Embed Size (px)
DESCRIPTION
1 TowhomcorrespondenceshouldbeaddressedatDepartmentofPhysiology,FacultyofMedicineandHealthSciences,UnitedArabEmiratesUniversity, POBox17666,AlAin,UnitedArabEmirates.Fax:þ97137671966.E-mail:[email protected]. TOXICOLOGICALSCIENCES 113(1),267–277(2010) doi:10.1093/toxsci/kfp222 AdvanceAccesspublicationOctober1,2009 ÓTheAuthor2009.PublishedbyOxfordUniversityPressonbehalfoftheSocietyofToxicology.Allrightsreserved. Forpermissions,pleaseemail:[email protected]
Citation preview

TOXICOLOGICAL SCIENCES 113(1), 267–277 (2010)
doi:10.1093/toxsci/kfp222
Advance Access publication October 1, 2009
Diesel Exhaust Particles in the Lung Aggravate Experimental AcuteRenal Failure
Abderrahim Nemmar,*,1 Suhail Al-Salam,† Shaheen Zia,* Javed Yasin,‡ Isehaq Al Husseni,§ and Badreldin H. Ali§
*Department of Physiology, †Department of Pathology, and ‡Department of Internal Medicine, Faculty of Medicine and Health Sciences, United Arab Emirates
University, Al Ain, United Arab Emirates 17666; and §Department of Pharmacology and Clinical Pharmacy, College of Medicine & Health Sciences, Sultan
Qaboos University, Muscat, 123 Sultanate of Oman
1 To whom correspondence should be addressed at Department of Physiology, Faculty of Medicine and Health Sciences, United Arab Emirates University,
PO Box 17666, Al Ain, United Arab Emirates. Fax: þ9713 7671966. E-mail: [email protected].
Received May 11, 2009; accepted September 14, 2009
Inhaled particles are associated with pulmonary and extrapulmo-
nary effects. Also, acute renal failure (ARF) is associated with
increased mortality, related to pulmonary complications. Here, we
tested the possible potentiating effect of diesel exhaust particles
(DEP) in an animal model of ARF induced by a single ip injection of
cisplatin (CP, 6 mg/kg) in rats. Six days later, the rats were
intratracheally instilled with either DEP (0.5 or 1 mg/kg) or saline
(control) and renal, systemic, and pulmonary variables were studied
24 h thereafter. CP increased the serum concentrations of urea and
creatinine and reduced glutathione (GSH) concentration and
superoxide dismutase activity in renal cortex. CP caused renal
tubular necrosis; increased urine volume, protein concentrations,
and N-acetyl-b-D-glucosaminidase (NAG) activity; and decreased
urine osmolality. The combination of DEP and CP aggravated the
CP-induced effects on serum urea and creatinine, urine NAG
activity, and renal GSH. The arterial O2 saturation and PO2 were
significantly decreased in CP1 DEP versus CP1 saline and CP1DEP versus DEP. The number of platelets was reduced in DEP
compared to saline-treated rats andCP1DEP versus DEP alone or
CP 1 saline. Increases in macrophage and neutrophils numbers in
bronchoalveolar lavage were found in DEP versus saline group and
CP 1 DEP versus CP. Histopathological changes in lungs of DEP-
treated rats were aggravated by the combination of CP 1 DEP.
These included marked interstitial cell infiltration and congestion.
We conclude that the presence of DEP in the lung aggravated the
renal, pulmonary, and systemic effects of CP-induced ARF.
Key Words: air pollution; diesel exhaust particles; lung
inflammation; acute renal failure.
Acute renal failure (ARF) is increasingly becoming more
frequent and is associated with high costs and adverse clinical
outcomes, including excess mortality, increased length of
hospital stay, and the requirement for chronic dialysis in
survivors (Hoste and Schurgers, 2008; Pannu et al., 2008).
Several studies have reported consistent association between
ARF and dysfunction of extrarenal organs, particularly the
lungs (Hoke et al., 2007; Pierson, 2006;). Experimentally, ARF
resulting from either ischemia or bilateral nephrectomy has
been reported to cause lung inflammation (Hoke et al., 2007).
Furthermore, it has been recently demonstrated that the kidney
plays an important role in the production and elimination of
mediators of pulmonary injury and that prolonged exposure to
these mediators contributes to pulmonary injury (Grigoryev
et al., 2008; Hoke et al., 2007).
Ischemia and toxicity are considered the main pathophysi-
ological factors that lead to the development of ARF (Ali and
Al Moundhri, 2006; Ali et al., 2007; Grigoryev et al., 2008;
Hoke et al., 2007). A substance that is well known to induce
toxic kidney injury is cisplatin (CP). CP is a potent anticancer
drug that is commonly used against multiple solid human
cancers, including testicular, cervical, ovarian, head, and neck
malignancies. The drug is bioactivated to a nephrotoxicant and
is also known to produce proximal tubular injury, which is
thought to be due to a combination of direct cytotoxicity,
intrarenal vasoconstriction, and oxidative stress (Ali and Al
Moundhri, 2006; Ali et al., 2007, 2008).
Inhaled particulate air pollution with particle diameter less than
2.5 lm contributes to respiratory and cardiovascular morbidity
and mortality (Kunzli et al., 2005; Pekkanen et al., 2002; Peters
et al., 2001; Pope et al., 2002). Diesel exhaust particles (DEP),
which are the major contributors to PM2.5 and ultrafine particles
(diameter� 0.1 lm) in cities, have been identified in a number of
epidemiological studies to cause adverse health effects, including
cardiorespiratory diseases, particularly in individuals with
preexisting disease (Atkinson et al., 2001; Pope et al., 1992).
Experimental exposure to DEP causes systemic and inflamma-
tory response in the airways and impairs the regulation of
vascular tone and endogenous fibrinolysis in healthy human
volunteers (Mills et al., 2005, 2007; Salvi et al., 1999). Moreover,
we (Nemmar and Inuwa, 2008; Nemmar et al., 2003a,b, 2004a,
2007) and others (Inoue et al., 2005, 2006) have reported that
exposure to DEP cause pulmonary inflammation and thrombotic
complication in hamsters and mice.
� The Author 2009. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved.For permissions, please email: [email protected]
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from

Interest in the nonpulmonary targets of particulate air
pollutants has been increasing since the demonstration that
inhaled ultrafine particles are able to translocate directly from
the lungs to extrapulmonary tissues and cause the release of
soluble inflammatory mediators into the systemic circulation,
which affect other organs, such as the liver, the heart, and even
the brain (Nemmar et al., 2004b; Oberdorster et al., 2005;
Peters et al., 2006; Vermylen et al., 2005). However, as far as
we are aware, the effect of particulate air pollution on ARF has
not been yet investigated.
Therefore, the aim of this study was to investigate, in vivo,
the possible aggravating effect of pulmonary exposure to DEP
in an animal model of ARF induced by CP, by measuring some
commonly used renal, systemic, and pulmonary variables.
MATERIALS AND METHODS
Animals. Male Wistar rats (Taconic Farms Inc., Germantown, NY), aged
10–12 weeks and initially weighing 258 ± 6 g, were given a standard laboratory
chow and water ad libitum. They were randomly divided into four groups and
individually housed in metabolic cages to facilitate urine collection, at
a temperature of 23 ± 2�C, relative humidity of 50–60%, and a 12-h dark-
light cycle. An acclimatization period of 4 days was allowed for the rats before
any experimentation. The rats were weighed at the beginning of the experiment
and just before sacrifice. Rats were cared for under a protocol approved by the
Animal Research Ethics Committee of our college and according to the
National Institutes of Health Guide for the Care and Use of Laboratory
Animals, NIH publication no. 85-23, 1985.
Intratracheal instillation. We used DEP (SRM 2975) from the National
Institute of Standards and Technology (Gaithersburg, MD). We have recently
(Nemmar et al., 2007) analyzed the size of DEP used in the present study by
transmission electron microscopy and found a substantial amount of ultrafine
(nano)-sized particle aggregates and larger particle aggregates (< 1 lm in
largest diameter). DEP were suspended in sterile normal saline (NaCl 0.9%)
containing Tween 80 (0.01%). To minimize aggregation, particle suspensions
were always sonicated (Clifton Ultrasonic Bath, Clifton, NJ) for 15 min and
vortexed before their dilution and prior to intratracheal (i.t.) administration.
Control animals received normal saline containing Tween 80 (0.01%).
Treatments. The ARF in rats was induced by a single ip injection of CP
(David Bull Laboratories, PTY Ltd, Victoria, Australia) at a dose of 6 mg/kg
(Ali et al., 2007, 2008). Control animals received similar volume of normal
saline ip. On day 6 of treatment, the animals were anesthetized with ip injection
of ketamine (75 mg/kg) and xylazine (10 mg/kg) and placed supine with
extended neck on an angled board. A Becton Dickinson 18 Gauge cannula
(Franklin Lakes, NJ) was inserted via the mouth into the trachea. DEP
suspension (0.5 or 1 mg/kg in 150 ll) or vehicle only were instilled (150 ll) via
a sterile syringe and followed by an air bolus of 100 ll.
The six groups were treated as follows (n ¼ 6–8 in each group):
� Group 1: single normal saline (control, 500 ll/rat) given ip, and on day 6
of the treatment, a single i.t. administration of saline (150 ll per rat);
� Group 2: single normal saline (control, 500 ll/rat) given ip, and on day 6
of the treatment, a single i.t. administration of DEP (0.5 mg/kg);
� Group 3: single normal saline (control, 500 ll/rat) given ip, and on day 6
of the treatment, a single i.t. administration of DEP (1 mg/kg);
� Group 4: single CP (6 mg/kg) given ip, and on day 6 of the treatment,
a single i.t. administration of saline (150 ll per rat);
� Group 5: single CP (6 mg/kg) given ip, and on day 6 of the treatment,
a single i.t. administration of DEP (0.5 mg/kg); and
� Group 6: single CP (6 mg/kg) given ip, and on day 6 of the treatment,
a single i.t. administration of DEP (1 mg/kg).
On day 6, immediately after i.t. administration of saline or DEP, rats were
placed in metabolic cages and urine of each rat was collected over a 24-h period
and the volume measured.
Blood collection and bronchoalveolar lavage. Twenty-four hour after the
i.t. administration of saline or DEP, the rats were anesthetized as described
above and blood was drawn from the inferior vena cava in EDTA (4%).
A sample was used for hematocrit measurement and platelets and white blood
cells counts using an ABX Micros 60 counter (ABX Diagnostics, Montpellier,
France). The remaining blood was left at room temperature for 2 h before it was
centrifuged at 900 3 g at 4�C for 15 min to separate serum. The serum obtained
was stored frozen at �80�C to await biochemical analyses.
The rats were then sacrificed with an overdose of ketamine. Bronchoalveolar
lavage (BAL) was then performed by cannulating the trachea, the left bronchus
was clamped. The bronchi and right lung were lavaged three times with 5 ml
sterile 0.9% NaCl. The BAL fluid was pooled in a plastic tube on ice. No
difference in the amount of recovered fluid was observed between the different
groups. BAL fluid was centrifuged (1000 3 g 3 10 min, 4�C). Cell counting
was performed in a hemocytometer after resuspension of the pellets and
staining with 1% gentian violet. The cell differentials were performed on
cytocentrifuge preparations fixed in methanol and stained with Diff Quick
(Dade Behring, Marburg, Germany). The supernatant was stored at �80�Cuntil further analysis.
Biochemical analysis and histopathology. Lungs and kidneys were
excised, washed with ice-cold saline, blotted with filter paper, and weighed.
Small pieces from the left lung and left kidney were fixed in 10% neutral
formalin, dehydrated in increasing concentrations of ethanol, cleared with
xylene, and embedded in paraffin. The cortex of the right kidney was excised
from the medulla and rapidly homogenized in ice-cold saline to produce
10 (wt/vol) tissue homogenate.
The concentrations of urea and creatinine in serum were spectrophotomet-
rically measured using commercial kits (BioMerieux, Marcy-l’Etoile, France).
Urine osmolality was measured by the freezing point depression method
(�70�C) using an osmometer (Roebling, Berlin, Germany) and N-acetyl-b-D-
glucosaminidase (NAG) activity by kits from Diazyme, General Atomics, San
Diego, CA. Urine protein concentration was measured spectrophotometrically
using a kit from BioMerieux. In renal cortex homogenates, glutathione (GSH)
concentration and superoxide dismutase (SOD) activity were measured
spectrophotometrically (Randox, Antrim, UK). The concentration of CP (as
platinum) in cortical tissue was measured by flameless atomic absorption
spectrophotometry (Perkin-Elmer, Vernon Hills, IL; 3300 DV ICP-OES
equipped with a cross-flow nebulizer, in addition to an ultrasonic nebulizer).
The procedure involved mineralization of the kidney cortex tissue with
a mixture of concentrated HNO3 and H2O2, followed by determination of
platinum in the extract, using inductively coupled plasma optical emission
spectrometry, at an emission wavelength of 265.945 nm.
Five-micrometer sections were prepared from left lung and left kidney
paraffin blocks and stained with hematoxylin and eosin. Staining for apoptosis
in the kidney sections has been performed using signal stain cleaved caspase-3
Immunohistochemical detection Kit (Cell Signaling Technology, Boston, MA).
This kit was used to detect the activation of caspase using avidin-biotin
immunoperoxidase method to detect intracellular caspase-3 protein. Staining
was performed on 5-lm paraffin sections from left kidney by standard
technique using rabbit anti-cleaved caspase-3 (clone Asp175, 1:50) (Vielhauer
et al., 2005). A known positive control sections for apoptosis were used. For
negative control, primary antibody was replaced with normal rabbit serum.
Blood gas measurements. Arterial blood gases were measured in separate
animals following the protocol described above. Immediately after the
anesthesia, arterial blood was obtained via cardiac puncture in EDTA. Analysis
was performed immediately after collection with a Roche blood gas analyzer
(Mannheim, Germany).
268 NEMMAR ET AL.
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
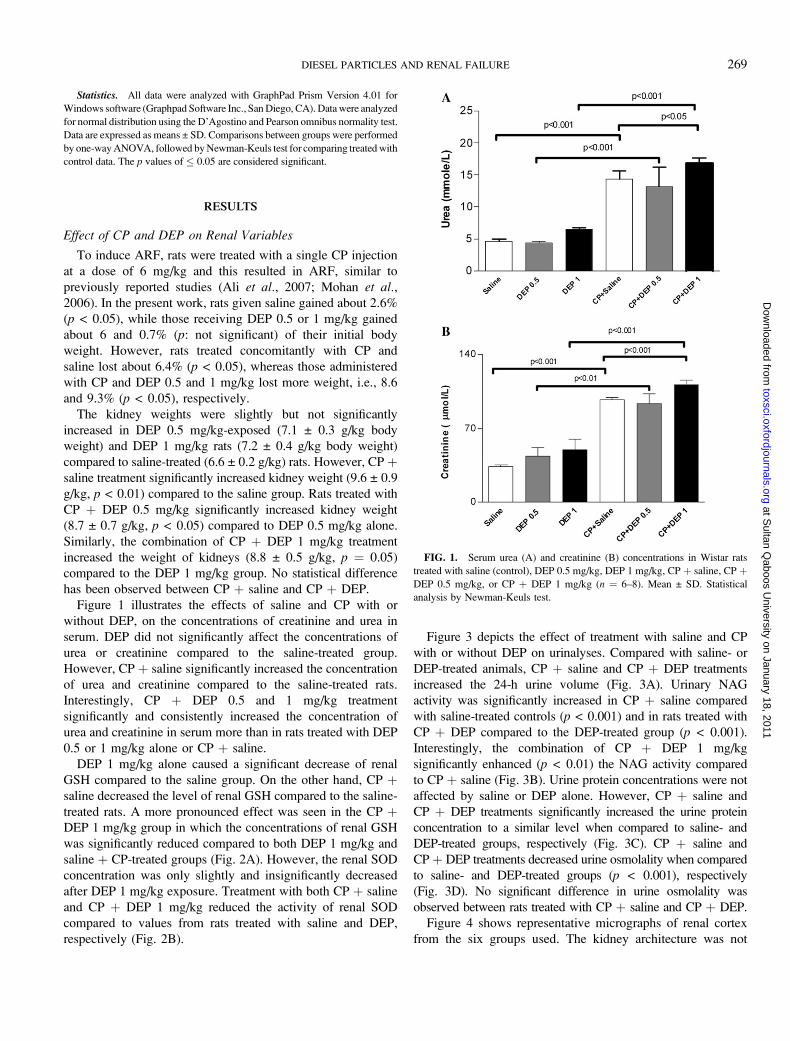
Statistics. All data were analyzed with GraphPad Prism Version 4.01 for
Windows software (Graphpad Software Inc., San Diego, CA). Data were analyzed
for normal distribution using the D’Agostino and Pearson omnibus normality test.
Data are expressed as means ± SD. Comparisons between groups were performed
by one-way ANOVA, followed by Newman-Keuls test for comparing treated with
control data. The p values of � 0.05 are considered significant.
RESULTS
Effect of CP and DEP on Renal Variables
To induce ARF, rats were treated with a single CP injection
at a dose of 6 mg/kg and this resulted in ARF, similar to
previously reported studies (Ali et al., 2007; Mohan et al.,2006). In the present work, rats given saline gained about 2.6%
(p < 0.05), while those receiving DEP 0.5 or 1 mg/kg gained
about 6 and 0.7% (p: not significant) of their initial body
weight. However, rats treated concomitantly with CP and
saline lost about 6.4% (p < 0.05), whereas those administered
with CP and DEP 0.5 and 1 mg/kg lost more weight, i.e., 8.6
and 9.3% (p < 0.05), respectively.
The kidney weights were slightly but not significantly
increased in DEP 0.5 mg/kg-exposed (7.1 ± 0.3 g/kg body
weight) and DEP 1 mg/kg rats (7.2 ± 0.4 g/kg body weight)
compared to saline-treated (6.6 ± 0.2 g/kg) rats. However, CP þsaline treatment significantly increased kidney weight (9.6 ± 0.9
g/kg, p < 0.01) compared to the saline group. Rats treated with
CP þ DEP 0.5 mg/kg significantly increased kidney weight
(8.7 ± 0.7 g/kg, p < 0.05) compared to DEP 0.5 mg/kg alone.
Similarly, the combination of CP þ DEP 1 mg/kg treatment
increased the weight of kidneys (8.8 ± 0.5 g/kg, p ¼ 0.05)
compared to the DEP 1 mg/kg group. No statistical difference
has been observed between CP þ saline and CP þ DEP.
Figure 1 illustrates the effects of saline and CP with or
without DEP, on the concentrations of creatinine and urea in
serum. DEP did not significantly affect the concentrations of
urea or creatinine compared to the saline-treated group.
However, CP þ saline significantly increased the concentration
of urea and creatinine compared to the saline-treated rats.
Interestingly, CP þ DEP 0.5 and 1 mg/kg treatment
significantly and consistently increased the concentration of
urea and creatinine in serum more than in rats treated with DEP
0.5 or 1 mg/kg alone or CP þ saline.
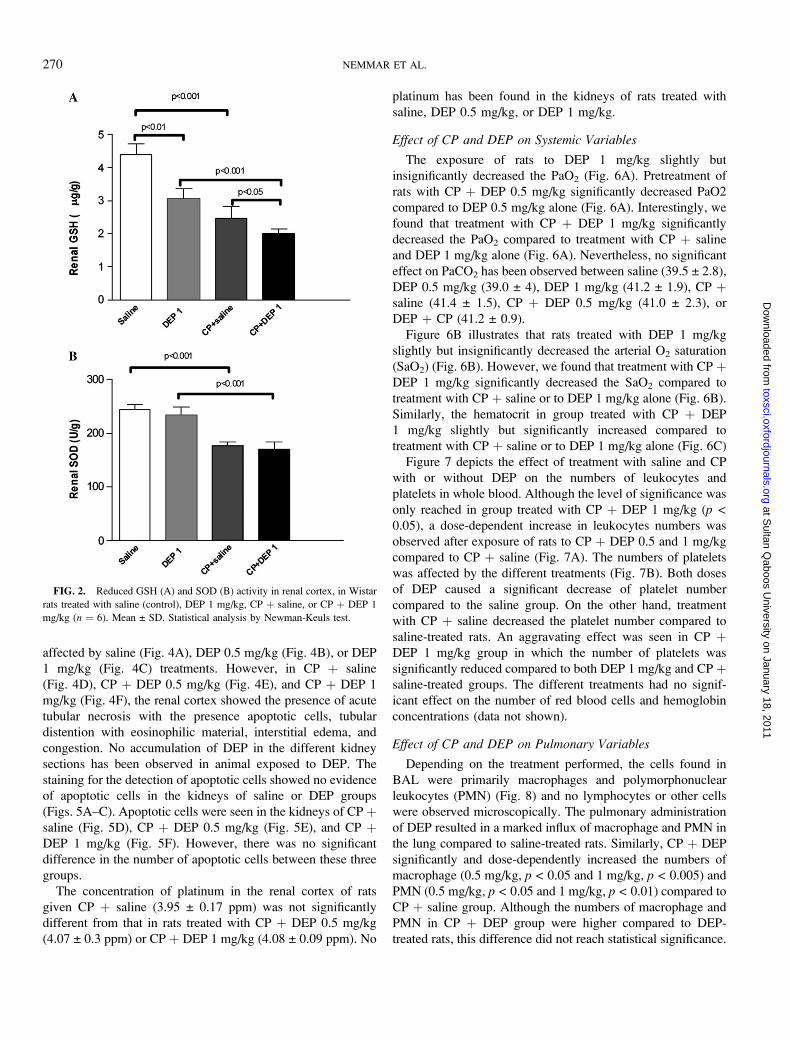
DEP 1 mg/kg alone caused a significant decrease of renal
GSH compared to the saline group. On the other hand, CP þsaline decreased the level of renal GSH compared to the saline-
treated rats. A more pronounced effect was seen in the CP þDEP 1 mg/kg group in which the concentrations of renal GSH
was significantly reduced compared to both DEP 1 mg/kg and
saline þ CP-treated groups (Fig. 2A). However, the renal SOD
concentration was only slightly and insignificantly decreased
after DEP 1 mg/kg exposure. Treatment with both CP þ saline
and CP þ DEP 1 mg/kg reduced the activity of renal SOD
compared to values from rats treated with saline and DEP,
respectively (Fig. 2B).
Figure 3 depicts the effect of treatment with saline and CP
with or without DEP on urinalyses. Compared with saline- or
DEP-treated animals, CP þ saline and CP þ DEP treatments
increased the 24-h urine volume (Fig. 3A). Urinary NAG
activity was significantly increased in CP þ saline compared
with saline-treated controls (p < 0.001) and in rats treated with
CP þ DEP compared to the DEP-treated group (p < 0.001).
Interestingly, the combination of CP þ DEP 1 mg/kg
significantly enhanced (p < 0.01) the NAG activity compared
to CP þ saline (Fig. 3B). Urine protein concentrations were not
affected by saline or DEP alone. However, CP þ saline and
CP þ DEP treatments significantly increased the urine protein
concentration to a similar level when compared to saline- and
DEP-treated groups, respectively (Fig. 3C). CP þ saline and
CP þ DEP treatments decreased urine osmolality when compared
to saline- and DEP-treated groups (p < 0.001), respectively
(Fig. 3D). No significant difference in urine osmolality was
observed between rats treated with CP þ saline and CP þ DEP.
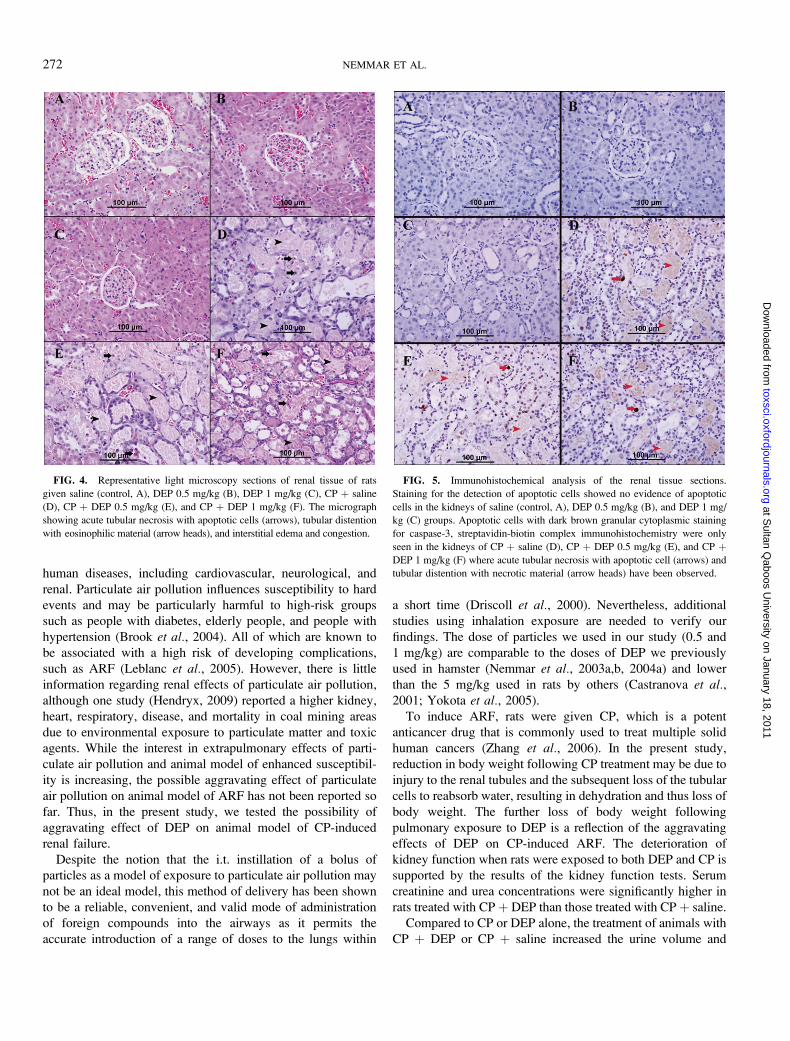
Figure 4 shows representative micrographs of renal cortex
from the six groups used. The kidney architecture was not
FIG. 1. Serum urea (A) and creatinine (B) concentrations in Wistar rats
treated with saline (control), DEP 0.5 mg/kg, DEP 1 mg/kg, CP þ saline, CP þDEP 0.5 mg/kg, or CP þ DEP 1 mg/kg (n ¼ 6–8). Mean ± SD. Statistical
analysis by Newman-Keuls test.
DIESEL PARTICLES AND RENAL FAILURE 269
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
affected by saline (Fig. 4A), DEP 0.5 mg/kg (Fig. 4B), or DEP
1 mg/kg (Fig. 4C) treatments. However, in CP þ saline
(Fig. 4D), CP þ DEP 0.5 mg/kg (Fig. 4E), and CP þ DEP 1
mg/kg (Fig. 4F), the renal cortex showed the presence of acute
tubular necrosis with the presence apoptotic cells, tubular
distention with eosinophilic material, interstitial edema, and
congestion. No accumulation of DEP in the different kidney
sections has been observed in animal exposed to DEP. The
staining for the detection of apoptotic cells showed no evidence
of apoptotic cells in the kidneys of saline or DEP groups
(Figs. 5A–C). Apoptotic cells were seen in the kidneys of CP þsaline (Fig. 5D), CP þ DEP 0.5 mg/kg (Fig. 5E), and CP þDEP 1 mg/kg (Fig. 5F). However, there was no significant
difference in the number of apoptotic cells between these three
groups.
The concentration of platinum in the renal cortex of rats
given CP þ saline (3.95 ± 0.17 ppm) was not significantly
different from that in rats treated with CP þ DEP 0.5 mg/kg
(4.07 ± 0.3 ppm) or CP þ DEP 1 mg/kg (4.08 ± 0.09 ppm). No
platinum has been found in the kidneys of rats treated with
saline, DEP 0.5 mg/kg, or DEP 1 mg/kg.
Effect of CP and DEP on Systemic Variables
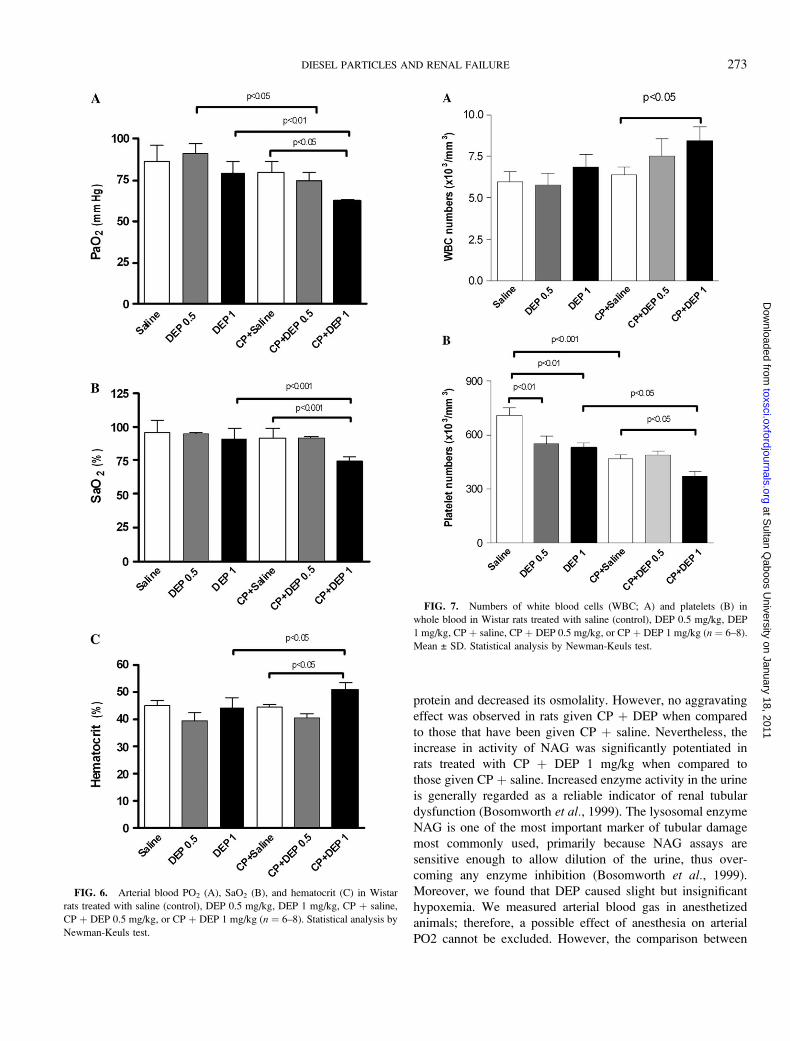
The exposure of rats to DEP 1 mg/kg slightly but
insignificantly decreased the PaO2 (Fig. 6A). Pretreatment of
rats with CP þ DEP 0.5 mg/kg significantly decreased PaO2
compared to DEP 0.5 mg/kg alone (Fig. 6A). Interestingly, we
found that treatment with CP þ DEP 1 mg/kg significantly
decreased the PaO2 compared to treatment with CP þ saline
and DEP 1 mg/kg alone (Fig. 6A). Nevertheless, no significant
effect on PaCO2 has been observed between saline (39.5 ± 2.8),
DEP 0.5 mg/kg (39.0 ± 4), DEP 1 mg/kg (41.2 ± 1.9), CP þsaline (41.4 ± 1.5), CP þ DEP 0.5 mg/kg (41.0 ± 2.3), or
DEP þ CP (41.2 ± 0.9).
Figure 6B illustrates that rats treated with DEP 1 mg/kg
slightly but insignificantly decreased the arterial O2 saturation
(SaO2) (Fig. 6B). However, we found that treatment with CP þDEP 1 mg/kg significantly decreased the SaO2 compared to
treatment with CP þ saline or to DEP 1 mg/kg alone (Fig. 6B).
Similarly, the hematocrit in group treated with CP þ DEP
1 mg/kg slightly but significantly increased compared to
treatment with CP þ saline or to DEP 1 mg/kg alone (Fig. 6C)
Figure 7 depicts the effect of treatment with saline and CP
with or without DEP on the numbers of leukocytes and
platelets in whole blood. Although the level of significance was
only reached in group treated with CP þ DEP 1 mg/kg (p <0.05), a dose-dependent increase in leukocytes numbers was
observed after exposure of rats to CP þ DEP 0.5 and 1 mg/kg
compared to CP þ saline (Fig. 7A). The numbers of platelets
was affected by the different treatments (Fig. 7B). Both doses
of DEP caused a significant decrease of platelet number
compared to the saline group. On the other hand, treatment
with CP þ saline decreased the platelet number compared to
saline-treated rats. An aggravating effect was seen in CP þDEP 1 mg/kg group in which the number of platelets was
significantly reduced compared to both DEP 1 mg/kg and CP þsaline-treated groups. The different treatments had no signif-
icant effect on the number of red blood cells and hemoglobin
concentrations (data not shown).
Effect of CP and DEP on Pulmonary Variables
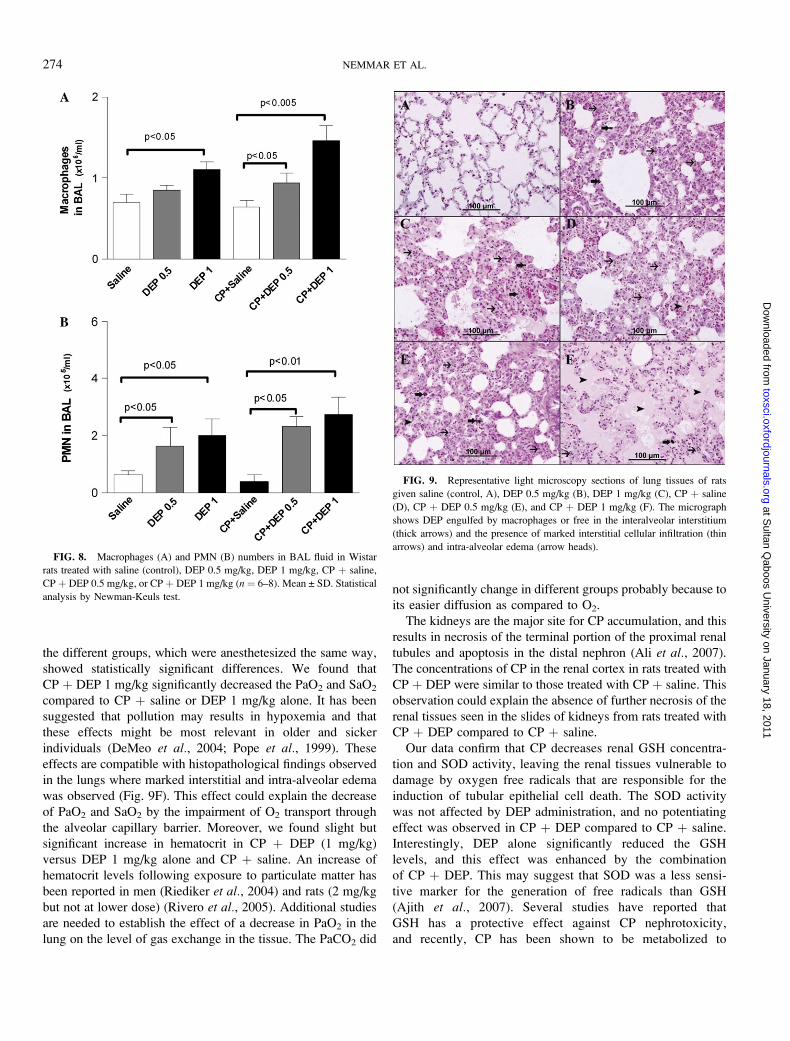
Depending on the treatment performed, the cells found in
BAL were primarily macrophages and polymorphonuclear
leukocytes (PMN) (Fig. 8) and no lymphocytes or other cells
were observed microscopically. The pulmonary administration
of DEP resulted in a marked influx of macrophage and PMN in
the lung compared to saline-treated rats. Similarly, CP þ DEP
significantly and dose-dependently increased the numbers of
macrophage (0.5 mg/kg, p < 0.05 and 1 mg/kg, p < 0.005) and
PMN (0.5 mg/kg, p < 0.05 and 1 mg/kg, p < 0.01) compared to
CP þ saline group. Although the numbers of macrophage and
PMN in CP þ DEP group were higher compared to DEP-
treated rats, this difference did not reach statistical significance.
FIG. 2. Reduced GSH (A) and SOD (B) activity in renal cortex, in Wistar
rats treated with saline (control), DEP 1 mg/kg, CP þ saline, or CP þ DEP 1
mg/kg (n ¼ 6). Mean ± SD. Statistical analysis by Newman-Keuls test.
270 NEMMAR ET AL.
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
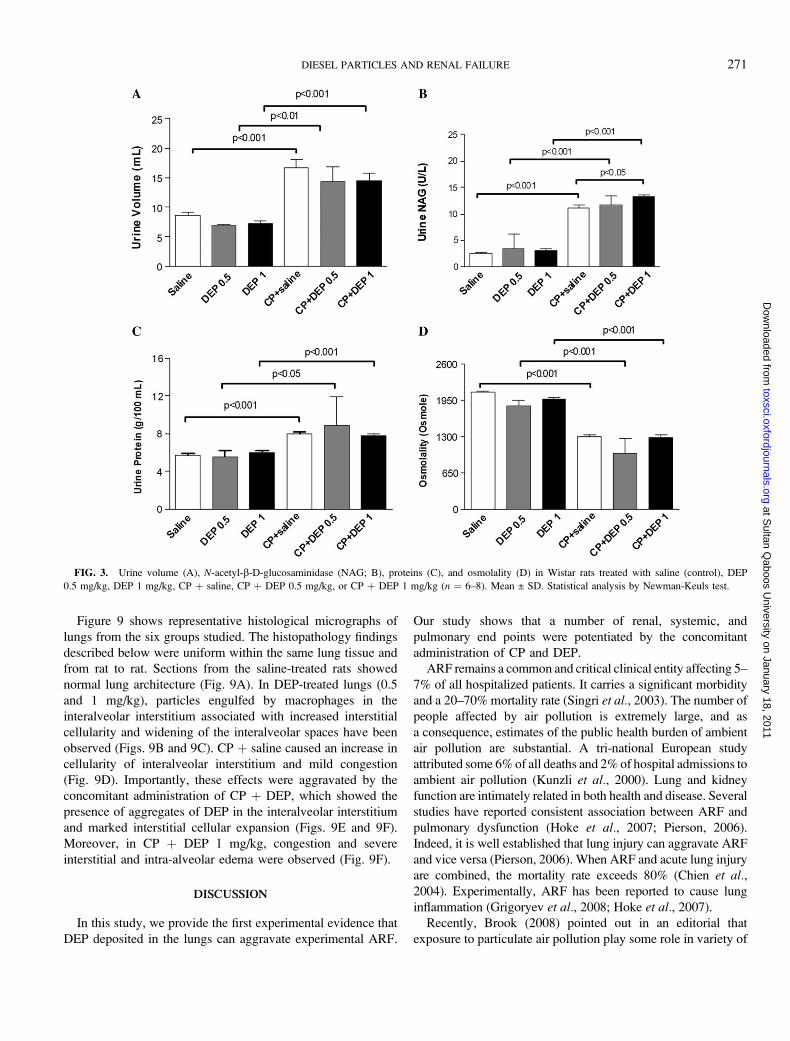
Figure 9 shows representative histological micrographs of
lungs from the six groups studied. The histopathology findings
described below were uniform within the same lung tissue and
from rat to rat. Sections from the saline-treated rats showed
normal lung architecture (Fig. 9A). In DEP-treated lungs (0.5
and 1 mg/kg), particles engulfed by macrophages in the
interalveolar interstitium associated with increased interstitial
cellularity and widening of the interalveolar spaces have been
observed (Figs. 9B and 9C). CP þ saline caused an increase in
cellularity of interalveolar interstitium and mild congestion
(Fig. 9D). Importantly, these effects were aggravated by the
concomitant administration of CP þ DEP, which showed the
presence of aggregates of DEP in the interalveolar interstitium
and marked interstitial cellular expansion (Figs. 9E and 9F).
Moreover, in CP þ DEP 1 mg/kg, congestion and severe
interstitial and intra-alveolar edema were observed (Fig. 9F).
DISCUSSION
In this study, we provide the first experimental evidence that
DEP deposited in the lungs can aggravate experimental ARF.
Our study shows that a number of renal, systemic, and
pulmonary end points were potentiated by the concomitant
administration of CP and DEP.
ARF remains a common and critical clinical entity affecting 5–
7% of all hospitalized patients. It carries a significant morbidity
and a 20–70% mortality rate (Singri et al., 2003). The number of
people affected by air pollution is extremely large, and as
a consequence, estimates of the public health burden of ambient
air pollution are substantial. A tri-national European study
attributed some 6% of all deaths and 2% of hospital admissions to
ambient air pollution (Kunzli et al., 2000). Lung and kidney
function are intimately related in both health and disease. Several
studies have reported consistent association between ARF and
pulmonary dysfunction (Hoke et al., 2007; Pierson, 2006).
Indeed, it is well established that lung injury can aggravate ARF
and vice versa (Pierson, 2006). When ARF and acute lung injury
are combined, the mortality rate exceeds 80% (Chien et al.,2004). Experimentally, ARF has been reported to cause lung
inflammation (Grigoryev et al., 2008; Hoke et al., 2007).
Recently, Brook (2008) pointed out in an editorial that
exposure to particulate air pollution play some role in variety of
FIG. 3. Urine volume (A), N-acetyl-b-D-glucosaminidase (NAG; B), proteins (C), and osmolality (D) in Wistar rats treated with saline (control), DEP
0.5 mg/kg, DEP 1 mg/kg, CP þ saline, CP þ DEP 0.5 mg/kg, or CP þ DEP 1 mg/kg (n ¼ 6–8). Mean ± SD. Statistical analysis by Newman-Keuls test.
DIESEL PARTICLES AND RENAL FAILURE 271
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
human diseases, including cardiovascular, neurological, and
renal. Particulate air pollution influences susceptibility to hard
events and may be particularly harmful to high-risk groups
such as people with diabetes, elderly people, and people with
hypertension (Brook et al., 2004). All of which are known to
be associated with a high risk of developing complications,
such as ARF (Leblanc et al., 2005). However, there is little
information regarding renal effects of particulate air pollution,
although one study (Hendryx, 2009) reported a higher kidney,
heart, respiratory, disease, and mortality in coal mining areas
due to environmental exposure to particulate matter and toxic
agents. While the interest in extrapulmonary effects of parti-
culate air pollution and animal model of enhanced susceptibil-
ity is increasing, the possible aggravating effect of particulate
air pollution on animal model of ARF has not been reported so
far. Thus, in the present study, we tested the possibility of
aggravating effect of DEP on animal model of CP-induced
renal failure.
Despite the notion that the i.t. instillation of a bolus of
particles as a model of exposure to particulate air pollution may
not be an ideal model, this method of delivery has been shown
to be a reliable, convenient, and valid mode of administration
of foreign compounds into the airways as it permits the
accurate introduction of a range of doses to the lungs within
a short time (Driscoll et al., 2000). Nevertheless, additional
studies using inhalation exposure are needed to verify our
findings. The dose of particles we used in our study (0.5 and
1 mg/kg) are comparable to the doses of DEP we previously
used in hamster (Nemmar et al., 2003a,b, 2004a) and lower
than the 5 mg/kg used in rats by others (Castranova et al.,2001; Yokota et al., 2005).
To induce ARF, rats were given CP, which is a potent
anticancer drug that is commonly used to treat multiple solid
human cancers (Zhang et al., 2006). In the present study,
reduction in body weight following CP treatment may be due to
injury to the renal tubules and the subsequent loss of the tubular
cells to reabsorb water, resulting in dehydration and thus loss of
body weight. The further loss of body weight following
pulmonary exposure to DEP is a reflection of the aggravating
effects of DEP on CP-induced ARF. The deterioration of
kidney function when rats were exposed to both DEP and CP is
supported by the results of the kidney function tests. Serum
creatinine and urea concentrations were significantly higher in
rats treated with CP þ DEP than those treated with CP þ saline.
Compared to CP or DEP alone, the treatment of animals with
CP þ DEP or CP þ saline increased the urine volume and
FIG. 4. Representative light microscopy sections of renal tissue of rats
given saline (control, A), DEP 0.5 mg/kg (B), DEP 1 mg/kg (C), CP þ saline
(D), CP þ DEP 0.5 mg/kg (E), and CP þ DEP 1 mg/kg (F). The micrograph
showing acute tubular necrosis with apoptotic cells (arrows), tubular distention
with eosinophilic material (arrow heads), and interstitial edema and congestion.
FIG. 5. Immunohistochemical analysis of the renal tissue sections.
Staining for the detection of apoptotic cells showed no evidence of apoptotic
cells in the kidneys of saline (control, A), DEP 0.5 mg/kg (B), and DEP 1 mg/
kg (C) groups. Apoptotic cells with dark brown granular cytoplasmic staining
for caspase-3, streptavidin-biotin complex immunohistochemistry were only
seen in the kidneys of CP þ saline (D), CP þ DEP 0.5 mg/kg (E), and CP þDEP 1 mg/kg (F) where acute tubular necrosis with apoptotic cell (arrows) and
tubular distention with necrotic material (arrow heads) have been observed.
272 NEMMAR ET AL.
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
protein and decreased its osmolality. However, no aggravating
effect was observed in rats given CP þ DEP when compared
to those that have been given CP þ saline. Nevertheless, the
increase in activity of NAG was significantly potentiated in
rats treated with CP þ DEP 1 mg/kg when compared to
those given CP þ saline. Increased enzyme activity in the urine
is generally regarded as a reliable indicator of renal tubular
dysfunction (Bosomworth et al., 1999). The lysosomal enzyme
NAG is one of the most important marker of tubular damage
most commonly used, primarily because NAG assays are
sensitive enough to allow dilution of the urine, thus over-
coming any enzyme inhibition (Bosomworth et al., 1999).
Moreover, we found that DEP caused slight but insignificant
hypoxemia. We measured arterial blood gas in anesthetized
animals; therefore, a possible effect of anesthesia on arterial
PO2 cannot be excluded. However, the comparison between
FIG. 6. Arterial blood PO2 (A), SaO2 (B), and hematocrit (C) in Wistar
rats treated with saline (control), DEP 0.5 mg/kg, DEP 1 mg/kg, CP þ saline,
CP þ DEP 0.5 mg/kg, or CP þ DEP 1 mg/kg (n ¼ 6–8). Statistical analysis by
Newman-Keuls test.
FIG. 7. Numbers of white blood cells (WBC; A) and platelets (B) in
whole blood in Wistar rats treated with saline (control), DEP 0.5 mg/kg, DEP
1 mg/kg, CP þ saline, CP þ DEP 0.5 mg/kg, or CP þ DEP 1 mg/kg (n ¼ 6–8).
Mean ± SD. Statistical analysis by Newman-Keuls test.
DIESEL PARTICLES AND RENAL FAILURE 273
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from
the different groups, which were anesthetesized the same way,
showed statistically significant differences. We found that
CP þ DEP 1 mg/kg significantly decreased the PaO2 and SaO2
compared to CP þ saline or DEP 1 mg/kg alone. It has been
suggested that pollution may results in hypoxemia and that
these effects might be most relevant in older and sicker
individuals (DeMeo et al., 2004; Pope et al., 1999). These
effects are compatible with histopathological findings observed
in the lungs where marked interstitial and intra-alveolar edema
was observed (Fig. 9F). This effect could explain the decrease
of PaO2 and SaO2 by the impairment of O2 transport through
the alveolar capillary barrier. Moreover, we found slight but
significant increase in hematocrit in CP þ DEP (1 mg/kg)
versus DEP 1 mg/kg alone and CP þ saline. An increase of
hematocrit levels following exposure to particulate matter has
been reported in men (Riediker et al., 2004) and rats (2 mg/kg
but not at lower dose) (Rivero et al., 2005). Additional studies
are needed to establish the effect of a decrease in PaO2 in the
lung on the level of gas exchange in the tissue. The PaCO2 did
not significantly change in different groups probably because to
its easier diffusion as compared to O2.
The kidneys are the major site for CP accumulation, and this
results in necrosis of the terminal portion of the proximal renal
tubules and apoptosis in the distal nephron (Ali et al., 2007).
The concentrations of CP in the renal cortex in rats treated with
CP þ DEP were similar to those treated with CP þ saline. This
observation could explain the absence of further necrosis of the
renal tissues seen in the slides of kidneys from rats treated with
CP þ DEP compared to CP þ saline.
Our data confirm that CP decreases renal GSH concentra-
tion and SOD activity, leaving the renal tissues vulnerable to
damage by oxygen free radicals that are responsible for the
induction of tubular epithelial cell death. The SOD activity
was not affected by DEP administration, and no potentiating
effect was observed in CP þ DEP compared to CP þ saline.
Interestingly, DEP alone significantly reduced the GSH
levels, and this effect was enhanced by the combination
of CP þ DEP. This may suggest that SOD was a less sensi-
tive marker for the generation of free radicals than GSH
(Ajith et al., 2007). Several studies have reported that
GSH has a protective effect against CP nephrotoxicity,
and recently, CP has been shown to be metabolized to
FIG. 8. Macrophages (A) and PMN (B) numbers in BAL fluid in Wistar
rats treated with saline (control), DEP 0.5 mg/kg, DEP 1 mg/kg, CP þ saline,
CP þ DEP 0.5 mg/kg, or CP þ DEP 1 mg/kg (n ¼ 6–8). Mean ± SD. Statistical
analysis by Newman-Keuls test.
FIG. 9. Representative light microscopy sections of lung tissues of rats
given saline (control, A), DEP 0.5 mg/kg (B), DEP 1 mg/kg (C), CP þ saline
(D), CP þ DEP 0.5 mg/kg (E), and CP þ DEP 1 mg/kg (F). The micrograph
shows DEP engulfed by macrophages or free in the interalveolar interstitium
(thick arrows) and the presence of marked interstitial cellular infiltration (thin
arrows) and intra-alveolar edema (arrow heads).
274 NEMMAR ET AL.
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from

a nephrotoxicant through a GSH-conjugate intermediate
(Hanigan and Devarajan, 2003; Zhang et al., 2006). Oxidative
stress in kidney plays an important role in CP-induced renal
damage, and several antioxidants and thiol compounds have
been shown to protect against CP nephrotoxicity (Ali and Al
Moundhri, 2006; Ali et al., 2007).
Studies in humans and in animal models have demonstrated
that ARF has a significant effect on the function of extrarenal
organs (Grigoryev et al., 2008; Hoke et al., 2007). In-
flammation is a major component of the initiation and
exacerbation of kidney injury, and local inflammation of
kidney tissues could be a source of the development of
inflammation and injury in extrarenal organs (Grigoryev et al.,2008). Therefore, in the present study, we sought to count the
number of circulatory cells, as a marker of systemic
inflammation. Although not statistically significant, we found
an increase of leukocyte numbers in DEP-treated group (1 mg/
kg) compared to saline group. We made a similar observation
in mice exposed to DEP (Nemmar et al., 2009). The fact that
CP þ DEP dose-dependently and significantly (at 1 mg/kg)
increased leukocyte numbers compared to CP þ saline
suggests that ARF exacerbates systemic inflammation. This
finding is in agreement with the concept that particulate air
pollution effects are aggravated in people with preexisting
diseases (Brook et al., 2004). We have shown that the
systemic administration of DEP increases the number of
leukocytes and decreases both the number of erythrocytes and
the hemoglobin concentration (Nemmar and Inuwa, 2008). In
contrast, DEP alone caused a significant decrease of platelet
number compared to the saline-treated group. Moreover,
a more pronounced reduction in platelet number was seen in
the CP þ DEP group, suggesting the occurrence of platelet
aggregation in vivo. These results are in agreement with
animal and human studies, which have reported a decrease in
platelet number following exposure to particulate air pollution
(Nemmar et al., 2008; Ruckerl et al., 2007). Moreover, we
have recently demonstrated a prothrombotic effect of DEP in
hamsters as well as an activation of platelets (Nemmar et al.,2003a,b, 2004a). It remains to be established whether these
effects are aggravated by direct effect of DEP that have
presumably translocated into blood and/or through pulmonary
release of proinflammatory cytokines, such as interleukin (IL)-
6, IL-8, or tumor necrosis factor-a. The fact that DEP could
not be found in kidney sections does not totally exclude the
direct effects of DEP on the kidneys. Recently, L’azou et al.(2008) reported that carbon black nanoparticles exert a cyto-
toxic effect on renal cells in vitro and suggested involvement
of particle internalization as well as activation of intracellular
mechanisms that might include generation of reactive oxygen
species.
Our study showed that exposure to DEP caused lung
inflammation characterized by an increase in the number of
macrophage and PMN in BAL. This finding corroborate with
our previous studies in hamsters, which showed influx of
inflammatory cells in BAL after pulmonary exposure to DEP
(0.05–5 mg/kg) (Nemmar et al., 2003a,b, 2004a). Moreover,
Rao et al. (2005) reported in rats a dose-dependent increase in
PMN in BAL 1, 7, and 30 days following the i.t. instillation
of three concentrations of DEP (5, 35, and 50 mg/kg body
weight). The histologic findings revealed the presence of
increased interstitial cellularity and widening of the inter-
alveolar spaces. These findings are in agreement with
previous studies, which reported the direct effect of DEP
on lung inflammation (Nemmar et al., 2004a). CP pre-
treatment did not affect the number of BAL macrophages
and PMN, but histologic examination showed an increase in
cellularity of the interalveolar interstitium and mild conges-
tion. A major finding of the present work is that the combined
administration of DEP and CP aggravated pulmonary
inflammation and caused interstitial congestion and severe
interstitial and intra-alveolar edema (observed at 1 mg/kg).
These results confirm earlier studies that showed that ARF
resulting from either ischemia or bilateral nephrectomy
causes lung inflammation (Grigoryev et al., 2008; Hoke
et al., 2007) and thus suggest that inhaled particulate air
pollution can potentiate ARF.
DEP consists of an elemental carbonaceous core onto which
various organic compounds are adsorbed. Consequently,
additional studies are needed to establish that constituents of
DEP are responsible for the observed effects and the potential
effect of cytochrome P450 activity in lung, liver, and kidney on
circulating levels of chemical constituents associated with
particles on kidney toxicity (Pratibha et al., 2006).
Our data provide novel evidence that pulmonary deposition
of DEP potentiates the renal, systemic, and pulmonary effects
of CP-induced ARF and highlight the importance of environ-
mental factors such as particulate air pollution in aggravating
ARF. Our findings provide a plausible explanation for both the
extrarenal effect of ARF and the extrapulmonary effects of
particulate air pollution.
FUNDING
Faculty of Medicine and Health Sciences grant (NP/09/04);
United Arab Emirates individual grant (02-05-8-11/09).
ACKNOWLEDGMENTS
We thank Mr M. H. Mansour (College of Agriculture, Sultan
Qaboos University), Ms M. Sudhadevi (Department of
Pathology, Faculty of Medicine and Health Sciences, UAE
University), and Mr S. Dhanasekaran (Department of Physi-
ology, Faculty of Medicine and Health Sciences, UAE
University) for their technical assistances. We are grateful to
Professor David Cook, Ontario, Canada, for his critical reading
of the manuscript.
DIESEL PARTICLES AND RENAL FAILURE 275
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from

REFERENCES
Ajith, T. A., Nivitha, V., and Usha, S. (2007). Zingiber officinale Roscoe alone
and in combination with alpha-tocopherol protect the kidney against
cisplatin-induced acute renal failure. Food Chem. Toxicol. 45, 921–927.
Ali, B. H., and Al Moundhri, M. S. (2006). Agents ameliorating or augmenting
the nephrotoxicity of cisplatin and other platinum compounds: a review of
some recent research. Food Chem. Toxicol. 44, 1173–1183.
Ali, B. H., Al Moundhri, M., Eldin, M. T., Nemmar, A., Al Siyabi, S., and
Annamalai, K. (2008). Amelioration of cisplatin-induced nephrotoxicity in
rats by tetramethylpyrazine, a major constituent of the Chinese herb
Ligusticum wallichi. Exp. Biol. Med. (Maywood) 233, 891–896.
Ali, B. H., Al Moundhri, M. S., Tag, E. M., Nemmar, A., and Tanira, M. O.
(2007). The ameliorative effect of cysteine prodrug L-2-oxothiazolidine-4-
carboxylic acid on cisplatin-induced nephrotoxicity in rats. Fundam. Clin.
Pharmacol. 21, 547–553.
Atkinson, R. W., Anderson, H. R., Sunyer, J., Ayres, J., Baccini, M.,
Vonk, J. M., Boumghar, A., Forastiere, F., Forsberg, B., Touloumi, G., et al.
(2001). Acute effects of particulate air pollution on respiratory admissions:
results from APHEA 2 project. Air pollution and health: a European
approach. Am. J. Respir. Crit. Care Med. 164, 1860–1866.
Bosomworth, M. P., Aparicio, S. R., and Hay, A. W. (1999). Urine N-acetyl-
beta-D-glucosaminidase—a marker of tubular damage? Nephrol. Dial.
Transplant. 14, 620–626.
Brook, R. D. (2008). Potential health risks of air pollution beyond triggering
acute cardiopulmonary events. JAMA 299, 2194–2196.
Brook, R. D., Franklin, B., Cascio, W., Hong, Y. L., Howard, G., Lipsett, M.,
Luepker, R., Mittleman, M., Samet, J., Smith, S. C., et al. (2004). Air
pollution and cardiovascular disease—a statement for healthcare professio-
nals from the expert panel on population and prevention science of the
American Heart Association. Circulation 109, 2655–2671.
Castranova, V., Ma, J. Y., Yang, H. M., Antonini, J. M., Butterworth, L.,
Barger, M. W., Roberts, J., and Ma, J. K. (2001). Effect of exposure to diesel
exhaust particles on the susceptibility of the lung to infection. Environ.
Health Perspect. 109(Suppl. 4), 609–612.
Chien, C. C., King, L. S., and Rabb, H. (2004). Mechanisms underlying
combined acute renal failure and acute lung injury in the intensive care unit.
Contrib. Nephrol. 144, 53–62.
DeMeo, D. L., Zanobetti, A., Litonjua, A. A., Coull, B. A., Schwartz, J., and
Gold, D. R. (2004). Ambient air pollution and oxygen saturation. Am.
J. Respir. Crit. Care Med. 170, 383–387.
Driscoll, K. E., Costa, D. L., Hatch, G., Henderson, R., Oberdorster, G.,
Salem, H., and Schlesinger, R. B. (2000). Intratracheal instillation as an
exposure technique for the evaluation of respiratory tract toxicity: uses and
limitations. Toxicol. Sci. 55, 24–35.
Grigoryev, D. N., Liu, M., Hassoun, H. T., Cheadle, C., Barnes, K. C., and
Rabb, H. (2008). The local and systemic inflammatory transcriptome after
acute kidney injury. J. Am. Soc. Nephrol. 19, 547–558.
Hanigan, M. H., and Devarajan, P. (2003). Cisplatin nephrotoxicity: molecular
mechanisms. Cancer Ther. 1, 47–61.
Hendryx, M. (2009). Mortality from heart, respiratory, and kidney disease in
coal mining areas of Appalachia. Int. Arch. Occup. Environ. Health 82,
243–249.
Hoke, T. S., Douglas, I. S., Klein, C. L., He, Z., Fang, W., Thurman, J. M.,
Tao, Y., Dursun, B., Voelkel, N. F., Edelstein, C. L., et al. (2007). Acute
renal failure after bilateral nephrectomy is associated with cytokine-mediated
pulmonary injury. J. Am. Soc. Nephrol. 18, 155–164.
Hoste, E. A., and Schurgers, M. (2008). Epidemiology of acute kidney injury:
how big is the problem? Crit. Care Med. 36, S146–S151.
Inoue, K., Takano, H., Sakurai, M., Oda, T., Tamura, H., Yanagisawa, R.,
Shimada, A., and Yoshikawa, T. (2006). Pulmonary exposure to diesel
exhaust particles enhances coagulatory disturbance with endothelial damage
and systemic inflammation related to lung inflammation. Exp. Biol. Med.
(Maywood) 231, 1626–1632.
Inoue, K., Takano, H., Yanagisawa, R., Ichinose, T., Shimada, A., and
Yoshikawa, T. (2005). Pulmonary exposure to diesel exhaust particles
induces airway inflammation and cytokine expression in NC/Nga mice.
Arch. Toxicol. 79, 595–599.
Kunzli, N., Jerrett, M., Mack, W. J., Beckerman, B., LaBree, L., Gilliland, F.,
Thomas, D., Peters, J., and Hodis, H. N. (2005). Ambient air pollution and
atherosclerosis in Los Angeles. Environ. Health Perspect. 113, 201–206.
Kunzli, N., Kaiser, R., Medina, S., Studnicka, M., Chanel, O., Filliger, P.,
Herry, M., Horak, F., Jr.., Puybonnieux-Texier, V., Quenel, P., et al. (2000).
Public-health impact of outdoor and traffic-related air pollution: a European
assessment. Lancet 356, 795–801.
L’azou, B., Jorly, J., On, D., Sellier, E., Moisan, F., Fleury-Feith, J.,
Cambar, J., Brochard, P., and Ohayon-Courtes, C. (2008). In vitro effects of
nanoparticles on renal cells. Part. Fibre. Toxicol. 5, 22.
Leblanc, M., Kellum, J. A., Gibney, R. T., Lieberthal, W., Tumlin, J., and
Mehta, R. (2005). Risk factors for acute renal failure: inherent and
modifiable risks. Curr. Opin. Crit. Care 11, 533–536.
Mills, N. L., Tornqvist, H., Gonzalez, M. C., Vink, E., Robinson, S. D.,
Soderberg, S., Boon, N. A., Donaldson, K., Sandstrom, T., Blomberg, A.,
et al. (2007). Ischemic and thrombotic effects of dilute diesel-exhaust
inhalation in men with coronary heart disease. N. Engl. J. Med. 357,
1075–1082.
Mills, N. L., Tornqvist, H., Robinson, S. D., Gonzalez, M., Darnley, K.,
MacNee, W., Boon, N. A., Donaldson, K., Blomberg, A., Sandstrom, T.,
et al. (2005). Diesel exhaust inhalation causes vascular dysfunction and
impaired endogenous fibrinolysis. Circulation 112, 3930–3936.
Mohan, I. K., Khan, M., Shobha, J. C., Naidu, M. U., Prayag, A.,
Kuppusamy, P., and Kutala, V. K. (2006). Protection against cisplatin-
induced nephrotoxicity by Spirulina in rats. Cancer Chemother. Pharmacol.
58, 802–808.
Nemmar, A., Al Maskari, S., Ali, B. H., and Al Amri, I. S. (2007).
Cardiovascular and lung inflammatory effects induced by systemically
administered diesel exhaust particles in rats. Am. J. Physiol. Lung Cell. Mol.
Physiol. 292, L664–L670.
Nemmar, A., Al Salam, S., Dhanasekaran, S., Sudhadevi, M., and Ali, B. H.
(2009). Pulmonary exposure to diesel exhaust particles promotes cerebral
microvessel thrombosis: protective effect of a cysteine prodrug l-2-
oxothiazolidine-4-carboxylic acid. Toxicology 263, 84–92.
Nemmar, A., Hoet, P. H., Dinsdale, D., Vermylen, J., Hoylaerts, M. F., and
Nemery, B. (2003a). Diesel exhaust particles in lung acutely enhance
experimental peripheral thrombosis. Circulation 107, 1202–1208.
Nemmar, A., Hoet, P. H. M., Vermylen, J., Nemery, B., and Hoylaerts, M. F.
(2004a). Pharmacological stabilization of mast cells abrogates late
thrombotic events induced by diesel exhaust particles in hamsters.
Circulation 110, 1670–1677.
Nemmar, A., Hoylaerts, M. F., Hoet, P. H., and Nemery, B. (2004b). Possible
mechanisms of the cardiovascular effects of inhaled particles: systemic
translocation and prothrombotic effects. Toxicol. Lett. 149, 243–253.
Nemmar, A., and Inuwa, I. M. (2008). Diesel exhaust particles in blood trigger
systemic and pulmonary morphological alterations. Toxicol. Lett. 176,
20–30.
Nemmar, A., Melghit, K., and Ali, B. H. (2008). The acute proinflammatory
and prothrombotic effects of pulmonary exposure to rutile TiO2 nanorods in
rats. Exp. Biol. Med. (Maywood) 233, 610–619.
Nemmar, A., Nemery, B., Hoet, P. H. M., Vermylen, J., and Hoylaerts, M. F.
(2003b). Pulmonary inflammation and thrombogenicity caused by diesel
particles in hamsters—role of histamine. Am. J. Respir. Crit. Care Med. 168,
1366–1372.
276 NEMMAR ET AL.
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from

Oberdorster, G., Oberdorster, E., and Oberdorster, J. (2005). Nanotoxicology:
an emerging discipline evolving from studies of ultrafine particles. Environ.
Health Perspect. 113, 823–839.
Pannu, N., Klarenbach, S., Wiebe, N., Manns, B., and Tonelli, M. (2008).
Renal replacement therapy in patients with acute renal failure: a systematic
review. J. A. M. A. 299, 793–805.
Pekkanen, J., Peters, A., Hoek, G., Tiittanen, P., Brunekreef, B., de Hartog, J.,
Heinrich, J., Ibald-Mulli, A., Kreyling, W. G., Lanki, T., et al. (2002).
Particulate air pollution and risk of ST-segment depression during repeated
submaximal exercise tests among subjects with coronary heart disease: the
Exposure and Risk Assessment for Fine and Ultrafine Particles in Ambient
Air (ULTRA) study. Circulation 106, 933–938.
Peters, A., Dockery, D. W., Muller, J. E., and Mittleman, M. A. (2001).
Increased particulate air pollution and the triggering of myocardial infarction.
Circulation 103, 2810–2815.
Peters, A., Veronesi, B., Calderon-Garciduenas, L., Gehr, P., Chen, L. C.,
Geiser, M., Reed, W., Rothen-Rutishauser, B., Schurch, S., and Schulz, H.
(2006). Translocation and potential neurological effects of fine and ultrafine
particles a critical update. Part. Fibre. Toxicol. 3, 13.
Pierson, D. J. (2006). Respiratory considerations in the patient with renal
failure. Respir. Care 51, 413–422.
Pope, C. A., III., Burnett, R. T., Thun, M. J., Calle, E. E., Krewski, D., Ito, K.,
and Thurston, G. D. (2002). Lung cancer, cardiopulmonary mortality, and
long-term exposure to fine particulate air pollution. JAMA 287, 1132–1141.
Pope, C. A., III., Schwartz, J., and Ransom, M. R. (1992). Daily mortality and
PM10 pollution in Utah Valley. Arch. Environ. Health 47, 211–217.
Pope, C. A., III., Verrier, R. L., Lovett, E. G., Larson, A. C., Raizenne, M. E.,
Kanner, R. E., Schwartz, J., Villegas, G. M., Gold, D. R., and
Dockery, D. W. (1999). Heart rate variability associated with particulate
air pollution. Am. Heart J. 138, 890–899.
Pratibha, R., Sameer, R., Rataboli, P. V., Bhiwgade, D. A., and Dhume, C. Y.
(2006). Enzymatic studies of cisplatin induced oxidative stress in hepatic
tissue of rats. Eur. J. Pharmacol. 532, 290–293.
Rao, K. M., Ma, J. Y., Meighan, T., Barger, M. W., Pack, D., and
Vallyathan, V. (2005). Time course of gene expression of inflammatory
mediators in rat lung after diesel exhaust particle exposure. Environ. Health
Perspect. 113, 612–617.
Riediker, M., Cascio, W. E., Griggs, T. R., Herbst, M. C., Bromberg, P. A.,
Neas, L., Williams, R. W., and Devlin, R. B. (2004). Particulate matter
exposure in cars is associated with cardiovascular effects in healthy young
men. Am. J. Respir. Crit. Care Med. 169, 934–940.
Rivero, D. H., Soares, S. R., Lorenzi-Filho, G., Saiki, M., Godleski, J. J.,
Antonangelo, L., Dolhnikoff, M., and Saldiva, P. H. (2005). Acute
cardiopulmonary alterations induced by fine particulate matter of Sao Paulo.
Brazil. Toxicol. Sci. 85, 898–905.
Ruckerl, R., Phipps, R. P., Schneider, A., Frampton, M., Cyrys, J.,
Oberdorster, G., Wichmann, H. E., and Peters, A. (2007). Ultrafine particles
and platelet activation in patients with coronary heart disease—results from
a prospective panel study. Part. Fibre. Toxicol. 4, 1.
Salvi, S., Blomberg, A., Rudell, B., Kelly, F., Sandstrom, T., Holgate, S. T.,
and Frew, A. (1999). Acute inflammatory responses in the airways and
peripheral blood after short-term exposure to diesel exhaust in healthy
human volunteers. Am. J. Respir. Crit. Care Med. 159, 702–709.
Singri, N., Ahya, S. N., and Levin, M. L. (2003). Acute renal failure. JAMA
289, 747–751.
Vermylen, J., Nemmar, A., Nemery, B., and Hoylaerts, M. F. (2005). Ambient
air pollution and acute myocardial infarction. J. Thromb. Haemost. 3,
1955–1961.
Vielhauer, V., Stavrakis, G., and Mayadas, T. N. (2005). Renal cell-expressed
TNF receptor 2, not receptor 1, is essential for the development of
glomerulonephritis. J. Clin. Invest. 115, 1199–1209.
Yokota, S., Seki, T., Furuya, M., and Ohara, N. (2005). Acute functional
enhancement of circulatory neutrophils after intratracheal instillation with
diesel exhaust particles in rats. Inhal. Toxicol. 17, 671–679.
Zhang, L., Cooper, A. J., Krasnikov, B. F., Xu, H., Bubber, P.,
Pinto, J. T., Gibson, G. E., and Hanigan, M. H. (2006). Cisplatin-
induced toxicity is associated with platinum deposition in mouse kidney
mitochondria in vivo and with selective inactivation of the alpha-
ketoglutarate dehydrogenase complex in LLC-PK1 cells. Biochemistry
45, 8959–8971.
DIESEL PARTICLES AND RENAL FAILURE 277
at Sultan Q
aboos University on January 18, 2011
toxsci.oxfordjournals.orgD
ownloaded from





























