Embed Size (px)
Citation preview

© 1995 Oxford University Press Human Molecular Genetics, 1995, Vol. 4, No. 1 51-58
DNA-based mutation analysis of Bruton'styrosine kinase gene in patients with X-linked agammaglobulinaemiaIgor Vorechovsky1'*, Mauno Vihinen2, Genevieve de Saint Basile3, Stanislava Honsovd4,Lennart Hammarstrom1'5, Susanne Muller1, Lennart Nilsson2, Alain Fischer3 andC.I.Edvard Smith1-5
1Center for BioTechnology and 2Center for Structural Biochemistry, Karolinska Institute at NOVUM, S-14157 Huddinge, Sweden,3INSERM U132, H6pital Necker-Enfants-Malades, F-75743 Paris Cedex 15, France, department of Clinical Immunology, FacultyHospital Motol, CZ-15085 Prague, Czech Republic and 5Department of Clinical Immunology, Huddinge Hospital, S-14186Huddinge, Sweden
Received October 3, 1994; Revised and Accepted November 8, 1994
The identification of the BTK (Bruton's tyrosine kinase) gene defective In human Immunoglobulln deficiencyX-linked agammaglobulinaemia (XLA) and characterisation of B7X exon intron boundaries has now allowedthe analysis of mutations and polymorphisms at the level of genomic DNA. Using Southern blot analysis andthe polymerase chain reaction single strand conformation polymorphism (PCR-SSCP) assay, amplifying all19 exons and the putative promoter region with a single annealllng temperature, mutations have been identifiedin 19 out of 24 unrelated patients diagnosed as having XLA. Apart from a large deletion involving exon 19,nine missense (F25S, R288W, I370M, M509V, R525P, N526K, R562W, A582V and G594R), two nonsense (E277Xand R525X), five frameshift and two splice site mutations have been found affecting most coding exons andall major enzyme domains. No mutations or polymorphisms were detected in the putative promoter region. Asingle nucleotlde deletion located in the last exon, resulting in a truncation of the eight C-termlnal residuesof Btk and a typical XLA phenotype, indicates structural and/or functional importance of Btk helix I In thecatalytic domain. Although allelic heterogeneity at the BTK locus may partly explain clinical variability Infamilies with XLA, compensatory and redundant mechanisms involved in B-cell development must play a rolein the phenotypic diversity of the disease.
INTRODUCTIONX-linked agammaglobulinaemia (XLA; MIM 300300) is a rareimmunoglobulin deficiency (1) characterised by a B-lineage-specific differentiation defect resulting in a decreased level orabsence of mature B-cells and immunoglobulins. Affectedmales suffer from recurrent bacterial infections starting typic-ally in early childhood and require immunoglobulin replace-ment therapy (2).
The XLA locus was mapped to the region Xq21.3—q22 bygenetic linkage analysis (3,4). In a positional cloning strategyinvolving direct cDNA selection, a gene was isolated whichmapped to the XLA locus, was expressed in B-cells andshowed deletions and point mutations in families with XLA(5). This gene, now designated BTK (Bruton's tyrosine kinase),was also identified in a search for B-cell-specific tyrosinekinases and was found to exhibit deficient mRNA and proteinexpression in XLA pre-B and B-cell lines (6). The BTK geneencodes a cytoplasmic protein-tyrosine kinase with a significantsimilarity to Tec (7,8), ltk/Tsk/Emt (9-11), Bmx (12) and Txk(13). Apart from the N-terminal pleckstrin-homology (PH)
domain (14,15) and Tec-homology (TH) domain (16,17) withyet unknown function(s), Btk contains Src-homology domain3 (SH3), likely to bind to proline-rich stretches, SH2 domaininvolved in phosphotyrosine interactions and the catalytickinase domain (SHI) (5,6).
The mouse BTK cDNA was shown to have a single aminoacid substitution in an immunodeficient (xid) strain CBA/Nand xid phenotype was mapped to the region homologous tothat of XLA (18,19). Further support that the deficient Btkcauses human XLA came from mutation studies analysingcDNA in patients with the disease (20-25) and observing pointmutations, deletions and insertions in the coding region. Theelucidation of the genomic organisation of human and murineBTK loci (26-28) has now made it possible to analyse mutationsand polymorphisms at the genomic level (26,27,29) and herewe report the results of such a DNA-based single strandconformation polymorphism (SSCP) analysis (30) in the largestEuropean study so far comprising 24 unrelated patients withXLA.
*To whom correspondence should be addressed
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
52 Human Molecular Genetics, 1995, Vol. 4, No. 1
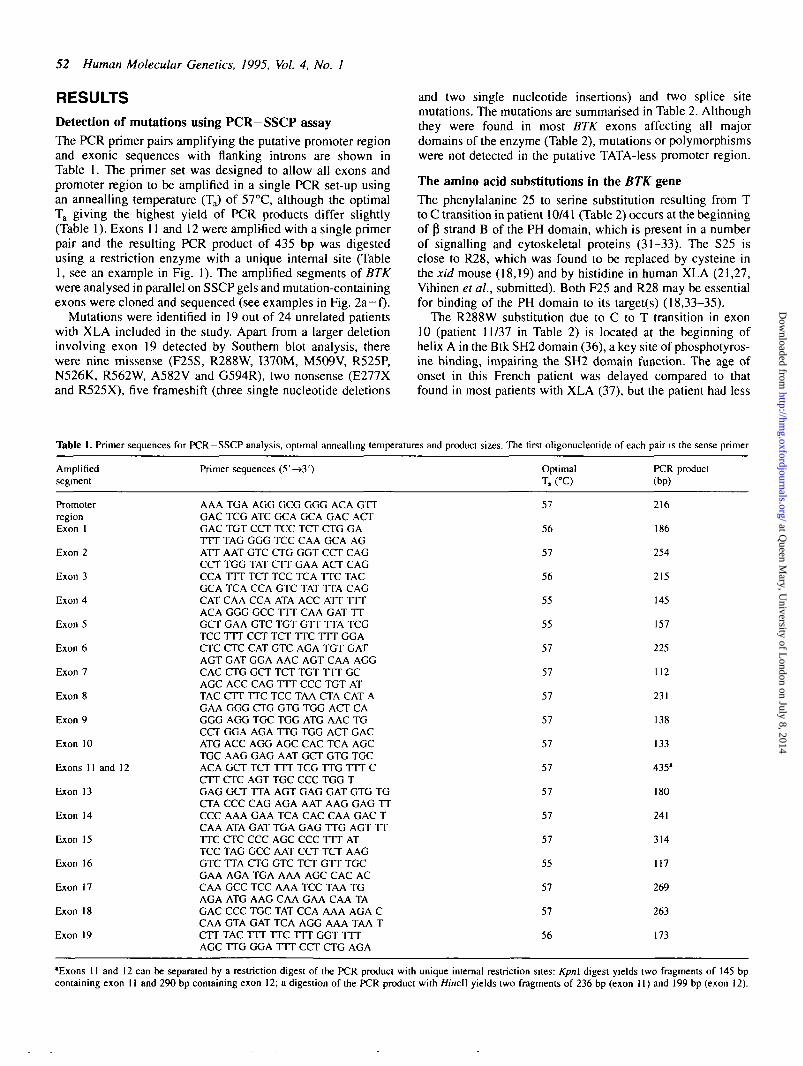
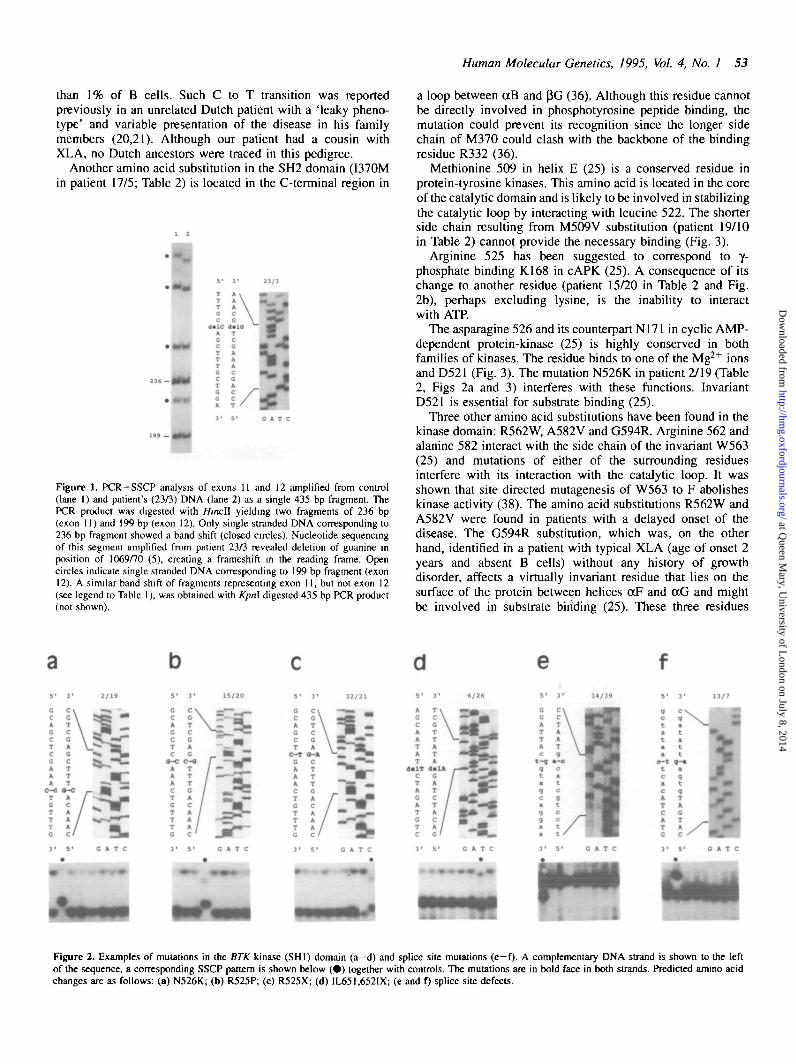
RESULTSDetection of mutations using PCR-SSCP assayThe PCR primer pairs amplifying the putative promoter regionand exonic sequences with flanking introns are shown inTable 1. The primer set was designed to allow all exons andpromoter region to be amplified in a single PCR set-up usingan annealling temperature (TJ of 57°C, although the optimalTa giving the highest yield of PCR products differ slightly(Table 1). Exons 11 and 12 were amplified with a single primerpair and the resulting PCR product of 435 bp was digestedusing a restriction enzyme with a unique internal site (Table1, see an example in Fig. 1). The amplified segments of BTKwere analysed in parallel on SSCP gels and mutation-containingexons were cloned and sequenced (see examples in Fig. 2a -0 -
Mutations were identified in 19 out of 24 unrelated patientswith XLA included in the study. Apart from a larger deletioninvolving exon 19 detected by Southern blot analysis, therewere nine missense (F25S, R288W, I370M, M509V, R525P,N526K, R562W, A582V and G594R), two nonsense (E277Xand R525X), five frameshift (three single nucleotide deletions
and two single nucleotide insertions) and two splice sitemutations. The mutations are summarised in Table 2. Althoughthey were found in most BTK exons affecting all majordomains of the enzyme (Table 2), mutations or polymorphismswere not detected in the putative TATA-less promoter region.
The amino acid substitutions in the BTK geneThe phenylalanine 25 to serine substitution resulting from Tto C transition in patient 10/41 (Table 2) occurs at the beginningof P strand B of the PH domain, which is present in a numberof signalling and cytoskeletal proteins (31-33). The S25 isclose to R28, which was found to be replaced by cysteine inthe xid mouse (18,19) and by histidine in human XLA (21,27,Vihinen et al., submitted). Both F25 and R28 may be essentialfor binding of the PH domain to its target(s) (18,33-35).
The R288W substitution due to C to T transition in exon10 (patient 11/37 in Table 2) is located at the beginning ofhelix A in the Btk SH2 domain (36), a key site of phosphotyros-ine binding, impairing the SH2 domain function. The age ofonset in this French patient was delayed compared to thatfound in most patients with XLA (37), but the patient had less
Table 1. Primer sequences for PCR—SSCP analysis, optimal annealling temperatures and product sizes. The first oligonucleotide of each pair is the sense primer
Amplifiedsegment
Primer sequences (5'—>3') OptimalT, (°C)
57
56
57
56
55
55
57
57
57
57
57
57
57
57
57
55
57
57
56
PCR product(bp)
216
186
254
215
145
157
225
112
231
138
133
4351
180
241
314
117
269
263
173
PromoterregionExon 1
Exon
Exon
Exon
Exon
Exon
Exon
Exon
Exon
Exon
Exon!
Exon
Exon
Exon
Exon
Exon
Exon
Exon
2
3
4
5
6
7
8
9
10
> 11 and 12
13
14
15
16
17
18
19
AAA TGA AGG GCG GGG ACA GTTGAC TCG ATC GCA GCA GAC ACTGAC TGT CCT TCC TCT CTG GATTT TAG GGG TCC CAA GCA AGATT AAT GTC CTG GGT CCT CAGCCT TGG TAT CTT GAA ACT CAGCCA TTT TCT TCC TCA TTC TACGCA TCA CCA GTC TAT TTA CAGCAT CAA CCA ATA ACC ATT TTTACA GGG GCC TTT CAA GAT TTGCT GAA GTC TGT GTT TTA TCGTCC TTT CCT TCT TTC TTT GGACTC CTC CAT GTC AGA TGT GATAGT GAT GGA AAC AGT CAA AGGCAC CTG GCT TCT TGT TTT GCAGC ACC CAG TTT CCC TGT ATTAC CTT TTC TCC TAA CTA CAT AGAA GGG CTG GTG TGG ACT CAGGG AGG TGC TGG ATG AAC TGCCT GGA AGA TTG TGG ACT GACATG ACC AGG AGC CAC TCA AGCTGC AAG GAG AAT GCT GTG TGCACA GCT TCT TTT TCG TTG TTT CCTT CTC AGT TGC CCC TGG TGAG GCT TTA AGT GAG GAT GTG TGCTA CCC CAG AGA AAT AAG GAG TTCCC AAA GAA TCA CAC CAA GAC TCAA ATA GAT TGA GAG TTG AGT TTTTC CTC CCC AGC CCC TTT ATTCC TAG GCC AAT CCT TCT AAGGTC TTA CTG GTC TCT GTT TGCGAA AGA TGA AAA AGC CAC ACCAA GCC TCC AAA TCC TAA TGAGA ATG AAG CAA GAA CAA TAGAC CCC TGC TAT CCA AAA AGA CCAA GTA GAT TCA AGG AAA TAA TCTT TAC TTT TTC TTT GGT TTTAGC TTG GGA TTT CCT CTG AGA
•Exons 11 and 12 can be separated by a restriction digest of the PCR product with unique internal restriction sites: Kpnl digest yields two fragments of 145 bpcontaining exon 11 and 290 bp containing exon 12; a digestion of the PCR product with HincW yields two fragments of 236 bp (exon 11) and 199 bp (exon 12).
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
Human Molecular Genetics, 1995, Vol. 4, No. 1 53
than 1% of B cells. Such C to T transition was reportedpreviously in an unrelated Dutch patient with a 'leaky pheno-type' and variable presentation of the disease in his familymembers (20,21). Although our patient had a cousin withXLA, no Dutch ancestors were traced in this pedigree.
Another amino acid substitution in the SH2 domain (I37OMin patient 17/5; Table 2) is located in the C-terminal region in
I 2
I 5' 3' 23/3
83 ' S 1 0 A T C
Figure 1. PCR —SSCP analysis of exons 11 and 12 amplified from control(lane 1) and patient's (23/3) DNA (lane 2) as a single 435 bp fragment. ThePCR product was digested with Hincll yielding two fragments of 236 bp(exon 11) and 199 bp (exon 12). Only single stranded DNA corresponding to236 bp fragment showed a band shift (closed circles). Nucleotide sequencingof this segment amplified from patient 23/3 revealed deletion of guanine inposition of 1069/70 (5), creating a frameshift in the reading frame. Opencircles indicate single stranded DNA corresponding to 199 bp fragment (exon12). A similar band shift of fragments representing exon 11, but not exon 12(see legend to Table 1), was obtained with Kpn\ digested 435 bp PCR product(not shown).
a loop between aB and pG (36). Although this residue cannotbe directly involved in phosphotyrosine peptide binding, themutation could prevent its recognition since the longer sidechain of M370 could clash with the backbone of the bindingresidue R332 (36).
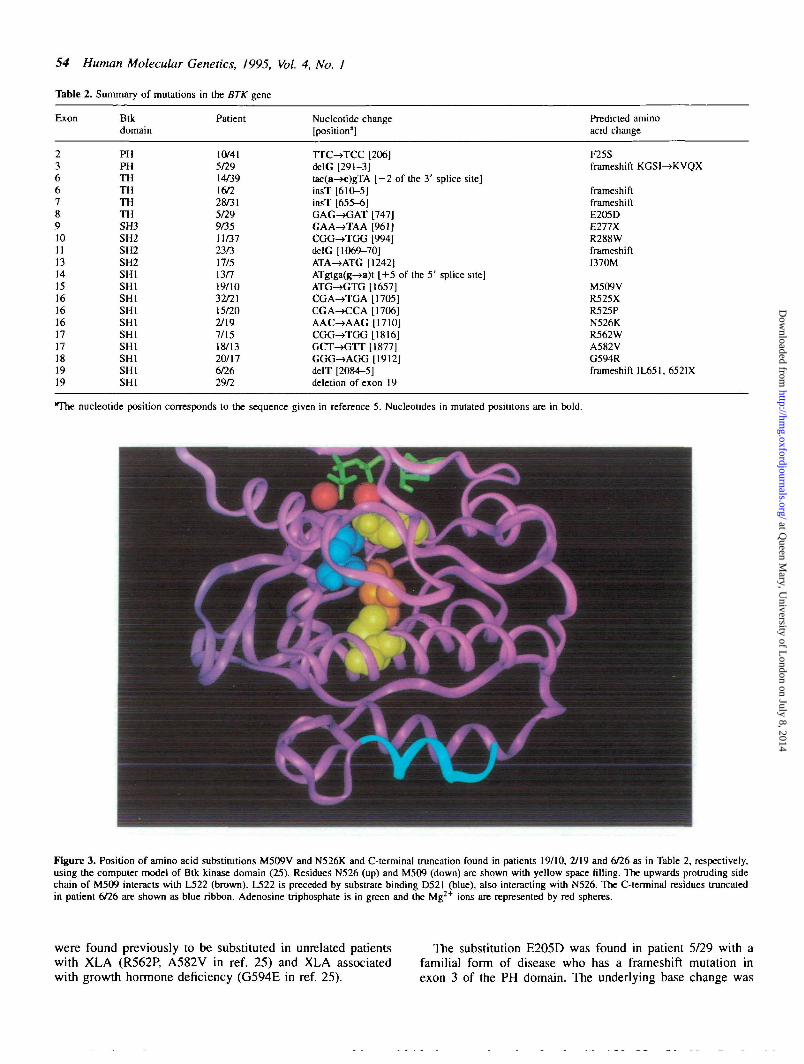
Methionine 509 in helix E (25) is a conserved residue inprotein-tyrosine kinases. This amino acid is located in the coreof the catalytic domain and is likely to be involved in stabilizingthe catalytic loop by interacting with leucine 522. The shorterside chain resulting from M509V substitution (patient 19/10in Table 2) cannot provide the necessary binding (Fig. 3).
Arginine 525 has been suggested to correspond to y-phosphate binding K168 in cAPK (25). A consequence of itschange to another residue (patient 15/20 in Table 2 and Fig.2b), perhaps excluding lysine, is the inability to interactwith ATP.
The asparagine 526 and its counterpart N171 in cyclic AMP-dependent protein-kinase (25) is highly conserved in bothfamilies of kinases. The residue binds to one of the Mg2+ ionsand D521 (Fig. 3). The mutation N526K in patient 2/19 (Table2, Figs 2a and 3) interferes with these functions. InvariantD521 is essential for substrate binding (25).
Three other amino acid substitutions have been found in thekinase domain: R562W, A582V and G594R. Arginine 562 andalanine 582 interact with the side chain of the invariant W563(25) and mutations of either of the surrounding residuesinterfere with its interaction with the catalytic loop. It wasshown that site directed mutagenesis of W563 to F abolisheskinase activity (38). The amino acid substitutions R562W andA582V were found in patients with a delayed onset of thedisease. The G594R substitution, which was, on the otherhand, identified in a patient with typical XLA (age of onset 2years and absent B cells) without any history of growthdisorder, affects a virtually invariant residue that lies on thesurface of the protein between helices aF and aG and mightbe involved in substrate binding (25). These three residues
e f15/20
3 - 5 ' G X T C
Cm*Figure 2. Examples of mutations in the BTK kinase (SHI) domain (a-d) and splice site mutations (e-f). A complementary DNA strand is shown to the leftof the sequence, a corresponding SSCP pattern is shown below ( • ) together with controls. The mutations are in bold face in both strands. Predicted amino acidchanges are as follows: (a) N526K; (b) R525P; (c) R525X; (d) IL651.652IX; (e and D splice site defects.
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
54 Human Molecular Genetics, 1995, Vol. 4, No. 1
Table 2. Summary of mutations in the BTK gene
Exon Btkdomain
Patient Nucleotide change[position0]
Predicted aminoacid change
236678910111314151616161717181919
PHPHTHTHTHTHSH3SH2SH2SH2SHISHISHISHISHISHISHISHISHISHI
10/415/2914/39167228/315/299/3511/3723/317/513/719/1032/2115/202/197/1518/1320/176/2629/2
TTC->TCC [206]delG [291-3]tac(a->c)gTA [ -2 of the 3' splice site]insT [610-5]insT [655-6]GAG->GAT [747]GAA->TAA [961]CGG->TGG [994]delG [1069-70]ATA->ATG [1242]ATgtga(g-»a)t [+5 of the 5' splice site]ATG->GTG [1657]CGA->TGA [1705]CGA-KXA [1706]AAC-»AAG [1710]CGG->TGG [1816]GCT->GTT[1877]GGG->AGG [1912]delT [2084-5]deletion of exon 19
F25Sframeshift KGSI->KVQX
frameshiftframeshiftE2O5DE277XR288WframeshiftI370M
M509VR525XR525PN526KR562WA582VG594Rframeshift IL651, 652IX
"The nucleotide position corresponds to the sequence given in reference 5. Nucleotides in mutated posititons are in bold.
Figure 3. Position of amino acid substitutions M509V and N526K and C-terminal truncation found in patients 19/10, 2/19 and 6/26 as in Table 2, respectively,using the computer model of Btk kinase domain (25). Residues N526 (up) and M509 (down) are shown with yellow space filling. The upwards protruding sidechain of M509 interacts with L522 (brown). L522 is preceded by substrate binding D521 (blue), also interacting with N526. The C-terminal residues truncatedin patient 6/26 are shown as blue ribbon. Adenosine triphosphate is in green and the Mg^+ ions are represented by red spheres.
were found previously to be substituted in unrelated patientswith XLA (R562P, A582V in ref. 25) and XLA associatedwith growth hormone deficiency (G594E in ref. 25).
The substitution E205D was found in patient 5/29 with afamilial form of disease who has a frameshift mutation inexon 3 of the PH domain. The underlying base change was
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Human Molecular Genetics, 1995, Vol. 4, No. 1 55
not detected by screening of 30 unrelated individuals usingour PCR-SSCP analysis (not shown).
Splice site mutations in the BTK geneThe guanine residue at position +5 of 5' splice sites, whichwas changed to adenine in patient 13/7 (Table 2 and Fig. 2f)is the third most conserved nucleotide in this splice junctionconsensus sequence with estimated nucleotide frequency of0.84 (39). The replacement of the G residue in our patientwould be predicted to reduce the stability of base-pairing ofthe 5' splice site with the 5' end of Ul small-nuclear RNA.The spontaneous G to A transitions in this position havebeen described so far in the genes for (J-thalassaemia (40),osteogenesis imperfecta type II (41), haemophilia B (42),acatalasaemia (43), Lesch-Nyhan syndrome (44), Tay-Sachsdisease (45) and protein C and S deficiencies (46,47), mostlyresulting in exon skipping. A recent case report showed a Gto A transition in position +5 of 5' splice site of BTK intron17 in a patient with XLA associated with growth hormonedeficiency (48). Our patient had typical XLA (total absenceof B cells, IgA 0.08 g/1, IgG 0.22 g/1 and IgM 0.25 g/1 atdiagnosis) with no history of growth hormone deficiency.
The second splice site mutation found at position - 2 of the3' splice site of intron 5 changes the invariant adenine tocytosine in patient 14/39 (Table 2 and Fig. 2e). One suchtransversion was found to result in skipping of exon 12 inthe mitochondrial acetoacetyl-coenzyme A thiolase (49). Thephenotypic consequences at the nucleotide level have not beenstudied in the second of two such transversions reported sofar (50).
Truncation of the eight C-terminal residues of Btk resultsin XLAThe single nucleotide deletion in exon 19 found in patient 6/26truncates the last eight residues (652-659) and removes thehelix I of the Btk C-terminus (Table 2, Figs 2d and 3).The mutation could loosen interactions in the C-terminus,potentially leading to protease susceptibility or a decrease ininteractions with the other domains of Btk. The importance ofleucine 562 in the middle of the helix I is supported bythe recently reported helix I-breaking substitution L562Passociated with a typical disease (29).
Southern blot analysis of the patients with XLASouthern blot analysis revealed an abnormal 2.3 kb Taq\fragment in two patients (15/20 and 32/21), resulting from theloss of Taql restriction site at position 174)4 of BTK cDNA (5)due to alterations of codon 525. In addition, a single deletionwas identified as a missing 5 kb //mdlll fragment (not shown)in a Czech patient with a typical XLA phenotype and foundto involve exon 19. Mutations in all three patients have alsobeen detected using PCR—SSCP assay as missing or aberrantPCR products (not shown).
In a patient with a mild phenotype diagnosed as commonvariable immune deficiency (CVID) and reported previously(51) as having a deletion at the 3' end of the BTK gene, wehave determined that this deletion comprises exons 17, 18 and19 (not shown). This finding is consistent with the length of thetruncated transcript being approximately 1.9 kb, as identified byNorthern blot analysis (51).
Polymorphisms in the BTK geneAlthough the oligonucleotides were designed to prime closeto exon—intron boundaries in order to minimize detectingintronic polymorphisms, one two-allele polymorphism (T toC) was found at position - 2 9 of the 3' splice site of intron14. The cytosine-containing alleles that were present in similarfrequency in both unaffected controls and XLA patients showfaster mobility on SSCP gels. Using SSCP analysis withprimers flanking exon 15 (Table 1), screening of 63 Xchromosomes in unrelated Scandinavian Caucasians gave anestimate of maximum likelihood heterozygosity of 0.20 in thislocus, the frequency of cytosine-containing alleles being 0.11.Our PCR-SSCP assay using primers flanking exon 18 alsodetected the previously reported polymorphism at position2031 (20,21) of BJTKCDNA (5), but the mutation in our patient20/17 (Table 2) was revealed by a distinct pattern on SSCPgels (not shown). Another polymorphic site located in the 3'untranslated region at position 2228 of BTK cDNA (52) liesdownstream of the most 3' amplified segment and is notdetected by the primer pairs shown in Table 1.
DISCUSSIONAlthough not all patients with XLA phenotype have beenfound to have a mutation in the BTK gene, a mutation wasidentified and found likely to interfere with enzyme functionin 19 out of 24 XLA cases analysed (80%). The proportion ofpatients in whom mutations were not found is comparable toother mutation, studies (20,26,29). A limited sensitivity ofthe PCR—SSCP assay, clinical variability of this immunedeficiency, in particular an overlapping phenotype with CVID(27,51), and an existence of intronic/regulatory mutations mayprovide an explanation.
Although we used SSCP conditions that were found to showa high sensitivity when screening a number of differentmutations in the cystic fibrosis gene and recommended for aninitial mutation analysis of any gene (53), it is possible thatdifferent conditions would be more sensitive in detecting abase change in a particular segment of the gene (54).
Clinical variability has been reported to exist both betweenand within the families with XLA (2,20,21,27,55-59). Aclassical XLA phenotype was described, together with a milderform of immunodeficiency in sibs (55,57), nephews (56,57)or more distant relatives (58,59). In a screening of a set ofmale patients referred with diagnosis of CVID using Southernblot analysis with BTK cDNA as a probe, one patient wasfound with a BTK deletion (51). This case was the firstmolecular evidence that a defect in the BTK gene can beassociated with a milder immunodeficiency and distinct clinicaldiagnosis. Since then, more patients referred with a diagnosisof CVID have been analysed and mutations in the BTK genereported (27 and H.D.Ochs, personal communication).
The phenotypes of most patients included in our study weretypical of XLA with less than \% of B cells; only one patienthad 2% CD 19+ cells (14/39 in Table 2). It is possible that inthis patient the underlying mutation in invariant adenine atposition - 2 of the 3' splice site of intron 5 leads to bothaberrant and normal mRNA species compensating the defect,but we could not study its phenotypic impact on mRNAbecause patients' RNA samples were not available to us.
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

56 Human Molecular Genetics, 1995, Vol. 4, No. 1
Although a locus heterogeneity could not yet be excluded,cDNA clones isolated from the region surrounding the BTKgene in Xq21.3-q22 have not been found to give an alteredpattern in Southern blot analyses in a limited set of patientswith isolated XLA (60). Genetic heterogeneity may also existin CVID with thus far unidentified autosomal defect(s) andcontribute to the clinical variability and overlap with XLAphenotype. This hypothesis would be consistent with occasionalfindings of females with a disorder phenotypically identical totypical XLA (61,62). However, three patients with familyhistory of XLA out of five patients in whom mutation was notfound in this study might point to a limited sensitivity ofmutation assay or existence of regulatory regions at a sitenot screened by PCR-SSCP analysis rather than geneticheterogeneity.
In summary, a rapid and convenient PCR-SSCP methodwas introduced for molecular diagnosis, mutation screeningand carrier status determination in the families with XLA andcandidate phenotypes, allowing the identification of mutationsin the majority of XLA patients within a day if their RNA/cDNA samples are not available. Mutation patterns of the BTKgene in a set of 19 unrelated patients were characterised andstructural implications of Btk defects due to predicted aminoacid substitutions were analysed based on previous computer-aided modelling of Btk domains.
MATERIAL AND METHODSPatientsTwenty-four unrelated French and Czech patients were included in the study.They were all males, aged between 2 and 23 years and diagnosed as havingXLA. Diagnosis was based on the absence or very low levels of circulatingB cells, low levels of immunoglobulins and susceptibility to infections earlyin childhood. The age of onset of infections varied between 1 and 16 years.Eleven patients had a history of a male family member affected with XLA,but the putative earners have not been available for analysis.
DNA preparation
DNA was prepared from peripheral blood cells using methods describedpreviously (63).
PCR amplification
The segments of the BTK gene were amplified from genomic DNA using aset of primer pairs flanking all exons and the putative promoter region basedon the sequence as in (28) The exons were numbered as in ref. 26 and 27.The primer sequences, optimal annealling temperatures and sizes of the PCRproducts are shown in Table 1. The oligonucleotides were designed using thecomputer programme Oligo, version 4.05 (National Biosciences, Inc.) to allowamplification of all BTK segments from a patient using an anneallingtemperature of 57°C in a single PCR. The PCR reaction volume was 20 \i\and contained 100 ng DNA, 0.25 nJM of each primer, 100 nM of each dNTP,1.5 mM MgCl2, 10 mM Tris-HCl (pH 8.3), 50 mM KC1, 0.01% gelatin, 0.2HCi [a-"P]dCTP (3,000 Ci/mmol) and 0.5 U Taq polymerase (Perkin-Elmer).The initial denaturation was for 4 min at 94°C. Amplification was for 32cycles at 94°C for 30 s, annealing at 57°C for 45 s and extension at 72°C for30 s in a Perkin-Elmer thermocycler (system 9600).
SSCP analysis
The samples were mixed with 95% formamide, 0.05% bromophenol blue,0.05% xylene cyanol, 50 mM NaOH, denatured at 95°C for 10 min andloaded onto 0.4 mm/30 cm/45 cm 5% polyacrylamide—10% glycerol gels(crosslinking 2.5%). The electrophoresis was conducted at 5 W at roomtemperature overnight. The gels were dried and autoradiographed for 1-2days at -70°C.
Cloning and sequencing of mutation-containing exons
BTK segments identified to contain polymorphisms/mutations were re-ampli-fied using the same conditions except for omission of labelled dCTP. The
PCR products were separated using 2% agarose gels in 1 XTBE. DNA waspurified using GeneClean (BIO 101) and subcloned into pCR-Script (SK+)vectors according to manufacturer's recommendations (Stratagene). At leasttwo insert-containing clones per patient were sequenced (64) in both directionsusing a Sequenase 2.0 sequencing kit (USB).
Southern blot analysis
DNA was digested with Taq\ and W/ndlll restriction enzymes, fragments wereseparated by eleclrophoresis in a 0.9% agarose gel and blotted onto HybondN membranes (Amersham). 32P-labelled probes were prepared using randomhexamer priming of BTKcDNA clone 14-6(5). Prehybridisation, hybridisation,washing and autoradiography were as described previously (60).
ACKNOWLEDGEMENTSThis work was supported by the Swedish Medical Research Council, theSwedish Cancer Society, the Axel and Margret Ax:son Johnson Foundation,the Swedish Natural Science Research Council, the Karolinska Institute, theAFM, INSERM and the European BIOMED concerted action 'PL 1321'.
ABBREVIATIONSBTK/Btk, Bruton's tyrosine kinase gene/protein; CVID, common variableimmune deficiency; PH, pleckstnn homology domain; SH1-3, Src-homologydomains 1-3; SSCP, single strand conformation polymorphism; TH, Tec-homology domain; XLA, X-hnked agammaglobulinaemia.
REFERENCES1. Bruton, O. C. (1952) Agammaglobulinemia. Pediatrics, 9, 722-727.2. Rosen, F. S., Cooper, M. and Wedgwood, R. J. P. (1984) The primary
immunodeficiencies. N. Engl. J. Med., 311, 235-242.3. Kwan, S.-P, Kunkel, L., Bruns, G., Wedgwood, R. J., Latt, S. and Rosen,
F. S. (1986) Mapping of the X-linked agammaglobulinemia locus by useof restriction fragment length polymorphism. J. Clin. Invest., 77, 649-652
4. Guioli, S., Arveiler, B., Bardoni, B., Notarangelo, L. D., Panina, P., Duse,M., Ugazio, A., Oberle, I., de Saint Basile, G., Mandel, J L. andCamerino, G. (1989) Close linkage of probe p212 (DXS178) to X-linkedagammaglobulinaemia. Hum. Genet., 84, 19-21.
5. Vetrie, D., Vofechovsky, I., Sideras, P., Holland, J., Davies, A., Flinter,F., Hammarstrom, L., Kinnon, C , Levinsky, R., Bobrow, M., Smith, C.I. E. and Bentley, D. (1993) The gene involved in X-linkedagammaglobulinaemia is a member of the ire family of protein-tyrosinekinases. Nature, 361, 226-233.
6. Tsukada, S., Saffran, D. C, Rawlings, D. J., Parolini, O., Allen, R. C ,Klisak, I., Sparkes, R. S., Kubagawa, H., Mohandas, T, Quan, S.,Belmont, J. W., Cooper, M. D., Conley, M. E. and Wine, O. N. (1993)Deficient expression of a B cell cytoplasmic tyrosine kinase in humanX-linked agammaglobulinemia. Cell, 72, 279-290.
7. Mano, H., Ishikawa, F., Nishida, J., Hirai, H., and Takaku, F. (1990). Anovel protein-tyrosine kinase, Tec, is preferentially expressed in liver.Oncogene, S, 1781-1786.
8. Mano, H., Mano, K., Tang, B., Koehler, M., Yi, T, Gilbert, DJ., Jenkins,N. A., Copeland, N. G., and Ihle, J. N. (1993) Expression of a novelform of Tec kinase in hematopoietic cells and mapping of the gene tochromosome 5 near Kit. Oncogene, 8, 417—421.
9. Sihciano, J. D., Morrow, T. A., and Desideno, S. V. (1992). itk, a T-cell-specific tyrosine kinase gene inducible by interleukin 2. Proc. Natl Acad,Sci. USA, 89, 11194-11198.
10. Heyeck, S. D. and Berg, L. J. (1993) Developmental regulation of amurine T-cell-specinc tyrosine kinase gene, Tsk. Proc. Natl Acad. Sci.USA 90, 669-673.
11. Yamada, N., Kawakami, Y., Kimura, H., Fukamachi, H., Baier, G., Altman,A., Kato, T., Inagaki, Y. and Kawakami, T. (1993) Structure and expressionof novel protein-tyrosine kinases, Emb and Emt, in hematopoietic cells.Biochem. Biophys. Res. Commun., 192, 231-237.
12. Tamagnone, L., Lahlinen, I., Mustonen, T., Virtaneva, K., Muscatelli, F.,Francis, F., Alitalo, R., Smith, C. I. E., Larsson, C. and Alitali, K. (1994)BMX, a novel cytoplasmic tyrosine kinase gene of the BTK/TTK/TECfamily located in chromosome Xp22.2. Oncogene, in press
13. Haire, R. N., Ohta, Y, Lewis, J. E., Fu, S. M., Kroisel, P. and Litman,G. W (1994) TXK, a novel human tyrosine kinase expressed in T cells
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Human Molecular Genetics, 1995, Vol. 4, No. 1 57
shares sequence identity with Tec family kinases and maps to 4pl2. Hum.Mol. Genet., 3, 897-901.
14. Musacchio, A., Gibson, T, Rice, P., Thompson, J. and Saraste M. (1993)The PH domain: a common piece in the structural patchwork of signallingproteins. Trends Biochem. Sci., 18, 343—348.
15. Shaw, G. (1993) Identification of novel pleckstrin homology(PH) domainsprovides a hypothesis for PH domain function. Biochem. Biophys. Res.Commun., 195, 1145-1151.
16. Smith, C. I. E., Islam, K. B., Vofechovsky, I., Olerup, O., Wallin,E., Rabbani, H., Baskin, B. and Hammarstrom, L. (1994) X-linkedagammaglobulinemia and other immunoglobulin deficiencies. Immunol.Rev., 138, 159-183.
17. Vihinen, M., Nilsson, L. and Smith C. I. E. (1994) Tec homology (TH)adjacent to the PH domain. FEBS Lett., 350, 263-265.
18. Thomas, J. D., Sideras, P., Smith, C. I. E., Vofechovsky, I., Chapman, V.and Paul, W. E. (1993) Colocalization of X-linked agammaglobulinemiaand X-linked immunodeficiency genes. Science, 261, 355-358.
19. Rawlings, D. J., Saffran, D. C, Tsukada, S., Largaespada, D. A., Grimaldi,J. C , Cohen, L., Mohr, R. N., Bazan, J. F., Howard, M., Copeland, N.G., Jenkins, N. A. and Witte, O. N. (1993) Mutation of unique region ofBruton's tyrosine kinase in immunodeficient XID mice. Science, 261,358-360
20. Bradley, L. A. D., Sweatman, A. K., Lovering, R. C . Jones, A. M.,Morgan, G., Levinsky, R. J., and Kinnon, C. (1994) Mutation detectionin the X-linked agammaglobulinemia gene, BTK, using single strandconformation polymorphism analysis. Hum. Mol. Genet. 3, 79-83.
21. de Weers, M., Mensink, R G. J., Kraakman, M. E. M., Schuurman, R.K. B., and Hendriks, R. W. (1994) Mutation analysis of the Bruton'styrosine kinase gene in X-linked agammaglobulinemia: identification ofa mutation which affects the same codon as is altered in immunodeficientxid mice. Hum. Mol. Genet. 3, 161-166.
22. Zhu, Q., Zhang, M., Rawlings, D. J., Vihinen, M., Hagemann, T, Saffran,D. C, Kwan, S.-P, Nilsson, L., Smith, C. I. E., Witte, O. N., Chen, S.-H. and Ochs, H. D. (1994) Deletion within the Src homology domain 3of Bruton's tyrosine kinase resulting in X-linked agammaglobulinemia(XLA). J. Exp. Med 180, 461^70.
23. Zhu, Q., Zhang, M., Winkelstein, J., Chen, S.-H. and Ochs, H. D. (1994)Unique mutations of Bruton's tyrosine kinase in fourteen unrelated X-1 inked agammaglobulinemia families. Hum. Mol. Genet. 3, 1899—1900.
24. Saffran, D. C, Parolini, O., Hilgenberg-Fitch, M. E., Rawlings, D. J.,Afar, D. E. H., Witte, O.N. and Conley, M. E. (1994) Brief report: apoint mutation in the SH2 domain of Bruton's tyrosine kinase in atypicalX-linked agammaglobulinemia. N. Engl. J. Med., 330, 1488-1491.
25. Vihinen, M., Vetrie, D., Maniar, H., Ochs, H., Zhu, Q., Vofechovsky, I.,Webster, A. D. B., Notarangelo, L., Nilsson, L., Sowadski, J. and Smith,C. I. E. (1994) Structural basis for X-linked agammaglobulinemia. Atyrosine kinase disease. Proc. Nat i Acad Sci. USA, 91, 12803-12807.
26. Hagemann, T. L., Chen, Y., Rosen, F. S. and Kwan, S.-P. (1994) Genomicorganization of the Btk gene and exon scanning for mutations in patientswith X-linked agammaglobulinemia. Hum. Mol. Genet., 3, 1743-1749.
27. Ohta, Y., Haire, R. N., Litman, R. T, Fu, S. M., Nelson, R. P., Kratz, J.,Kornfeld, S. J., De La Morena, M., Good, R. A. and Litman, G. W.(1994) Genomic organization and structure of Brutonagammaglobulinemia tyrosine kinase: localization of mutations associatedwith varied clinical presentations and course in X-chromosome linkedagammaglobulinemia. Proc. Nail Acad. Sci. USA, 91, 9062-9066.
28. Sideras, P., Muller, S., Shiels, H., Jin, H., Khan, W., Nilsson, L., Parkinson,E., Thomas, J. D., Branden, L., Larsson, I., Paul, W. E., Rosen, F. S.,Alt, F. W.t Vetrie, D., Smith, C. I. E. and Xanthopoulos, K. G. (1994)Genomic organization of mouse and human Bruton'sagammaglobulinemia tyrosine kinase (Btk) loci. J. Immunol., in press.
29. Conley, M. E., Fitch-Hilgenberg, M. E., Cleveland, J. L., Parolini, O. andRohrer, J. (1994) Screening of genomic DNA to identify mutations inthe gene for Bruton's tyrosine kinase. Hum. Mol. Genet., 3, 1751-1756.
30. Orita, M., Suzuki, Y, Sekiya, T. and Hayashi, K. (1989) Rapid andsensitive detection of point mutations and DNA polymorphisms usingthe polymerase chain reaction. Genomics, 5, 874—879.
31. Mayer, B. J., Ren, R., Clark, K. L. and Baltimore, D. (1993) A putativemodular domain present in diverse signaling proteins. Cell, 73, 629-630.
32. Haslam, R. J., Kolde, H. B. and Hemmings, B. A. (1993) Pleckstrinhomology domain. Nature, 363, 309-310.
33. Gibson, T. J., Hyvonen, M., Musacchio, A. and Saraste, M. (1994) PHdomain: the first anniversary. Trends Biochem. Sci., 19, 349-353.
34. Yao, L., Kawakami, Y. and Kawakami, T. (1994) The pleckstrin homologydomain of Bruton tyrosine kinase interacts with protein kinase C. Proc.
Nail Acad. Sci. USA, 91, 9175-9179.35. Harlan, J. E., Hajduk, P. J., Yoon, H. S. and Fesik, S. W. (1994) Pleckstrin
homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature,371, 168-170.
36. Vihinen, M., Nilsson, L. and Smith, C. I. E. (1994) Structural basis forSH2 domain mutations in X-linked agammaglobulinaemia. Biochem.Biophys. Res. Commun., in press.
37. Lederman, H. M. and Winkelstein, J. A. (1985) X-linkedagammaglobulinemia: an analysis of % patients. Medicine, 64, 145-156.
38. Maniar, S. H., Vihinen, M., Webster, A. D. B., Nilsson, L. and Smith, C.I. E (1994) Structural basis for X-linked agammaglobulinemia (XLA).Mutations at interacting residues R562, W563 and A582. Clin. Immunol,lmmunopathol., in press.
39. Shapiro, M. B. and Senapathy, P. (1987) RNA splice junctions of differentclasses of eukaryotes: sequence statistics and functional implications ingene expression. Nucleic Acids Res., 15, 7155-7174.
40. Lapoumeroulie, C , Pagnier, J., Bank, A., Labie, D. and Krishnamoorthy,R. (1986) P thalassemia due to a novel mutation in IVS 1 sequence donorsite consensus sequence creating a restriction site. Biochem. Biophys.Res. Commun., 139, 709-713.
41. Bonadio, J., Ramirez, F. and Barr, M. (1990) An mtron mutation in thehuman a 1(1) collagen gene alters the efficiency of pre-mRNA splicingand is associated with osteogenesis imperfecta type II. J. Biol. Chem.,265, 2262-2268.
42. Green, P. M., Montandon, A. J., Ljung, R., Bentley, D. R., Nilsson, I.-M., KJing, S. and Giannelli, F. (1991) Haemophilia B mutations in acomplete Swedish population sample, a test of new strategy for thegenetic counselling of diseases with high mutational heterogeneity. Br. J.Haematoi, 78, 390-397.
43. Wen, J. K., Osumi, T., Hashimoto, T. and Ogata, M. (1990) Molecularanalysis of human acatalasemia. Identification of a splicing mutation. /.Mol. Biol., 211, 383-393.
44. Gibbs, R. A., Hguyen, P. N., Edwards, A., Civitello, A. B. and Caskey,C. T. (1990) Multiplex DNA deletion detection and exon sequencingof the hypoxanthine phosphonbosyltransferase gene in Lesch—Nyhanfamilies. Genomics, 7, 235-244.
45. Akli, S., Chelly, J., Lacorte, J.-M., Poenaru, L. and Kahn, A. (1991)Seven novel Tay-Sachs mutations detected by chemical mismatchcleavage of PCR-amplified cDNA fragments. Genomics, 11, 124—134.
46. Reitsma, P. H , Poort, S. R., Allan, C. F., Briet, E. and Bertina R. M.(1991) The spectrum of genetic defects in a panel of 40 Dutch familieswith symptomatic protein C deficiency type I: heterogeneity and foundereffects. Blood, 78, 890-894.
47. Cooper, D. N. and Krawczak, M. (1993) Human Gene Mutation. BIOSScientific Publications Ltd., London.
48. Duriez, B., Duquesnoy, P., Dastot, F., Bougneres, P., Amselem, S. andGoossens, M. (1994) An exon-skipping mutation in the btk gene of apatient with X-linked agammaglobulinemia and isolated growth hormonedeficiency. FEBS Lett., 346, 165-170.
49. Fukao, T, Yamaguchi, S., Orii, T, Schutgens, R. B., Osumi, T. andHashimoto, T. (1992) Identification of three mutant alleles of the genefor mitochondria] acetoacetyl-coenzyme A thiolase. J. Clin. Invest., 89,474^79.
50. Padanilam, B. J. and Huisman, T. H. (1986) The f3°-thalassemia in anAmerican black family is due to a single nucleotide substitution in theacceptor splice junction of the second intervening sequence. Am. J.Hematol, 22, 259-263.
51. Vofechovsky, I., Zhou, J.-N., Vetrie, D., Bentley, D. R,, Bjorkander, J.,HammarstrOm, L. and Smith, C. I. E. (1993) Molecular diagnosis of X-linked agammaglobulinaemia Lancet, 341, 1153.
52. Vofechovsky, I. (1994) SSCP at the BTK locus. Hum Mol. Genet., 3, 1031.53. Ravnik-Glavaf, M., Glavai, D. and Dean, M. (1994) Sensitivity of single-
strand conformation polymorphism and heteroduplex method for mutationdetection in the cystic fibrosis gene. Hum. Mol. Genet., 3, 801-807.
54. Glavad, D. and Dean, M. (1993) Optimization of the single-strandconformation polymorphism (SSCP) technique for detection of pointmutation. Hum. Mutat., 2, 404-414.
55. Buckley, R. and Sidbury, J. B. Jr. (1968) Hereditary alterations inthe immune response: coexistence of 'agammaglobulinemia', acquiredhypogammaglobulinemia and selective immunoglobulin deficiency in asibship. Pediat. Res., 2, 72-84.
56. Wedgwood, R. J. and Ochs, H. D. (1980) Variability in the expression ofX-linked agammaglobulinemia: the co-existence of classic XLA (Brutontype) and 'common variable immunodeficiency' in the same families. InSeligmann, M. and Hitzig, W. H. (eds), Primary immunodeficiencies,
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

5* Human Molecular Genetics, 1995, Vol. 4, No. 1
INSERM Symposium No 16. Amsterdam. Elsevier, pp. 69-78.57. Goldblum, R. M, Lord, R. A., Cooper, M. D., Gathings, W. E. and
Goldman, A. S. (1974) X-linkcd B lymphocyte deficiency. J. Pediat., 85,188-191.
58. Leickley, F. E. and Buckley, R. (1986) Variability in B cell maturationand differentiation in X-linked agammaglobulinemia. Clin. Exp. Immunol.,65, 90-99.
59. Vofechovsky, I., Litzman, J., Lokaj, J. and Sobotkovd, R. (1991) Familystudies in common variable immunodeficiency. J. Hyg. Epid. Microb.Immunol, 35, 17-26.
60. Vofechovsky, I., Vetrie, D., Holland, J., Bentley, D. R., Thomas, K., Zhou,J.-N., Notarangelo, L., Plebani, A., Fontan, G., Ochs, H., Hammarstrom,L., Sideras, P., and Smith, C. I. E. (1994) Isolation of cosmid and cDNAclones in the region surrounding the BTK gene at Xq21.3-q22. Genomics,21,517-524.
61. Hoffman, T, Winchester, R., Schulkind, M., Frias, J. L., Ayoub, E. M.and Good, R. (1977) Hypogammaglobulinemia with normal T cellfunction in female siblings. Clin. Immunol. Immunopathoi, 7, 364—371.
62. Conley, M. E. and Sweinberg, S. K. (1992) Females with a disoderphenotypically identical to X-linked agammaglobulinemia. J. Clin.Immunol, 12, 139-143.
63. Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989) Molecular Cloning:A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press,Cold Spring Harbor, NY.
64. Sanger, F, Nicklen, S. and Coulson, A. R. (1977) DNA sequencing withchain-terminating inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463-5467.
at Queen M
ary, University of L
ondon on July 8, 2014http://hm
g.oxfordjournals.org/D
ownloaded from





























