una alícuota solo se le agregó xil 0.5% y se uso como control. A los 120 min (A) o 240 min (B) se tomaron
muestras y se plaquearon en placas de LB.
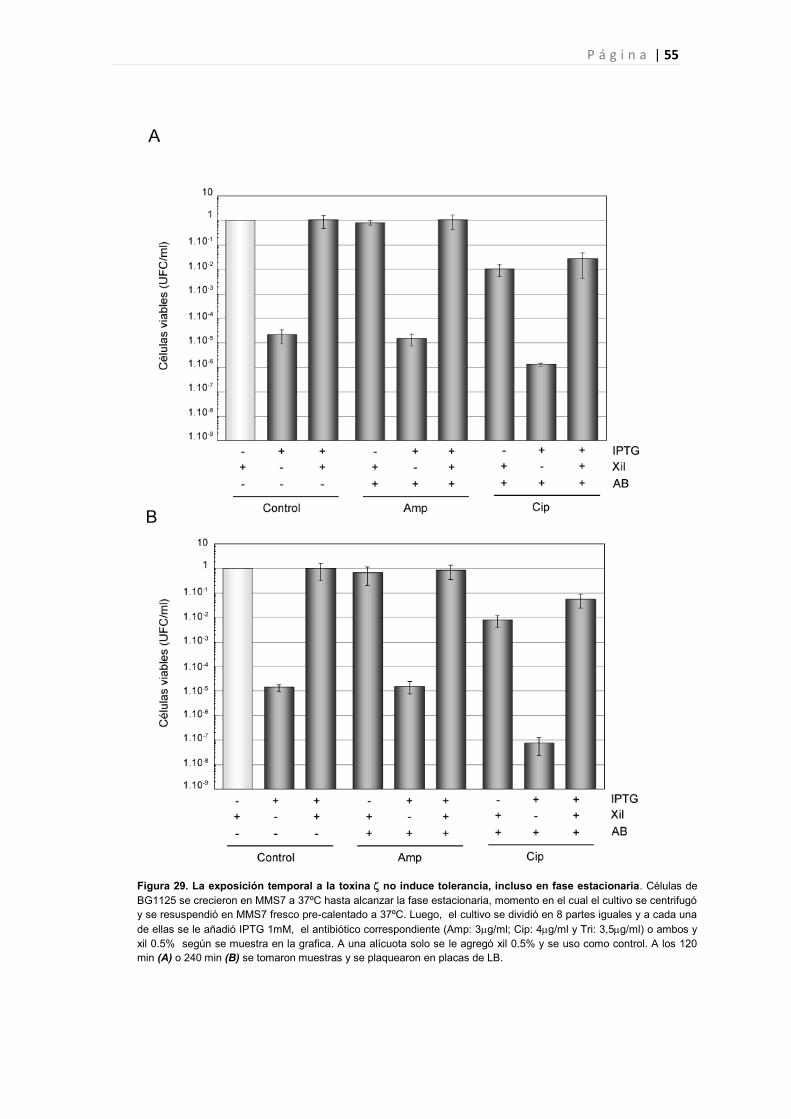
P á g i n a | 55
Figura 29. La exposición temporal a la toxina no induce tolerancia, incluso en fase estacionaria. Células de
BG1125 se crecieron en MMS7 a 37ºC hasta alcanzar la fase estacionaria, momento en el cual el cultivo se centrifugó
y se resuspendió en MMS7 fresco pre-calentado a 37ºC. Luego, el cultivo se dividió en 8 partes iguales y a cada una
de ellas se le añadió IPTG 1mM, el antibiótico correspondiente (Amp: 3g/ml; Cip: 4g/ml y Tri: 3,5g/ml) o ambos y
xil 0.5% según se muestra en la grafica. A una alícuota solo se le agregó xil 0.5% y se uso como control. A los 120
min (A) o 240 min (B) se tomaron muestras y se plaquearon en placas de LB.

P á g i n a | 56
Parte V. El sistema como diana antimicrobiana.
El sistema de TA se encuentra entre otros en el cromosoma de bacterias patógenas como
S. pnemoniae, E. coli A7 o en plásmidos que otorgan resistencia a vancomicina en enterococos
(Van Melderen & Saavedra De Bast, 2009, Moritz & Hergenrother, 2007). Como se demostró
en la sección anterior, a diferencia de otras toxinas como HipA o RelE, la toxina no produce
un aumento en las células tolerantes a los antibióticos, sino que por el contrario aumenta la
eficiencia de los mismos. Sumado a esto, estudios anteriores permitieron determinar que la
expresión de la toxina no incrementaba la tasa de mutación celular, y que su diana debe
tener un carácter esencial para la célula ya que no ha sido posible aislar mutantes resistentes
al efecto tóxico de . Por estos motivos se seleccionó al sistema como un firme candidato
para desarrollar nuevas dianas antimicrobianas.
5.1 Identificación de los aminoácidos esenciales
para la interacción del complejo ∙
En colaboración con el Laboratorio Salvat (Barcelona), se utilizó modelado molecular de la
estructura cristalina del complejo para buscar una molécula que fuera capaz de romper la
interacción T∙A, de forma tal que liberase a la toxina y le permitiese reconocer su diana. Para
esto primero era necesario conocer los residuos más importantes en la interacción T∙A, lo que
permitiría modelar una molécula que interaccione con esos residuos y romper así la interacción
T∙A.
El complejo forma un heterotetrámero en donde un dímero de la antitoxina se encuentra
entre dos moléculas de la toxina (Figura 12). Para determinar qué aminoácido era más
importante en la interacción ∙, se llevaron a cabo estudios de modelado molecular. El análisis
mostró un total de 32 interacciones, donde 15 residuos pertenecían a y 17 a . Se
encontraron dos de las interacciones más importantes en la hélice A de la toxina , en donde
el residuo D18 interacciona con los aminoácidos Y22, K36 y K51 de y el residuo E22
interacciona con K51 y K54 de (Figura 30). También el estudio de modelado molecular dio
como resultado otras dos interacciones posiblemente menos importantes en la -hélice G y
fueron los residuos R158 y R165 de (Lioy et al., 2009). El primer paso para estudiar la
relevancia de estos residuos fue realizar mutaciones in silico en donde los residuos
seleccionados se cambiaron por una alanina (A). Así, se obtuvieron los complejos TA en donde
los aminoácidos D18, E22, R158 y R165 fueron cambiados por una A (D18A, E22A,
R158A y R165A, respectivamente). Como se puede ver en la Figura 31, el cambio del
aminoácido D18A, generó inestabilidad en el complejo ∙. Esta inestabilidad se traduce no
solo en un incremento en la distancia entre los carbonos (C de los residuos de Y22 y
D18A (Figura 31A, línea rosa), sino también entre los C de otros residuos ubicados en la
lejanía del residuo analizado como son los residuos 27N y R15 (Figura 31B, línea rosa). Sin
embargo, si se estudian las distancias entre los C de estos residuos en una simulación con el
aminoácido D18 silvestre (Figura 31, línea azul) o en una simulación donde se cambia el
residuo no relevante S48 por una A (Figura 31, línea amarilla), se ve que no se observa un
aumento en la distancia entre el par Y22 y D18 o entre 27N y R15, indicando la relevancia
del resido D18 para la estabilidad del complejo . Los mismos estudios realizados en el
resto de residuos identificados como relevantes, indicaron que solo los residuos D18 y E22
podían llegar a ser esenciales para la estabilidad de (Lioy et al., 2009).
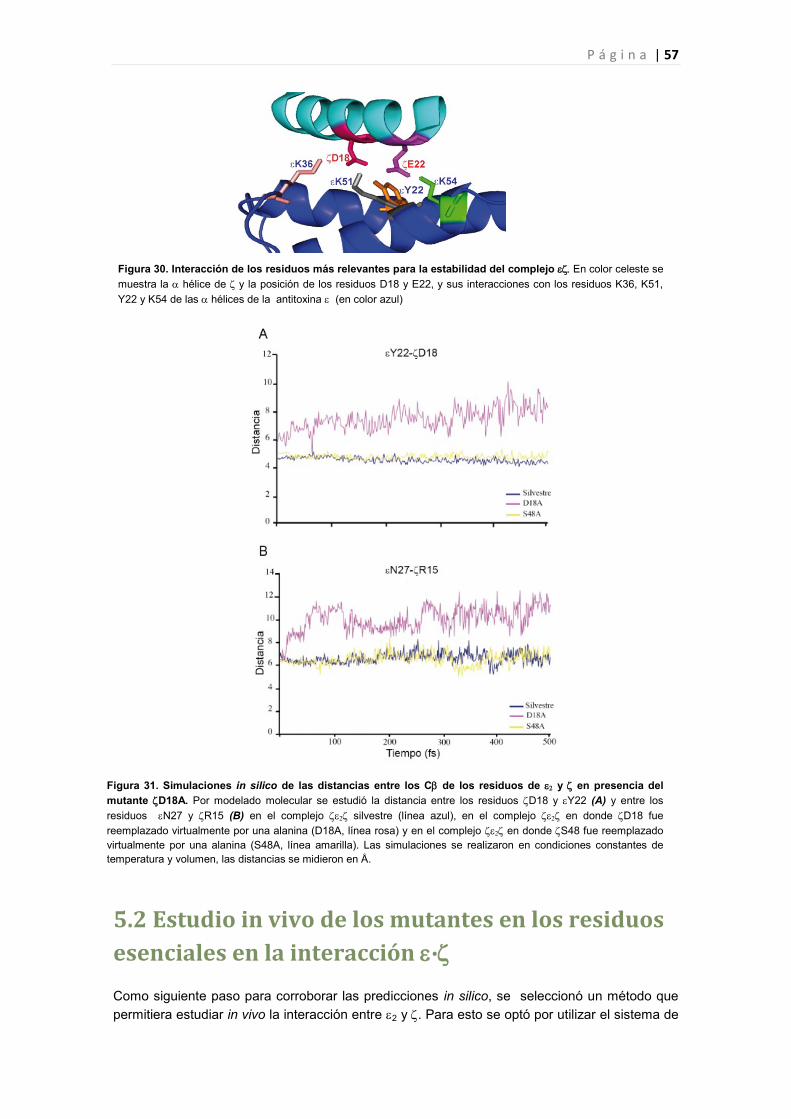
P á g i n a | 57
5.2 Estudio in vivo de los mutantes en los residuos
esenciales en la interacción ∙
Como siguiente paso para corroborar las predicciones in silico, se seleccionó un método que
permitiera estudiar in vivo la interacción entre 2 y . Para esto se optó por utilizar el sistema de
Figura 30. Interacción de los residuos más relevantes para la estabilidad del complejo En color celeste se
muestra la hélice de y la posición de los residuos D18 y E22, y sus interacciones con los residuos K36, K51,
Y22 y K54 de las hélices de la antitoxina (en color azul)
Figura 31. Simulaciones in silico de las distancias entre los C de los residuos de y en presencia del
mutante D18A. Por modelado molecular se estudió la distancia entre los residuos D18 y Y22 (A) y entre los
residuos N27 y R15 (B) en el complejo silvestre (línea azul), en el complejo en donde D18 fue
reemplazado virtualmente por una alanina (D18A, línea rosa) y en el complejo en donde S48 fue reemplazado
virtualmente por una alanina (S48A, línea amarilla). Las simulaciones se realizaron en condiciones constantes de
temperatura y volumen, las distancias se midieron en Å.

P á g i n a | 58
transferencia de energía de bioluminiscencia (BRET). La transferencia de energía se produce
cuando una proteína bioluminiscente, en este caso la Luciferasa de Renilla (Luc), en presencia
de un substrato es capaz de generar luz que luego será utilizada por una molécula aceptora
para fluorescer, en este caso la proteína verde fluorescente o GFP (del inglés- green
fluorescence protein). Las proteínas Luc y GFP no interaccionan naturalmente a menos que se
fusionen a ellas proteínas que si lo hagan. Si las proteínas fusionadas interaccionan, y Luc y
GFP se encuentran lo suficientemente cercanas (hasta 100 Å de distancia), Luc puede excitar
a GFP y ésta ultima emitir fluorescencia. De esta forma, se realizaron fusiones
transcripcionales de la proteína GFP a , por el extremo carboxilo terminal (:GFP) y de la
proteína Luc a por el extremo amino terminal (:Luc). Para estudiar si los aminoácidos D18 y
E22 de eran relevantes en la interacción con la antitoxina , se decidió mutar estos residuos a
alanina. Para esto se seleccionó una variante no tóxica de , la K46A, en donde el residuo
lisina 46 fue cambiado por una alanina. La lisina 46 se encuentra en el motivo Walker A que es
esencial para la toxicidad de pero no para la interacción con 2 (Meinhart et al., 2003). Fue
vital realizar el estudio de las interacciones entre 2 y con esta variante no tóxica, dado que
mutaciones en los residuos relevantes para la estabilidad del complejo TA podrían dejar libre a
la toxina y ésta, inhibir el crecimiento celular. Se construyeron los mutantes D18A-K46A:GFP y
E22A-K46A:GFP, así como también :Luc, en donde tanto el gen de :luc como el de :gfp o
sus variantes estaban bajo el control del promotor de la proteína .
Como primer paso para saber si la fusión :GFP no afectaba la actividad de la proteína, se
clonó este gen bajo el control de un promotor constitutivo (Pcat, promotor de cloranfenicol), y al
gen de la antitoxina bajo el control de un promotor inducible por IPTG (Plac). Como se muestra
en la tabla 7 cuando células de E. coli en crecimiento exponencial que habían crecido en
presencia de IPTG (+), se lavaron y resuspendieron en medio fresco sin IPTG se observó que
más del 99 % de las células perdían la capacidad de formar colonias tanto si se utilizaba la
proteína silvestre o la variante :GFP. Esto demostró que la actividad de la toxina fusionada a
la proteína verde fluorescente era la misma que la de la toxina silvestre, indicando que la
fusión a GFP no afectaba el plegamiento de , ni su actividad.
A continuación se evaluó la interacción de los complejos formados por las proteínas
:Luc,:GFP (); :LucK46A:GFP(-K46A); :Luc, K46A-D18A:GFP (- D18A-K46A);
:LucE22A-K46A:GFP (-E22A-K46A) células de E. coli (Figura 32), por medio de la
cuantificación de señal de BRET (relación 515/410, ver materiales y métodos). Una mutación
en un aminoácido relevante para la interacción ∙ daría una caída en la señal de BRET. Como
se esperaba, la mutación D18A resultó en una caída dramática de la señal de BRET, sin
Genes codificados en los plásmidos
Presencia de IPTG
UFC/ml (%)
+ Si (+) 100
+ No (-) 0,04
+ :gfp Si (+) 100
+ :gfp No (-) 0,03
Tabla 7. Efecto de :GFP inhibe la proliferación celular en E. coli. Las celulas se crecieron en presencia de
IPTG (+) y cuando llegaron a fase exponencial el cultivo se lavó y resuspendió en medio fresco con o sin IPTG (+
y –). A las dos horas se tomaron muestras y se plaquearon en LB.
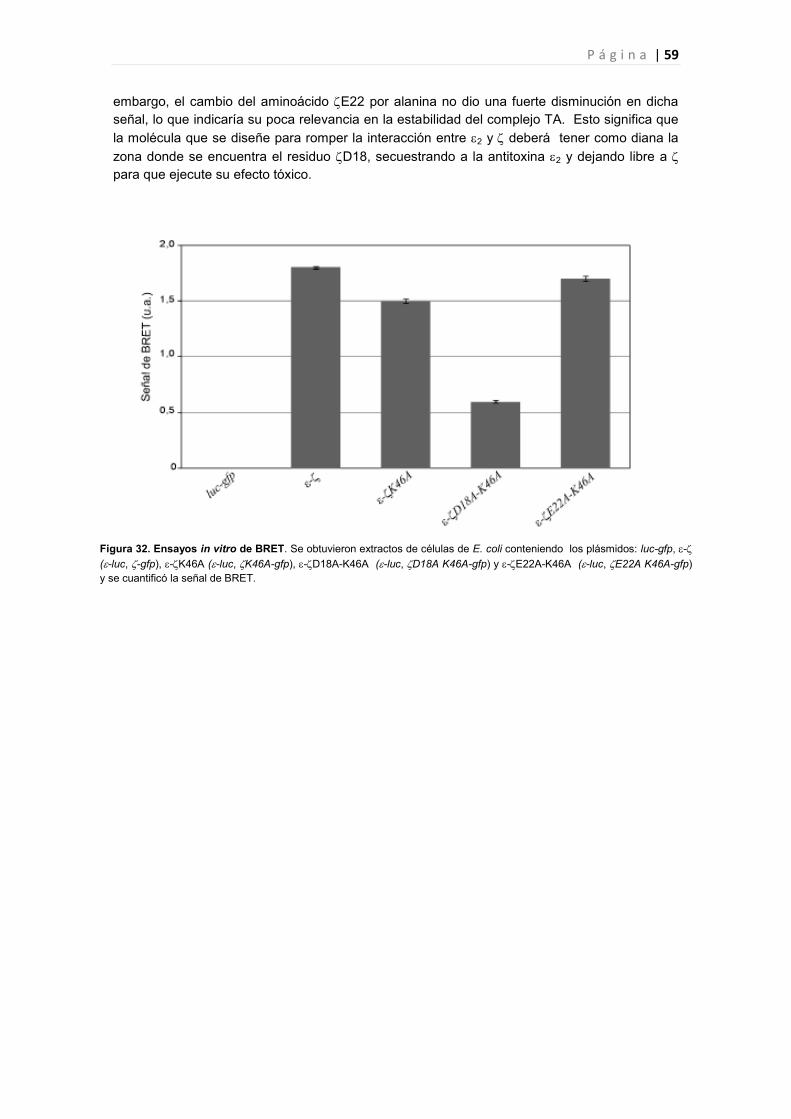
P á g i n a | 59
embargo, el cambio del aminoácido E22 por alanina no dio una fuerte disminución en dicha
señal, lo que indicaría su poca relevancia en la estabilidad del complejo TA. Esto significa que
la molécula que se diseñe para romper la interacción entre 2 y deberá tener como diana la
zona donde se encuentra el residuo D18, secuestrando a la antitoxina 2 y dejando libre a
para que ejecute su efecto tóxico.
Figura 32. Ensayos in vitro de BRET. Se obtuvieron extractos de células de E. coli conteniendo los plásmidos: luc-gfp, -
(-luc, -gfp), -K46A (-luc, K46A-gfp), -D18A-K46A (-luc, D18A K46A-gfp) y -E22A-K46A (-luc, E22A K46A-gfp)
y se cuantificó la señal de BRET.

P á g i n a | 60

Discusión


P á g i n a | 61
1. La segregación del plásmido pSM19035 es muy eficiente
El plásmido pSM19035 es de amplio rango de hospedador dentro del Filo Firmicutes, de bajo
número de copia (1 a 3 copias por célula) y se propaga manera muy estable. Esta estabilidad
se debe al uso de 4 estrategias: (i) Control de la replicación de manera ajustada, (ii)
maximización del número de copias para su distribución al azar, (iii) utilización de un sistema
de partición activa y (iv) Utilización de un sistema de toxina antitoxina. Estas estrategias se dan
de manera organizada y coordinada en la célula, siendo la proteína 2 la coordinadora principal
de estas funciones. En la actualidad, se encuentran ampliamente documentadas las funciones
de 2 en el control de la replicación o el número de copia del plásmido, así como también el
papel en la correcta segregación del sistema de partición activa (Pratto et al., 2008, de la Hoz
et al., 2000), sin embargo, no se había caracterizado hasta el momento si existía una conexión
entre el sistema de partición activa y el sistema de TA. La idea de que los sistemas de
estabilidad de los plásmidos funcionan en conjunto minimizando el coste biológico de tener un
producto tóxico no es nueva, ya que se propuso por primera vez para el sistema de partición
activa compuesto por las proteínas ParA y ParB de P1 y el sistema de TA MvpAT aislado del
plásmido de virulencia de Shigella flexneri (Brendler et al., 2004). En este trabajo se demostró
que células de E. coli que tenían un plásmido con el sistema de TA tenían un tiempo de
duplicación más largo que aquellas que además de tener le sistema MvpAT tenían el sistema
de partición activa. Esto se debía a que el sistema de partición activa aseguraba la correcta
segregación del plásmido de manera que todas las células hijas tenían una copia del mismo y
el sistema TA no se activaba. En el presente trabajo, se ha demostrado una interacción directa
entre el sistema de partición activa, , y el de TA, En este caso, se observó que la proteína
ParA () aumentaba la afinidad de la proteína ParB (2), y que lo hacia de manera
independiente de la unión a nucleótido y ADN. El hecho de que la actividad ATPasa de la
proteína ParA sea necesaria para la correcta segregación del plásmido (Pratto et al., 2008) y
no para la interacción con la proteína ParB indica que estas dos actividades son
independientes. De esta manera, se pueden proponer 2 funciones al sistema de partición
activa. La primera, asegurando la correcta segregación del plásmido, haciendo que cada célula
hija reciba al menos una copia del mismo, y evitando así, la inhibición del crecimiento mediada
por . La segunda, asegurando un control estricto de la concentración de toxina y antitoxina de
manera que no se produzca la inhibición del crecimiento de células que tienen una copia del
plásmido. Sin embargo, a pesar de estos resultados, aun es necesario conocer más detalles
acerca de la regulación del sistema TA, entre otras cosas determinar si existe una interacción
proteína-proteína entre el complejo inocuo y 2 o 2 y determinar si el sistema de TA regula
de alguna manera el promotor P, ya que se demostró que 2 no se encarga de su regulación
(de la Hoz et al., 2000)
2. Caracterización fisiológica del efecto tóxico de
Los experimentos realizados para caracterizar la función de in vivo, demostraron que la
toxina es capaz de inhibir más de 10.000 veces la UFC, con la muerte de una fracción de la
población. Recientemente se demostró que cuando el sistema MazEF se induce como
consecuencia de la acción del antibiótico espectinomicina, no solo se inhiben la síntesis de
proteínas como consecuencia de la actividad tóxica de MazF, sino que también se induce la
síntesis de proteínas de bajo peso molecular. Entre las proteínas inducidas se encuentran
proteínas de muerte (ClpP, SlyD o YfiD) y proteínas de superviviencia (SoxS, SoxR o DeoC).

P á g i n a | 62
De esta forma se propuso que una fracción de células sobrevive a expensas de la muerte de
otra parte de la población (Amitai et al., 2009). En presencia de la toxina se observó una
disminución en la síntesis de ADN, ARN y proteínas. Sin embargo, esta disminución no fue
total, ya que en los ensayos realizados se observó que aun había incorporación de 3H-timidina,
3H-uracilo y
3H-leucina después de 60 min bajo la exposición de la toxina (Figura 19). Más aún,
el análisis del proteoma de B. subtilis no demostró la inhibición selectiva de ninguna proteína
(Figura 18). Estos resultados nos llevaron a estudiar el transcriptoma de B. subtilis, para poder
identificar si existía alguna ruta metabólica especialmente inducida o reprimida. Los resultados
obtenidos a partir del análisis de genes reprimidos en respuesta a la toxina Y83C mostraron
genes involucrados principalmente en tres tipos de rutas metabólicas: en el metabolismo de
carbohidratos, en el metabolismo de nucleótidos y en el metabolismo de lípidos. Los productos
de los genes reprimidos están involucrados en procesos esenciales para la bacteria, sin
embargo, no son suficientes para inducir muerte celular de toda la población. La represión de
genes involucrados en la glícolisis tales como yqeC, mtlD, glpK, glpD, yvkC, gutP, gntK, gntR y
gntZ (Figura 33),
podría ser una estrategia de la célula para minimizar el gasto energético que supone el
metabolismo de carbohidratos, dejando a las células con un metabolismo mínimo o de
mantenimiento hasta que la situación de estrés se revierta. Es importante destacar que el
producto del gen yqeC forma parte de la vía de las pentosas fosfato. Esta vía es esencial para
la síntesis de nucleótidos, indicando que es probable que esté reprimida. El producto de los
genes reprimidos que estaban involucrados en el metabolismo de ácidos nucleídos formaban
parte de un operón involucrado en la síntesis de pirimidinas (dra, pdp y nupC, algunos de los
productos de estos genes se puede ver en la Figura 34). Sorprendentemente, el represor de
este operón también estaba reprimido, el producto del gen deoR. La represión de DeoR no es
Figura 33. Principales vías involucradas en la glicolisis. En rojo se muestran las enzimas que son codificadas por
genes esenciales y en azul aquellas cuyos genes que las codifican no son esenciales. Con una cruz roja se
representan los productos de los genes que fueron reprimidos por la toxina Y83C

P á g i n a | 63
contradictoria con la represión del operón, ya que cuando disminuyen los niveles de DeoR, el
operón está sometido a la represión catabólica mediada por las proteínas CcpA y HPr-
fosforilada que se unen a las regiones CRE que se encuentran en el promotor del operón (Zeng
et al., 2000). Esta represión seria una manera indirecta de confirmar que los niveles de UTP, al
igual que los de GTP y ATP están disminuidos (Figura 21 y Figura 34), ya que el
producto de los genes nupC y pdp están involucrados en la síntesis de pirimidinas (la proteína
NupC se encarga de transportar timidina y uridina a la célula). Los genes involucrados en el
metabolismo de lípidos forman parte de las rutas de biosíntesis de ácidos grasos y fosfolípidos
y mucho de ellos son esenciales (Figura 35). Esto podría indicar que la muerte de una fracción
de las células se dé por medio de la pérdida del potencial de membrana, explicando así que
una parte de la población tratada con Y83C se tiña con IP (Figura 16B y Tabla 5). En cuanto a
los genes inducidos, a diferencia de MazF, no se han encontrado indicios de que sus productos
estén involucrados en la muerte celular. No se encontró aumentada la síntesis de proteínas
involucradas en ROS, lo que confirma nuevamente que a diferencia de MazF, la toxina no
induce senescencia ni inducción del sistema SOS (Lioy et al., 2006). De los genes inducidos
por la toxina los más importantes de destacar son aquellos involucrados en la síntesis de
nucleótidos pyrR, pyrP, pyrAB, pyrB y pyrE (alguno de ellos pueden verse en la Figura 34).
Según los niveles de uridina y guanosina PyrR es capaz de reprimir o des-reprimir la expresión
de los genes del operón pir. Se demostró en B. subtilis que GTP es probablemente el
modulador fisiológico más importante, seguido de UTP. De esta forma los niveles de GTP y
UTP son una señal que utiliza la célula para determinar que el resto de factores necesarios
para la síntesis del ADN y ARN se encuentran normales, es decir, que la fuente de carbono,
nitrógeno y fuentes de energía son suficientes.
Figura 34. Principales vías involucradas en la síntesis de pirimidinas. En rojo se muestran las enzimas que son
codificadas por genes esenciales y en azul aquellas cuyos genes que las codifican no son esenciales. Con una cruz roja
se representan los productos de los genes que fueron reprimidos por la toxina y con una flecha verde aquellos que
fueron inducidos por Y83C
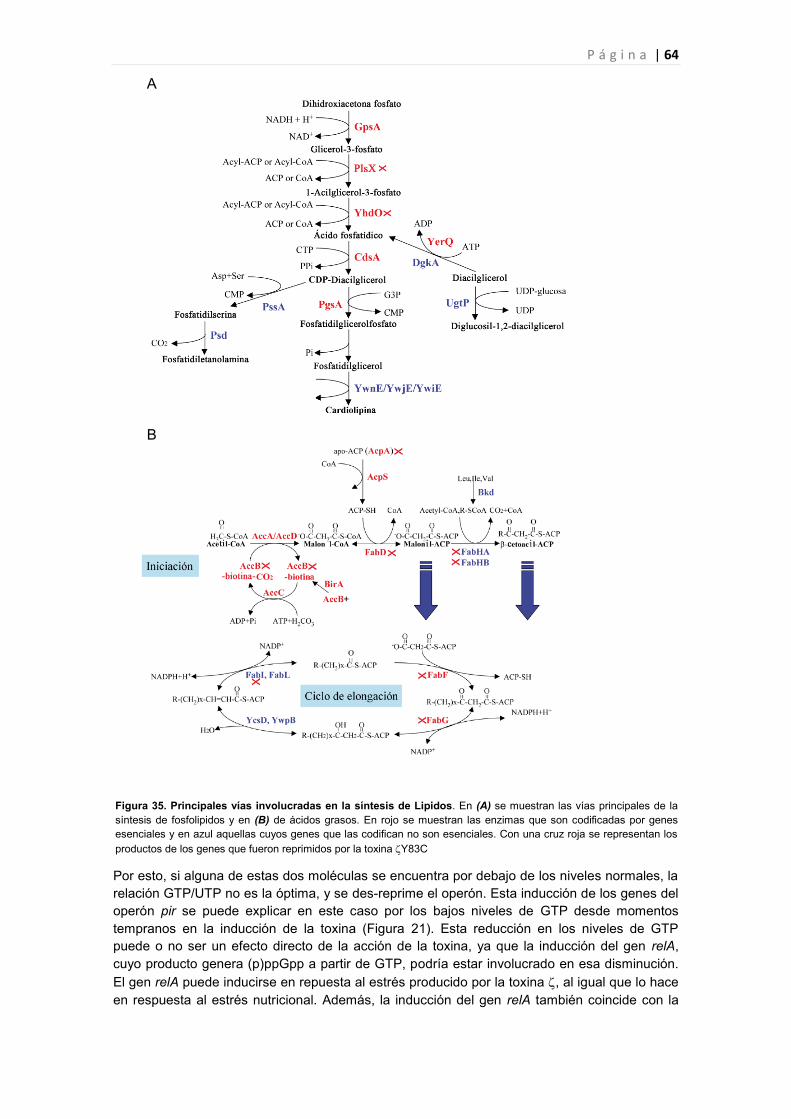
P á g i n a | 64
Por esto, si alguna de estas dos moléculas se encuentra por debajo de los niveles normales, la
relación GTP/UTP no es la óptima, y se des-reprime el operón. Esta inducción de los genes del
operón pir se puede explicar en este caso por los bajos niveles de GTP desde momentos
tempranos en la inducción de la toxina (Figura 21). Esta reducción en los niveles de GTP
puede o no ser un efecto directo de la acción de la toxina, ya que la inducción del gen relA,
cuyo producto genera (p)ppGpp a partir de GTP, podría estar involucrado en esa disminución.
El gen relA puede inducirse en repuesta al estrés producido por la toxina , al igual que lo hace
en respuesta al estrés nutricional. Además, la inducción del gen relA también coincide con la
Figura 35. Principales vías involucradas en la síntesis de Lipidos. En (A) se muestran las vías principales de la
síntesis de fosfolipidos y en (B) de ácidos grasos. En rojo se muestran las enzimas que son codificadas por genes
esenciales y en azul aquellas cuyos genes que las codifican no son esenciales. Con una cruz roja se representan los
productos de los genes que fueron reprimidos por la toxina Y83C
A
B

P á g i n a | 65
represión del gen homólogo a mazG, yvdC. La función principal de MazG en E. coli es la de
disminuir los niveles de (p)ppGpp aumentando el período de supervivencia de las células bajo
estrés nutricional. Robert Switzer y colaboradores explicaron que si los niveles de GTP están
reducidos, y no los de UTP, se produciría una mayor represión del operón pir. Por lo que estos
resultados indicarían que los niveles de UTP también se encuentran reducidos, ya que se
observa una inducción del operón (Jorgensen et al., 2008).
3. La antitoxina 2es capaz de recuperar la actividad metabólica de las células que estaban bajo el efecto de .
Si se compara el efecto de la toxina sobre las células con el que ejercen otros sistemas TA,
se puede ver que el fenotipo producido es similar al observado para las toxinas de los sistemas
RelBE y MazEF, ya que al igual que en estos sistemas, la antitoxina 2 es capaz de revertir el
efecto tóxico producido por (Pedersen et al., 2002). Esta reversión del efecto tóxico confirma
que las células se encontraban en un estado bacteriostático, a la espera de que la situación de
estrés se revierta. Estos resultados son indicativos de que la interacción de la toxina con su
diana debe ser reversible. La reversión del efecto tóxico se observo tanto en células de E. coli
como de B. subtilis. Sin embargo, el grupo de Anna Engelberg-Kulka demostró que la reversión
del efecto tóxico de MazF era limitada, llegando a un punto de no retorno en donde la toxicidad
mediada por la expresión de MazF no podía ser revertida por MazE (Amitai et al., 2004).
Resultados utilizando la cepa BG1125, que presenta alto niveles de expresión de la toxina ,
demostraronn que el efecto tóxico podía ser revertido hasta 4 hs post inducción de la toxina
(Figura 23), aunque había una fracción de la población que no podía recuperarse. En E. coli, se
demostró que esta ventana de tiempo aumentaba hasta 5 hs cuando se utilizaba un promotor
constitutivo, expresando menores niveles de la toxina (Figura 24) (Lioy et al., 2006). Todavía no
se ha demostrado, por problemas metodológicos, si ese punto de “no retorno” se puede
detectar en presencia de concentraciones fisiológicas de la toxina .
4. La toxina podría modificar el pool de nucleótidos induciendo un estado bacteriostático.
Todos los resultados expuestos en este trabajo, junto con el análisis de la estructura cristalina
de la toxina, así como también el estudio de mutantes en el sitio catalítico, indicarían que
estaría modificando el pool de nucleótidos. Esta modificación se podría dar por la fosforilación o
desfosforilación del sustrato, generando variantes de nucleótidos que sean tóxicos para la
célula. Esto, explicaría porque sucesivos intentos de encontrar la diana ya sea por medio de la
obtención de mutantes resistentes a la toxina o a través de ensayos de cross-linking no fueron
exitosos. De esta forma, la toxina modificaría los nucleótidos afectando desde la replicación
hasta la síntesis de carbohidratos, generando un efecto pleitropico en la célula. Sin embargo
esta hipótesis aun debe ser confirmada. Por lo tanto, el efecto producido por la toxina es tal
que induce un estado bacteriostático en gran parte de la población. Este estado se ve
caracterizado por una disminución en el metabolismo, priorizando solo la síntesis de rutas
metabólicas de mantenimiento. La síntesis de ADN, ARN y proteínas se encuentra detenida
esperando que la situación revierta por la síntesis de novo de 2.

P á g i n a | 66
5. La toxina induce un estado bacteriostático y podría disparar la muerte de una pequeña fracción de la población, que asegurará la supervivencia del resto, pero no parece inducir senescencia
Como se expuso en la parte II de resultados, los sistemas de TA fueron relacionados con
diversas teorías (sistemas de muerte programada, sistemas que inducen células durmientes o
persistentes o sistemas que inducen senescencia). En este trabajo se llevaron a cabo una serie
de experimentos para determinar cuál era el rol que la toxina realizaba en in vivo. Los
resultados obtenidos permitieron descartar la hipótesis de que estaba involucrada en la
generación de senescencia, ya que no se pudo demostrar que la toxina estuviera involucrada
en la formación de ROS. En cuanto a su papel como parte de un sistema de muerte celular
programada, se puede decir que si bien la toxina induce la muerte de una fracción de la
población (lisis del 20 % de las células junto con una fracción que no es capaz de revertir el
efecto tóxico), no se puede descartar que esto sea debido a los niveles de toxina con los que
se realiza el experimento, ya que por dificultades técnicas, no es posible trabajar con el sistema
en condiciones fisiológicas. Por lo tanto, es probable que el papel de dentro de la fisiología
celular es el de inducir un estado bacteriostático. Este estado, se revertiría (o al menos la
mayor parte de la población sería capaz de hacerlo) en presencia de la síntesis de novo de 2.
Fisiológicamente, esto significa que si alguno de los otros sistemas de estabilidad del plásmido
fallase, sería el sistema TA quien detendría el crecimiento celular, dejando a las bacterias en
un estado de arresto o estasis. Este estado, se perpetuará tanto como sea necesario, hasta
que luego de un tiempo en arresto inevitablemente las células morirán, si no logran recuperar el
plásmido u obtener, al menos, una fuente de síntesis de novo de 2. Este modelo contrastaría
con la idea de que los sistemas TA de plásmidos inducen la muerte post-segregacional de las
bacterias que lo pierden, aunque esto no es así. El hecho de parar el crecimiento a una célula
que ha perdido el plásmido aportará una gran ventaja al resto de células que no lo perdieron,
de forma tal que en unas pocas duplicaciones pueden llegar a colonizar el nicho. Las células
que entran en arresto y no son capaces de captar nuevo ADN que sintetice la antitoxina2 de
novo, serán utilizadas como fuente de nutrientes por el resto de bacterias que seguirán
creciendo.
6. La toxina como diana antimicrobiana
6.1 La toxina aumenta la eficacia de los antibióticos sin
producir células persistentes.
Las células persistentes son células que no crecen, ya que se encuentran en un estado
“durmiente”, y cuyo fenotipo no pasa de generación en generación. Recientemente varios
trabajos han involucrando a los sistemas TA en la generación de células persistentes o
tolerantes a los antibióticos (Keren et al., 2004). Esta hipótesis se demostró para algunas
toxinas, pero en especial para el sistema HipBA, que fue el primero en describirse como el
responsable de producir un fenotipo híper persistente en E. coli (Moyed & Bertrand, 1983,
Korch et al., 2003, Correia et al., 2006). Se propuso que el modo de generación de células

P á g i n a | 67
persistentes se producía cuando, por variaciones estocásticas, los niveles de toxina
aumentaban. De esta manera, cuando la toxina se unía a su diana, afectaba la diana del
antibiótico, de forma que se volvía no accesible al mismo. Por ej., las toxinas HipA ó RelB
inhiben la traducción, de forma que los antibióticos que también actúan sobre este proceso
celular no tendrían efecto y darían lugar a células tolerantes al antibiótico (Figura 5). Además la
inhibición de la traducción produce también una inhibición en la síntesis de ADN y en la síntesis
de la pared celular, explicando porque la gran mayoría de los antibióticos dejan de ser eficaces.
Como la inhibición de la síntesis de proteínas mediadas por la toxina no es del 100%,
nuevamente debido a un proceso estocástico, se vuelve a sintetizar la antitoxina. De esta forma
se revierte el efecto tóxico de la toxina y se logra la supervivencia de un mayor número de
células, que habían sido insensibles al efecto del antibiótico (Keren et al., 2004). Algunos años
más tarde se encontró que no solo el efecto de las toxinas producían un aumento en la
persistencia a antibióticos, sino que también la sobre-expresión del producto de otros genes
tales como plsB y glpD (síntesis de fosfolipidos), así como también los niveles de glicerol-3-
fosfato estaban involucrados en la formación de células persistentes (Spoering et al., 2006). Sin
embargo, los ensayos realizados para determinar si la toxina participaba en la producción de
este tipo de células demostraron que por el contrario, no solo no induce tolerancia a los
antibióticos sino que además aumenta la eficacia de los mismos. Estos resultados, también se
confirmaron con el patrón de genes afectado por la toxina, que fueron completamente distintos
a los que se observaron en las células persistentes. De hecho, los genes plsX (homólogo a
plsB de E. coli) y glpD se encuentran reprimidos en presencia de la toxina . Esto indica que si
bien la toxina induce un estado bacteriostático en la mayoría de la población, éste es
diferente al que manifiestan las células persistentes. Esta diferencia puede deberse a la
naturaleza de su diana, ya que los sistemas en los que se describió el fenotipo de persistentes
tienen como diana la inhibición de la síntesis de proteínas (aunque a través de mecanismos
distintos). La diana de la toxina es aun desconocida, pero gracias a los estudios realizados en
este trabajo se sabe que no es la traducción, ni la transcripción ni la replicación (al menos no
directamente). Por esto, se puede proponer que el mecanismo para el aumento de la
sensibilidad a los antibióticos mediado por se debe a que la diana de es distinta a la del
antibiótico utilizado, y por esto no genera tolerancia (Figura 36). No obstante, aunque se hayan
probado representantes de las familias más usadas de antibióticos (un inhibidor de la síntesis
de la pared celular, un inhibidor de la síntesis de lípidos, un inhibidor de la síntesis de proteínas
y un inhibidor de la replicación) no puede descartarse el hecho poco probable de que el uso de
otros antibióticos en conjunto con la toxina produzcan persistencia, ya que existe una gran
diversidad de antibióticos con distintas dianas pero que inhiben los mismos procesos
biológicos, que aun no fueron probados. Una vez que la diana de sea conocida, es necesario
volver a evaluar esta actividad, utilizando antibióticos que inhiban el crecimiento celular de la
misma manera que lo hace la toxina.
Figura 36: Modelo de sobre la generación de células sensibles a antibióticos. En condiciones normales, se
sintetizan suficientes cantidades de y , que forman un complejo inocuo muy estable. Cuando por razones estocásticas
se produce un exceso de toxina (situación señalada por un rayo), la toxina queda libre y une su diana. En presencia de
un antibiótico, el aumento de la sensibilidad al mismo puede explicarse si las dianas de y del antibiótico son

P á g i n a | 68
6.2 Diseño de nuevas dianas antimicrobianas.
Muchos autores han propuesto el uso de los sistemas TA como noveles dianas
antimicrobianas. Siempre se ha manifestado el interés de identificar los aminoácidos más
importantes en la interacción T∙A para desarrollar moléculas que sean capaces de romper
dicha interacción. La idea se basa en dejar libre a la toxina de forma que aun pueda
interaccionar con su diana y de esta forma inhibir el crecimiento de las bacterias que lleven el
plásmido. Esta estrategia también puede extenderse a aquellos sistemas TA que se
encuentran en el cromosoma (Nieto et al., 2006, Moritz & Hergenrother, 2007, Williams &
Hergenrother, 2008). En este, trabajo, a partir del modelado molecular del cristal se
identificaron las interacciones más importantes para la estabilidad del complejo. De esta
manera se predijeron 4 residuos de como los más importantes para la interacción con
(D18, E22, R158 y R165). De estas 4 interacciones se demostró por medio de ensayos de
BRET que la del residuo D18 con los aminoácidos K36 y K51 de era la más importante
(Figura 31 y 32). Este ensayo no solo permitió validar las predicciones in silico, sino que
también permitió desarrollar un sistema capaz de permitir el procesamiento y la evaluación de
un gran número de moléculas con potencial antimicrobiano usando HTS (del inglés- High
throughput screening). De esta manera, la molécula que se diseñe para inhibir la interacción de
la toxina con la antitoxina , debera “imitar” a la toxina, de forma tal que el nuevo producto
que se forme (antitoxina-molécula) deje libre a la toxina y ésta pueda ejecutar su efecto tóxico.
Para esto, la molécula deberá imitar la región cercana al resido D18 y E22, interaccionando
con los residuos de K36, K51, Y22 y K54 (Figura 30). En colaboración con el Laboratorio
Salvat (Barcelona) se evaluaron más de 49.000.000 de péptidos diseñados específicamente
para desestabilizar al complejo . Si bien el complejo pudo desestabilizarse,
lamentablemente no se logró identificar un péptido responsable de dicha desestabilización, sino
que se observó que un grupo de dos o más péptidos fueron responsables de la
desestabilización del complejo (Lioy et al., 2009). Estos resultados demostraron que aunque el
complejo puede ser desestabilizado in vitro, se necesitan evaluar aun mas compuestos
hasta que se pueda identificar una única molécula que tenga éxito en la desestabilización del
complejo TA. Una vez que se tenga esa molécula se podrá trabajar para aumentar la eficiencia
de los antibióticos.

Conclusiones


C o n c l u s i o n e s | 69
Conclusiones
Las conclusiones que se extrajeron de este trabajo son las siguientes:
1. La proteína 2 del sistema de partición activa, trabaja en conjunto con la proteína 2 para
asegurar la correcta regulación del operón , de forma tal que la proteína 2 es capaz de
aumentar la afinidad de la proteína 2 por la región promotora del operón más de 4
veces y lo hace de manera independiente de la unión a ADN y nucleótido. Esta actividad no
solo minimiza el coste biológico de tener un sistema de TA sino que también permite una
correcta regulación de la expresión del sistema TA.
2. La expresión de la toxina en células de B. subtilis y E. coli inhibe la proliferación celular,
induciendo un estado bacteriostático, junto con la lisis de una pequeña fracción de la
población.
3. El efecto tóxico de se caracteriza por la inhibición de la síntesis ADN, ARN, proteínas y
purinas. El análisis de los genes cuya expresión se encuentran modificados por la toxina
mostró que 8 genes son esenciales y forman parte de rutas biosintéticas de lípidos. También
se encuentran modificados genes involucrados en la biosíntesis de carbohidratos y ácidos
nucleicos. Alguno de los genes inducidos por la toxina sugiere que los niveles de UTP
también se encuentran disminuidos.
4. El efecto inducido por la toxina puede ser revertido por la síntesis de novo de la
antitoxina tanto en B. subtilis como en E. coli. Esta reversión depende de la concentración
de toxina, ya que si los niveles de toxina son bajos la ventana de tiempo durante el cual el
efecto es reversible es mayor.
5. La toxina no induce un aumento de células persistentes en B. subtilis, sino por el
contrario es capaz de aumentar la sensibilidad al antibiótico.
6. Por medio de modelado molecular se identificaron los residuos D18 y E22 como los
más importantes para la estabilidad del complejo . Por medio de ensayos de BRET se
validó la predicción demostrando que los mutantes D18A y E22A forman un complejo
inestable con la antitoxina

C o n c l u s i o n e s | 70

Bibliografía


P á g i n a | 71
Bibliografía
Aizenman, E., H. Engelberg-Kulka & G. Glaser, (1996) An Escherichia coli chromosomal "addiction module" regulated by guanosine [corrected] 3',5'-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci U S A 93: 6059-6063.
Alonso, J. C., S. Ayora, I. Canosa, F. Weise & F. Rojo, (1996) Site-specific recombination in gram-positive theta-replicating plasmids. FEMS Microbiol Lett 142: 1-10.
Amitai, S., I. Kolodkin-Gal, M. Hananya-Meltabashi, A. Sacher & H. Engelberg-Kulka, (2009) Escherichia coli MazF leads to the simultaneous selective synthesis of both "death proteins" and "survival proteins". PLoS Genet 5: e1000390.
Amitai, S., Y. Yassin & H. Engelberg-Kulka, (2004) MazF-mediated cell death in Escherichia coli: a point of no return. J Bacteriol 186: 8295-8300.
Bahassi, E. M., M. H. O'Dea, N. Allali, J. Messens, M. Gellert & M. Couturier, (1999) Interactions of CcdB with DNA gyrase. Inactivation of Gyra, poisoning of the gyrase-DNA complex, and the antidote action of CcdA. J Biol Chem 274: 10936-10944.
Bernard, P. & M. Couturier, (1992) Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J Mol Biol 226: 735-745.
Bernard, P., K. E. Kezdy, L. Van Melderen, J. Steyaert, L. Wyns, M. L. Pato, P. N. Higgins & M. Couturier, (1993) The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. J Mol Biol 234: 534-541.
Bernhardt, J., K. Buttner, C. Scharf & M. Hecker, (1999) Dual channel imaging of two-dimensional electropherograms in Bacillus subtilis. Electrophoresis 20: 2225-2240.
Birnboim, H. C. & J. Doly, (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7: 1513-1523.
Black, D. S., B. Irwin & H. S. Moyed, (1994) Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 176: 4081-4091.
Black, D. S., A. J. Kelly, M. J. Mardis & H. S. Moyed, (1991) Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 173: 5732-5739.
Bott, K. F. & G. A. Wilson, (1968) Metabolic and nutritional factors influencing the development of competence for transfection of Bacillus subtilis. Bacteriol Rev 32: 370-378.
Brantl, S., D. Behnke & J. C. Alonso, (1990) Molecular analysis of the replication region of the conjugative Streptococcus agalactiae plasmid pIP501 in Bacillus subtilis. Comparison with plasmids pAM beta 1 and pSM19035. Nucleic Acids Res 18: 4783-4790.
Brantl, S. & E. G. Wagner, (1997) Dual function of the copR gene product of plasmid pIP501. J Bacteriol 179: 7016-7024.
Brendler, T., L. Reaves & S. Austin, (2004) Interplay between plasmid partition and postsegregational killing systems. J Bacteriol 186: 2504-2507.
Camacho, A. G., R. Misselwitz, J. Behlke, S. Ayora, K. Welfle, A. Meinhart, B. Lara, W. Saenger, H. Welfle & J. C. Alonso, (2002) In vitro and in vivo stability of the epsilon2zeta2 protein complex of the broad host-range Streptococcus pyogenes pSM19035 addiction system. Biol Chem 383: 1701-1713.
Canosa, I., G. Lopez, F. Rojo, M. R. Boocock & J. C. Alonso, (2003) Synapsis and strand exchange in the resolution and DNA inversion reactions catalysed by the beta recombinase. Nucleic Acids Res 31: 1038-1044.
Canosa, I., R. Lurz, F. Rojo & J. C. Alonso, (1998) beta Recombinase catalyzes inversion and resolution between two inversely oriented six sites on a supercoiled DNA substrate and only inversion on relaxed or linear substrates. J Biol Chem 273: 13886-13891.
Canosa, I., F. Rojo & J. C. Alonso, (1996) Site-specific recombination by the beta protein from the streptococcal plasmid pSM19035: minimal recombination sequences and crossing over site. Nucleic Acids Res 24: 2712-2717.
Ceglowski, P., A. Boitsov, S. Chai & J. C. Alonso, (1993a) Analysis of the stabilization system of pSM19035-derived plasmid pBT233 in Bacillus subtilis. Gene 136: 1-12.
Ceglowski, P., A. Boitsov, N. Karamyan, S. Chai & J. C. Alonso, (1993b) Characterization of the effectors required for stable inheritance of Streptococcus pyogenes pSM19035-derived plasmids in Bacillus subtilis. Mol Gen Genet 241: 579-585.

P á g i n a | 72
Correia, F. F., A. D'Onofrio, T. Rejtar, L. Li, B. L. Karger, K. Makarova, E. V. Koonin & K. Lewis, (2006) Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J Bacteriol 188: 8360-8367.
Christensen, S. K., M. Mikkelsen, K. Pedersen & K. Gerdes, (2001) RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A 98: 14328-14333.
Dao-Thi, M. H., L. Van Melderen, E. De Genst, H. Afif, L. Buts, L. Wyns & R. Loris, (2005) Molecular basis of gyrase poisoning by the addiction toxin CcdB. J Mol Biol 348: 1091-1102.
de la Hoz, A. B., S. Ayora, I. Sitkiewicz, S. Fernandez, R. Pankiewicz, J. C. Alonso & P. Ceglowski, (2000) Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc Natl Acad Sci U S A 97: 728-733.
de la Hoz, A. B., F. Pratto, R. Misselwitz, C. Speck, W. Weihofen, K. Welfle, W. Saenger, H. Welfle & J. C. Alonso, (2004) Recognition of DNA by omega protein from the broad-host range Streptococcus pyogenes plasmid pSM19035: analysis of binding to operator DNA with one to four heptad repeats. Nucleic Acids Res 32: 3136-3147.
DeNap, J. C. & P. J. Hergenrother, (2005) Bacterial death comes full circle: targeting plasmid replication in drug-resistant bacteria. Org Biomol Chem 3: 959-966.
Diago-Navarro, E., M. B. Kamphuis, R. Boelens, A. Barendregt, A. J. Heck, R. H. van den Heuvel & R. Diaz-Orejas, (2009a) A mutagenic analysis of the RNase mechanism of the bacterial Kid toxin by mass spectrometry. FEBS J 276: 4973-4986.
Diago-Navarro, E., L. Mora, R. H. Buckingham, R. Diaz-Orejas & M. Lemonnier, (2009b) Novel Escherichia coli RF1 mutants with decreased translation termination activity and increased sensitivity to the cytotoxic effect of the bacterial toxins Kid and RelE. Mol Microbiol 71: 66-78.
Dwyer, D. J., M. A. Kohanski, B. Hayete & J. J. Collins, (2007) Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol 3: 91.
Engelberg-Kulka, H., S. Amitai, I. Kolodkin-Gal & R. Hazan, (2006) Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet 2: e135.
Gerdes, K., (2000) Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J Bacteriol 182: 561-572.
Gerdes, K., F. W. Bech, S. T. Jorgensen, A. Lobner-Olesen, P. B. Rasmussen, T. Atlung, L. Boe, O. Karlstrom, S. Molin & K. von Meyenburg, (1986) Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J 5: 2023-2029.
Gerdes, K., S. K. Christensen & A. Lobner-Olesen, (2005) Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3: 371-382.
Gerdes, K. & E. G. Wagner, (2007) RNA antitoxins. Curr Opin Microbiol 10: 117-124. Gotfredsen, M. & K. Gerdes, (1998) The Escherichia coli relBE genes belong to a new toxin-
antitoxin gene family. Mol Microbiol 29: 1065-1076. Gross, M., I. Marianovsky & G. Glaser, (2006) MazG -- a regulator of programmed cell death in
Escherichia coli. Mol Microbiol 59: 590-601. Hanahan, D., (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:
557-580. Hazan, R. & H. Engelberg-Kulka, (2004) Escherichia coli mazEF-mediated cell death as a
defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics 272: 227-234.
Hazan, R., B. Sat, M. Reches & H. Engelberg-Kulka, (2001) Postsegregational killing mediated by the P1 phage "addiction module" phd-doc requires the Escherichia coli programmed cell death system mazEF. J Bacteriol 183: 2046-2050.
Heidrich, N. & S. Brantl, (2007) Antisense RNA-mediated transcriptional attenuation in plasmid pIP501: the simultaneous interaction between two complementary loop pairs is required for efficient inhibition by the antisense RNA. Microbiology 153: 420-427.
Jiang, Y., J. Pogliano, D. R. Helinski & I. Konieczny, (2002) ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44: 971-979.
Jorgensen, C. M., C. J. Fields, P. Chander, D. Watt, J. W. Burgner, 2nd, J. L. Smith & R. L. Switzer, (2008) pyr RNA binding to the Bacillus caldolyticus PyrR attenuation protein -

P á g i n a | 73
characterization and regulation by uridine and guanosine nucleotides. FEBS J 275: 655-670.
Kamada, K., F. Hanaoka & S. K. Burley, (2003) Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol Cell 11: 875-884.
Kamphuis, M. B., A. M. Bonvin, M. C. Monti, M. Lemonnier, A. Munoz-Gomez, R. H. van den Heuvel, R. Diaz-Orejas & R. Boelens, (2006) Model for RNA binding and the catalytic site of the RNase Kid of the bacterial parD toxin-antitoxin system. J Mol Biol 357: 115-126.
Kamphuis, M. B., M. C. Monti, R. H. van den Heuvel, J. Lopez-Villarejo, R. Diaz-Orejas & R. Boelens, (2007a) Structure and function of bacterial kid-kis and related toxin-antitoxin systems. Protein Pept Lett 14: 113-124.
Kamphuis, M. B., M. C. Monti, R. H. van den Heuvel, S. Santos-Sierra, G. E. Folkers, M. Lemonnier, R. Diaz-Orejas, A. J. Heck & R. Boelens, (2007b) Interactions between the toxin Kid of the bacterial parD system and the antitoxins Kis and MazE. Proteins 67: 219-231.
Keren, I., D. Shah, A. Spoering, N. Kaldalu & K. Lewis, (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186: 8172-8180.
Khoo, S. K., B. Loll, W. T. Chan, R. L. Shoeman, L. Ngoo, C. C. Yeo & A. Meinhart, (2007) Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J Biol Chem 282: 19606-19618.
Kohanski, M. A., D. J. Dwyer, B. Hayete, C. A. Lawrence & J. J. Collins, (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130: 797-810.
Kolodkin-Gal, I. & H. Engelberg-Kulka, (2008) The extracellular death factor: physiological and genetic factors influencing its production and response in Escherichia coli. J Bacteriol 190: 3169-3175.
Kolodkin-Gal, I., R. Hazan, A. Gaathon, S. Carmeli & H. Engelberg-Kulka, (2007) A linear pentapeptide is a quorum-sensing factor required for mazEF-mediated cell death in Escherichia coli. Science 318: 652-655.
Kolodkin-Gal, I., B. Sat, A. Keshet & H. Engelberg-Kulka, (2008) The communication factor EDF and the toxin-antitoxin module mazEF determine the mode of action of antibiotics. PLoS Biol 6: e319.
Korch, S. B., T. A. Henderson & T. M. Hill, (2003) Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol Microbiol 50: 1199-1213.
Korch, S. B. & T. M. Hill, (2006) Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J Bacteriol 188: 3826-3836.
Kunst, F., N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessieres, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S. K. Choi, J. J. Codani, I. F. Connerton, A. Danchin & et al., (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390: 249-256.
Lee, S., M. H. Kim, B. S. Kang, J. S. Kim, G. H. Kim, Y. G. Kim & K. J. Kim, (2008) Crystal structure of Escherichia coli MazG, the regulator of nutritional stress response. J Biol Chem 283: 15232-15240.
Lewis, K., (2007) Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5: 48-56. Lioy, V. S., M. T. Martin, A. G. Camacho, R. Lurz, H. Antelmann, M. Hecker, E. Hitchin, Y.
Ridge, J. M. Wells & J. C. Alonso, (2006) pSM19035-encoded zeta toxin induces stasis followed by death in a subpopulation of cells. Microbiology 152: 2365-2379.
Lioy, V. S., O. Rey, D. Balsa, T. Pellicer & J. C. Alonso, (2009) A toxin-antitoxin module as a target for antimicrobial development. Plasmid.
Madl, T., L. Van Melderen, N. Mine, M. Respondek, M. Oberer, W. Keller, L. Khatai & K. Zangger, (2006) Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J Mol Biol 364: 170-185.
Maki, S., S. Takiguchi, T. Miki & T. Horiuchi, (1992) Modulation of DNA supercoiling activity of Escherichia coli DNA gyrase by F plasmid proteins. Antagonistic actions of LetA (CcdA) and LetD (CcdB) proteins. J Biol Chem 267: 12244-12251.

P á g i n a | 74
Meinhart, A., J. C. Alonso, N. Strater & W. Saenger, (2003) Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc Natl Acad Sci U S A 100: 1661-1666.
Miki, T., J. A. Park, K. Nagao, N. Murayama & T. Horiuchi, (1992) Control of segregation of chromosomal DNA by sex factor F in Escherichia coli. Mutants of DNA gyrase subunit A suppress letD (ccdB) product growth inhibition. J Mol Biol 225: 39-52.
Monti, M. C., A. M. Hernandez-Arriaga, M. B. Kamphuis, J. Lopez-Villarejo, A. J. Heck, R. Boelens, R. Diaz-Orejas & R. H. van den Heuvel, (2007) Interactions of Kid-Kis toxin-antitoxin complexes with the parD operator-promoter region of plasmid R1 are piloted by the Kis antitoxin and tuned by the stoichiometry of Kid-Kis oligomers. Nucleic Acids Res 35: 1737-1749.
Moritz, E. M. & P. J. Hergenrother, (2007) Toxin-antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc Natl Acad Sci U S A 104: 311-316.
Moyed, H. S. & K. P. Bertrand, (1983) hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155: 768-775.
Moyed, H. S. & S. H. Broderick, (1986) Molecular cloning and expression of hipA, a gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 166: 399-403.
Munoz-Gomez, A. J., M. Lemonnier, S. Santos-Sierra, A. Berzal-Herranz & R. Diaz-Orejas, (2005) RNase/anti-RNase activities of the bacterial parD toxin-antitoxin system. J Bacteriol 187: 3151-3157.
Munoz-Gomez, A. J., S. Santos-Sierra, A. Berzal-Herranz, M. Lemonnier & R. Diaz-Orejas, (2004) Insights into the specificity of RNA cleavage by the Escherichia coli MazF toxin. FEBS Lett 567: 316-320.
Nieto, C., T. Pellicer, D. Balsa, S. K. Christensen, K. Gerdes & M. Espinosa, (2006) The chromosomal relBE2 toxin-antitoxin locus of Streptococcus pneumoniae: characterization and use of a bioluminescence resonance energy transfer assay to detect toxin-antitoxin interaction. Mol Microbiol 59: 1280-1296.
Nystrom, T., (2003) Conditional senescence in bacteria: death of the immortals. Mol Microbiol 48: 17-23.
Ogura, T. & S. Hiraga, (1983) Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 80: 4784-4788.
Overgaard, M., J. Borch & K. Gerdes, (2009) RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J Mol Biol 394: 183-196.
Pedersen, K., S. K. Christensen & K. Gerdes, (2002) Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol Microbiol 45: 501-510.
Pedersen, K., A. V. Zavialov, M. Y. Pavlov, J. Elf, K. Gerdes & M. Ehrenberg, (2003) The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112: 131-140.
Pimentel, B., M. A. Madine & G. de la Cueva-Mendez, (2005) Kid cleaves specific mRNAs at UUACU sites to rescue the copy number of plasmid R1. EMBO J 24: 3459-3469.
Pratto, F., A. Cicek, W. A. Weihofen, R. Lurz, W. Saenger & J. C. Alonso, (2008) Streptococcus pyogenes pSM19035 requires dynamic assembly of ATP-bound ParA and ParB on parS DNA during plasmid segregation. Nucleic Acids Res 36: 3676-3689.
Rojo, F. & J. C. Alonso, (1994) A novel site-specific recombinase encoded by the Streptococcus pyogenes plasmid pSM19035. J Mol Biol 238: 159-172.
Ruiz-Echevarria, M. J., A. Berzal-Herranz, K. Gerdes & R. Diaz-Orejas, (1991a) The kis and kid genes of the parD maintenance system of plasmid R1 form an operon that is autoregulated at the level of transcription by the co-ordinated action of the Kis and Kid proteins. Mol Microbiol 5: 2685-2693.
Ruiz-Echevarria, M. J., G. de Torrontegui, G. Gimenez-Gallego & R. Diaz-Orejas, (1991b) Structural and functional comparison between the stability systems ParD of plasmid R1 and Ccd of plasmid F. Mol Gen Genet 225: 355-362.
Sat, B., R. Hazan, T. Fisher, H. Khaner, G. Glaser & H. Engelberg-Kulka, (2001) Programmed cell death in Escherichia coli: some antibiotics can trigger mazEF lethality. J Bacteriol 183: 2041-2045.

P á g i n a | 75
Sat, B., M. Reches & H. Engelberg-Kulka, (2003) The Escherichia coli mazEF suicide module mediates thymineless death. J Bacteriol 185: 1803-1807.
Saxild, H. H., L. N. Andersen & K. Hammer, (1996) Dra-nupC-pdp operon of Bacillus subtilis: nucleotide sequence, induction by deoxyribonucleosides, and transcriptional regulation by the deoR-encoded DeoR repressor protein. J Bacteriol 178: 424-434.
Scherrer, R. & H. S. Moyed, (1988) Conditional impairment of cell division and altered lethality in hipA mutants of Escherichia coli K-12. J Bacteriol 170: 3321-3326.
Schumacher, M. A., K. M. Piro, W. Xu, S. Hansen, K. Lewis & R. G. Brennan, (2009) Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323: 396-401.
Shah, D., Z. Zhang, A. Khodursky, N. Kaldalu, K. Kurg & K. Lewis, (2006) Persisters: a distinct physiological state of E. coli. BMC Microbiol 6: 53.
Sletvold, H., P. J. Johnsen, I. Hamre, G. S. Simonsen, A. Sundsfjord & K. M. Nielsen, (2008) Complete sequence of Enterococcus faecium pVEF3 and the detection of an omega-epsilon-zeta toxin-antitoxin module and an ABC transporter. Plasmid 60: 75-85.
Spoering, A. L., M. Vulic & K. Lewis, (2006) GlpD and PlsB participate in persister cell formation in Escherichia coli. J Bacteriol 188: 5136-5144.
Stulke, J., R. Hanschke & M. Hecker, (1993) Temporal activation of beta-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J Gen Microbiol 139: 2041-2045.
Takagi, H., Y. Kakuta, T. Okada, M. Yao, I. Tanaka & M. Kimura, (2005) Crystal structure of archaeal toxin-antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat Struct Mol Biol 12: 327-331.
Tam, J. E. & B. C. Kline, (1989) The F plasmid ccd autorepressor is a complex of CcdA and CcdB proteins. Mol Gen Genet 219: 26-32.
Tozzi, M. G., M. Camici, L. Mascia, F. Sgarrella & P. L. Ipata, (2006) Pentose phosphates in nucleoside interconversion and catabolism. FEBS J 273: 1089-1101.
Van Melderen, L., P. Bernard & M. Couturier, (1994) Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol Microbiol 11: 1151-1157.
Van Melderen, L. & M. Saavedra De Bast, (2009) Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet 5: e1000437.
Wang, J. D., G. M. Sanders & A. D. Grossman, (2007) Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 128: 865-875.
Weihofen, W. A., A. Cicek, F. Pratto, J. C. Alonso & W. Saenger, (2006) Structures of omega repressors bound to direct and inverted DNA repeats explain modulation of transcription. Nucleic Acids Res 34: 1450-1458.
Welfle, K., F. Pratto, R. Misselwitz, J. Behlke, J. C. Alonso & H. Welfle, (2005) Role of the N-terminal region and of beta-sheet residue Thr29 on the activity of the omega2 global regulator from the broad-host range Streptococcus pyogenes plasmid pSM19035. Biol Chem 386: 881-894.
Wilbaux, M., N. Mine, A. M. Guerout, D. Mazel & L. Van Melderen, (2007) Functional interactions between coexisting toxin-antitoxin systems of the ccd family in Escherichia coli O157:H7. J Bacteriol 189: 2712-2719.
Williams, J. J. & P. J. Hergenrother, (2008) Exposing plasmids as the Achilles' heel of drug-resistant bacteria. Curr Opin Chem Biol 12: 389-399.
Zeng, X., A. Galinier & H. H. Saxild, (2000) Catabolite repression of dra-nupC-pdp operon expression in Bacillus subtilis. Microbiology 146 ( Pt 11): 2901-2908.
Zhang, J., Y. Zhang, L. Zhu, M. Suzuki & M. Inouye, (2004) Interference of mRNA function by sequence-specific endoribonuclease PemK. J Biol Chem 279: 20678-20684.
Zhang, Y., J. Zhang, K. P. Hoeflich, M. Ikura, G. Qing & M. Inouye, (2003) MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 12: 913-923.
Zielenkiewicz, U. & P. Ceglowski, (2005) The toxin-antitoxin system of the streptococcal plasmid pSM19035. J Bacteriol 187: 6094-6105.

P á g i n a | 76

Anexo I

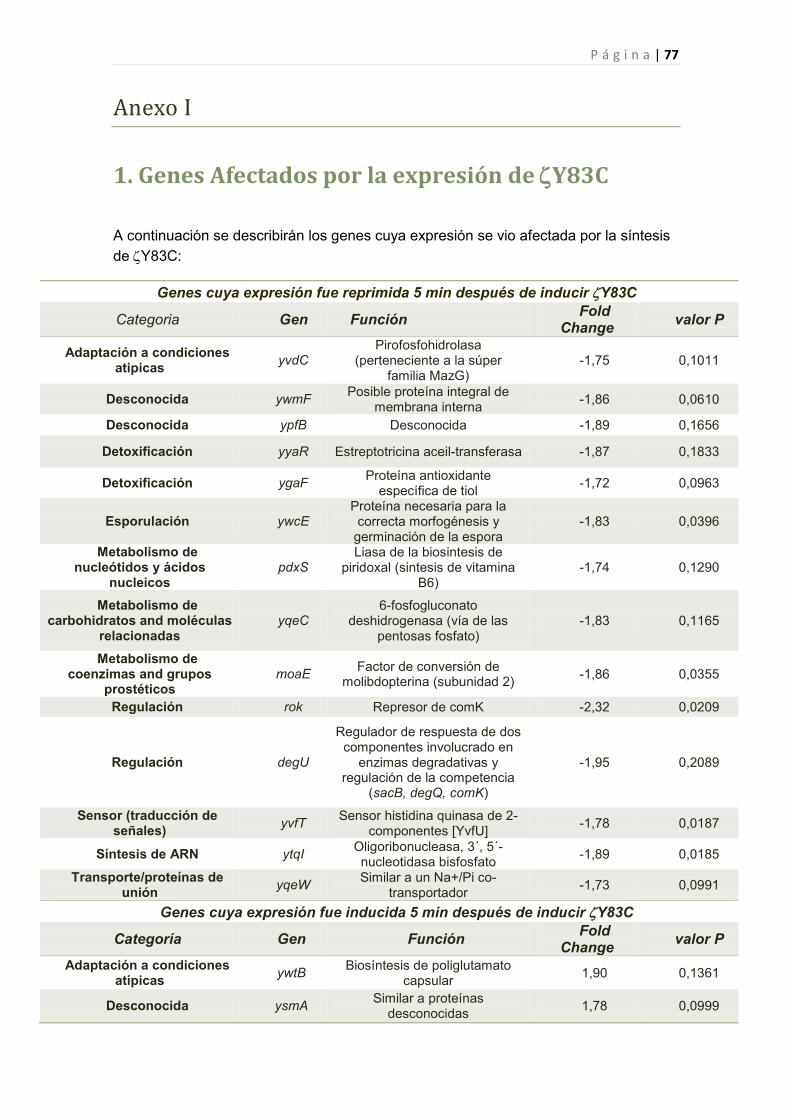
P á g i n a | 77
Anexo I
1. Genes Afectados por la expresión de Y83C
A continuación se describirán los genes cuya expresión se vio afectada por la síntesis
de Y83C:
Genes cuya expresión fue reprimida 5 min después de inducir Y83C
Categoria Gen Función Fold
Change valor P
Adaptación a condiciones atipicas
yvdC Pirofosfohidrolasa
(perteneciente a la súper familia MazG)
-1,75 0,1011
Desconocida ywmF Posible proteína integral de
membrana interna -1,86 0,0610
Desconocida ypfB Desconocida -1,89 0,1656
Detoxificación yyaR Estreptotricina aceil-transferasa -1,87 0,1833
Detoxificación ygaF Proteína antioxidante
específica de tiol -1,72 0,0963
Esporulación ywcE Proteína necesaria para la correcta morfogénesis y
germinación de la espora -1,83 0,0396
Metabolismo de nucleótidos y ácidos
nucleicos pdxS
Liasa de la biosintesis de piridoxal (sintesis de vitamina
B6) -1,74 0,1290
Metabolismo de carbohidratos and moléculas
relacionadas yqeC
6-fosfogluconato deshidrogenasa (vía de las
pentosas fosfato) -1,83 0,1165
Metabolismo de coenzimas and grupos
prostéticos moaE
Factor de conversión de molibdopterina (subunidad 2)
-1,86 0,0355
Regulación rok Represor de comK -2,32 0,0209
Regulación degU
Regulador de respuesta de dos componentes involucrado en
enzimas degradativas y regulación de la competencia
(sacB, degQ, comK)
-1,95 0,2089
Sensor (traducción de señales)
yvfT Sensor histidina quinasa de 2-
componentes [YvfU] -1,78 0,0187
Síntesis de ARN ytqI Oligoribonucleasa, 3´, 5´-nucleotidasa bisfosfato
-1,89 0,0185
Transporte/proteínas de unión
yqeW Similar a un Na+/Pi co-
transportador -1,73 0,0991
Genes cuya expresión fue inducida 5 min después de inducir Y83C
Categoría Gen Función Fold
Change valor P
Adaptación a condiciones atípicas
ywtB Biosíntesis de poliglutamato
capsular 1,90 0,1361
Desconocida ysmA Similar a proteínas
desconocidas 1,78 0,0999
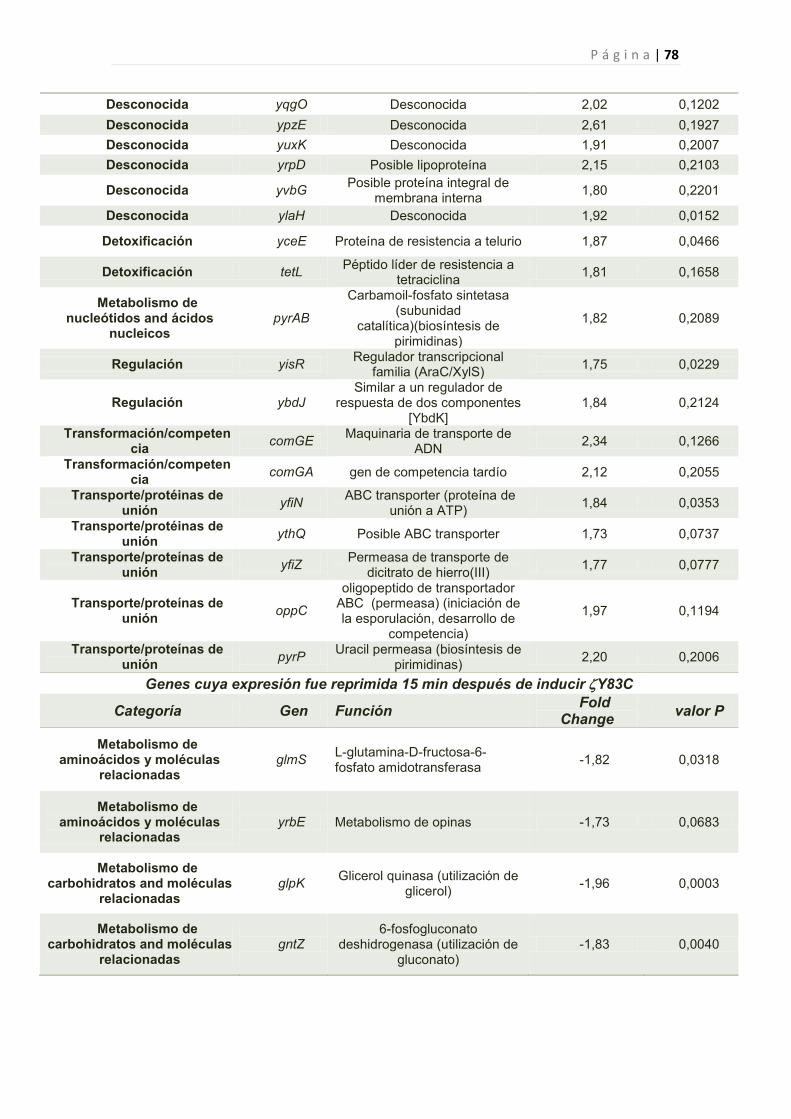
P á g i n a | 78
Desconocida yqgO Desconocida 2,02 0,1202
Desconocida ypzE Desconocida 2,61 0,1927
Desconocida yuxK Desconocida 1,91 0,2007
Desconocida yrpD Posible lipoproteína 2,15 0,2103
Desconocida yvbG Posible proteína integral de
membrana interna 1,80 0,2201
Desconocida ylaH Desconocida 1,92 0,0152
Detoxificación yceE Proteína de resistencia a telurio 1,87 0,0466
Detoxificación tetL Péptido líder de resistencia a
tetraciclina 1,81 0,1658
Metabolismo de nucleótidos and ácidos
nucleicos pyrAB
Carbamoil-fosfato sintetasa (subunidad
catalítica)(biosíntesis de pirimidinas)
1,82 0,2089
Regulación yisR Regulador transcripcional
familia (AraC/XylS) 1,75 0,0229
Regulación ybdJ Similar a un regulador de
respuesta de dos componentes [YbdK]
1,84 0,2124
Transformación/competencia
comGE Maquinaria de transporte de
ADN 2,34 0,1266
Transformación/competencia
comGA gen de competencia tardío 2,12 0,2055
Transporte/protéinas de unión
yfiN ABC transporter (proteína de
unión a ATP) 1,84 0,0353
Transporte/protéinas de unión
ythQ Posible ABC transporter 1,73 0,0737
Transporte/proteínas de unión
yfiZ Permeasa de transporte de
dicitrato de hierro(III) 1,77 0,0777
Transporte/proteínas de unión
oppC
oligopeptido de transportador ABC (permeasa) (iniciación de la esporulación, desarrollo de
competencia)
1,97 0,1194
Transporte/proteínas de unión
pyrP Uracil permeasa (biosíntesis de
pirimidinas) 2,20 0,2006
Genes cuya expresión fue reprimida 15 min después de inducir Y83C
Categoría Gen Función Fold
Change valor P
Metabolismo de aminoácidos y moléculas
relacionadas glmS
L-glutamina-D-fructosa-6-fosfato amidotransferasa
-1,82 0,0318
Metabolismo de aminoácidos y moléculas
relacionadas yrbE Metabolismo de opinas -1,73 0,0683
Metabolismo de carbohidratos and moléculas
relacionadas glpK
Glicerol quinasa (utilización de glicerol)
-1,96 0,0003
Metabolismo de carbohidratos and moléculas
relacionadas gntZ
6-fosfogluconato deshidrogenasa (utilización de
gluconato) -1,83 0,0040
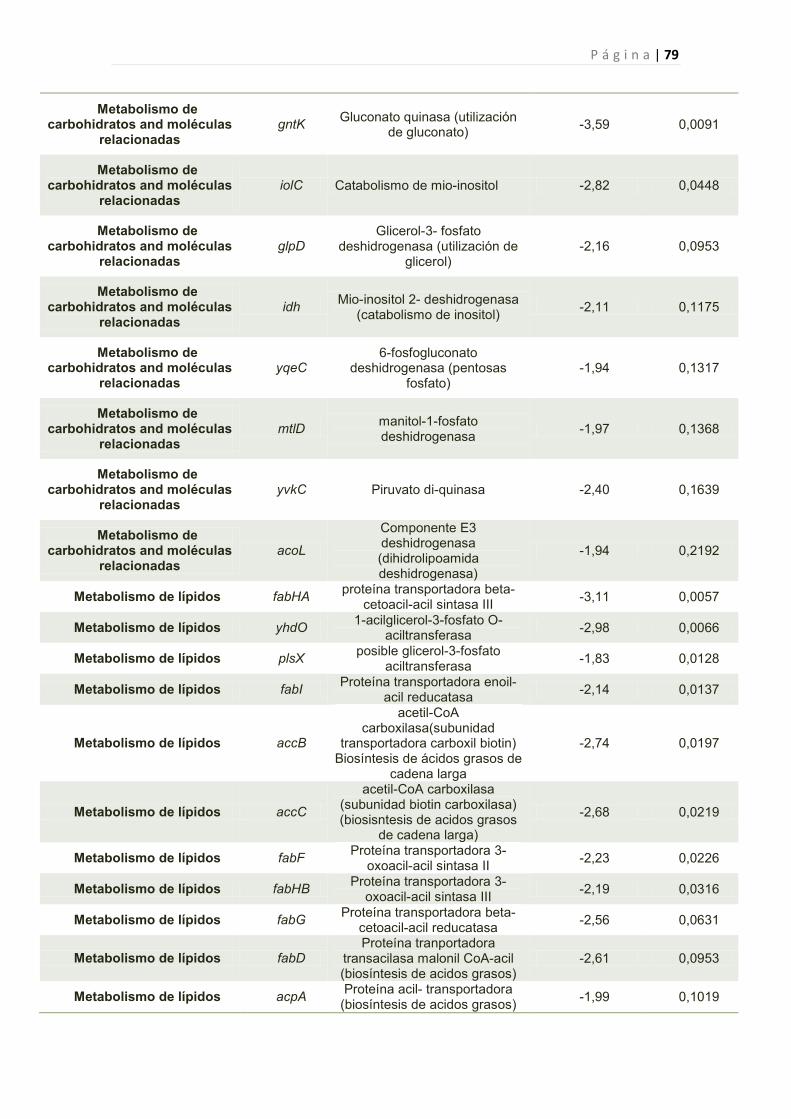
P á g i n a | 79
Metabolismo de carbohidratos and moléculas
relacionadas gntK
Gluconato quinasa (utilización de gluconato)
-3,59 0,0091
Metabolismo de carbohidratos and moléculas
relacionadas iolC Catabolismo de mio-inositol -2,82 0,0448
Metabolismo de carbohidratos and moléculas
relacionadas glpD
Glicerol-3- fosfato deshidrogenasa (utilización de
glicerol) -2,16 0,0953
Metabolismo de carbohidratos and moléculas
relacionadas idh
Mio-inositol 2- deshidrogenasa (catabolismo de inositol)
-2,11 0,1175
Metabolismo de carbohidratos and moléculas
relacionadas yqeC
6-fosfogluconato deshidrogenasa (pentosas
fosfato) -1,94 0,1317
Metabolismo de carbohidratos and moléculas
relacionadas mtlD
manitol-1-fosfato deshidrogenasa
-1,97 0,1368
Metabolismo de carbohidratos and moléculas
relacionadas yvkC Piruvato di-quinasa -2,40 0,1639
Metabolismo de carbohidratos and moléculas
relacionadas acoL
Componente E3 deshidrogenasa (dihidrolipoamida deshidrogenasa)
-1,94 0,2192
Metabolismo de lípidos fabHA proteína transportadora beta-
cetoacil-acil sintasa III -3,11 0,0057
Metabolismo de lípidos yhdO 1-acilglicerol-3-fosfato O-
aciltransferasa -2,98 0,0066
Metabolismo de lípidos plsX posible glicerol-3-fosfato
aciltransferasa -1,83 0,0128
Metabolismo de lípidos fabI Proteína transportadora enoil-
acil reducatasa -2,14 0,0137
Metabolismo de lípidos accB
acetil-CoA carboxilasa(subunidad
transportadora carboxil biotin) Biosíntesis de ácidos grasos de
cadena larga
-2,74 0,0197
Metabolismo de lípidos accC
acetil-CoA carboxilasa (subunidad biotin carboxilasa) (biosisntesis de acidos grasos
de cadena larga)
-2,68 0,0219
Metabolismo de lípidos fabF Proteína transportadora 3-
oxoacil-acil sintasa II -2,23 0,0226
Metabolismo de lípidos fabHB Proteína transportadora 3-
oxoacil-acil sintasa III -2,19 0,0316
Metabolismo de lípidos fabG Proteína transportadora beta-
cetoacil-acil reducatasa -2,56 0,0631
Metabolismo de lípidos fabD Proteína tranportadora
transacilasa malonil CoA-acil (biosíntesis de acidos grasos)
-2,61 0,0953
Metabolismo de lípidos acpA Proteína acil- transportadora
(biosíntesis de acidos grasos) -1,99 0,1019
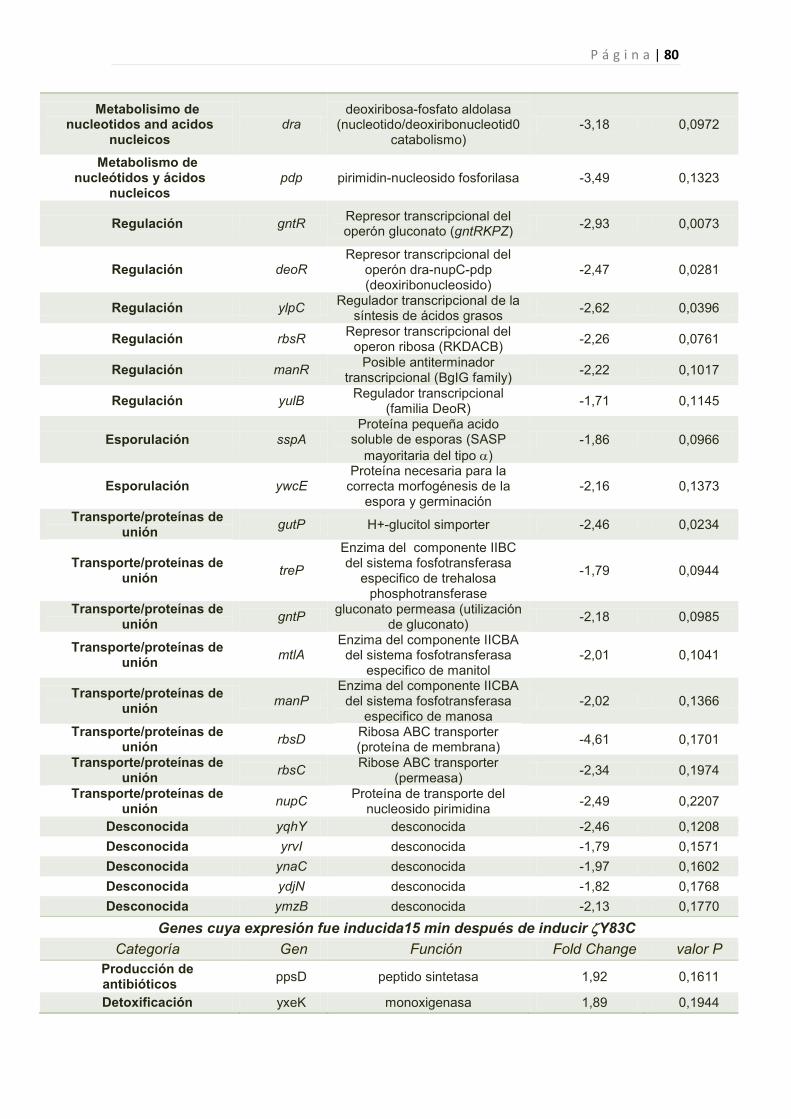
P á g i n a | 80
Metabolisimo de nucleotidos and acidos
nucleicos dra
deoxiribosa-fosfato aldolasa (nucleotido/deoxiribonucleotid0
catabolismo) -3,18 0,0972
Metabolismo de nucleótidos y ácidos
nucleicos pdp pirimidin-nucleosido fosforilasa -3,49 0,1323
Regulación gntR Represor transcripcional del operón gluconato (gntRKPZ)
-2,93 0,0073
Regulación deoR Represor transcripcional del
operón dra-nupC-pdp (deoxiribonucleosido)
-2,47 0,0281
Regulación ylpC Regulador transcripcional de la
síntesis de ácidos grasos -2,62 0,0396
Regulación rbsR Represor transcripcional del
operon ribosa (RKDACB) -2,26 0,0761
Regulación manR Posible antiterminador
transcripcional (BgIG family) -2,22 0,1017
Regulación yulB Regulador transcripcional
(familia DeoR) -1,71 0,1145
Esporulación sspA Proteína pequeña acido
soluble de esporas (SASP
mayoritaria del tipo )
-1,86 0,0966
Esporulación ywcE Proteína necesaria para la
correcta morfogénesis de la espora y germinación
-2,16 0,1373
Transporte/proteínas de unión
gutP H+-glucitol simporter -2,46 0,0234
Transporte/proteínas de unión
treP
Enzima del componente IIBC del sistema fosfotransferasa
especifico de trehalosa phosphotransferase
-1,79 0,0944
Transporte/proteínas de unión
gntP gluconato permeasa (utilización
de gluconato) -2,18 0,0985
Transporte/proteínas de unión
mtlA Enzima del componente IICBA
del sistema fosfotransferasa especifico de manitol
-2,01 0,1041
Transporte/proteínas de unión
manP Enzima del componente IICBA
del sistema fosfotransferasa especifico de manosa
-2,02 0,1366
Transporte/proteínas de unión
rbsD Ribosa ABC transporter (proteína de membrana)
-4,61 0,1701
Transporte/proteínas de unión
rbsC Ribose ABC transporter
(permeasa) -2,34 0,1974
Transporte/proteínas de unión
nupC Proteína de transporte del
nucleosido pirimidina -2,49 0,2207
Desconocida yqhY desconocida -2,46 0,1208
Desconocida yrvI desconocida -1,79 0,1571
Desconocida ynaC desconocida -1,97 0,1602
Desconocida ydjN desconocida -1,82 0,1768
Desconocida ymzB desconocida -2,13 0,1770
Genes cuya expresión fue inducida15 min después de inducir Y83C
Categoría Gen Función Fold Change valor P
Producción de antibióticos
ppsD peptido sintetasa 1,92 0,1611
Detoxificación yxeK monoxigenasa 1,89 0,1944
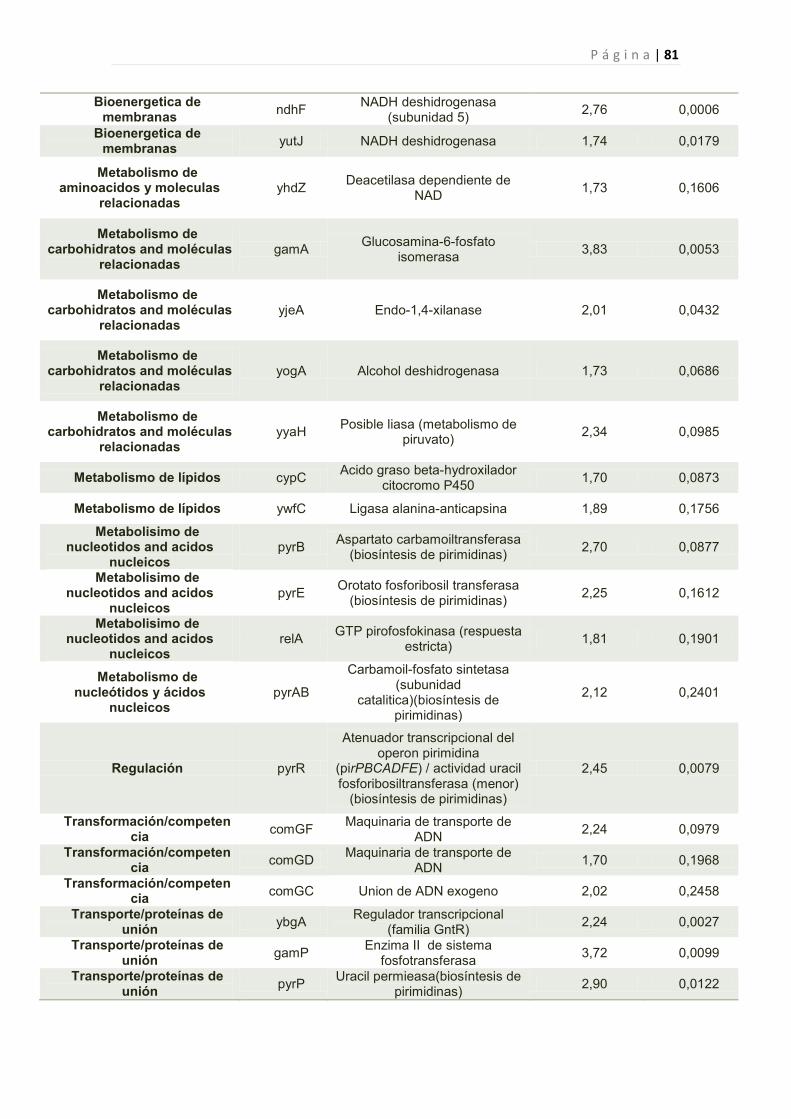
P á g i n a | 81
Bioenergetica de membranas
ndhF NADH deshidrogenasa
(subunidad 5) 2,76 0,0006
Bioenergetica de membranas
yutJ NADH deshidrogenasa 1,74 0,0179
Metabolismo de aminoacidos y moleculas
relacionadas yhdZ
Deacetilasa dependiente de NAD
1,73 0,1606
Metabolismo de carbohidratos and moléculas
relacionadas gamA
Glucosamina-6-fosfato isomerasa
3,83 0,0053
Metabolismo de carbohidratos and moléculas
relacionadas yjeA Endo-1,4-xilanase 2,01 0,0432
Metabolismo de carbohidratos and moléculas
relacionadas yogA Alcohol deshidrogenasa 1,73 0,0686
Metabolismo de carbohidratos and moléculas
relacionadas yyaH
Posible liasa (metabolismo de piruvato)
2,34 0,0985
Metabolismo de lípidos cypC Acido graso beta-hydroxilador
citocromo P450 1,70 0,0873
Metabolismo de lípidos ywfC Ligasa alanina-anticapsina 1,89 0,1756
Metabolisimo de nucleotidos and acidos
nucleicos pyrB
Aspartato carbamoiltransferasa (biosíntesis de pirimidinas)
2,70 0,0877
Metabolisimo de nucleotidos and acidos
nucleicos pyrE
Orotato fosforibosil transferasa (biosíntesis de pirimidinas)
2,25 0,1612
Metabolisimo de nucleotidos and acidos
nucleicos relA
GTP pirofosfokinasa (respuesta estricta)
1,81 0,1901
Metabolismo de nucleótidos y ácidos
nucleicos pyrAB
Carbamoil-fosfato sintetasa (subunidad
catalitica)(biosíntesis de pirimidinas)
2,12 0,2401
Regulación pyrR
Atenuador transcripcional del operon pirimidina
(pirPBCADFE) / actividad uracil fosforibosiltransferasa (menor)
(biosíntesis de pirimidinas)
2,45 0,0079
Transformación/competencia
comGF Maquinaria de transporte de
ADN 2,24 0,0979
Transformación/competencia
comGD Maquinaria de transporte de
ADN 1,70 0,1968
Transformación/competencia
comGC Union de ADN exogeno 2,02 0,2458
Transporte/proteínas de unión
ybgA Regulador transcripcional
(familia GntR) 2,24 0,0027
Transporte/proteínas de unión
gamP Enzima II de sistema
fosfotransferasa 3,72 0,0099
Transporte/proteínas de unión
pyrP Uracil permieasa(biosíntesis de
pirimidinas) 2,90 0,0122
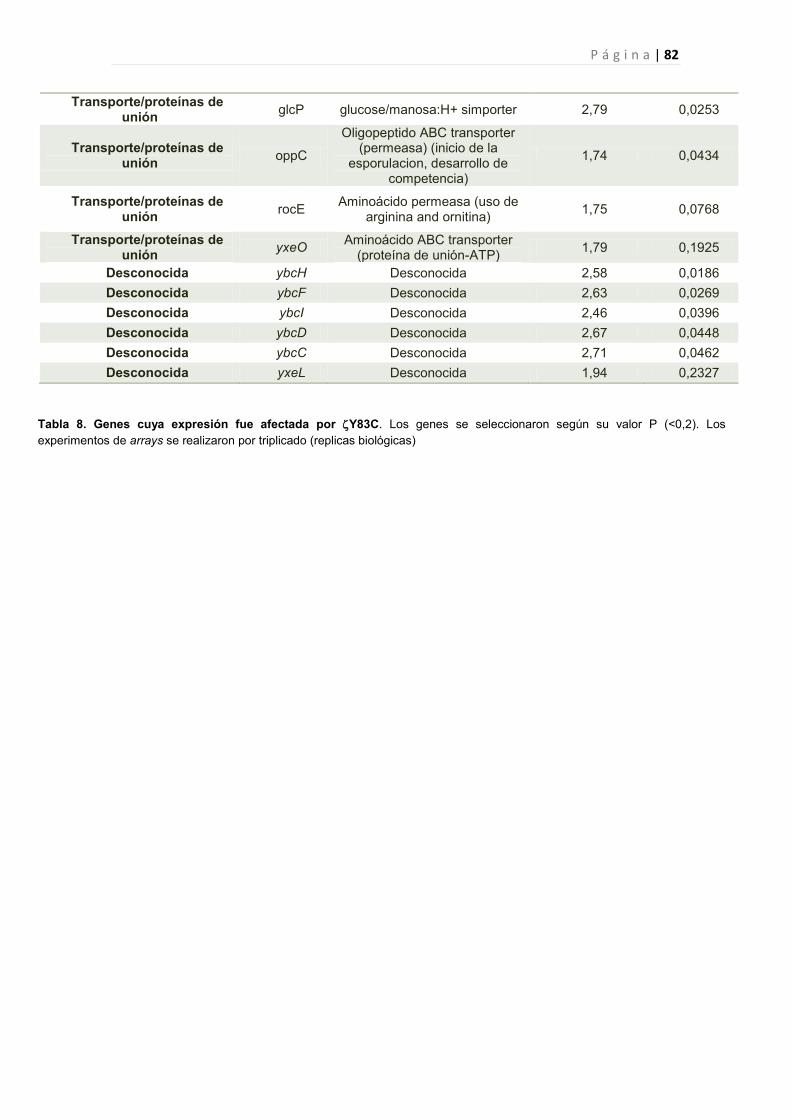
P á g i n a | 82
Transporte/proteínas de unión
glcP glucose/manosa:H+ simporter 2,79 0,0253
Transporte/proteínas de unión
oppC
Oligopeptido ABC transporter (permeasa) (inicio de la
esporulacion, desarrollo de competencia)
1,74 0,0434
Transporte/proteínas de unión
rocE Aminoácido permeasa (uso de
arginina and ornitina) 1,75 0,0768
Transporte/proteínas de unión
yxeO Aminoácido ABC transporter
(proteína de unión-ATP) 1,79 0,1925
Desconocida ybcH Desconocida 2,58 0,0186
Desconocida ybcF Desconocida 2,63 0,0269
Desconocida ybcI Desconocida 2,46 0,0396
Desconocida ybcD Desconocida 2,67 0,0448
Desconocida ybcC Desconocida 2,71 0,0462
Desconocida yxeL Desconocida 1,94 0,2327
Tabla 8. Genes cuya expresión fue afectada por Y83C. Los genes se seleccionaron según su valor P (<0,2). Los
experimentos de arrays se realizaron por triplicado (replicas biológicas)

Anexo II


P á g i n a | 83
Anexo II
Los resultados obtenidos durante el desarrollo de esta tesis forman parte de las
siguientes publicaciones.
Lioy, V. S., M. T. Martin, A. G. Camacho, R. Lurz, H. Antelmann, M. Hecker, E. Hitchin, Y. Ridge, J. M. Wells & J. C. Alonso, (2006) pSM19035-encoded zeta toxin induces stasis followed by death in a subpopulation of cells. Microbiology 152: 2365-2379. Lioy, V. S., O. Rey, D. Balsa, T. Pellicer & J. C. Alonso, (2009) A toxin-antitoxin module as a target for antimicrobial development. Plasmid, en prensa También formará parte de las siguientes publicaciones:
Pratto F1, Lioy VS1, Soberón NE, Volante A and Alonso JC, Molecular anatomy of the
Streptococcus pyogenes pSM19035 partition complex. (en preparación) 1 Los autores contribuyeron de igual manera al trabajo.
Lioy VS, Ayora S, González-Pastor JE and Alonso JC, The streptococcal toxin
increases multidrug sensitivity. (en preparación)
Lioy VS, Pratto F, de la Hoz A, Ayora S and Alonso JC. Stable inheritance of plasmid
pSM19035. Review. (en preparación)
Lioy VS, Soberón N, Austin S and Alonso JC. Accurate redistribution of pSM19035 by
dynamic plasmid pairing and separation. (en preparación)

P á g i n a | 84





























