Embed Size (px)
Citation preview

El maravilloso mundo de la estadística en la industria farmacéutica: instrucciones, interacciones y contraindicaciones
Xavier Núñez,CStat Senior Statistician

Introduction to:
• CRO and Clinical Trial: definitions• TFS Company & Organisation• Global Biometrics• Data Management working flow• Statistics working flow • Regulatory guidelines• Type of clinical trials • Statistical Analyses vs. Clinical trials• Examples of clinical trials• Day-to-day example• Conclusions

What is a CRO?
Chief Risk Officer
Cathode Ray Oscilloscope
Cro-Magnons
Clinical Research Organization: a service organization that provides support to the pharmaceutical and biotechnology industries in the form of outsourced pharmaceutical research services (for both drugs and medical devices)
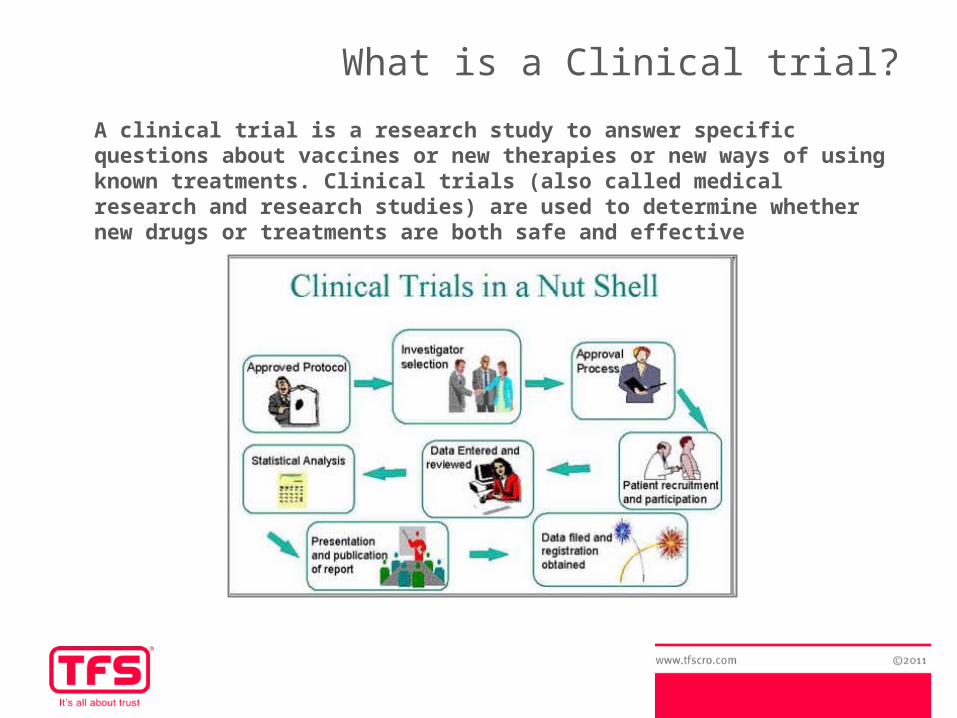
What is a Clinical trial?
A clinical trial is a research study to answer specific questions about vaccines or new therapies or new ways of using known treatments. Clinical trials (also called medical research and research studies) are used to determine whether new drugs or treatments are both safe and effective

TFS -Introduction
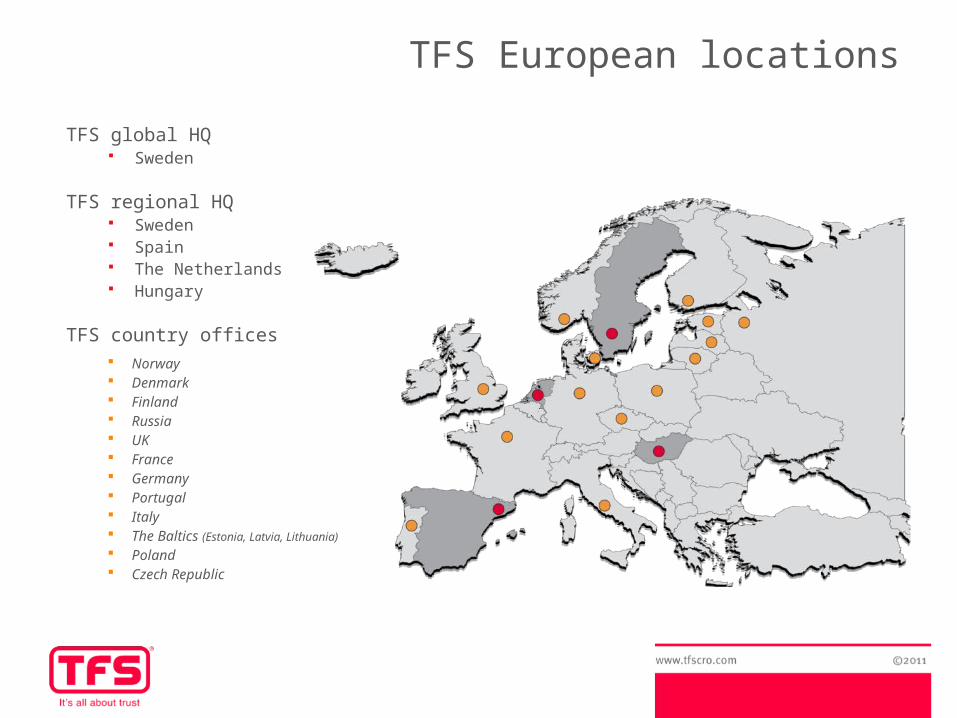
TFS global HQ Sweden
TFS regional HQ Sweden Spain The Netherlands Hungary
TFS country offices Norway Denmark Finland Russia UK France Germany Portugal Italy The Baltics (Estonia, Latvia, Lithuania)
Poland Czech Republic
TFS European locations
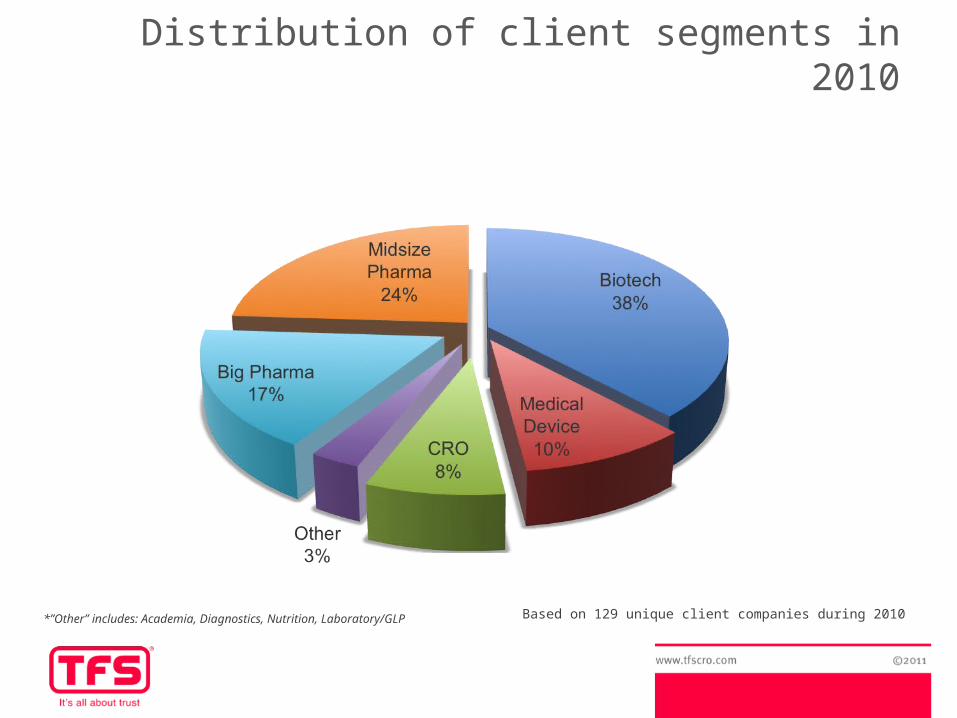
Based on 129 unique client companies during 2010*”Other” includes: Academia, Diagnostics, Nutrition, Laboratory/GLP
Distribution of client segments in 2010
20 largest customers in 2010
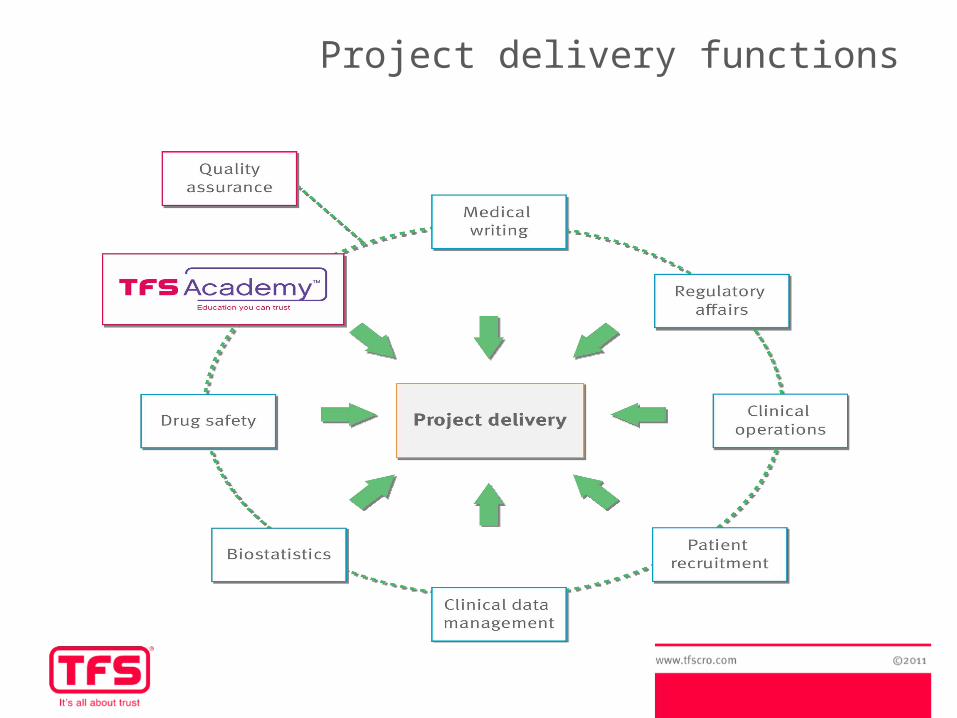
Project delivery functions
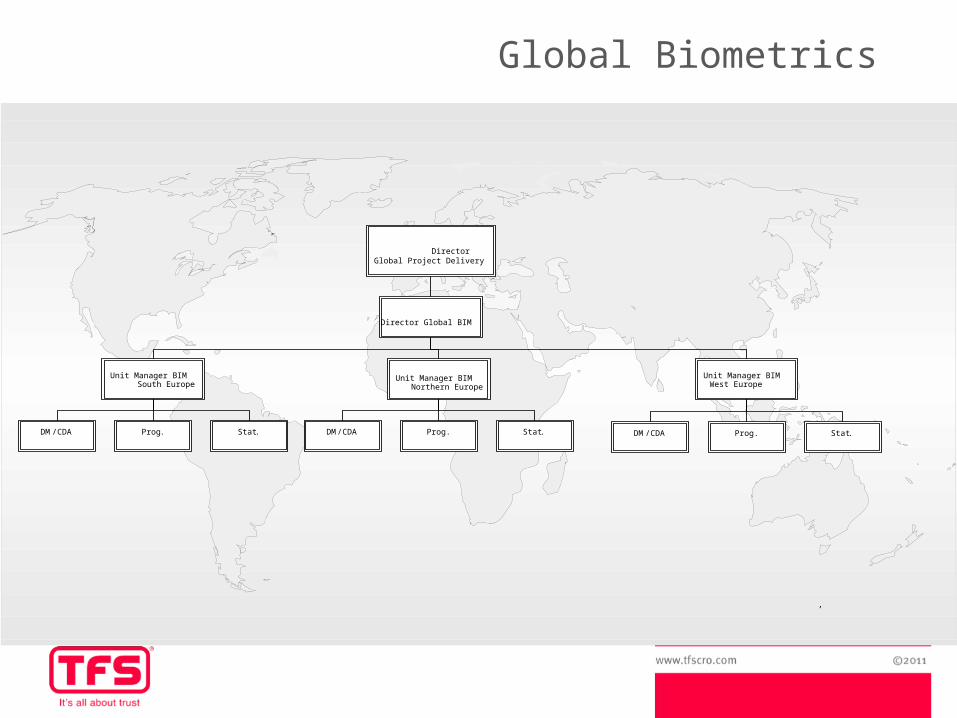
DirectorGlobal Project Delivery
Director Global BIM
Unit Manager BIMWest Europe
Unit Manager BIMNorthern Europe
,
Prog . Stat .DM /CDA Prog . Stat .DM /CDA
Unit Manager BIMSouth Europe
Prog . Stat .DM /CDA
Global Biometrics

Global Biometrics - Services
TFS Global Biometrics offer:Biostatistics, Programming and Clinical Data Management
Currently 40 employees working in Global Biometrics (Spain, Sweden, Netherlands and Denmark)
Support for Life Science projects• Clinical trials, phases 1-4• Evaluation of Medical device, diagnostic test• Non-interventional studies
Software: SAS, SPSS, Minitab, Access, NQuery, Ene...
By establishing a sound approach to clinical biostatistics and clinical data management during the planning stages of the clinical development program we:
• Improve the quality of submissions• Accelerate timelines• Decrease costs• Reduce risks

Global Biometrics - Services
Clinical Data Management- Case report form (CRF) / eCRF design- Database and Data Entry solutions
Statistical services & consultations- Input to study design- Randomisations- Statistical analysis plan (SAP)- SAS programming: tables, figures and listings (TFLs), statistical analyses,
standard macros...- Statistical analysis and report- Support with publications & clinical study report (CSR)
Support with CDISC standards SDTM and ADaM formats Training via TFS Academy CPS (contract placement services)
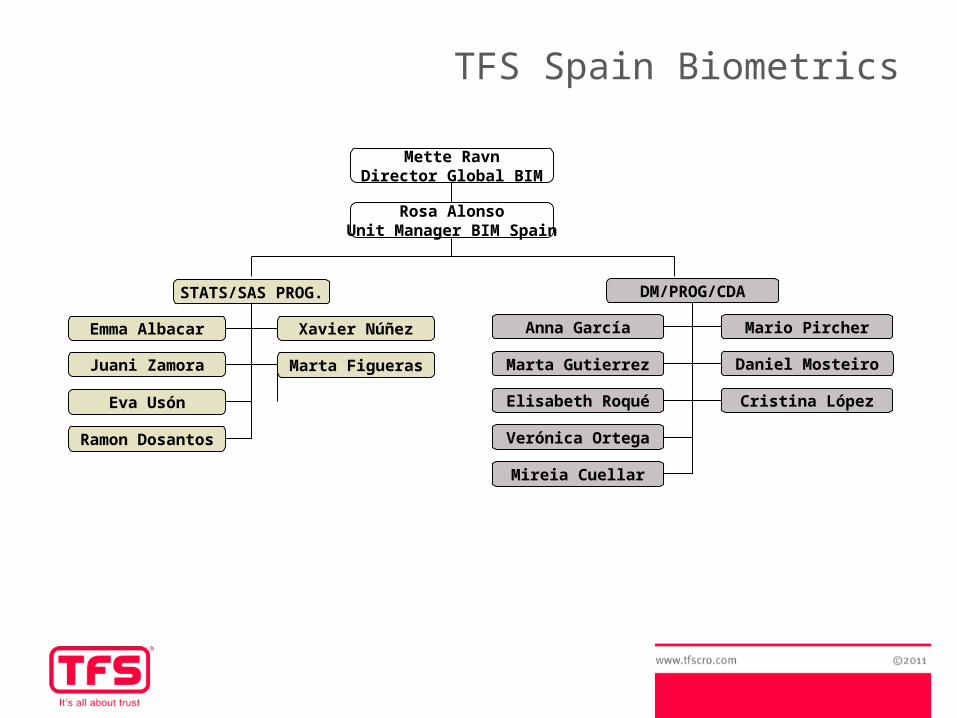
DM/PROG/CDA
Anna García Mario Pircher
Marta Gutierrez Daniel Mosteiro
Elisabeth Roqué Cristina López
Verónica Ortega
Mireia Cuellar
STATS/SAS PROG.
Emma Albacar Xavier Núñez
Juani Zamora Marta Figueras
Eva Usón
Ramon Dosantos
Mette RavnDirector Global BIM
Rosa AlonsoUnit Manager BIM Spain
TFS Spain Biometrics
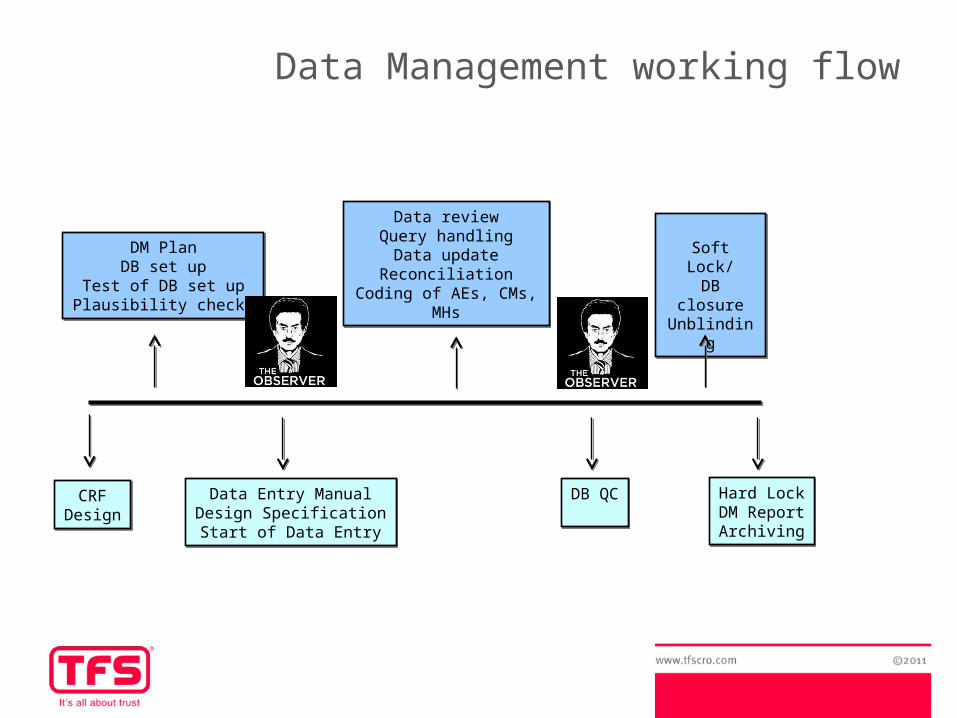
DM PlanDB set up
Test of DB set upPlausibility checks
DM PlanDB set up
Test of DB set upPlausibility checks
Data reviewQuery handling
Data updateReconciliation
Coding of AEs, CMs, MHs
Data reviewQuery handling
Data updateReconciliation
Coding of AEs, CMs, MHs
Soft Lock/DB closureUnblinding
Soft Lock/DB closureUnblinding
Data Entry ManualDesign SpecificationStart of Data Entry
Data Entry ManualDesign SpecificationStart of Data Entry
DB QCDB QC Hard LockDM ReportArchiving
Hard LockDM ReportArchiving
Data Management working flow
CRFDesignCRF
Design
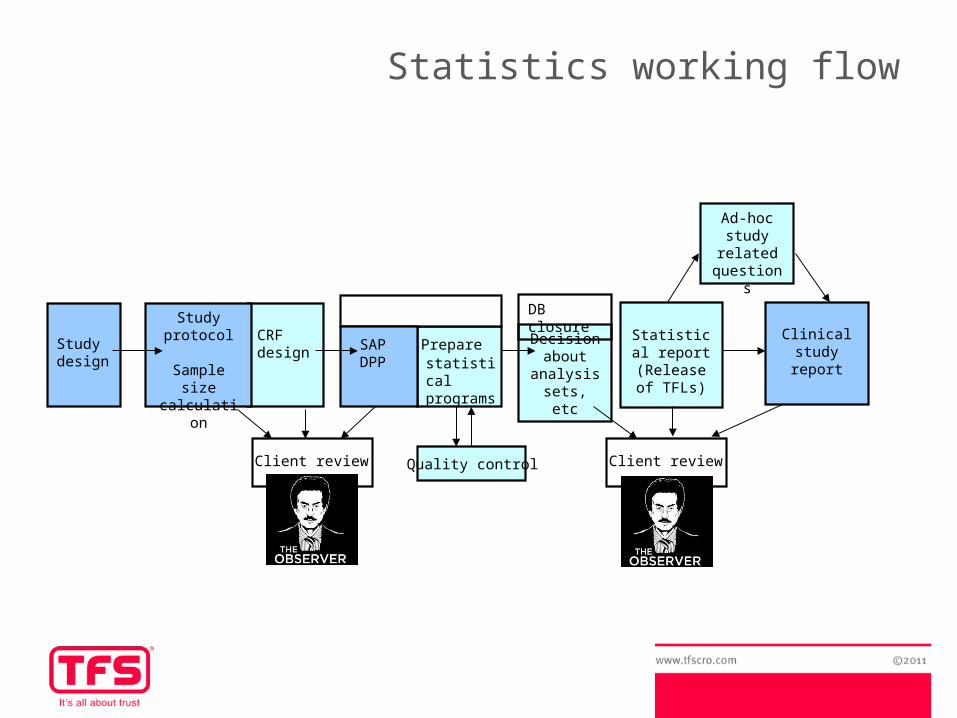
Statistics working flow
SAPDPP
Statistical report
(Release of TFLs)
CRF design
Study protocol
Sample size calculation
Clinical study
report
Study design
Preparestatistical programs
Decision about
analysis sets, etc
DB closure
Quality controlClient review Client review
Ad-hoc study
related questions

Medical research - Regulations
Good Clinical Practice (GCP)
An international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects
The most important sources for GCP-compliant guidelines referring to the EU are the following: - Declaration of Helsinki (1964) - ICH GCP –E6 (1996) - EU Directive 2001/20/EC - EU Directive 2005/28/EC

Medical research - Regulations
Additional guidelines refer to specific statistical or DM regulations or to other recommendations, such as - ICH –E9: Statistical principles for clinical trials - ICH –E3: Structure and contents of clinical study reports - Good Clinical Data Management Practices - CDISC Clinical Data Interchange Standards Consortium,
Operational Data model (ODM)

Specific FDA Issues
The FDA is the US Government regulatory office for registration of Pharmaceutical products. Here especially the Code of Federal Regulations (CFR) applies, which is the codification of the general and permanent rules published in the Federal Register by the agencies of the Federal Government. FDA regulation is relevant for EU projects in development of drugs considered for possible registration in the US.
However, it must be clarified, that in the EU it is not the FDA regulations which are governing, but the national implementations of EU directives or the EMEA/EMA implementations of EU Regulations.
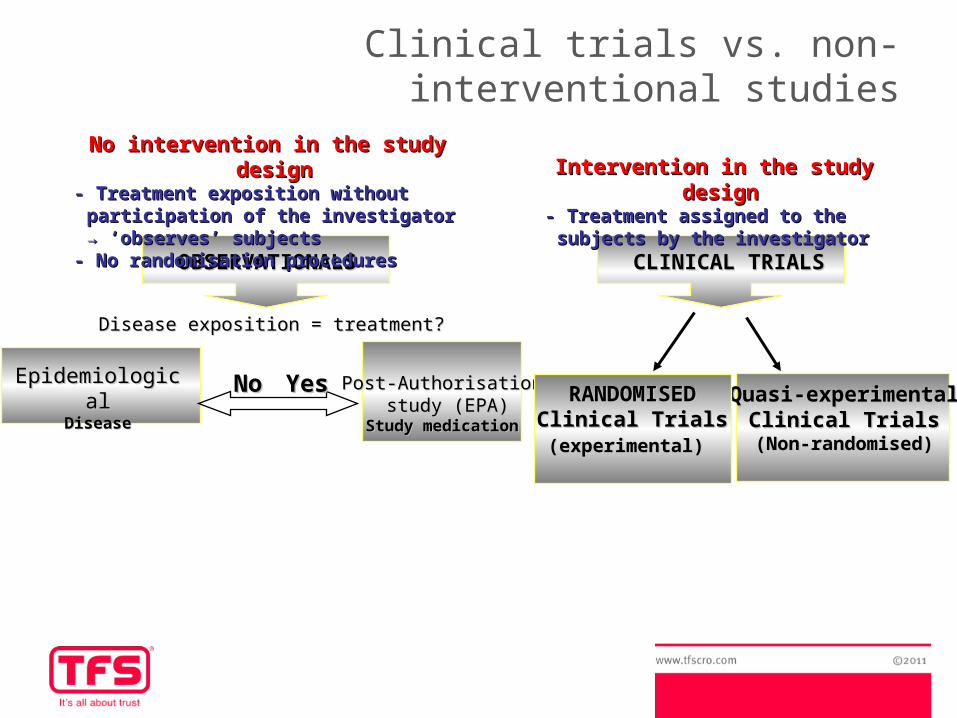
Clinical trials vs. non-interventional studies
OBSERVATIONALSOBSERVATIONALS CLINICAL TRIALSCLINICAL TRIALS
Intervention in the study designIntervention in the study design- Treatment - Treatment assigned to the subjects assigned to the subjects
by the investigatorby the investigator
EpidemiologicalEpidemiologicalDiseaseDisease
Post-AuthorisationPost-Authorisation study (EPA)study (EPA)
Study medicationStudy medication
Disease exposition = treatment? Disease exposition = treatment?
NoNo YesYes
No intervention in the study designNo intervention in the study design- Treatment exposition without - Treatment exposition without
participation of the investigator → participation of the investigator → ‘observes’ subjects‘observes’ subjects
- No randomisation procedures- No randomisation procedures
Quasi-experimentalQuasi-experimentalClinical TrialsClinical Trials
(Non-randomised)(Non-randomised)
RANDOMISED RANDOMISED Clinical TrialsClinical Trials(experimental)(experimental)

Type of clinical trials
Phase I - Healthy volunteers
- Small sample size (6-30 subjects)
- Usually FTIH
- Objectives: safety (adverse events), dose range, PK/PD
Phase II- Healthy volunteers / Patients
- Larger sample size (20-300 subjects)
- Objectives: efficacy, safety, dose-response
Phase III- Patients
- Huge sample size, multicentre (1000-3000 subjects)
- Objectives: confirm efficacy –superiority?, no safety issues
Phase IV (post-authorisation)- Patients
- Objectives: optimal use of treatment, risk-benefit, marketing, etc.

By the awareness of treatment administered
- Open-label: both investigators and subjects know which treatment is being administered
- Single-blinded: investigator is aware of the treatment administered, but the subject is not
- Double-blinded: neither investigators nor subjects know which treatment is being administered
By time of observation- Retrospective: data from past records is collected in a unique visit, with no follow-up
- Cross-sectional: all present data from subjects is collected at a defined time-point
- Prospective: subjects are followed over a period of time, collecting data in different visits
By sequence of treatments- Parallel : subjects are randomly assigned to a unique treatment throughout the study
- Cross-over: subjects are randomly assigned to a sequence of treatments
Type of clinical trials

Type of clinical trials
By nature of comparator treatment- Placebo-controlled: a group of subjects receives a ‘placebo’ treatment, which is
specifically designed to have no real effect → sometimes is not ethical!
- Active-control: the experimental treatment is compared to an existing treatment → that is clearly better than doing nothing for the subject
By type of comparison
- Superiority: the clinical objective of efficacy is to show that the response to the experimental treatment is superior to the comparator treatment → usually superiority to placebo
- Equivalence or non-inferiority: the clinical objective of efficacy is to show that the response to the experimental treatment is at least as good, or not clinically inferior, to the comparator treatment → usually non-inferiority to active control

Statistical analyses vs. clinical trials
Phase I - Graphical tools (individual PK graphs –Cmax, AUC,...)
- Descriptive analysis
Phase II- Descriptive and statistical procedures for efficacy- Oncology: survival analysis (Kaplan-Meier, Cox regression)- Dose-response models
Phase III- Modelling techniques for efficacy: adjustment for covariates, multicentre
studies, treatment of missing data, multiple comparisons...
Phase IV (post-authorisation)- Explicative models, correlations and interactions, graphical display (bar chart,
pie chart, map areas...)

Examples of clinical trials
- A prospective, open-label, non-randomized, clinical trial to determine if xxxx improves ambulatory measures in relapsing-remitting multiple sclerosis (RRMS) patients → phase IV - Pharmacokinetic study of single doses of xxxx, 75 mg and 300 mg, in healthy subjects → open-label, two-treatment crossover, phase I - A multicenter, randomized, parallel, double-blind, dose ranging, placebo-controlled study to compare antiviral effect, safety, tolerability and pharmacokinetics of xxxx monotherapy vs. placebo over 10 days in HIV-1 Infected Adults → phase IIA- Efficacy, safety and tolerability of split-dose of xxxx compared to yyyy solution for colonoscopy preparation: a randomized, controlled trial → phase III - xxxx plus radiotherapy and Induction Chemotherapy in patients with head and neck cancer → phase II - phase III

Day-to-day example
1. A client contacts me in order to ask me about the sample size calculation and statistical input of a new clinical trial
Dear Xavier,
I hope you are well. Please find attached a draft version of the SEA Protocol, this is an open-label, randomised, multicentre phase III study in patients with colorectal cancer. The primary endpoint of the trial is the progression free survival. Could you please give us advice on the sample size and the statistical sections of the protocol (the mentioned paragraphs are highlighted in yellow).
Looking forward to hearing from you soon,
Best wishes,
Llorenç Badiella

Day-to-day example
2. The statistician reads the protocol, look for references about the disease and clinical variables/endpoints used for those specific area, checks the study assumptions and primary endpoint, and from these information, estimates the sample size and writes the statistical section of the protocol
Dear Llorenç,
Thank you for your email. Please find attached the SEA Protocol with my input. The sample size calculation resulted in the following: to achieve a 80% power to detect differences in the contrast of the null hypothesis Ho (Equality of the progression-free survival curves between groups) through a Log-Rank test for two independent samples bilaterally, with a significance level of 5% and assuming that the probability of PFS at 24 months will be 30% for the reference group, and 45% for the experimental group, a total of 454 subjects (227 in each group) will be required.
Best regards,
Xavier

Day-to-day example
3. Sometimes, the client gets back to the statistician as the sample size estimated is too high for- The company resources, or- The recruitment expectation
In this situations, new strategies are required, which normally imply to - Increase the expected clinical difference, or- Change the primary endpoint

Conclusions
Instructions:
Become a statistician: open-minded and objective in the assumptions; precise and analytical in the results
“They want to believe”: be responsible, our work is key in the outcome of a clinical trial ; the client will listen to you and act from the results you present
Teach and be taught, and share your knowledge with your colleagues
Recycle yourself: statistics are a dynamic matter, self-study, training courses and new guidelines are a must do
Follow GCPs, regulatory requirements and company’s SOPs

Interactions:
Work closely with your team: you need the study input from the project leader, the clinical expertise from the medical writer, the knowledge of the data from the CRA and CDA, and the DB experience from the DM
“One step forward, three steps back”: do not move on without the OK from the client: sometimes it can turn against you
“Statisticians seem to talk double Dutch”: make yourself and the results understandable to any person with no knowledge of statistics at all
Conclusions

Contraindications:
Learn to say NO: sometimes it is not possible to do everything the client ask us to do
“You don’t know the power of the dark side”: if your study is underpowered or you carry out statistical analysis of secondary endpoints, beware of the conclusions: the results do not ‘conclude that’ but the ‘suggest that’
Conclusions

Some remarks to end...
-Biostatisticians are always talking about power but do not have any
-Statisticians expect the average but on average people do not expect statisticians
-An idiot with a computer is often more powerful than a statistician with a pencil
-Statisticians worry about interactions and this often makes them lonely
-Even if you have a significant relationship with a statistician you may not find it relevant
Guernsey McPearson http://www.senns.demon.co.uk/Confuseus.htm
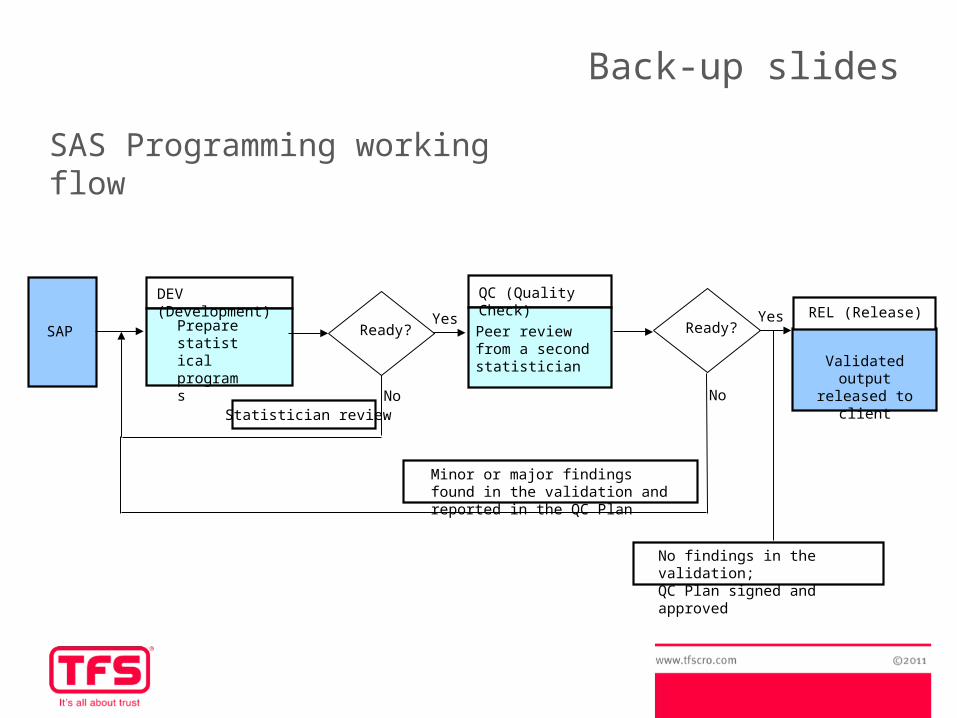
Prepare statistical programs
DEV (Development)
SAP Peer review from a second statistician
QC (Quality Check)
Ready?Yes
No
Minor or major findings found in the validation and reported in the QC Plan
NoStatistician review
Ready?Yes
Validated output released to client
REL (Release)
No findings in the validation; QC Plan signed and approved
Back-up slides
SAS Programming working flow
Back-up slides
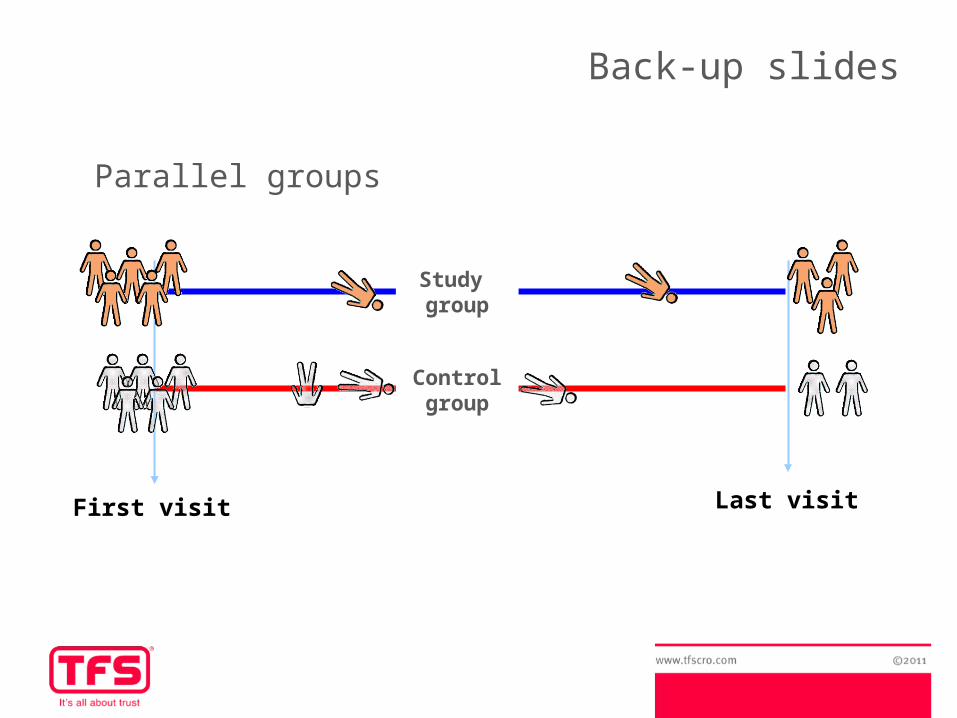
First visit
Study group
Control group
Last visit
Parallel groups
Back-up slides
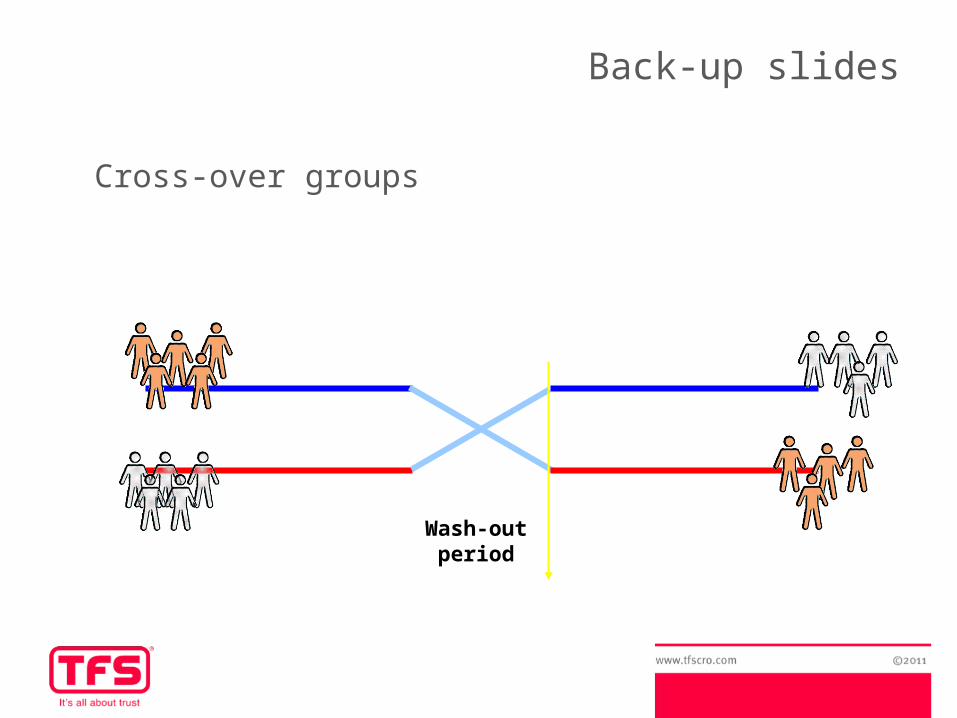
Cross-over groups
Wash-out period

Back-up slides
Advantages of Cross-over groups:- Reduction of variability → each subject is his own control –no within-subject variability- Study design is more efficient, allows for a smaller sample size
Inconvenients:- Wash-out period may not exist or may be difficult to calculate
Back-up slides
First visit
A + B
A + placebo
Last visit
B + placebo
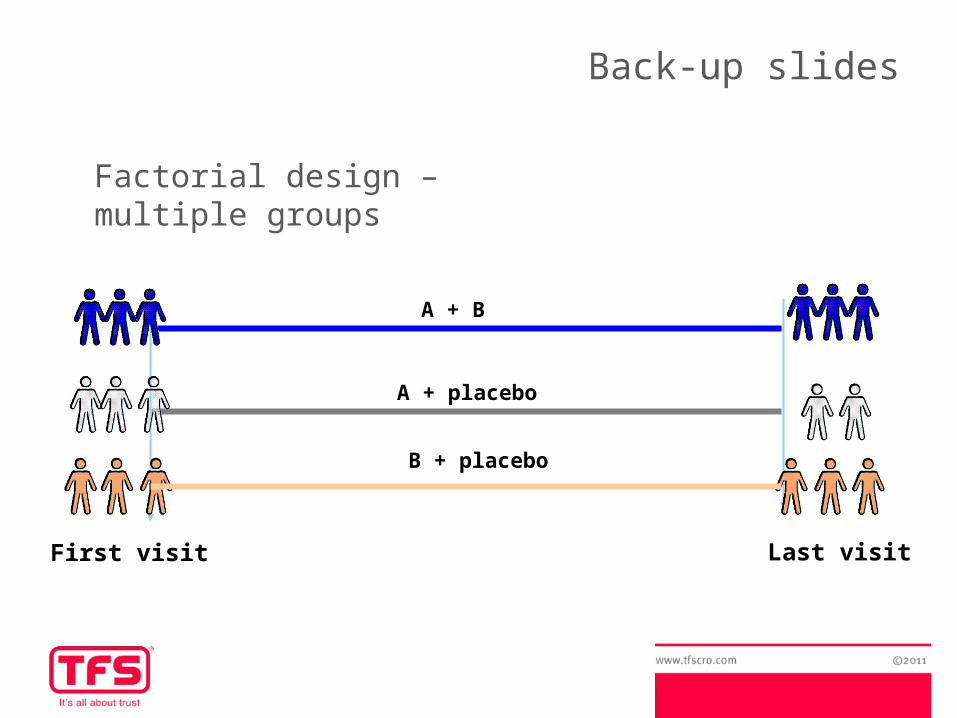
Factorial design – multiple groups

Back-up slides
Advantages of factorial designs:- Efficiency of study design → allows to respond two or more questions in the same trial
-Inconvenients:- Complex design, difficulty of treatment-compliance and follow-up- Study power is sometimes underestimated






























