Embed Size (px)
Citation preview

9
Colonies of bacterial cell clones containing recombinant DNA molecules.
C H A P T E R
277
Deconstructing the Genome: DNA at High Resolution
The vivid red color of our blood stems from its life-sustainingability to carry oxygen. This ability, in turn, derives from bil-
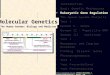
lions of red blood cells suspended in proteinaceous solution, eachone packed with close to 280 million copies of the protein pig-ment known as hemoglobin (Fig. 9.1a). The hemoglobin picks upoxygen in the lungs and transports it to tissues throughout thebody, where, after its release, the oxygen contributes to a multi-tude of metabolic reactions. A normal adult hemoglobin moleculeconsists of four polypeptide chains, two alpha (�) and two beta(�) globins, each surrounding an iron-containing small molecularstructure known as a heme group (Fig. 9.1b). The iron atomwithin the heme sustains a reversible interaction with oxygen,binding it firmly enough to hold it on the trip from lungs to bodytissue but loosely enough to release it where needed. The intri-cately folded � and � chains protect the iron-containing hemesfrom molecules in the cell’s interior. Each hemoglobin moleculecan carry up to four oxygen atoms, one per heme; and it is theseoxygenated hemes that impart a scarlet hue to the pigment mol-ecules and thus to the blood cells that carry them.
Interestingly, the genetically determined molecular composi-tion of hemoglobin changes several times during human devel-opment, enabling the molecule to adapt its oxygen-transportfunction to the varying environments of the embryo, fetus, new-born, and adult (Fig. 9.1c). In the first five weeks after conception,the red blood cells carry embryonic hemoglobin, which consists oftwo alphalike zeta (�) chains and two betalike epsilon (�) chains.Thereafter, throughout the rest of gestation, the cells contain fetalhemoglobin, composed of two bona fide � chains and two �-likegamma (�) chains. Then, shortly before birth, production of adulthemoglobin, composed of two � and two � chains, begins toclimb. By the time an infant reaches three months of age, almostall of his or her hemoglobin is of the adult type.
The various forms of hemoglobin evolved in response to thedifferent environments that exist at different stages of humandevelopment. The early embryo, which is not yet associated with afully functional placenta, has the least access to oxygen in thematernal circulation. The fetus, which is associated with a fully func-tional placenta, has access to the gases dissolved in the maternal
278 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
bloodstream, but the amount of oxygen in the maternal circulationis less than that found in the air that enters our lungs after birth.Both embryonic and fetal hemoglobin evolved to bind oxygenmore tightly than adult hemoglobin would at the lower levels ofoxygen found in the embryonic and fetal environments. All thehemoglobins readily release their oxygen to cells, which have aneven lower level of oxygen than any source of the gas. After birth,when oxygen is abundantly available in the lungs, adult hemoglo-bin, with its more relaxed kinetics of oxygen binding, allows for themost efficient pickup and delivery of the vital gas.
What structure and arrangement of hemoglobin genes facil-itate the crucial switches in the type of protein produced? Andwhat can go wrong with these genes to generate hemoglobin dis-orders? Such disorders are the most common genetic diseases inthe world and include sickle-cell anemia, which arises from analtered � chain, and thalassemia, which results from decreases inthe amount of either �- or �-chain production.
To answer these questions, researchers must have a way oflooking at the hemoglobin genes of individuals with normal andabnormal phenotypes. But these genes lie buried in a diploidhuman genome containing 6 billion base pairs distributed among46 different strings of DNA (the chromosomes) that range in sizefrom 60 million to 360 million base pairs each. In this chapter, wedescribe the powerful tools of modern molecular analysis thatmedical researchers now use to search through these enormouslylong strings of information for genes that may be only severalthousand base pairs in length. Ideally suited to the analysis ofcomplex biological systems, these tools take advantage of
enzymes and biochemical reactions that occur naturally withinthe simplest life-forms, bacterial cells. Researchers refer to thewhole kit of modern tools and reactions as recombinant DNAtechnology. The most important enzymes in this tool kit areones that operate on the DNA molecule itself: restriction enzymesthat cut DNA, ligases that join two molecules together, and poly-merases that synthesize new DNA strands. The most importantuncatalyzed biochemical reaction is hybridization: the bindingtogether of two DNA strands with complementary nucleotidesequences. Hybridization results from the natural propensity ofcomplementary single-stranded molecules to form stable doublehelixes. A final tool in the recombinant DNA kit is the naturalprocess of DNA replication, which occurs within growing coloniesof cells.
Developed during a technological revolution that began inthe mid-1970s, recombinant DNA technology makes it possibleto characterize DNA molecules directly, rather than indirectlythrough the phenotypes they produce. Geneticists use recombi-nant DNA techniques to gather information unobtainable in anyother way, or to analyze the results of breeding and cytologicalstudies with greater speed and accuracy than ever before.
Two general themes recur in our discussion. First, the newmolecular tools of genetic analysis emerged from a knowledge ofDNA structure and function. For example, complementary basepairing between similar or identical stretches of DNA is the basisof hybridization techniques that use labeled DNA probes to iden-tify related DNA molecules. Second, the speed, sensitivity, andaccuracy of the new tools make it possible to answer questions
(a)
(b) (c)0 6 12 18 24 30 36 Birth 6 12 18 24 30 36 42 48
0
10
20
30
40
50
Weeks of development
Am
ount
of h
emog
lobi
n pr
otei
n w
ith d
iffer
ent t
ypes
of c
hain
s
α
βα α
β β γ
εζ
δ
Heme group
Figure 9.1 Hemoglobin is composed of four polypeptide chains that changeduring development. (a) Scanning electron micrograph of adult human red blood cellsloaded with one kind of protein—hemoglobin. (b) Adult hemoglobin consists almostentirely of two � and two � polypeptide chains. (c) The hemoglobin, carried by red bloodcells, switches slowly during human development from an embryonic form containing two�-like � chains and two �-like � chains, to a fetal form containing two � chains and two �-like � chains, and finally to the adult form containing two � and two � chains. In a smallpercentage of adult hemoglobin molecules, a �-like � chain replaces the actual � chain.

that were impossible to resolve just a short time ago. In onerecent study, for instance, researchers used a protocol for makingmany copies of a specific DNA segment to trace the activity of theAIDS virus and found that, contrary to prior belief, the virus, afterinfecting a person, does not become latent inside all the cells itenters; it remains active in certain cells of the lymph nodes andadenoids.
As we illustrate the power of the modern tools for analyzing aseemingly simple biological system—the hemoglobins—that turnedout to be surprisingly complex, we describe how researchers userecombinant DNA technology to carry out five basic operations:
■ Cut the enormously long strings of DNA into much smallerfragments with scissorslike restriction enzymes; and separate
the small fragments according to size through gelelectrophoresis.
■ Isolate, amplify, and purify the fragments through molecularcloning.
■ Use purified DNA fragments as hybridization probes toidentify the presence of similar sequences in libraries ofclones or in complex mixtures of DNA or RNA molecules.
■ Rapidly isolate and amplify previously defined genomic ormRNA sequences from new individuals or cell sourcesthrough the polymerase chain reaction (PCR).
■ Determine the precise sequence of bases within isolatedDNA fragments.
Fragmenting Complex Genomes into Bite-Size Pieces for Analysis 279
■■■
Fragmenting Complex Genomes into Bite-Size Pieces for Analysis
Every intact diploid human body cell, including the precursorsof red blood cells, carries two nearly identical sets of 3 billionbase pairs of information that, when unwound, extend 2 m inlength and contain two copies of 40,000–60,000 genes. If youcould enlarge the cell nucleus to the size of a basketball, theunwound DNA would have the diameter of a fishing line and alength of 200 km. This is much too much material and infor-mation to study as a whole. To reduce its complexity,researchers first cut the genome into “bite-size” pieces.
Restriction Enzymes Fragment the Genome at Specific SitesResearchers use restriction enzymes to cut the DNAreleased from the nuclei of cells at specific sites. These well-defined cuts generate fragments suitable for manipulationand characterization. A restriction enzyme recognizes aspecific sequence of bases anywhere within the genome andthen severs two covalent bonds (one in each strand) in thesugar-phosphate backbone at particular positions within ornear that sequence. The fragments generated by restrictionenzymes are referred to as restriction fragments, and theact of cutting is often called digestion.
Restriction enzymes originate in and can be purified frombacterial cells. In these prokaryotic cells, the enzymes protectagainst invasion by viruses through the digestion of viral DNA.Bacteria shield their own DNA from digestion by indigenousrestriction enzymes through the selective addition of methylgroups (–CH3) to the restriction recognition sites in their DNA.In the test tube, restriction enzymes from bacteria recognizetarget sequences of 4–8 bp in DNA isolated from any otherorganism, and cut the DNA at or near these sites. Table 9.1 liststhe names, recognition sequences, and microbial origins of just10 of the more than 100 commonly used restriction enzymes.
TABLE 9.1 Ten Commonly Used RestrictionEnzymes
Sequence of Enzyme Recognition Site Microbial Origin
TaqI Thermus aquaticus YTI
RsaI Rhodopseudomonas sphaeroides
Sau3AI Staphylococcus aureus 3A
EcoRl Escherichia coli
BamHI Bacillusamyloliquefaciens H.
HindIII Haemophilus influenzae
KpnI Klebsiella pneumoniaeOK8
ClaI Caryophanon latum
BssHII Bacillusstearothermophilus
NotI Nocardia otitidiscaviarum
5'
5'
3'
3'
5'
5'
3'
3'
5'
5'
3'
3'
5'
5'
3'
3'
5'
5'
3'
3'
5'
5'
3'
5'
3'
3'
5'
3'
5'
5'
3'
3'
C
G
A
T A
T
C
G
C
G
A
TA
T
C
G
C
G C
GA
T
A
TA
T
A
T
C
G
C
G C
G
C
GA
TA
T
C
G C
G A
T
A
TA
T
A
T
C
G
C
G
C
G
C
G
C
G C
G
C
G
C
G
C
G
C
G
C
G
C
G
C
GC
G
C
G
C
G
C
G
C
GA
T A
T
C
GC
G A
T
A
TA
T
A
T
C
G
A
T A
T
C
G
5'
5'
3'
3'
5'
5'
3'
3'

280 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Most of the tools and techniques for cloning and analyzing DNAfragments emerged from studies of bacteria and the viruses thatinfect them. Molecular biologists had observed, for example, thatviruses able to grow abundantly on one strain of bacteria grewpoorly on a closely related strain of the same bacteria. Whileexamining the mechanisms of this discrepancy, they discoveredrestriction enzymes.
To follow the story, one must know that researchers comparerates of viral proliferation in terms of plating efficiency: the fractionof viral particles that enter and replicate inside a host bacterialcell, causing the cell to lyse and release viral progeny, which goon to infect and replicate inside neighboring cells, which lyse andrelease further virus particles, which continue this process. Whena petri plate is coated with a continuous “lawn” of bacterial cells,an active viral infection can be observed as a visibly cleared spot,or plaque, where bacteria have been eliminated (see Fig. 7.18).The plating efficiency of lambda virus grown on E. coli C is nearly1.0. This means that 100 original virus particles will cause close to100 plaques on a lawn of E. coli C bacteria.
The plating efficiency of the same virus grown on E. coli K12is only 1 in 104 or 0.0001. The ability of a bacterial strain to pre-vent the replication of an infecting virus, in this case the growthof lambda on E. coli K12, is called restriction.
Interestingly, restriction is rarely absolute. Although lambdavirus grown on E. coli K12 produces almost no progeny (virusesinfect cells but can’t replicate inside them), a few viral particles domanage to proliferate. If their progeny are then tested on E. coliK12, the plating efficiency is nearly 1.0. The phenomenon inwhich growth on a restricting host modifies a virus so that suc-ceeding generations grow more efficiently on that same host isknown as modification.
What mechanisms account for restriction and modifica-tion? Studies following viral DNA after bacterial infection foundthat during restriction, the viral DNA is broken into pieces anddegraded (Fig. A). When the enzyme responsible for the initialbreakage was isolated, it was found to be an endonuclease, anenzyme that breaks the phosphodiester bonds in the viral DNAmolecule, usually making double-strand cuts at a specificsequence in the viral chromosome. Because this breakagerestricts the biological activity of the viral DNA, researcherscalled the enzymes that accomplish it restriction enzymes.Subsequent studies showed that the small percentage of viralDNA that escapes digestion and goes on to generate new viralparticles has been modified by the addition of methyl groupsduring its replication in the host cell. Researchers named theenzymes that add methyl groups to specific DNA sequencesmodification enzymes.
In nature, pairs of restriction-modification enzymes protectbacterial cells from foreign DNA. Unmodified DNA of any typefalls prey to digestion by restriction enzymes. But as described inthe Fast Forward box in Chapter 6 on pp. 170, a bacterial cell’sown DNA is newly methylated and protected from the same
Serendipity in Science: The Discovery of Restriction Enzymes
Genetics and Society
E. coli K12—rare cell
E. coli CLambdavirusparticle
E. coli K12—most cells
DNA modification
me me
me me
DNA restriction
Replication
Replication
Lysis—bacterium dies
Lysis—bacterium dies
Bacterium lives. Noviruses produced
Figure A Operation of the restriction enzyme/modificationsystem in nature. E. coli C does not have a functional restrictionenzyme/modification system and is susceptible to infection by thelambda phage. In contrast, E. coli K12 generally resists infection by theviral particles produced during an E. coli C-phage infection.This isbecause E. coli K12 makes several restriction enzymes, including EcoRI,which cut the lambda DNA molecule before it can express itself.However, in rare K12 cells, the lambda DNA replicates before restrictionenzymes can attack it, and the newly replicated lambda DNA is thenmodified by an enzyme that protects its recognition sites from attack bythe host cell’s restriction enzymes.This lambda DNA can now generatephage particles, which eventually destroy the bacterial cell.

Fragmenting Complex Genomes into Bite-Size Pieces for Analysis 281
For the majority of these enzymes, the recognition site contains4–6 base pairs and exhibits a kind of palindromic symmetry inwhich the base sequences of each of the two DNA strands areidentical when read in the 5′-to-3′ direction. Because of this,base pairs on either side of a central line of symmetry are mir-ror images of each other. Each enzyme always cuts at the sameplace relative to its specific recognition sequence, and mostenzymes make their cuts in one of two ways: either straightthrough both DNA strands right at the line of symmetry to pro-duce fragments with blunt ends, or displaced equally in oppo-site directions from the line of symmetry by one or more basesto generate fragments with single-stranded ends (Fig. 9.2).Geneticists often refer to these protruding single strands assticky ends. They are considered “sticky” because they are freeto base pair with a complementary sequence from the DNA ofany organism cut by the same restriction enzyme. (TheGenetics and Society box on p. 280 of this chapter and the FastForward box on p. 170 of Chapter 6 contain more informationabout restriction enzymes.)
Different Restriction Enzymes ProduceFragments of Different LengthsFor each restriction enzyme, it is possible to calculate the aver-age length of the fragments the enzyme will generate and thenuse that information to estimate the approximate number anddistribution of recognition sites in a genome. The estimatedepends on two simplifying assumptions: First, that each of thefour bases occurs in equal proportions such that a genome iscomposed of 25% A, 25% T, 25% G, and 25% C; second, thatthe bases are randomly distributed in the DNA sequence.Although these assumptions are never precisely valid, theyenable us to determine the average distance between recogni-
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
A A
T T
C
G
C
G C
G
C
G
A
T
C
G
A
T A
T
C
G
C
G C
G
A
T
A
TA
T A
TA
T
A
T
C
G
C
G C
G
A
T
A
TA
T
A
T
A
T
C
G
C
G
A
T
C
GC
G
C
G
A
T A
T A
T
A
T C
G
C
G
A
T
C
GC
G
C
G
A
T A
T A
T
A
T
RsaI
EcoRI
KpnI
(a) Blunt ends (RsaI)
(c) Sticky 3' ends (KpnI)
(b) Sticky 5' ends (EcoRI)
Sugar-phosphatebackbone
5' overhangs
3' overhangs
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
5'
3'
C
G
A
T
A
T
C
GC
G
C
G
C
G
A
T
A
T A
T
Figure 9.2 Restriction enzymes cut DNA molecules atspecific locations to produce restriction fragments witheither blunt or sticky ends. (a) The restriction enzyme RsaIproduces blunt-ended restriction fragments. (b) EcoRI produces stickyends with a 5′ overhang. (c) KpnI produces sticky ends with a 3′overhang.
digestive fate by modification enzymes that operate with eachround of replication. Modification enzymes also ensure the sur-vival of those few viral DNA molecules that happen to evadedigestion. These viral DNA molecules, modified by methylation,then use the host cell’s machinery to produce new viral particles.
Biologists have identified a large number of restriction-modification systems in a variety of bacterial strains. Purificationof the systems has yielded a mainstay of recombinant DNA tech-nology: the battery of restriction enzymes used to cut DNA invitro for cloning, mapping, and ligation (see Table 9.1).
This example of serendipity in science sheds some light on thedebate between administrators who distribute and oversee researchfunding and scientists who carry out the research. Microbial investi-gators did not set out to find restriction enzymes; they could nothave known that these enzymes would be one of their finds. Rather,they sought to understand the mechanisms by which viruses infectand proliferate in bacteria. Along the way, they discovered restric-tion enzymes and how they work. Their insights have had broad
applications in genetic engineering and biotechnology. DNAcloning, for example, has produced, among many other medicallyuseful products, human insulin for diabetics, which provokes farfewer allergic reactions than the pig insulin that preceded it; tissueplasminogen activator (TPA), a protein that helps dissolve bloodclots; and gamma interferon, a protein produced by the humanimmune system that protects laboratory-grown human cells frominfection by herpes and hepatitis viruses. The politicians and admin-istrators in charge of allocating funds often want to direct researchspending to urgent health or agricultural problems, assuming thatmuch of the basic research upon which such studies depend hasbeen completed. The scientists in charge of laboratory research callfor a broad distribution of funds to all projects investigating inter-esting biological phenomena, arguing that we still know relativelylittle about molecular biology and that the insights necessary forunderstanding disease may emerge from studies on any organism.The validity of both views suggests the need for a balancedapproach to the funding of research activities.
tion sites of any length by the general formula 4n, where n isthe number of bases in the site (Fig. 9.3).
The Size of the Recognition Site Is the PrimaryDeterminant of Fragment LengthAccording to the 4n formula, RsaI, which recognizes the four-base-sequence GTAC, will cut on average once every 44, or

every 256 base pairs (bp), creating fragments averaging 256 bpin length. By comparison, the enzyme EcoRI, which recognizesthe six-base-sequence GAATTC, will cut on average onceevery 46, or 4096 bp; since 1000 base pairs � 1 kilobase pair,researchers often round off this large number to roughly 4.1kilobase pairs, abbreviated 4.1 kb. Similarly, an enzyme such asNotI, which recognizes the eight bases GCGGCCGC, will cuton average every 48 bp, or every 65.5 kb. Note, however, thatbecause the actual distances between restriction sites for anyenzyme vary considerably, very few of the fragments producedby the three enzymes mentioned here will be precisely 65.5 kb,4.1 kb, or 256 bp in length.
The Timing of Exposure to a Restriction Enzyme Helps Determine Fragment SizeGeneticists often need to produce DNA fragments of a particu-lar length—larger ones to study the organization of a chromo-somal region, smaller ones to examine a whole gene, and onesthat are smaller still for DNA sequence analysis (that is, for thedetermination of the precise order of bases in a DNA fragment).
If their goal is 4 kb fragments, they have a range of six-base-cutter enzymes to choose from. By exposing the DNA to a six-base cutter for a long enough time, they give the restrictionenzyme ample opportunity for digestion. The result is a com-plete digest in which the DNA has been cut at every one of therecognition sites it contains. But what if the researchers need 20kb fragments—a length in between that produced by a six-basecutter and an eight-base cutter? The answer is a partial digest:a cutting obtained by controlling the amount of enzyme or theamount of time the DNA is exposed to the restriction enzyme.With EcoRI, for example, exposing the DNA just long enoughfor the enzyme to cut on average only one of five target siteswould produce fragments averaging 20 kb in length (Fig. 9.4).Researchers determine the correct timing by trial and error.Complete or partial digestion with different enzymes producesfragments of average lengths that are appropriate for differentexperimental uses.
Different Restriction Enzymes ProduceDifferent Numbers of Fragments from the Same GenomeWe have seen that the four-base cutter RsaI cuts the genome onaverage every 44 (256) bp. If you exposed the haploid humangenome with its 3 billion bp to RsaI for a sufficient time underappropriate conditions, you would ensure that all of the recog-nition sites in the genome that can be cleaved will be cleaved,and you would get
By comparison, the six-base cutter EcoRI cuts the DNA onaverage every 46 (4096) bp, or every 4.1 kb. If you exposed thehaploid human genome with its 3 billion bp, or 3 million kb, toEcoRI in the proper way, you would get
3,000,000,000bp
~4100bp�
~700,000 fragments that are~4.1 kb in average length
3,000,000,000bp
~256bp�
~12,000,000 fragments that are~256 bp in average length
282 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Intact human DNA
Enzyme cuts at one random site in five.
EcoRI sites
Figure 9.4 Partial digests. By reducing the time allotted for thereaction, you can ensure that an enzyme actually cuts only a subset ofthe total number of recognition sites within a DNA sample. Thisdiagram shows three copies of the same DNA sequence. The chosenreaction time allowed for the cutting of only 20% of all the EcoRI sites.Since the cutting is at random, the particular 20% of sites at which cutsoccur is different, even on the identical DNA molecules shown here.
2. Probability that a six-base recognition site will be found =
1. Probability that a four-base recognition site will be found in a genome =
(a)
(b)
1. Four-base
RsaI sites
EcoRI sites
NotI sites
2. Six-base
3. Eight-base
1 kb
1/4 1/4 1/4 1/4 = 1/256
1/4 1/4 1/4 1/4 1/4 1/4 = 1/4,096
Intact Human DNA
Figure 9.3 The number of base pairs in a restriction enzymerecognition site determines the average distance betweenrestriction sites in a genome and thus the size of fragmentsproduced. RsaI recognizes and cuts at a 4 bp site, EcoRI cuts at a 6 bpsite, and Not I cuts at an 8 bp site. The actual distances betweenrestriction sites in the genome are variable.

And if you exposed the same haploid human genome tothe eight-base cutter NotI, which cuts on average every 48
(65,536) bp, or 65.5 kb, you would obtain
Clearly, the larger the recognition site, the smaller thenumber of fragments generated by enzymatic digestion.
Restriction enzymes were first used to study the very smallgenomes of viruses like bacteriophage lambda (), whosegenome has a length of approximately 48.5 kb, and the animaltumor virus SV40, whose genome has a length of 5.2 kb. Wenow know that the six-base cutter EcoRI digests lambda DNAinto 5 fragments, and the four-base cutter RsaI digests SV40into 12 fragments. But when molecular biologists first usedrestriction enzymes to digest these viral genomes, they alsoneeded a tool that could distinguish the different fragments in agenome from each other and determine their sizes. That toolwas gel electrophoresis.
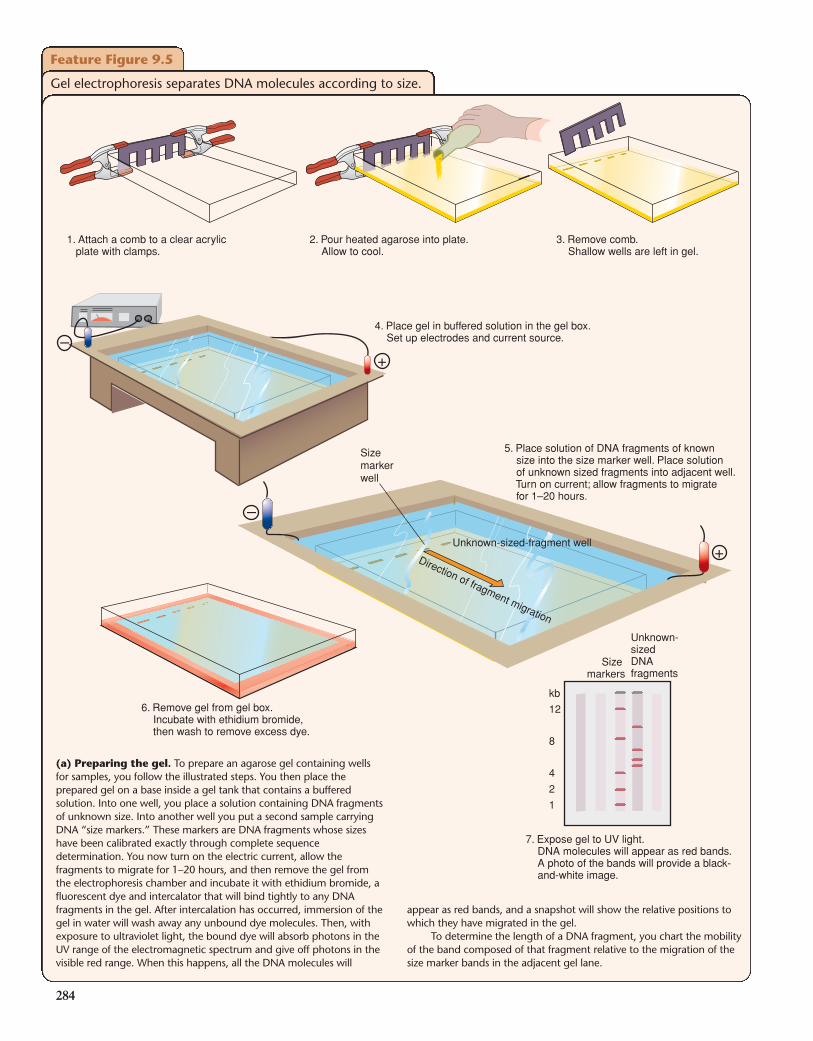
Gel Electrophoresis Distinguishes DNAFragments According to SizeElectrophoresis is the movement of charged molecules in anelectric field. Biologists use it to separate many different typesof molecules, for example, DNA of one length from DNA ofother lengths, DNA from protein, or one kind of protein fromanother. In this discussion, we focus on its application to theseparation of DNA fragments of varying length in a gel (Fig.9.5). To carry out such a separation, you place a solution ofDNA molecules into indentations called wells at one end of aporous gel-like matrix. When you then place the gel in abuffered aqueous solution and set up an electric field betweenelectrodes affixed at either end, the electric field causes allcharged molecules in the wells to migrate in the direction of theelectrode having an opposite charge. Since all of the phosphategroups in the backbone of DNA carry a net negative charge ina solution near neutral pH, DNA molecules are pulled througha gel toward the positive electrode.
Several variables determine the rate at which DNA mole-cules (or any other molecules) move during electrophoresis.These variables are the strength of the electric field appliedacross the gel, the composition of the gel, the charge per unit vol-ume of molecule (known as charge density), and the physicalsize of the molecule. The only one of these variables that actuallydiffers among any set of linear DNA fragments migrating in aparticular gel is size. This is because all molecules placed in awell are subjected to the same electric field and the same gelmatrix; and all DNA molecules have the same charge density(because the charge of all nucleotide pairs is nearly identical). Asa result, only differences in size cause different linear DNA mol-ecules to migrate at different speeds during electrophoresis.
With linear DNA molecules, differences in size are pro-portional to differences in length: the longer the molecule, thelarger the volume it will occupy as a random coil. The larger thevolume a molecule occupies, the less likely it is to find a pore
3,000,000,000bp
~65,500bp�
~46,000 fragments that are~65.5 kb in average length
in the matrix big enough to squeeze through and the more oftenit will bump into the matrix. And the more often the moleculebumps into the matrix, the lower its rate of migration (alsoreferred to as its mobility). With this background, you can fol-low the steps of Fig. 9.5a to determine the length of the restric-tion fragments in the DNA under analysis.
When electrophoresis is completed, the gel is incubatedwith a fluorescent DNA-binding dye called ethidium bromide.After the unbound dye has been washed away, it is easy to visu-alize the DNA by placing the gel under an ultraviolet light. Theactual size of restriction fragments observed on gels is alwaysdetermined by comparison to migration distances of knownmarker fragments that are subjected to electrophoresis in anadjacent lane of the gel.
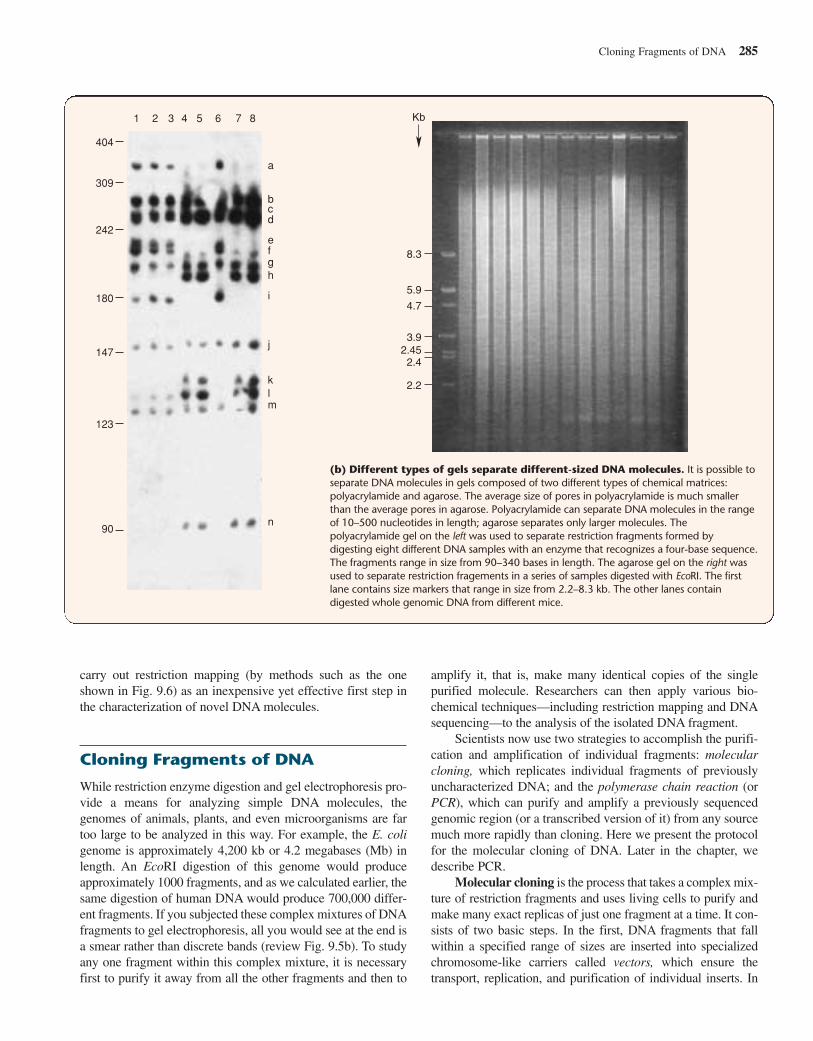
DNA molecules range in size from small fragments of lessthan 10 bp to whole human chromosomes that have an averagelength of 130,000,000 bp. No one sizing procedure has thecapacity to separate molecules throughout this enormous range.To detect DNA molecules in different size ranges, researchersuse a variety of protocols based mainly on two kinds of gels:polyacrylamide (formed by covalent bonding between acryl-amide monomers), which is good for distinguishing smallerDNA fragments; and agarose (formed by the noncovalent asso-ciation of agarose polymers), which is suitable for looking atlarger fragments. Figure 9.5b illustrates these differences.
Restriction Maps Provide a Rough Roadmap of Virus Genomes and Other Purified DNA FragmentsResearchers can use restriction enzymes not only as molecularscissors to create DNA fragments of different sizes but also asan analytic tool to create maps of viral genomes and other puri-fied DNA fragments. These maps, called restriction maps,show the relative order and distances between multiple restric-tion sites, which thus act as landmarks along a DNA molecule.
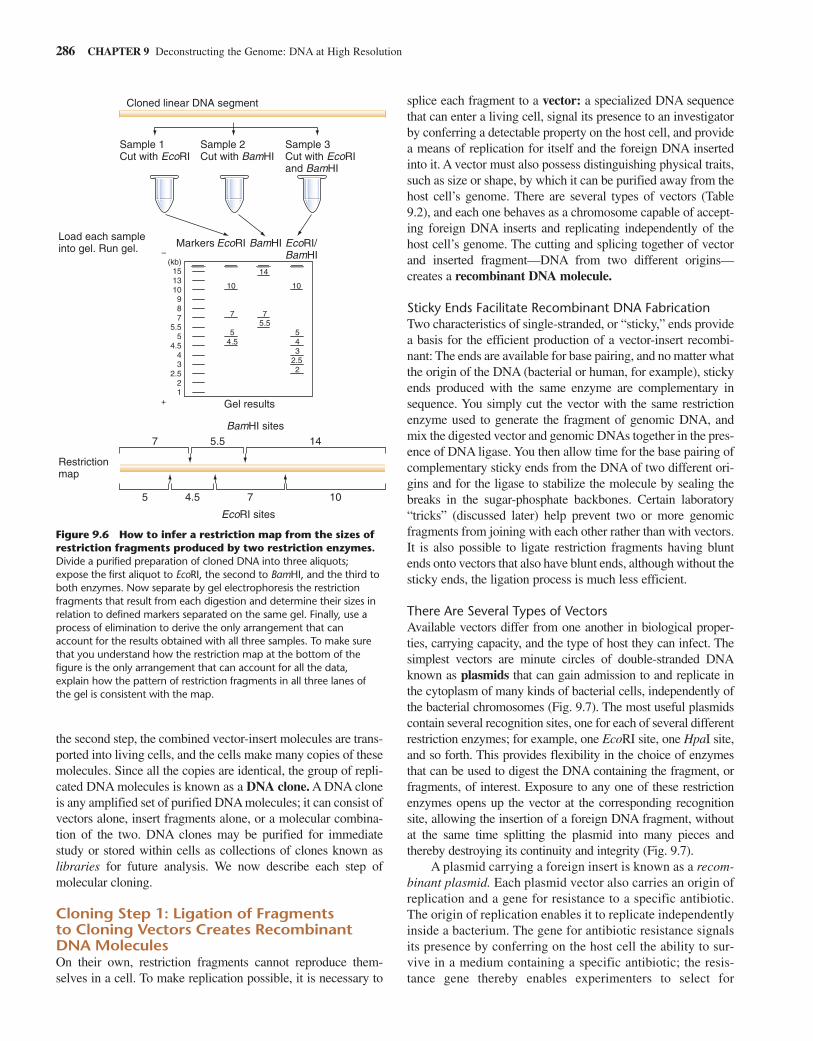
There are numerous approaches to the derivation of a restric-tion map. One of the most commonly used methods involvesdigestion with multiple restriction enzymes—alone or mixedtogether—followed by gel electrophoresis to visualize the frag-ments produced. If the relative arrangement of sites for the vari-ous restriction enzymes employed does not create too manyfragments, the data obtained can provide enough information topiece together a map showing the position of each restriction site.Figure 9.6 shows how a process of elimination allows you to inferthe arrangement of restriction sites consistent with the results ofdigestion, using either of two enzymes alone or both enzymessimultaneously. This is one way to make a restriction map.
Today, molecular biologists can use automated DNAsequencing (along with computer analysis) to rapidly obtaindetailed information on the size and sequence of any DNA mol-ecule in the size range of SV40 or bacteriophage ; we describesuch sequencing techniques later in this chapter. Although thesequence information obtained in this way can immediatelypinpoint restriction sites for enzymes whose recognitionsequences are known (see Table 9.1), many investigators still
Fragmenting Complex Genomes into Bite-Size Pieces for Analysis 283
Feature Figure 9.5
Gel electrophoresis separates DNA molecules according to size.
+
+
–
–
1. Attach a comb to a clear acrylic plate with clamps.
2. Pour heated agarose into plate. Allow to cool.
3. Remove comb. Shallow wells are left in gel.
4. Place gel in buffered solution in the gel box. Set up electrodes and current source.
5. Place solution of DNA fragments of known size into the size marker well. Place solution of unknown sized fragments into adjacent well. Turn on current; allow fragments to migrate for 1–20 hours.
6. Remove gel from gel box. Incubate with ethidium bromide, then wash to remove excess dye.
7. Expose gel to UV light. DNA molecules will appear as red bands. A photo of the bands will provide a black- and-white image.
Size marker well
Unknown-sized-fragment well
Direction of fragment migration
kb
12
8
4
2
1
Size markers
Unknown-sizedDNAfragments
(a) Preparing the gel. To prepare an agarose gel containing wellsfor samples, you follow the illustrated steps. You then place theprepared gel on a base inside a gel tank that contains a bufferedsolution. Into one well, you place a solution containing DNA fragmentsof unknown size. Into another well you put a second sample carryingDNA “size markers.” These markers are DNA fragments whose sizeshave been calibrated exactly through complete sequencedetermination. You now turn on the electric current, allow thefragments to migrate for 1–20 hours, and then remove the gel fromthe electrophoresis chamber and incubate it with ethidium bromide, afluorescent dye and intercalator that will bind tightly to any DNAfragments in the gel. After intercalation has occurred, immersion of thegel in water will wash away any unbound dye molecules. Then, withexposure to ultraviolet light, the bound dye will absorb photons in theUV range of the electromagnetic spectrum and give off photons in thevisible red range. When this happens, all the DNA molecules will
appear as red bands, and a snapshot will show the relative positions towhich they have migrated in the gel.
To determine the length of a DNA fragment, you chart the mobilityof the band composed of that fragment relative to the migration of thesize marker bands in the adjacent gel lane.
284
carry out restriction mapping (by methods such as the oneshown in Fig. 9.6) as an inexpensive yet effective first step inthe characterization of novel DNA molecules.
Cloning Fragments of DNA
While restriction enzyme digestion and gel electrophoresis pro-vide a means for analyzing simple DNA molecules, thegenomes of animals, plants, and even microorganisms are fartoo large to be analyzed in this way. For example, the E. coligenome is approximately 4,200 kb or 4.2 megabases (Mb) inlength. An EcoRI digestion of this genome would produceapproximately 1000 fragments, and as we calculated earlier, thesame digestion of human DNA would produce 700,000 differ-ent fragments. If you subjected these complex mixtures of DNAfragments to gel electrophoresis, all you would see at the end isa smear rather than discrete bands (review Fig. 9.5b). To studyany one fragment within this complex mixture, it is necessaryfirst to purify it away from all the other fragments and then to
Cloning Fragments of DNA 285
1
404
a
bcd
efgh
i
j
klm
n
309
242
180
147
123
90
2 3 4 5 6 7 8 Kb
8.3
5.9
4.7
3.92.452.4
2.2
(b) Different types of gels separate different-sized DNA molecules. It is possible toseparate DNA molecules in gels composed of two different types of chemical matrices:polyacrylamide and agarose. The average size of pores in polyacrylamide is much smallerthan the average pores in agarose. Polyacrylamide can separate DNA molecules in the rangeof 10–500 nucleotides in length; agarose separates only larger molecules. Thepolyacrylamide gel on the left was used to separate restriction fragments formed bydigesting eight different DNA samples with an enzyme that recognizes a four-base sequence.The fragments range in size from 90–340 bases in length. The agarose gel on the right wasused to separate restriction fragements in a series of samples digested with EcoRI. The firstlane contains size markers that range in size from 2.2–8.3 kb. The other lanes containdigested whole genomic DNA from different mice.
amplify it, that is, make many identical copies of the singlepurified molecule. Researchers can then apply various bio-chemical techniques—including restriction mapping and DNAsequencing—to the analysis of the isolated DNA fragment.
Scientists now use two strategies to accomplish the purifi-cation and amplification of individual fragments: molecularcloning, which replicates individual fragments of previouslyuncharacterized DNA; and the polymerase chain reaction (orPCR), which can purify and amplify a previously sequencedgenomic region (or a transcribed version of it) from any sourcemuch more rapidly than cloning. Here we present the protocolfor the molecular cloning of DNA. Later in the chapter, wedescribe PCR.
Molecular cloning is the process that takes a complex mix-ture of restriction fragments and uses living cells to purify andmake many exact replicas of just one fragment at a time. It con-sists of two basic steps. In the first, DNA fragments that fallwithin a specified range of sizes are inserted into specializedchromosome-like carriers called vectors, which ensure thetransport, replication, and purification of individual inserts. In
the second step, the combined vector-insert molecules are trans-ported into living cells, and the cells make many copies of thesemolecules. Since all the copies are identical, the group of repli-cated DNA molecules is known as a DNA clone. A DNA cloneis any amplified set of purified DNA molecules; it can consist ofvectors alone, insert fragments alone, or a molecular combina-tion of the two. DNA clones may be purified for immediatestudy or stored within cells as collections of clones known aslibraries for future analysis. We now describe each step of molecular cloning.
Cloning Step 1: Ligation of Fragments to Cloning Vectors Creates RecombinantDNA MoleculesOn their own, restriction fragments cannot reproduce them-selves in a cell. To make replication possible, it is necessary to
splice each fragment to a vector: a specialized DNA sequencethat can enter a living cell, signal its presence to an investigatorby conferring a detectable property on the host cell, and providea means of replication for itself and the foreign DNA insertedinto it. A vector must also possess distinguishing physical traits,such as size or shape, by which it can be purified away from thehost cell’s genome. There are several types of vectors (Table9.2), and each one behaves as a chromosome capable of accept-ing foreign DNA inserts and replicating independently of thehost cell’s genome. The cutting and splicing together of vectorand inserted fragment—DNA from two different origins—creates a recombinant DNA molecule.
Sticky Ends Facilitate Recombinant DNA FabricationTwo characteristics of single-stranded, or “sticky,” ends providea basis for the efficient production of a vector-insert recombi-nant: The ends are available for base pairing, and no matter whatthe origin of the DNA (bacterial or human, for example), stickyends produced with the same enzyme are complementary insequence. You simply cut the vector with the same restrictionenzyme used to generate the fragment of genomic DNA, andmix the digested vector and genomic DNAs together in the pres-ence of DNA ligase. You then allow time for the base pairing ofcomplementary sticky ends from the DNA of two different ori-gins and for the ligase to stabilize the molecule by sealing thebreaks in the sugar-phosphate backbones. Certain laboratory“tricks” (discussed later) help prevent two or more genomicfragments from joining with each other rather than with vectors.It is also possible to ligate restriction fragments having bluntends onto vectors that also have blunt ends, although without thesticky ends, the ligation process is much less efficient.
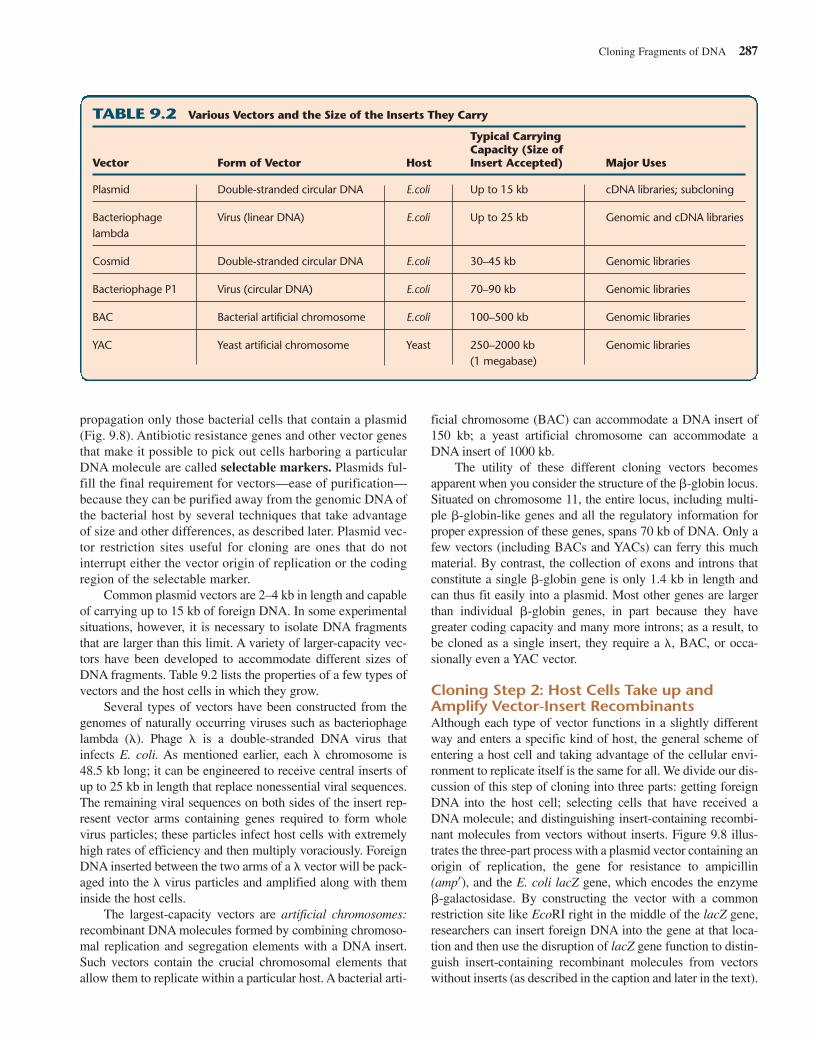
There Are Several Types of VectorsAvailable vectors differ from one another in biological proper-ties, carrying capacity, and the type of host they can infect. Thesimplest vectors are minute circles of double-stranded DNAknown as plasmids that can gain admission to and replicate inthe cytoplasm of many kinds of bacterial cells, independently ofthe bacterial chromosomes (Fig. 9.7). The most useful plasmidscontain several recognition sites, one for each of several differentrestriction enzymes; for example, one EcoRI site, one HpaI site,and so forth. This provides flexibility in the choice of enzymesthat can be used to digest the DNA containing the fragment, orfragments, of interest. Exposure to any one of these restrictionenzymes opens up the vector at the corresponding recognitionsite, allowing the insertion of a foreign DNA fragment, withoutat the same time splitting the plasmid into many pieces andthereby destroying its continuity and integrity (Fig. 9.7).
A plasmid carrying a foreign insert is known as a recom-binant plasmid. Each plasmid vector also carries an origin ofreplication and a gene for resistance to a specific antibiotic.The origin of replication enables it to replicate independentlyinside a bacterium. The gene for antibiotic resistance signalsits presence by conferring on the host cell the ability to sur-vive in a medium containing a specific antibiotic; the resis-tance gene thereby enables experimenters to select for
286 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Load each sampleinto gel. Run gel.
Sample 1Cut with EcoRI
Sample 2Cut with BamHI
Sample 3Cut with EcoRI and BamHI
Cloned linear DNA segment
EcoRI sites
5 4.5 7 10
7 5.5 14
Restrictionmap
BamHI sites
–(kb)
151310987
5.55
4.543
2.521
+
Markers EcoRI BamHI EcoRI/BamHI
10 10
5
7
4.5 43
2.52
5
7
14
5.5
Gel results
Figure 9.6 How to infer a restriction map from the sizes ofrestriction fragments produced by two restriction enzymes.Divide a purified preparation of cloned DNA into three aliquots;expose the first aliquot to EcoRI, the second to BamHI, and the third toboth enzymes. Now separate by gel electrophoresis the restrictionfragments that result from each digestion and determine their sizes inrelation to defined markers separated on the same gel. Finally, use aprocess of elimination to derive the only arrangement that canaccount for the results obtained with all three samples. To make surethat you understand how the restriction map at the bottom of thefigure is the only arrangement that can account for all the data,explain how the pattern of restriction fragments in all three lanes ofthe gel is consistent with the map.
propagation only those bacterial cells that contain a plasmid(Fig. 9.8). Antibiotic resistance genes and other vector genesthat make it possible to pick out cells harboring a particularDNA molecule are called selectable markers. Plasmids ful-fill the final requirement for vectors—ease of purification—because they can be purified away from the genomic DNA ofthe bacterial host by several techniques that take advantageof size and other differences, as described later. Plasmid vec-tor restriction sites useful for cloning are ones that do notinterrupt either the vector origin of replication or the codingregion of the selectable marker.
Common plasmid vectors are 2–4 kb in length and capableof carrying up to 15 kb of foreign DNA. In some experimentalsituations, however, it is necessary to isolate DNA fragmentsthat are larger than this limit. A variety of larger-capacity vec-tors have been developed to accommodate different sizes ofDNA fragments. Table 9.2 lists the properties of a few types ofvectors and the host cells in which they grow.
Several types of vectors have been constructed from thegenomes of naturally occurring viruses such as bacteriophagelambda (). Phage is a double-stranded DNA virus thatinfects E. coli. As mentioned earlier, each chromosome is48.5 kb long; it can be engineered to receive central inserts ofup to 25 kb in length that replace nonessential viral sequences.The remaining viral sequences on both sides of the insert rep-resent vector arms containing genes required to form wholevirus particles; these particles infect host cells with extremelyhigh rates of efficiency and then multiply voraciously. ForeignDNA inserted between the two arms of a vector will be pack-aged into the virus particles and amplified along with theminside the host cells.
The largest-capacity vectors are artificial chromosomes:recombinant DNA molecules formed by combining chromoso-mal replication and segregation elements with a DNA insert.Such vectors contain the crucial chromosomal elements thatallow them to replicate within a particular host. A bacterial arti-
ficial chromosome (BAC) can accommodate a DNA insert of150 kb; a yeast artificial chromosome can accommodate aDNA insert of 1000 kb.
The utility of these different cloning vectors becomesapparent when you consider the structure of the �-globin locus.Situated on chromosome 11, the entire locus, including multi-ple �-globin-like genes and all the regulatory information forproper expression of these genes, spans 70 kb of DNA. Only afew vectors (including BACs and YACs) can ferry this muchmaterial. By contrast, the collection of exons and introns thatconstitute a single �-globin gene is only 1.4 kb in length andcan thus fit easily into a plasmid. Most other genes are largerthan individual �-globin genes, in part because they havegreater coding capacity and many more introns; as a result, tobe cloned as a single insert, they require a , BAC, or occa-sionally even a YAC vector.
Cloning Step 2: Host Cells Take up andAmplify Vector-Insert RecombinantsAlthough each type of vector functions in a slightly differentway and enters a specific kind of host, the general scheme ofentering a host cell and taking advantage of the cellular envi-ronment to replicate itself is the same for all. We divide our dis-cussion of this step of cloning into three parts: getting foreignDNA into the host cell; selecting cells that have received aDNA molecule; and distinguishing insert-containing recombi-nant molecules from vectors without inserts. Figure 9.8 illus-trates the three-part process with a plasmid vector containing anorigin of replication, the gene for resistance to ampicillin(ampr), and the E. coli lacZ gene, which encodes the enzyme�-galactosidase. By constructing the vector with a commonrestriction site like EcoRI right in the middle of the lacZ gene,researchers can insert foreign DNA into the gene at that loca-tion and then use the disruption of lacZ gene function to distin-guish insert-containing recombinant molecules from vectorswithout inserts (as described in the caption and later in the text).
Cloning Fragments of DNA 287
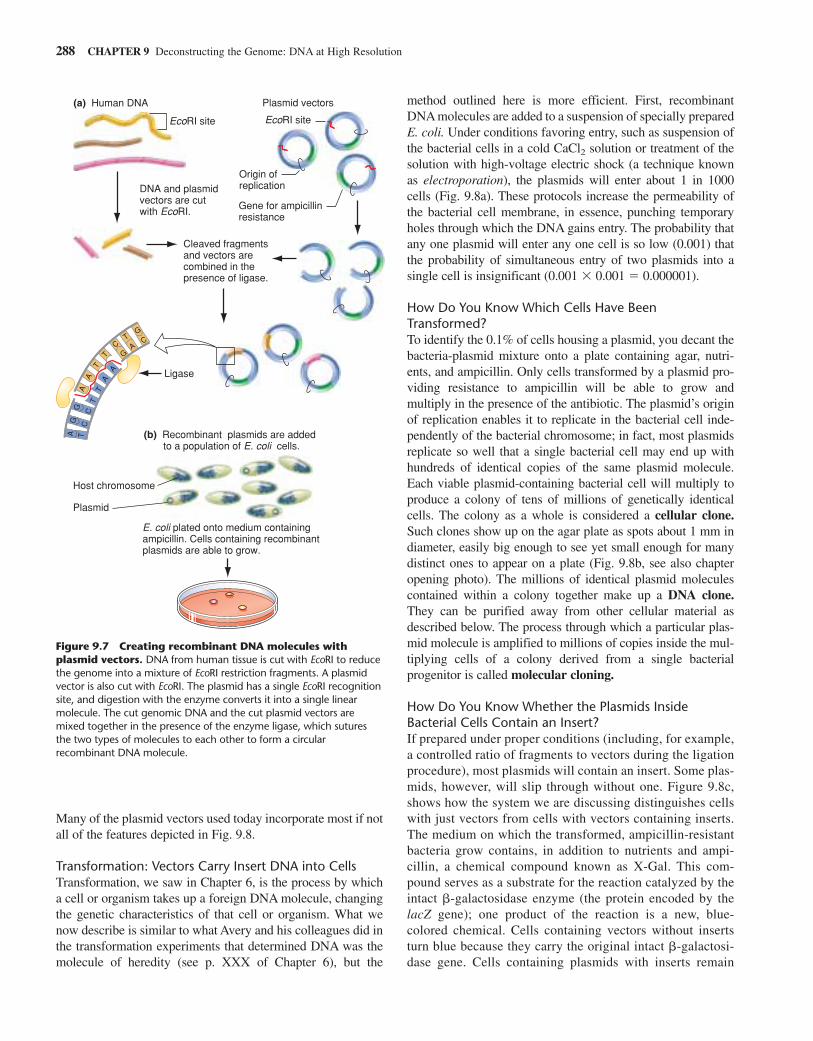
TABLE 9.2 Various Vectors and the Size of the Inserts They Carry
Typical Carrying Capacity (Size of
Vector Form of Vector Host Insert Accepted) Major Uses
Plasmid Double-stranded circular DNA E.coli Up to 15 kb cDNA libraries; subcloning
Bacteriophage Virus (linear DNA) E.coli Up to 25 kb Genomic and cDNA librarieslambda
Cosmid Double-stranded circular DNA E.coli 30–45 kb Genomic libraries
Bacteriophage P1 Virus (circular DNA) E.coli 70–90 kb Genomic libraries
BAC Bacterial artificial chromosome E.coli 100–500 kb Genomic libraries
YAC Yeast artificial chromosome Yeast 250–2000 kb Genomic libraries(1 megabase)
Many of the plasmid vectors used today incorporate most if notall of the features depicted in Fig. 9.8.
Transformation: Vectors Carry Insert DNA into CellsTransformation, we saw in Chapter 6, is the process by whicha cell or organism takes up a foreign DNA molecule, changingthe genetic characteristics of that cell or organism. What wenow describe is similar to what Avery and his colleagues did inthe transformation experiments that determined DNA was themolecule of heredity (see p. XXX of Chapter 6), but the
method outlined here is more efficient. First, recombinantDNA molecules are added to a suspension of specially preparedE. coli. Under conditions favoring entry, such as suspension ofthe bacterial cells in a cold CaCl2 solution or treatment of thesolution with high-voltage electric shock (a technique knownas electroporation), the plasmids will enter about 1 in 1000cells (Fig. 9.8a). These protocols increase the permeability ofthe bacterial cell membrane, in essence, punching temporaryholes through which the DNA gains entry. The probability thatany one plasmid will enter any one cell is so low (0.001) thatthe probability of simultaneous entry of two plasmids into asingle cell is insignificant (0.001 0.001 � 0.000001).
How Do You Know Which Cells Have BeenTransformed?To identify the 0.1% of cells housing a plasmid, you decant thebacteria-plasmid mixture onto a plate containing agar, nutri-ents, and ampicillin. Only cells transformed by a plasmid pro-viding resistance to ampicillin will be able to grow andmultiply in the presence of the antibiotic. The plasmid’s originof replication enables it to replicate in the bacterial cell inde-pendently of the bacterial chromosome; in fact, most plasmidsreplicate so well that a single bacterial cell may end up withhundreds of identical copies of the same plasmid molecule.Each viable plasmid-containing bacterial cell will multiply toproduce a colony of tens of millions of genetically identicalcells. The colony as a whole is considered a cellular clone.Such clones show up on the agar plate as spots about 1 mm indiameter, easily big enough to see yet small enough for manydistinct ones to appear on a plate (Fig. 9.8b, see also chapteropening photo). The millions of identical plasmid moleculescontained within a colony together make up a DNA clone.They can be purified away from other cellular material asdescribed below. The process through which a particular plas-mid molecule is amplified to millions of copies inside the mul-tiplying cells of a colony derived from a single bacterialprogenitor is called molecular cloning.
How Do You Know Whether the Plasmids InsideBacterial Cells Contain an Insert?If prepared under proper conditions (including, for example,a controlled ratio of fragments to vectors during the ligationprocedure), most plasmids will contain an insert. Some plas-mids, however, will slip through without one. Figure 9.8c,shows how the system we are discussing distinguishes cellswith just vectors from cells with vectors containing inserts.The medium on which the transformed, ampicillin-resistantbacteria grow contains, in addition to nutrients and ampi-cillin, a chemical compound known as X-Gal. This com-pound serves as a substrate for the reaction catalyzed by theintact �-galactosidase enzyme (the protein encoded by thelacZ gene); one product of the reaction is a new, blue-colored chemical. Cells containing vectors without insertsturn blue because they carry the original intact �-galactosi-dase gene. Cells containing plasmids with inserts remain
288 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
(b) Recombinant plasmids are added to a population of E. coli cells.
Host chromosome
Ligase
Cleaved fragmentsand vectors arecombined in thepresence of ligase.
DNA and plasmidvectors are cutwith EcoRI.
Plasmid vectors
Origin ofreplication
EcoRI siteEcoRI site
(a) Human DNA
E. coli plated onto medium containingampicillin. Cells containing recombinantplasmids are able to grow.
Gene for ampicillinresistance
Plasmid
GG C
TA
C
AA
TT
G
TA
C
T GC A
T
A
Figure 9.7 Creating recombinant DNA molecules withplasmid vectors. DNA from human tissue is cut with EcoRI to reducethe genome into a mixture of EcoRI restriction fragments. A plasmidvector is also cut with EcoRI. The plasmid has a single EcoRI recognitionsite, and digestion with the enzyme converts it into a single linearmolecule. The cut genomic DNA and the cut plasmid vectors aremixed together in the presence of the enzyme ligase, which suturesthe two types of molecules to each other to form a circularrecombinant DNA molecule.

colorless, because the interrupted lacZ gene does not allowproduction of functional �-galactosidase enzyme.
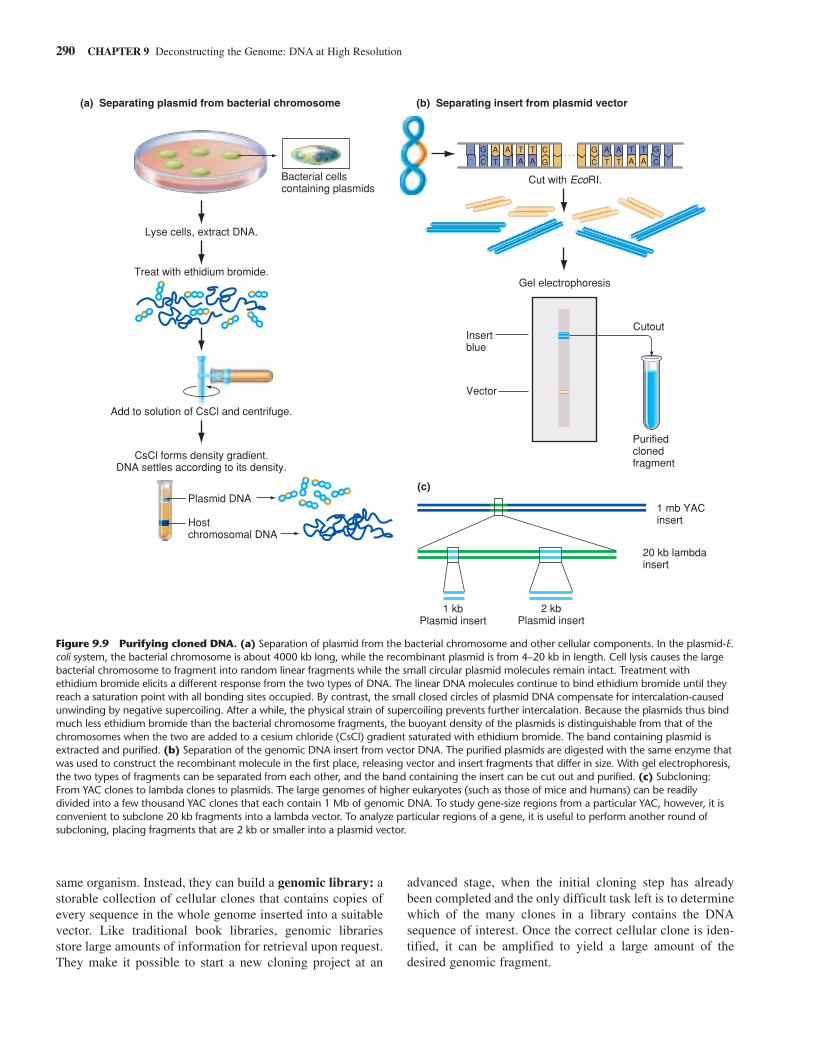
To Purify Cloned DNA, You SeparateRecombinant Plasmid Vector from HostDNA, and DNA Insert from VectorTo obtain a test tube filled with copies of just the cloned DNAinsert, you first purify the cloned vector-insert recombinantmolecule away from the host cell chromosome and other cellu-lar components. You then purify the DNA insert away fromvector DNA. The purification of cloned DNA away from thehost chromosome depends on the unique physical characteris-tics that distinguish the two types of DNA. The cloned mole-cule is always much smaller and sometimes different in form(circular versus linear) from the host chromosome. Figure 9.9aillustrates the protocol that exploits these physical differencesto separate cloned plasmids from bacterial chromosomes.
To separate genomic inserts from plasmid vectors, yousimply cut the purified vector-insert molecules with the samerestriction enzyme that you used to make the recombinants inthe first place. You then subject the resulting mixture of insertDNA disengaged from vector DNA to gel electrophoresis (Fig.9.9b). At the completion of electrophoresis, the region of thegel that contains the insert is cut out and the DNA within it puri-fied. Molecular cloning ultimately yields a large amount ofpurified fragments—billions of identical copies of the originalsmall piece of genomic DNA—that are now ready for study.
Libraries Are Collections of Cloned FragmentsMoving step by step from the DNA of any organism to a sin-gle purified DNA fragment is a long and tedious process.Fortunately, scientists do not have to return to step 1 everytime they need to purify a new genomic fragment from the
Cloning Fragments of DNA 289
DisruptedlacZ gene
lacZ gene
lacZ gene intact
lacZ gene transcript
lacZ
(c) Distinguishing cells carrying insert-containing recombinant molecules from cells carrying vectors without inserts
lacZ gene split byforeign DNA insert
No product
Intact vector,no insert
Vector with insert
X-Gal Blue pigment
Disrupted lacZ gene
Foreign DNA insert
Origin ofreplication
Recombinantplasmid
EcoRIsites
AmpR
(a) Transformation: foreign DNA enters the host cell
(b) Selecting cells that have received a plasmid
Figure 9.8 How to identify transformed bacterial cells containing plasmids with DNA inserts. Under proper conditions (including,for example, exposure to phosphatase and a controlled ratio of fragments to vectors), most plasmids will accept an insert. Some, however, will slipthrough without one. To identify such interlopers, plasmid vectors are constructed so that they contain the lacZ gene with a restriction site right inthe middle of the gene. If the vector reanneals to itself without inclusion of an insert, the lacZ gene will remain uninterrupted; if it accepts an insert,the gene will be interrupted. (a) Transformation: When added to a culture of bacteria, plasmids enter about 1 in 1000 cells. (b) Only cellstransformed by a plasmid carrying a gene for ampicillin resistance will grow. (c) Cells containing vectors that have reannealed to themselveswithout the inclusion of an insert will express the uninterrupted lacZ gene that they carry. The polypeptide product of the gene is �-galactosidase.Reaction of this enzyme with a substrate known as X-Gal produces a molecule that turns the cell blue. By contrast, if the opened ends of theplasmid vector have reannealed to a fragment of foreign DNA, this insert will interrupt and inactivate the lacZ gene on the plasmid, rendering itunable to produce a functional polypeptide. As a result of this process, called insertional inactivation, any cells containing recombinant plasmids willnot generate active �-galactosidase and will therefore not turn blue. It is thus possible to visually distinguish colorless colonies that contain insertsfrom blue colonies that do not.
same organism. Instead, they can build a genomic library: astorable collection of cellular clones that contains copies ofevery sequence in the whole genome inserted into a suitablevector. Like traditional book libraries, genomic librariesstore large amounts of information for retrieval upon request.They make it possible to start a new cloning project at an
advanced stage, when the initial cloning step has alreadybeen completed and the only difficult task left is to determinewhich of the many clones in a library contains the DNAsequence of interest. Once the correct cellular clone is iden-tified, it can be amplified to yield a large amount of thedesired genomic fragment.
290 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Gel electrophoresis
Insert blue
Cutout
Purifiedclonedfragment
(c)
Vector
(a) Separating plasmid from bacterial chromosome
Bacterial cells containing plasmids
Lyse cells, extract DNA.
Treat with ethidium bromide.
Add to solution of CsCl and centrifuge.
CsCl forms density gradient.DNA settles according to its density.
Plasmid DNA
Host chromosomal DNA
A
T
A
T
C
G C
GA
T AT
ATA
TAT
ATA
T
A
T C
GG
C
(b) Separating insert from plasmid vector
Cut with EcoRI.
1 mb YAC insert
20 kb lambda insert
1 kbPlasmid insert
2 kbPlasmid insert
Figure 9.9 Purifying cloned DNA. (a) Separation of plasmid from the bacterial chromosome and other cellular components. In the plasmid-E.coli system, the bacterial chromosome is about 4000 kb long, while the recombinant plasmid is from 4–20 kb in length. Cell lysis causes the largebacterial chromosome to fragment into random linear fragments while the small circular plasmid molecules remain intact. Treatment withethidium bromide elicits a different response from the two types of DNA. The linear DNA molecules continue to bind ethidium bromide until theyreach a saturation point with all bonding sites occupied. By contrast, the small closed circles of plasmid DNA compensate for intercalation-causedunwinding by negative supercoiling. After a while, the physical strain of supercoiling prevents further intercalation. Because the plasmids thus bindmuch less ethidium bromide than the bacterial chromosome fragments, the buoyant density of the plasmids is distinguishable from that of thechromosomes when the two are added to a cesium chloride (CsCl) gradient saturated with ethidium bromide. The band containing plasmid isextracted and purified. (b) Separation of the genomic DNA insert from vector DNA. The purified plasmids are digested with the same enzyme thatwas used to construct the recombinant molecule in the first place, releasing vector and insert fragments that differ in size. With gel electrophoresis,the two types of fragments can be separated from each other, and the band containing the insert can be cut out and purified. (c) Subcloning:From YAC clones to lambda clones to plasmids. The large genomes of higher eukaryotes (such as those of mice and humans) can be readilydivided into a few thousand YAC clones that each contain 1 Mb of genomic DNA. To study gene-size regions from a particular YAC, however, it isconvenient to subclone 20 kb fragments into a lambda vector. To analyze particular regions of a gene, it is useful to perform another round ofsubcloning, placing fragments that are 2 kb or smaller into a plasmid vector.

How to Compile a Genomic LibraryIf you digested the genome of a single cell with a restrictionenzyme and ligated every fragment to a vector with 100% effi-ciency, and then transformed all of these recombinant DNAmolecules into host cells with 100% efficiency, the resulting setof clones would represent the entire genome in a fragmentedform. A hypothetical collection of cellular clones that includesone copy—and one copy only—of every sequence in the entiregenome would be a complete genomic library.
How many clones will be present in this hypotheticallibrary? If you started with the 3,000,000 kb of DNA from ahaploid human sperm and reliably cut it into a series of 150 kbrestriction fragments, you would generate 3,000,000/150 �20,000 genomic fragments. If you placed each and every one ofthese fragments into BAC cloning vectors that were then trans-formed into E. coli host cells, you would create a perfect libraryof 20,000 clones that collectively carry every locus in thegenome. The number of clones in this perfect library defines agenomic equivalent. For any library, to find the number ofclones that constitute one genomic equivalent, you simplydivide the length of the genome (here, 3,000,000 kb) by theaverage size of the inserts carried by the library’s vector (in thiscase, 150 kb).
In real life, it is impossible to obtain a perfect library. Eachstep of cloning is far from 100% efficient, and the DNA of asingle cell does not supply sufficient raw material for theprocess. Researchers must thus harvest DNA from the millionsof cells in a particular tissue or organism. If you make agenomic library with this DNA by collecting only one genomicequivalent (20,000 clones for a human library in BAC vectors),the uncertainties of random sampling of DNA fragments wouldmean that some human DNA fragments would appear morethan once, while others might not be present at all. To increasethe chance that a library contains at least one clone with a par-ticular genomic fragment, the number of clones in the librarymust exceed one genomic equivalent. Including four to fivegenomic equivalents produces an average of four to five clonesfor each locus, and a 95% probability that any individual locusis present at least once.
For many types of molecular genetics experiments, it isadvantageous to use a vector like a BAC that allows the cloningof large fragments of DNA. This is because the number ofclones in a genomic equivalent is much smaller than in librariesconstructed with a vector that accepts only smaller fragments,so investigators have to search through fewer clones to find aparticular clone of interest. For example, a genomic equivalentof BAC clones with 150 kb inserts contains only 20,000 clones,while a genomic equivalent of plasmid clones with 10 kbinserts would contain 300,000 clones.
But BACs have a disadvantage: It takes a lot more work tocharacterize the larger inserts they carry. Researchers mustoften subclone smaller restriction fragments from a BAC cloneto look more closely at a particular region of genomic DNA(Fig. 9.9c). Sometimes, it is useful to reduce complexity onestep at a time, subcloning a BAC DNA fragment into a lambda
vector and then subcloning fragments of the lambda clone intoa plasmid vector. This process of subcloning establishessmaller and smaller DNA clones that are enriched for thesequences of interest.
cDNA Libraries Carry Information from the RNATranscripts Present in a Particular TissueOften, only the information in a gene’s coding sequence is ofexperimental interest, and it would be advantageous to limitanalysis to the gene’s exons without having to determine thestructure of the introns as well. Because coding sequencesaccount for a very small percentage of genomic DNA in highereukaryotes, however, it is inefficient to look for them ingenomic libraries. The solution is to generate cDNA libraries,which store sequences copied into DNA from all the RNA tran-scripts present in a particular cell type, tissue, or organ.Because they are obtained from RNA transcripts, thesesequences carry only exon information. As we saw in Chapter8, exons contain all the protein-coding information of a gene,as well as sequences that will appear in the 5′ and 3′ untrans-lated regions at the ends of corresponding mRNAs.
To produce DNA clones from mRNA sequences,researchers rely on a series of in vitro reactions that mimic sev-eral stages in the life cycle of viruses known as retroviruses.Retroviruses, which include among their ranks the HIV virusthat causes AIDS, carry their genetic information in moleculesof RNA. As part of their gene-transmission kit, retroviruses alsocontain the unusual enzyme known as RNA-dependent DNApolymerase, or simply reverse transcriptase (review theGenetics and Society box in Chapter 8, p. 252). After infectinga cell, a retrovirus uses reverse transcriptase to copy its singlestrand of RNA into a mirror-image-like strand of complemen-tary DNA, often abbreviated as cDNA. The reverse transcrip-tase, which can also function as a DNA-dependent DNApolymerase, then makes a second strand of DNA complemen-tary to this first cDNA strand (and equivalent in sequence to theoriginal RNA template). Finally, this double-stranded copy ofthe retroviral RNA chromosome integrates into the host cell’sgenome. Although the designation cDNA originally meant a sin-gle strand of DNA complementary to an RNA molecule, it nowrefers to any DNA—single- or double-stranded—derived fromRNA. In 1975, researchers began to use purified reverse tran-scriptase to convert mRNA sequences from any tissue in anyorganism into cDNA sequences for cloning.
Suppose you were interested in studying the structure of amutant �-globin protein. You have already analyzed hemoglo-bin obtained from a patient carrying this mutation and foundthat the alteration affects the amino-acid structure of the proteinitself and not its regulation, so you now need only look at thesequence of the mutant gene’s coding region to understand the primary genetic defect. To establish a library enriched for themutant gene sequence and lacking all the extraneous informa-tion from genomic introns and nontranscribed regions, youwould first obtain mRNA from the cytoplasm of the patient’sred blood cell precursors (Fig. 9.10a). About 80% of the total
Cloning Fragments of DNA 291
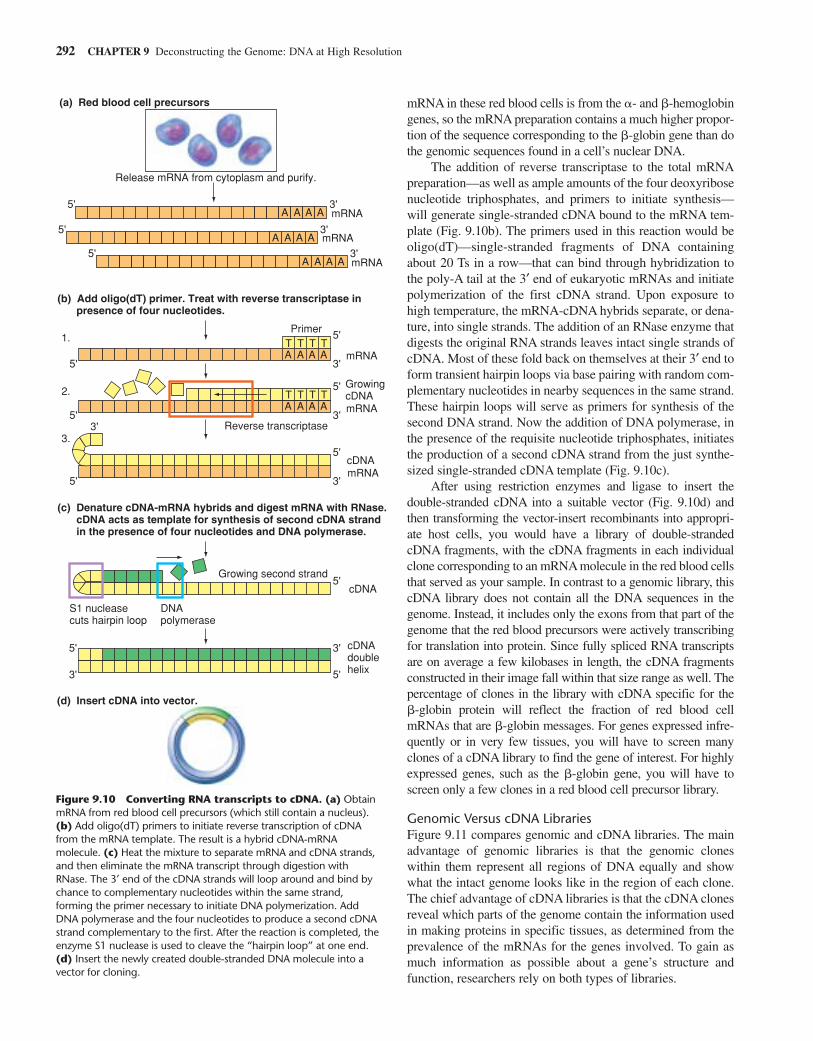
mRNA in these red blood cells is from the �- and �-hemoglobingenes, so the mRNA preparation contains a much higher propor-tion of the sequence corresponding to the �-globin gene than dothe genomic sequences found in a cell’s nuclear DNA.
The addition of reverse transcriptase to the total mRNApreparation—as well as ample amounts of the four deoxyribosenucleotide triphosphates, and primers to initiate synthesis—will generate single-stranded cDNA bound to the mRNA tem-plate (Fig. 9.10b). The primers used in this reaction would beoligo(dT)—single-stranded fragments of DNA containingabout 20 Ts in a row—that can bind through hybridization tothe poly-A tail at the 3′ end of eukaryotic mRNAs and initiatepolymerization of the first cDNA strand. Upon exposure tohigh temperature, the mRNA-cDNA hybrids separate, or dena-ture, into single strands. The addition of an RNase enzyme thatdigests the original RNA strands leaves intact single strands ofcDNA. Most of these fold back on themselves at their 3′ end toform transient hairpin loops via base pairing with random com-plementary nucleotides in nearby sequences in the same strand.These hairpin loops will serve as primers for synthesis of thesecond DNA strand. Now the addition of DNA polymerase, inthe presence of the requisite nucleotide triphosphates, initiatesthe production of a second cDNA strand from the just synthe-sized single-stranded cDNA template (Fig. 9.10c).
After using restriction enzymes and ligase to insert the double-stranded cDNA into a suitable vector (Fig. 9.10d) andthen transforming the vector-insert recombinants into appropri-ate host cells, you would have a library of double-strandedcDNA fragments, with the cDNA fragments in each individualclone corresponding to an mRNA molecule in the red blood cellsthat served as your sample. In contrast to a genomic library, thiscDNA library does not contain all the DNA sequences in thegenome. Instead, it includes only the exons from that part of thegenome that the red blood precursors were actively transcribingfor translation into protein. Since fully spliced RNA transcriptsare on average a few kilobases in length, the cDNA fragmentsconstructed in their image fall within that size range as well. Thepercentage of clones in the library with cDNA specific for the �-globin protein will reflect the fraction of red blood cellmRNAs that are �-globin messages. For genes expressed infre-quently or in very few tissues, you will have to screen manyclones of a cDNA library to find the gene of interest. For highlyexpressed genes, such as the �-globin gene, you will have toscreen only a few clones in a red blood cell precursor library.
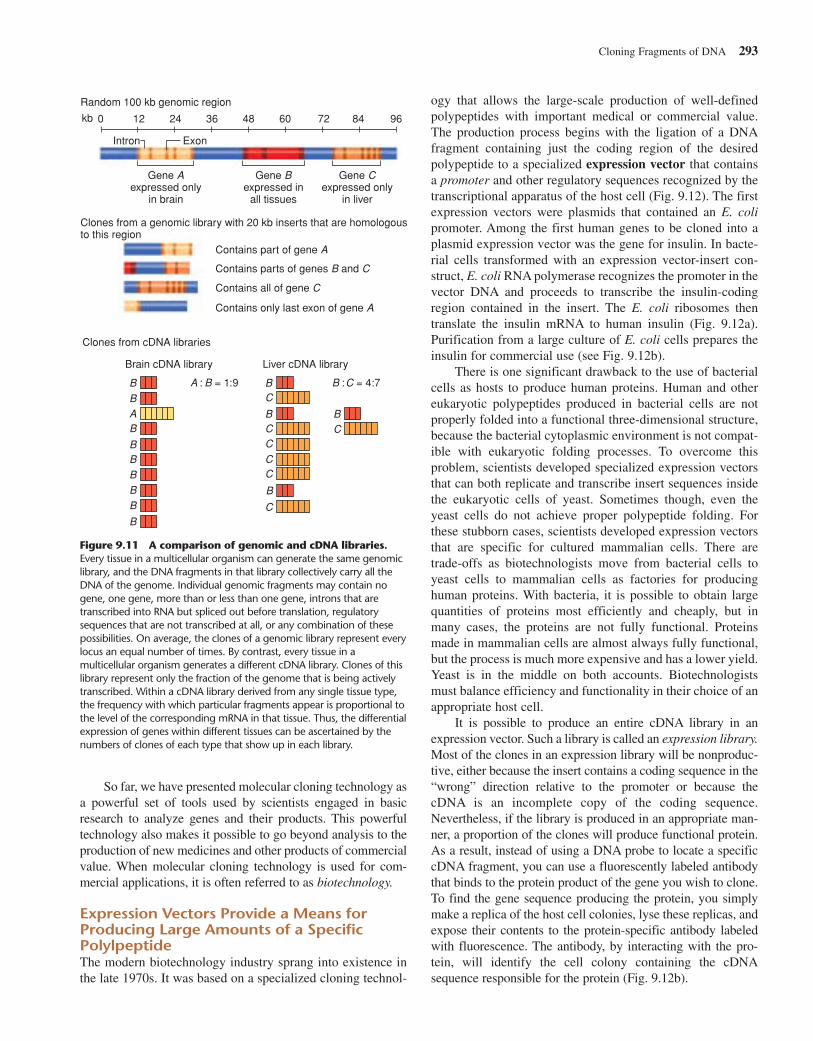
Genomic Versus cDNA LibrariesFigure 9.11 compares genomic and cDNA libraries. The mainadvantage of genomic libraries is that the genomic cloneswithin them represent all regions of DNA equally and showwhat the intact genome looks like in the region of each clone.The chief advantage of cDNA libraries is that the cDNA clonesreveal which parts of the genome contain the information usedin making proteins in specific tissues, as determined from theprevalence of the mRNAs for the genes involved. To gain asmuch information as possible about a gene’s structure andfunction, researchers rely on both types of libraries.
292 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
T T T T
T T T T
(a) Red blood cell precursors
Release mRNA from cytoplasm and purify.
5'
5'
3'
5' 3'
5'3'
mRNA
mRNA
mRNA
(b) Add oligo(dT) primer. Treat with reverse transcriptase in presence of four nucleotides.
1.
2.
3.
Primer
mRNA
mRNA
GrowingcDNA
cDNA
cDNAdoublehelix
Reverse transcriptase
(c) Denature cDNA-mRNA hybrids and digest mRNA with RNase. cDNA acts as template for synthesis of second cDNA strand in the presence of four nucleotides and DNA polymerase.
Growing second strand
S1 nucleasecuts hairpin loop
DNApolymerase
(d) Insert cDNA into vector.
A A A A
5' 3'A A A A
5' 3'A A A A
mRNA5' 3'
A A A A
5'
5'3'
3'A A A A
5'
5' 3'
cDNA5'
Figure 9.10 Converting RNA transcripts to cDNA. (a) ObtainmRNA from red blood cell precursors (which still contain a nucleus).(b) Add oligo(dT) primers to initiate reverse transcription of cDNAfrom the mRNA template. The result is a hybrid cDNA-mRNAmolecule. (c) Heat the mixture to separate mRNA and cDNA strands,and then eliminate the mRNA transcript through digestion withRNase. The 3′ end of the cDNA strands will loop around and bind bychance to complementary nucleotides within the same strand,forming the primer necessary to initiate DNA polymerization. AddDNA polymerase and the four nucleotides to produce a second cDNAstrand complementary to the first. After the reaction is completed, theenzyme S1 nuclease is used to cleave the “hairpin loop” at one end.(d) Insert the newly created double-stranded DNA molecule into avector for cloning.
So far, we have presented molecular cloning technology asa powerful set of tools used by scientists engaged in basicresearch to analyze genes and their products. This powerfultechnology also makes it possible to go beyond analysis to theproduction of new medicines and other products of commercialvalue. When molecular cloning technology is used for com-mercial applications, it is often referred to as biotechnology.
Expression Vectors Provide a Means forProducing Large Amounts of a SpecificPolylpeptideThe modern biotechnology industry sprang into existence inthe late 1970s. It was based on a specialized cloning technol-
ogy that allows the large-scale production of well-definedpolypeptides with important medical or commercial value.The production process begins with the ligation of a DNAfragment containing just the coding region of the desiredpolypeptide to a specialized expression vector that containsa promoter and other regulatory sequences recognized by thetranscriptional apparatus of the host cell (Fig. 9.12). The firstexpression vectors were plasmids that contained an E. colipromoter. Among the first human genes to be cloned into aplasmid expression vector was the gene for insulin. In bacte-rial cells transformed with an expression vector-insert con-struct, E. coli RNA polymerase recognizes the promoter in thevector DNA and proceeds to transcribe the insulin-codingregion contained in the insert. The E. coli ribosomes thentranslate the insulin mRNA to human insulin (Fig. 9.12a).Purification from a large culture of E. coli cells prepares theinsulin for commercial use (see Fig. 9.12b).
There is one significant drawback to the use of bacterialcells as hosts to produce human proteins. Human and othereukaryotic polypeptides produced in bacterial cells are notproperly folded into a functional three-dimensional structure,because the bacterial cytoplasmic environment is not compat-ible with eukaryotic folding processes. To overcome thisproblem, scientists developed specialized expression vectorsthat can both replicate and transcribe insert sequences insidethe eukaryotic cells of yeast. Sometimes though, even theyeast cells do not achieve proper polypeptide folding. Forthese stubborn cases, scientists developed expression vectorsthat are specific for cultured mammalian cells. There aretrade-offs as biotechnologists move from bacterial cells toyeast cells to mammalian cells as factories for producinghuman proteins. With bacteria, it is possible to obtain largequantities of proteins most efficiently and cheaply, but inmany cases, the proteins are not fully functional. Proteinsmade in mammalian cells are almost always fully functional,but the process is much more expensive and has a lower yield.Yeast is in the middle on both accounts. Biotechnologistsmust balance efficiency and functionality in their choice of anappropriate host cell.
It is possible to produce an entire cDNA library in anexpression vector. Such a library is called an expression library.Most of the clones in an expression library will be nonproduc-tive, either because the insert contains a coding sequence in the“wrong” direction relative to the promoter or because thecDNA is an incomplete copy of the coding sequence.Nevertheless, if the library is produced in an appropriate man-ner, a proportion of the clones will produce functional protein.As a result, instead of using a DNA probe to locate a specificcDNA fragment, you can use a fluorescently labeled antibodythat binds to the protein product of the gene you wish to clone.To find the gene sequence producing the protein, you simplymake a replica of the host cell colonies, lyse these replicas, andexpose their contents to the protein-specific antibody labeledwith fluorescence. The antibody, by interacting with the pro-tein, will identify the cell colony containing the cDNAsequence responsible for the protein (Fig. 9.12b).
Cloning Fragments of DNA 293
Random 100 kb genomic region
kb
Intron Exon
Gene Aexpressed only
in brain
Gene Bexpressed in
all tissues
Gene Cexpressed only
in liver
Clones from a genomic library with 20 kb inserts that are homologousto this region
Contains part of gene A
Contains parts of genes B and C
Contains all of gene C
Contains only last exon of gene A
Clones from cDNA libraries
Brain cDNA library Liver cDNA library
A : B = 1:9 B :C = 4:7
0 12 24 36 48 60 72 84 96
A
B B
BB
B
B
B
BBBBB
B
C
CCCC
C
C
Figure 9.11 A comparison of genomic and cDNA libraries.Every tissue in a multicellular organism can generate the same genomiclibrary, and the DNA fragments in that library collectively carry all theDNA of the genome. Individual genomic fragments may contain nogene, one gene, more than or less than one gene, introns that aretranscribed into RNA but spliced out before translation, regulatorysequences that are not transcribed at all, or any combination of thesepossibilities. On average, the clones of a genomic library represent everylocus an equal number of times. By contrast, every tissue in amulticellular organism generates a different cDNA library. Clones of thislibrary represent only the fraction of the genome that is being activelytranscribed. Within a cDNA library derived from any single tissue type,the frequency with which particular fragments appear is proportional tothe level of the corresponding mRNA in that tissue. Thus, the differentialexpression of genes within different tissues can be ascertained by thenumbers of clones of each type that show up in each library.

Hybridization Is Used to IdentifySimilar DNA Sequences
While you can “screen” an expression library with an antibodythat recognizes the protein product of the gene that you want toclone, the DNA inserts in all other types of libraries do not sig-nal their presence in such a straightforward fashion. So, onceyou have collected the hundreds of thousands of human DNAfragments in a genomic or a nonexpressing cDNA library, howdo you find the gene you wish to study, the proverbial “needlein the haystack”? For example, how would you go about find-ing a genomic clone that contained just a portion of the �-globin gene? One way is to take advantage of hybridization—thenatural propensity of complementary single-stranded moleculesto form stable double helixes. If a clone containing any portionof a �-globin sequence is already available—perhaps as a previ-ously obtained cDNA clone—it can be denatured (separated intosingle strands), linked with a radioactive or fluorescent tag, andthen used to probe a whole genome library that is spread out as aseries of colonies on one or more petri plates. The tagged DNAprobe will hybridize with denatured DNA from the genomicclones that contain a complementary sequence. After nonhy-bridizing probe is washed away, only the tiny number of �-globin-containing clones (among the hundreds of thousands inthe library) are tagged by virtue of their hybridization to theprobe. Individual cellular clones can then be retrieved from thelibrary and put into culture to produce larger amounts of materialin preparation for recovery of the purified DNA insert.
Hybridization has a single critical requirement: The regionof complementarity between two single strands must be suffi-ciently long and accurate to produce a large enough number ofhydrogen bonds to generate a cohesive force. Accuracy refersto the percentage of bases within the complementary regionsthat are actual complements of each other (C–G or A–T). If theoverall region is at least several hundred bases long, strandsthat show only 80% complementarity can still form hybrids.The cohesive force formed by adding together large numbers ofhydrogen bonds will counteract the thermal forces that tend todisrupt the double helix. Individual hydrogen bonds are veryweak, and the two (A:T) or three (G:C) hydrogen bonds thatform between individual pairs of bases are far too weak to pre-vent thermal forces from pulling them apart. But if two singlestrands form hydrogen bonds between 25 or more contiguousbase pairs, the combined force is sufficient to counteract thethermal forces that operate even at room temperature.Hybridization can occur between any two single strands ofnucleic acid: DNA/DNA, DNA/RNA, or RNA/RNA. In thissection, we focus on DNA/DNA hybridization.
We now describe how to screen a library with a DNA probe.
Preparing the LibraryBefore you can use a DNA probe to screen a library, you mustdistribute the library’s clones on petri plates and place a piece ofnitrocellulose paper over each plate, allowing some, but not all,of the cells from each colony to transfer to the nitrocellulose diskreplica (Fig. 9.13). You now lyse the bacteria on the disk with an
294 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
(a) An expression vector allows production of specific polypeptide
E. coli expressionvector
E. coli promoter
TransformE. coli
Insulin mRNA
Insulin cDNA +
Insulin mRNARNA polymerase
Host chromosome
Antibody
Binding sites
Fluorescent tag
Overlay platewith nitrocellulosepaper. Pick up cells.
UV
Wash filter. Expose to UV light and identify flourescent spots.Compare with original plate in order to find bacterial clone containing human insulin gene.
Lyse cell. Treatwith NaOH. Proteins adhere to paper.
Incubate paper in solution of labeled insulin antibody. Antibodies will bind to insulin protein.
Bacterial cell
Insulin protein
(b) Screening for insulin gene expression
Figure 9.12 An expression vector can be used to produce adesired polypeptide. (a) A cDNA fragment containing just thecoding region of insulin is ligated to a plasmid vector containing an E.coli promoter next to the restriction site used to cut it open. Therecombinant DNA molecule is transformed into E. coli cells, where it isrecognized by RNA polymerase and transcribed into insulin mRNA.When the host-cell translational machinery sees the transcripts, ittranslates them into insulin protein. (b) Screening an expressionlibrary with an antibody probe. Clones of an expression library arepartially transferred to a nitrocellulose paper disk. The disk is removedfrom the plate and exposed to a solution that lyses the cells andreleases the proteins, which are attached to the disk through baking.The disk is then exposed to a solution containing a fluorescentlylabeled antibody to insulin. Antibody molecules bind to protein withinspots formed by clones expressing the insulin gene. After washingaway unbound antibody, exposure to UV light allows the identificationof colonies expressing the gene product recognized by the antibody.The recombinant DNA clones in these colonies must carry the DNAsequence that encodes the protein recognized by the antibody.
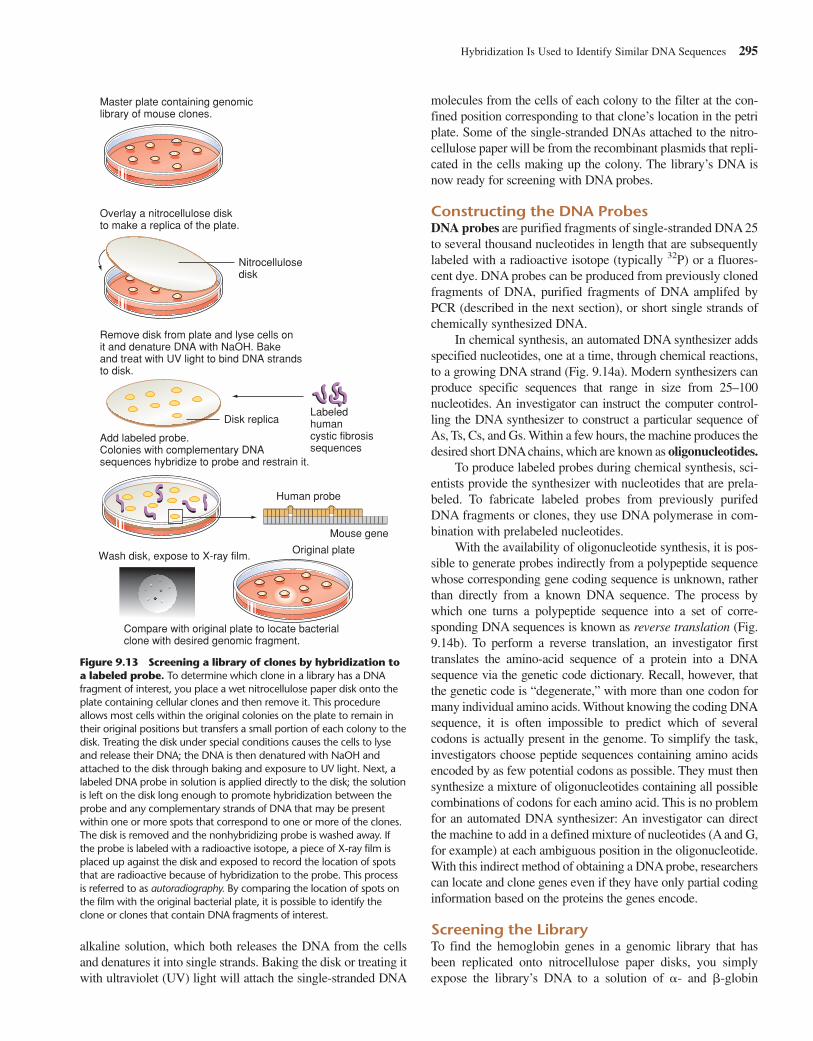
molecules from the cells of each colony to the filter at the con-fined position corresponding to that clone’s location in the petriplate. Some of the single-stranded DNAs attached to the nitro-cellulose paper will be from the recombinant plasmids that repli-cated in the cells making up the colony. The library’s DNA isnow ready for screening with DNA probes.
Constructing the DNA ProbesDNA probes are purified fragments of single-stranded DNA 25to several thousand nucleotides in length that are subsequentlylabeled with a radioactive isotope (typically 32P) or a fluores-cent dye. DNA probes can be produced from previously clonedfragments of DNA, purified fragments of DNA amplifed byPCR (described in the next section), or short single strands ofchemically synthesized DNA.
In chemical synthesis, an automated DNA synthesizer addsspecified nucleotides, one at a time, through chemical reactions,to a growing DNA strand (Fig. 9.14a). Modern synthesizers canproduce specific sequences that range in size from 25–100nucleotides. An investigator can instruct the computer control-ling the DNA synthesizer to construct a particular sequence ofAs, Ts, Cs, and Gs. Within a few hours, the machine produces thedesired short DNAchains, which are known as oligonucleotides.
To produce labeled probes during chemical synthesis, sci-entists provide the synthesizer with nucleotides that are prela-beled. To fabricate labeled probes from previously purifedDNA fragments or clones, they use DNA polymerase in com-bination with prelabeled nucleotides.
With the availability of oligonucleotide synthesis, it is pos-sible to generate probes indirectly from a polypeptide sequencewhose corresponding gene coding sequence is unknown, ratherthan directly from a known DNA sequence. The process bywhich one turns a polypeptide sequence into a set of corre-sponding DNA sequences is known as reverse translation (Fig.9.14b). To perform a reverse translation, an investigator firsttranslates the amino-acid sequence of a protein into a DNAsequence via the genetic code dictionary. Recall, however, thatthe genetic code is “degenerate,” with more than one codon formany individual amino acids. Without knowing the coding DNAsequence, it is often impossible to predict which of severalcodons is actually present in the genome. To simplify the task,investigators choose peptide sequences containing amino acidsencoded by as few potential codons as possible. They must thensynthesize a mixture of oligonucleotides containing all possiblecombinations of codons for each amino acid. This is no problemfor an automated DNA synthesizer: An investigator can directthe machine to add in a defined mixture of nucleotides (A and G,for example) at each ambiguous position in the oligonucleotide.With this indirect method of obtaining a DNA probe, researcherscan locate and clone genes even if they have only partial codinginformation based on the proteins the genes encode.
Screening the LibraryTo find the hemoglobin genes in a genomic library that hasbeen replicated onto nitrocellulose paper disks, you simplyexpose the library’s DNA to a solution of �- and �-globin
Hybridization Is Used to Identify Similar DNA Sequences 295
Master plate containing genomiclibrary of mouse clones.
Overlay a nitrocellulose diskto make a replica of the plate.
Remove disk from plate and lyse cells on it and denature DNA with NaOH. Bakeand treat with UV light to bind DNA strandsto disk.
Add labeled probe. Colonies with complementary DNAsequences hybridize to probe and restrain it.
Compare with original plate to locate bacterialclone with desired genomic fragment.
Original plate
Human probe
Mouse gene
Disk replicaLabeledhumancystic fibrosissequences
Nitrocellulose disk
Wash disk, expose to X-ray film.
Figure 9.13 Screening a library of clones by hybridization toa labeled probe. To determine which clone in a library has a DNAfragment of interest, you place a wet nitrocellulose paper disk onto theplate containing cellular clones and then remove it. This procedureallows most cells within the original colonies on the plate to remain intheir original positions but transfers a small portion of each colony to thedisk. Treating the disk under special conditions causes the cells to lyseand release their DNA; the DNA is then denatured with NaOH andattached to the disk through baking and exposure to UV light. Next, alabeled DNA probe in solution is applied directly to the disk; the solutionis left on the disk long enough to promote hybridization between theprobe and any complementary strands of DNA that may be presentwithin one or more spots that correspond to one or more of the clones.The disk is removed and the nonhybridizing probe is washed away. Ifthe probe is labeled with a radioactive isotope, a piece of X-ray film isplaced up against the disk and exposed to record the location of spotsthat are radioactive because of hybridization to the probe. This processis referred to as autoradiography. By comparing the location of spots onthe film with the original bacterial plate, it is possible to identify theclone or clones that contain DNA fragments of interest.
alkaline solution, which both releases the DNA from the cellsand denatures it into single strands. Baking the disk or treating itwith ultraviolet (UV) light will attach the single-stranded DNA

probes labeled with radioactivity or fluorescence (see Fig.9.13). After a sufficient period of incubation with radioactivelylabeled probe, you wash away all probe molecules that have nothybridized to complementary DNA strands stuck to the disk,place a large piece of X-ray film in direct contact with the paperdisk to record the radioactive emissions from the hybridizingprobe, and then develop the X-ray film to see the dark spotsrepresenting the sites of hybridization. An X-ray film that hasbeen processed in this way to identify the locations of hybridiz-ing radioactive probes is called an autoradiograph, and theprocess of creating an autoradiograph is called autoradiogra-phy. If you use fluorescence-labeled probes, you can see thehybridizing clones directly by shining ultraviolet light on thedisks. The spots revealed by hybridization correspond togenomic clones containing sequences complementary to the �-and �-globin probes. Knowing the position on the disk that
gave rise to these spots, you can return to the original masterpetri plates and pick out the corresponding colonies containingthe cloned inserts of interest. You can then harvest and purifythe cloned DNA identified by the globin probes and proceedwith further analysis. The DNA clones recovered with thisscreening procedure would lead to the discovery that thehuman genome carries one gene for � globin, but two similar�-globin genes back to back, named �1 and �2.
Of great importance to geneticists is the fact thathybridization can occur between single strands that are notcompletely complementary, including related sequences fromdifferent species. In general, two single DNA strands that arelonger than 50–100 bp will hybridize so long as the extent oftheir complementarity is more than 80%, even though mis-matches may appear throughout the resulting hybrid molecule.Imperfect hybrids are less stable than perfect ones, but geneti-cists can exploit this difference in stability to evaluate the sim-ilarity between molecules from two different sources.Hybridization, for example, occurs between the mouse andhuman genes for the cystic fibrosis protein. Researchers canthus use the human genes to identify and isolate the corre-sponding mouse sequences, and then use these sequences todevelop a mouse containing a defective cystic fibrosis gene.Such a mouse provides a model for cystic fibrosis in a speciesthat, unlike humans, is amenable to experimental analysis.
Gel Electrophoresis Combined withHybridization Provides a Tool for MappingDNA FragmentsWe have just seen how researchers use hybridization to screen alibrary of thousands of clones for particular clones carrying DNAsequences complementary to specific probes. Hybridization canalso provide information about sequences related to a gene in acloned DNA fragment or a whole genome. The protocol foraccomplishing this task combines gel electrophoresis (reviewdiscussion on p. 283) with the hybridization of DNA probes toDNA targets immobilized within nitrocellulose paper.
Suppose you had a clone of a gene called H2K from themouse major histocompatibility complex (or MHC). The H2Kgene is known to play a critical role in the body’s ability tomount an immune response to foreign cells. You want to knowwhether there are other genes in the mouse genome that aresimilar to H2K and, by extrapolation, also play a role in theimmune response. To get an estimate of the number of H2K-like genes that exist in the genome, you could turn to ahybridization technique called the Southern blot, named forEdward Southern, the British scientist who developed it. Figure9.15 illustrates the details of the technique.
Southern blotting can identify individual H2K-like DNAsequences within the uncloned expanse of DNA present in amammalian genome. Cutting the total genomic DNAwith EcoRIproduces about 700,000 fragments. When you separate thesefragments by gel electrophoresis and stain them with ethidiumbromide, all you see is a smear, because it is impossible to dis-tinguish 700,000 fragments spread over a distance of some 10cm (Fig. 9.15). But you can still blot the smear of fragments to a
296 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
(b) Synthesizing DNA probes based on reverse translation
(a)
Protein sequence
Sequences that must bepresent in the probe
G
G
G GGGA A
GAA
AAA TT
A TT
T
GGT
T
GAT
T
GAT
G G GGGA
GGA
AAA TTT TT
AG G GGGA AAA TTT TC
GG G GGGA AAA TTT TC
AG G GGGA AAA TTT CT
GG G GGGA AAA TTT CT
AG G GGGA AAA TTT CC
GG G GGGA AAA TTT CC
GACDegenerate coding sequences
TAC
Glu Asp Met Trp Tyr
Figure 9.14 How to make oligonucleotide probes forscreening a library. (a) A DNA synthesizer is a machine thatautomates the addition through chemical reactions of specifiednucleotides to the growing DNA chains, known as oligonucleotides.Four of the five smaller bottles at the top contain solutions of each ofthe four different nucleotide triphosphates (A, T, C, and G), and thefifth bottle contains a mixture of the four nucleotides for situationswhere an investigator wants to create an oligonucleotide with arandom base in that position. The other bottles contain variousreagents used in the various chemical reactions that the synthesizercarries out. (b) Reverse translation. An amino-acid sequence can be“reverse translated” into a degenerate DNA sequence, which can beprogrammed into a DNA synthesizer to create a set of oligonucleotidesthat must include the one present in the actual genomic DNA.

nitrocellulose filter paper and probe the resulting Southern blotwith a labeled H2K clone, which picks out the bands containingthe H2K-like gene sequences. The result shown in Fig. 9.15 is apattern of approximately two dozen fragments that constitute aseries of related genes within the mouse MHC. The Southernblot thus makes it possible to start with a very complex mixtureand find the small number of fragments among hundreds of thou-sands that are related to your original clone.
Southern blotting can also determine the location of onecloned sequence (such as a 1.4 kb human �-globin cDNAsequence from a plasmid vector) within a larger clonedsequence (such as 30 kb cosmid containing the �-globin locusand surrounding genome). Suppose you want to use these twoclones to discover the location of the gene within the cosmid aswell as to learn which parts of the full gene are exons andwhich parts are introns. To answer both of these questions in avery straightforward fashion you would turn to Southern blot-ting. First you would use gel electrophoresis of restriction-enzyme digested cosmid DNA, and staining with ethidiumbromide, to construct a restriction map of the cosmid. Next,you would transfer the restriction fragments from the gel ontoa filter paper, and probe the filter by hybridization to the labeled�-globin cDNA clone. An autoradiograph will then show theprecise cosmid restriction fragments that carry coding regionsof the �-globin gene. With very high resolution restrictionmaps of cosmid subclones and high-resolution gel elec-trophoresis of small restriction fragments (produced with four-cutter restriction enzymes), it would be possible to distinguishrestriction fragments containing exons from the in-betweenfragments that contain the introns of the �-globin gene.
The Polymerase Chain ReactionProvides a Rapid Method forIsolating DNA Fragments
Genes are rare targets in a complex genome: the �-globin gene,for example, spans only about 1400 of the 3,000,000,000nucleotide pairs in the haploid human genome. Cloning over-comes the problem of studying such rarities by amplifyinglarge amounts of a specific DNA fragment in isolation. Butcloning is a tedious, labor-intensive process. Once a sequenceis known, or even partially known, molecular biologists nowuse an alternative method to recover versions of the samesequence from any source material, including relatedsequences from different species, from different individualswithin the same species, and from sequences in RNA tran-scripts or whole genomes. This alternative method is called thepolymerase chain reaction, or PCR. First developed in 1985,PCR is faster, less expensive, and more flexible in applicationthan cloning. From a complex mixture of DNA—like that pres-ent in a person’s blood sample—PCR can isolate a purifiedDNA fragment in just a few hours.
PCR is also extremely efficient. We have seen that in cre-ating a genomic or cDNA library, it is necessary to use a largenumber of cells from one or more tissues as the source of DNA
or mRNA. By contrast, the single copy of a genome present inone sperm cell, or the minute amount of severely degradedDNA recovered from the bone marrow of a 30,000-year-oldNeanderthal skeleton, provides enough material for PCR tomake 100 million or more copies of a target DNA sequence inan afternoon.
How PCR Achieves the ExponentialAccumulation of Target DNAThe polymerase chain reaction is a kind of reiterative loop inwhich an operation is repeatedly applied to the products of ear-lier rounds of the same operation. You can liken it to the oper-ation of an imaginary generously paying automatic slotmachine. You start the machine by inserting a quarter, at whichpoint the handle cranks, and the machine pays out two quarters;it then reinserts those two coins, cranks, and produces fourquarters; reinserts the four, cranks, and spits out eight coins,and so on. By the twenty-second round, this fantasy machinedelivers more than 4 million quarters.
The PCR operation brings together and exploits themethod of DNA hybridization described earlier in this chapterand the essential features of DNA replication described inChapter 6. Once a specific genomic region (which may be any-where from a few dozen base pairs to 25 kb in length) has beenchosen for amplification, an investigator uses knowledge of thesequence (obtained from previous sequencing based on themethod described later in this chapter) to synthesize twooligonucleotides that correspond to the two ends of the targetregion. One oligonucleotide is complementary to one strand ofDNA at one end of the region; the other oligonucleotide is com-plementary to the other strand at the other end of the region.The process of amplification is initiated by the hybridization ofthese oligonucleotides to denatured DNA molecules within thesample. The oligonucleotides act as primers directing DNApolymerase to create new strands of DNA complementary toboth strands between the two primed sections (Fig. 9.16).
This initial replication is followed by subsequent rounds inwhich both the starting DNA and the copies synthesized in pre-vious steps become templates for further replication, resultingin an exponential increase by doubling the number of copies ofthe replicated region with each step. Figure 9.16 diagrams thesteps of the PCR operation, showing how you could use it toobtain many copies of a small portion of the �-globin gene forfurther study.
PCR Products Can Be Used Just Like ClonedRestriction FragmentsWhen properly executed, PCR provides all the highly enrichedDNA you could want for unambiguous analyses of many types.PCR products can be labeled to produce hybridization probesfor detecting related clones within libraries and relatedsequences within whole genomes spread out on Southern blots.PCR products can be sequenced (as described in the next sec-tion) to determine the exact genetic information they contain.Because the products are obtained without cloning, it is possibleto amplify and learn the sequence of a specific DNA segment in
The Polymerase Chain Reaction Provides a Rapid Method for Isolating DNA Fragments 297
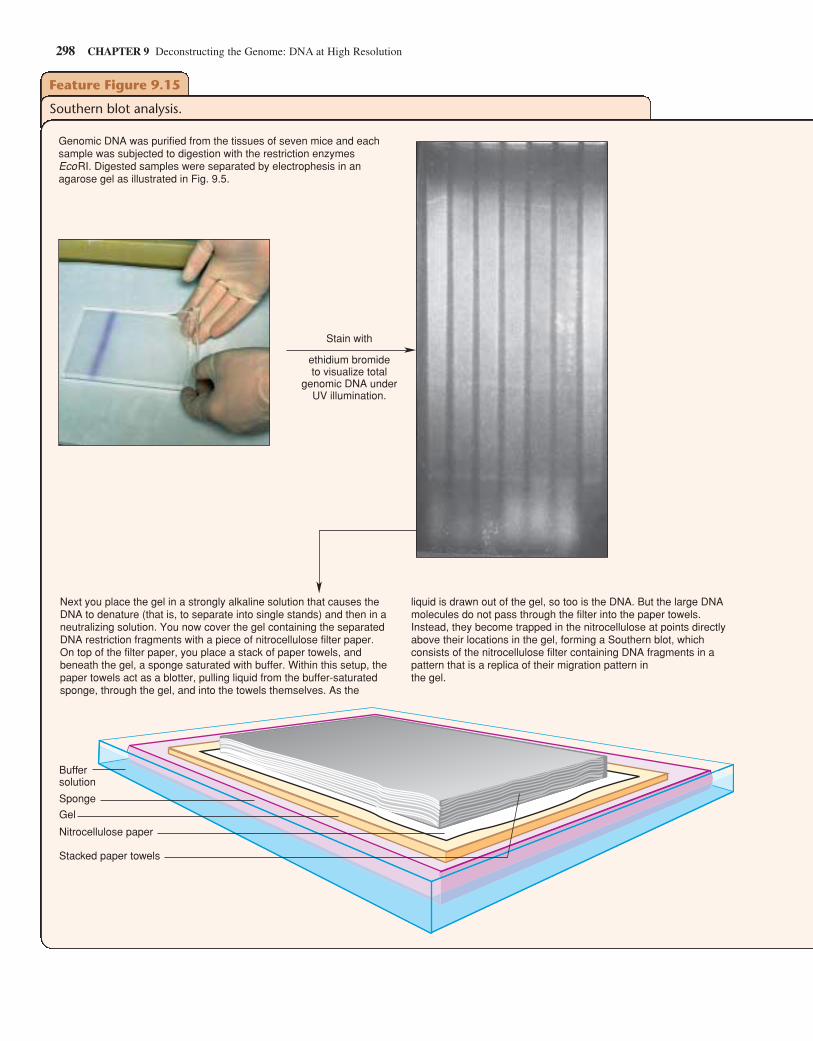
298 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Feature Figure 9.15
Southern blot analysis.
Buffersolution
Sponge
Gel
Nitrocellulose paper
Stacked paper towels
Stain with
ethidium bromideto visualize total
genomic DNA underUV illumination.
Genomic DNA was purified from the tissues of seven mice and each sample was subjected to digestion with the restriction enzymes EcoRI. Digested samples were separated by electrophesis in an agarose gel as illustrated in Fig. 9.5.
Next you place the gel in a strongly alkaline solution that causes the DNA to denature (that is, to separate into single stands) and then in a neutralizing solution. You now cover the gel containing the separated DNA restriction fragments with a piece of nitrocellulose filter paper. On top of the filter paper, you place a stack of paper towels, and beneath the gel, a sponge saturated with buffer. Within this setup, the paper towels act as a blotter, pulling liquid from the buffer-saturated sponge, through the gel, and into the towels themselves. As the
liquid is drawn out of the gel, so too is the DNA. But the large DNA molecules do not pass through the filter into the paper towels. Instead, they become trapped in the nitrocellulose at points directly above their locations in the gel, forming a Southern blot, which consists of the nitrocellulose filter containing DNA fragments in a pattern that is a replica of their migration pattern in the gel.
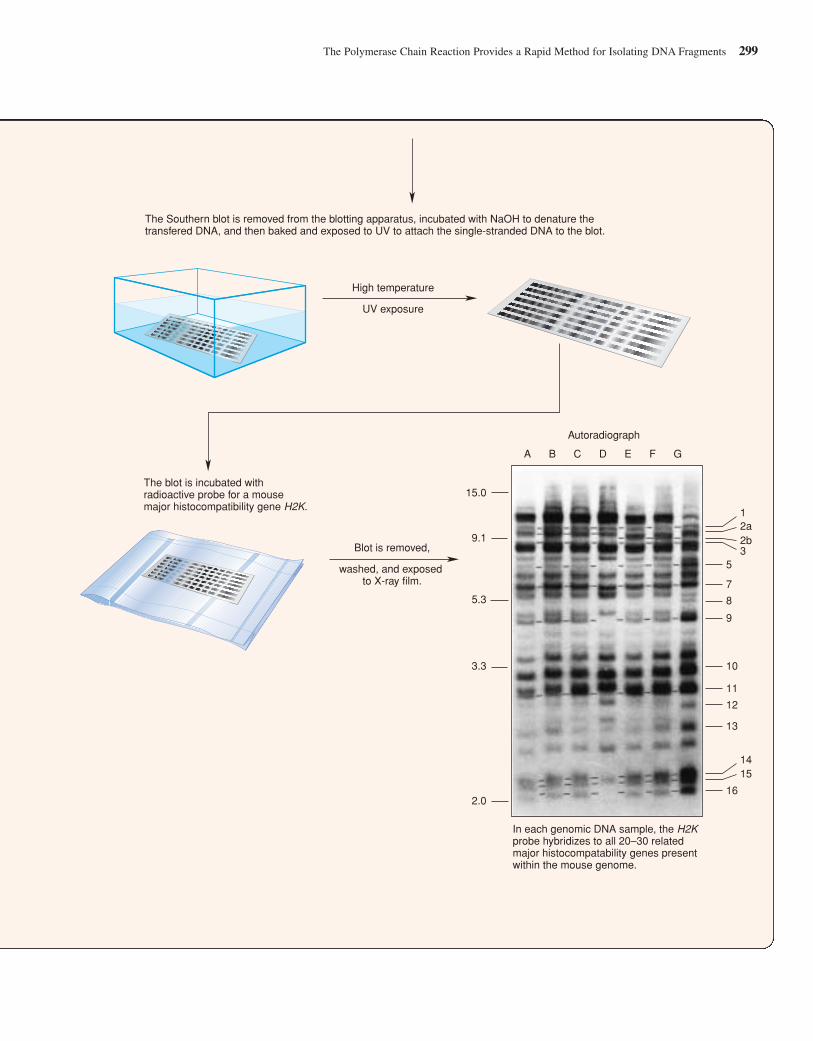
The Polymerase Chain Reaction Provides a Rapid Method for Isolating DNA Fragments 299
High temperature
UV exposure
The Southern blot is removed from the blotting apparatus, incubated with NaOH to denature the transfered DNA, and then baked and exposed to UV to attach the single-stranded DNA to the blot.
The blot is incubated with radioactive probe for a mouse major histocompatibility gene H2K.
Blot is removed,
washed, and exposed to X-ray film.
Autoradiograph
A B C D E F G
12a
1415
2b3
5
7
8
9
10
11
12
13
16
15.0
9.1
5.3
3.3
2.0
In each genomic DNA sample, the H2K probe hybridizes to all 20–30 related major histocompatability genes present within the mouse genome.
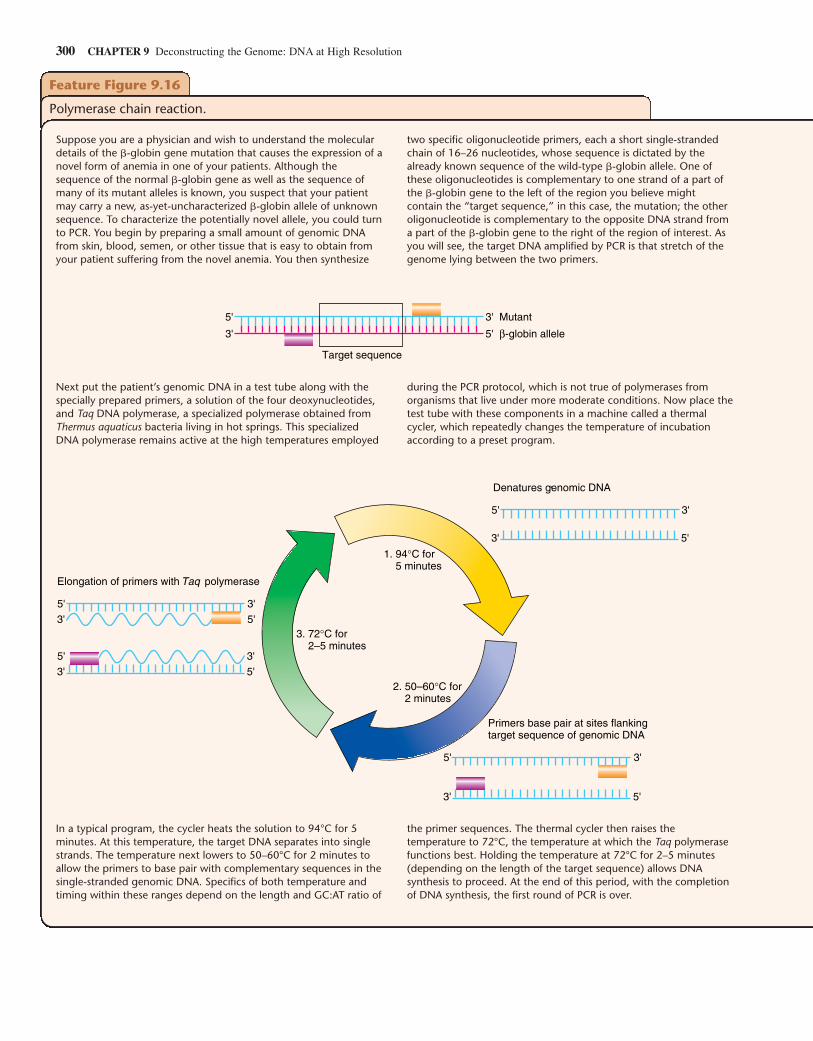
300 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Feature Figure 9.16
Polymerase chain reaction.
Suppose you are a physician and wish to understand the moleculardetails of the �-globin gene mutation that causes the expression of anovel form of anemia in one of your patients. Although thesequence of the normal �-globin gene as well as the sequence ofmany of its mutant alleles is known, you suspect that your patientmay carry a new, as-yet-uncharacterized �-globin allele of unknownsequence. To characterize the potentially novel allele, you could turnto PCR. You begin by preparing a small amount of genomic DNAfrom skin, blood, semen, or other tissue that is easy to obtain fromyour patient suffering from the novel anemia. You then synthesize
two specific oligonucleotide primers, each a short single-strandedchain of 16–26 nucleotides, whose sequence is dictated by thealready known sequence of the wild-type �-globin allele. One ofthese oligonucleotides is complementary to one strand of a part ofthe �-globin gene to the left of the region you believe mightcontain the “target sequence,” in this case, the mutation; the otheroligonucleotide is complementary to the opposite DNA strand froma part of the �-globin gene to the right of the region of interest. Asyou will see, the target DNA amplified by PCR is that stretch of thegenome lying between the two primers.
Target sequence
3' 5' β-globin allele
5' 3' Mutant
Next put the patient’s genomic DNA in a test tube along with thespecially prepared primers, a solution of the four deoxynucleotides,and Taq DNA polymerase, a specialized polymerase obtained fromThermus aquaticus bacteria living in hot springs. This specializedDNA polymerase remains active at the high temperatures employed
during the PCR protocol, which is not true of polymerases fromorganisms that live under more moderate conditions. Now place thetest tube with these components in a machine called a thermalcycler, which repeatedly changes the temperature of incubationaccording to a preset program.
3' 5'
5' 3'
3' 5'
5' 3'
3. 72°C for 2–5 minutes
2. 50–60°C for 2 minutes
1. 94°C for 5 minutes
Elongation of primers with Taq polymerase
5' 3'
3' 5'
Primers base pair at sites flankingtarget sequence of genomic DNA
5' 3'
3' 5'
Denatures genomic DNA
In a typical program, the cycler heats the solution to 94°C for 5minutes. At this temperature, the target DNA separates into singlestrands. The temperature next lowers to 50–60°C for 2 minutes toallow the primers to base pair with complementary sequences in thesingle-stranded genomic DNA. Specifics of both temperature andtiming within these ranges depend on the length and GC:AT ratio of
the primer sequences. The thermal cycler then raises thetemperature to 72°C, the temperature at which the Taq polymerasefunctions best. Holding the temperature at 72°C for 2–5 minutes(depending on the length of the target sequence) allows DNAsynthesis to proceed. At the end of this period, with the completionof DNA synthesis, the first round of PCR is over.
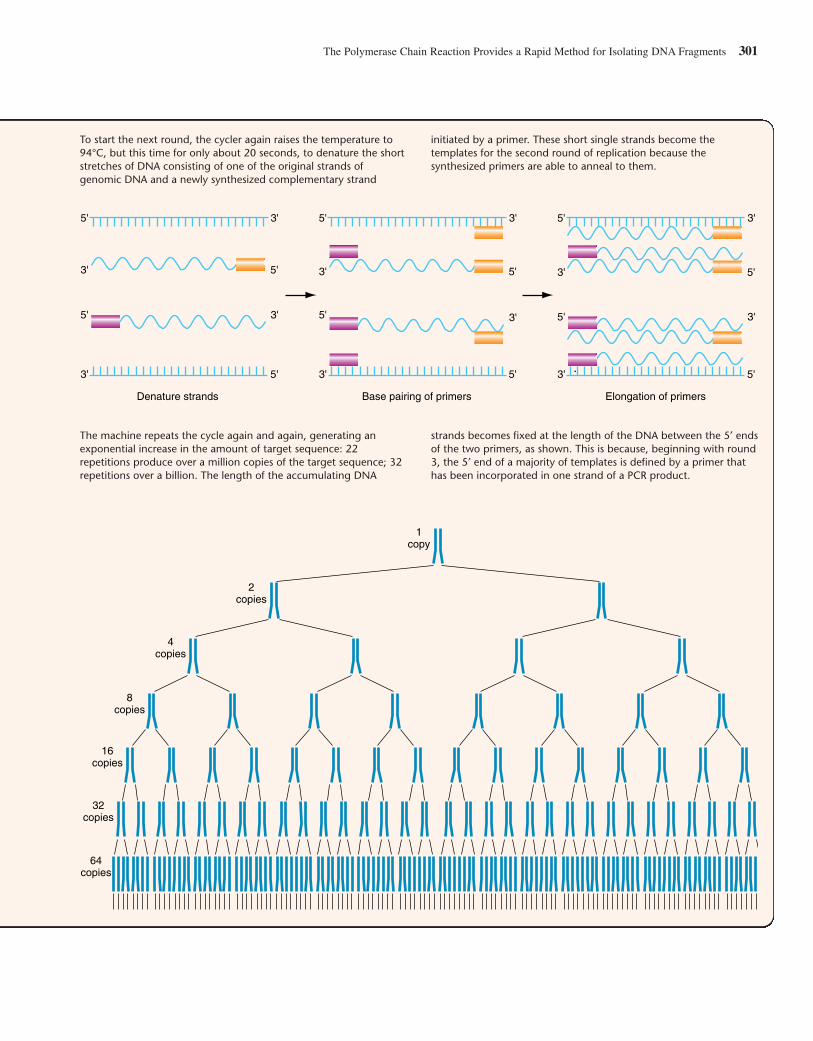
The Polymerase Chain Reaction Provides a Rapid Method for Isolating DNA Fragments 301
Denature strands Base pairing of primers Elongation of primers
5' 3'
5' 3'
3' 5'
3' 5'
5' 3'
5' 3'
3' 5'
3' 5'
5' 3'
5' 3'
3' 5'
3' 5'
To start the next round, the cycler again raises the temperature to94°C, but this time for only about 20 seconds, to denature the shortstretches of DNA consisting of one of the original strands ofgenomic DNA and a newly synthesized complementary strand
initiated by a primer. These short single strands become thetemplates for the second round of replication because thesynthesized primers are able to anneal to them.
The machine repeats the cycle again and again, generating anexponential increase in the amount of target sequence: 22repetitions produce over a million copies of the target sequence; 32repetitions over a billion. The length of the accumulating DNA
strands becomes fixed at the length of the DNA between the 5′ endsof the two primers, as shown. This is because, beginning with round3, the 5′ end of a majority of templates is defined by a primer thathas been incorporated in one strand of a PCR product.
64copies
32copies
16copies
8copies
4copies
2copies
1copy

a very short time. In fact, in checking for a particular hemoglo-bin mutation, one could start with a blood sample and determinea DNA sequence within two days.
As an analytic tool, PCR has several advantages overcloning. First, it provides the ultimate in sensitivity: The mini-mum input for the polymerase chain reaction to proceed is asingle DNA molecule. Second, as we have seen, it is very fast,requiring no more than a few hours to generate enough ampli-fied DNA for analysis; by comparison, the multiple steps ofcloning usually require at least a week to complete. Third, inthe sometimes highly competitive research world, it is an agentof democracy: Once the base sequence of the oligonucleotideprimers that define a particular target sequence appears in print,anyone with the relatively small amount of funds needed tosynthesize or buy these primers can reproduce the reaction.PCR is nevertheless unsuitable in certain situations. Becausethe protocol only copies DNA fragments up to 25 kb in length,it cannot amplify larger regions of interest. And because thesynthesis of PCR primers depends on sequence informationfrom the target region, the protocol cannot serve as the startingpoint for the analysis of genes or genomic regions that have notyet been cloned and sequenced.
PCR Has Many UsesPCR is one of the most powerful techniques in molecular biol-ogy. Its originator, Kary Mullis, received the 1993 Nobel Prizein chemistry for his 1985 invention of this tool for geneticanalysis. By making it possible to amplify a specific sequencedirectly from complex DNAs (either genomic or cDNA), PCRhas made molecular analysis an essential component of geno-type detection and gene mapping; we describe its applicationsin these areas in Chapters 10 and 11. By making it possible toanalyze traces of partially degraded DNA from ancient samples(based on related sequences from living organisms) and tocompare homologous sequences from a variety of sources,PCR has revolutionized evolutionary studies, enablingresearchers to analyze sequences from both living and extinctorganisms and to determine the relatedness between theseorganisms with greater accuracy than ever before (Chapter 21discusses evolution at the molecular level). By making it possi-ble to analyze the same sequence in hundreds or thousands ofindividual samples from humans, other animals, or plants with-out having to make hundreds or thousands of libraries, PCR hasfacilitated the study of gene diversity at the nucleotide level inpopulations and greatly simplified the process of monitoringgenetic changes in a group over time (see Chapter 20 for thedetails of population genetics). Finally, PCR has helped bringmolecular genetics to many fields outside of traditional genet-ics. The following example of its use in diagnosing infectiousdisease provides an inkling of its potential impact on medicine.
AIDS, like other viral diseases, though not inherited, is inone sense a genetic disease, because it is caused by the activityof foreign DNA inside a subgroup of somatic cells. HIV, thevirus associated with AIDS, gains entry to a person’s bodythrough the bloodstream or lymphatic system, then docks atspecific membrane receptors on a few types of white blood
cells, fuses with the cell membrane, and releases its RNA chro-mosome along with several copies of reverse transcriptase intothe cell (see the Genetics and Society box on p. 252 in Chapter8). Once inside the cell, the reverse transcriptase copies theRNA to cDNA. The double-stranded DNA copy of the viralgenome then integrates itself into the host genome where,known as a provirus, it can lie latent for up to 10 years orbecome active at any time. When activated, it directs the cellu-lar machinery to make more viral particles.
Standard tests for HIV detect antibodies to the virus, but itmay take several months for the antibodies produced by aninfected person’s immune system to reach levels that are mea-surable in the blood. Then, in another few months, when ongo-ing viral activity inside many types of circulating white bloodcells subsides, most of the antibodies may disappear from thecirculation. This is because once the viral particles have enteredthe latent state, they are literally in hiding and able to avoiddetection by the immune system.
With PCR, it is possible to detect small amounts of viruscirculating in the blood or lymph very soon after infection,before antibody production is in full swing. PCR can alsodetect viral DNA incorporated in the genome of any cell, pick-ing up as little as 1–10 copies of viral DNA per million cells.Thus, with PCR, it becomes possible to confirm and begintreating HIV infection during the critical period before anti-bodies reach measurable levels. It also becomes possible to fol-low the progress of each person’s HIV infection and tailortherapies accordingly, using a large dose of certain drugs tocombat a large amount of viral activity but small doses of per-haps other drugs to prevent a small number of cells fromemerging from latency.
In summary, once the base sequence of a particular locushas been determined, it becomes possible to develop a PCRassay capable of picking out any allelic version of that locusthat is present in any DNA sample. This is because if the primertemplates find a match, PCR will amplify any sequencebetween the primers, even those with a slightly different orderof bases. Automation of the PCR procedure makes it possibleto screen thousands of samples in a short time.
DNA Sequence Analysis
The definitive description of any DNA molecule—from a shortfragment in a DNA clone to a whole human chromosome—isthe sequence of its bases. The DNA sequence of a gene pro-vides a staggering amount of practical information. Restrictionenzyme recognition sites useful for manipulating a gene areimmediately visible from the DNA sequence. The codons inopen reading frames allow scientists to predict the amino-acidsequence of the protein product of the gene. Once the protein’samino-acid sequence is known, computer programs can beinstructed to model the three-dimensional shape of the protein,thereby projecting something about its function. Comparison ofgenomic and cDNA sequences immediately shows how a geneis divided into exons and introns and may suggest whether
302 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution

alternative splicing of the gene’s primary transcript occurs.Compilations of the DNA sequences of many genes whoseexpression is turned on by the same environmental cues, aswell as the sequences of the genes’ surrounding regions, cansupply clues about the DNA signals that determine this kind ofgene regulation. Even an exploration of the DNA sequencesbetween genes can provide important information about theevolution of genomes.
No matter how large the ultimate goal—up to and includ-ing the determination of the sequence of the whole humangenome—all sequencing projects depend on the protocoldescribed next. It allows the sequence determination of 800bases at a time from a DNA fragment that has been isolatedand amplified by cloning or PCR. For larger sequencing proj-ects, the protocol is repeated on hundreds, thousands, or evenmillions of individually purified DNA fragments. The finalextended sequence is then pieced together like a puzzleaccording to computer programs that search for overlaps in thesequence information generated by each application of theprotocol.
The protocol currently used for determining DNAsequence derives from one of two sequencing technologiesdeveloped in the 1970s. One technology, developed by AlanMaxam and Walter Gilbert, is based on the chemical cleavageof DNA molecules at specific nucleotide types. The secondtechnology, developed by Fred Sanger, is based on the enzy-matic extension of DNA strands to a defined terminating base.Maxam, Gilbert, and Sanger all won the Nobel Prize for theircontribution to DNA sequencing technology. Their techniqueshave a similar accuracy, which approaches 99.9%. But only theSanger technique is readily amenable to automation, and it isonly with automation that DNA sequencing reaches its fullpotential.
General Principles of the ProcedureThere are two steps to the Sanger method of sequencing, whoseobject is to reveal the order of base pairs in an isolated DNAmolecule. The first step is the generation of a complete seriesof single-stranded subfragments complementary to a portion ofthe DNA template under analysis. (Although both strands of aDNA fragment are present in a typical DNA sample, only oneis used as a template for sequencing.) Each subfragment differsin length by a single nucleotide from the preceding and suc-ceeding fragments; and the graduated set of fragments isknown as a nested array. A critical feature of the subfragmentsis that each one is distinguishable according to its terminal 3′base. Thus, each subfragment has two defining attributes—rel-ative length and one of four possible terminating nucleotides.In the second step of the sequencing process, biologists analyzethe mixture of DNA subfragments through polyacrylamide gelelectrophoresis, under conditions that allow the separation ofDNA molecules differing in length by only a single nucleotide.
The original Sanger sequencing procedure (illustrated inFig. 9.17) begins with the denaturing into single strands of theDNA to be sequenced. The single strands are then mixed insolution with DNA polymerase, the four deoxynucleotide
triphosphates (dATP, dCTP, dGTP, and dTTP), and a radioac-tively labeled oligonucleotide primer complementary to DNAadjacent to the 3′ end of the template strand under analysis.(Typically, sequencing is performed on purified DNA clones or amplified PCR products. For sequencing DNA clones, thevector-insert recombinant molecule is retained intact, and aprimer is used that is complementary to the vector sequenceimmediately adjacent to the insert DNA. For sequencing PCRproducts, the PCR primer can also serve as the sequencingprimer.) The solution is next divided into four aliquots. To eachone, an invesitgator adds a small amount of a single type ofdeoxynucleotide triphosphate lacking the 3′-hydroxyl groupthat is critical for the formation of the phosophodiester bondsthat lead to chain extension (review Fig. 6.7); this nucleotideanalogue is called a dideoxynucleotide and it comes in fourforms: ddTTP, ddATP, ddGTP, or ddCTP (abbreviated fromhere on as ddT, ddA, ddG, and ddC).
In each sample reaction tube, the oligonucleotide primerwill hybridize at the same location on the template DNAstrand. As a primer, it will supply a free 3′ end for DNA chainextension by DNA polymerase. The polymerase will addnucleotides to the growing strand that are complementary tothe nucleotides of the sample’s template strand (that is, theactual DNA strand under analysis). The addition ofnucleotides will continue until, by chance, a dideoxynu-cleotide is incorporated instead of a normal nucleotide. Theabsence of a 3′-hydroxyl group in the dideoxynucleotide pre-vents the DNA polymerase from forming a phosphodiesterbond with any other nucleotide, ending the polymerization forthat new strand of DNA. Next, after allowing enough time forthe polymerization of all molecules to reach completion, aninvestigator separates the templates from the newly synthe-sized strands by denaturing the DNA at high temperature.Each sample tube now holds a whole collection of single-stranded radioactive DNA chains as well as the nonradioactivesingle strands of the template DNA. The lengths of theradioactive chains reflect the distance from the 5′ end of theoligonucleotide primer to the position in the sequence at whichthe specific dideoxynucleotide present in that particular tubewas incorporated into the growing chain. (Recall that con-struction of this chain, which is complementary to the templateDNA under analysis, was initiated by a radioactive primer.)
The samples in the four tubes are now electrophoresed inadjacent lanes on a polyacrylamide gel, and the gel is subjectedto autoradiography. Because the template strands are notlabeled, they do not show up on the autoradiograph. The inves-tigator reads out the sequence of the radioactive strand by start-ing at the bottom and moving up the autoradiograph,determining which lane carries each subsequent band in theascending series, as shown in Fig. 9.17. Recall that as youascend the radiograph, each step pictures a chain that is onenucleotide longer than the chain of the step below. Once thesequence of the newly synthesized DNA is known, it is a sim-ple matter to convert this sequence (on paper or by computer)into the complementary sequence of the template strand underanalysis.
DNA Sequence Analysis 303
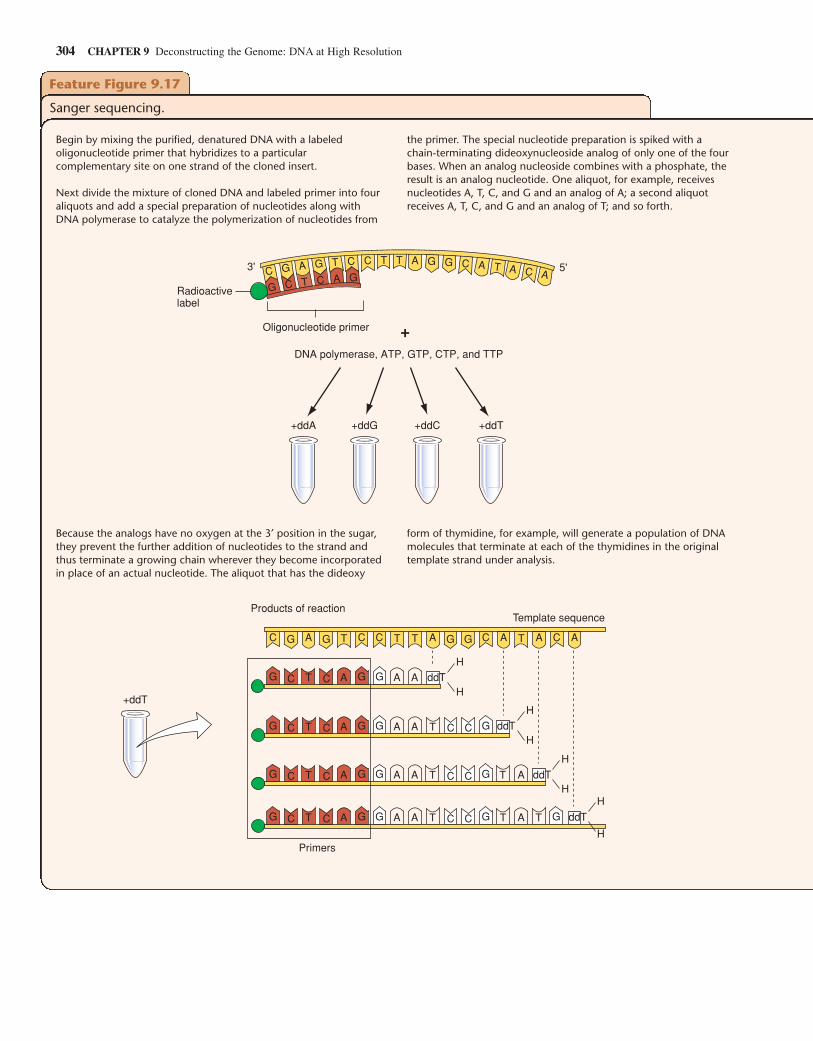
304 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Feature Figure 9.17
Sanger sequencing.
Begin by mixing the purified, denatured DNA with a labeledoligonucleotide primer that hybridizes to a particularcomplementary site on one strand of the cloned insert.
Next divide the mixture of cloned DNA and labeled primer into fouraliquots and add a special preparation of nucleotides along withDNA polymerase to catalyze the polymerization of nucleotides from
DNA polymerase, ATP, GTP, CTP, and TTP
+ddA +ddG +ddC +ddT
C G A G T C C T T A G G C A T A C AG C T C A G
3' 5'
Oligonucleotide primer
Radioactivelabel
+
the primer. The special nucleotide preparation is spiked with achain-terminating dideoxynucleoside analog of only one of the fourbases. When an analog nucleoside combines with a phosphate, theresult is an analog nucleotide. One aliquot, for example, receivesnucleotides A, T, C, and G and an analog of A; a second aliquotreceives A, T, C, and G and an analog of T; and so forth.
Because the analogs have no oxygen at the 3′ position in the sugar,they prevent the further addition of nucleotides to the strand andthus terminate a growing chain wherever they become incorporatedin place of an actual nucleotide. The aliquot that has the dideoxy
form of thymidine, for example, will generate a population of DNAmolecules that terminate at each of the thymidines in the originaltemplate strand under analysis.
C C C C CA A A AAT T T TG G GG
C C A A AG G GT ddT
C C C CA A AG G GGT
C C A A A AG G GT
T
C C GT T
C C G GT T TC C A A A AG G GT
ddT
H
H
ddT
ddT
+ddT
Products of reactionTemplate sequence
Primers
H
H
H
HH
H

DNA Sequence Analysis 305
Then use electrophoresis on a polyacrylamide gel to separate thefragments in each of the four aliquots and arrange them by size. Theresolution of the gel is such that you can distinguish DNA molecules
that differ in length by only a single base. The appearance of a DNAfragment of a particular length demonstrates the presence of aparticular nucleotide at that position in the strand.
Suppose, for example, that the aliquot polymerized in the presenceof dideoxythymidine shows fragments 32, 35, and 39 bases inlength. These fragments indicate that thymidine is present at thosepositions in the strand of nucleotides. In practice, one does notindependently determine the exact lengths of each fragment.
Instead, one starts at the bottom of the gel, looks at which of thefour lanes has a band in it, records that base, then moves up oneposition and determines which lane has the next band, and so on.In this way, it is possible to read several hundred bases from a singleset of reactions.
Gel analysis of fragments Sequence ofsynthesizedDNA
Sequence oftemplateDNA
T
G
G
G
C
C
A
A
A
T
T
T
C
C
C
G
G
T
T
T
A
A
A
A
5'
3'
ddA ddG ddC ddT
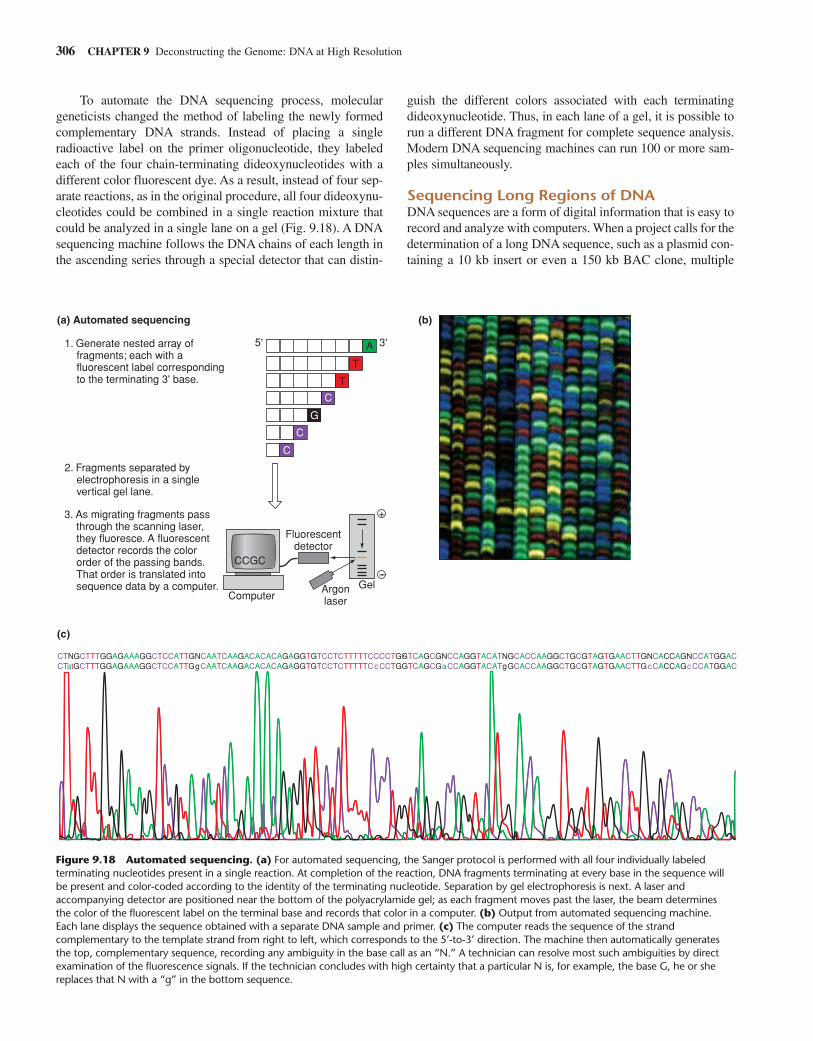
To automate the DNA sequencing process, moleculargeneticists changed the method of labeling the newly formedcomplementary DNA strands. Instead of placing a singleradioactive label on the primer oligonucleotide, they labeledeach of the four chain-terminating dideoxynucleotides with adifferent color fluorescent dye. As a result, instead of four sep-arate reactions, as in the original procedure, all four dideoxynu-cleotides could be combined in a single reaction mixture thatcould be analyzed in a single lane on a gel (Fig. 9.18). A DNAsequencing machine follows the DNA chains of each length inthe ascending series through a special detector that can distin-
guish the different colors associated with each terminatingdideoxynucleotide. Thus, in each lane of a gel, it is possible torun a different DNA fragment for complete sequence analysis.Modern DNA sequencing machines can run 100 or more sam-ples simultaneously.
Sequencing Long Regions of DNADNA sequences are a form of digital information that is easy torecord and analyze with computers. When a project calls for thedetermination of a long DNA sequence, such as a plasmid con-taining a 10 kb insert or even a 150 kb BAC clone, multiple
306 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
(c)
(a) Automated sequencing (b)
1. Generate nested array offragments; each with afluorescent label correspondingto the terminating 3' base.
2. Fragments separated byelectrophoresis in a singlevertical gel lane.
3. As migrating fragments passthrough the scanning laser,they fluoresce. A fluorescentdetector records the colororder of the passing bands.That order is translated intosequence data by a computer.
3'5'
G
C
C
C
A
T
T
CCGC
Fluorescentdetector
Argonlaser
Gel
+
–
Computer
CTatGCTTTGGAGAAAGGCTCCATTGgCAATCAAGACACACAGAGGTGTCCTCTTTTTCcCCTGGTCAGCGaCCAGGTACATgGCACCAAGGCTGCGTAGTGAACTTGcCACCAGcCCATGGACCTNGCTTTGGAGAAAGGCTCCATTGNCAATCAAGACACACAGAGGTGTCCTCTTTTTCCCCTGGTCAGCGNCCAGGTACATNGCACCAAGGCTGCGTAGTGAACTTGNCACCAGNCCATGGAC
Figure 9.18 Automated sequencing. (a) For automated sequencing, the Sanger protocol is performed with all four individually labeledterminating nucleotides present in a single reaction. At completion of the reaction, DNA fragments terminating at every base in the sequence willbe present and color-coded according to the identity of the terminating nucleotide. Separation by gel electrophoresis is next. A laser andaccompanying detector are positioned near the bottom of the polyacrylamide gel; as each fragment moves past the laser, the beam determinesthe color of the fluorescent label on the terminal base and records that color in a computer. (b) Output from automated sequencing machine.Each lane displays the sequence obtained with a separate DNA sample and primer. (c) The computer reads the sequence of the strandcomplementary to the template strand from right to left, which corresponds to the 5′-to-3′ direction. The machine then automatically generatesthe top, complementary sequence, recording any ambiguity in the base call as an “N.” A technician can resolve most such ambiguities by directexamination of the fluorescence signals. If the technician concludes with high certainty that a particular N is, for example, the base G, he or shereplaces that N with a “g” in the bottom sequence.

rounds of sequencing must be performed to obtain data that thecomputer can combine into a single final sequence.
There are two basic strategies for sequencing the relativelylarge DNA inserts of a clone: primer walking and shotgunsequencing.
Primer walking begins with the use of two vector-basedprimers, flanking the two ends of an insert. Each primer makesit possible to sequence into one strand of the unknown insert for500–800 nucleotides. The information obtained at the end ofthese sequences can be used to program an automatedsequencer to synthesize new primers for the next round ofsequencing. An investigator repeats this process until he or shehas sequenced both strands of the entire insert. Although thetwo strands are complementary to each other, and thus defineeach other entirely, it is useful to have two independentlyobtained sequences to confirm the accuracy of the dataobtained.
The primer-walking protocol is a reasonable approach toobtaining a sequence for a relatively small clone of perhaps 5kb in length. But for a 150 kb BAC clone, under ideal condi-tions, primer walking would require 300 sequential rounds ofDNA sequencing and primer synthesis. This could take a yearto complete.
Shotgun sequencing provides a much faster way to deter-mine the DNA sequence of a larger clone. To carry out shotgunsequencing, you begin by fragmenting the BAC clone, or otherDNA starting material, at random into a large number ofsmaller pieces. Partial digestion of the DNA with a four-basecutter or through intense shearing with high-frequency, high-intensity sound waves (Fig. 9.19) will generate these smallerfragments. You can then clone and sequence them using anoligonucleotide primer complementary to a vector sequencedirectly adjacent to the unknown insert. If you ascertain thesequences of a sufficient number of random fragments (thenumber is determined by the total size of the original clone orgenome), the likelihood that you have obtained complete cov-erage of the original DNA fragment is high. You now enter thesequences into a computer programmed to look for sequenceoverlaps. If the number of sequenced fragments is largeenough, it is possible to find overlaps of sequences extendingacross the entire original clone. Any gaps can be filled in by theprimer-walking protocol described previously.
The shotgun approach thus relies on redundancy—youhave to gather sequence information on three to four times theactual number of base pairs in the original clone to make itwork. Even so, it is an efficient method of sequencing. Thedownside of shotgun sequencing is that only large laboratoriesequipped with many automated DNA synthesizers able to oper-ate simultaneously can carry it out.
Understanding the Genes for Hemoglobin: A ComprehensiveExample
Geneticists have used the tools of recombinant DNA technol-ogy to analyze the clusters of related genes that make up the �-and �-globin loci. They have isolated mRNA from red bloodcell precursors and with the use of reverse transcriptase, pro-duced cDNA libraries. They easily probed these libraries for �-globin and �-globin cDNA clones because 80% of thetranscripts within red blood cell precursors code for thesepolypeptides, and the proportion of �-globin and �-globincDNA clones in the libraries was equivalently high. They havesequenced individual cDNA clones to identify those that car-ried coding sequences for either �-globin or �-globin polypep-tides. They have used PCR to amplify for sequencing theglobin genes present in many individuals, both normal and diseased.
Geneticists have probed genomic libraries with the �- and�-globin cDNA clones and identified genomic clones carryingglobin genes and their surrounding regions. They have alsoused cDNA clones to probe Southern blots containing restric-tion fragments of the genomic clones to determine the numberand location of coding sequences within each genomic locus.Finally, they have sequenced the entire chromosomal regionsthat contain the �-globin or �-globin genes.
Fundamental insights from these studies have helpedexplain how the linear information of DNA encompasses all the
Understanding the Genes for Hemoglobin: A Comprehensive Example 307
Individualsequences
Sequences derived from overlapping fragments
Original fragment
Random cut with four-base recognition restriction enzyme
Clone fragments
Enter data into computer; look for overlaps.
Sequence from vector-based primer
Primer Chain extension
Figure 9.19 Sequencing a long DNA molecule. In shotgunsequencing, the long DNA molecule is reduced into a large number ofsmaller fragments that are cloned and individually sequenced. Theindividual sequences are aligned by computer to generate a singlecontinuous sequence that spans the entire length of the originalmolecule.

instructions for the development of the hemoglobin system,including the changes in globin expression during normaldevelopment. The studies have also clarified how the globingenes evolved, and how a large number of different mutationsproduce the phenotypic permutations that give rise to a range ofglobin-related disorders.
The Genes Encoding Hemoglobin Occur in Two Clusters on Two SeparateChromosomesThe �-globin gene cluster contains three functional genes andspans about 28 kb on chromosome 16 (Fig. 9.20a). All thegenes in the �-gene cluster point in the same direction; that is,they all use the same strand of DNA as the template for tran-scription. Moving in the 5′-to-3′ direction along the RNA-likestrand, the � or �-like genes appear in the order �, �2, �1. Thegenes in the �-gene cluster, like those in the �-gene cluster, allpoint in the same direction. The �-globin gene cluster covers50 kb on chromosome 11 and contains five functional genes inthe order �, G�, A�, �, and � (Fig. 9.20b). Geneticists refer tothe chromosomal region carrying all of the �-globin-like genes
as the �-globin locus and the region containing the �-globin-like genes as the �-globin locus. Note that the term locus sig-nifies a location on a chromosome; that location may be assmall as a single nucleotide or as large as a cluster of relatedgenes.
The Succession of Genes in Each Cluster Correlateswith the Sequence of Their Expression DuringDevelopmentThe linear organization of the genes in the �- and �-gene clus-ters reflects the order in which they are expressed during devel-opment. For the �-like chains, that temporal order is � duringthe first five weeks of embryonic life, followed by the two �chains during fetal and adult life. For the �-like chains, theorder is � during the first five weeks of embryonic life; then thetwo � chains during fetal life; and finally, within a few monthsof birth, mostly � but also some � chains (see Fig. 9.1 on p. 278and Fig. 9.20).
The fact that the order of genes on the chromosomesreflects the order of their expression during development sug-gests that whatever mechanism turns these genes on and offtakes advantage of their relative positions. We now understandwhat that mechanism is: A locus control region (or LCR) asso-ciated with specialized DNA binding proteins at the 5′ end ofeach locus works its way down the locus, bending the chro-matin back on itself to turn genes on and off in order. Wedescribe this regulatory mechanism in more detail on p. XXXof Chapter 17.
One consequence of a master regulatory element that con-trols an entire gene complex is seen in a rare medical conditionwith a surprising prognosis. In some adults, the red blood cellprecursors express neither � or � chains. Although this shouldbe a lethal situation, these adults remain healthy. Cloning andsequence analysis of the �-globin locus from affected adultsshow that they have a deletion extending across the � and �genes. Because of this deletion, the master regulatory controlcan’t switch at birth, as it normally would, from �-globin pro-duction to �- and �-globin production (Fig. 9.20c). People withthis rare condition continue to produce large amounts of fetal �globin throughout adulthood, and that � globin is sufficient tomaintain a near-normal level of health.
A Variety of Mutations Account for the Diverse Symptoms of Globin-Related DiseasesBy comparing DNA sequences from affected individuals withthose from normal individuals, researchers have learned thatthere are two general classes of disorders arising from alter-ations in the hemoglobin genes. In one class, mutations changethe amino-acid sequence and thus the three-dimensional struc-ture of the �- or �-globin chain, and these structural changesresult in an altered protein whose malfunction causes thedestruction of red blood cells. Diseases of this type are knownas hemolytic anemias (Fig. 9.21a). An example is sickle-cellanemia, caused by an A-to-T substitution in the sixth codon ofthe �-globin chain. This simple change in DNA sequence alters
308 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
X
(a) α-globin genes on human chromosome 16
(b) β-globin genes on human chromosome 11
kb
kb
30 20 10 0
60 50 40 30 20 10 0
ζ ψζ ψα1 α2 α1
ψβ2 ε Gγ Aγ ψβ1 δ β
Expressed genes Pseudogenes
(c) Mutant deleted form of β-globin locus
β-δ deletion
ψβ2 ε Gγ Aγ ψβ1 δ β
LCR
LCR
LCR
Figure 9.20 The genes for the polypeptide components ofhuman hemoglobin are located in two genomic clusters ontwo different chromosomes. (a) Schematic representation of the�-globin gene cluster on chromosome 16. The cluster contains threefunctional genes and two pseudogenes (designated by the symbol �);pseudogenes are sequences that look like but do not function asgenes. (b) Schematic representation of the �-globin gene cluster onchromosome 11; this cluster has five functional genes and twopseudogenes. Upstream from both the �- and �-globin gene clusterslies the locus control region (LCR), which interacts with and activatessequential genes in each complex during human development. (c) Inthis example of a mutant chromosome, the adult �-globin genes havebeen deleted; as a result, the LCR cannot switch from activating thefetal genes to activating the adult genes, and the fetal genes remainactive in the adult.
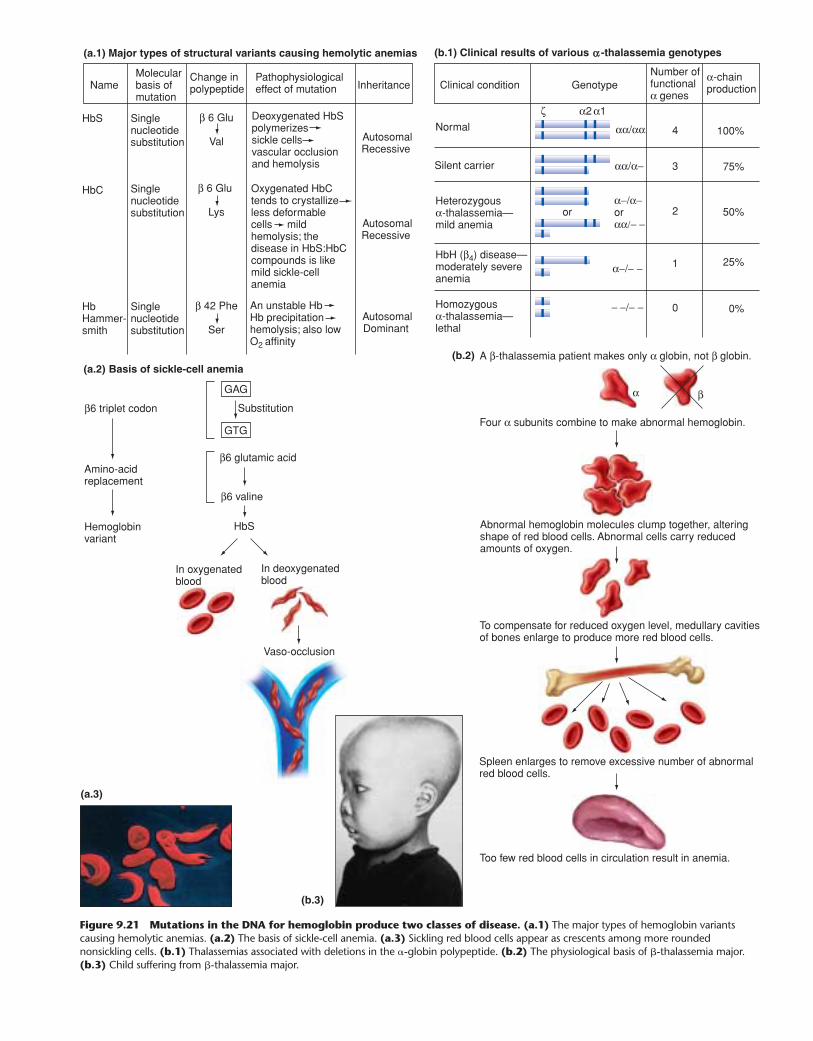
Basis of sickle-cell anemia
GAG
GTG
(b.1) Clinical results of various -thalassemia genotypes
Clinical condition
Normal
A β-thalassemia patient makes only α globin, not β globin.
Four α subunits combine to make abnormal hemoglobin.
Abnormal hemoglobin molecules clump together, alteringshape of red blood cells. Abnormal cells carry reduced amounts of oxygen.
Silent carrier
Heterozygous α-thalassemia—mild anemia
HbH (β4) disease—moderately severe anemia
Homozygous α-thalassemia—lethal
GenotypeNumber of functional α genes
α-chain production
αα/αα
α2ζ α1
αα/α−
4 100%
3 75%
α−/α−orαα/− −
α−/− −
− −/− −
2
1
0
50%
25%
0%
To compensate for reduced oxygen level, medullary cavitiesof bones enlarge to produce more red blood cells.
Spleen enlarges to remove excessive number of abnormalred blood cells.
Too few red blood cells in circulation result in anemia.
or
α β
(a.1) Major types of structural variants causing hemolytic anemias
NameMolecular basis of mutation
Change inpolypeptide
Pathophysiologicaleffect of mutation Inheritance
β6 triplet codon Substitution
Amino-acidreplacement
Hemoglobinvariant
HbS
β6 glutamic acid
In oxygenatedblood
In deoxygenatedblood
Vaso-occlusion
β6 valine
Single nucleotide substitution
Single nucleotide substitution
Single nucleotide substitution
Deoxygenated HbS polymerizes sickle cells vascular occlusion and hemolysis
Oxygenated HbC tends to crystallize less deformable cells mild hemolysis; the disease in HbS:HbC compounds is like mild sickle-cell anemia
An unstable Hb Hb precipitation hemolysis; also low O2 affinity
AutosomalRecessive
AutosomalRecessive
AutosomalDominant
β 6 Glu
Val
β 6 Glu
Lys
β 42 Phe
Ser
HbS
HbC
HbHammer-smith
(a.3)
(b.3)
(b.2)(a.2)
Figure 9.21 Mutations in the DNA for hemoglobin produce two classes of disease. (a.1) The major types of hemoglobin variantscausing hemolytic anemias. (a.2) The basis of sickle-cell anemia. (a.3) Sickling red blood cells appear as crescents among more roundednonsickling cells. (b.1) Thalassemias associated with deletions in the �-globin polypeptide. (b.2) The physiological basis of �-thalassemia major.(b.3) Child suffering from �-thalassemia major.

the sixth amino acid in the chain from glutamic acid to valine,which, in turn, modifies the form and function of the affectedhemoglobin molecules. Red blood cells carrying these alteredmolecules often have abnormal shapes that cause them to blockblood vessels or be degraded.
The second major class of hemoglobin-related genetic dis-eases arises from DNA mutations that reduce or eliminate theproduction of one of the two globin polypeptides. The diseasestate resulting from such mutations is known as thalassemia,from the Greek words thalassa meaning “sea” and emia mean-ing “blood”; the name arose from the observation that a rela-tively high rate of this blood disease occurs among people wholive near the Mediterranean Sea. Several different types ofmutation can cause thalassemia, including those that delete anentire � or � gene, those that alter the sequence in regions thatare outside the gene but necessary for its regulation, or thosethat alter the sequence within the gene such that no protein canbe produced. The consequence of these changes in DNAsequence is the total absence or a deficient amount of one or theother of the normal hemoglobin chains. Because there are two� genes (�1 and �2) that see roughly equal expression begin-ning a few weeks after conception, individuals carrying dele-tions within the �-globin locus may be missing anywhere fromone to four copies of the �-globin genes (Fig. 9.21b). A personlacking only one would be a heterozygote for the deletion ofone of two � genes; a person missing all four would be ahomozygote for deletions of both � genes. The range of muta-tional possibilities explains the range of phenotypes seen in �-thalassemia. Individuals missing only one of four possiblecopies of the � genes are normal; those lacking two of the fourhave a mild anemia, and those without all four die before birth.The fact that the � chains are expressed early in fetal lifeexplains why the �-thalassemias are detrimental in utero. Bycontrast, �-thalassemia major, the disease occurring in peoplewho are homozygotes for most deletions of the single � gene,also usually results in death, but not until early childhood.
Comparisons of the altered DNA sequences from affectedindividuals with wild-type sequences from normal individualshave helped illuminate the sequences necessary for normalhemoglobin expression. In some �-thalassemia patients, forexample, disease symptoms arise from the alteration of a fewnucleotides adjacent to the 5′ end of the coding region for the� chain. Data of this type have defined sequences that areimportant for expression of the �-globin locus. One such seg-ment is the TATA box, a sequence found in many eukaryoticpromoters (Fig. 9.22a; see p. XXX in Chapter 17 for a moredetailed discussion). In other thalassemia patients, the entire �-globin locus and adjacent regulatory segments, including theTATA box, are intact, but a mutation has altered the LCR foundfar to the 5′ side of all the �-like genes. This LCR is necessaryfor a high level of tissue-specific expression of all �-like genesin red blood cell progenitors (Fig. 9.22b). Mutations in theTATA box or the locus control region, depending on how dis-ruptive they are, produce �- or �-thalassemias of varyingseverity.
The �- and �-Globin Loci House Multiple Genes That Evolved from One Ancestral GeneFrom sequencing the DNA of all the human globin genes,researchers can see that the genes form a closely related group,or family, of genes that evolved by duplication and divergencefrom one ancestral gene (Fig. 9.23). All gene sequences evolvecontinuously. The two separate products of a duplication,
310 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
start3'5'
DNAof β-globin gene
mRNA
LCR
60 30 20
2
10 05kb
ζ α2α-globin cluster
α1 θ
β-globinprotein Met • • • • • •Val His Leu
3'
5'
5'
TATA box 25–30bp Transcription
Translation
A
T A
T
A
T
A
T
A
T
A
T A
T A
T
A
T
A
T
A
T
A
T
A U G G U UG GC A C C
Figure 9.22 Mutations in globin regulatory regions canaffect globin gene activity and result in thalassemia.(a) Mutations in the TATA box associated with the �-globin gene caneliminate transcription and cause �-thalassemia. (b) A locus controlregion is present 25–50 kb upstream of the �-globin gene cluster. Thefunction of the LCR is to open up the chromatin domain associatedwith the complete cluster of �-globin genes. In contrast, theregulatory regions next to each particular gene fine-tune geneexpression during development. Mutations in the LCR can preventexpression of all the �-globin genes, resulting in severe �-thalassemia.
Ancestral α and β gene
ExonIntron
β-globin genes, β-like genes, and pseudogenes
α-globin genes, α-like genes, and pseudogenes
α-globin gene α-like gene β-globin gene β-like gene
Ancestral α-globin gene Ancestral β-globin gene
Further duplications and divergences
Duplication and divergence
Duplication and divergence Duplication and divergence
Further duplications and divergences
Figure 9.23 Evolution of the globin gene family. Duplicationof an ancestral gene followed by divergence of the separateduplication products established the �- and �-globin lineages. Furtherrounds of duplication and divergence within the separate lineagesgenerated the two sets of genes and pseudogenes of the globin genefamily.
(b)
(a)

to the � lineage. Each lineage then underwent further duplica-tions to generate the present array of three �-like and five �-like genes in humans.
Interestingly, the duplications also produced genes thateventually lost the ability to function. Molecular geneticistsmade this last deduction from data showing two additional �-like sequences within the � locus and one �-like sequencewithin the � locus that no longer have the capacity for properexpression. The reading frames are interrupted by frameshifts,missense mutations, and nonsense codons, while regionsneeded to control the expression of the genes have lost keyDNA signals. Sequences that look like, but do not function as,a gene are known as pseudogenes; they occur throughout allhigher eukaryotic genomes.
Essential Concepts 311
which start out identical, eventually diverge because theirsequences evolve independently of each other. The members ofa gene family may be grouped together on one chromosome(like the very closely related �-globin genes) or dispersed ondifferent chromosomes (like the less closely related �- and �-globin genes). All the �-like genes are exactly the same lengthand have two introns at exactly the same positions. All the �-like genes also have two introns at exactly the same positions,but these positions are different from those of the � genes. Thesequences of all the �-like genes are more similar to each otherthan they are to the �-like sequences, and vice versa. Thesecomparisons suggest that a single ancestral globin gene dupli-cated, and one copy moved to another chromosome. With time,one of the two gene copies gave rise to the � lineage, the other
C O N N E C T I O N S
The tools of recombinant DNA technology grew out of anunderstanding of the DNA molecule and its interaction with theenzymes that operate on DNA in normal cells. Geneticists usethe tools singly or in combination to look at DNA directly.Through cloning, hybridization, PCR, and sequencing, theyhave been able to isolate the genes that encode, for example,the hemoglobin proteins; identify sequences near the genes thatregulate their expression; determine the complete nucleotidesequence of each gene; and discover the changes in sequenceproduced by the hundreds of mutations that affect hemoglobinproduction. The results give a fascinating and detailed pictureof how the nucleotides along a DNA molecule determine pro-tein structure and function and how mutations in sequence pro-duce far-ranging and varied effects on human health. Themethods of classical genetics that we examined in Chapters 2–5complement those of recombinant DNA technology to producean integrated picture of genes and genomes at many levels.
In Chapter 10, we describe how the use of recombinantDNA technology has expanded from the analysis of singlegenes and gene complexes to the sequencing and examinationof whole genomes. Through the automation of sequencing andhigh-powered computer analysis of the data, scientific teamshave determined the DNA sequence of the entire humangenome and the genomes of many other organisms as well.DNA arrays, another recently developed technology, allow sci-entists to follow the patterns of expression from all the genes inthe genome as a tissue develops normally or undergoes car-cinogenic transformation. With these new technologies, geneti-cists are entering an era where they will be able to look at allthe parts of a biological system simultaneously. This new capa-bility for global analysis will allow them to better understandthe normal processes of differentiation and development andhow these processes can go awry.
E S S E N T I A L C O N C E P T S
1. An intact eukaryotic genome is too complex for mosttypes of analysis. Geneticists have appropriated theenzymes that normally operate on the DNA moleculeinside a bacterial cell and used them in the test tube tocreate the tools of recombinant DNA technology.Restriction enzymes cut DNA into pieces of a definedsize, ligase splices the pieces together, DNA polymerasemakes DNA copies, and reverse transcriptase copiesRNA into DNA.
2. Restriction enzymes cut DNA into restriction fragmentsat specific sites that are 4–8 bp in length. Enzymes that
recognize a 4 bp site digest DNA into fragments thataverage 256 bp in length; enzymes that recognize 6 bpdigest DNA into fragments that average 4 kb in length;enzymes that recognize 8 bp digest DNA into fragmentsthat average 64 kb in length. Complete digestion atevery site recognized by a particular enzyme can occurunder optimal reaction conditions. Under less thanoptimal conditions (of temperature, enzymeconcentration, or duration of reaction), a partialdigestion occurs in which only a random fraction ofrestriction sites are digested. Partial digestion enablesinvestigators to obtain a sample of DNA fragments

312 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
having an average size in-between those obtainable bycomplete digestion with enzymes that recognize 4, 6, or8 bp sites. Most restriction enzymes produce DNAmolecules with sticky ends, but some produce DNAmolecules with blunt ends.
3. Gel electrophoresis provides a method for separatingDNA fragments according to their size. When biologistssubject a viral genome, plasmid, or small chromosometo restriction digestion and gel electrophoresis, they canobserve the resulting DNA fragments by ethidiumbromide staining and size the fragments by comparingtheir migration within the gel with the migration ofknown marker fragments.
4. Cloning DNA fragments:a. Restriction fragments and cloning vectors with
matching sticky ends can be spliced together toproduce recombinant DNA molecules. A cloningvector is a DNA sequence that can enter a host cell,make its presence known, and provide a means ofreplicating and purifying both itself and any DNA towhich it is spliced. Different types of vectors carrydifferent-size inserts, which can serve as the basis fordifferent levels of analysis.
b. Once inside a living cell, vector-insert recombinantsare replicated during each cell cycle (just as the cell’sown chromosomes are) in a process known ascloning. The millions of cells arising from a singlecell by consecutive divisions make up a cellularclone. The vector-insert recombinant moleculesinside the cells of a clone, often referred to as DNAclones, can be purified by procedures that separaterecombinant molecules from host DNA. The insertcan then be cut away from the vector by restrictionenzymes and purified.
c. Genomic libraries are random collections of vector-insert recombinants containing the DNA fragments of
a given species. The most useful libraries carry fourto five genomic equivalents. cDNA libraries carryDNA copies of the RNA transcripts produced in aparticular tissue. The clones in a cDNA libraryrepresent only that part of the genome transcribedand spliced into mRNA in the cells of the specifictissue, organ, or organism.
5. Hybridization is the natural propensity ofcomplementary DNA strands to form stable doublehelixes. Hybridization makes it possible to usepreviously purified DNA fragments as labeled probes.Biologists use such probes to identify clones containingidentical or similar sequences within genomic or cDNA libraries. Hybridization can also be used incombination with gel electrophoresis as part of ananalytic technique called Southern blotting. Southernblot hybridization allows an investigator to determinethe numbers and positions of complementary sequenceswithin isolated DNA fragments or whole genomes ofany complexity.
6. The polymerase chain reaction (or PCR) is a methodfor the rapid purification and amplification of a singleDNA fragment from a complex mixture such as thewhole human genome. The DNA fragment to beamplified is defined by a pair of oligonucleotideprimers complementary to either end on oppositestrands. The PCR procedure operates through areiterative loop that amplifies the sequence between theprimers in an exponential manner. PCR is used in placeof cloning to purify DNA fragments whenever sequenceinformation for primers is already available.
7. Sequencing provides the ultimate description of acloned fragment. Automation has increased the speedand scope of sequencing.
1. A grocery chain in the Midwest has been receiving milkfrom the same dairy supplier for many years. Recently,that supplier, as well as many of the dairy farmers in theMidwest, have begun to give their cows bovinesomatotropin hormone (BST), a peptide that is mass-produced using a cloned BST gene. Treatment with thisgrowth hormone results in a 10% to 25% increase in milkproduction. Because milk production per cow isincreased, the price of the milk is lower. The peptide isinjected directly into cows and is undetectable in the milk.Cows treated with the hormone do have an increased
incidence of the relatively common disease mastitis,which is treated with antibiotics. A consequence ofincreased mastitis is that small amounts of antibiotics maybe more often found in the milk. Some people worry thatthis exposure to antibiotics could cause an increase inantibiotic-resistant bacteria in humans, while manyexperts argue that this risk is negligible. The grocerychain would like to carry this milk because it has a lowerprice. Does it have a responsibility to customers to informthem about the conditions under which the milk wasproduced? What proportion of customers at a typical
S O C I A L A N D E T H I C A L I S S U E S

grocery store are likely to have the knowledge baserequired to understand the process sufficiently to make aninformed decision as to whether to accept milk from BST-injected cows? There are several ways to deal withcomplicated scientific information that could potentiallyaffect society. Which do you think makes the most sense?(a) Consumers should always be provided with thescientific information even if they are unable to evaluateits significance. (b) An independent commission shouldevaluate the scientific information and inform consumersof its significance. (c) The government should be trustedto protect the health and well-being of its citizens.
2. Kerry Mullis, while working at Cetus Corporation,developed the polymerase chain reaction technique, oneof the most powerful new tools of molecular biology. Asan employee of the company, he had previouslyrelinquished much of his right to royalties from the patentand received a compensation that is small relative to thevalue of the technique he developed. Should an employeebe required to give most of the patent rights to thecompany? How should these rights be handled in a
research university? If the idea belonged to one person,should that person alone profit? Is it possible to determineaccurately the relative contributions of individualresearchers and of the institutions with which they areassociated to the development of new ideas that becomecommercially viable?
3. A new biotechnology company that has an interest incloning genes involved in the immune response hasapproached a scientist at the university and wants tosupport some of her research. A stipulation of receivingthe money is that the research cannot be published injournals until papers have been filed to cover anypotential patent. A postdoctoral fellow who has beendoing much of the research on this project is currentlylooking for a job and wants to present this research in hisseminars at job interviews. His boss has warned him thatthis may go against the scientist’s contract with thecompany. What should the postdoctoral fellow do? Isindustry support of research in the university compatiblewith academic life in the university?
Solved Problems 313
I. The following map of the plasmid cloning vector pBR322shows the locations of the ampicillin (amp) andtetracycline (tet) resistance genes as well as two uniquerestriction enzyme recognition sites, one for EcoRI andone for BamHI. You digested this plasmid vector withboth EcoRI and BamHI enzymes and purified the largeEcoRI-BamHI vector fragment. You also digested thecellular DNA that you want to insert into the vector withboth EcoRI and BamHI. After mixing the plasmid vectorand the fragments together and ligating, you transformedan ampicillin-sensitive strain of E. coli and selected forampicillin-resistant colonies. If you test all of yourselected ampicillin-resistant transformants for tetracyclineresistance, what result do you expect and why?
AnswerThis problem requires an understanding of vectors and theprocess of combining DNAs using sticky ends generatedby restriction enzymes.
The plasmid must be circular to replicate in E. coliand in this case a circular molecule will be formed only ifthe insert fragment joins with the cut vector DNA. Thecut vector will not be able to re-ligate without an insertedfragment because the BamHI and EcoRI sticky ends arenot complementary and cannot base pair. All ampicillin-resistant colonies therefore contain a BamHI-EcoRIfragment ligated to the BamHI-EcoRI sites of the vector.Fragments cloned at the BamHI-EcoRI site interrupt andtherefore inactivate the tetracycline resistance gene. Allampicillin-resistant clones will be tetracycline sensitive.
II. The gene for the human peptide hormone somatostatin(encoding nine amino acids) is completely contained onan EcoRI (5′ G ^AA T T C 3′) fragment, which can be cutout of the larger fragment shown here. (The ^ symbolindicates the site where the sugar-phosphate backbone iscut by the restriction enzyme.)
S O L V E D P R O B L E M S
BamHIEcoRI
tetamp
5′GCCGAATTCGATCCTATCAACACGAAGTGAAAGTCTTACAACCCATGAATTCGATTCG 3′
3′CGGCTTAAGCTAGGATAGTTGTGCTTCACTTTCAGAATGTTGGGTACTTAAGCTAAGC 5′^
^
^
^

a. What is the amino-acid sequence of humansomatostatin?
b. Indicate the direction of transcription of this gene.
c. The first step in synthesizing large amounts of humansomatostatin for pharmacological treatments involvesconstructing a so-called fusion gene. In this fusionconstruct, the N terminus of the protein encoded by thefusion gene consists of the N-terminal half of the lacZgene (encoding �-galactosidase), while the remainderof the product of the fusion gene is humansomatostatin. A family of three plasmid vectors for theconstruction of such a fusion gene has been created.All of these vectors have an ampicillin resistance geneand part of the lacZ gene encoding the first 583 aminoacids of the �-galactosidase protein. The EcoRIfragment (that is, the fragment produced by cuttingwith EcoRI) containing human somatostatin can beinserted into the single EcoRI restriction site on thevectors. The sequence of three vectors in the vicinityof the EcoRI site is shown here. The numbers refer toamino acids in the �-galactosidase protein with the N-terminal amino acid being number 1. The DNAsequence presented is the same as that of the lacZmRNA (with Ts replacing the Us found in RNA). Inwhich of these three vectors must the EcoRI fragmentcontaining human somatostatin be inserted to generatea fusion protein with an N-terminal region from �-galactosidase and a C-terminal region from humansomatostatin?
AnswerThis problem requires an understanding of the sticky endsformed by restriction enzyme digestion and therequirement of appropriate reading frames for theproduction of proteins.
a. The only complete open reading frame (ATG startcodon to a stop codon) is found on the bottom strand(underlined on the following figure). The amino-acidsequence is met-gly-cys-lys-thr-phe-thr-ser-cys.
b. Based on the amino-acid sequence determined for parta, the gene must be transcribed from right to left.
c. The cut site for EcoRI is after the G at the 5′ end of theEcoRI recognition sequence on each strand. For eachof the three vectors, the cut will be shifted relative tothe reading frame of the lacZ gene by one base. TheEcoRI fragment containing somatostatin can ligate tothe vector in two possible orientations, but since weknow the sequence on the bottom strand codes for theprotein, a fusion protein will be produced only if thefragment is inserted with that coding sequence on thesame strand as the vector coding sequences. Consideronly this orientation to determine which vector willproduce the fusion protein. The EcoRI fragment to beinserted into the vector next to the lacZ gene has fivenucleotides that precede the first codon of thesomatostatin gene (see following figure) and thereforerequires one more nucleotide to match the readingframe of the vector. For pWR590-1, the cut results inan in-frame end; pWR590-2 has one base extra beyondthe reading frame; pWR590-3 has a two-baseextension past the reading frame. The EcoRI fragmentmust be inserted in the vector pWR590-2 to getsomatostatin protein produced.
314 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
582 583Gly Asn
pWR590-1 5′ GGCAACCGGGCGAGCTCGAATTCG
582 583Gly Asn
pWR590-2 5′ GGCAACCCGGGCGAGCTCGAATTC
582 583Gly Asn
pWR590-3 5′ GGCAACGGGGCAGCTCGAATTCGA
EcoRI
EcoRI
EcoRI
5′GCCGAATTCGATCCTATCAACACGAAGTGAAAGTCTTACAACCCATGAATTCGATTCG 3′
3′CGGCTTAAGCTAGGATAGTTGTGCTTCACTTTCAGAATGTTGGGTACTTAAGCTAAGC5′^ ^
^^

III. Imagine you have cloned a 14.7 kb piece of DNA, whichcontains restriction sites as shown here.
AnswerThis problem deals with partial and complete digests andradioactive labeling of fragments.
a. Only the left-most fragment would be seen aftercomplete digestion with any of the three enzymessince only the left end contains radioactivity.Radioactive bands seen after digestion with BamHI: 2kb; EcoRI: 3.4 kb; and HindIII: 6.9 kb.
b. Partial digestions result in all possible combinations ofadjacent fragments but again only those containing theleft end of the fragment will be radioactive. Forexample, partial digestion with BamHI results in thefollowing fragments (asterisks indicate fragments thatare radioactive). BamHI: 2*, 7.9, 1.2, 9.9*, 12.7, and14.7*. The radioactive fragments are BamHI: 2, 9.9,14.7 kb; EcoRI: 3.4, 10.7, 14.7 kb; and HindIII: 6.9,14.7 kb.
Problems 315
P R O B L E M S
9-1 Match each of the terms in the left column to the best-fitting phrase from the right column.
a. oligonucleotide 1. a DNA molecule used for transporting,replicating, and purifying a DNA fragment
b. vector 2. a collection of the DNA fragments of agiven species, inserted into a vector
c. sticky ends 3. DNA copied from RNA by reversetranscriptase
d. recombinant DNA 4. stable binding of single-stranded DNAmolecules to each other
e. reverse translation 5. efficient and rapid technique for amplifyingthe number of copies of a DNA fragment
f. genomic library 6. computational method for determining thepossible sequence of base pairs associatedwith a particular region of a polypeptide
g. genomic equivalent 7. contains genetic material from twodifferent organisms
h. cDNA 8. the number of DNA fragments that aresufficient in aggregate length to contain theentire genome of a specified organism
i. PCR 9. short single-stranded sequences found atthe ends of many restriction fragments
j. hybridization 10. a short DNA fragment that can besynthesized by a machine
9-2 Approximately how many restriction fragments wouldresult from the complete digestion of the human genome(3 109 bases) with the following restriction enzymes?(The recognition sequence for each enzyme is given inparentheses.)a. Sau3A (^GATC)b. BamHI (G^GATCC)c. SfiI (GGCCNNNN^NGGCC)
9-3 What purpose do selectable markers serve in vectors?
9-4 What is an artificial chromosome, and what purpose doesit serve for molecular geneticists?
9-5 A plasmid vector pBS281 is cleaved by the enzymeBamHI (G^GATCC), which recognizes only one site inthe DNA molecule. Human DNA is partially digestedwith the enzyme MboI (^GATC), which recognizes manysites in human DNA. These two digested DNAs are nowligated together. Consider only those molecules in whichthe pBS281 DNA has been joined with a fragment ofhuman DNA. Answer the following questions concerningthe junction between the two different kinds of DNA.
B E H B E
2 1.4 3.5 3 0.8 4 kb
B = BamHI site, E = EcoRI site, H = HindIII site
Numbers under the segments represent the sizes of theregions in kilobases (kb). You have labeled the left end ofthe molecule with 32P.
a. What radioactive bands would you expect to seefollowing electrophoresis if you did a completedigestion with BamHI? EcoRI? HindIII?
b. What radioactive bands would you expect to seefollowing electrophoresis if you did a partial digestionwith BamHI? EcoRI? HindIII?

a. What proportion of the junctions between pBS281 andall possible human DNA fragments can be cleavedwith MboI?
b. What proportion of the junctions between pBS281 andall possible human DNA fragments can be cleavedwith BamHI?
c. What proportion of the junctions between pBS281 andall possible human DNA fragments can be cleavedwith XorII (C^GATCG)?
d. What proportion of the junctions between pBS281 andall possible human DNA fragments can be cleavedwith EcoRII (Pu Pu A ^ T Py Py)? (Pu and Py stand forpurine and pyrimidine, respectively.)
e. What proportion of all possible junctions that can becleaved with BamHI will result from cases in whichthe cleavage site in human DNA was not a BamHI sitein the human chromosome?
9-6 Consider four different kinds of human libraries: agenomic library, a brain cDNA library, a liver cDNAlibrary, and a unique genomic sequence (no repetitiveDNA) library.a. Assuming inserts of approximately equal size, which
would contain the greatest number of different clones?b. Would you expect any of these not to overlap the
others at all in terms of the sequences it contains?Explain.
c. How do these four libraries differ in terms of thestarting material for constructing the clones in thelibrary?
9-7 As a molecular biologist and horticulturist specializing insnapdragons, you have decided that you need to make agenomic library to characterize the flower color genes ofsnapdragons.a. How many genomic equivalents would you like to
have represented in your library to be 95% confidentof having a clone containing each gene in your library?
b. How do you determine the number of clones thatshould be isolated and screened to guarantee thisnumber of genomic equivalents?
9-8 Imagine that you are a molecular geneticist studying aparticular gene in which mutations cause a serious humandisease. The gene, including its flanking regulatorysequences, spans 200 kb of DNA. The distance from thefirst to the last coding base is 140 kb, which is dividedamong 10 exons and 9 introns. The exons contain a totalof 38.7 kb, and the introns contain 101.3 kb of DNA. Youwould like to obtain the following for your work: (a) anintact clone of the whole gene, including flankingsequences; (b) a clone containing the entire codingsequences but no noncoding sequences; and (c) a clone of
exon 3, which is the site of the most common disease-causing mutation in this gene. Based on the size of yourinserts, what vectors would you use to obtain each ofthese clones and why?
9-9 Listed here are steps you would take to clone theidiosynchratase gene from the hoot owl. Put these intotheir appropriate order (there are multiple correctanswers).a. Mix together BAC vector DNA and hoot owl DNA
with ligase.b. Expose nitrocellulose paper disks to UV radiation
and baking.c. Extract genomic DNA from hoot owl cells.d. Perform autoradiography.e. Produce a labeled DNA probe to the idiosynchratase
gene.f. Completely digest BAC vector DNA with the
restriction enzyme HindIII.g. Place nitrocellulose paper disks onto the agar surface
to transfer colonies.h. Incubate DNA probe with nitrocellulose paper disks.i. Distribute bacteria onto a petri plate containing agar
and nutrients and allow growth into colonies.j. Partially digest hoot owl genomic DNA with the
restriction enzyme HindIII.k. Transform bacteria.
9-10 Expression vectors containing sequences from both theanimal virus SV40 and a bacterial plasmid can be used toproduce polypeptide products in both E. coli and animalcells.a. Give the general characteristics that are necessary for
this type of plasmid to be used as a vector in twovery different types of cells.
b. What is required for E. coli expression of a humanprotein?
9-11 Why do longer DNA molecules move more slowly thanshorter ones during electrophoresis?
9-12 You have a circular plasmid containing 9 kb of DNA,and you wish to map its EcoRI and BamHI sites. Whenyou digest the plasmid with EcoRI and run the resultingDNA on a gel, you observe a single band at 9 kb. You getthe same result when you digest the DNA with BamHI.When you digest with a mixture of both enzymes, youobserve two bands, one 6 kb and the other 3 kb in size.Explain these results. Draw a map of the restriction sites.
9-13 A 49 bp EcoRI fragment containing the somatostatingene was inserted into the vector pWR590 shown here.The sequence of the inserted fragment is:
316 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
5′ AATTCGATCCTATCAACACGAAGTGAAAGTCTTACAACCCATGAATTCG 3′3′ GCTAGGATAGTTGTGCTTCACTTTCAGAATGTTGGGTACTTAAGCTTAA 5′
Distances between adjacent restriction sites are shown.What are the patterns of restriction digests with EcoRI(G^AATTC) or with MboI (^GATC) before and aftercloning the somatostatin gene into the vector? (E � EcoRI, M � MboI)
9-16 The following fragments were found after digestion of acircular plasmid with restriction enzymes as noted. Drawa restriction map of the plasmid.
EcoRI: 7.0 kbSalI: 7.0 kbHindIII: 4.0, 2.0, 1.0 kbSalI � HindIII: 2.5, 2.0, 1.5, 1.0 kbEcoRI � HindIII: 4.0, 2.0, 0.6, 0.4 kbEcoRI � SalI: 2.9, 4.3 kb
9-17 You need to create a restriction map of a 7 kb circularplasmid you are studying. The following figure showsthe results of digesting the plasmid DNA with variousrestriction endonucleases. The figure represents a photoof a gel on which the DNA products of the digestionswere subjected to electrophoresis. The numbers acrossthe top of the blot represent the lanes. The DNAs in eachlane were treated in the following way: lane 1: EcoRI;lane 2: HindIII; lane 3: PstI; lane 4: BamHI; lane 5:EcoRI � HindIII; lane 6: BamHI � HindIII; lane 7:BamHI � EcoRI; lane 8: PstI � EcoRI; and lane 9: PstI� BamHI. The numbers down the left edge of the gelrepresent the molecular sizes of the various bands, inkilobases. Draw a map of the plasmid indicatingrestriction sites and their spatial relation to each other.
Problems 317
E
M
M M
0.7 kb0.9 kb
0.5 kb
0.3 kb
9-14 The Notch gene, involved in Drosophila development, iscontained within a restriction fragment of Drosophilagenomic DNA produced by cleavage with the enzymeSalI. The restriction map of this Drosophila fragment forseveral enzymes (SalI, PstI, and XhoI) is shown here;numbers indicate the distances between adjacentrestriction sites. This fragment is cloned by sticky-endligation into the single SalI site of a bacterial plasmidvector that is 5.2 kb long. The plasmid vector has norestriction sites for PstI or XhoI enzymes.
S P P X P X S
3.0 2.0 2.5 kb
P = PstI; S = Sal I; X = XhoI
0.2 3.34.0
Make a sketch of the expected patterns seen afteragarose gel electrophoresis and staining of a SalI digest(alone), of a PstI digest (alone), of a XhoI digest(alone), of the plasmid containing vector andDrosophila fragment. Indicate the fragment sizes inkilobases.
9-15 The linear bacteriophage genomic DNA has at eachend a single-strand extension of 20 bases. (These are“sticky ends” but are not, in this case, produced byrestriction enzyme digestion.) These sticky ends can be ligated to form a circular piece of DNA. In a seriesof separate tubes, either the linear or circular forms ofthe DNA are digested to completion with EcoRI,BamHI, or a mixture of the two enzymes. The resultsare shown here.a. Which of the samples (A or B) represents the circular
form of the DNA molecule?b. What is the total length of the linear form of the
DNA molecule?c. What is the total length of the circular form of the
DNA molecule?d. Draw a restriction map of the linear form of the DNA
molecule. Label all restriction enzyme sites as EcoRIor BamHI.
Sample A Sample B
EcoRI BamHI Eco + Bam EcoRI BamHI Eco + Bam7.3
2.7
5
4
3.5
3
2
1.81.5
1.2
1
0.5
2.2
1 2 3 4 5 6 7 8 9
7
54.532.521.50.5

9-18 You have purified an interesting protein from mousebrains that influences the ability of animals to taste sweetsubstances. You name the protein SWT. You now want tostudy the gene that encodes this protein, which you’vedetermined is a single polypeptide monomer.a. Assuming that you have unlimited quantities of
purified SWT, describe two approaches that youcould use to obtain a cDNA clone of the codingsequence for this polypeptide.
You have been successful in cloning the full-lengthcDNA of the SWT transcript as a 2 kb insert in a 4 kbplasmid vector. You use the insert from this cDNA cloneto probe a genomic library of BAC clones producedfrom an inbred (totally homozygous) mouse liver, andyou recover a SWT-gene-containing clone with a 150 kbgenomic insert. You now take purified DNA from(1) mouse liver, (2) the BAC clone containing the SWTgene, and (3) the SWT cDNA clone and digest all threesamples with the restriction enzyme (NotI), which has an8 bp recognition site. The three samples of digestedDNA are subjected to gel electrophoresis. After thecompletion of electrophoresis, the gel is stained withethidium bromide.b. Compare the patterns of staining that you are likely
to see in the different lanes of the gel.
You now subject the same three DNA samples (genomic,BAC, and plasmid) to digestion with an enzyme (RsaI)that recognizes a 4 bp recognition site and then performgel electrophoresis and ethidium bromide staining.c. Compare the staining patterns that you are likely to
see in the different lanes of the gel.
You now subject both gels to Southern blot analysis witha radioactive probe made from the purified insert of thecDNA clone.d. Compare the results that you are likely to see in the
different lanes of both gels.
9-19 A fragment of human DNA with an EcoRI site at oneend and a HindIII site at the other end is labeled with 32Pat the EcoRI end. This molecule is now partially digestedwith either restriction enzyme HhaI or Sau3A, and thepartial digests are fractionated on an agarose gel. Anautoradiograph of this gel is presented here.
a. How large is the EcoRI to HindIII fragment in basepairs?
b. Draw a map of this fragment showing the locationsof the HhaI and Sau3A recognition sites and thedistances of sites from one end.
c. The same fragment, labeled with 32P in the samefashion, is now digested to completion with HhaI,and the products are separated on an agarose gel.Sketch the expected pattern for the ethidium bromidestained gel and autoradiography of this gel.
9-20 a. Given the following restriction map of a cloned 10kb piece of DNA, what size fragments would you seeafter digesting this linear DNA fragment with each ofthe enzymes or combinations of enzymes listed:(1) EcoRI, (2) BamHI, (3) EcoRI � HindIII,(4) BamHI � PstI, and (5) EcoRI � BamHI.
b. What fragments in the last three double digestswould hybridize with a probe made from the 4 kbBamHI fragment?
318 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
Hhal Sau3A
1.83 kb
1.41 kb1.32 kb
0.39 kb
0.13 kb
1.83 kb
1.57 kb
0.93 kb
0.58 kb0.48 kb
0.24 kb
E H H E B P B E
1.5 0.6 1.0 1.2 2.1 1.9 1.7 kb
9-21 Professor Ozone is hard at work trying to produce hugequantities of the Ozonase enzyme to reduce smog levelsin his home town of Los Angeles. The protein isproduced naturally in small quantities by an elusivebacterium called Phew. Professor Ozone has alreadydetermined the amino-acid sequence of the protein andnow he wants to clone the gene that encodes the protein.Describe two different approaches that he could use to

create and identify clones of the gene. Describe how heshould go about producing large quantities of theenzyme.
9-22 a. If you are presented with the following sequencinggel pattern, what can you say about the sequence ofthe template strand used in these sequencingreactions?
b. If the template for sequencing is the strand thatresembles the mRNA, is this portion of the genomelikely to be within an coding region? Explain youranswer.
you obtained the following results with electrophoresisand autoradiography.What would be the mRNA sequence of the mutant?Where does it differ from the wild-type sequence?
Problems 319
G A T C
9-23 You have isolated a trp E. coli mutant and to study thenature of the mutation, you isolate DNA from thismutant and determine the sequence of the DNA in the trpgene. After carrying out a set of sequencing reactions,
G A T C
9-24 You read the following sequence directly from a gel.
5′ TCTAGCCTGAACTAATGC 3′
a. Make a drawing that reproduces the gel patternfrom which this sequence was read.
b. Assuming this sequence is from an exon in themiddle of a gene, does this newly synthesizedstrand or the template strand have the samesequence as the mRNA for the gene (except thatTs are present instead of Us)? Justify youranswer.
c. Using the genetic code table, give the amino-acid sequence of the hexapeptide (six aminoacids) translated from the 18-base message.Indicate which is the amino terminal end of thepeptide.

9-25 Which of the following set(s) of primers could you useto amplify the target DNA sequence indicated by thedots?
320 CHAPTER 9 Deconstructing the Genome: DNA at High Resolution
5′ GGCTAAGATCTGAATTTTCCGAG . . . TTGGGCAATAATGTAGCGCCTT 3′3′ CCGATTCTAGACTTAAAAGGCTC . . . AACCCGTTATTACATCGCGGAA 5′
a. 5′ GGAAAATTCAGATCTTAG 3′; 5′ TGGGCAATAATGTAGCG 3′b. 5′ GCTAAGATCTGAATTTTC 3′; 3′ ACCCGTTATTACATCGC 5′c. 3′ GATTCTAGACTTAAAGGC 5′; 3′ ACCCGTTATTACATCGC 5′d. 5′ GCTAAGATCTGAATTTTC 3′; 5′ TGGGCAATAATGTAGCG 3′
R E F E R E N C E S
Baltimore, D. (1970). RNA-dependent DNA polymerase invirions of RNA tumor viruses. Nature 226:1209–11.
Cohen, S. N., Chang, A. C., Boyer, H. W., and Helling, R. B.(1973). Construction of biologically functional bacterialplasmids in vitro. Proc Natl Acad Sci U S A. 70:3240–44.
Maniatis, T., Hardison, R. C., Lacy, E., Lauer, J., O’Connell,C., Quon, D., Sim, G. K., and Efstratiadis, A. (1978). Theisolation of structural genes from libraries of eukaryotic DNA.Cell 2:687–701.
Maxam, A. M., and Gilbert W., (1977). A new method forsequencing DNA. Proc Natl Acad Sci U S A. 74:560–64.
Nathans, D., and Smith, H. O. (1975). Restrictionendonucleases in the analysis and restructuring of DNAmolecules. Ann. Rev. Biochem. 44:273–93.
Saiki, R. K., Scharf, S., Faloona, F., Mullis, K. B., Horn, G. T.,Erlich, H. A., and Arnheim, N. (1985). Enzymaticamplification of beta-globin genomic sequences andrestriction site analysis for diagnosis of sickle cell anemia.Science 230:1350–54.
Sanger F., and Coulson, A. R. (1975). A rapid method fordetermining sequences in DNA by primed synthesis withDNA polymerase. J Mol Biol. 94:441–48.
Southern, E. M. (1975). Detection of specific sequencesamong DNA fragments separated by gel electrophoresis. J Mol Biol. 98:503–17.
Temin, H. M., and Mizutani, S. (1970). RNA-dependent DNApolymerase in virions of Rous sarcoma virus. Nature226:1211–13.


![Genome-Wide Analysis of the Core DNA Replication ......Genome Analysis Genome-Wide Analysis of the Core DNA Replication Machinery in the Higher Plants Arabidopsis and Rice1[W][OA]](https://img.pdfslide.us/doc/110x75/5f05685d7e708231d412d002/genome-wide-analysis-of-the-core-dna-replication-genome-analysis-genome-wide.jpg)