Embed Size (px)
Citation preview
Direct Synthesis of Controlled Poly(styrene-co-acrylic
acid)s of Various Compositions by Nitroxide-Mediated
Random Copolymerization
Laurence Couvreur,1 Bernadette Charleux,*1 Olivier Guerret,2 Stephanie Magnet2
1 Laboratoire de Chimie des Polymeres, UMR 7610, associee au CNRS, Universite Pierre et Marie Curie, Tour 44, 1er etage,4, place Jussieu – 75252 PARIS Cedex 05, FranceFax: 0033 1 44 27 70 89; E-mail: [email protected]
2 ATOFINA, Groupement de Recherches de Lacq, B.P. n8 34, 64170 LACQ, France
Received: May 12, 2003; Revised: September 3, 2003; Accepted: September 5, 2003; DOI: 10.1002/macp.200350065
Keywords: copolymerization; living polymerization; nitroxide; radical polymerization
Introduction
With the advent of controlled radical polymerization
(CRP),[1–3] the field of free-radical polymerization has con-
siderably extended towards the synthesis of well-defined
homopolymers and copolymers with various architec-
tures.[4–6] Because the free-radical polymerization process
is particularly attractive from the industrial viewpoint, the
expectation is that the CRP technique might replace, to
some extent, the ionic polymerizations that are the current
methods of choice to elaborate precise structures, but which
require drastic synthetic conditions.[7] More interestingly,
some monomers can only polymerize via a free-radical
process. This is particularly the case with hydrophilic and
ionic monomers, which find significant applications in the
synthesis of water-soluble and amphiphilic (co)polymers.
Well-defined macromolecular structures derived from such
monomers cannot be obtained by a direct ionic polymer-
ization but by various indirect synthetic strategies, inclu-
ding protection of the polar functionality or chemical
modification of a hydrophobic (co)polymer precursor.
These procedures are rather expensive and cannot always
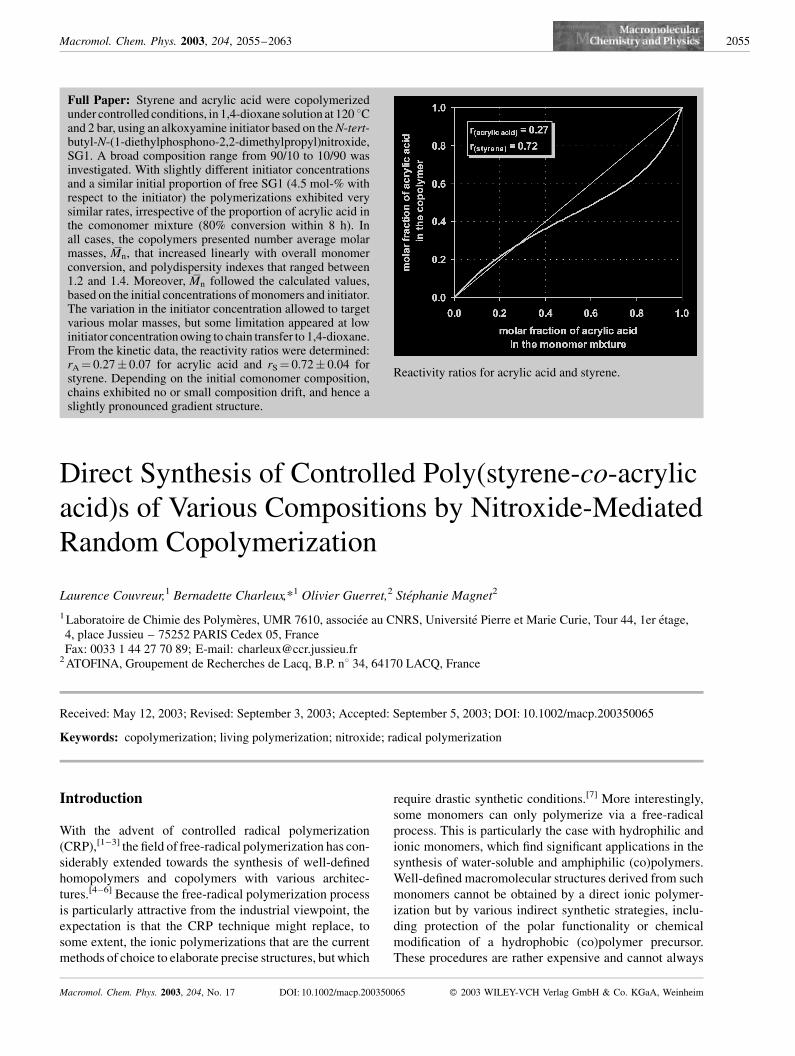
Full Paper: Styrene and acrylic acid were copolymerizedunder controlled conditions, in 1,4-dioxane solution at 120 8Cand 2 bar, using an alkoxyamine initiator based on the N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl)nitroxide,SG1. A broad composition range from 90/10 to 10/90 wasinvestigated. With slightly different initiator concentrationsand a similar initial proportion of free SG1 (4.5 mol-% withrespect to the initiator) the polymerizations exhibited verysimilar rates, irrespective of the proportion of acrylic acid inthe comonomer mixture (80% conversion within 8 h). Inall cases, the copolymers presented number average molarmasses, Mn, that increased linearly with overall monomerconversion, and polydispersity indexes that ranged between1.2 and 1.4. Moreover, Mn followed the calculated values,based on the initial concentrations of monomers and initiator.The variation in the initiator concentration allowed to targetvarious molar masses, but some limitation appeared at lowinitiator concentration owing to chain transfer to 1,4-dioxane.From the kinetic data, the reactivity ratios were determined:rA¼ 0.27� 0.07 for acrylic acid and rS¼ 0.72� 0.04 forstyrene. Depending on the initial comonomer composition,chains exhibited no or small composition drift, and hence aslightly pronounced gradient structure.
Reactivity ratios for acrylic acid and styrene.
Macromol. Chem. Phys. 2003, 204, 2055–2063 2055
Macromol. Chem. Phys. 2003, 204, No. 17 DOI: 10.1002/macp.200350065 � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

be applied. Thus, when possible, the best strategy remains
the direct (co)polymerization of the selected monomer(s).
This is now theoretically achievable with CRP, because
free-radicals are tolerant to a great variety of functionalities,
including ionic and polar ones.[8,9]
An example of monomer that is particularly important
for industry is acrylic acid (A), for which only free-radical
polymerization is appropriate. The incorporation of poly-
(acrylic acid) sequences in a controlled copolymer (i.e.
predictable molar mass, narrow molar mass distribution and
precise architecture) was initially done by polymerizing
tert-butyl acrylate anionically, followed by hydrolysis of
the ester groups.[10] Although this might appear unneces-
sary, such an approach was also applied with CRP.[11–15]
Indeed, early works on CRP of acrylic acid showed that this
monomer was actually rather reluctant in both ATRP (atom
transfer radical polymerization) and NMP (nitroxide-
mediated polymerization).[8] With ATRP, some incompat-
ibility with the transition metal complex catalyst was
suspected;[16] with NMP, the acidic group was supposed to
be involved in side reactions with the nitroxide,[17] although
no mechanism has ever been suggested. Even though
unwanted chemical reactions were usually invoked, it is
worth noting that acrylic acid also exhibits a very high rate
constant of propagation in free-radical polymerization[18]
together with a propensity for self-initiation,[19] both cap-
able of ruining the quality of control.
Nevertheless, successful CRP of this monomer was
reported in a small number of papers[8] with the synthesis of
well defined homopolymers and/or various types of
copolymer architectures.
So far, RAFT (reversible addition-fragmentation trans-
fer)[20] was the only CRP technique able to yield controlled
homopolymers of acrylic acid as well as block copolymers
with poly(acrylic acid) sequences. The first example of
homopolymerization, performed in dimethylformamide
solution at 60 8C, was reported by Chiefari et al.,[20] but
no detailed information was provided. They also briefly
reported the synthesis of poly(butyl acrylate)-block-poly-
(acrylic acid) block copolymers by the same technique.[21]
Ladaviere et al.[22] examined a series of RAFT agents to
control the free-radical polymerization of acrylic acid in
alcohol or water solution, and they came to the conclusion
that phenoxyxanthates and trithiocarbonates were particu-
larly well-suited. Recently, the same group completed this
work and showed, as an application, that low polydispersity
poly(acrylic acid)s were very efficient dispersants of inor-
ganic particles.[23] They also described the application of
RAFT to the synthesis of controlled poly(butyl acrylate)-
block-poly(acrylic acid) block copolymers, and their use as
stabilizers in emulsion polymerization.[24] The controlled
homopolymerization of acrylic acid was also performed at
room temperature in the presence of dibenzyltrithiocar-
bonate, under 60Co irradiation.[25] The MADIX process
(macromolecular design via the interchange of xanthates)
was successfully used in aqueous solution to prepare a well-
defined poly(acrylic acid) homopolymer and hydrophilic
copolymers based on acrylamide and acrylic acid.[26] A few
other examples of copolymers bearing a poly(acrylic acid)
sequence and synthesized using the RAFT technique can be
found in the literature.[27–30]
Graft copolymers with poly(sodium acrylate) branches
were prepared via cyanoxyl-mediated free-radical poly-
merization of sodium acrylate starting from polystyrene
precursors with pendant arene-diazonium salts.[31]
Random copolymers of acrylic acid and butyl acrylate
were prepared in bulk via nitroxide-mediated polymeriza-
tion using the 2,2,5-trimethyl-4-phenyl-3-azahexane-3-
nitroxide.[32] The copolymerizations were controlled below
50 mol-% of the acidic comonomer, whereas for larger
proportions the control became quite poor. For instance, the
polydispersity index increased from 1.26 to 1.55 when the
proportion of Awas raised from 20 to 50 mol-%. Also using
NMP, sodium acrylate was polymerized in water-solution,
using poly(sodium 4-styrenesulfonate) macroinitiators
end-capped with water-soluble nitroxides, but the con-
trolled behavior was not demonstrated.[33]
The purpose of this article is to demonstrate that, under
selected conditions, acrylic acid can be copolymerized with
styrene (S) in a controlled way, using nitroxide-mediated
polymerization. The nitroxide used was the N-tert-butyl-
N-(1-diethylphosphono-2,2-dimethylpropyl), also called
SG1, which proved to be better suited than TEMPO
(2,2,6,6-tetramethylpiperidinyl-1-oxy) to control the poly-
merization of monomers other than styrene and deriva-
tives.[34,35] A broad composition range was investigated
from 10/90 to 90/10 A/S molar ratios. The effect of
composition on control over molar mass and distribution
and on kinetics was studied and discussed. It is the first time
that such controlled copolymers with broad composition
range are synthesized in a direct way, which is very pro-
mising for a variety of applications. This study is part of a
more general work devoted to SG1-mediated polymeriza-
tion of acrylic acid and we recently reported the controlled
homopolymerization and its limitation.[36,37]
Experimental Part
Materials
Styrene (S, Aldrich, 99% purity) was distilled under vacuumbefore use. Acrylic acid (A, purest grade, Atofina, stabilizedwith 200 ppm of hydroquinone) was stored at roomtemperature and used without further purification. Thealkoxyamine initiator (SG1-based alkoxyamine derived frommethyl acrylate, CH3–O–C( O)–CH(CH3)–SG1, Monams,96% purity) and the N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide (SG1, 86.5% purity) were kindlysupplied by Atofina. The solvent 1,4-dioxane (from SDS,synthesis grade) was used as received. The methylation agent,
2056 L. Couvreur, B. Charleux, O. Guerret, S. Magnet
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

trimethylsilyldiazomethane (2 M solution in hexane) wassupplied by Aldrich and used as received.
Solution Copolymerizations of Styrene and Acrylic Acid
The copolymerization reactions were carried out in a Parrreactor of 300 mL, thermostatted at 120 8C, under a 2 barnitrogen atmosphere. The stirring rate was 300 rpm. In a typicalrecipe (Expt. 3), the Monams (1.731 g, 4.5� 10�3 mol) andSG1 (0.0610 g, 2.1� 10�4 mol, 4.5 mol-% with respect toMonams) were dissolved in styrene (30.01 g, 0.289 mol) andsolvent (1,4-dioxane, 143 mL). Then, acrylic acid (20.70 g,0.287 mol) was added to the mixture. Deoxygenation wasperformed by nitrogen bubbling for 30 min. Afterwards, thepolymerization solution was transferred into the reactor(already heated at 120 8C) and a 2 bar pressure of nitrogenwas applied. Time zero of the reaction was arbitrarily setwhen the mixture reached 110 8C. Samples were periodicallywithdrawn (on the top of the reactor) and cooled in an icedwater bath to stop the polymerization. Polymers were reco-vered by drying in a ventilated oven at 35 8C for 2 d. Aftermodification (next section) they were analyzed by sizeexclusion chromatography (SEC) in THF solution. Allperformed experiments are collected in Table 1.
Chemical Modification of the Copolymers
For size exclusion chromatography, the polymers weremodified by methylation of the carboxylic acid groups, usingtrimethylsilyldiazomethane.[38] In this way, 50 mg of eachsample was dissolved in a mixture of THF and water (overallvolume¼ 10 mL; the proportion of water was increased withthe amount of acid functions, to get solubilization at roomtemperature). The yellow solution of trimethylsilyldiazo-methane was added dropwise at room temperature into thecopolymer solution. Upon addition, bubbles appeared and thesolution became slowly colorless. Addition of the methylationagent was continued until the solution remained yellow andstopped bubbling. Then, an excess of methylation agent wasadded and the solution was stirred for 3 more hours at roomtemperature. As verified by 1H NMR analysis of the copoly-mers, methyl ester formation was always quantitative.
Analytical Techniques
The overall conversions were determined by gravimetry usingan automatic thermic balance (Mettler Toledo, HG53) at125 8C (evaporation till constant weight, approximately for15 min). This technique gave access to a weight conversion(xwt), that can be written as a function of the individualconversions xA and xS and of the weight fraction of eachmonomer in the initial mixture (wA0 and wS0 for acrylic acidand styrene respectively) (Equation (1)).
xwt ¼ xA � wA0 þ xS � wS0 ð1Þ
For the largest conversions however (above approximately50%) the samples were never perfectly dry even when dryingconditions were improved (remaining monomers were alwaysdetected by NMR); therefore only weight conversions obtainedfrom gravimetry below 50% were used in this work.
The raw polymerization media with added deuteratedacetone were analyzed by proton NMR spectroscopy at regulartime intervals. Analyses were performed in 5 mm tubes at roomtemperature using a Bruker AC200 apparatus, operating at afrequency of 200 MHz. The chemical shift scale was calibratedon the basis of the solvent peak (deuterated acetone at2.04 ppm). The overall molar conversions (xmol) and theindividual conversions of each monomer (xA and xS) weredetermined by integrating the peaks corresponding to thevinyl protons of the monomers, using the broad peak between6.5 and 7.5 ppm as an internal reference (5 aromatic H forstyrene and polystyrene, and 1 vinylic H for the styrenemonomer that was subtracted before calculation). The overallmolar conversion is a function of the individual conversions xA
and xS and of the molar fraction of each monomer in the initialmixture (fA0 and fS0 for acrylic acid and styrene respectively)(Equation (2)).
xmol ¼ xA � fA0 þ xS � fS0 ð2Þ
Overall molar conversions from NMR were used for kineticanalysis (for instance conversion versus time plots). Molarmasses and polydispersity indices were always plotted as afunction of the overall weight conversion. The latter wasderived from the direct gravimetric analysis (below 50%) orwas calculated from the NMR individual conversions (overthe whole conversion range) using Equation (1). This leads to
Table 1. Experimental conditions for the SG1-mediated copolymerizations of styrene and acrylic acid in 1,4-dioxane solution at 120 8C.
Expt. Symbol [S]0 [A]0 fA0a) [Monams]0 [SG1]0 rb) hkpi � [P�]
mol �L�1 mol �L�1 mol �L�1 mol �L�1 s�1
1 * 2.70 0.30 0.101 0.0274 0.0012 0.044 8.8� 10�5
2 ~ 2.25 0.77 0.255 0.0262 0.0012 0.044 1.1� 10�4
3 & 1.47 1.47 0.499 0.0235 0.0011 0.045 1.4� 10�4
4 ^ 0.75 2.25 0.749 0.0218 0.0010 0.046 1.5� 10�4
5 ~ 0.30 2.69 0.899 0.0205 0.0009 0.046 1.2� 10�4
6 * 1.48 1.47 0.498 0.0117 0.0006 0.047 –7 & 1.49 1.47 0.497 0.0058 0.0003 0.045 –
a) fA0: molar fraction of A in the initial monomer feed.b) r¼ [SG1]0/[Monams]0.
Direct Synthesis of Controlled Poly(styrene-co-acrylic acid)s . . . 2057
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

linear theoretical relationships between the number averagemolar mass, Mn, and conversion (ideally, for wA0 in Equation(1) we should consider the additional contribution of themethyl ester group introduced by methylation, but the diffe-rence was usually small enough to be neglected).
Composition of the comonomer mixture (fA, the molarproportion of acrylic acid) was also determined from 1HNMR analysis at regular time intervals, by integrating thepeaks corresponding to the vinyl protons of the residualmonomers.
The molar mass and molar mass distribution of themethylated copolymers were obtained by SEC in THF with a1 mL �min�1 flow rate. The apparatus is composed of adegasser (Viscotek, VE7510 GPC), a Waters 515 HPLC pump,an auto-sampler (Viscotek,VE5200 GPC), 3 linear columns(3 PSS SDV linear) in an oven (Croco-cilTM) thermostatted at40 8C and two detectors: differential RI (LDC Analytical,refractoMonitor IV) and UV at 254 nm (Waters 484). Themolar masses were derived from a calibration curve based onpolystyrene standards (162 to 1 090 000 g �mol�1, fromPolymer Standards Service).
Results and Discussion
Selection of Suitable Experimental Conditions
SG1-mediated copolymerization of styrene and acrylic acid
was carried out using a SG1-based unimolecular alkox-
yamine initiator, CH3–O–C( O)–CH(CH3)–SG1, also
called Monams. Because of too fast polymerization and
poorly controlled behavior, bulk copolymerization was
discarded. Consequently, an important point was the choice
of an appropriate solvent, in which the monomers and
the copolymers remained soluble throughout the reaction.
The purpose here was to work under homogeneous condi-
tions: any macrophase separation would actually alter the
quality of control by changing the local concentrations.
Preliminary experiments were performed in tert-butylben-
zene and in toluene. The copolymerization medium
remained homogeneous when the acrylic acid molar ratio
was rather low (10 mol-% for instance). However, phase
separation occurred when this ratio was raised above
25 mol-%. A solvent that actually met the required con-
ditions was 1,4-dioxane, in which the copolymers were
fully soluble, irrespective of the composition and of the
monomer conversion. In this work, the initial monomer
concentration was kept constant and equal to 3 mol �L�1;
only the relative proportions of styrene and acrylic acid
were changed. For Expts. 1–5, the initial concentration of
Monams was adjusted in order to target molar masses
close to 11 000 g �mol�1 (in the acidic form). In two other
examples (Expts. 6 and 7) Monams concentration was
divided by a factor of respectively 2 and 4, to achieve larger
molar masses. A small initial concentration of free SG1 was
introduced (r¼ 4.5 mol-% with respect to Monams), as was
previously proposed for butyl acrylate, to moderate the
polymerization rate and reach the best compromise
between fast reaction and quality of control.[35] Indeed, it
should be kept in mind, as mentioned above, that acrylic
acid, similarly to butyl acrylate, exhibits a very high pro-
pagation rate constant.[18] Finally, the polymerization
temperature was 120 8C, which is the optimal value for
Monams decomposition as well as for styrene homopoly-
merization.[39] Details on the experimental parameters can
be found in Table 1.
Effect of Monomer Feed Ratio
Molar Mass and Molar Mass Distribution
One of the features that a polymer should exhibit
when synthesized under controlled conditions is the
linear increase of molar mass with monomer conversion,
along with the decrease of the polydispersity index.[1–3]
They reveal that polymerization takes place with a cons-
tant chain concentration and that all chains grow simulta-
neously with the same rate of monomer incorporation.
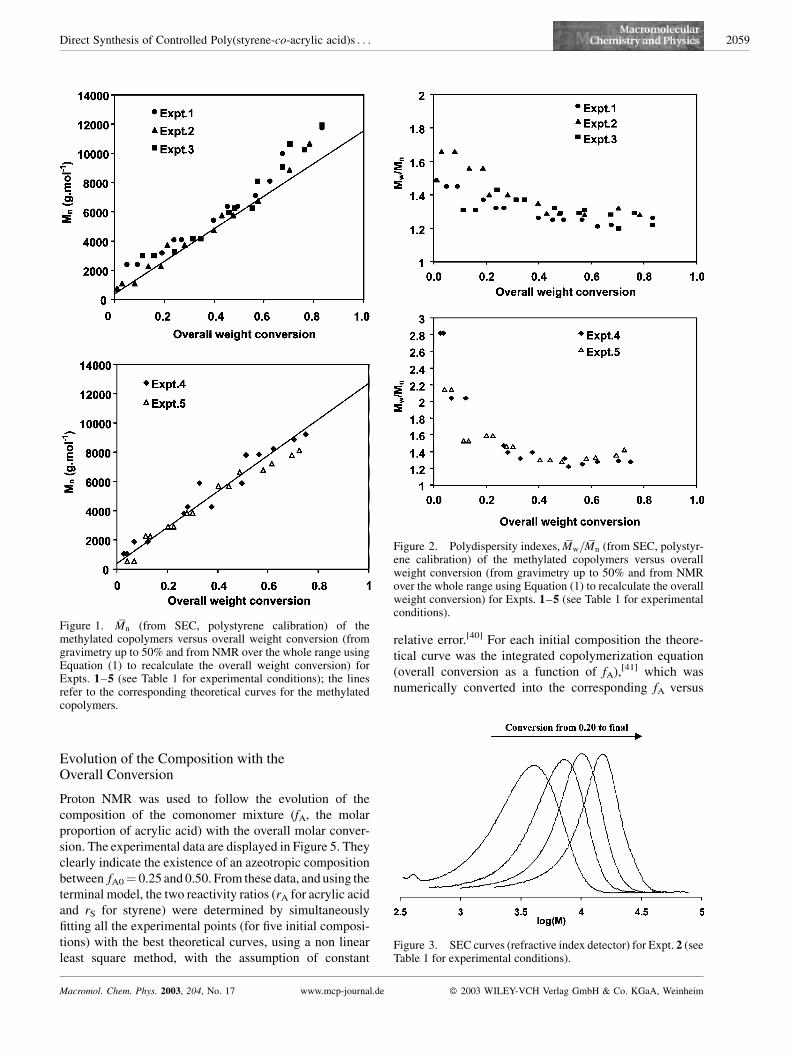
Figure 1 and Figure 2 show that these criteria were
actually fulfilled by the copolymerizations 1 to 5, over the
whole conversion range, indicating that they actually took
place via a nitroxide-mediated controlled free radical
process.
The Mn values of the methylated copolymers followed
the theoretical line (calculated from the initial concentra-
tions of monomers and initiator), pointing out that all
growing chains were indeed generated by the alkoxyamine
initiator (i.e. concentration of chains equals the initial con-
centration of Monams). Simultaneously, the polydispersity
indices decreased with increasing conversion from approxi-
mately 1.6 to 1.2 for Expt. 1 to 3. Such final values indicate a
narrow molar mass distribution that could not be reached
under classical free-radical copolymerization conditions.
This behavior is illustrated in Figure 3, which displays the
continuous shift of the SEC curves towards the higher molar
masses, obtained for the methylated copolymers of Expt. 2.
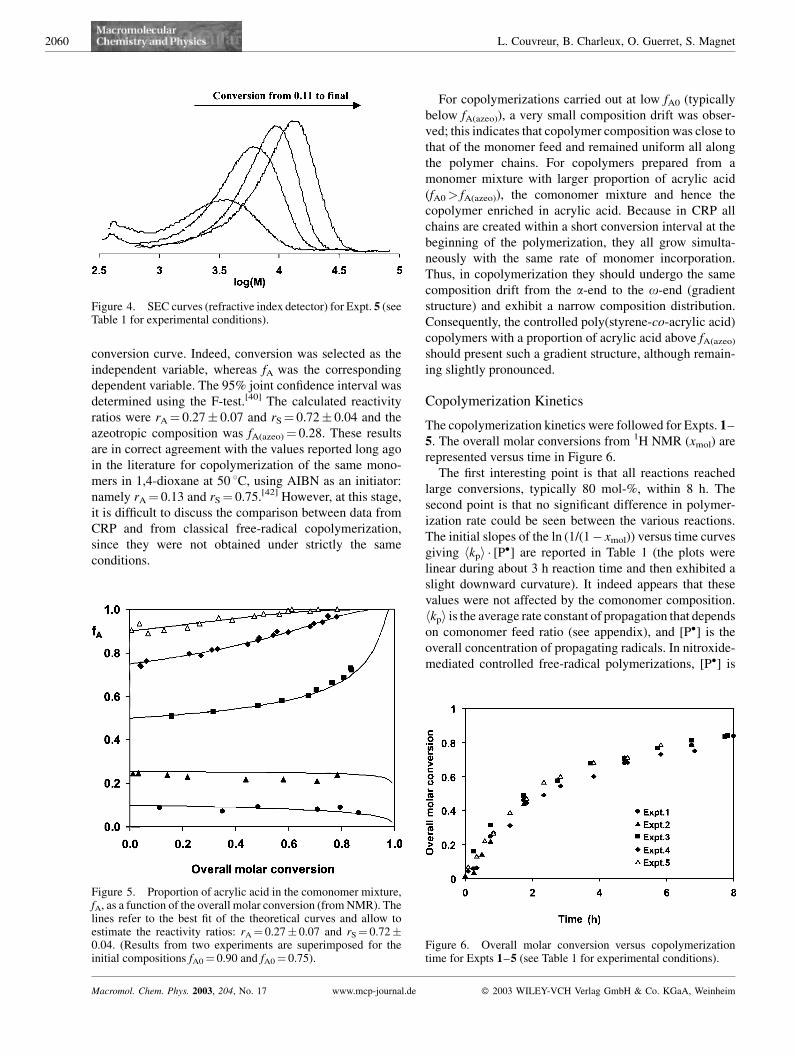
The situation was slightly different when the proportion of
acrylic acid was increased; all the other parameters remain-
ing the same (Expt. 4 with 75 mol-% of A; Expt. 5 with
90 mol-% of A). At first, the initial polydispersity indices
were far above the 1.6 value observed for the previous
experiments, but decreased down to 1.2–1.3 in the 40–
60 wt.-% conversion range, and slightly increased at larger
conversions, particularly for the experiment with fA0¼0.90. The shift of the molar mass distribution with
conversion can be clearly seen in Figure 4 for Expt. 5, but
in contrast to the previous experiments with a lower
proportion of acrylic acid, a tailing was observed on the low
molar mass side, as an indication of the possible existence
of irreversibly terminated chains (by bimolecular macro-
radical termination or more likely by chain transfer to
solvent as previously shown for SG1-mediated homopoly-
merization of acrylic acid).[36,37]
2058 L. Couvreur, B. Charleux, O. Guerret, S. Magnet
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Evolution of the Composition with theOverall Conversion
Proton NMR was used to follow the evolution of the
composition of the comonomer mixture (fA, the molar
proportion of acrylic acid) with the overall molar conver-
sion. The experimental data are displayed in Figure 5. They
clearly indicate the existence of an azeotropic composition
between fA0 ¼ 0.25 and 0.50. From these data, and using the
terminal model, the two reactivity ratios (rA for acrylic acid
and rS for styrene) were determined by simultaneously
fitting all the experimental points (for five initial composi-
tions) with the best theoretical curves, using a non linear
least square method, with the assumption of constant
relative error.[40] For each initial composition the theore-
tical curve was the integrated copolymerization equation
(overall conversion as a function of fA),[41] which was
numerically converted into the corresponding fA versus
Figure 1. Mn (from SEC, polystyrene calibration) of themethylated copolymers versus overall weight conversion (fromgravimetry up to 50% and from NMR over the whole range usingEquation (1) to recalculate the overall weight conversion) forExpts. 1–5 (see Table 1 for experimental conditions); the linesrefer to the corresponding theoretical curves for the methylatedcopolymers.
Figure 2. Polydispersity indexes, Mw=Mn (from SEC, polystyr-ene calibration) of the methylated copolymers versus overallweight conversion (from gravimetry up to 50% and from NMRover the whole range using Equation (1) to recalculate the overallweight conversion) for Expts. 1–5 (see Table 1 for experimentalconditions).
Figure 3. SEC curves (refractive index detector) for Expt. 2 (seeTable 1 for experimental conditions).
Direct Synthesis of Controlled Poly(styrene-co-acrylic acid)s . . . 2059
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
conversion curve. Indeed, conversion was selected as the
independent variable, whereas fA was the corresponding
dependent variable. The 95% joint confidence interval was
determined using the F-test.[40] The calculated reactivity
ratios were rA¼ 0.27� 0.07 and rS¼ 0.72� 0.04 and the
azeotropic composition was fA(azeo) ¼ 0.28. These results
are in correct agreement with the values reported long ago
in the literature for copolymerization of the same mono-
mers in 1,4-dioxane at 50 8C, using AIBN as an initiator:
namely rA¼ 0.13 and rS ¼ 0.75.[42] However, at this stage,
it is difficult to discuss the comparison between data from
CRP and from classical free-radical copolymerization,
since they were not obtained under strictly the same
conditions.
For copolymerizations carried out at low fA0 (typically
below fA(azeo)), a very small composition drift was obser-
ved; this indicates that copolymer composition was close to
that of the monomer feed and remained uniform all along
the polymer chains. For copolymers prepared from a
monomer mixture with larger proportion of acrylic acid
(fA0> fA(azeo)), the comonomer mixture and hence the
copolymer enriched in acrylic acid. Because in CRP all
chains are created within a short conversion interval at the
beginning of the polymerization, they all grow simulta-
neously with the same rate of monomer incorporation.
Thus, in copolymerization they should undergo the same
composition drift from the a-end to the o-end (gradient
structure) and exhibit a narrow composition distribution.
Consequently, the controlled poly(styrene-co-acrylic acid)
copolymers with a proportion of acrylic acid above fA(azeo)
should present such a gradient structure, although remain-
ing slightly pronounced.
Copolymerization Kinetics
The copolymerization kinetics were followed for Expts. 1–
5. The overall molar conversions from 1H NMR (xmol) are
represented versus time in Figure 6.
The first interesting point is that all reactions reached
large conversions, typically 80 mol-%, within 8 h. The
second point is that no significant difference in polymer-
ization rate could be seen between the various reactions.
The initial slopes of the ln (1/(1� xmol)) versus time curves
giving hkpi � [P�] are reported in Table 1 (the plots were
linear during about 3 h reaction time and then exhibited a
slight downward curvature). It indeed appears that these
values were not affected by the comonomer composition.
hkpi is the average rate constant of propagation that depends
on comonomer feed ratio (see appendix), and [P�] is the
overall concentration of propagating radicals. In nitroxide-
mediated controlled free-radical polymerizations, [P�] is
Figure 4. SEC curves (refractive index detector) for Expt. 5 (seeTable 1 for experimental conditions).
Figure 5. Proportion of acrylic acid in the comonomer mixture,fA, as a function of the overall molar conversion (from NMR). Thelines refer to the best fit of the theoretical curves and allow toestimate the reactivity ratios: rA¼ 0.27� 0.07 and rS¼ 0.72�0.04. (Results from two experiments are superimposed for theinitial compositions fA0 ¼ 0.90 and fA0¼ 0.75).
Figure 6. Overall molar conversion versus copolymerizationtime for Expts 1–5 (see Table 1 for experimental conditions).
2060 L. Couvreur, B. Charleux, O. Guerret, S. Magnet
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

given by the activation-deactivation equilibrium relation-
ship (Equation 3; see Scheme 1).[43]
½P�� ¼ Kh i � ½P--SG1�½SG1� ¼ Kh i � ½Monams�0
½SG1� ð3Þ
In this equation the concentration of SG1-capped macro-
molecular chains, [P–SG1], was replaced by the initial
concentration of Monams (owing to the good match
between experimental and calculated Mn’s, which indicates
a fast and complete initiation). The concentration of free
SG1 is a function of the initial concentration and of the
amount released owing to the persistent radical effect.[44]
With fairly high initial concentration of SG1 (close to
10�3 mol �L�1 for Expts. 1–5) one can simplify Equation 3
and consider that [SG1] remained close to the initial
concentration [SG1]0 (Equation 4).
½P�� ¼ Kh i � ½Monams�0½SG1�0
¼ Kh ir
ð4Þ
In contrast to homopolymerization where the activation-
deactivation equilibrium constant K only depends upon
temperature for a given monomer (Scheme 1), in copoly-
merization hKi should be an apparent value that also
depends upon comonomer composition.
From the kinetic analysis and Equation (4), it is then
possible to estimate hkpi � hKi¼ hkpi � [P�] � r. However, it
should be kept in mind that using r in this equation might
lead to underestimated values (use of [SG1]0 instead of
‘‘true’’ [SG1]). Nevertheless, we do not intend to give
accurate data, but simply discuss the results qualitatively.
The consequence of a rather constant hkpi � [P�] with an
increasing proportion of acrylic acid in the comonomer
mixture, together with similar r values, is that hkpi � hKiremained rather constant too. However, the propagation
rate constant was reported to be much larger for acrylic acid
than for styrene. Indeed, the propagation rate constant of
acrylic acid, kpA, is in the order of 105 L �mol�1 � s�1,[18]
whereas it is kpS¼ 2 000 L �mol�1 � s�1 for styrene at
120 8C.[45] As a consequence, hkpi is likely to increase with
the increase of fA0 (see appendix) and hence, hKi should
decrease, leading to a decrease of the overall concentration
of propagating radicals. This trend indicates that the
activation-deactivation equilibrium constant for the SG1-
mediated homopolymerization of acrylic acid, KA, might
be significantly lower than that for styrene homopoly-
merization, KS. Indeed, the latter was reported to be
KS¼ 6� 10�9 in bulk at 120 8C,[39] whereas KA was shown
to be close to 10�10 or even below, as derived from our
kinetic study of SG1-mediated homopolymerization of
acrylic acid.[36,37]
Effect of the Alkoxyamine Initiator Concentration
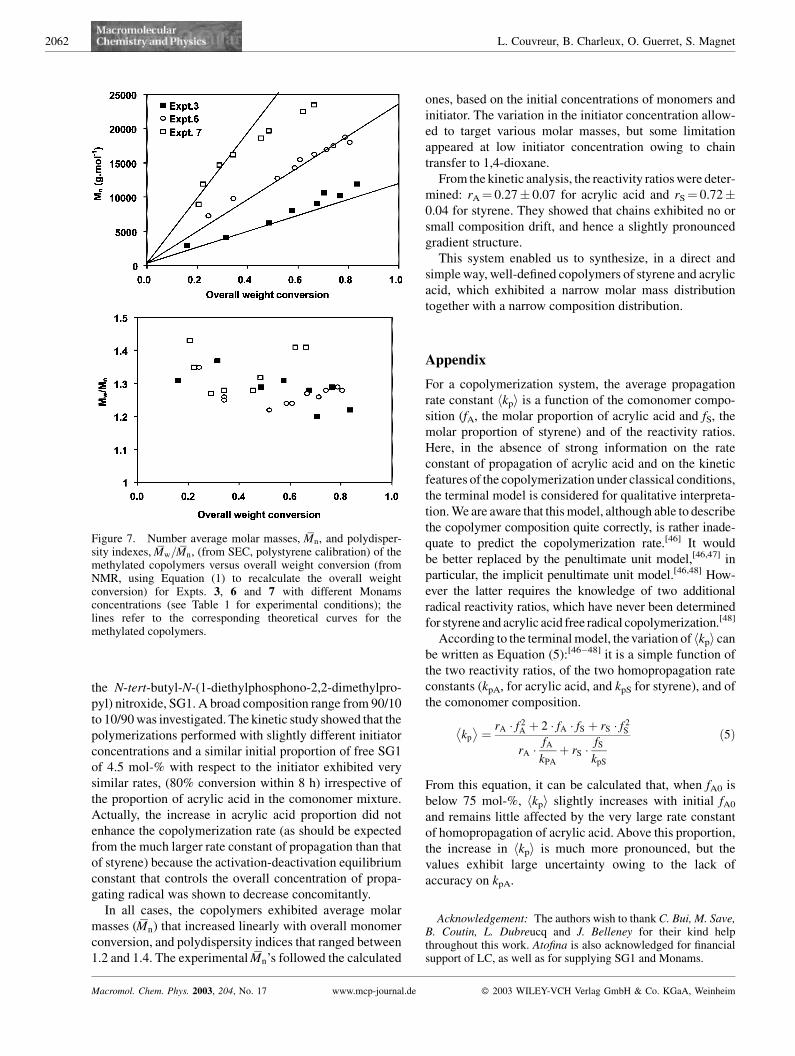
For a given molar composition of the comonomer feed, the
variation in alkoxyamine initiator concentration should
lead to a change in the average molar mass. As reported
in Table 1, two copolymerizations similar to Expt. 3(fA0 ¼ 0.5) were carried out, with Monams concentration
divided by a factor of 2 (Expt. 6) and by a factor of 4
(Expt.7). The initial concentration of free SG1 was adjusted
so as to keep r constant. As anticipated, the change in the
experimental conditions affected the average molar masses
as displayed in Figure 7. Indeed, it appeared that the
experimental Mn’s followed the theoretical lines, and that
the polydispersity indices remained in the 1.2–1.4 range.
These results fully support the controlled character of the
copolymerization reaction. However, for the lowest initia-
tor concentration ([Monams]0¼ 0.0058 mol �L�1, Expt. 7),
the variation of Mn with conversion did not remain linear
throughout the reaction but deviated from linearity above
�40% conversion. At this stage the polydispersity indices
were slightly above those found for the other two experi-
ments (i.e. 1.4 instead of 1.2–1.3). This behavior was
assigned to chain transfer to 1,4-dioxane, which creates new
short chains, as was also found for SG1-mediated homo-
polymerization of acrylic acid,[36,37] as well as for the
RAFT polymerizations performed in the same solvent.[23]
This result shows that our system actually undergoes molar
mass limitation at low initiator concentration, which is
however less pronounced in copolymerization of A with
styrene than in homopolymerization.[36,37] Additionally, it
is likely that the chain transfer constant (and hence the
impact of transfer on molar mass) depends on the como-
nomer composition.
Conclusion
Free-radical copolymerization of styrene and acrylic acid
was carried out under controlled conditions in 1,4-dioxane
solution at 120 8C, using an alkoxyamine initiator based on
Scheme 1. Activation (rate constant kd) – deactivation (rateconstant kc) equilibrium in SG1-mediated controlled free-radicalpolymerization. In homopolymerization, the equilibrium constantis K ¼ kd
kc¼ ½P���½SG1�
½P�SG1� , where P� represents the propagating macro-radical and P-SG1 is the dormant chain with alkoxyamine end-group.
Direct Synthesis of Controlled Poly(styrene-co-acrylic acid)s . . . 2061
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
the N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpro-
pyl) nitroxide, SG1. A broad composition range from 90/10
to 10/90 was investigated. The kinetic study showed that the
polymerizations performed with slightly different initiator
concentrations and a similar initial proportion of free SG1
of 4.5 mol-% with respect to the initiator exhibited very
similar rates, (80% conversion within 8 h) irrespective of
the proportion of acrylic acid in the comonomer mixture.
Actually, the increase in acrylic acid proportion did not
enhance the copolymerization rate (as should be expected
from the much larger rate constant of propagation than that
of styrene) because the activation-deactivation equilibrium
constant that controls the overall concentration of propa-
gating radical was shown to decrease concomitantly.
In all cases, the copolymers exhibited average molar
masses (Mn) that increased linearly with overall monomer
conversion, and polydispersity indices that ranged between
1.2 and 1.4. The experimental Mn’s followed the calculated
ones, based on the initial concentrations of monomers and
initiator. The variation in the initiator concentration allow-
ed to target various molar masses, but some limitation
appeared at low initiator concentration owing to chain
transfer to 1,4-dioxane.
From the kinetic analysis, the reactivity ratios were deter-
mined: rA¼ 0.27� 0.07 for acrylic acid and rS¼ 0.72�0.04 for styrene. They showed that chains exhibited no or
small composition drift, and hence a slightly pronounced
gradient structure.
This system enabled us to synthesize, in a direct and
simple way, well-defined copolymers of styrene and acrylic
acid, which exhibited a narrow molar mass distribution
together with a narrow composition distribution.
Appendix
For a copolymerization system, the average propagation
rate constant hkpi is a function of the comonomer compo-
sition (fA, the molar proportion of acrylic acid and fS, the
molar proportion of styrene) and of the reactivity ratios.
Here, in the absence of strong information on the rate
constant of propagation of acrylic acid and on the kinetic
features of the copolymerization under classical conditions,
the terminal model is considered for qualitative interpreta-
tion. We are aware that this model, although able to describe
the copolymer composition quite correctly, is rather inade-
quate to predict the copolymerization rate.[46] It would
be better replaced by the penultimate unit model,[46,47] in
particular, the implicit penultimate unit model.[46,48] How-
ever the latter requires the knowledge of two additional
radical reactivity ratios, which have never been determined
for styrene and acrylic acid free radical copolymerization.[48]
According to the terminal model, the variation of hkpi can
be written as Equation (5):[46–48] it is a simple function of
the two reactivity ratios, of the two homopropagation rate
constants (kpA, for acrylic acid, and kpS for styrene), and of
the comonomer composition.
kp
� �¼ rA � f 2
A þ 2 � fA � fS þ rS � f 2S
rA � fA
kPA
þ rS �fS
kpS
ð5Þ
From this equation, it can be calculated that, when fA0 is
below 75 mol-%, hkpi slightly increases with initial fA0
and remains little affected by the very large rate constant
of homopropagation of acrylic acid. Above this proportion,
the increase in hkpi is much more pronounced, but the
values exhibit large uncertainty owing to the lack of
accuracy on kpA.
Acknowledgement: The authors wish to thank C. Bui, M. Save,B. Coutin, L. Dubreucq and J. Belleney for their kind helpthroughout this work. Atofina is also acknowledged for financialsupport of LC, as well as for supplying SG1 and Monams.
Figure 7. Number average molar masses, Mn, and polydisper-sity indexes, Mw=Mn, (from SEC, polystyrene calibration) of themethylated copolymers versus overall weight conversion (fromNMR, using Equation (1) to recalculate the overall weightconversion) for Expts. 3, 6 and 7 with different Monamsconcentrations (see Table 1 for experimental conditions); thelines refer to the corresponding theoretical curves for themethylated copolymers.
2062 L. Couvreur, B. Charleux, O. Guerret, S. Magnet
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim

[1] ‘‘Controlled Radical Polymerization’’, in: ACS Symp. Ser.,Vol. 685, K. Matyjaszewski, Ed., Washington 1998.
[2] ‘‘Controlled/Living Radical Polymerization: Progress inATRP, NMP, and RAFT’’, in: ACS Symp. Ser., Vol. 768,K. Matyjaszewski, Ed., Washington 2000.
[3] ‘‘Advances in Controlled/Living Radical Polymerization’’,in: ACS Symp. Ser., Vol. 854, K. Matyjaszewski, Ed.,Washington 2003.
[4] C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 2001,101, 3661.
[5] K. Matyjaszewski, J. Xia, Chem. Rev. 2001, 101, 2921.[6] M. Kamigaito, T. Ando, M. Sawamoto, Chem. Rev. 2001,
101, 3689.[7] N. Hadjichristidis, M. Pitsikalis, S. Pispas, H. Iatrou, Chem.
Rev. 2001, 101, 3747.[8] J. Qiu, B. Charleux, K. Matyjaszewski, Prog. Polym. Sci.
2001, 26, 2083.[9] A. B. Lowe, C. L. McCormick, Aust. J. Chem. 2002, 55, 367.
[10] A large number of references are available. See for instance:[10a] F. Zeng, M. Yang, J. Zhang, S. K. Varshney, J. Polym.Sci., Part A: Polym. Chem. 2002, 40, 4387; [10b] X. F.Zhong, S. K. Varshney, A. Eisenberg, Macromolecules 1992,25, 7160.
[11] K. A. Davis, K. Matyjaszewski, Macromolecules 2000, 33,4039.
[12] K. A. Davis, B. Charleux, K. Matyjaszewski, J. Polym. Sci.,Part A: Polym. Chem. 2000, 38, 2274.
[13] S. Hou, E. L. Chaikof, D. Taton, Y. Gnanou, Macromolecules2003, 36, 3874.
[14] O. A. Otmakhova, S. A. Kuptsov, R. V. Talroze, T. E. Patten,Macromolecules 2003, 36, 3432.
[15] M. Zhang, T. Breiner, H. Mori, A. H. E. Muller, Polymer2003, 44, 1449.
[16] T. E. Patten, K. Matyjaszewski, Adv. Mater. 1998, 10, 901.[17] P. G. Odell, R. P. N. Veregin, L. M. Michalak, D. Brousmiche,
M. K. Georges, Macromolecules 1995, 38, 8453.[18] I. Lacik, S. Beuermann, M. Buback, Macromolecules 2001,
34, 6224.[19] S. Ali-Miraftab, A. Chapiro, Z. Mankowski, Eur. Polym. J.
1981, 17, 1197.[20] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery,
T. P. T. Le, R. T. A. Mayadunne, G. G. Meijs, C. L. Moad,G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998,31, 5559.
[21] Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo, S. H. Thang,Macromolecules 1999, 32, 2071.
[22] C. Ladaviere, N. Dorr, J. P. Claverie, Macromolecules 2001,34, 5370.
[23] J. Loiseau, N. Dorr, J. M. Suau, J. B. Egraz, M. F. Llauro, C.Ladaviere, J. Claverie, Macromolecules 2003, 36, 3066.
[24] N. Gaillard, A. Guyot, J. Claverie, J. Polym. Sci., Part A:Polym. Chem. 2003, 41, 684.
[25] C.-Y. Hong, Y.-Z. You, R.-K. Bai, C.-Y. Pan, G. Borjihan, J.Polym. Sci., Part A: Polym. Chem. 2001, 39, 3934.
[26] D. Taton, A.-Z. Wilczewska, M. Destarac, Macromol. RapidCommun. 2001, 22, 1497.
[27] H. T. Lord, M. R. Whittaker, J. F. Quinn, C. Barner-Kowollik, M. H. Stenzel, J. P. A. Heuts, T. P. Davis, Polym.Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 2002, 43(2),118.
[28] J. T. Lai, D. Filla, R. Shea, Macromolecules 2002, 35, 6754.[29] C. M. Schilli, A. H. E. Muller, E. Rizzardo, S. H. Thang, Y. K.
Chong, ACS Symp. Ser. 2003, 854, 603.[30] C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R.
G. Gilbert, A. K. Serelis, C. H. Such, Macromolecules 2002,35, 9243.
[31] Y. Gnanou, D. Grande, R. Guerrero, Polym. Prepr. (Am.Chem. Soc., Div. Polym. Chem.) 1999, 40(2), 99.
[32] D. Benoit, V. Chaplinski, R. Braslau, C. J. Hawker, J. Am.Chem. Soc. 1999, 121, 3904.
[33] W. Huang, B. Charleux, R. Chiarelli, L. Marx, A. Rassat, J. P.Vairon, Macromol. Chem. Phys. 2002, 203, 1715.
[34] S. Grimaldi, J. P. Finet, F. Le Moigne, A. Zeghdaoui, P.Tordo, D. Benoit, M. Fontanille, Y. Gnanou, Macromole-cules 2000, 33, 1141.
[35] P. Lacroix-Desmazes, J. F. Lutz, F. Chauvin, R. Severac, B.Boutevin, Macromolecules 2001, 34, 8866.
[36] L. Couvreur, Ph.D. thesis, University Pierre and MarieCurie, Paris, France, 2003.
[37] L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret,S. Magnet, Macromolecules 2003, in press.
[38] H. Norio, A. Toyohiro, S. Takayuki, Chem. Pharm. Bull.1981, 29, 1475.
[39] D. Benoit, S. Grimaldi, S. Robin, J. P. Finet, P. Tordo, Y.Gnanou, J. Am. Chem. Soc. 2000, 122, 5929.
[40] A. M. van Herk, T. Droge, Macromol. Theory Simul. 1997, 6,1263.
[41] G. Odian, ‘‘Principles of Polymerization’’, 3rd edition, JohnWiley and Sons, Inc., New York 1991.
[42] R. Kerber, Makromol. Chem. 1966, 96, 30.[43] T. Fukuda, A. Goto, K. Ohno, Macromol. Rapid Commun.
2000, 21, 151.[44] H. Fischer, Chem. Rev. 2001, 101, 3581.[45] M. Buback, R. G. Gilbert, R. A. Hutchinson, B. Klumper-
mann, F.-D. Kuchta, B. G. Manders, K. F. O’Driscoll, G. T.Russell, J. Schweer, Macromol. Chem. Phys. 1995, 196, 3267.
[46] T. Fukuda, Y.-D. Ma, H. Inagaki, Macromolecules 1985, 18,17.
[47] M. L. Coote, T. P. Davis, Prog. Polym. Sci. 1999, 24, 1217.[48] S. Beuermann, M. Buback, Prog. Polym. Sci. 2002, 27, 191.
Direct Synthesis of Controlled Poly(styrene-co-acrylic acid)s . . . 2063
Macromol. Chem. Phys. 2003, 204, No. 17 www.mcp-journal.de � 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim





![709 ' # '6& *#7 & 8 · 2018-03-27 · of natural rubber. The synthetic rubbers were produced through radical copolymerization of styrene and butadiene [1-5]. Today, emulsion polymerization](https://img.pdfslide.us/doc/110x75/5f0f5b8d7e708231d443c296/709-6-7-8-2018-03-27-of-natural-rubber-the-synthetic-rubbers.jpg)









![4,800 122,000 135M · 2020. 5. 16. · chloride (Et3NHCl-AlCl3) as an initiator for the cationic copolymerization of 1,3-penta‐ diene with styrene [14]. The best results were obtained](https://img.pdfslide.us/doc/110x75/61229fa7c428bf369828ea8c/4800-122000-135m-2020-5-16-chloride-et3nhcl-alcl3-as-an-initiator-for-the.jpg)





![NovelComplexPolymerswithCarbazoleFunctionalityby … · 2011. 9. 27. · NMP, whereas the copolymerization with styrene under the same conditions proceeded in a living fashion [46]](https://img.pdfslide.us/doc/110x75/60ae5f4263e17345284cde18/novelcomplexpolymerswithcarbazolefunctionalityby-2011-9-27-nmp-whereas-the.jpg)
![Styrene butadiene anionic copolymerization · 3419 Smallman Street, Pittsburgh, PA 15201 412-682-5882 ] v ] Ì Pressure Chemical Co. (PCC) has experience in styrene butadiene](https://img.pdfslide.us/doc/110x75/5b2a072c7f8b9acb148b6ffd/styrene-butadiene-anionic-3419-smallman-street-pittsburgh-pa-15201-412-682-5882.jpg)






