Embed Size (px)
Citation preview

Determinants of Rapamycin Sensitivity in Breast Cancer Cells
Woo-Chul Noh,1 Wallace H. Mondesire,2
Junying Peng,2 Weiguo Jian,2 Haixia Zhang,2
JinJiang Dong,2 Gordon B. Mills,3
Mien-Chie Hung,2,4 and Funda Meric-Bernstam2
1Korea Cancer Center Hospital, Nowon-gu, Seoul, Korea, andDepartments of 2Surgical Oncology, 3Molecular Therapeutics, and4Molecular and Cellular Oncology, The University of Texas M. D.Anderson Cancer Center, Houston, Texas
ABSTRACTPurpose: Rapamycin inhibits the serine-threonine ki-
nase mammalian target of rapamycin (mTOR), blockingphosphorylation of p70 S6 kinase (S6K1) and 4E-bindingprotein 1 (4E-BP1) and inhibiting protein translation andcell cycle progression. Rapamycin and its analogues arecurrently being tested in clinical trials as novel-targetedanticancer agents. Although rapamycin analogues show ac-tivity in clinical trials, only some of the treated patientsrespond. The purpose of this study is to identify determi-nants of rapamycin sensitivity that may assist the selectionof appropriate patients for therapy.
Experimental Design: Breast cancer cell lines represent-ing a spectrum of aberrations in the mTOR signaling path-way were tested for rapamycin sensitivity. The expressionand phosphorylation state of multiple components of thepathway were tested by Western blot analysis, in the pres-ence and absence of rapamycin.
Results: Cell proliferation was significantly inhibited inresponse to rapamycin in 12 of 15 breast cancer cell lines.The ratio of total protein levels of 4E-BP1 to its bindingpartner eukaryotic initiation factor 4E did not predictrapamycin sensitivity. In contrast, overexpression of S6K1,and phosphorylated Akt independent of phosphatase andtensin homologue deleted from chromosome 10 status, wereassociated with rapamycin sensitivity. Targeting S6K1 andAkt with small interfering RNA and dominant-negative con-structs, respectively, decreased rapamycin sensitivity. Rapa-mycin inhibited the phosphorylation of S6K1, ribosomal S6protein, and 4E-BP1 in rapamycin-resistant as well as -sen-sitive cells, indicating that its ability to inhibit the mTOR
pathway is not sufficient to confer sensitivity to rapamycin.In contrast, rapamycin treatment was associated with de-creased cyclin D1 levels in the rapamycin-sensitive cells butnot in rapamycin-resistant cells.
Conclusions: Overexpression of S6K1 and expression ofphosphorylated Akt should be evaluated as predictors ofrapamycin sensitivity in breast cancer patients. Further-more, changes in cyclin D1 levels provide a potential phar-macodynamic marker of response to rapamycin.
INTRODUCTIONRapamycin, a macrolide fungicide, was first isolated from
Streptomyces hygroscopicus in the early 1970s and initiallydeveloped clinically for its immunosuppressant properties. Sub-sequently, rapamycin became of significant interest as a poten-tial anticancer drug. Rapamycin inhibits the serine threoninekinase mammalian target of rapamycin (mTOR) by binding toone of the immunophilin family of FK 506-binding proteins,FKBP 12 (1, 2). The inhibition of mTOR decreases the phos-phorylation and activation of S6K1 and 4E-binding protein 1(4E-BP1), and this in turn inhibits the translation of criticalmRNAs that are involved in the cell cycle progression and cellproliferation that are hallmarks of carcinogenesis (1, 2). Clini-cally, rapamycin analogues with improved stability and phar-macological properties have been well tolerated by patients inPhase I trials, and the agents have shown a promising antitumoreffect in several types of refractory tumors, including breastcancer (3–5). However, only a minority of patients in eachtumor lineage appear to respond to rapamycin analogues. Thus,there is an urgent need to identify markers of rapamycin sensi-tivity to allow prospective selection of patients likely to respondto rapamycin analogues in clinical trials.
mTOR (also known as RAFT1, RAPT1, and FRAP) mod-ulates at least two separate downstream pathways that are con-jectured to control the translation of specific subsets of mRNAs(6). In one pathway, mTOR directly phosphorylates eukaryoticinitiation factor 4E-BP1, which triggers additional phosphoryl-ation events that cause hyperphosphorylated 4E-BP1 to disso-ciate from eukaryotic initiation factor 4E (eIF4E), thereby in-creasing the availability of functional eIF4E. eIF4E is thought tobe the rate-limiting component for cap-dependent translation.The increase in free eIF4E levels thus leads to more efficientcap-dependent translation initiation, increasing the translation ofmRNAs with long, highly structured 5�-untranslated regions,such as cyclin D1 and c-myc (7, 8). The second downstreamtarget of mTOR is S6K1. It is unclear whether mTOR activatesS6K1 by direct phosphorylation or the inhibition of a phospha-tase (9). Regardless, on phosphorylation/activation by mTOR,S6K1 phosphorylates the 40S ribosomal protein S6. This phos-phorylation of the S6 protein enhances the translation of mR-NAs with a 5� terminal oligopyrimidine tract, such as elongationfactor-1� and ribosomal proteins (10).
S6K1 and 4E-BP1 are also regulated by phosphatidylinosi-tol 3�-kinase (PI3-K) and its downstream target Akt. In vitro,
Received 8/14/03; revised 10/27/03; accepted 10/31/03.Grant support: The Gillson Longenbaugh Foundation, The Universityof Texas Physician-Scientist Program, and NIH Grant 1K08-CA-91895-01 (to F. M-B.) and NIH Grant 5T32CA09599 (to W. H. M.).The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.Requests for reprints: Funda Meric-Bernstam, The University ofTexas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box444, Houston, TX 77030. Phone: (713) 745-4453; Fax: (713) 745-4926;E-mail: [email protected].
1013Vol. 10, 1013–1023, February 1, 2004 Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Akt can phosphorylate mTOR on Ser2448, and this site is alsophosphorylated on Akt activation in vivo (11, 12). However,although mTOR appears to be directly downstream of PI3-K/Akt in a linear pathway, Akt may also regulate mTOR in a morecomplex fashion, because mTOR is inhibited by the tuberoussclerosis gene products TSC1 and TSC2, which are also targetsfor Akt (13). S6K1 is activated by several different stimuli as aresult of phosphorylation of multiple different sites. The PI3-Kinhibitors Wortmannin and rapamycin, however, cause a de-crease in S6K1 phosphorylation, particularly at phosphorylationsites shown to be dependent on the PI3-K pathway (14). Inter-estingly, a �2–46/�CT104 mutant of S6K1 is inhibited byWortmannin but not rapamycin (15), suggesting that the PI3-Kpathway can exhibit effects on S6K1 independent of regulationof mTOR activity.
The PI3-K/Akt/mTOR signaling pathway is regulated bythe tumor suppressor gene product phosphatase and tensinhomologue deleted from chromosome 10 (PTEN; Ref. 16).Germ-line mutations in the PTEN tumor suppressor gene lead toabnormal activation of the PI3-K/Akt pathway. Germ-line mu-tations in PTEN are responsible for Cowden’s syndrome, whichpredisposes to breast cancer (17). Although PTEN mutations arerare in sporadic cases of breast cancer, PTEN is at a site offrequent allelic imbalance, and the PTEN protein is absent ordecreased in a significant number of breast cancers (16, 18, 19).
Increased signaling through the PI3-K pathway as a con-sequence of deletion of PTEN has been proposed as an indicatorof sensitivity of rapamycin (2, 20, 21). However, Yu et al. (22)reported that, of eight breast cancer cell lines they tested, twowere PTEN deficient, but six were sensitive to rapamycin ana-logue CCI-779. This suggests that sensitivity to mTOR inhibi-tors is not limited to PTEN-deficient breast cancers. The geneticand molecular abnormalities that render cells sensitive to rapa-mycin, particularly in breast cancer, remain unclear.
Here, we report an in vitro study of the effects of rapamy-cin in a panel of breast cancer cell lines representing a spectrumof aberrations in the PI3-K mTOR pathway. The results showthat rapamycin induces G1 cell cycle arrest of breast cancer celllines expressing high levels of phospho-Akt, irrespective of theirPTEN status, as well as cells highly expressing phospho-S6K1.Targeting Akt and S6K1, with dominant-negative constructs andsmall interfering RNA, respectively, decreases rapamycin sen-sitivity. Our results demonstrate that phospho-Akt and overex-pression of S6K1 confer rapamycin sensitivity and may beuseful predictive markers of sensitivity. Furthermore, our resultssuggested that cyclin D1 may be useful as a pharmacodynamicmarker of response to rapamycin and its analogues in breastcancer patients.
MATERIALS AND METHODSCell Lines and Cultures. The following human breast
cancer cell lines were obtained from the American Type CultureCollection: MCF-7, BT-20, BT-549, SK-BR-3, MDA-MB-231,MDA-MB-361, MDA-MB-468, BT-474, BT-483, T-47D,MDA-MB-453, and ZR-75-1. MDA-MB-435 and MDA-MB-330 were obtained from the M. D. Anderson Breast CancerResearch Program Core Laboratory Cell Line Depository. TheNCI/ADR-RES cells were obtained from the National Cancer
Institute Division of Cancer Treatment and Diagnosis TumorRepository. The HER2/neu-transformed NIH3T3 (HER2/neu-3T3), DN-Akt (kinase-dead) transfectants of the HER2/neu-3T3cells, and DN-Akt transfectants of MDA-MB-453 cells havebeen described previously (23). All cell lines were cultured inDMEM/F12 supplemented with 10% fetal bovine serum, 2 mM
glutamine, and 1% penicillin–streptomycin at 37°C and humid-ified 5% CO2.
Reagents. Rapamycin and antibodies against phospho-Akt (Ser473), mTOR, phospho-mTOR (Ser2448), phosphoS6K1 (Thr389), total S6K1, phospho-S6 ribosomal protein(Ser235/236), phospho-4E-BP1 (Ser65, Thr46), phospho-eIF4E(Ser209), and phospho-eIF2� (Ser51) were purchased from CellSignal Technology, Inc. (Beverly, MA). Antibodies againstPTEN, total 4E-BP1, and eIF4E were purchased from CascadeBioscience (Winchester, MA), Santa Cruz Biotechnology, Inc.(Santa Cruz, CA) and BD Bioscience (San Jose, CA), respec-tively. Antibodies against cyclin D1, eEF-1�, and c-myc wereobtained from NeoMarkers, Inc. (Freemont, CA), Upstate Bio-technology (Waltham, MA), and Oncogene Research Products(San Diego, CA), respectively. All other chemicals were pur-chased from Sigma Chemical Co. (St. Louis, MO).
Cell Treatment and Cell Proliferation Assays. To testthe effect of rapamycin on cell proliferation, cells were platedinto 96-well, flat-bottomed plates at 2–4 � 103 cells/100 �l/well, with the density determined on the basis of the growthcharacteristics of each cell line. After the overnight incubation,triplicate wells were treated with varying concentrations ofrapamycin ranging from 1 to 100 nM for 4 days. Relativepercentage of metabolically active cells relative to untreatedcontrols was then determined on the basis of the mitochondrialconversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazo-lium bromide to formazine. The amount of 3-(4,5-dimethylthia-zol-2-yl)-2,5-diphenyltetrazolium bromide that is converted toformazine indicates the number of viable cells. The results wereassessed in a 96-well format plate reader by measuring theabsorbance at a wavelength of 540 nM (A540 nm). The percentageof metabolically active cells was compared with the percentageof control cells growing in the absence of rapamycin in the sameculture plate. The IC50s were determined by nonlinear regres-sion analysis using the equation for a sigmoid plot. The rates ofDNA synthesis were determined by the percentage of cellsshowing [3H]thymidine incorporation into DNA. In brief, afterthe cells were treated with rapamycin in the same manner as inthe 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bro-mide assay, 0.5 �Ci of [3H] thymidine was added to each well,and the cells were incubated for an additional 16 h before beingharvested. The incorporation of [3H] thymidine was measuredby liquid scintillation counting. The rates of DNA synthesis inthe treated cells were compared with the rates seen for controlcells not treated with rapamycin in the same culture plate.
Cell Cycle Analysis and Determination of ApoptoticCells. Cells were incubated with or without 100 nM rapamycinfor 4 days and harvested when they reached a confluency of50–70%. After the cells were washed with PBS, they were fixedwith 75% ethanol overnight at 4°C. The cells were then washedtwice with PBS and resuspended in hypotonic propidium iodidesolution (10 �g of propidium iodide, 10 �g of RNase A, and0.5% Tween 20 in 1 ml of PBS) for 1 h at room temperature and
1014 Rapamycin Sensitivity in Breast Cancer Cells
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

kept in the dark at 4°C before analysis. Cell cycle distributionwas determined by analyzing 1000–20,000 cells using a FAC-Scan flow cytometer and Cell Quest software (Becton Dickin-son, San Jose, CA). The percentage of apoptotic cells wasdetermined by the subG1 peak in the DNA histogram.
Western Blot Analysis. Cultured cells were washed withcold PBS and lysed in lysis buffer as described elsewhere (15).To test the effect of rapamycin on the expression of mTOR andits downstream molecules, cells were treated with 100 nM rapa-mycin for 24 h before lysis. Cell lysates containing 50 �g ofprotein were separated by SDS-PAGE with 7–12.5% gel, de-pending on their molecular weight, and transferred to a 0.2-�mpolyvinylidene difluoride membrane (Bio-Rad Laboratories,Hercules, CA). Membranes were blocked with 5% nonfat drymilk in Tris-buffered saline with 0.02% Tween 20 and immu-noblotted with antibodies, as specified in the Reagents section.The immunoblots were visualized by an enhanced chemilumi-nescence detection system (Amersham Life Sciences, ArlingtonHeights, IL).
Small Interfering RNA (siRNA). To silence S6K1, asingle transfection of siRNA duplex was performed using Li-pofectamine reagent according to manufacturer’s protocol (LifeTechnologies, Inc.). A 21-mer double stranded RNA with d(TT)overhang was selected for ability to silence S6K1 expression.The siRNA target sequence was AAUGUAUGACAUGCU-GACUGGTT. The siRNA was synthesized by Dharmacon Re-search (Lafayette, CO). Nonspecific control siRNA (Duplex X)was purchased from Dharmacon Research.
Statistical Analysis. Results were presented as means �SD for three separate experiments. For comparison betweengroups, data were analyzed by Student’s t test. Differencesbetween groups were considered statistically significant at P �0.05.
RESULTSExpression of mTOR and its Downstream Molecules.
To determine the baseline activity of the PI3-K/Akt/mTORsignaling pathway in breast cancer cell lines, we measured thebasal expression level of mTOR and related molecules by West-ern blot analysis (Fig. 1). The tumor suppressor gene PTEN,which negatively regulates the PI3-K/Akt/mTOR signalingpathway by counteracting PI3-K, was not detected in the BT-549 and MDA-MB-468 cell lines, consistent with previousreports of PTEN mutations in these cells (19). Phospho-Akt, adownstream component of the PI3-K pathway, was present infour of the eight cell lines, including the two PTEN-deficientcell lines, indicating that Akt was strongly activated in thesecells. The direct target of rapamycin, mTOR, and its phospho-rylated/activated form was present at relatively similar levels inall eight cell lines. Indeed, there was no obvious correlationbetween phospho-Akt and phosho-mTOR levels in these lines.
S6K1 is one of the major downstream components of themTOR signaling pathway. Phospho-S6K1 (Thr389) was presentat high levels in MCF-7 and MDA-MB-361 cells, indicating thatS6K1 was highly activated in these cells. The 40S ribosomalprotein S6 is phosphorylated by activated S6K1; phospho-S6protein (Ser235/236) was present at similar levels in all eightcell lines. Once again, phospho-S6K1 did not directly correlate
with phospho-S6 protein, suggesting additional modes of regu-lation. In addition to S6K1, mTOR can phosphorylate 4E-BP1.We used three antibodies against 4E-BP1 to detect the total4E-BP1 protein levels, Ser65 phospho-4E-BP1 and Thr46 phos-pho-4E-BP1. Although Thr46 has been shown to be a directtarget site of phosphorylation by mTOR, Ser65 may be phos-phorylated by additional kinases. The band intensities of total4E-BP1 differed, but the amounts of phospho-4E-BP1 roughlycorrelated with those of the total 4E-BP1, indicating that theproportions of phosphorylated 4E-BP1 were similar in all celllines. eIF4E was expressed almost equally in all eight cell lines,but phospho-eIF4E, which has an enhanced affinity for mRNA(24, 25), was detected in only three, SK-BR-3, MDA-MB-361,and MDA-MB-435. Once again, levels of phospho-eIF4E do notexhibit a simple relationship with the phosphorylation status ofAkt, mTOR, S6K1, S6, or 4E-BP1.
In summary, phosphorylated mTOR is ubiquitously ex-pressed. Although only two cell lines were PTEN negative, Aktwas phosphorylated in four of the eight cell lines. mTOR as wellas its targets, 4E-BP1 and S6K1, were phosphorylated in alleight cell lines. Thus, the mTOR signaling pathway is activated
Fig. 1 The expression of mammalian target of rapamycin (mTOR) andits upstream and downstream molecules in a panel of breast cancer celllines cultured in the presence of 10% fetal bovine serum. The sameamounts (50 �g) of protein were prepared from the indicated cell linesat 60–70% confluence, loaded onto an SDS-PAGE gel, and immuno-blotted with antibodies against phosphatase and tensin homologue de-leted from chromosome 10 (PTEN), phospho-Akt (Ser473), total mTOR,phospho-mTOR (Ser473), phospho S6K1 (Thr389), phospho-S6 protein(Ser235/236), total 4E-BP1, phospho-4E-BP1 (Thr46), phospho-4E-BP1 (Ser65), total eIF4E, phospho-eIF4E (Ser209), and actin (from toplane to bottom).
1015Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

in all of the breast cancer cell lines tested. However, the prox-imal mediators of the activation of mTOR and its substratesappear to vary between the different lines.
Sensitivity of Breast Cancer Cell Lines to Rapamycin.To examine the sensitivity of each cell line to rapamycin, cellswere treated with rapamycin at different concentrations for 4days, and cell proliferation was measured by a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay(Fig. 2A) and DNA synthesis by thymidine incorporation (Fig.2B). All of the cell lines, with the exception of MDA-MB-231and MDA-MB-435, were inhibited by rapamycin in both of theassays, indicating that the decrease in cell number was accom-panied by a decrease in S phase progression (thymidine incor-poration). BT-20, BT-549, SK-BR3, and MDA-MB-468 cells,which express high levels of phospho-Akt, were significantlyinhibited by rapamycin. However, two cell lines, MCF-7 andMDA-MB-361 cells, which did not exhibit high levels of phos-phorylated Akt, were also inhibited by rapamycin. Thus, al-though high Akt activity may confer sensitivity to rapamycin, itis not required for rapamycin sensitivity. Intriguingly, MCF-7and MDA-MB-361 cells highly express phospho-S6K1 and areknown to have an amplification of the S6K1 gene (26), poten-tially conferring sensitivity to rapamycin. MDA-MB-231 and
MDA-MB-435 cell lines, which express PTEN, do not haveactivated Akt, and do not highly express phospho-S6K1, wereresistant to growth inhibition induced by rapamycin. Thus, anyof the three aberrations, high AKT phosphorylation, PTENabsence, or amplification of S6K1, is sufficient to engendersensitivity to rapamycin.
Mechanism of Rapamycin-Mediated Growth Inhibi-tion. The decreased cell growth induced by rapamycin couldbe attributable to decreased cell cycle progression or increasedapoptosis. As shown in Fig. 3, A and B, after 4 days of treatmentwith 100 nM rapamycin, the percentage of cells in the G1 phasewas increased in the rapamycin-sensitive cell lines. In contrast,
Fig. 2 Inhibition of cell growth and DNA synthesis by rapamycin. InA, breast cancer cell lines were treated with rapamycin in the indicatedconcentration for 4 days, and cell viability was determined by a standard3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. InB, cells were treated with rapamycin for 4 days and incubated for anadditional 16 h with [3H]thymidine. The data are presented as the meanof three separate experiments at each treatment dose. The SDs were allwithin 5% of the respective means. �, P � 0.05 by Student’s t test.
Fig. 3 Effect of rapamycin on cell cycle progression and apoptosis. InA, rapamycin inhibited cell cycle progression from the G1 to S phase inMCF-7 cells. MCF-7 cells were incubated with or without 100 nM
rapamycin for 4 days and then fixed with 75% ethanol and stained withpropidium iodide. The histogram showing the relative DNA content wasdetermined by flow cytometry. In B, breast cancer cell lines wereincubated with or without 100 nM rapamycin for 4 days, and percentageof cells in G1, S, and G2-M phases of the cell cycle was determined byflow cytometry. In C, cells were incubated with or without 100 nM
rapamycin for 4 days and then prepared and stained as described in A.The histogram shows the percentage of cells in the subG1 peak, whichis believed to indicate apoptotic cells. Each column represents the meanof the results of three separate experiments � SD.
1016 Rapamycin Sensitivity in Breast Cancer Cells
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

the percentage of apoptotic cells in the subG1 peak did notincrease after treatment with the same concentration of rapamy-cin (Fig. 3C). These results indicated that rapamycin inhibitedcell proliferation by arresting the cell cycle in the G1 phase butdid not induce apoptosis in the breast cancer cell lines studiedunder the conditions and time course analyzed.
The 4E-BP1:eIF4E Ratio As a Predictor of the IntrinsicSensitivity to Rapamycin in Breast Cancer Cell Lines. Wenext sought additional molecular markers that can predict sen-sitivity to rapamycin in breast cancer cells on the basis ofrapamycin’s mechanism of action, which is to reduce the phos-phorylation of 4E-BP1, thereby enhancing the binding affinityof 4E-BP1 to eIF4E and decreasing the availability of eIF4E. Onthe basis of this mechanism, the ratio of the total protein levelsof 4E-BP1:eIF4E has been suggested to reflect acquired orintrinsic resistance to rapamycin in rhabdomyosarcoma andcolon carcinoma cells (27). This contention prompted us toexamine whether the 4E-BP1:eIF4E ratios correlated with rapa-mycin sensitivity in breast cancer cell lines. As shown in Fig. 4,however, this ratio did not correlate with the intrinsic sensitivityto rapamycin in breast cancer cell lines.
Phospho-Akt and S6K1 As Predictors of RapamycinSensitivity. On the basis of our breast cancer cell line sensi-tivity pattern, we hypothesized that phospho-Akt and overex-pression of S6K1 are predictors of rapamycin sensitivity. Wetherefore determined the rapamycin sensitivity of an additionaldifferent panel of breast cancer cell lines to validate the efficacyof these predictive markers. We treated breast cancer cells withescalating doses of rapamycin and determined the IC50s ofseven other breast cancer cell lines, BT-474, T-47D, ZR-75-1,BT-483, MDA-MB-453, MDA-MB-330, NCI/ADR-RES, aswell as the rapamycin-resistant MDA-MB-231 cell line. West-ern blot analysis was performed to determine the expressionof PTEN, phospho-Akt, phospho-S6K1, total S6K1, and actin(Fig. 5).
Six of the eight cell lines tested in this panel were sensitiveto rapamycin. High levels of phospho-Akt were detected in allsix rapamycin-sensitive cell lines, whereas only one cell line,BT-483, did not express PTEN. These results confirm that
expression of phospho-Akt is a predictor of rapamycin sensitiv-ity, independent of PTEN status.
Two of the cell lines, BT-474 and ZR-75-1, overexpressedtotal S6K1. These two cell lines were among the most rapamy-cin-sensitive cells within our panel. These results confirm thatS6K1 overexpression is associated with rapamycin sensitivity.
Similar to MDA-MB-231 and MDA-MB-435, which areresistant to rapamycin (see Figs. 1 and 2), NCI/ADR-RES didnot express high levels of phospho-AKT or phospho-S6K1 andexpressed wild-type levels of PTEN. This supports the conten-tion that the absence of all three of these aberrations correlateswith resistance to rapamycin.
The Effect of Modulation of S6K1 and Akt on Rapa-mycin Sensitivity. To test whether S6K1 overexpression con-fers rapamycin sensitivity, we used siRNA oligonucleotides tosilence the expression of S6K1. The siRNA sequence we se-lected markedly decreases total S6K1 protein levels 72 h aftertransfection as demonstrated in Fig. 6A. MCF-7 cells weretransfected with either S6K1 siRNA or a nonspecific controlsiRNA. After transfection (48 h), the cells were incubated withor without rapamycin. S6K1 siRNA-transfected MCF-7 cellswere less sensitive to the effects of rapamycin (Fig. 6, B and C).
Fig. 4 The 4E-binding protein 1:eukaryotic initiation factor 4E (4E-BP1: eIF4E) ratio in breast cancer cell lines. The histogram shows theratio of the band intensity of the total protein levels of 4E-BP1:eIF4Eseen in the Western blot analysis in Fig. 1, as determined by a scanninglaser densitometer. Rapamycin-sensitive cells were denoted with �.
Fig. 5 Predictors of rapamycin sensitivity. Cell lysates from BT-474,T-47D, ZR-75-1, BT-483, MDA-MB-453, MDA-MB-330, NCI/ADR-RES, and MDA-MB-231 cell lines (in order of decreasing rapamycinsensitivity) were immunoblotted with antibodies against phosphataseand tensin homologue deleted from chromosome 10, phospho-Akt(Ser473), phospho S6K1 (Thr389), total S6K1, and actin (from top laneto bottom). The IC50s of each cell line for rapamycin 4 days of treatmentare depicted in the bar graph below.
1017Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
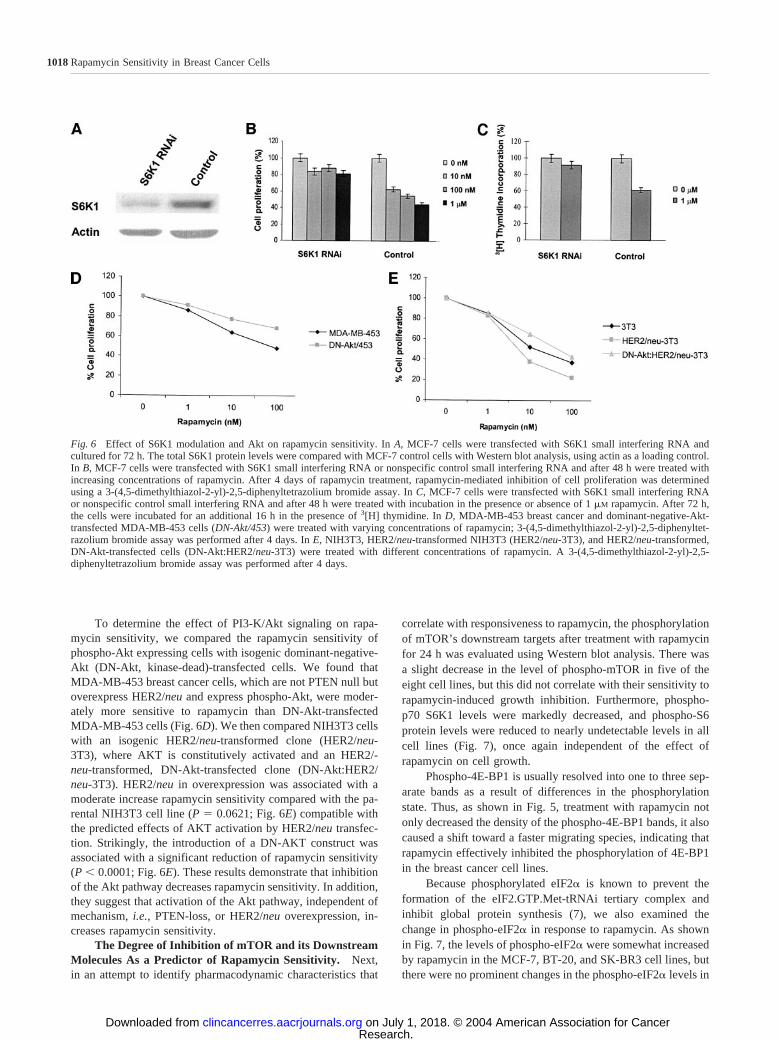
To determine the effect of PI3-K/Akt signaling on rapa-mycin sensitivity, we compared the rapamycin sensitivity ofphospho-Akt expressing cells with isogenic dominant-negative-Akt (DN-Akt, kinase-dead)-transfected cells. We found thatMDA-MB-453 breast cancer cells, which are not PTEN null butoverexpress HER2/neu and express phospho-Akt, were moder-ately more sensitive to rapamycin than DN-Akt-transfectedMDA-MB-453 cells (Fig. 6D). We then compared NIH3T3 cellswith an isogenic HER2/neu-transformed clone (HER2/neu-3T3), where AKT is constitutively activated and an HER2/-neu-transformed, DN-Akt-transfected clone (DN-Akt:HER2/neu-3T3). HER2/neu in overexpression was associated with amoderate increase rapamycin sensitivity compared with the pa-rental NIH3T3 cell line (P � 0.0621; Fig. 6E) compatible withthe predicted effects of AKT activation by HER2/neu transfec-tion. Strikingly, the introduction of a DN-AKT construct wasassociated with a significant reduction of rapamycin sensitivity(P � 0.0001; Fig. 6E). These results demonstrate that inhibitionof the Akt pathway decreases rapamycin sensitivity. In addition,they suggest that activation of the Akt pathway, independent ofmechanism, i.e., PTEN-loss, or HER2/neu overexpression, in-creases rapamycin sensitivity.
The Degree of Inhibition of mTOR and its DownstreamMolecules As a Predictor of Rapamycin Sensitivity. Next,in an attempt to identify pharmacodynamic characteristics that
correlate with responsiveness to rapamycin, the phosphorylationof mTOR’s downstream targets after treatment with rapamycinfor 24 h was evaluated using Western blot analysis. There wasa slight decrease in the level of phospho-mTOR in five of theeight cell lines, but this did not correlate with their sensitivity torapamycin-induced growth inhibition. Furthermore, phospho-p70 S6K1 levels were markedly decreased, and phospho-S6protein levels were reduced to nearly undetectable levels in allcell lines (Fig. 7), once again independent of the effect ofrapamycin on cell growth.
Phospho-4E-BP1 is usually resolved into one to three sep-arate bands as a result of differences in the phosphorylationstate. Thus, as shown in Fig. 5, treatment with rapamycin notonly decreased the density of the phospho-4E-BP1 bands, it alsocaused a shift toward a faster migrating species, indicating thatrapamycin effectively inhibited the phosphorylation of 4E-BP1in the breast cancer cell lines.
Because phosphorylated eIF2� is known to prevent theformation of the eIF2.GTP.Met-tRNAi tertiary complex andinhibit global protein synthesis (7), we also examined thechange in phospho-eIF2� in response to rapamycin. As shownin Fig. 7, the levels of phospho-eIF2� were somewhat increasedby rapamycin in the MCF-7, BT-20, and SK-BR3 cell lines, butthere were no prominent changes in the phospho-eIF2� levels in
Fig. 6 Effect of S6K1 modulation and Akt on rapamycin sensitivity. In A, MCF-7 cells were transfected with S6K1 small interfering RNA andcultured for 72 h. The total S6K1 protein levels were compared with MCF-7 control cells with Western blot analysis, using actin as a loading control.In B, MCF-7 cells were transfected with S6K1 small interfering RNA or nonspecific control small interfering RNA and after 48 h were treated withincreasing concentrations of rapamycin. After 4 days of rapamycin treatment, rapamycin-mediated inhibition of cell proliferation was determinedusing a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. In C, MCF-7 cells were transfected with S6K1 small interfering RNAor nonspecific control small interfering RNA and after 48 h were treated with incubation in the presence or absence of 1 �M rapamycin. After 72 h,the cells were incubated for an additional 16 h in the presence of 3[H] thymidine. In D, MDA-MB-453 breast cancer and dominant-negative-Akt-transfected MDA-MB-453 cells (DN-Akt/453) were treated with varying concentrations of rapamycin; 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltet-razolium bromide assay was performed after 4 days. In E, NIH3T3, HER2/neu-transformed NIH3T3 (HER2/neu-3T3), and HER2/neu-transformed,DN-Akt-transfected cells (DN-Akt:HER2/neu-3T3) were treated with different concentrations of rapamycin. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was performed after 4 days.
1018 Rapamycin Sensitivity in Breast Cancer Cells
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

the other five cell lines. Once again, there was no correlationwith effects of rapamycin on cell growth.
In summary, the phosphorylation of 4E-BP1, S6K1, andthe S6 ribosomal protein was effectively inhibited by rapamycinin both rapamycin-sensitive and -resistant cell lines. In addition,although rapamycin reduced the level of phospho-mTOR and itsdownstream molecules, the extent to which phosphorylation ofmTOR’s targets were inhibited did not correlate with the extentto which cell growth was inhibited by rapamycin. This indicatedthat phospho-S6K1 and -4E-BP1 are not likely to prove to beuseful monitors of the effectiveness of rapamycin.
Rapamycin-Induced Inhibition of Cyclin D1 Expressionin Rapamycin-Sensitive Cell Lines. We next evaluated theeffect of rapamycin on the expression of mRNAs that are knownto be under translational control of mTOR and its downstreamtargets. First, we determined the protein levels of cyclin D1 andc-myc, whose translation is regulated by eIF4E, and elongationfactor-1�, whose translation is thought to be regulated by S6K1.Western blot results were quantified by densitometry, and pro-tein levels were determined after normalizing for actin levels.
Baseline cyclin D1 expression level was higher in therapamycin-sensitive cells, MCF-7 and MDA-MB-468, with 2-fold reduction in these levels after rapamycin treatment (Fig. 8).In contrast, the baseline cyclin D1 expression level was 20-foldlower in the MDA-MB-435 and MDA-MB-231 cells, with noreduction, but rather a slight increase, in cyclin D1 expression inresponse to rapamycin. c-myc levels showed 26% decrease inthe MCF-7 cells with treatment but were unchanged in the othercells. Rapamycin treatment was associated with 2-fold reduc-tion in the elongation factor-1� levels in MCF-7 cells, but thelevels remained unchanged in the other cell lines after treatment.Thus, the presence of high basal levels of cyclin D1 followed bya decrease in cyclin D1 expression correlated with responsive-ness to rapamycin.
To verify that the lack of rampamycin-induced cyclin D1downregulation is a reproducible finding in rapamycin-resistant
breast cancer cells, we determined the effect of rapamycin in ourthird rapamycin-resistant cell line NCI/ADR-RES (Fig. 8B). Wefound that unlike MDA-MB-231 and MDA-MB-435 cells, thatNCI/ADR-RES had moderate levels of basal cyclin D1 expression.However, rapamycin treatment of this cell line did not reducecyclin D1 levels. These results demonstrate that expression ofcyclin D1 levels with rapamycin treatment may correlate withresponse and holds potential as a pharmacodynamic marker.
DISCUSSIONRapamycin and its analogues are considered among the
most promising drugs in the anticancer pipeline (3). Rapamycinand its analogues CCI-779 (Wyeth-Ayerst Research, Colle-geville, PA) and RAD001 (Novartis, Basel, Switzerland) arenow being evaluated as anticancer agents in multiple clinicaltrials (3, 4). Thus far, in preclinical studies, human prostatecancer, small cell lung cancer, glioblastoma, T-cell leukemia,and breast cancer have been among the most sensitive cancers torapamycin (1).
PTEN-deficient tumors are considered the best candidatesfor rapamycin treatment, because the PI3-K/Akt pathway isconstitutively activated in these tumor cells. This was the viewtaken as a result of studies in PTEN/ and PTEN�/� mousecells, transformed cells of PTEN�/� mice, and human prostatecancer and multiple myeloma cells with a defined PTEN statusshowing that the growth of PTEN-deficient cancer cells ispreferentially blocked when mTOR is inhibited (2, 20–22, 28).Extrapolating from this, we reasoned that, because PTEN mu-tations in sporadic cases of breast cancer are not very common,with a reported incidence of 4% (18), if rapamycin was onlyeffective in tumors with PTEN mutations, only a small portionof breast tumors might respond to rapamycin analogues. Incontrast, we found that rapamycin effectively inhibited thegrowth of most of the breast cancer cell lines tested. Similarly,Yu et al. (22) reported that, of the eight breast cancer cell lines
Fig. 7 Effects of rapamycin on mammalian tar-get of rapamycin (mTOR) and its downstreammolecules in a panel of breast cancer cell lines.Rapamycin-sensitive cells were denoted with �.Cells were treated with 100 nM rapamycin for 24 hin the presence of 10% fetal bovine serum. Fiftymicrograms of total protein were prepared fromthe indicated cell lines and immunoblotted withantibodies against phospho-mTOR (Ser473),phospho-p70 S6K1 (Thr389), phospho-S6 protein(Ser235/236), phospho-4E-BP1 (Thr46), phospho-4E-BP1 (Ser65), phospho-eIF2� (ser51), and actin(�, without rapamycin; , with rapamycin).
1019Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

they tested, two were PTEN deficient, but six were sensitive tothe rapamycin analogue CCI-779. Thus, although PTEN-defi-cient cells are clearly responsive to rapamycin, additional mo-lecular aberrations can also render cells sensitive to rapamycin.Therefore, to ensure that responsive patients are offered treat-ment with rapamycin or its analogues, it is important to identifyadditional markers or characteristics that can predict the sensi-tivity to rapamycin in breast cancer patients.
Recently, Dilling et al. (27) reported that the 4E-BP1:eIF4E ratio was reduced in rhabdomyosarcoma cell lines withan acquired rapamycin resistance and in several colon carci-noma cell lines with an intrinsic resistance to rapamycin. Theysuggested that the 4E-BP1:eIF4E ratio might therefore be adeterminant of rapamycin sensitivity. However, in our study,sensitivity to rapamycin did not correlate with the 4E-BP1phosphorylation status or 4E-BP1:eIF4E ratio, suggesting thatthe predictive markers for rapamycin sensitivity may not beuniversally applicable to different types of cancer.
Our study also showed that all breast cancer cell lines withhigh baseline levels of phospho-Akt, irrespective of their PTENstatus, were sensitive to rapamycin. This suggested that theactivation status of PI3-K/Akt, irrespective of the mechanism ofthis activation, was associated with rapamycin sensitivity. Thisagreed with the finding of Neshat et al. (20) that the enhancedtumor growth caused by constitutive activation of Akt inPTEN/ prostate cancer cells can be reversed by CCI-779.This is an especially intriguing finding, because signal trans-duction pathways that are activated in breast cancer, such as theHer2/neu pathway, activate Akt (23, 29). Supporting this hy-pothesis, we have demonstrated that HER2/neu overexpressionin NIH3T3 cells enhances rapamycin sensitivity, with a signif-icant reduction in rapamycin sensitivity in DN-Akt-transfectedcells. Our results are consistent with those of Hermanto et al.(30) who found that the forced expression of HER2/neu inMDA-MB-231 cells sensitized the cells to inhibition of colonyformation by rapamycin. These results taken together suggestthat activation of the PI3-K/Akt pathway, regardless of mecha-nism (i.e., PTEN loss or HER2/neu overexpression), is associ-ated with increased mTOR signaling and rapamycin sensitivity.Of particular importance, these findings also mean that manymore breast tumors may respond to rapamycin analogues thanwould have been the case if rapamycin’s effects were limited toPTEN-null tumors.
We also observed that two cell lines, MCF-7 and MDA-MB-361, which do not have high levels of phospho-Akt buthighly express phospho-S6K1, were also sensitive to rapamycin.Interestingly, both of these cell lines have a genomic amplifi-cation of the S6K1 gene located on chromosome 17q23 (31).Furthermore, the two cell lines with phospho-Akt expressionthat also overexpressed phospho-S6K1 and total S6K1, BT-474,and ZR-75–1 were among the most rapamycin-sensitive celllines within our panel. Both of these cells have been shownpreviously to have an amplification of the S6K1 gene (32). Thissuggested that S6K1 amplification/overexpression could be apredictor of rapamycin sensitivity. We found that reduction inS6K1 levels mediated by siRNA leads to a decrease in rapamy-cin sensitivity, confirming that S6K1 overexpression is causallylinked to rapamycin sensitivity. These findings are especiallyimportant because S6K1 gene is amplified in 8.8% of primarybreast cancers, and the S6K1 gene is overexpressed at the RNAlevel in 38% of breast tumors (26, 33). Such S6K1 amplificationhas been associated with a poorer prognosis (26). Rapamycinanalogues thus may be an especially useful molecularly targetedtherapy in this subgroup of patients.
Phospholipase D-dependent accumulation of phosphatidicacid is required for activation of mTOR signaling, and it hasbeen suggested that rapamycin competes with phosphatidic acidfor the FKBP12-rapamycin-binding domain in mTOR (34).Recently, Chen et al. (35) have reported that increased phos-pholipase D activity predicts rapamycin resistance in breastcancer cell lines. Indirectly assessing phospholipase D activityby determining total and phosphorylated phospholipase D ex-pression, along with assessing predictors of rapamycin sensitiv-ity identified in our study, phospho-Akt, and S6K1, may furtheroptimize selection of patients that will derive the most benefitfrom rapamycin and its analogues.
Treatment with the rapamycin analogues have been shown
Fig. 8 Differential effects of rapamycin in rapamycin-sensitive and-resistant cell lines. The rapamycin-sensitive cell lines MCF-7 andMDA-MB-468 and rapamycin-resistant cell lines MDA-MB-435and MDA-MB-231 were incubated in the absence or presence of100 nM rapamycin for 4 days. The expression of cyclin D1, c-myc,elongation factor (EF)-1�, and actin was determined by immuno-blotting. In B, Rapamycin-resistant cell lines MDA-MB-435 and NCI-ADR-RES were incubated in the abscence or presence of 100 nMrapamycin for 4 days. The expression of cylin D1 and actin wasdetermined by immunoblotting.
1020 Rapamycin Sensitivity in Breast Cancer Cells
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

to inhibit S6K1 activity in tumors, skin, and peripheral lympho-cytes in animal models (36, 37). In fact, it has been proposedthat peripheral blood lymphocytes may be used as a surrogatebiomarker when planning dosing regimens. Alternatively, Dud-kin et al. (38) found that when 4E-BP1 phosphorylation (Thr70)was inhibited by CCI-779, this correlated with the growth inhi-bition of prostate, glioma, and ovarian carcinoma xenografts byCCI-779, prompting them to propose that phospho-4E-BP1 sta-tus may be useful for determining whether mTOR activity isinhibited in tumor specimens. In our study, 4E-BP1 phospho-rylation and S6 phosphorylation were inhibited in both rapamy-cin-sensitive and -resistant cell lines. Our results are thereforesimilar to those from studies of prostate cancer and multiplemyeloma cells (20, 28) and suggest that although these mea-surements may indicate that a biologically relevant dose ofrapamycin is present, these will not be useful in predictingwhich patients will respond to the drug. Taken together, theseresults showed that the differential sensitivity to rapamycin isnot explained by differential ability to inhibit the mTOR path-way. Thus, although phospho-S6K1 and phospho-4E-BP1 maybe useful to monitor whether the drug levels are sufficientlyhigh to achieve adequate target inhibition, phospho-S6K1 andphospho-4E-BP1 do not appear to be useful for predictingwhether breast tumors will respond to the inhibitor.
One explanation for this finding is that blockade of mTORmay potentially inhibit cell growth by mechanisms other thanthrough its known effects on 4E-BP1 and S6K1. A secondexplanation is that, although the mTOR signaling pathway isinhibited in all cell lines, the effect on proliferation may be moredramatic in some cell lines, such as those with an activatedPI3-K/Akt pathway, which may not only activate mTOR butmay also potentiate its effects. A third explanation is that inhi-bition of the phospho-mTOR’s downstream targets 4E-BP1 andS6K1 has different downstream effects in specific cell lines,leading to differences in the gene expression and translationalprofile and thus alterations in the rapamycin-mediated growthresponse. Alternatively, although the mTOR pathway is acti-vated in particular tumors, it may not be obligatory for contin-ued cell cycle progression, with this process being mediated byalternative, rapamycin-insensitive pathways. Finally, mTORmay not be the only target of rapamycin, or S6K1 and 4E-BP1may not be mTOR’s critical downstream effectors.
Rapamycin’s effects on cellular physiology is thought to beat least in part mediated by alterations in the translation ofmRNAs important for cell growth and proliferation. It effec-tively inhibits S6K1 phosphorylation, which is thought to de-crease the translation of 5� terminal oligopyrimidine mRNAs,such as elongation factor-1� (10). In our experiments, althoughS6 phosphorylation was dramatically inhibited in all cell lines,there was only a 2-fold reduction in the elongation factor-1�protein level in MCF-7 cells and no change in the rapamycin-sensitive MDA-MB-468 cells and rapamycin-resistant cells.This finding is especially interesting in light of the recentfinding of Stolovich et al. (39) that the complete inhibition ofS6K1 by rapamycin in various cell lines only mildly repressedthe translation of terminal oligopyrimidine mRNAs. The inhi-bition of terminal oligopyrimidine mRNA translation may there-fore not be an important contributor to the ability of rapamycinto inhibit cell growth or alternatively the effects of rapamycin
may be more prominent in cell lines highly expressing phospho-S6K1. Alternately, S6K1 inhibition may affect cell growththrough additional targets.
Rapamycin’s other translational effect is through 4E-BP1.Specifically, rapamycin decreases the hyperphosphorylation of4E-BP1, thereby increasing 4E-BP1 binding to eIF4E and inturn inhibiting cap-dependent translation. The resultant de-creased availability of eIF4E would be expected to selectivelydecrease the translation of mRNA with highly structured 5�un-translated region, such as cyclin D1 and c-myc. Indeed, wefound that cyclin D1 expression, which is regulated by a cap-dependent translation, was decreased in rapamycin-sensitivebreast cancer cells MCF-7 and MDA-MB-468 but not in any ofthe rapamycin-resistant breast cancer cell lines we identified.Thus, alterations in cyclin D1 may play an important role in thecell cycle regulatory effects of rapamycin and be a valuablepredictor of response to therapy. Furthermore, the higher base-line expression of cyclin D1 in the rapamycin-sensitive cellssuggests that cyclin D1 plays a relatively important role in theproliferation of these cells. However, the moderate expressionof cyclin D1 in rapamycin-resistant cell lines NCI/ADR-RESdemonstrates that the expression of cyclin D1 alone is notsufficient to confer rapamycin-sensitivity, but rather that down-regulation of expressed cyclin D1 may be an indicator of sen-sitivity.
In contrast to cyclin D1, c-myc was only minimally mod-ulated in our experiments, with only a slight decrease in thec-myc levels in MCF-7 cells with treatment but no change in theother cells. This finding may be explained by the fact that c-mycmRNA has an internal ribosome entry site (40), which mayallow the c-myc mRNA to be translated although cap-mediatedtranslation is inhibited by rapamycin. Rapamycin may alsoregulate cyclin D1 by additional mechanisms. In fact, Hashe-molhosseini et al. reported that NIH3T3 cells rapamycin inhibitscyclin D1 expression by decreasing cyclin D1 mRNA andprotein stability (41). Thus, further work is needed to determinethe mechanism of cyclin D1 regulation in breast cancer cells.
Interestingly, our findings in breast cancer cells differ fromthose observed with rhabdomyosarcoma cells and multiple my-eloma cells. In particular, Hosoi et al. (42) found that rapamycininhibited c-myc induction by serum and that the failure ofrapamycin to inhibit c-myc induction correlated with rapamycinresistance in rhabdomyosarcoma cells. Shi et al. (28) reportedthat CCI-779 inhibited the expression of c-myc in CCI-sensitivemyeloma cells but not CCI-resistant cells. In contrast, cyclin D1expression was not altered in either sensitive or resistant cells.Thus, the modulation of mTOR’s downstream targets may differin different tumor types. Alternately, cyclin D1 may not bedownregulated in all rapamycin-sensitive cell lines; in that sce-nario downregulation of cyclin D1 would be able to predictresponse, but lack of downregulation would not necessarilypredict resistance. Our results suggest that cyclin D1 may provesuperior to c-myc as a pharmacodynamic marker of the rapa-mycin response in breast cancer. However, monitoring the pro-teomic profile rather than individual genes may provide agreater degree of predictive power.
Previous studies in other cell types have shown that themajor mechanism by which rapamycin suppressed tumor cellgrowth was by inhibiting of cell cycle progression in the G1
1021Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

phase (41, 43). However, it was reported that rapamycin alsoinduced apoptosis in certain types of tumors (44, 45), e.g., Hosoiet al. (45) and Huang et al. (44, 45) reported that rapamycininduced apoptosis in rhabdomyosarcoma cell lines with defi-cient p53 function as a consequence of continued cell cycleprogression during mTOR inhibition. In addition, Shi et al. (46)found that rapamycin increased interleukin-2 deprivation-induced apoptosis in an interleukin-2-dependent mouse T-cellline. Furthermore, the reports that rapamycin enhances the cy-totoxicity of chemotherapeutic agents (46–48) support the hy-pothesis that rapamycin induces or enhances apoptosis in certaintumor types or conditions. In our study, rapamycin was notsufficient to induce apoptosis of tumor cells, irrespective of theirp53 status (e.g., MCF-7 is p53 wild type; MDA-MB-468 is p53mutant), but rather inhibited tumor cell proliferation by produc-ing G1 cell cycle arrest. However, the preliminary results ofclinical trials with rapamycin analogues suggest that some ofpatients indeed did have tumor regression, consistent with acytotoxic response (5). Thus rapamycin analogues indeed maybe cytotoxic in a clinical setting, and further work is needed tobetter select the patients that are most likely to benefit fromthese therapies.
REFERENCES1. Hidalgo, M., and Rowinsky, E. K. The rapamycin-sensitive signaltransduction pathway as a target for cancer therapy. Oncogene, 19:6680–6686, 2000.2. Mills, G. B., Lu, Y., and Kohn, E. C. Linking molecular therapeuticsto molecular diagnostics: inhibition of the FRAP/RAFT/TOR compo-nent of the PI3K pathway preferentially blocks PTEN mutant cells invitro and in vivo. Proc. Natl. Acad. Sci. USA, 98: 10031–10033, 2001.3. Garber, K. Rapamycin’s resurrection: a new way to target the cancercell cycle. J. Natl. Cancer Inst. (Bethesda), 93: 1517–1519, 2001.4. Hidalgo, M., Rowinsky, E., Erlichman, C., Drengler, R., Marshall,B., Adjei, A., et al. Phase I pharmacological study of CCI-779, a cellcycle inhibitor. Clin. Cancer Res., 6 (Suppl.): 4548s, 2000.5. Chan, S., Scheulen, M. E., Johnston, S., Mross, K., Piccart, M., Hess,D., et al. Phase 2 study of two dose levels of CCI-779 in locallyadvanced or metastatic breast cancer (MBC) failing prior anthracyclineand/or taxane regimens. Proc. Am. Soc. Clin. Oncol. Annu. Meet., 22:193, 2003.6. Meric, F., and Hunt, K. K. Translation initiation in cancer: a noveltarget for therapy. Mol. Cancer Ther., 1: 971–979, 2002.7. Clemens, M. J., and Bommer, U. A. Translational control: the cancerconnection. Int. J. Biochem. Cell Biol., 31: 1–23, 1999.8. Dufner, A., Andjelkovic, M., Burgering, B. M., Hemmings, B. A.,and Thomas, G. Protein kinase B localization and activation differen-tially affect S6 kinase 1 activity and eukaryotic translation initiationfactor 4E-binding protein 1 phosphorylation. Mol. Cell. Biol., 19: 4525–4534, 1999.9. Peterson, R. T., Desai, B. N., Hardwick, J. S., and Schreiber, S. L.Protein phosphatase 2A interacts with the 70-kDa S6 kinase and isactivated by inhibition of FKBP12-rapamycin-associated protein. Proc.Natl. Acad. Sci. USA, 96: 4438–4442, 1999.10. Jefferies, H. B., Fumagalli, S., Dennis, P. B., Reinhard, C., Pearson,R. B., and Thomas, G. Rapamycin suppresses 5�TOP mRNA translationthrough inhibition of p70s6k. EMBO J., 16: 3693–3704, 1997.11. Sekulic, A., Hudson, C. C., Homme, J. L., Yin, P., Otterness, D. M.,Karnitz, L. M., et al. A direct linkage between the phosphoinositide3-kinase-AKT signaling pathway and the mammalian target of rapamy-cin in mitogen-stimulated and transformed cells. Cancer Res., 60:3504–3513, 2000.12. Reynolds, T. H. T., Bodine, S. C., and Lawrence, J. C., Jr. Controlof Ser2448 phosphorylation in the mammalian target of rapamycin by
insulin and skeletal muscle load. J. Biol. Chem., 277: 17657–17662,2002.
13. Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. TSC2 isphosphorylated and inhibited by Akt and suppresses mTOR signalling.Nat. Cell Biol., 4: 648–657, 2002.
14. Weng, Q. P., Kozlowski, M., Belham, C., Zhang, A., Comb, M. J.,and Avruch, J. Regulation of the p70 S6 kinase by phosphorylation invivo. Analysis using site-specific anti-phosphopeptide antibodies.J. Biol. Chem., 273: 16621–16629, 1998.
15. Weng, Q. P., Andrabi, K., Klippel, A., Kozlowski, M. T., Williams,L. T., and Avruch, J. Phosphatidylinositol 3-kinase signals activation ofp70 S6 kinase in situ through site-specific p70 phosphorylation. Proc.Natl. Acad. Sci. USA, 92: 5744–5748, 1995.
16. Mills, G. B., Lu, Y., Fang, X., Wang, H., Eder, A., Mao, M., et al.The role of genetic abnormalities of PTEN and the phosphatidylinositol3-kinase pathway in breast and ovarian tumorigenesis, prognosis, andtherapy. Semin. Oncol., 28: 125–141, 2001.
17. Nelen, M. R., van Staveren, W. C., Peeters, E. A., Hassel, M. B.,Gorlin, R. J., Hamm, H., et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum. Mol. Genet., 6:1383–1387, 1997.
18. Rhei, E., Kang, L., Bogomolniy, F., Federici, M. G., Borgen, P. I.,and Boyd, J. Mutation analysis of the putative tumor suppressor genePTEN/MMAC1 in primary breast carcinomas. Cancer Res., 57: 3657–3659, 1997.
19. Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I., etal. PTEN, a putative protein tyrosine phosphatase gene mutated inhuman brain, breast, and prostate cancer. Science (Wash. DC), 275:1943–1947, 1997.
20. Neshat, M. S., Mellinghoff, I. K., Tran, C., Stiles, B., Thomas, G.,Petersen, R., et al. Enhanced sensitivity of PTEN-deficient tumors toinhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA, 98: 10314–10319, 2001.
21. Podsypanina, K., Lee, R. T., Politis, C., Hennessy, I., Crane, A.,Puc, J., et al. An inhibitor of mTOR reduces neoplasia and normalizesp70/S6 kinase activity in Pten/� mice. Proc. Natl. Acad. Sci. USA,98: 10320–10325, 2001.
22. Yu, K., Toral-Barza, L., Discafani, C., Zhang, W. G., Skotnicki, J.,Frost, P., et al. mTOR, a novel target in breast cancer: the effect ofCCI-779, an mTOR inhibitor, in preclinical models of breast cancer.Endocr. Relat. Cancer, 8: 249–258, 2001.
23. Zhou, B. P., Hu, M. C., Miller, S. A., Yu, Z., Xia, W., Lin, S. Y.,et al. HER-2/neu blocks tumor necrosis factor-induced apoptosis via theAkt/NF-kappaB pathway. J. Biol. Chem., 275: 8027–8031, 2000.
24. De Benedetti, A., and Harris, A. L. eIF4E expression in tumors: itspossible role in progression of malignancies. Int. J. Biochem. Cell Biol.,31: 59–72, 1999.
25. McKendrick, L., Pain, V. M., and Morley, S. J. Translation initia-tion factor 4E. Int. J. Biochem. Cell Biol., 31: 31–35, 1999.
26. Barlund, M., Forozan, F., Kononen, J., Bubendorf, L., Chen, Y.,Bittner, M. L., et al. Detecting activation of ribosomal protein S6 kinaseby complementary DNA and tissue microarray analysis. J. Natl. CancerInst. (Bethesda), 92: 1252–1259, 2000.
27. Dilling, M. B., Germain, G. S., Dudkin, L., Jayaraman, A. L.,Zhang, X., Harwood, F. C., et al. 4E-binding proteins, the suppressorsof eukaryotic initiation factor 4E, are down-regulated in cells withacquired or intrinsic resistance to rapamycin. J. Biol. Chem., 277:13907–13917, 2002.
28. Shi, Y., Gera, J., Hu, L., Hsu, J. H., Bookstein, R., Li, W., et al.Enhanced sensitivity of multiple myeloma cells containing PTEN mu-tations to CCI-779. Cancer Res., 62: 5027–5034, 2002.
29. Meric, F., Hung, M. C., Hortobagyi, G. N., and Hunt, K. K.HER2/neu in the management of invasive breast cancer. J. Am. Coll.Surg., 194: 488–501, 2002.
30. Hermanto, U., Zong, C. S., and Wang, L. H. ErbB2-overexpressinghuman mammary carcinoma cells display an increased requirement for
1022 Rapamycin Sensitivity in Breast Cancer Cells
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

the phosphatidylinositol 3-kinase signaling pathway in anchorage-inde-pendent growth. Oncogene, 20: 7551–7562, 2001.
31. Hyman, E., Kauraniemi, P., Hautaniemi, S., Wolf, M., Mousses, S.,Rozenblum, E., et al. Impact of DNA amplification on gene expressionpatterns in breast cancer. Cancer Res., 62: 6240–6245, 2002.
32. Barlund, M., Monni, O., Kononen, J., Cornelison, R., Torhorst, J.,Sauter, G., et al. Multiple genes at 17q23 undergo amplification andoverexpression in breast cancer. Cancer Res., 60: 5340–5344, 2000.
33. Wu, G. J., Sinclair, C. S., Paape, J., Ingle, J. N., Roche, P. C., James,C. D., et al. 17q23 amplifications in breast cancer involve the PAT1,RAD51C, PS6K, and SIGma1B genes. Cancer Res., 60: 5371–5375,2000.
34. Fang, Y., Vilella-Bach, M., Bachmann, R., Flanigan, A., and Chen,J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling.Science (Wash. DC), 294: 1942–1945, 2001.
35. Chen, Y., Zheng, Y., and Foster, D. A. Phospholipase D confersrapamycin resistance in human breast cancer cells. Oncogene, 22:3937–3942, 2003.
36. Boulay, A., Zumstein-Mecker, S., Beuvink, I., Zilbermann, F.,Stephan, C., Haller, R., et al. Prolonged effect of the rapamycin deriv-ative RAD001 on p70S6 kinase activity in tumors, skin and peripheralblood lymphocytes derived from a syngeneic rat pancreatic tumor mod-el; correlation with efficacy of intermittent dosing schedules. Proc.Annu. Meet. Am. Assoc. Cancer Res., 43: 602, 2002.
37. Peralba, J. M., DeGraffenried, L., Friedrichs, W., Fulcher, L.,Grunwald, V., Weiss, G., et al. Pharmacodynamic evaluation of CCI-779, an inhibitor of mTOR, in cancer patients. Clin. Cancer Res., 9:2887–2892, 2003.
38. Dudkin, L., Dilling, M. B., Cheshire, P. J., Harwood, F. C., Holl-ingshead, M., Arbuck, S. G., et al. Biochemical correlates of mTORinhibition by the rapamycin ester CCI-779 and tumor growth inhibition.Clin. Cancer Res., 7: 1758–1764, 2001.
39. Stolovich, M., Tang, H., Hornstein, E., Levy, G., Cohen, R., Bae,S. S., et al. Transduction of growth or mitogenic signals into transla-tional activation of TOP mRNAs is fully reliant on the phosphatidyli-
nositol 3-kinase-mediated pathway but requires neither S6K1 nor rpS6phosphorylation. Mol. Cell. Biol., 22: 8101–8113, 2002.40. Nanbru, C., Lafon, I., Audigier, S., Gensac, M. C., Vagner, S.,Huez, G., et al. Alternative translation of the proto-oncogene c-myc byan internal ribosome entry site. J. Biol. Chem., 272: 32061–32066,1997.41. Hashemolhosseini, S., Nagamine, Y., Morley, S. J., Desrivieres, S.,Mercep, L., and Ferrari, S. Rapamycin inhibition of the G1 to Stransition is mediated by effects on cyclin D1 mRNA and proteinstability. J. Biol. Chem., 273: 14424–14429, 1998.42. Hosoi, H., Dilling, M. B., Liu, L. N., Danks, M. K., Shikata, T.,Sekulic, A., et al. Studies on the mechanism of resistance to rapamycinin human cancer cells. Mol. Pharmacol., 54: 815–824, 1998.43. Kawamata, S., Sakaida, H., Hori, T., Maeda, M., and Uchiyama, T.The upregulation of p27Kip1 by rapamycin results in G1 arrest inexponentially growing T-cell lines. Blood, 91: 561–569, 1998.44. Huang, S., Liu, L. N., Hosoi, H., Dilling, M. B., Shikata, T., andHoughton, P. J. p53/p21(CIP1) cooperate in enforcing rapamycin-in-duced G(1) arrest and determine the cellular response to rapamycin.Cancer Res., 61: 3373–3381, 2001.45. Hosoi, H., Dilling, M. B., Shikata, T., Liu, L. N., Shu, L., Ashmun,R. A., et al. Rapamycin causes poorly reversible inhibition of mTORand induces p53-independent apoptosis in human rhabdomyosarcomacells. Cancer Res., 59: 886–894, 1999.46. Shi, Y., Frankel, A., Radvanyi, L. G., Penn, L. Z., Miller, R. G., andMills, G. B. Rapamycin enhances apoptosis and increases sensitivity tocisplatin in vitro. Cancer Res., 55: 1982–1988, 1995.47. Geoerger, B., Kerr, K., Tang, C. B., Fung, K. M., Powell, B.,Sutton, L. N., et al. Antitumor activity of the rapamycin analogueCCI-779 in human primitive neuroectodermal tumor/medulloblastomamodels as single agent and in combination chemotherapy. Cancer Res.,61: 1527–1532, 2001.48. Grunwald, V., DeGraffenried, L., Russel, D., Friedrichs, W. E.,Ray, R. B., and Hidalgo, M. Inhibitors of mTOR reverse doxorubicinresistance conferred by PTEN status in prostate cancer cells. CancerRes., 62: 6141–6145, 2002.
1023Clinical Cancer Research
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

2004;10:1013-1023. Clin Cancer Res Woo-Chul Noh, Wallace H. Mondesire, Junying Peng, et al. CellsDeterminants of Rapamycin Sensitivity in Breast Cancer
Updated version
http://clincancerres.aacrjournals.org/content/10/3/1013
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/10/3/1013.full#ref-list-1
This article cites 47 articles, 33 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/10/3/1013.full#related-urls
This article has been cited by 47 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/10/3/1013To request permission to re-use all or part of this article, use this link
Research. on July 1, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

![Rapamycin Enhances Apoptosis and ... - Cancer Research · [CANCER RESEARCH 55, 1982-1988, May 1, 1995] Rapamycin Enhances Apoptosis and Increases Sensitivity to Cisplatin in Vitro1](https://img.pdfslide.us/doc/110x75/5fc155478e57503b59573a1a/rapamycin-enhances-apoptosis-and-cancer-research-cancer-research-55-1982-1988.jpg)





![Target of Rapamycin Signaling in Plant Stress …Update on Target of Rapamycin Signaling in Plant Stress Responses Target of Rapamycin Signaling in Plant Stress Responses1[OPEN] Liwen](https://img.pdfslide.us/doc/110x75/5f05e4b57e708231d4153f1e/target-of-rapamycin-signaling-in-plant-stress-update-on-target-of-rapamycin-signaling.jpg)





















