Embed Size (px)
Citation preview

dzd
Archives of Biochemistry and BiophysicsVol. 382, No. 2, October 15, pp. 253–261, 2000doi:10.1006/abbi.2000.2027, available online at http://www.idealibrary.com on
Effect of a Prolonged Superoxide Fluxon Transferrin and Ferritin
Thomas Paul1
Steacie Institute for Molecular Sciences, National Research Council of Canada,100 Sussex Drive, Ottawa, Ontario, K1A 0R6 Canada
Received April 17, 2000, and in revised form July 20, 2000
The involvement of “free” iron in damage caused byoxidative stress is well recognized. Superoxide gener-ated in a short burst and at a relatively high flux by thexanthine/xanthine oxidase couple is known to releaseiron from ferritin in the presence of phenanthrolinederivatives as iron chelators. However, superoxidegeneration via xanthine oxidase is accompanied bythe simultaneous direct generation of hydrogen per-oxide and, in the presence of ferritin, there is also asuperoxide-independent release of iron. In this studyit was found that the iron chelator employed attenu-ates superoxide formation from the xanthine/xanthineoxidase couple. The reaction of ferritin and trans-ferrin with a clean chemical source of superoxide,di(4-carboxybenzyl)hyponitrite (SOTS-1) was there-fore investigated. The efficiency of superoxide-in-duced iron release from ferritin increases dramati-cally as the superoxide flux is decreased, reaching ashigh as 0.5 Fe per O2
•2. Treatment of ferritin for 16 hwith SOTS-1 yielded as many as 130 Fe atoms/ferritinmolecule, which greatly exceeds the amount of possi-ble “contaminating” iron absorbed on the proteinshell. © 2000 Academic Press
Key Words: superoxide; ferritin; transferrin; “free”iron; reactive oxygen species; oxidative stress.
The importance of the superoxide radical anion(O2
•2)2 as a reactive oxygen species (ROS) in living
1 Correspondence and reprint requests should be addressed to theauthor. Fax: (613) 952 0068. E-Mail: [email protected].
2 Abbreviations used: CAT, catalase; Ferrozine, 3-(2-pyridyl)-5,6-di-phenyl-1,2,4-triazine-p,p9-disulfonic acid monosodium salt monohy-drate; HO•, hydroxyl radical; H2O2, hydrogen peroxide; HX, hypoxan-thine; O2
•2, superoxide; ROS, reactive oxygen species; SOD, superoxideismutase; SOTS-1, first superoxide thermal source (di(4-carboxyben-yl)hyponitrite); Tiron, 4,5-dihydroxy-1,3-benzenedisulfonic acid diso-
ium salt monohydrate; X, xanthine; XO, xanthine oxidase.0003-9861/00 $35.00Copyright © 2000 by Academic PressAll rights of reproduction in any form reserved.
organisms is well recognized and its participation in alarge number of pathobiological processes is well es-tablished (1, 2). The superoxide dismutases (SODs),which catalyze the dismutation of superoxide to hydro-gen peroxide (H2O2) and dioxygen (reaction 1) (3, 4),provide a first line of defense against O2
•2-induced dam-age.
2 O2•2 1 2 H1O¡
SODH2O2 1 O2 [1]
Superoxide has a very low reactivity in typical freeradical reactions such as hydrogen atom abstractions(5, 6). However, it is a source of more reactive ROS,which contribute to or cause damage to biological sys-tems (7–11), for example, the very reactive hydroxylradical (HO•) may be generated by the Fenton reaction(reaction 2).
Fe21 1 H2O2 3 HO• 1 HO2 1 Fe31 [2]
Living organisms minimize this iron-mediated for-mation of HO• by two strategies: (i) H2O2, the productof superoxide dismutation (reaction 1), is converted towater and dioxygen by catalase (CAT) and reduced towater by glutathione peroxidase and (ii) the amount of“free” iron (often called low molecular weight iron butexcluding heme iron and iron incorporated into theprosthetic groups of enzymes) is minimized by seques-tration into special storage (e.g., ferritin) and transport(e.g., transferrin) proteins. However, these proteinsare, at least, potential sources of “free” iron.
Ferritin is a large spherical protein (450 kDa), whichhas been claimed on theoretical grounds to be able tosequester up to 4500 Fe atoms in a ferric oxide core(12). However, actual preparations of ferritin containmuch less iron, i.e., usually ,3000 atoms of iron per
ferritin (13). It is reported that reducing agents such as253

t
X(
Ft
254 THOMAS PAUL
dihydroflavins (14) and paraquat (15, 16) as well assome iron chelators (17, 18) are able to release Fe fromferritin. More interestingly, in vitro experiments haverevealed that O2
•2 is also an effective reducing agent(19–25) and consequently it has been suggested thatiron released from ferritin is involved in O2
•2 associatedtissue injury (8, 10, 11, 26, 27). On the other hand, ithas also been argued that the Fe detected in the O2
•2/ferritin reaction originates from contaminating Fe onthe surface of the protein rather than from the ferricoxide core (28).
The major carrier of iron in serum is transferrinwhich can transport two tightly bound Fe31 ions (29).The ability of superoxide to release iron from holo-transferrin, the iron-loaded form of transferrin, is con-troversial (30–35).
Studies of the reaction of O2•2 with ferritin and trans-
ferrin have mainly employed high fluxes of superoxidegenerated for relatively short times. Most frequentlythe superoxide has been generated together with H2O2
via the aerobic hypoxanthine (HX)/xanthine oxidase(XO) or xanthine (X)/XO couples. Little is known aboutthe more biologically relevant effect of a prolonged butlow flux of O2
•2 on these iron containing proteins al-though there is some evidence that the efficiency of Ferelease from ferritin increases as the O2
•2 flux de-creases (20, 36). However, the role of iron chelators andthe possible influence of the buffer during the reductivemobilization of iron from ferritin has not been studiedin any detail.
This paper presents results obtained when ferritinand transferrin were subjected to low, steady fluxes ofO2
•2 from a novel superoxide thermal source (di(4-car-boxybenzyl)hyponitrite; SOTS-1), which decomposes inaqueous solution at 37°C (pH 6.5–8), with a half-life of1.3 h and an efficiency for superoxide production of 40mol% (reaction 3) (37). The transient free radicalsformed during the decomposition of SOTS-1, whicheventually lead to the formation of O2
•2 are all veryshort lived with lifetimes in aqueous aerated solutionsthat are insufficient for them to react with other com-pounds, their only fate being to produce O2
•2 from di-oxygen (38).
With SOTS-1 it is possible to generate a well definedand slow flux of superoxide with no simultaneous anddirect generation of H2O2 (as is the case for the X/XO
couple). bMATERIALS AND METHODS
Chemicals. SOTS-1 was synthesized as described previously (38).SOD (from bovine erythrocytes, EC 1.15.1.1), XO (from cow milk, EC1.1.3.22), and ATP and CAT (from beef liver, EC 1.11.1.6) werepurchased from Boehringer-Mannheim. 3-(2-Pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p9-disulfonic acid monosodium salt hydrate (Fer-rozine) and 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium saltmonohydrate (Tiron) were from Aldrich. Ammonium acetate waspurchased from Fisher and trisodium citrate and ascorbic acid fromBDH. PD-10 columns came from Pharmacia Biotech and histidinewas from Research Organics. Ferritin (from human liver) was pur-chased from Calbiochem. All other chemicals, proteins, and enzymeswere from Sigma. For all reactions, a Na/K phosphate buffer (50 mM,pH 7.5) or Tris buffer (50 mM, pH 7.5) were used which had beenstored for at least for 48 h over Chelex 100. In these buffers no ironwas detectable (using Ferrozine as a probe, meaning that the Feconcentration must be ,0.4 mM). The proteins and enzymes wereused as received except for horse spleen ferritin which was passedover a PD-10 column prior use. (The human liver ferritin was tooexpensive for us to purchase a sufficient quantity for PD-10 columnpurification.) UV/Vis measurements were performed at 37°C with aVarian Cary 3 UV/Visible spectrophotometer equipped with a ther-mostatically controlled multicell holder or a Hewlett-Packard ther-mostatically controlled 8452 A diode array spectrophotometer. TheUV/Vis measurements were performed in 1 cm polystyrene cuvettes.
Proteins. Protein assays were performed with Bradford reagentpurchased from Sigma using a bovine serum albumin (fraction V)solution for calibration (39). The iron content of proteins was mea-sured following a procedure described by Fish (40): apoferritin (fromhorse spleen), #0.3 mg Fe/mg protein (#3 Fe atoms/molecule); holo-ransferrin (bovine), 1.75 mg Fe/mg protein (2.4 Fe atoms/molecule);
ferritin (from horse spleen, type 1) ca. 900–1000 Fe atoms/molecule(depending on the batch of ferritin) and ferritin (from human liver)460 Fe atoms/molecule.
O2•2 production from X/XO. The superoxide production from the
/XO couple at 37°C was quantified using cytochrome c as a probelmax 5 550 nm, e 5 21,000 M21 cm21) (41). The reaction solutions
contained cytochrome c (150 mM), CAT (2600 U/ml) to prevent sidereactions of simultaneously generated H2O2, X (300 mM) and anappropiate amount of XO (0.2–2 mU/ml). The absorption change at550 nm was measured in the presence and absence of SOD (75 U/ml)or Ferrozine (1 mM) (Fig. 1A). It should be noted that the buffer used(Tris or phosphate) had a significant effect on superoxide production.
O2•2 production from SOTS-1. Superoxide production from
SOTS-1 (120 mM) at 37°C was followed for 4 h, which corresponds toca. 3 half-lives of SOTS-1 (;90% decomposition of SOTS-1), usingcytochrome c (100 mM) as a probe. The possiblity of a transitionmetal-catalyzed decomposition of SOTS-1 was tested by addingFeSO4 or FeCl3 (50 mM) either alone or together with differentchelators (Ferrozine (150 mM), Tiron (150 mM), EDTA (100 mM),DTPA (100 mM), and nitrilotriacetic acid (150 mM)) to the assaysolution containing SOTS-1 and cytochrome c in Tris buffer (Fig. 1B).In all cases, the total yield of superoxide as well as the kinetics of the550 nm absorption build-up were identical within experimental error(610%). Also, the addition of Ferrozine (1 mM) alone to an assaysolution containing SOTS-1 and cytochrome c did not change thetotal yield of superoxide. SOTS-1 decay was also followed in deuter-ated phosphate buffer (pD 7.4) in the presence of FeSO4 by 1H-NMRspectroscopy at 37°C. The calculated rate constant of (1.4 3 1024 s21)is identical within experimental error to the rate constant forSOTS-1 decomposition previously reported in the absence of transi-tion metals (37).
Fe21 release from ferritin and holotransferrin in the presence oferrozine. Solutions with different amounts of the iron-loaded pro-ein (ferritin or holotransferrin), Ferrozine and SOTS-1 were incu-
ated for 4 h at 37°C. In some experiments, a second potentially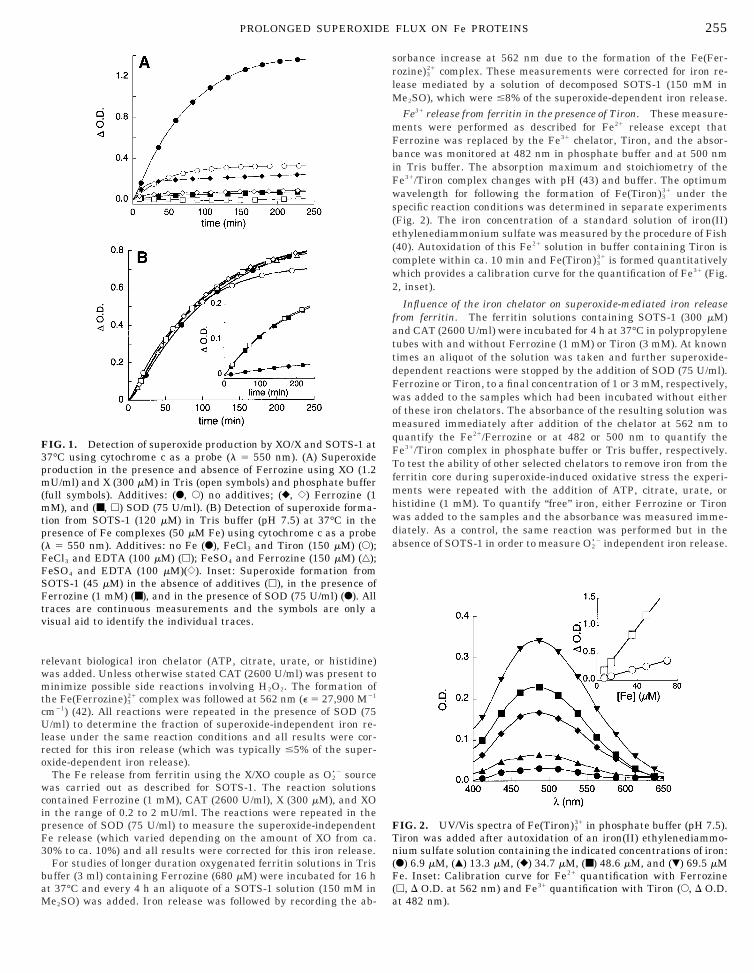
wc
FbiF
3pm
F
(
255PROLONGED SUPEROXIDE FLUX ON Fe PROTEINS
relevant biological iron chelator (ATP, citrate, urate, or histidine)was added. Unless otherwise stated CAT (2600 U/ml) was present tominimize possible side reactions involving H2O2. The formation ofthe Fe(Ferrozine)3
21 complex was followed at 562 nm (e 5 27,900 M21
cm21) (42). All reactions were repeated in the presence of SOD (75U/ml) to determine the fraction of superoxide-independent iron re-lease under the same reaction conditions and all results were cor-rected for this iron release (which was typically #5% of the super-oxide-dependent iron release).
The Fe release from ferritin using the X/XO couple as O2•2 source
as carried out as described for SOTS-1. The reaction solutionsontained Ferrozine (1 mM), CAT (2600 U/ml), X (300 mM), and XO
in the range of 0.2 to 2 mU/ml. The reactions were repeated in thepresence of SOD (75 U/ml) to measure the superoxide-independentFe release (which varied depending on the amount of XO from ca.30% to ca. 10%) and all results were corrected for this iron release.
For studies of longer duration oxygenated ferritin solutions in Trisbuffer (3 ml) containing Ferrozine (680 mM) were incubated for 16 hat 37°C and every 4 h an aliquote of a SOTS-1 solution (150 mM in
FIG. 1. Detection of superoxide production by XO/X and SOTS-1 at7°C using cytochrome c as a probe (l 5 550 nm). (A) Superoxideroduction in the presence and absence of Ferrozine using XO (1.2U/ml) and X (300 mM) in Tris (open symbols) and phosphate buffer
(full symbols). Additives: (F, E) no additives; (}, {) Ferrozine (1mM), and (■, h) SOD (75 U/ml). (B) Detection of superoxide forma-tion from SOTS-1 (120 mM) in Tris buffer (pH 7.5) at 37°C in thepresence of Fe complexes (50 mM Fe) using cytochrome c as a probe(l 5 550 nm). Additives: no Fe (F), FeCl3 and Tiron (150 mM) (E);FeCl3 and EDTA (100 mM) (h); FeSO4 and Ferrozine (150 mM) (‚);
eSO4 and EDTA (100 mM)({). Inset: Superoxide formation fromSOTS-1 (45 mM) in the absence of additives (h), in the presence ofFerrozine (1 mM) (■), and in the presence of SOD (75 U/ml) (F). Alltraces are continuous measurements and the symbols are only avisual aid to identify the individual traces.
Me2SO) was added. Iron release was followed by recording the ab-
sorbance increase at 562 nm due to the formation of the Fe(Fer-rozine)3
21 complex. These measurements were corrected for iron re-lease mediated by a solution of decomposed SOTS-1 (150 mM inMe2SO), which were #8% of the superoxide-dependent iron release.
Fe31 release from ferritin in the presence of Tiron. These measure-ments were performed as described for Fe21 release except that
errozine was replaced by the Fe31 chelator, Tiron, and the absor-ance was monitored at 482 nm in phosphate buffer and at 500 nmn Tris buffer. The absorption maximum and stoichiometry of thee31/Tiron complex changes with pH (43) and buffer. The optimum
wavelength for following the formation of Fe(Tiron)331 under the
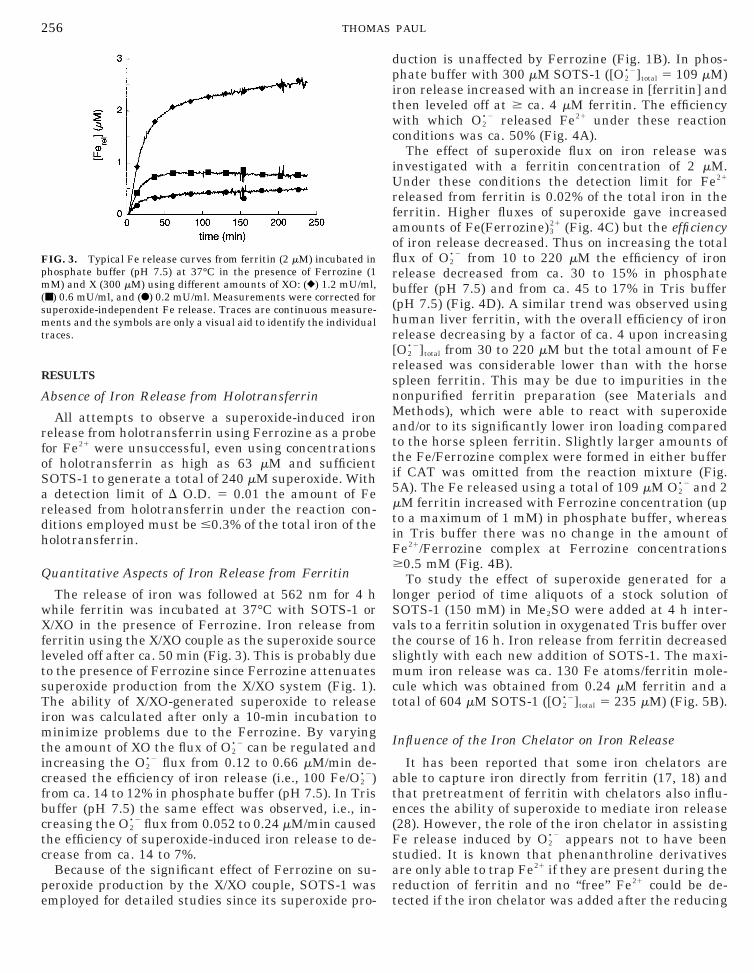
specific reaction conditions was determined in separate experiments(Fig. 2). The iron concentration of a standard solution of iron(II)ethylenediammonium sulfate was measured by the procedure of Fish(40). Autoxidation of this Fe21 solution in buffer containing Tiron iscomplete within ca. 10 min and Fe(Tiron)3
31 is formed quantitativelywhich provides a calibration curve for the quantification of Fe31 (Fig.2, inset).
Influence of the iron chelator on superoxide-mediated iron releasefrom ferritin. The ferritin solutions containing SOTS-1 (300 mM)and CAT (2600 U/ml) were incubated for 4 h at 37°C in polypropylenetubes with and without Ferrozine (1 mM) or Tiron (3 mM). At knowntimes an aliquot of the solution was taken and further superoxide-dependent reactions were stopped by the addition of SOD (75 U/ml).Ferrozine or Tiron, to a final concentration of 1 or 3 mM, respectively,was added to the samples which had been incubated without eitherof these iron chelators. The absorbance of the resulting solution wasmeasured immediately after addition of the chelator at 562 nm toquantify the Fe21/Ferrozine or at 482 or 500 nm to quantify theFe31/Tiron complex in phosphate buffer or Tris buffer, respectively.To test the ability of other selected chelators to remove iron from theferritin core during superoxide-induced oxidative stress the experi-ments were repeated with the addition of ATP, citrate, urate, orhistidine (1 mM). To quantify “free” iron, either Ferrozine or Tironwas added to the samples and the absorbance was measured imme-diately. As a control, the same reaction was performed but in theabsence of SOTS-1 in order to measure O2
•2 independent iron release.
FIG. 2. UV/Vis spectra of Fe(Tiron)331 in phosphate buffer (pH 7.5).
Tiron was added after autoxidation of an iron(II) ethylenediammo-nium sulfate solution containing the indicated concentrations of iron:(F) 6.9 mM, (Œ) 13.3 mM, (}) 34.7 mM, (■) 48.6 mM, and (�) 69.5 mMFe. Inset: Calibration curve for Fe21 quantification with Ferrozineh, D O.D. at 562 nm) and Fe31 quantification with Tiron (E, D O.D.
at 482 nm).
o
ardh
Q
wXfltsTimti
fbc
it
U
lS
pm(smt
256 THOMAS PAUL
RESULTS
Absence of Iron Release from Holotransferrin
All attempts to observe a superoxide-induced ironrelease from holotransferrin using Ferrozine as a probefor Fe21 were unsuccessful, even using concentrationsf holotransferrin as high as 63 mM and sufficient
SOTS-1 to generate a total of 240 mM superoxide. Withdetection limit of D O.D. 5 0.01 the amount of Fe
eleased from holotransferrin under the reaction con-itions employed must be #0.3% of the total iron of theolotransferrin.
uantitative Aspects of Iron Release from Ferritin
The release of iron was followed at 562 nm for 4 hhile ferritin was incubated at 37°C with SOTS-1 or/XO in the presence of Ferrozine. Iron release from
erritin using the X/XO couple as the superoxide sourceeveled off after ca. 50 min (Fig. 3). This is probably dueo the presence of Ferrozine since Ferrozine attenuatesuperoxide production from the X/XO system (Fig. 1).he ability of X/XO-generated superoxide to release
ron was calculated after only a 10-min incubation toinimize problems due to the Ferrozine. By varying
he amount of XO the flux of O2•2 can be regulated and
ncreasing the O2•2 flux from 0.12 to 0.66 mM/min de-
creased the efficiency of iron release (i.e., 100 Fe/O2•2)
rom ca. 14 to 12% in phosphate buffer (pH 7.5). In Trisuffer (pH 7.5) the same effect was observed, i.e., in-reasing the O2
•2 flux from 0.052 to 0.24 mM/min causedthe efficiency of superoxide-induced iron release to de-crease from ca. 14 to 7%.
Because of the significant effect of Ferrozine on su-peroxide production by the X/XO couple, SOTS-1 was
FIG. 3. Typical Fe release curves from ferritin (2 mM) incubated inhosphate buffer (pH 7.5) at 37°C in the presence of Ferrozine (1M) and X (300 mM) using different amounts of XO: (}) 1.2 mU/ml,
■) 0.6 mU/ml, and (F) 0.2 mU/ml. Measurements were corrected foruperoxide-independent Fe release. Traces are continuous measure-ents and the symbols are only a visual aid to identify the individual
races.
employed for detailed studies since its superoxide pro- t
duction is unaffected by Ferrozine (Fig. 1B). In phos-phate buffer with 300 mM SOTS-1 ([O2
•2]total 5 109 mM)ron release increased with an increase in [ferritin] andhen leveled off at $ ca. 4 mM ferritin. The efficiency
with which O2•2 released Fe21 under these reaction
conditions was ca. 50% (Fig. 4A).The effect of superoxide flux on iron release was
investigated with a ferritin concentration of 2 mM.nder these conditions the detection limit for Fe21
released from ferritin is 0.02% of the total iron in theferritin. Higher fluxes of superoxide gave increasedamounts of Fe(Ferrozine)3
21 (Fig. 4C) but the efficiencyof iron release decreased. Thus on increasing the totalflux of O2
•2 from 10 to 220 mM the efficiency of ironrelease decreased from ca. 30 to 15% in phosphatebuffer (pH 7.5) and from ca. 45 to 17% in Tris buffer(pH 7.5) (Fig. 4D). A similar trend was observed usinghuman liver ferritin, with the overall efficiency of ironrelease decreasing by a factor of ca. 4 upon increasing[O2
•2]total from 30 to 220 mM but the total amount of Fereleased was considerable lower than with the horsespleen ferritin. This may be due to impurities in thenonpurified ferritin preparation (see Materials andMethods), which were able to react with superoxideand/or to its significantly lower iron loading comparedto the horse spleen ferritin. Slightly larger amounts ofthe Fe/Ferrozine complex were formed in either bufferif CAT was omitted from the reaction mixture (Fig.5A). The Fe released using a total of 109 mM O2
•2 and 2mM ferritin increased with Ferrozine concentration (upto a maximum of 1 mM) in phosphate buffer, whereasin Tris buffer there was no change in the amount ofFe21/Ferrozine complex at Ferrozine concentrations$0.5 mM (Fig. 4B).
To study the effect of superoxide generated for aonger period of time aliquots of a stock solution ofOTS-1 (150 mM) in Me2SO were added at 4 h inter-
vals to a ferritin solution in oxygenated Tris buffer overthe course of 16 h. Iron release from ferritin decreasedslightly with each new addition of SOTS-1. The maxi-mum iron release was ca. 130 Fe atoms/ferritin mole-cule which was obtained from 0.24 mM ferritin and atotal of 604 mM SOTS-1 ([O2
•2]total 5 235 mM) (Fig. 5B).
Influence of the Iron Chelator on Iron Release
It has been reported that some iron chelators areable to capture iron directly from ferritin (17, 18) andthat pretreatment of ferritin with chelators also influ-ences the ability of superoxide to mediate iron release(28). However, the role of the iron chelator in assistingFe release induced by O2
•2 appears not to have beenstudied. It is known that phenanthroline derivativesare only able to trap Fe21 if they are present during thereduction of ferritin and no “free” Fe21 could be de-
ected if the iron chelator was added after the reducing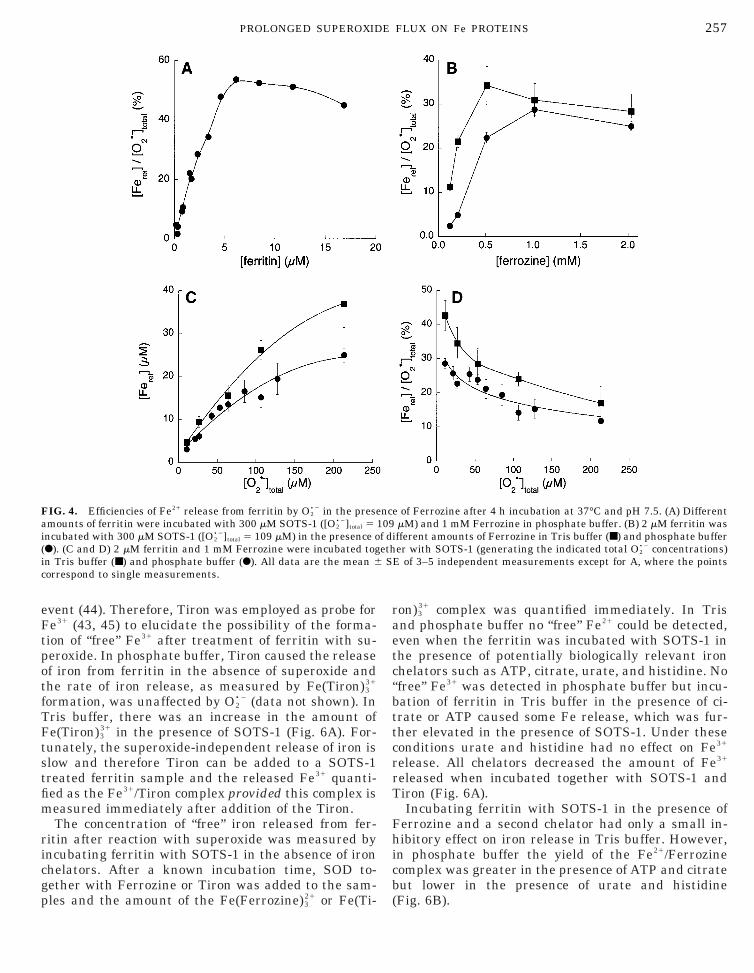
bttc
i
257PROLONGED SUPEROXIDE FLUX ON Fe PROTEINS
event (44). Therefore, Tiron was employed as probe forFe31 (43, 45) to elucidate the possibility of the forma-tion of “free” Fe31 after treatment of ferritin with su-peroxide. In phosphate buffer, Tiron caused the releaseof iron from ferritin in the absence of superoxide andthe rate of iron release, as measured by Fe(Tiron)3
31
formation, was unaffected by O2•2 (data not shown). In
Tris buffer, there was an increase in the amount ofFe(Tiron)3
31 in the presence of SOTS-1 (Fig. 6A). For-tunately, the superoxide-independent release of iron isslow and therefore Tiron can be added to a SOTS-1treated ferritin sample and the released Fe31 quanti-fied as the Fe31/Tiron complex provided this complex ismeasured immediately after addition of the Tiron.
The concentration of “free” iron released from fer-ritin after reaction with superoxide was measured byincubating ferritin with SOTS-1 in the absence of ironchelators. After a known incubation time, SOD to-gether with Ferrozine or Tiron was added to the sam-
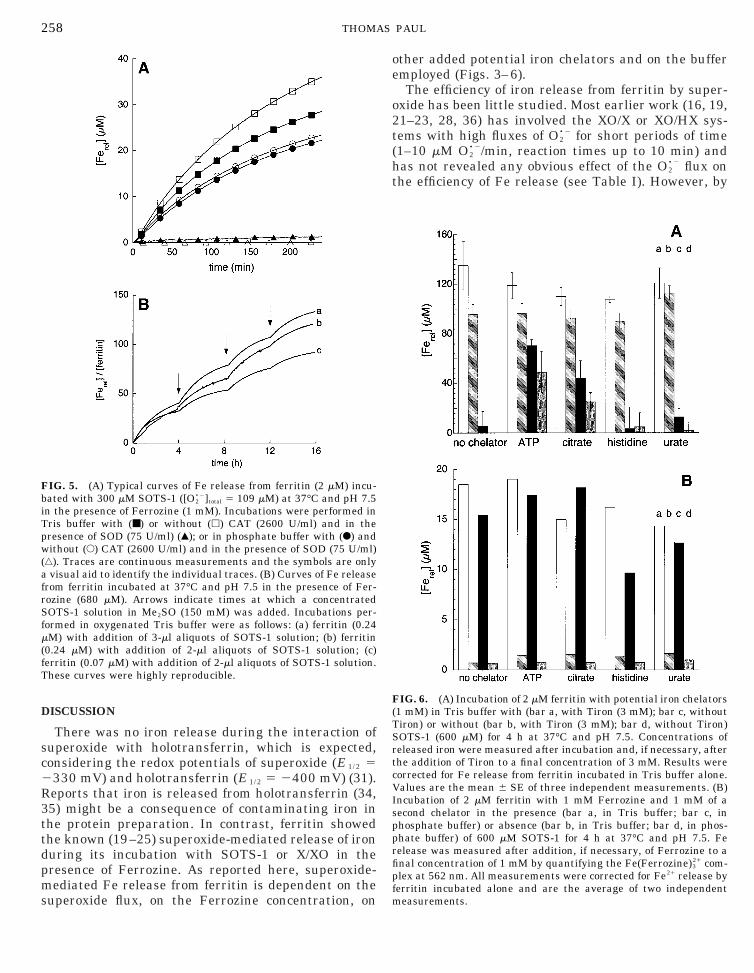
FIG. 4. Efficiencies of Fe21 release from ferritin by O2•2 in the prese
amounts of ferritin were incubated with 300 mM SOTS-1 ([O2•2]total 5
ncubated with 300 mM SOTS-1 ([O2•2]total 5 109 mM) in the presence o
(F). (C and D) 2 mM ferritin and 1 mM Ferrozine were incubated togin Tris buffer (■) and phosphate buffer (F). All data are the mean 6correspond to single measurements.
ples and the amount of the Fe(Ferrozine)321 or Fe(Ti-
ron)331 complex was quantified immediately. In Tris
and phosphate buffer no “free” Fe21 could be detected,even when the ferritin was incubated with SOTS-1 inthe presence of potentially biologically relevant ironchelators such as ATP, citrate, urate, and histidine. No“free” Fe31 was detected in phosphate buffer but incu-ation of ferritin in Tris buffer in the presence of ci-rate or ATP caused some Fe release, which was fur-her elevated in the presence of SOTS-1. Under theseonditions urate and histidine had no effect on Fe31
release. All chelators decreased the amount of Fe31
released when incubated together with SOTS-1 andTiron (Fig. 6A).
Incubating ferritin with SOTS-1 in the presence ofFerrozine and a second chelator had only a small in-hibitory effect on iron release in Tris buffer. However,in phosphate buffer the yield of the Fe21/Ferrozinecomplex was greater in the presence of ATP and citratebut lower in the presence of urate and histidine
e of Ferrozine after 4 h incubation at 37°C and pH 7.5. (A) DifferentmM) and 1 mM Ferrozine in phosphate buffer. (B) 2 mM ferritin was
ifferent amounts of Ferrozine in Tris buffer (■) and phosphate bufferer with SOTS-1 (generating the indicated total O2
•2 concentrations)E of 3–5 independent measurements except for A, where the points
nc109f dethS
(Fig. 6B).
R3ttdpms
b
S
f
258 THOMAS PAUL
DISCUSSION
There was no iron release during the interaction ofsuperoxide with holotransferrin, which is expected,considering the redox potentials of superoxide (E 1/ 2 52330 mV) and holotransferrin (E 1/ 2 5 2400 mV) (31).
eports that iron is released from holotransferrin (34,5) might be a consequence of contaminating iron inhe protein preparation. In contrast, ferritin showedhe known (19–25) superoxide-mediated release of ironuring its incubation with SOTS-1 or X/XO in theresence of Ferrozine. As reported here, superoxide-ediated Fe release from ferritin is dependent on the
FIG. 5. (A) Typical curves of Fe release from ferritin (2 mM) incu-ated with 300 mM SOTS-1 ([O2
•2]total 5 109 mM) at 37°C and pH 7.5in the presence of Ferrozine (1 mM). Incubations were performed inTris buffer with (■) or without (h) CAT (2600 U/ml) and in thepresence of SOD (75 U/ml) (Œ); or in phosphate buffer with (F) andwithout (E) CAT (2600 U/ml) and in the presence of SOD (75 U/ml)(‚). Traces are continuous measurements and the symbols are onlya visual aid to identify the individual traces. (B) Curves of Fe releasefrom ferritin incubated at 37°C and pH 7.5 in the presence of Fer-rozine (680 mM). Arrows indicate times at which a concentrated
OTS-1 solution in Me2SO (150 mM) was added. Incubations per-formed in oxygenated Tris buffer were as follows: (a) ferritin (0.24mM) with addition of 3-ml aliquots of SOTS-1 solution; (b) ferritin(0.24 mM) with addition of 2-ml aliquots of SOTS-1 solution; (c)erritin (0.07 mM) with addition of 2-ml aliquots of SOTS-1 solution.
These curves were highly reproducible.
uperoxide flux, on the Ferrozine concentration, on
other added potential iron chelators and on the bufferemployed (Figs. 3–6).
The efficiency of iron release from ferritin by super-oxide has been little studied. Most earlier work (16, 19,21–23, 28, 36) has involved the XO/X or XO/HX sys-tems with high fluxes of O2
•2 for short periods of time(1–10 mM O2
•2/min, reaction times up to 10 min) andhas not revealed any obvious effect of the O2
•2 flux onthe efficiency of Fe release (see Table I). However, by
FIG. 6. (A) Incubation of 2 mM ferritin with potential iron chelators(1 mM) in Tris buffer with (bar a, with Tiron (3 mM); bar c, withoutTiron) or without (bar b, with Tiron (3 mM); bar d, without Tiron)SOTS-1 (600 mM) for 4 h at 37°C and pH 7.5. Concentrations ofreleased iron were measured after incubation and, if necessary, afterthe addition of Tiron to a final concentration of 3 mM. Results werecorrected for Fe release from ferritin incubated in Tris buffer alone.Values are the mean 6 SE of three independent measurements. (B)Incubation of 2 mM ferritin with 1 mM Ferrozine and 1 mM of asecond chelator in the presence (bar a, in Tris buffer; bar c, inphosphate buffer) or absence (bar b, in Tris buffer; bar d, in phos-phate buffer) of 600 mM SOTS-1 for 4 h at 37°C and pH 7.5. Ferelease was measured after addition, if necessary, of Ferrozine to afinal concentration of 1 mM by quantifying the Fe(Ferrozine)3
21 com-plex at 562 nm. All measurements were corrected for Fe21 release byferritin incubated alone and are the average of two independent
measurements.
tlt
th
cO
w
H
it
XXXX
L
S
259PROLONGED SUPEROXIDE FLUX ON Fe PROTEINS
using stimulated leukocytes to generate O2•2 at rates as
low as 0.33 mM/min, Koster and coworkers (20) ob-served a higher efficiency of Fe release at lower O2
•2
fluxes. The present study confirms the observations ofKoster et al. and is important because the O2
•2 flux willbe very low in vivo. The total amount of iron releasedincreases with higher O2
•2 production but efficiency ofhe Fe release from ferritin (horse spleen and humaniver) decreases. By using SOTS-1 (Table I) several ofhe problems associated with O2
•2 generation by X/XOand HX/XO were avoided. These problems include thehigh fluxes of O2
•2 generated under the reaction condi-ions reported which lead to increased dismutation andence to increased H2O2 formation with consequent
increased reoxidation of Fe21 unless CAT is present(20, 21, 23, 36) and the O2
•2 independent XO-inducedrelease of Fe, a phenomenon which appears to berather irreproducible (0–30%) (15, 21, 22). Another,and so far overlooked, problem comes from the use ofFerrozine (and probably other phenanthroline deri-vates) as an iron chelator since this compound has beenfound to attenuate superoxide production from theX/XO system (Fig. 1). This observation cannot be ex-
TABLE I
Efficiencies of Fe21 Release from Ferritin by Superoxidein the Presence of an Iron(II) Chelator
(Phenanthroline Derivative)
O2•2 source
Rate of O2•2
productiona
[mM/min]Reaction time
[min]
Fe releaseper O2
•2a
[%] Ref.
XO/X 10 1 4 21XO/X 10 1 6 28XO/X 6.7 10 7–0b 22
O/HX 6 1 2 16O/X 1.4 1 25 19O/X 1 2 3 23O/HX 1.7 10 2 36
6.8 10 211.9 10 1
eukocytes 0.33 15 45 201.1 15 204.5 15 8
OTS-1 0.044c,d 240 29 6 1g —h
0.53c,d 240 15 6 3g —h
0.044c,e 240 43 6 4g —h
0.53c,e 240 17 6 5g —h
0.044c,f 240 47 6 6g —h
0.53c,f 240 24 6 4g —h
a When necessary calculated from graphs in the publications.b Dependent of the ferritin source.c Average rate of O2
•2 production over the 4-h incubations.d Phosphate buffer with CAT.e Tris buffer with CAT.f Tris buffer without CAT.g Mean 6 SE of four independent measurements.h This work.
plained by a direct reaction of Ferrozine with O2•2 be-
cause the amount of O2•2 derived from SOTS-1 is not
influenced by Ferrozine. It is unclear whether Fer-rozine deactivates XO or is reduced directly by XOduring enzyme turnover. In most previous work, O2
•2
production from XO was quantified in the absence ofthe iron chelator that was used with the ferritin and,therefore, O2
•2 production in the actual ferritin reactionsystem is likely to have been lower than estimated.Quantification of the efficiency of Fe release from fer-ritin by XO-generated O2
•2 is simply not possible andhence earlier reports are of only qualitative value.With SOTS-1, in contrast, there is no direct formationof H2O2 and the dismutation of O2
•2 will be a very minorprocess under the reaction conditions employed (videinfra). It should also be noted that the rates of SOTS-1decomposition and O2
•2 production are not influencedby added iron complexes or Ferrozine, which demon-strates that SOTS-1 is a clean and much more reliablesuperoxide source than XO under the reaction condi-tions which were employed. The fact that CAT pro-duces a small decrease in Fe(Ferrozine)3
21 formationan probably be attributed to its direct reaction with
2•2 (46).It is unlikely that increased self-dismutation of O2
•2
at higher SOTS-1 concentrations accounts for the de-creasing efficiency of Fe release. The generation of O2
•2
from SOTS-1 is slow and the rate constant for dismu-tation of O2
•2 at pH 7.5 is only ca. 2 3 105 M21 s21 (5),which is 10 times slower than the reported rate con-stant of 2 3 106 M21 s21 for the reaction of O2
•2 withferritin (47). In the absence of ferritin the only possibledecomposition pathway for O2
•2 is its dismutation andthe steady state concentration of superoxide ([O2
•2]ss)ill be substantial, e.g., using SOTS-1 (300 mM, gen-
erating 10.3 mM O2•2 in 10 min), [O2
•2]ss will be 0.29 mM.owever, in the presence of ferritin (2 mM) most O2
•2
reacts with the ferritin and [O2•2]ss decreases to only
0.004 mM. This large decrease in the [O2•2]ss calculated
ndicates that under the reaction conditions used inhis study very little of the O2
•2 generated can dismu-tate to H2O2 and O2. Under other conditions such aslower SOTS-1 concentration or after reaction timeslonger than 10 min the O2
•2 reaction with ferritin be-comes even more favorable (because dismutation ofO2
•2 is second order in O2•2 and will be much slower at
lower rates of superoxide formation).It is also unlikely that superoxide assists the incor-
poration of Fe into ferritin because there was no obvi-ous difference in the loading of apoferritin with Fe31
solutions in the presence or absence of SOTS-1 (datanot shown). I therefore tentatively suggest that reoxi-dation of Fe21 by O2
•2 is the reason for the decreasedefficiency of Fe release at higher O2
•2 fluxes. If this iscorrect, ferritin would be a SOD-mimic under thesereaction conditions. The up to ca. 50% efficiencies of Fe
release by O2•2 are among the highest reported (Table

iTti
oibmmsdt
d
c
r1t
260 THOMAS PAUL
I). The treatment of ferritin (5 mM) with SOTS-1 (300mM 5̂ [O2
•2]total 5 109 mM) for 4 h at 37°C induces anron release of ca. 30 Fe atoms per ferritin molecule.he amount of iron released was further increased byhe addition of “fresh” SOTS-1. This was shown byncubating a ferritin solution (0.24 mM, 3 ml) for a total
of 16 h and adding 3-ml aliquots of a SOTS-1 solution(150 mM in Me2SO) every 4 h (Fig. 5B). Each singleaddition of this SOTS-1 solution generated during each4-h incubation a total of ca. 59 mM O2
•2, which sequen-tially cause the release of 40, 37, 30, and 26 (average33) Fe atoms/ferritin. Thus, after a 16-h incubation atotal of 235 mM O2
•2 has been generated and ca. 130 Featoms/ferritin molecule have been released. There isevidence that ferritin can carry up to 12 Fe31 on theutside of the protein shell, i.e., 12 Fe31 that are notncorporated in the ferric oxide core (48, 49). It haseen reported that O2
•2 (generated by the X/XO couple)ediates the release of only 1 to 2 Fe atoms/ferritinolecule and that this iron originates from the protein
hell (28). However, the present results provide evi-ence that superoxide can, indeed, release iron fromhe ferric oxide core of ferritin.
The reoxidation of Fe21 seems to play an importantrole in studies of reductive iron release from ferritin.Ferrozine added to a ferritin solution immediately af-ter a short burst of O2
•2 (generated by pulse radiolysis)id not yield detectable amounts of the Fe21/Ferrozine
complex (44). Also I was unable to detect “free” Fe21
after incubation of ferritin with SOTS-1 and addition ofFerrozine immediately before the UV/Vis measure-ment. Even incubation of ferritin with SOTS-1 in thepresence of ATP, citrate, urate, and histidine did notyield any “free” Fe21 detectable with Ferrozine. How-ever, there is a buffer effect on O2
•2-mediated Fe21
release from ferritin since much more Fe/Ferrozinecomplex was formed in Tris buffer than in phosphatebuffer. Similar results have been obtained using ascor-bate as the reductant (10) and the need for a largeexcess of Ferrozine to detect all the Fe21 released hasalso been noted. Incubating ferritin with SOTS-1 andFerrozine together with a second iron chelator did notincrease the yield of Fe21 significantly. All these resultsindicate that the reoxidation of Fe21 is fast and that itcan be only detected when it is immediately “captured”by stabilizing ligands. It is likely that oxygen-atomcontaining ligands (such as those provided by phos-phate or citrate both of which are known to stabilizeFe31) compete with Ferrozine for Fe21 by enhancing itsreoxidation to Fe31. No chelator has yet been identifiedwhich might stabilize Fe21 in vivo. All results implythat “free” Fe21 is not released from ferritin despite thefact that it is detected when chelating agents whichstabilize Fe21 are present.
There is, of course, the possibility that “free” Fe21 is
released and rapidly oxidized to Fe31. Tiron, a dihy- tdroxybenzene derivative, was employed as a probe forFe31. As known for other dihydroxybenzene derivatives(16, 50), Tiron is able to release iron directly from theferritin. However, this process is slow and the additionof Tiron after incubating ferritin with SOTS-1 gavenegligible yields of the Fe/Tiron complex. Thus, wehave been unable to detect O2
•2-generated “free” ironfrom ferritin as either Fe21 or Fe31. The addition ofbiologically relevant iron chelators (ATP, citrate,urate, and histidine) did not yield detectable “free” Fe31
in phosphate buffer but in Tris buffer there was anelevated level of “free” Fe31 in the presence of citrate orATP. The effect of the buffer substance on the amountof Fe31 detected is probably due to the above mentionedfaster reoxidation of Fe21 in the presence of phosphatethan in the presence of Tris. These results show that“free” Fe31 really can be generated in the reaction offerritin with O2
•2 provided suitable iron chelators arepresent. The amount of “free” Fe31 produced was foundto be 22 mM using ATP as chelator and 19 mM usingcitrate. These yields are not much smaller than the O2
•2
mediated Fe21 release in the presence of Ferrozineunder similar conditions, viz., 25 mM in phosphatebuffer and 35 mM in Tris buffer.
It has been reported that ferritins from differentorgans and vertebrates show different sensitivity tosuperoxide-mediated iron release and that human liverferritin is a particular sensitive ferritin (22). However,the X/XO couple was employed which I have shown isnot an easily quantifiable superoxide source under thereaction conditions which were employed. In thepresent experiments with SOTS-1 and human liverferritin the same trend was observed as for horsespleen ferritin, i.e., superoxide generated at slow ratesis more efficient in releasing iron than when generatedat high rates. This result together with the reportedhigher sensitivity of human liver ferritin toward thesuperoxide-mediated iron release may be important forthe generation of “free” iron under pathobiological con-ditions. However, a detailed study will be necessary toelucidate the true sensitivity of human ferritin to lowfluxes of superoxide.
In summary, this study has shown that previouslyreported low efficiencies of O2
•2-induced formation ofFe21/phenanthroline from ferritin were artifactual be-ause of the high O2
•2 fluxes generated from XO and theattenuation of superoxide production from XO in thepresence of phenanthroline derivatives. Low fluxes ofO2
•2 should better reflect in vivo situations. I havefound that, at low fluxes, O2
•2 generated from a cleanchemical source is remarkably efficient (up to ca. 50%)at releasing iron from ferritin (detected as Fe21/Fer-ozine). In 16-h experiments an iron release of up to30 atoms Fe/ferritin molecule provide evidence thathe iron can be released from the ferritin core because
his amount of iron exceeds any possible amount of
111
1
1
1
1
1
1
1
2
22
2
2
2
22
2
2
3
33
3
3
3
3
3
3
34444
4
4
4
4
4
4
5
261PROLONGED SUPEROXIDE FLUX ON Fe PROTEINS
“contaminating” iron absorbed on the protein shell.However, in the absence of suitable Fe21 chelatingligands the reoxidation is probably sufficient fast thatno free Fe21 will be released from ferritin.
ACKNOWLEDGMENTS
This work was partly supported by the National Foundation forCancer Research. I thank Drs. Keith U. Ingold and George R. Hodgesfor helpful discussions and Shuqiong Lin for synthesis of SOTS-1.
REFERENCES
1. Fridovich, I. (1976) in Free Radicals in Biology (Pryor, W. A.,Ed.), Vol. 1, pp. 239–277, Academic Press, New York.
2. Halliwell, B., and Gutteridge, J. M. C. (1999) Free Radicals inBiology and Medicine, 3rd ed., Oxford Univ. Press, Oxford.
3. McCord, J. M., and Fridovich, I. (1969) J. Biol. Chem. 244,6049–6055.
4. Fridovich, I. (1995) Annu. Rev. Biochem. 64, 97–112.5. Bielski, B. H. J., Cabelli, D. E., Arudi, R. L., and Ross, A. B.
(1985) J. Phys. Chem. Ref. Data 14, 1041–1100.6. Bielski, B. H. J., and Cabelli, D. E. (1991) Int. J. Radiat. Biol. 59,
291–319.7. Halliwell, B., and Gutteridge, J. M. C. (1986) Arch. Biochem.
Biophys. 246, 501–514.8. Halliwell, B. (1992) J. Neurochem. 59, 1609–1623.9. Herbert, V., Shaw, S., Jayatilleke, E., and Stopler-Kasdan, T.
(1994) Stem Cells 12, 289–303.0. Reif, D. W. (1992) Free Rad. Biol. Med. 12, 417–427.1. Aust, S. D. (1995) Toxicol. Lett. 82/83, 941–944.2. Harrison, P. M., and Arosio, P. (1996) Biochim. Biophys. Acta
1275, 161–203.3. de Silva, D., Guo, J.-H., and Aust, S. D. (1993) Arch. Biochem.
Biophys. 303, 451–455.4. Jones, T., Spencer, R., and Walsh, C. (1978) Biochemistry 17,
4011–4017.5. Thomas, C. E., and Aust, S. D. (1986) J. Biol. Chem. 261, 13064–
13070.6. Monteiro, H. P., Vile, G. F., and Winterbourn, C. C. (1989) Free
Rad. Biol. Med. 6, 587–591.7. O’Connell, M. J., Ward, R. J., Baum, H., and Peters, T. J. (1989)
Biochem. J. 260, 903–907.8. Pape, L., Multani, J. S., Stitt, C., and Saltman, P. (1968) Bio-
chemistry 7, 613–616.9. Biemond, P., Swaak, A. J. G., Beindorff, C. M., and Koster, J. F.
(1986) Biochem. J. 239, 169–173.0. Biemond, P., Eijk, H. G. v., Swaak, A. J. G., and Koster, J. F.
(1984) J. Clin. Invest. 73, 1576–1579.1. Bolann, B. J., and Ulvik, R. J. (1987) Biochem. J. 243, 53–59.2. Harris, L. R., Cake, M. H., and Macey, D. J. (1994) Biochem. J.
301, 385–389.
3. Thomas, C. E., Morehouse, L. A., and Aust, S. D. (1985) J. Biol.Chem. 260, 3275–3280.
4. Thomas, C. E., and Aust, S. D. (1986) Free Rad. Biol. Med. 1,293–300.
5. Yoshida, T., Tanaka, M., Sotomatsu, A., and Hirai, S. (1995)Neurosci. Lett. 190, 21–24.
6. Lipinski, P., and Drapier, J.-C. (1997) JBIC 2, 559–566.7. de Silva, D. M., and Aust, S. D. (1993) Can. J. Physiol. Pharma-
col. 71, 715–720.8. Bolann, B. J., and Ulvik, R. J. (1990) Eur. J. Biochem. 193,
899–904.9. Aisen, P. (1980) in Iron in Biochemistry and Medicine II (Jacobs,
A., and Worwood, M., Eds.), pp. 87–130, Academic Press, Lon-don.
0. Baldwin, D. A., Jenny, E. R., and Aisen, P. (1984) J. Biol. Chem.259, 13391–13394.
1. Buettner, G. R. (1987) J. Biol. Chem. 262, 11995–11998.2. Aruoma, O. I., and Halliwell, B. (1987) Biochem. J. 241, 273–
278.3. Saito, M., Morehouse, L. A., and Aust, S. D. (1986) J. Free Rad.
Biol. Med. 2, 99–105.4. Brieland, J. K., and Fantone, J. C. (1991) Arch. Biochem. Bio-
phys. 284, 78–83.5. Brieland, J. K., Clarke, S. J., Karmiol, S., Phan, S. H., and
Fantone, J. C. (1992) Arch. Biochem. Biophys. 294, 265–270.6. Monteiro, H. P., and Winterbourn, C. C. (1988) Biochem. J. 256,
923–928.7. Ingold, K. U., Paul, T., Young, M. J., and Doiron, L. (1997) J. Am.
Chem. Soc. 119, 12364–12365.8. Konya, K. G., Paul, T., Lin, S., Lusztyk, J., and Ingold, K. U.
(2000) J. Am. Chem. Soc. 122, 7518–7527.9. Bradford, M. M. (1976) Anal. Biochem. 72, 248–254.0. Fish, W. W. (1988) Methods Enzymol. 158, 357–364.1. Massey, V. (1959) Biochim. Biophys. Acta 34, 255–256.2. Stookey, L. L. (1970) Anal. Chem. 42, 779–781.3. Harvey, Jr., A. E., and Manning, D. L. (1950) J. Am. Chem. Soc.
72, 4488–4493.4. Reif, D. W., Schubert, J., and Aust, S. (1988) Arch. Biochem.
Biophys. 264, 238–243.5. Endo, M., and Abe, S. (1997) Fresenius J. Anal. Chem. 358,
546–547.6. Gebicka, L., Metodiewa, D., and Gebicki, J. L. (1989) Int. J.
Radiat. Biol. 55, 45–50.7. Buettner, G. R., Saran, M., and Bors, W. (1987) Free Rad. Res.
Commun. 2, 369–372.8. Yang, C., Meagher, A., Huynh, B. H., Sayers, D. E., and Theil,
E. C. (1987) Biochemistry 26, 497–503.9. Bakker, G. R., and Boyer, R. F. (1986) J. Biol. Chem. 261,
13182–13185.0. Boyer, R. F., Grabill, T. W., and Petrovich, R. M. (1988) Anal.
Biochem. 174, 17–22.





























