Embed Size (px)
Citation preview

257
EL LIPOPOLISACARIDO DE HELICOBACTER PYLORI COMO MODULADOR DE LA EXPRESIÓN DE CLAUDINAS EN CÉLULAS
EPITELIALES.
HELICOBACTER PYLORI LIPOPOLYSACARIDE AS MODULATOR OF CLAUDIN EXPRESSION IN EPITHELIAL CELLS.
Erika Patricia Rendón Huerta, Christian Oscar Chavarría Velázquez y Luis Felipe
Montaño Estrada.
Laboratorio de Inmunobiología, Departamento de Biología Celular y Tisular, Facultad de Medicina, UNAM.
[email protected], chavarrí[email protected], [email protected] Teléfono: 5623-2191.
Resumen
La estructura y función de las uniones estrechas, se encuentra
frecuentemente alterada en diferentes tipos de cáncer, lo que promueve la pérdida
de la polaridad y la adhesión celular, mecanismos importantes en el progreso del
cáncer. A nivel mundial, el cáncer gástrico ocupa el cuarto lugar entre los cánceres
más comunes y se considera la tercera causa de muerte por cáncer en nuestro
país. En México, más del 80% de la población está infectada por Helicobacter
Pylori y para eliminar a esta bacteria se recurre al tratamiento con antibióticos, sin
embargo, la recurrencia a la infección aumenta las probabilidades de desarrollar
cáncer gástrico. Por ello es importante estudiar los aspectos moleculares que
modifica la bacteria en el huésped, con el fin de encontrar marcadores tempranos
que refuercen los métodos de diagnóstico actuales. En esta revisión se describe la
Cárdenas Monroy C, González Andrade M, Guevara Flores A, Lara Lemus R, Matuz Mares D, Molina Jijón E, Torres Durán PV. Mensaje Bioquímico, Vol. XL, 257-280, Depto. de Bioquímica, Facultad de Medicina, Universidad Nacional Autónoma de México. Cd. Universitaria, CDMX.,MÉXICO.,(2016). (http://bioq9c1.fmedic.unam.mx/TAB)
(ISSN-0188-137X)

MENSAJE BIOQUÍMICO, VOL. XL (2016)
258
importancia que tiene el LPS de H. pylori como regulador de proteínas de las
uniones estrechas y el papel de éstas en la fisiología de células epiteliales.
Palabras clave: Lipopolisacárido, Helicobacter pylori, receptores tipo Toll (TLR),
claudinas.
Abstract
The structure and function of tight junctions, is frequently altered in different
cancers which promotes the loss of cell polarity and adhesion, important
mechanisms in cancer progression. Globally, gastric cancer is the fourth among
the most common cancers and is considered the third leading cause of cancer
death in our country. In Mexico, more than 80% of the population is infected with
Helicobacter pylori and treatment with antibiotics are used to eliminate this
bacteria, but recurrence of the infection increases the chances of developing
gastric cancer. Therefore, is important to study the molecular aspects the bacteria
modify in the host in order to find early markers that reinforce (support) the current
diagnostic methods. We describe the importance of H. pylori LPS as a regulator of
tight junction proteins and their role in epithelial cells physiology.
Keywords: Lipopolysacaride, Helicobacter pylori, Toll-like receptors (TLR),
claudins.
Helicobacter pylori.
Hace tres décadas, el patólogo Barry Marshall y el gastroenterólogo Robin
Warren, aislaron la bacteria Helicobacter pylori (H. pylori) a partir de una biopsia
de estómago humano [1]. H. pylori es un bacilo Gram negativo, microaerofílico que
coloniza selectivamente la mucosa gástrica humana. Tiene una morfología
espiralada cuando se encuentra en la mucosa gástrica y menos espiralada cuando
crece en medios artificiales. Mide de 0,5 a 1,0 micras de ancho, 3 micras de largo
y presenta de 3 a 5 flagelos monopolares [2]. Posee enzimas específicas como la
oxidasa, ureasa y catalasa, las cuales son utilizadas en los métodos de
diagnóstico para su identificación. H. pylori vive en el estómago y el duodeno de
los seres humanos y por medio de la enzima ureasa cataliza la hidrólisis de la urea
en amonio y dióxido de carbono. De esta manera genera un pH neutro a su
alrededor evadiendo la acidez del estómago para mantenerse en la mucosa
gástrica [3].

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
259
Helicobacter pylori y cáncer gástrico.
Estudios genéticos indican que la relación entre H. pylori y el humano
comenzó hace alrededor de 100.000 años. Simulaciones filogenéticas predicen
que la bacteria se propago desde el este de África [4]. Aproximadamente la mitad
de la población mundial está infectada por H. pylori y la mayoría de los infectados
desarrollan inflamación crónica. El 10% de los infectados se ha asociado con un
mayor riesgo para el desarrollo de patologías gastroduodenales (gastritis crónica,
gastritis inflamatoria, enfermedad úlcero péptica), reportandose que alrededor del
1-3% de los infectados desarrollan adenocarcinoma gástrico y el 0.1 % desarrolla
linfoma de tejido linfoide asociado a mucosa (MALT) [5 y 6]. Se ha observado que
del 70 al 90% de los pacientes con úlcera gástrica o duodenal se encuentran
infectados con H. pylori. La infección persiste a menudo durante toda la vida del
huésped a menos que reciba tratamiento. El tratamiento combinado con
antibióticos (amoxicilina y claritomicina) y un bloqueador de las bombas de
protones (omeprazol) produce la erradicación del microorganismo en un 90% de
los casos [7] sin embargo, la resistencia es cada vez más común teniendo que
administrar triple o cuádruple terapia [8].
Las infecciones son muy comunes en los países poco desarrollados,
probablemente debido a la contaminación del agua y a las condiciones de vida
poco higiénicas.
A nivel mundial, el cáncer gástrico (CG) ocupa el cuarto lugar dentro de los
cánceres más diagnosticados y es la primera causa de muerte en países como
Japón. La asociación entre H. pylori y el CG es de aproximadamente el 63%, por
lo que la Agencia Internacional para la Investigación sobre el Cáncer lo ha
clasificado como carcinógeno tipo I desde 1994 [8, 9].
Patogenicidad de Helicobacter pylori.
Se han propuesto mecanismos para la patogenicidad de H. pylori, como
cambios en la expresión génica en el huésped, incremento en la proliferación
celular, elongación celular, pérdida de la polaridad, alteración de las uniones
célula-célula, y la disminución de la secreción de ácido gástrico. La patogenicidad
de H. pylori se atribuye en gran parte a sus diferentes factores de virulencia:
flagelina, toxina vacuolante VacA, el gen asociado a la citotoxina A en la isla de
patogenicidad (cagPAI) y el lipopolisácarido (LPS), un potente activador de la
respuesta inflamatoria [10] (Figura 1).

MENSAJE BIOQUÍMICO, VOL. XL (2016)
260
Figura 1. Principales factores de patogenicidad de H. Pylori. (Cover, T.L. and Blaser, M.J. (2009). Gastroenterology. 136 (6), 1863–1873).
Lipopolisacárido. La composición de la envoltura celular de H. pylori es similar a la de otras
bacterias Gram negativas. Se compone de una membrana interior citoplasmática,
una pared delgada de peptidoglicano y una membrana externa conformada por
fosfolípidos y lipopolisacárido (LPS). Entre la membrana interna y externa se
localiza el espacio periplásmico [11]. El LPS es considerado un componente tóxico
de las bacterias Gram negativas, con potentes propiedades inmunomoduladoras e
inmunoestimulantes [12] (Figura 2). Presenta una estructura de tres regiones
principales: un lípido A, un núcleo de oligosacáridos, y una capa externa de
polisacáridos o cadena lateral O [11]. El lípido A es el responsable de la actividad
inmunológica, la cual es baja en H. pylori, comparado con otras bacterias Gram
negativas, debido a una modificación en su estructura (bajo grado de fosforilación
y acilación), lo que explica la capacidad que tiene H. pylori para evadir la
respuesta inmunológica del huésped [13]. Al inducir una baja respuesta
inmunológica, la infección por H. pylori puede persistir durante más tiempo que
aquellas causadas por bacterias más agresivas, produciendo una infección crónica
[14]. Se ha obtenido LPS de alto peso molecular (fenotipo liso) con una cadena
lateral O a partir de cepas de H. pylori directamente obtenidas de muestras
clínicas; sin embargo, si se realizan cultivos in vitro de las bacterias, se obtienen
variantes de LPS (fenotipo rugoso) sin la cadena lateral O [15]. La cadena lateral
O del LPS de H. pylori puede ser fucosilada e imitar a antígenos del grupo
sanguíneo Lewis ayudando al mimetismo molecular de antígenos del huésped y a
la evasión inmune [16]. Cuando esto sucede, se dice que son cepas que expresan

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
261
los antígenos de Lewis y su expresión está asociada a patologías más severas. La
expresión del antígeno Lewis mejora la internalización bacteriana entre las células
epiteliales afectando potencialmente la respuesta inmune innata [17].
Figura 2. Componentes principales de la pared de bacterias Gram negativas. (Modificada de Hsing-Ju, W. (2008) Current Opinion in Chemical Biology. 12, 93-101).
Inmunidad innata.
En mamíferos la defensa frente a los patógenos es a través de dos tipos de
inmunidad: la innata y la adaptativa. La respuesta innata es generalmente un
primer proceso, “no específico”, en donde sus funciones son censar, reconocer y
discriminar entre moléculas de microorganismos patógenos y no patógenos [18].
Por el contrario, la respuesta adaptativa, es específica, generando anticuerpos
contra antígenos específicos en las bacterias conduciendo a la activación de
células T y B de memoria [19]. Las funciones del sistema inmune innato dependen
en gran medida de los Receptores de Reconocimiento de Patrones (PRRs), los
cuales reconocen componentes moleculares de agentes patógenos (bacterias,
hongos, virus, etc), esenciales para la supervivencia del patógeno y que se
conocen como patrones moleculares asociados a patógenos (PAMPs) [20].
Receptores tipo toll (TLR).
Entre los PRRs, los receptores tipo Toll (TLRs) fueron los primeros en
descubrirse y son los mejor caracterizados. El creciente interés en la inmunidad
innata desencadenó en 1991 el descubrimiento de los TLRs, por la descripción de
la homología entre la secuencia de una proteína transmembranal denominada Toll
involucrada en la embriogénesis en la mosca de la fruta Drosophila y el receptor
de interleucina-1 humana (IL-1) [21]. Tras la clonación y el mapeo cromosómico de
un TLR homólogo de mamíferos (TLR4), se confirmó su papel inmunológico, tanto
para la proteína Toll de Drosophila como de los TLRs de mamíferos [22].

MENSAJE BIOQUÍMICO, VOL. XL (2016)
262
Los TLRs discriminan específicamente entre componentes del huésped,
microorganismos comensales y componentes microbianos patógenos a través del
reconocimiento de los PAMPs. En diferentes compartimentos celulares, los TLRs
provocan la inducción controlada de la respuesta inmune para la defensa del
huésped a través de cuatro vías: 1) reconociendo patrones moleculares en los
patógenos, 2) expresándose en la interfaz con el medio ambiente externo 3)
activando vías efectoras antimicrobianas y 4) induciendo la secreción de cito-y
quimiocinas pro-o anti-inflamatorias (como IL-1β, IL-8, IL-17, TGF-α) e interferones
tipo I (INF-α), que enlazan y controlan la respuesta inmune adaptativa [23].
Además, los TLRs desempeñan un papel importante en las respuestas de
reparación de tejidos, manteniendo así la homeostasis de la mucosa [24]. Los
estudios iniciales sobre TLRs se centraron exclusivamente en las células de linaje
mieloide y se creía que su expresión era restringida en esta población celular.
Debido a su papel en la vigilancia inmunológica, los TLRs se expresan
principalmente en niveles más altos en los tejidos expuestos al ambiente externo
(por ejemplo, pulmón y tracto gastrointestinal) así como en sitios
inmunológicamente importantes que incluyen leucocitos de sangre periférica y
bazo [25]. Se ha observado que los TLRs son expresados por diferentes tipos de
células en todo el tracto gastrointestinal incluyendo a las células epiteliales del
intestino delgado y el colon [25], las células gástricas pit, las células intestinales
fetales [26], macrófagos intestinales de la lámina propia [27], así como los
hepatocitos [28] y células de Kupffer en el hígado. Se expresan principalmente en
las células presentadoras de antígeno (APC), tales como células dendríticas (DC)
y monocitos [29].
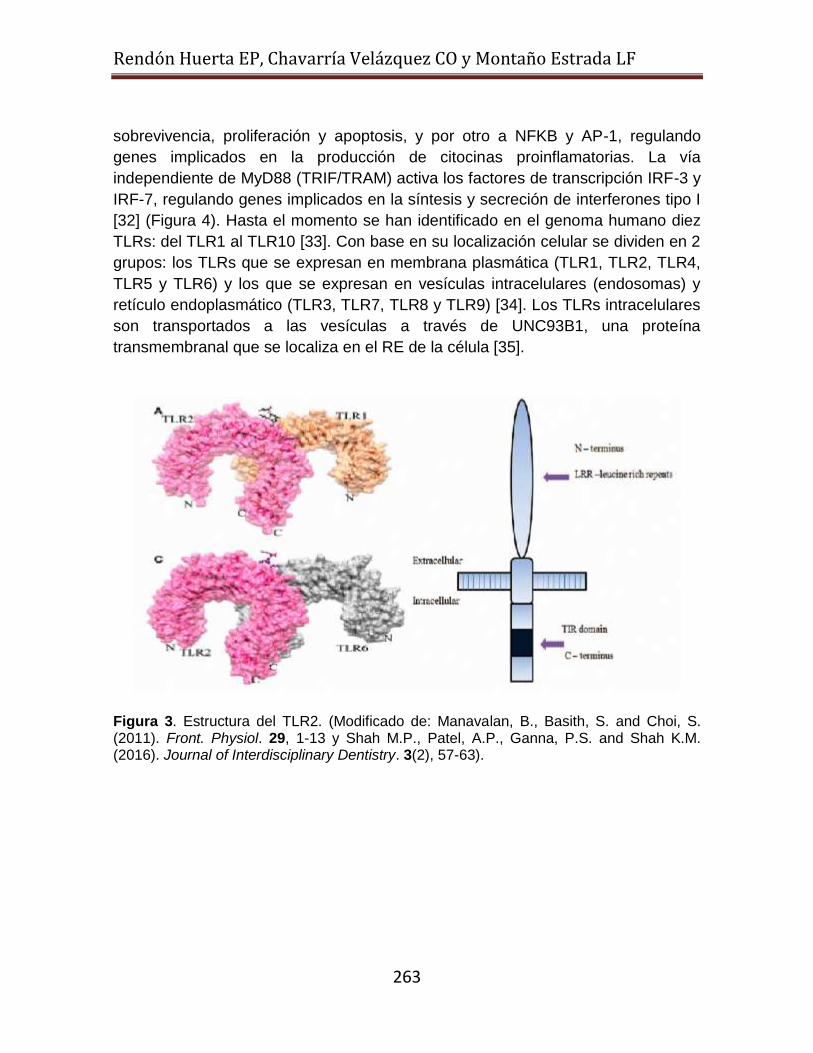
Los TLRs son proteínas transmembranales tipo I trimodulares (Figura 3). Su
estructura consta de tres dominios: 1) un dominio N- terminal extracelular, en
donde se localiza un ectodominio que se compone de aproximadamente 16 a 28
repeticiones ricas en leucina (LRR) y cada LRR consta de 20-30 aminoácidos con
un motivo conservado “LxxLxLxxN " que media el reconocimiento de PAMPs, 2) un
dominio transmembranal y 3) un dominio C- terminal citoplasmático. El dominio
citoplasmático muestra una gran similitud al receptor de IL-1, y como tal, es
conocido como el receptor Toll/IL-1 (TIR) [30, 31]. El dominio TIR se requiere para
la interacción y el reclutamiento de diferentes moléculas adaptadoras para activar
vías de señalización y promover la interacción entre los TLRs y PAMPs. Los TLRs
activados inician 2 vías de transducción de señales, interactuando diferentes
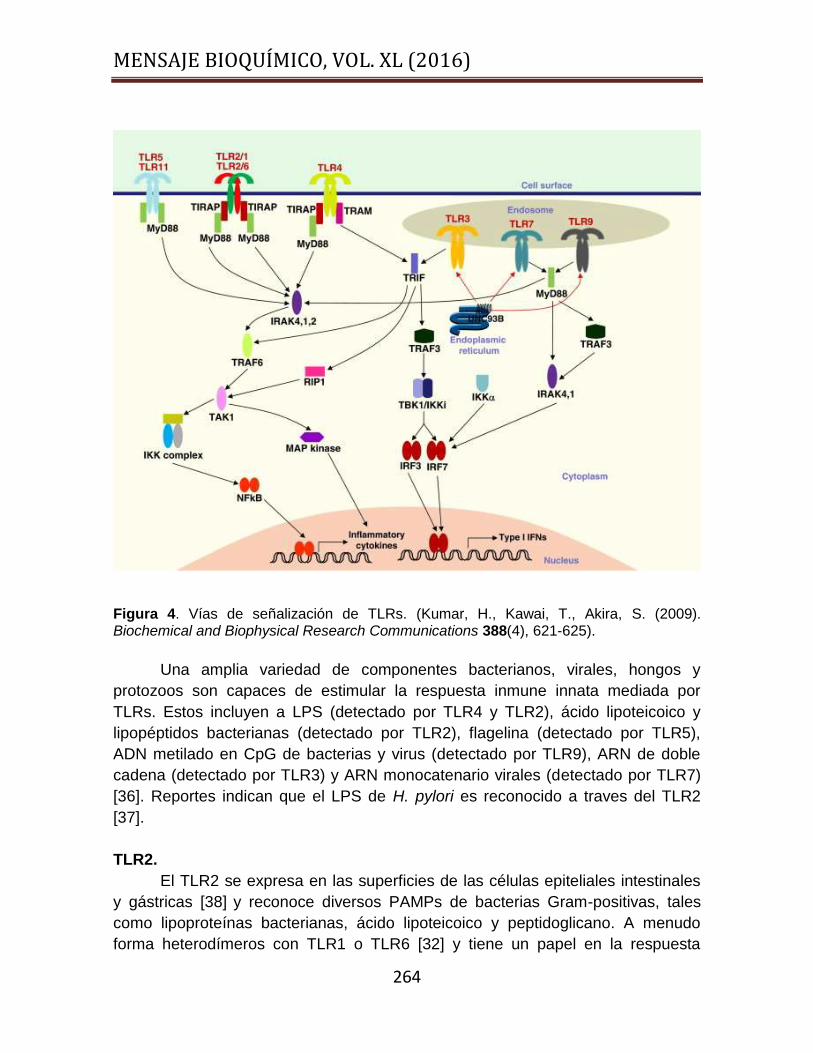
combinaciones de proteínas adaptadoras. La vía dependiente de MyD88
(TIRAP/MyD88) conduce por un lado la activación de MAPKs (ERK, JNK y P38),
Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
263
sobrevivencia, proliferación y apoptosis, y por otro a NFKB y AP-1, regulando
genes implicados en la producción de citocinas proinflamatorias. La vía
independiente de MyD88 (TRIF/TRAM) activa los factores de transcripción IRF-3 y
IRF-7, regulando genes implicados en la síntesis y secreción de interferones tipo I
[32] (Figura 4). Hasta el momento se han identificado en el genoma humano diez
TLRs: del TLR1 al TLR10 [33]. Con base en su localización celular se dividen en 2
grupos: los TLRs que se expresan en membrana plasmática (TLR1, TLR2, TLR4,
TLR5 y TLR6) y los que se expresan en vesículas intracelulares (endosomas) y
retículo endoplasmático (TLR3, TLR7, TLR8 y TLR9) [34]. Los TLRs intracelulares
son transportados a las vesículas a través de UNC93B1, una proteína
transmembranal que se localiza en el RE de la célula [35].
Figura 3. Estructura del TLR2. (Modificado de: Manavalan, B., Basith, S. and Choi, S. (2011). Front. Physiol. 29, 1-13 y Shah M.P., Patel, A.P., Ganna, P.S. and Shah K.M. (2016). Journal of Interdisciplinary Dentistry. 3(2), 57-63).
MENSAJE BIOQUÍMICO, VOL. XL (2016)
264
Figura 4. Vías de señalización de TLRs. (Kumar, H., Kawai, T., Akira, S. (2009). Biochemical and Biophysical Research Communications 388(4), 621-625).
Una amplia variedad de componentes bacterianos, virales, hongos y
protozoos son capaces de estimular la respuesta inmune innata mediada por
TLRs. Estos incluyen a LPS (detectado por TLR4 y TLR2), ácido lipoteicoico y
lipopéptidos bacterianas (detectado por TLR2), flagelina (detectado por TLR5),
ADN metilado en CpG de bacterias y virus (detectado por TLR9), ARN de doble
cadena (detectado por TLR3) y ARN monocatenario virales (detectado por TLR7)
[36]. Reportes indican que el LPS de H. pylori es reconocido a traves del TLR2
[37].
TLR2.
El TLR2 se expresa en las superficies de las células epiteliales intestinales
y gástricas [38] y reconoce diversos PAMPs de bacterias Gram-positivas, tales
como lipoproteínas bacterianas, ácido lipoteicoico y peptidoglicano. A menudo
forma heterodímeros con TLR1 o TLR6 [32] y tiene un papel en la respuesta

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
265
inmune a espiroquetas, micobacterias y hongos, y se ha implicado en la respuesta
al virus de la hepatitis C, herpes simple y el citomegalovirus. Al igual que TLR4,
TLR2 también reconoce ligandos endógenos liberados en los momentos de estrés
celular, tales como las proteínas de choque térmico [39].
Varios estudios han sugerido que el TLR2 desempeña un papel en el
reconocimiento de H. pylori y la subsiguiente inducción de cascadas de
señalización intracelular que activan procesos de inflamación induciendo la
producción de IL12 e IL23 en células presentadoras de antígeno [40]. Células
HEK293 transfectadas con TLR2 responden a diferentes cepas de H. pylori
mediante la activación de NF-kB, mientras que las células transfectadas con TLR4
no activan NF-kB [41]. En este mismo estudio se encontró que las células
epiteliales gástricas transfectadas con una mutante negativa para TLR2 (pero no
TLR4), tuvieron una respuesta atenuada a H. pylori. Sin embargo se determinó
que, mientras derivados de LPS de H. pylori estimulan a TLR4, TLR2 sólo
responde a bacterias intactas [41].
Otros estudios han demostrado que la infección por cepas de H. pylori
positivas a CagA, un factor de virulencia asociado con altas tasas de úlceras y
cáncer gástrico, promueve la secreción de altas concentraciones de IL-8 de
manera dependiente de TLR2 [42].
Reconocimiento de H.pylori por TLRs en células epiteliales gástricas.
La evidencia creciente de la expresión de TLRs en células no
pertenecientes al sistema inmune, sugiere un papel más amplio para estos
receptores en la respuesta de los tejidos infectados o lesionados. Estos hallazgos
apoyan al modelo que explica que no sólo el inicio de la respuesta inmune es
importante para la resolución de los estados anormales que amenazan la
integridad del hospedero, sino también el desarrollo de una serie de respuestas
metabólicas y de comportamiento [43].
Los mecanismos de defensa de las superficies epiteliales son muy
importantes por varias razones: i) Todas las infecciones invasivas se inician al
atravesar la barrera epitelial, ii) Muchas superficies corporales están densamente
colonizadas por una microflora normal de modo que la diferenciación entre
microorganismos comensales y patógenos supone un problema para el epitelio y
iii) La gran mayoría de retos microbianos hacia el hospedero inician por rupturas
menores de las superficies epiteliales generadas por lesiones traumáticas en los

MENSAJE BIOQUÍMICO, VOL. XL (2016)
266
que los microorganismos son rápidamente atacados por los mecanismos de
defensa local sin una activación de la respuesta sistémica [44].
Para poder responder ante el reto de la flora comensal y la patógena, el
epitelio requiere receptores como los TLRs. Es importante tener en cuenta que el
tracto respiratorio inferior, mantiene estériles sus superficies epiteliales de modo
que la presencia de PAMPs es indicativa de infección y por tanto debe activar los
mecanismos de defensa para eliminarla y mantener la función del órgano. Por el
contrario, la mayoría de superficies corporales tales como la piel, el tracto
respiratorio superior y el tracto gastrointestinal, están permanentemente
colonizadas por una variedad de microbios [44, 45].
Aunque algunas bacterias comensales no producen señales estimuladoras,
otras sí lo hacen y en ese caso es necesario que las células epiteliales sean
capaces de diferenciar los microbios comensales de los patógenos mediante
mecanismos aún desconocidos. Actualmente no es claro el mecanismo que regula
la tolerancia del epitelio a la flora normal y la respuesta a los patógenos invasores.
Sin embargo, con base en algunos estudios, se propone que la tolerancia puede
deberse a la expresión compartamentalizada de los TLRs en el epitelio y de
coreceptores como MD-2 [46, 47].
Las bacterias patógenas son capaces de depositar directamente sus
componentes tóxicos, tales como sus LPS, en la superficie apical del epitelio
intestinal para ser internalizado, reciclado, almacenado o transclocado desde el
polo apical al polo basolateral del epitelio [48].
Es crucial que los TLRs no se activen continuamente en respuesta a
PAMPs de las bacterias comensales y a la vez, que puedan tener la capacidad de
activar las vías de señalización en respuesta a patógenos potenciales. Se ha
demostrado que esto se puede lograr regulando a la baja a TLRs específicos,
tales como TLR2 y TLR4, en las superficies de las células epiteliales humanas de
colon e intestino [49, 50]. Trabajos previos sugieren que TLR2 o TLR4 sirven como
mediadores de la respuestas a LPS in vitro e in vivo [51-54]. La función del TLR2
es controversial ya que algunos estudios muestran que el TLR2 media la
señalización intracelular inducida por LPS, mientras que otros estudios mencionan
que no es esencial para responder al LPS [55].
A la fecha, muchos de los estudios sobre la respuesta inmune innata contra
H. pylori en las células epiteliales se han centrado en el TLR4, identificado como la

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
267
molécula de señalización responsable de las respuestas celulares al LPS de
bacterias Gram negativas [51]. Las células del epitelio gástrico pueden estar
parcialmente desensibilizadas o ser tolerantes al LPS para limitar la activación de
las células inmunes adyacentes ante la exposición constante de LPS en la
superficie apical [56]. Las respuestas al LPS a través del TLR4 requieren de los
coreceptores MD-2, CD14 y LBP (proteína de unión de lipopolisacarido) [57]. El
IFN-α e IFN-γ incrementan la expresión de TLR4 y de MD-2 respectivamente, lo
que sugiere que aunque la expresión de los TLRs sea baja en estado basal o en
presencia del LPS, ante un estímulo inflamatorio o infeccioso que genere
respuestas adaptativas Th1, su nivel y el de otros co-receptores puede
incrementarse para responder adecuadamente [57].
La principal barrera en el tracto gastrointestinal contra patógenos es la
monocapa de células epiteliales. Los TLRs, además de ser efectores importantes
en las células inmunitarias, se expresan en la superficie de células epiteliales
gastrointestinales. Sin embargo, el papel preciso que desempeñan en la respuesta
inmune a H. pylori sigue siendo controvertido [58]. Debido a la continua presencia
de microorganismos en el tracto gastrointestinal, el equilibrio entre la expresión de
TLRs y su activación está estrictamente regulado dentro de este nicho. Por lo que,
de acuerdo con esta hipótesis, el epitelio intestinal que es tolerante a los
comensales, puede integrarse a la respuesta inflamatoria sólo tardíamente cuando
el sistema inmune adaptativo requiere de su ayuda para eliminar a los patógenos
[58]. H. pylori se une a las células epiteliales a través de los TLRs y como
consecuencia modifica la estructura de las uniones intercelulares.
Uniones intercelulares.
Las células epiteliales y endoteliales forman barreras continuas que
protegen y mantienen a los órganos, cavidades y canales del organismo en
subcompartimentos funcionales. Este aislamiento y compartamentalización es
crucial para la función de los órganos en todos los organismos multicelulares ya
que forman la barrera de difusión paracelular, el aislamiento del ambiente externo,
el ensamblaje y organización de la lámina basal del epitelio, el mantenimiento de
la integridad epitelial durante la contracción y migración celular y el mantenimiento
de compartimientos celulares. Las células controlan estas barreras selectivas
regulando el movimiento de agua, iones y otras proteínas a través de las
monocapas, generando así la polaridad y función celular. El movimiento de iones y
moléculas a través del espacio intercelular es conocido como permeabilidad
paracelular y es regulada por las proteínas que se localizan en los sitios de
contactos célula-célula o uniones intercelulares [59].

MENSAJE BIOQUÍMICO, VOL. XL (2016)
268
Las uniones intercelulares están lejos de ser estructuras estáticas que
mantienen la barrera simplemente por la unión de células. De acuerdo a la
abundancia proteica que se expresa en las membranas laterales, la disposición de
estas uniones y la comunicación con el citoesqueleto puede variar
sustancialmente. El aumento o disminución en el número de proteínas asociadas a
las uniones está directamente relacionado con la regulación de la función celular.
La constante remodelación de estos contactos permite la extrusión de células en
apoptosis, así como la incorporación de nuevas células epiteliales diferenciadas
derivadas de células progenitoras sin la pérdida de la función de barrera. Durante
la cicatrización de heridas, las células epiteliales pueden experimentar
movimientos coordinados y proliferar para cerrar la herida y establecer nuevos
contactos célula-célula [61].
El complejo de uniones intercelulares, es una estructura formada por cuatro
principales estructuras dependiendo de su función: las Uniones comunicantes
(UC), los Desmosomas y hemidesmosomas, las Uniones adherentes (UA) y las
Uniones estrechas (UE) [62].
Uniones estrechas
La función de barrera en la mucosa gástrica es esencial para prevenir el
libre acceso de elementos potencialmente dañinos como microorganismos
patógenos, presentes en el lumen gástrico y que ingresan hacia la mucosa
gástrica [63, 64]. Esta función de barrera es controlada por uniones apicales
denominadas Uniones Estrechas (UE) y su alteración se asocia con una variedad
de enfermedades humanas, incluyendo cánceres del tracto gastrointestinal [65,
66]. Componentes específicos de H. pylori participan en la desregulación de las
UE. De estas bacterias, existen dos poblaciones, aquellas que se encuentran en
forma libre dentro de la capa protectora de la mucosa del estómago y aquellas que
se adhieren a las células epiteliales gástricas. Estas últimas representan
aproximadamente el 20% [67]. Se sabe que H. pylori se adhiere preferentemente
en las proximidades de las UE pudiendo alterar la localización de proteínas que
constituyen a estos complejos [68-70]. H. pylori ha sido detectada dentro de los
espacios intercelulares intraepiteliales directamente debajo de las uniones
estrechas, lo que lleva a la hipótesis de que la unión estrecha puede ser una
puerta de entrada de la bacteria, que permite el acceso a nutrientes esenciales
[71] (Figura 5).

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
269
Figura 5. Alteración de las Uniones Estrechas por H. pylori. (Wroblewski L.E., Peek Jr., R.M. (2011). Cell Communication and Signaling 9, 1:29).
Las UE forman parte de los complejos de uniones intercelulares. Se
encuentran localizadas rodeando la región apical de las células epiteliales y
endoteliales, formando un cinturón continuo que actúa como barrera fisiológica
para restringir el libre movimiento de proteínas y lípidos, así como regular el
transporte paracelular de agua, iones, pequeñas moléculas no iónicas, en el plano
de las membranas entre las superficies basolateral y apical, manteniendo la
polaridad celular [72]. Esto permite conservar la diferencia de gradientes entre la
membrana basal y lateral y previene el intercambio libre de la mayoría de los
solutos entre el lumen y el espacio intersticial a lo largo de la ruta paracelular [73].
Las UE también participan en cascadas de señalización, regulación
transcripcional, control del ciclo celular y tráfico vesicular. Debido a la gran
variedad de funciones que regulan las UE, su estructura y composición resultan
más complejas que otras uniones intercelulares [74].
Las UE están formadas por aproximadamente 50 diferentes tipos de
proteínas. Dentro de las cuales se encuentran a) proteínas transmembranales

MENSAJE BIOQUÍMICO, VOL. XL (2016)
270
(claudinas, ocludina, tricelulina y JAM [de sus siglas en inglés junction adhesión
molecule]), b) proteínas citoplásmaticas, que cumplen papeles de anclaje al
citoesqueleto (ZO-1, ZO-2, ZO-3, cingulina), y c) proteínas asociadas a vías de
transducción celular (MAPK) y factores transcripcionales ZONAB, ASIP, Afadin,
ZAK y GEF-H1. Mientras que las proteínas citoplasmáticas, también denominadas
proteínas adaptadoras, regulan el ensamblaje y la función de la UE, así como la
proliferación y la diferenciación celular, las proteínas transmembranales
interactúan con sus homologas en las célula adyacente para regular la función de
barrera [75, 76].
Claudinas.
El nombre claudina deriva de la palabra en latín claudere que significa
cerrado. Las claudinas forman una familia de proteínas integrales de membrana
consideradas como el esqueleto de las uniones estrechas y las principales
determinantes del flujo paracelular de solutos a través de epitelios y endotelios
[77]. A la fecha se han reportado alrededor de 24 isoformas en mamíferos y sus
patrones de expresión son tejido-específico. Tienen un peso molecular de 20 a 27
KDa y se clasifican en dos grupos: las claudinas “clásicas” y las “no clásicas”, de
acuerdo al análisis de similitud en sus secuencias. Presentan cuatro dominios
transmembranales (TMD-1, TMD-2, TMD-3 y TMD-4), dos asas extracelulares y su
región amino y carboxilo terminal están localizados en el citosol (Figura 6). La
región carboxilo terminal es variable en tamaño y secuencia, típicamente tiene de
21 a 63 residuos de aminoácidos, es altamente conservada y contiene un motivo
de unión a PDZ, que es un dominio de interacción entre proteínas, y que permite a
las claudinas interaccionar con proteínas citosólicas de andamiaje como ZO-1,
ZO-2, ZO-3, MUPP-1 y PATJ, todas ellas involucradas en diferentes vías de
señalización [78]. Así mismo esta región es blanco de varias modificaciones post-
traduccionales, como la fosforilación en residuos de serina /treonina y tirosina y la
palmitoilación que pueden alterar la localización, estabilidad y función en las
claudinas como la permeabilidad paracelular y resistencia transepitelial [79]. La
fosforilación afecta la localización en la membrana plasmática y da lugar a
cambios en la permeabilidad paracelular.

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
271
FIGURA 6. Estructura de un monómero de claudina. (Günzel D. and Yu A.S.L. (2013). Physiol. Rev. 93, 525-569).
La fosforilación de las claudinas ocurre por acción de la proteína cinasa A
(PKA), la proteína cinasa C (PKC) y las proteínas activadas por mitógenos (MAPK)
[80, 81, 82]. Se ha reportado la participación del factor de transcripción CDX-2 en
la expresión de claudina 2 en células AGS infectadas con H. Pylori [83] y la
regulación de CDX2 vía la activación de ERK1/2 [84]. Por otro lado el factor de
transcripción STAT3 se ha reportado como regulador en la expresión de claudina
4 y 2 [85]. STAT3 es activado a través de TLR2 en procesos inflamatorios
asociados a cáncer [86].
La primera asa extracelular de las claudinas (aprox. 52 residuos de
aminoácidos) contiene aminoácidos cargados, creando un filtro con selectividad
electrostática que controla la resistencia general y la selectividad de carga del flujo
paracelular. También contiene dos residuos de cisteínas altamente conservadas
que forman un enlace disulfuro intramolecular promoviendo la estabilidad de la
conformación proteica. Se ha reportado que la primera asa extracelular de

MENSAJE BIOQUÍMICO, VOL. XL (2016)
272
claudina 1 funciona como co-receptor para el anclaje del virus de la hepatitis C
[87].
La segunda asa extracelular (aprox. 16 a 33 residuos de aminoácidos)
puede formar dímeros entre claudinas de membranas celulares adyacentes, a
través de interacciones hidrofobicas. En las claudinas-3 y -4, esta asa funciona
como receptor para la enterotoxina de Clostridium perfringens (CPE) [88].
Las claudinas mantienen interacciones tipo “cis” con claudinas a lo largo de
la membrana plasmática de la misma célula, e interacciones tipo “trans” con
claudinas de la membrana de la célula adyacente. La interacción de una claudina
con otra de su mismo tipo se conoce como interacción homotípica (cldn4-cldn4) y
cuando la interacción se lleva a cabo con una claudina de otro tipo se le conoce
como interacción heterotípica (cldn4-cldn1) [75].
Claudinas y Cáncer.
El cáncer se define como el crecimiento tisular producido por la proliferación
continua y desordenada de células, insensibles a estímulos apoptóticos y capaces
de invadir y destruir otros tejidos. Existen diversos tipos de cáncer, dependiendo
del tejido del cual se originan. Los carcinomas proceden de tejidos epiteliales y
constituyen aproximadamente el 90% de todos los tumores.
Durante la transformación celular ocurren alteraciones en las uniones
intercelulares, entre ellas las UE. La evidencia de la alteración de las UE en
epitelios cancerosos se conoce desde hace poco más de 30 años. Inicialmente se
pensaba que la pérdida de estas uniones era consecuencia de las
transformaciones celulares. Sin embargo, esta idea ha ido cambiado y ahora se
sugiere que las modificaciones observadas en las UE constituyen los primeros
pasos para dar lugar a la transformación celular [89].
Observaciones en tumores han mostrado la disminución o la pérdida de la
función de las UE, permitiendo el libre paso de moléculas de la membrana
basolateral a la membrana apical y viceversa provocando un aumento en la
permeabilidad paracelular, disminución en la TER, y discontinuidades y
reducciones en el número de hebras de UE dando lugar a la pérdida de la
polaridad celular [89]. En conjunto, estos resultados muestran una reducción de la
función de la barrera epitelial de las células cancerosas promoviendo la pérdida de
las UE, un paso esencial en el desarrollo del cáncer [90]. Reportes indican que la
interrupción de las funciones de las UE se asocia con el desarrollo de varios tipos

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
273
de cáncer, incluyendo el cáncer gástrico, mama, colon, páncreas, próstata, útero y
ovario. Estos cambios se asocian con los fenotipos más invasivos de diferentes
tipos de cáncer y, por ende, con un mal pronóstico para los pacientes [91].
Así mismo, la pérdida de la función de compuerta de las UE, en particular
debida a cambios en la expresión de claudinas, permite el paso descontrolado de
factores de crecimiento y nutrientes a sus receptores promoviendo la proliferación
de células cancerosas. Del mismo modo se ha relacionado la alteración en la
expresión de claudinas en etapas tempranas y tardías de varios tipos de cáncer
con el incremento de la invasividad, el potencial metastásico, el mal pronóstico y la
recurrencia del tumor [89].
Se ha reportado la deslocalización de claudinas de la membrana celular, lo
que parece ser muy frecuente en las células transformadas. Asi mismo se ha
mostrado que, mientras una claudina en particular se encuentra silenciada en
ciertos tipos de cáncer, la misma proteína se encuentra sobreexpresada en otros
carcinomas, lo que sugiere que la función de cada claudina es tejido-específica y
puede depender de un circuito molecular en particular de la célula [92]. Sin
embargo, la relación entre la expresión de claudinas y su función en la progresión
tumoral sigue sin ser comprendida por completo.
Existen diversos estudios en los que se han evaluado los niveles de
expresión de claudinas en tumores primarios de humano. Claudinas 1, 2, 3, 4, y 7
se han observado que disminuyen o elevan su expresión, o se deslocalizan en las
células tumorales en comparación con las células adyacentes normales [91, 93,
94]. Por ejemplo, claudina 1 muestra una elevación consistente en carcinoma de
colon y se asocia con el estado de disociación celular en células de cáncer
pancreático a través de la activación de la proteína cinasa 2 activada por
mitogenos (MAPK2) [91].
Claudina 7 se reduce o se encuentra ausente en carcinomas ductales
invasivos de cáncer de mama metastásico y carcinoma de células escamosas de
cabeza y cuello [91]. Claudinas 3 y 4 se encuentran frecuentemente sobre-
reguladas en carcinomas de ovario, mama, próstata, cérvix, gástrico y pancreático,
mientras que claudina 2 se encuentra sub-regulada en cáncer de mama y próstata
[91]. Claudinas 6, 7 y 9 aumentan su expresión en biopsias de pacientes con
adenocarcinoma gástrico positivos a H. pylori y su sobreexpresión en células AGS
promueve la migración e invasividad celular [95, 96]. Estudios previos en nuestro
laboratorio han mostrado que co-cultivos de H. pylori con células AGS inducen la

MENSAJE BIOQUÍMICO, VOL. XL (2016)
274
secreción de IL-1 y 8 y modifican la expresión de claudinas 4, 5 y 7 [97]. A la fecha
no hay reportes de la posible asociación del efecto del LPS de H. pylori sobre la
expresión de claudinas y/o el desarrollo de cáncer gástrico.
Claudinas y TLRs.
Existen pocos reportes de la asociación de TLRs y claudinas. En células
epiteliales intestinales, la activación de TLR2 induce diferentes vías de
señalización, provocando respuestas inmunes que promueven la protección de la
función de barrera. En ensayos con monocapas de células IEC derivadas de ileo
de rata, la estimulación de TLR2 aumenta selectivamente la resistencia
transepitelial (TER, una medida esencial de la integridad de la barrera). Ensayos
utilizando antagonistas selectivos para la proteína cinasa C (PKC), mostraron que
éste cinasa esta involucrada en dicha regulación [98]. El aumento en la TER
correlaciona con el sellado apical de la membrana en el que participa ZO-1, una
proteína citosólica que forma parte de las UE y que sirve de andamio para otras
proteínas, a través de PKCα /δ [98]. Ratones deficientes en la expresión de TLR2
generan pérdida de la función de barrera en el epitelio intestinal, siendo
susceptibles a desarrollar enfermedad inflamatoria intestinal[99]. Por otra parte en
un modelo de in vitro con células Calu-3 derivadas de epitelio bronquial, se
demostró el reforzamiento de la función de barrera en la uniones estrechas,
debido a un aumento en la expresión de Claudina 1 y ZO-1 a través de la
estimulación con un agonista del TLR2 [99].
Conclusión.
La continuidad funcional de las células epiteliales en diferentes órganos y
tejidos depende de que la homeostasis se preserve. Cuando agentes extraños o
sus productos invaden un nicho que no les corresponde, se activan mecanismos
de discriminación que incluyen entre muchos, la respuesta inflamatoria que como
hemos mencionado modifica la expresión de proteínas necesarias para mantener
la función y estructura de los órganos, y en consecuencia se favorecen procesos
de carcinogénesis.

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
275
Referencias. 1. Warren, J. R., and Marshall, B.J. (1983) Lancet. 1, 1311–1315. 2. Eaton, K.A., Kersulyte, D., Mefford, M., Danon, S.J., Krakowka, S., and
Berg, D.E. (2001) Infect. Immun. 69, 2902–2908. 3. Weeks, D. L., Eskandari, S., Scott, D.R., and Sachs, G. (2000) Science.
287, 482–485.
4. Linz, B., Balloux, F., and Moodley, Y. (2007) Nature. 445, 915–918.
5. Peek, R.M., Jr., and Blaser, M.J. (2002) Nat. Rev. Cancer. 2, 28–37. 6. Kusters, G., van Vliet, A.H.M., and Kuipers, E.J. (2006) Clinical Microbiology
Reviews. 19, 449–490. 7. Vu, C., and Ng, Y. (2000) Singapore Med. J. 41, 478-481. 8. Ahmad, A., Govil, Y., Barbara, B., and Frank, M. (2003) The American
journal of gastroenterology. 98, 975-986. 9. Uemura, N., Okamoto, S., and Yamamoto, S. (2001) New England Journal
of Medicine. 345, 784–789, 10. Johannes, G., Kusters, van Vliet, A.H.M., and Kuipers, E.J. (2006) Clinical
Microbiology Reviews. 19, 449–490. 11. Raetz, C.R., and Whitfield, C. (2002) Annu. Rev. Biochem. 71, 635–700. 12. Erridge, C., Bennett-Guerrero, E., and Poxton, I.R. (2002) Microbes and
Infection. 4, 837–851. 13. Muotiala, A., Helander, I.M., Pyhälä, L., Kosunen, T.U., and Moran, A.P.
(1992) Infect. Immun, 60, 1714-1717. 14. Salaün, L., Ayraud, S., and Saunders, N.J. (2005) Microbiology. 151, 917-
23. 15. Moran, A. P. (2007) International journal of medical microbiology . 297, 307–
319. 16. Martin, S.L., Edbrooke, M.R., Hodgman, T.C., van den Eijnden, D.H., and
Bird, M.I. (1997) J Biol Chem. 272, 21349-21356. 17. Lozniewski, A., Haristoy, X., Rasko, D.A., Hatier, R., Plénat, F., Taylor, D.E.,
and Angioi-Duprez, K. (2003) Infect Immun. 5, 2902-2906. 18. Barton, G.M., and Kagan, J.C. (2009) Nature Reviews Immunology. 9, 535–
542. 19. Iwasaki, A., and Medzhitov, R. (2004) Nat. Immunol. 5, 987–995. 20. Akira, S., Uematsu, S., and Takeuchi, O. (2006) Cell. 124, 783–801. 21. Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J.M., and Hoffmann, J.A.
(1996) Cell. 86, 973–983. 22. Medzhitov, R., Preston-Hurlburt, P., and Janeway, C.A. (1997) Nature. 388,
394–397. 23. Medzhitov, R., and Janeway, C.A. (2002) Science. 296, 298–300. 24. Cario, E., Brown, D., McKee, M., Lynch-Devaney, K., Gerken, G., and
Podolsky, D.K. (2002) Am J Pathol. 160, 165–173. 25. Zarember, K.A., and Godowski, P.J. (2002) J Immunol 168, 554–561. 26. Fusunyan, R.D., Nanthakumar, N.N., Baldeon, M.E., and Walker W.A.
(2001) Pediatric Res. 49, 589–593.

MENSAJE BIOQUÍMICO, VOL. XL (2016)
276
27. Smith, P.D., Smythies, L.E., Mosteller-Barnum, M., Sibley D.A., Russell, M.W., Merger, M., Sellers, M.T. Orenstein J.M., Shimada, T., Graham, M.F., and Kubagawa, H. (2001) J Immunol. 167, 2651–2656.
28. Liu, S., Salyapongse, A.N., Geller, D.A., Vodovotz, Y., and Billiart, T.R. (2000) Shock. 14, 361–365.
29. Corbett, N.P., Blimkie, D., Ho, K.C., Cai, B., Sutherland, D.P., Kallos, A., Crabtree, J., Rein-Weston, A., Lavoie, P.M., Turvey, S.E., Hawkins, N.R., Self, S.G., Wilson, C.B., Hajjar, A.M., Fortuno, E.S., and Kollmann, T.R. (2010) PLoS One. 11, 15041.
30. O’Neill, L. A., and Bowie, A. G. (2007) Nat. Rev. Immunol. 7, 353–364. 31. O’Neill L. (2000) Biochem Soc Trans. 28, 557–56. 32. West, A.P., Koblansky, A.A., and Ghosh, S. (2006) Annu Rev Cell Dev Biol
22, 409–437. 33. Beutler, B. (2004) Nature. 430, 257–263. 34. Akira, S., Uematsu, S., and Takeuchi, O. (2006) Cell. 124, 783–801. 35. Kim, Y.M., Brinkmann, M.M., Paquet, M.E., and Ploegh H.L. (2008) Nature.
452, 234–238. 36. Kumar, H. Kawai, T., and Akira, S. (2009) Biochemical and Biophysical
Research Communications. 388, 621–625. 37. Uno, K., Kato, K., and Shimosegawa, T. (2014) World J Gastroenterol. 18,
5244-5251. 38. Cario, E., Rosenberg, I.M., Brandwein, S.L., Beck, P.L., Reinecker, H.C.,
and Podolsky, D.K. (2000) J Immunol 164, 966–972. 39. Kawai, T., and Akira, S. (2011) Immunity 34, 637-650. 40. Amedei, A., Cappon, A., Codolo, G., Cabrelle, A., Polenghi, A., Benagiano,
M., Tasca, E., Azzurri, A., D'Elios, M.M., Del Prete, G., and de Bernard M. (2006) J Clin Invest 4, 1092-1101.
41. Smith, M.F. Jr., Mitchell, A., Li, G., Ding, S., Fitzmaurice, A. M., Ryan, K., Crowe, S., and Goldberg, J.B. (2003) The Journal of biological chemistry. 278, 32552–60.
42. Fischer, W., Püls, J., Buhrdorf, R., Gebert, B., Odenbreit, S., and Haas, R. (2001) Mol Microbiol. 5, 1337-48.
43. Mogensen, T.H. (2009) Clinical Microbiology Reviews 2, 240–273. 44. Janeway, C.A., and Medzhitov, R. (2002) Annu Rev Immunol 20, 197-216. 45. Medzhitov, R. (2001) Nat Rev Immunol 1, 135-145. 46. Ishihara, S., Rumi, M.A.K., Kadowaki, Y., Ortega-Cava, C.F., Yuki, T.,
Yoshino, N., Miyaoka, Y., Kazumori, H., Ishimura, N., Amano, Y., and Kinoshita, Y. (2004) The Journal of Immunology. 173, 1406-1416.
47. Akashi, S., Shimazu, R., Ogata, H., Nagai, Y., Takeda, K., Kimoto, M., and Miyake, K. (2000) J. Immunol. 164, 3471.
48. Beatty, W. L., and Sansonetti, P. J. (1997) Infect. Immun 65, 4395. 49. Abreu, M.T., Vora, P., Faure, E., Thomas, L.S., Arnold, E.T., and Arditi, M.
(2001) J Immunol 167, 1609-1616. 50. Furrie, E., Macfarlane, S., Thomson, G., and Macfarlane, G.T. (2005)
Immunology. 115, 565–574.

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
277
51. Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Huffel, C. V., Du, X., Birdwell, D., Alejos, E.,Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., and Beutler, B. (1998) Science. 282, 2085–2088.
52. Hoshino, K., Takeuchi, O., Kawai, T., Sanjo, H., Ogawa, T., Takeda, Y., Takeda, K., and Akira, S. (1999) J. Immunol. 162, 3749.
53. Yang, R. B., Mark, M.R., Gray, A., Huang, A., Xie, M.H., Zhang, M., Goddard, A., Wood, W.I., Gurney, A.L., and Godowski, P. J. (1998) Nature. 395, 284.
54. Chow, J. C., Young, D.W., Golenbock, D.T., Christ, W.J., and Gusovsky, F. (1999) J. Biol. Chem. 274, 10689.
55. Yang, R. B., Mark, M.R., Gray, A., Huang, A., Xie, M.H., Zhang, M., Goddard, A., Wood, W.I., Gurney, A.L., and Godowski, P.J. (1998) Nature. 395, 284.
56. Su, B., Ceponis, P. J. M., Lebel, S., Huynh, H., and Sherman, P. M. (2003) Infect. Immun. 71, 3496-3502.
57. Abreu, M.T., Arnold, E.T., Thomas, L.S., Gonsky, R., Zhou, Y., Hu, B., and Arditi, M. (2002) J Biol Chem. 277, 20431-20437.
58. Richard, M., Peek, Jr., Fiske, C., and Wilson, K.T. (2010) Physiol Rev. 90, 831–858.
59. Findley, M.K., and Kpval, M. (2009) IUBMB Life. 61, 431-437. 60. Prakash, S., and Swaminathan, U. (2015) J Oral Maxillofac Pathol. 19, 230–
238. 61. Brennan, K., Offiah G., McSherry, E.A., and Hookins, A.M. (2010) J Biomed
Biotechnol. 2010, 1-16. 62. Glick, A. B., and Yusp, S. H. (2005) Semin. Cancer. 15, 75-83. 63. Mullin, J.M. (2004) Epithelial barriers, compartmentation, and cancer. Sci.
STKE. 216, 1-4. 64. Turksen, K., and Troy, T.C. (2004) Journal of Cell Science. 117, 2435-2447. 65. Findley, M.K.,and Koval. K. (2009) IUBMB Life. 61, 431–437. 66. Oliveira, S. S., and Morgado-Díaz, J. A. (2007) Cell. Mol. Life Sci. 64, 17–
28. 67. Guruge, J. L., Falk, P. G., Lorenz, R. G., Dans, M., Wirth, H. P., Blaser, M.
J., Berg, D. E., and Gordon. J. I. (1998) Proc. Natl. Acad. Sci. U. S. A. 95, 3925–3930.
68. Fedwick, J. P., Lapointe, T. K., Meddings, J. B., Sherman, P. M., and Buret A. G. (2005) Infect. Immun. 73, 7844–7852.
69. Krueger, S., Hundertmark, T., Kuester, D., Kalinski, T., Peitz, U., and Roessner, A. (2007) Pathol. Res. Pract. 203, 433–444.
70. Wroblewski, L. E., Shen, L., Ogden, S., Romero-Gallo, J., Lapierre, L. A., Israel, D. A., Turner, J. R., and Peek, Jr. R. M. (2009) Gastroenterology 136, 236–246.
71. Necchi, V., Candusso, M. E., Tava, F., Luinetti, O., Ventura, U., Fiocca, R., Ricci, V., and Solcia, E. (2007) Gastroenterology 132, 1009–1023.

MENSAJE BIOQUÍMICO, VOL. XL (2016)
278
72. Cereijido, M., Valdes, J., Shoshani, L., and Contreras, R. G. (1998) Role of tight junctions in establishing and maintaining cell polarity. Annu. Rev. Physiol. 60, 161–177.
73. Schneeberger, E. E., and Lynch, R. D. (2004) Cell Physiol. 286, 1213–1228. 74. Tsukita, S., Yamazaki, Y., Katsuno, T.,Tamura, A., and Tsukita S. (2008)
Oncogene 27, 6930–6938. 75. Cereijido, M., Contreras, R.G., Flores-Benitez, D. Flores-Maldonado, C.,
Larre, I., Ruiz, A., and Shoshani, L. (2007) Archives of Medical Research 38, 465-478.
76. Cereijido, M., Contreras, R.G., Shoshani, L., Flores-Benitez, D., and Larre, I. (2008) Biochimica et Biophysica Acta 8, 770–793.
77. Ghislin, S., Obino,D., Middendorp, S., Boggetto, N., Alcaide-Loridan, C., and Deshayes, F. (2011) Pigment Cell Res. 24, 504-511.
78. Krause, G., Winkler, L., Mueller, S.L., Haseloff, F.R., Piontek, J., and Blasig, I.E. (2008) Biochimica et Biophysica Acta 1778, 631-645.
79. Lal-Nag, M., and Morin P.J. (2009) Genome Biology. 10, 235. 80. Van Itallie CM, and Anderson, J. M. (2006) Annu Rev Physiol. 68, 403-429. 81. Chiba, H., Osanai, M., Murata, M., Kojima, T., and Sawada, N. (2008)
Biochim. Biophys. Acta. 1778, 588-600. 82. Furuse, M., and Tsukita, S. (2006) Trends Cell Biol. 16, 181-188. 83. Song, X., Chen, H. X., Wang X. Y., Deng, X. Y., Xi, Y. X., He, Q., Peng, T.
L., Chen, J., Chen, W., Wong, B. C., and Chen, M. H. (2013) Cellular Immunology. 286, 22-30.
84. Lara C., Pera, M., Garrido, M., Iglesias, M., and de Bolós. C. (2014) 1839, 785-792.
85. García-Hernández V., Flores-Maldonado, C., Rincon-Heredia, R., Verdejo-Torres, O., Bonilla-Delgado, J., Meneses-Morales, I., Gariglio, P., and Contreras, R.G. (2015) J. Cell. Physiol. 230, 105–115.
86. Tye, H., Kennedy, C.L., Najdovska, M., McLeod, L., McCormack, W., Hughes, N., Dev, A., Sievert, W., Ooi, C. H., Ishikawa, T. O., Oshima, H., Bhathal, P. S., Parker, A.E., Oshima, M., Tan, P., and Jenkins, B. J. (2012) Cancer Cell. 22, 466–478.
87. Franke, W. W. (2009) A Historical View. Cold Spring Harb Perspect Biol. 26 1-36.
88. Angelow, S., Ahlstrom, R., and Yu. A. S. L. (2008) AJP: Renal Physiology. F867-F876.
89. Berx, G., and Van Roy, F. (2001) Breast Cancer Research. 3, 289–293 90. Gonzalez- Mariscal, L., Lechuga, S., and Garay, E. (2007) Prog. Histochem.
Cytochem. 42, 1-57. 91. Soini, Y. (2005) Histopathology 46, 551-560. 92. Singh A. B., Sharma A., and Dhawan, P. (2010) An Overview. Journal of
Oncology. 10, 1-11. 93. Kominsky, S. L., Argani, P., Korz, D., Evron, E., Raman, V., Garret, E., Rein,
A., Sauter, G., Kallioniemi, O. P., and Sukumar, S. (2003) Oncogene 22, 2021-2033.

Rendón Huerta EP, Chavarría Velázquez CO y Montaño Estrada LF
279
94. Cheung, S. T., Leung, K. L., Ip, Y. C., Chen, X., Fong, D. Y., Ng, I. O., Fan, S. T., and So, S. (2005) Clin. Cancer Res. 11, 551-556.
95. Rendón-Huerta E., Fortoul T., Gorráez M. T., García-Samper X., Álvarez-Fernández G., Zavala-Zendejas V., and Montaño L. F. (2010) J Gastrointest Cancer. 41, 52-59.
96. Zavala-Zendejas V.E., Torres-Martínez A.C., Salas-Morales B., Fortoul T., Montaño L.F., and Rendón-Huerta E.P. (2011) Cancer Investigation 29, 1-11.
97. Paola Isabel Andrade Alvarez. (2011) Tesis de licenciatura, Fac. Ciencias, UNAM, 1-85.
98. Cario, E., Gerken, G., and Podolsky, D. K. (2004) Gastroenterology 127, 224 – 238.
99. Mitic L. L., and Anderson, J. M. (1998) Annu. Rev. Physiol 60, 121 – 142.

MENSAJE BIOQUÍMICO, VOL. XL (2016)
280
Semblanza de la Dra. Erika Patricia Rendón Huerta.
La Dra. Rendón es Bióloga
egresada de la Facultad de Ciencias de
la UNAM. Realizó sus estudios de
Maestría y Doctorado en Investigación
Biomédica Básica en la UNAM.
Posteriormente, realizó una estancia
Posdoctoral en el Lankenau Institute for
Medical Research, PA en donde inició
los proyectos básico-clínicos.
Actualmente es Profesor de tiempo
completo en el Departamento de Biología Celular y Tisular de la Facultad de
Medicina, UNAM y es miembro del Sistema Nacional de Investigadores. Sus
proyectos están enfocados al estudio de la participación de claudinas en el inicio y
progreso del cáncer y las vías de transducción involucradas.





























