Embed Size (px)
Citation preview

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r. com/ loca te /yexc r
Research Article
CXCR4 chemokine receptor engagement modifies integrindependent adhesion of renal carcinoma cells
Jon Jones, Dana Marian, Eva Weich, Tobias Engl, Steffen Wedel, Borna Relja,Dietger Jonas, Roman A. Blaheta⁎
Klinik für Urologie und Kinderurologie, Zentrum der Chirurgie, Johann Wolfgang Goethe-Universität, Frankfurt am Main, Germany
A R T I C L E I N F O R M A T I O N
⁎ Corresponding author. J. W. Goethe-UniveLaborgebäude, Chirurgische Forschung, Haus
E-mail address: [email protected]
0014-4827/$ – see front matter © 2007 Elsevidoi:10.1016/j.yexcr.2007.07.001
A B S T R A C T
Article Chronology:Received 7 May 2007Revised version received22 June 2007Accepted 2 July 2007Available online 10 July 2007
The mechanisms leading to renal cell carcinoma (RCC) metastasis are incompletelyunderstood. Although evidence shows that the chemokine receptor CXCR4 and its ligandCXCL12 may regulate tumor dissemination, their role in RCC is not clearly defined. Weexamined CXCR4 expression and functionality on RCC cell lines, and explored CXCL12-triggered tumor adhesion to human endothelium (HUVEC) or extracellular matrix proteins.Functional CXCR4 was expressed on A498 tumor cells, enabling them to migrate towards aCXCL12 gradient. CXCR4 engagement by CXCL12 induced elevated cell adhesion to HUVEC,to immobilized fibronectin, laminin or collagen. Anti-CXCR4 antibodies or CXCR4 knockdown by siRNA applied prior to CXCL12 stimulation impaired CXCL12-triggered tumoradhesion. However, blocking CXCR4 subsequent to CXCL12 stimulation did not. This pointedto an indirect control of tumor cell adhesion by CXCR4. In fact, CXCR4 engagement byCXCL12 also induced alterations of receptors of the integrin family, notably alpha3, alpha5,beta1 and beta3 subunits, and blocking beta1 integrins with a function-blocking antibodyprevented CXCL12-induced A498 adhesion. Focal adhesion kinase (total and activated) andintegrin-linked kinase significantly increased in CXCL12-treated A498 cells, accompanied bya distinct up-regulation of ERK1/2, JNK and p38 phosphorylation. Therefore, CXCR4 may becrucial in controlling adhesion of A498 cells via cross talking with integrin receptors. Thesedata show that CXCR4 receptors contribute to RCC dissemination and may provide a novellink between CXCR4 chemokine receptor expression and integrin triggered RCC adhesion tothe vascular wall and subendothelial matrix components.
© 2007 Elsevier Inc. All rights reserved.
Keywords:Renal cell carcinomaCXCL12CXCR4IntegrinsAdhesionChemotaxis
Introduction
The chemokines are a superfamily of low molecular weight (8–10 kDa) peptides, initially characterized by their ability to inducemigrationof leukocytes. Sincechemokine receptorswere found tobe expressed in cancers, it was concluded that chemokine/receptor pairs may also control the migration of tumor cells
rsitätsklinik, Klinik für25, Zi 204, Theodor-Sternt.de (R.A. Blaheta).
er Inc. All rights reserved
through the endothelial vessel wall and extracellular matrix.Among the chemokines and chemokine receptors identified todate, themembranous CXCchemokine receptor 4 (CXCR4) and itsligand stromal-derived factor-1 (SDF-1, syn. CXCL12) are thoughtto play a central role in regulating metastasis of many solid tu-mors including thoseof the lung,headandneck, esophagus, bone,breast, bladder, ovary, colon,pancreas, skin, prostateandbrain [1].
Urologie und Kinderurologie, Interdisziplinäres Forschungs-und-Kai 7, D-60590 Frankfurt amMain, Germany. Fax: +49 69 6301 7108.
.

4052 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
Recent evidence suggests that CXCR4might also be involvedin renal cell carcinoma (RCC). Based onDNAmicroarray studies,we found the CXCR4 gene to be more highly expressed in clearcellRCC than innormal tissue [2]. Accordingly, real-timeRT–PCR
analysis of RCC samples and respective adjacent normal kidneytissuedisclosed a distinct and divergent up-regulation of CXCR4in primary tumor tissue [3]. Nevertheless, CXCR4 gene data donot allow conclusions about the surface expression of this

4053E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
receptor. In fact, immunohistochemistryhas revealed thatover-expression of CXCR4 takes place within the cytosolic andnuclear compartment of the tumor cells but not on the cellmembrane [4]. Pan and coworkers have demonstrated that theexpression of CXCR4 on human RCC correlated with theirmetastatic ability in vivo in SCIDmouse models of human RCC[5]. However, up-regulated expression of the CXCR4 gene in RCCcells induced by hypoxia did not always result in an increasedchemotactic responsiveness of the tumor cells to CXCL12 [5].
Therefore, the relevance of CXCR4 in RCC dissemination isfar from being completely understood. In particular, theselective process of hematogenous spread to distant organsites requires intense cross talk between the tumor cells andthe vascular border. Consequently, if the hypothesis of CXCR4-driven RCC migration holds true, functionally active CXCR4receptors along the tumor cell membrane, participating in theinteraction of RCC cells with endothelium and extracellularmatrix proteins should be expected.
Based on an RCC cell culture model, we provide for the firsttime a detailed insight into how CXCR4 may contribute to thehematogenous phase of RCC transmigration. CXCR4 was ex-pressed on the cell surface on A498 cells to a low extent, butsufficient to guarantee CXCL12 responsiveness. CXCR4 en-gagement triggered RCC cell adhesion to endothelial cells, aswell as to extracellular matrix proteins. CXCR4 did not act asan anchoring molecule allowing firm cellular attachment butrather as a signalling receptor. CXCL12 altered subunits of theintegrin receptor family and modified focal adhesion kinase(total and activated) and integrin-linked kinase. The process ofCXCR4 stimulation by CXCL12 was accompanied by a distinctup-regulation of ERK1/2 and JNK phosphorylation and mod-erate activation of p38 MAPK.
Materials and methods
Chemokines and antibodies
Human CXCL12 (SDF-1) was purchased from Strathmann(Amsterdam, The Netherlands), phycoerythrin (PE)-conjugat-ed monoclonal antibody against CXCR4 (IgG2a, clone 12G5)was purchased from R&D Systems (Wiesbaden, Germany).Anti-ERK1 (clone MK12), phospho-specific anti ERK1/2 (pT202/pY204; clone 20A), anti-JNK (clone 37), phospho-specific anti-JNK (pT183/pY185; clone 41) anti-p38 (clone 27), phospho-
Fig. 1 – Expression of CXCR4 in A498, Caki-I and KTC-26 cells. (A) PA498, andmoderate CXCR4mRNA level in Caki-I and KTC-26 cellsreaction was performed by running parallel reaction mixtures wThe figure shows one representative from three separate experiKTC-26 cells are also shown. Themonoclonal antibody clone 12Gcontrol. One representative experiment of three is shown. Densianalysis of CXCR4 surface expression. A phycoerythrin (PE)-conjuto analyze CXCR4 level. A mouse IgG2a-PE served as the isotypecytometer, and a histogramplot (FL2-Height) was generated to shoof CXCR4 distribution in A498 cells. Unconjugated monoclonal anIndocarbocyanine (Cy3™) conjugated goat–anti-mouse IgG was aCXCR4 expression at the intercellular boundaries (arrows). Scale
specific anti-p38 (pT180/pY182; clone 30), anti-integrin-linkedkinase (ILK; clone 3), anti-focal adhesion kinase (Fak; clone 77)and phospho-specific Fak (pY397; clone 18) monoclonalantibodies were obtained from BD Biosciences (Heidelberg,Germany). Anti-β-actin monoclonal antibody was obtainedfrom Sigma (Taufenkirchen, Germany). Anti-alpha1 (IgG1;clone SR84), anti-alpha2 (IgG2a; clone 12F1-H6), anti-alpha3(IgG1; clone C3II.1), anti-alpha4 (IgG1; clone 9F10), anti-alpha5(IgG1; clone IIA1), anti-alpha6 (IgG2a; clone GoH3), anti-beta1(IgG1; clone MAR4), anti-beta3 (IgG1; clone VI-PL2) or anti-beta4 (IgG2a; clone 439-9B) integrins were all from BDBiosciences. Mouse IgG1-PE (MOPC-21) or IgG2a-PE (G155–178) isotype controls were purchased from BD Biosciences.Beta1 function-blocking antibody (clone 6S6) was from Che-micon (Hofheim, Germany).
Cell cultures
Kidney carcinoma Caki-I and KTC-26 cells were purchasedfrom LGC Promochem (Wesel, Germany). A498 cells werederived from CLS (Heidelberg, Germany). Tumor cells weregrown and subcultured in RPMI 1640 medium (Seromed,Berlin, Germany) supplemented with 10% FCS, 100 IU/mlpenicillin and 100 μg/ml streptomycin at 37 °C in a 5% CO2-humidified incubator.
Human endothelial cells (HUVEC) were isolated fromhumanumbilical veins and harvested by enzymatic treatmentwith chymotrypsin. HUVEC were grown in Medium 199 (M199;Biozol, Munich, Germany), supplemented with 10% fetal calfserum (FCS), 10% pooled human serum, 20 μg/ml endothelialcell growth factor (Boehringer, Mannheim, Germany), 0.1%heparin, 100 ng/ml gentamycin and 20 mM HEPES buffer(pH 7.4). Subcultures from passages 2 to 6 were selected forexperimental use.
Transfection of tumor cells with siRNA
siRNA was constructed directed against CXCR4 (GenBankAccession No. NM003467). Sense: r(GCA GUC CAU GUC AUCUAC A)dTdT, antisense: r(UGU AGA UGA CAU GGA CUG C)dCdT. A498 cells were transfected at 70% confluencewith 8 nMsiRNA using RNAiFect transfection reagent (Qiagen, Hilden,Germany). Optimum transfection was achieved in RPMI1640medium supplemented with 5% FCS and a siRNA/RNAiFectratio of 1:6. Viability of tumor cells was assessed by propidium
CR analysis demonstrates strong CXCR4mRNAexpression in(fragment length: 346 bp). The internal control for the RT–PCRith the housekeeping gene GAPDH (fragment length: 509 bp).ments. Western blot analyses of CXCR4 in A498, Caki-I and5was used to recognize CXCR4.β-Actin served as the internaltometry data are given below each diagram. (B) Fluorescencegatedmonoclonal antibody anti-CXCR4, clone 12G5,was usedcontrol. Fluorescence was analyzed using a FACScan flowwPE-fluorescence. The lower right refers to confocal analysistibody clone 12G5 was used to analyze CXCR4.dded as the secondary antibody. The figure shows distinct=10 μM. ×100/1.3 oil immersion objective.

4054 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
iodide dsDNA-intercalation or quantitative fluorescence anal-ysis of enzyme-catalyzed fluorescein–diacetate metabolism.
Tumor cell adhesion and migration
To analyze tumor cell adhesion, HUVEC were transferred to 6-well multiplates (Falcon Primaria; BD Biosciences) in completeHUVEC-medium. When confluency was reached, Caki-I, KTC-26 or A498 cells (activated with CXCL12 vs. non-activated)were detached from the culture flasks by accutase treatment(PAA Laboratories, Cölbe, Germany) and 0.5×106 cells werethen added to theHUVECmonolayer for 60min. Subsequently,non-adherent tumor cells were washed off using warmed(37 °C) Medium 199. The remaining cells were fixed with 1%glutaraldehyde.
Cell migration toward CXCL12 was examined using 6-wellTranswell chambers (Greiner, Frickenhausen, Germany) with8-μm pores. Caki-I, KTC-26 or A498 cells were removed fromthe culture flasks and resuspended at 0.5×106 cells/ml in
Fig. 2 – CXCR4 expressed onA498 cells is functionally active. Tumchamber assay. A498, Caki-I or KTC-26 cells were seeded in the uwere placed in the lower well. Cells which migrated to the lowerand were counted. In control experiments, medium without CXCfunctional activity, CXCR4 receptors of A498 cells were blocked bytowards 500 ng/ml CXCL12 (SDF; upper right). IgGmonoclonal antof six experiments is shown. *Significant difference to controls. #
CXCL12.
serum free culture medium. CXCL12 (0–500 ng/ml) was placedin the lower wells. Test cells were then placed in the upperchamber for 60 min. After incubation, the upper surface of theTranswell membrane was wiped gently with a cotton swab toremovenon-migrating cells. Cellswhichmigrated to the lowersurface of the membrane were stained using hematoxylin.
In each experimental setting, adherent or migrated tumorcells were counted in five different fields of a defined size(5×0.25 mm2) using a phase contrast microscope and themean cellular adhesion/migration rate was calculated.
Attachment to extracellular matrix components
6-well plates were coated with collagen G (extracted fromcalfskin, consisting of 90% collagen type I and 10% collagentype III; Seromed; diluted to 100 μg/ml in PBS), laminin (derivedfrom the Engelbreth–Holm–Swarm mouse tumor; BD Bios-ciences; diluted to 50 μg/ml in PBS), or fibronectin (derivedfrom human plasma; BD Biosciences; diluted to 50 μg/ml in
or cellmigration towards CXCL12was assessed in a transwellpper chamber and different concentrations of CXCL12 (SDF-1)surface of the membrane were stained using hematoxylinL12 was used. To demonstrate CXCR4 dependence andmonoclonal antibodies and cells were then allowed to move
ibodies were used in control experiments. One representative#Significant difference to non-treated cells moving towards

4055E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
PBS) overnight. Plastic dishes served as the backgroundcontrol. Unspecific cell binding was evaluated by cultureplates treated with poly-D-lysine (Nunc, Wiesbaden, Ger-many). Plates were washed with 1% BSA (bovine serumalbumin) in PBS to block nonspecific cell adhesion. Thereafter,0.5×106 tumor cells (activated with CXCL12 vs. non-activated)were added to each well for 60 min. Subsequently, non-adherent tumor cells were washed off, the remaining adher-ent cells were fixed with 1% glutaraldehyde and countedmicroscopically. The mean cellular adhesion rate, defined byadherent cellscoated well−adherent cellsbackground, was calculat-ed from five different observation fields.
Neutralization studies
For neutralization studies, cells were pretreated with 20 μg/mlbeta1 function-blocking antibody or with 20 μg/ml anti-human CXCR4 monoclonal antibody for 60 min, or tumorcells were transfected with CXCR4 siRNA and collected 24 hlater. Cells were then applied for the adhesion and migrationexperiments.
Flow cytometry
CXCR4 surface expressionCaki-I, KTC-26 or A498 cells were washed in blocking solution(PBS, 0.5% BSA) and then incubated for 60 min at 4 °C with thePE-labelled anti-CXCR4 monoclonal antibody. CXCR4 expres-sion on tumor cells was then measured using a FACScan (FL-2H (log) channel histogram analysis; 1×104 cells/scan) andexpressed as mean fluorescence units (MFU). Mouse IgG2a-PEwas used as an isotype control.
Fig. 3 – Adhesion of RCC cells to HUVEC depends on CXCR4.A498, Caki-I or KTC-26 cells were activated with 500 ng/mlCXCL12 (SDF; control: untreated) and thenaddedat a density of0.5×106 cells/well to HUVEC monolayers for 60 min.Non-adherent tumor cellswerewashedoff in each sample, theremaining cells were fixed and counted in five different fields(5×0.25mm2) using a phase contrastmicroscope.Mean valueswere calculated from five counts. Mean adhesion capacity isdepicted as counted cells/mm2. One representative of sixexperiments is shown. *Significant difference to controls.
Integrin expressionTo analyze integrin surface expression, A498 tumor cells werewashed in blocking solution (PBS, 0.5% BSA) and thenincubated for 60 min at 4 °C with PE-conjugated monoclonalantibodies anti-alpha1, anti-alpha2, anti-alpha3, anti-alpha4,anti-alpha5, anti-alpha6, anti beta1, anti-beta3 or anti-beta4.Integrin expression of tumor cells was then measured using aFACScan (Becton Dickinson; FL-2H (log) channel histogramanalysis; 1×104 cells/scan) and expressed as mean fluores-cence units (MFU). A mouse IgG1-PE or IgG2a-PE was used asan isotype control.
To analyze intracellular integrin expression, cells werefixed with 5 ml cold (−20 °C) methanol/acetone (1:1 v/v) for15 min. They were then incubated with the monoclonalantibodies as indicated.
Confocal microscopy
To explore CXCR4 localization, tumor cells were transferredto round cover slips which were placed in a 24-well multi-plate. Upon reaching confluency, cell cultures were washedtwo times with PBS (with Ca2+ and Mg2+) and then fixed incold (−20 °C) methanol/acetone (60:40 v/v). Subsequently,cells were washed again with PBS (without Ca2+ and Mg2+),and afterwards once with blocking buffer (0.5% BSA in PBSwithout Ca2+ and Mg2+). After removing the washing buffer,cells were incubated for 60 min with PE-conjugated anti-CXCR4 monoclonal antibody. To prevent photobleaching ofthe fluorescent dye, cover glasses with stained cells weretaken out of the wells and the residual liquid was removed.The cells were then embedded in an antifade reagent/moun-ting medium mixture (ProLong™ Antifade Kit, MoBiTec,Göttingen, Germany) and mounted on slides. The slides wereviewed using a confocal laser scanning microscope (LSM 10;Zeiss, Jena, Germany) with a plan-neofluar ×100/1.3 oil im-mersion objective.
Western blotting
CXCR4Total CXCR4 content was evaluated by Western blot analysis:Caki-I, KTC-26 or A498 cell lysates were applied to a 7%polyacrylamide gel and electrophoresed for 90 min at 100 V.The protein was then transferred to nitrocellulose membranes.After blockingwith non-fat drymilk for 1 h, themembraneswereincubated overnight with the anti-CXCR4-antibody (dilution1:100). HRP-conjugated goat–anti-mouse IgG (Upstate Biotechnol-ogy, LakePlacid,NY,USA;dilution1:5000) servedas thesecondaryantibody. The membranes were briefly incubated with ECLdetection reagent (ECL™, Amersham) to visualize the proteinsand exposed to an X-ray film (Hyperfilm™ EC™, Amersham).
Integrins and signalling proteinsCell lysates were prepared from unstimulated cells, or afterstimulationwith 500 ng/ml CXCL12.Western blot analysiswasperformed using following monoclonal antibodies: anti-integ-rin alpha1, anti-integrin alpha2, anti-integrin alpha3, anti-integrin alpha4, anti-integrin alpha5, anti-integrin alpha6,anti-integrin beta1, anti-integrin beta3, or anti-integrin beta4.Integrin activation was analyzed using the monoclonal
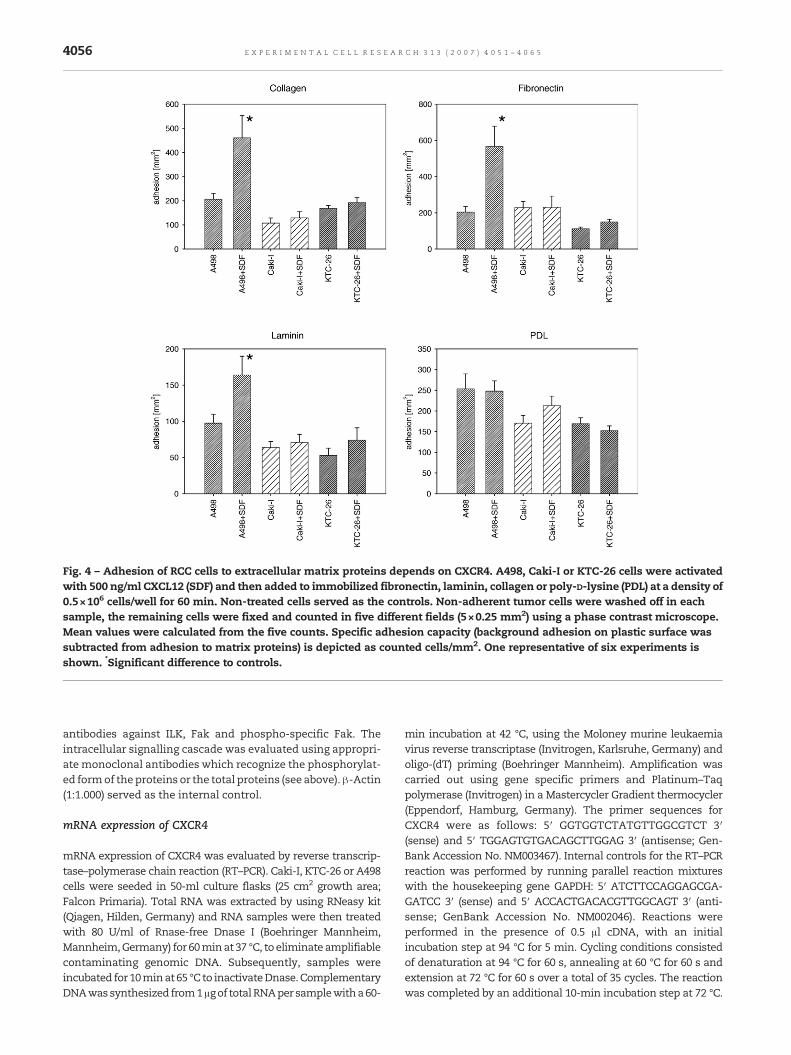
Fig. 4 – Adhesion of RCC cells to extracellular matrix proteins depends on CXCR4. A498, Caki-I or KTC-26 cells were activatedwith 500 ng/ml CXCL12 (SDF) and then added to immobilized fibronectin, laminin, collagen or poly-D-lysine (PDL) at a density of0.5×106 cells/well for 60 min. Non-treated cells served as the controls. Non-adherent tumor cells were washed off in eachsample, the remaining cells were fixed and counted in five different fields (5×0.25 mm2) using a phase contrast microscope.Mean values were calculated from the five counts. Specific adhesion capacity (background adhesion on plastic surface wassubtracted from adhesion to matrix proteins) is depicted as counted cells/mm2. One representative of six experiments isshown. *Significant difference to controls.
4056 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
antibodies against ILK, Fak and phospho-specific Fak. Theintracellular signalling cascade was evaluated using appropri-ate monoclonal antibodies which recognize the phosphorylat-ed formof theproteins or the total proteins (see above).β-Actin(1:1.000) served as the internal control.
mRNA expression of CXCR4
mRNA expression of CXCR4 was evaluated by reverse transcrip-tase–polymerase chain reaction (RT–PCR). Caki-I, KTC-26 or A498cells were seeded in 50-ml culture flasks (25 cm2 growth area;Falcon Primaria). Total RNA was extracted by using RNeasy kit(Qiagen, Hilden, Germany) and RNA samples were then treatedwith 80 U/ml of Rnase-free Dnase I (Boehringer Mannheim,Mannheim,Germany) for 60minat 37 °C, to eliminate amplifiablecontaminating genomic DNA. Subsequently, samples wereincubated for 10minat65 °C to inactivateDnase.ComplementaryDNAwassynthesized from1μgof totalRNAper samplewitha60-
min incubation at 42 °C, using the Moloney murine leukaemiavirus reverse transcriptase (Invitrogen, Karlsruhe, Germany) andoligo-(dT) priming (Boehringer Mannheim). Amplification wascarried out using gene specific primers and Platinum–Taqpolymerase (Invitrogen) in a Mastercycler Gradient thermocycler(Eppendorf, Hamburg, Germany). The primer sequences forCXCR4 were as follows: 5′ GGTGGTCTATGTTGGCGTCT 3′(sense) and 5′ TGGAGTGTGACAGCTTGGAG 3′ (antisense; Gen-Bank Accession No. NM003467). Internal controls for the RT–PCRreaction was performed by running parallel reaction mixtureswith the housekeeping gene GAPDH: 5′ ATCTTCCAGGAGCGA-GATCC 3′ (sense) and 5′ ACCACTGACACGTTGGCAGT 3′ (anti-sense; GenBank Accession No. NM002046). Reactions wereperformed in the presence of 0.5 μl cDNA, with an initialincubation step at 94 °C for 5 min. Cycling conditions consistedof denaturation at 94 °C for 60 s, annealing at 60 °C for 60 s andextension at 72 °C for 60 s over a total of 35 cycles. The reactionwas completed by an additional 10-min incubation step at 72 °C.

Fig. 5 – Control of CXCR4 knock down. A498 cells weretreated with specific siRNA as indicated in materials andmethods or with scrambled siRNA. Knocking down wascontrolled 24 h after treatment by RT–PCR (left panel of thefigure) and Western blot (right panel of the figure). Theinternal control for the RT–PCR reaction was performed byrunning parallel reaction mixtures with the housekeepinggene GAPDH (fragment length: 509 bp). β-Actin served as theinternal control in Western blotting. One representative ofthree experiments is shown.
4057E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
ThePCRproductsweresubjected toelectrophoresis in2%agarosegel and visualized by ethidium bromide.
Statistical analysis
All experiments were performed 3–6 times. Statistical signif-icance was investigated by the Wilcoxon–Mann–Whitney U-test. Differences were considered statistically significant at a pvalue less than 0.05.
Results
CXCR4 expression profile in Caki-I, KTC-26 and A498 cells
To follow the expression pattern of CXCR4 in RCC cells, theCXCR4 “route”was traced, including CXCR4 encodingmRNAaswell as cytoplasmic accumulation of CXCR4 proteins andmembrane presentation of CXCR4 receptors. Strong CXCR4mRNA activity was observed in A498 cells, whereas onlymoderate CXCR4 mRNA activity was noted in KTC-26 or Caki-I cells (Fig. 1A).Westernblot assay revealeddistinct amounts ofCXCR4 proteins in A498 and Caki-I cells and a lower CXCR4protein content inKTC-26 cells (Fig. 1A). Thenext step involvedexamining the CXCR4 surface expression level on these celllines. Histogramplots revealed no CXCR4 specific fluorescencein KTC-26 or Caki-I cells. In contrast, fluorescence intensitywas clearly enhanced over background values in A498 cells(Fig. 1B). Confocal microscopy of A498 cells showed intracel-lular localization of CXCR4 but also a distinct receptoraccumulation at the cell surface membrane (Fig. 1B; arrows).
Functionality of the CXCR4 receptor
Migration experiments were carried out to test whether theCXCR4 receptors detected on the A498 cell membrane arefunctionally active. Dose–response analysis revealed a strongchemotactic activity of A498 cells, which was maximal when500 ng/ml CXCL12 was applied (Fig. 2). We therefore used thisconcentration in subsequent studies. No response to CXCL12was seen in Caki-I or KTC-26 cells, which both did not expressCXCR4 along the cell surface.
The number of A498 cells migrating in response toCXCL12 was significantly higher than for cells not exposedto CXCL12 as a chemoattractant. CXCL12-dependent chemo-taxis was neutralized by treatment with an anti-CXCR4 anti-body, but not by unspecific anti-IgG antibody (Fig. 2, upperright). These experiments demonstrated that CXCR4 is func-tionally active on A498 cells and that CXCL12 specifically actson CXCR4.
CXCR4 is involved in adhesion regulation of RCC cells
A sufficient number of unstimulated A498, KTC-26 or Caki-Icells attached to HUVEC after 60 min (Fig. 3). CXCL12stimulation significantly enhanced the amount of adherent,CXCR4-positive, A498 cells, but did not influence bindingbehaviour of KTC-26 or Caki-I cells.
The attachment of RCC cell lines to immobilized laminin,collagen, or fibronectin was then investigated, since RCCtransmigration includes both adhesion to endothelial cellsand subendothelial matrix components. In doing so, all RCCcells attached well to the coated culture dishes, whereby thepercentage of adherent cells differed according to the matrixprotein. Maximum adhesion capacity was measured onfibronectin and collagen coated plates, a lower binding ratewas seenwhen the culture plateswere precoatedwith laminin(Fig. 4). CXCR4 activation by CXCL12 induced elevated bindingof A498 to thematrix proteins, but not of KTC-26 or Caki-I cells.Control experiments with poly-D-lysine-coated culture dishesdid not reveal any adhesion differences between CXCL12-treated and non-treated RCC cells.
To further elucidate the role of the CXCR4–CXCL12 axis inA498 cell adhesion, CXCR4 mRNA was knocked down in A498cells (Fig. 5) and binding assays repeated. In doing so, CXCL12significantly increased the adhesion rate of A498 control cellsand of A498 cells treated with scrambled siRNA. However,A498, whose CXCR4 was knocked down did not respond toCXCL12 stimulation and the amount of attached A498 CXCR4knock down cells was similar to the amount of attached A498cells not activated with CXCL12 (Fig. 6). The effect was seen inHUVEC- aswell as in collagen-, fibronectin- or laminin-bindingassays. Together, the data indicate that altered adhesioncapacity of RCC cells to endothelium or matrix is mediated byCXCR4.
CXCR4 receptors serve as signal transmitters
The adhesion experiments demonstrated that CXCR4 parti-cipates in the interaction of RCC cells with endothelium orextracellular matrix. The next step was to assess whetherCXCR4 serves as a mechanistic binding element or as a signal
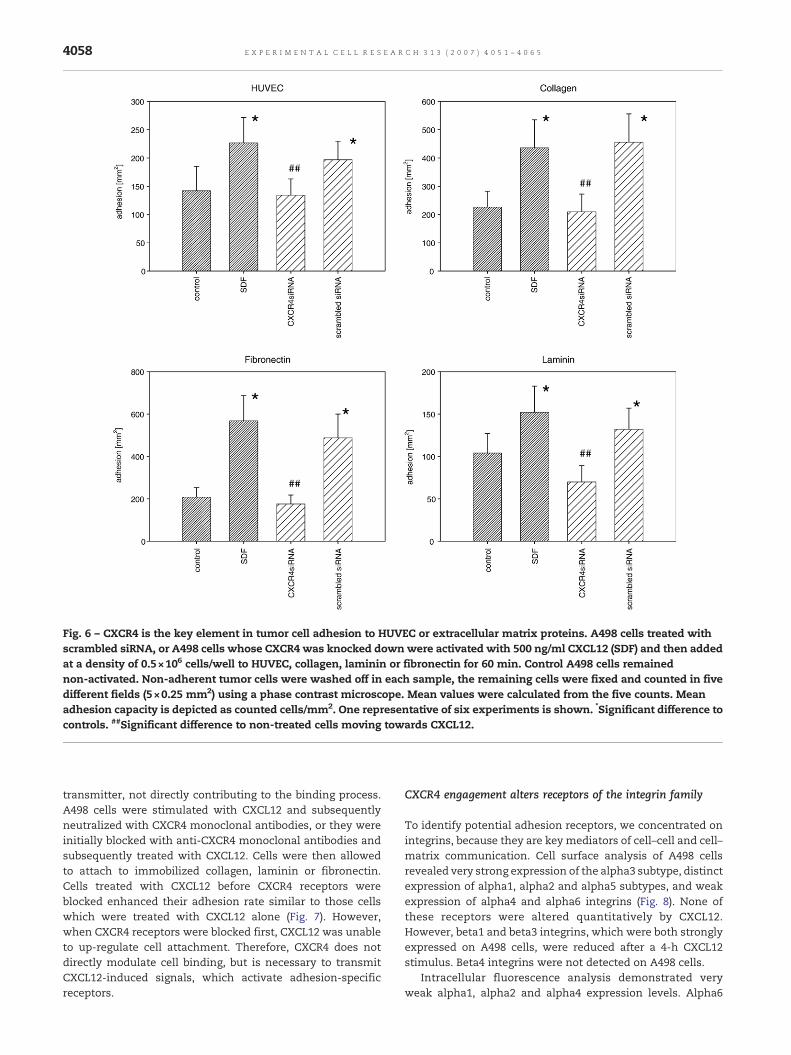
Fig. 6 – CXCR4 is the key element in tumor cell adhesion to HUVEC or extracellular matrix proteins. A498 cells treated withscrambled siRNA, or A498 cells whose CXCR4 was knocked downwere activated with 500 ng/ml CXCL12 (SDF) and then addedat a density of 0.5×106 cells/well to HUVEC, collagen, laminin or fibronectin for 60 min. Control A498 cells remainednon-activated. Non-adherent tumor cells were washed off in each sample, the remaining cells were fixed and counted in fivedifferent fields (5×0.25 mm2) using a phase contrast microscope. Mean values were calculated from the five counts. Meanadhesion capacity is depicted as counted cells/mm2. One representative of six experiments is shown. *Significant difference tocontrols. ##Significant difference to non-treated cells moving towards CXCL12.
4058 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
transmitter, not directly contributing to the binding process.A498 cells were stimulated with CXCL12 and subsequentlyneutralized with CXCR4 monoclonal antibodies, or they wereinitially blocked with anti-CXCR4 monoclonal antibodies andsubsequently treated with CXCL12. Cells were then allowedto attach to immobilized collagen, laminin or fibronectin.Cells treated with CXCL12 before CXCR4 receptors wereblocked enhanced their adhesion rate similar to those cellswhich were treated with CXCL12 alone (Fig. 7). However,when CXCR4 receptors were blocked first, CXCL12 was unableto up-regulate cell attachment. Therefore, CXCR4 does notdirectly modulate cell binding, but is necessary to transmitCXCL12-induced signals, which activate adhesion-specificreceptors.
CXCR4 engagement alters receptors of the integrin family
To identify potential adhesion receptors, we concentrated onintegrins, because they are key mediators of cell–cell and cell–matrix communication. Cell surface analysis of A498 cellsrevealed very strong expression of the alpha3 subtype, distinctexpression of alpha1, alpha2 and alpha5 subtypes, and weakexpression of alpha4 and alpha6 integrins (Fig. 8). None ofthese receptors were altered quantitatively by CXCL12.However, beta1 and beta3 integrins, which were both stronglyexpressed on A498 cells, were reduced after a 4-h CXCL12stimulus. Beta4 integrins were not detected on A498 cells.
Intracellular fluorescence analysis demonstrated veryweak alpha1, alpha2 and alpha4 expression levels. Alpha6

Fig. 7 – CXCR4 does not anchor A498 cells to matrix proteins but transmits signals after receptor engagement by CXCL12 whichthen allows cell adhesion. A498 cells were activated with 500 ng/ml CXCL12 (SDF) or remained untreated (controls).Additionally, A498 cells were activated with 500 ng/ml CXCL12 and CXCR4 was blocked thereafter by monoclonal antibodies(SDF+CXCR4ab). In a further experiment, A498 cells were first treated with anti-CXCR monoclonal antibodies and thenactivated with 500 ng/ml CXCL12 (CXCR4ab+SDF). Blocking with IgG antibodies (IgG+SDF) was done to control the specificity ofCXCR4 blockade. In all settings, A498 cells were then added to immobilized laminin, collagen or fibronectin. Mean adhesionfrom five different fields (5×0.25 mm2) was evaluated after 60 min. Adhesion capacity was strongly reduced in A498 cells ifCXCR4 was blocked before CXCL12 was applied. Adhesion was not reduced when tumor cells were activated with CXCL12 andreceptor blockade was initiated thereafter. One representative of six experiments is shown. *Significant difference to controls.##Significant difference to cells activated with CXCL12.
4059E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
integrins were not detected. None of these receptors werealtered quantitatively by CXCL12. Remarkably, intracellularalpha5, which was moderately expressed, and intracellu-lar alpha3, which was strongly expressed, became up-regulated after a 4-h CXCL12 stimulus (Fig. 9). The samewas true for beta1 and beta3 receptors (beta4 was notdetected fluorometrically), which both were enhanced byCXCL12.
Western blot data confirmed these findings, as alpha3,beta1 and beta3 proteins were all found to be enhanced inA498 cells after 4 h CXCL12 stimulation (Fig. 10). Surprisingly, aslight beta4 protein band was detected in lysates of CXCL12-treated A498 cells, compared to controls.
The relevance of integrin receptors in adhesion events wasdemonstrated in further studies, since CXCL12 induced adhe-
sion of A498 cells to fibronectin, laminin, or collagen wassignificantly inhibited by beta1-blocking antibodies (Fig. 11).
CXCR4 triggers integrin-related intracellular signalling
Quantitative alterations of the integrin expression level maynot be coupled to altered integrin activation. Consequently,in a further step CXCL12-induced intracellular signalling inA498 cells was analyzed. Integrin-linked kinase (ILK) andfocal adhesion kinase (FAK) were enhanced 4 h afterCXCL12 addition to the cell cultures. Phosphorylation ofFAK already took place after a 10-min incubation period(Fig. 12). No difference was observed between the totalamount of ERK1, p38 and JNK proteins (CXCL12-activated vs.untreated control cells). However, ERK1/2 and JNK became

Fig. 8 – FACS analysis of integrin surface expression on A498 cells. A498 cells were incubated with 500 ng/ml CXCL12 (SDF) for4 h or remained untreated (control). Cells were then washed in blocking solution and stained with specific monoclonalantibodies as listed inmaterials andmethods. Amouse IgG1-PE or IgG2a-PE was used as the isotype control. Fluorescence wasanalyzed using a FACScan flow cytometer, and a histogram plot was generated to show PE-fluorescence. Alpha subtypeexpressionwas not changed by CXCL12; histograms are therefore related to untreated cells. However, CXCL12 altered beta1 andbeta3 integrin surface expression which is indicated in the figure (control vs. SDF-treated). One of three independentexperiments.
4060 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
strongly activated after 4 h or 10 min, respectively. Onlyslight differences were seen with respect to p38 phosphor-ylation, 10 min and 4 h after adding CXCL12 (Fig. 12).CXCR4, therefore, not only influences integrin expressionbut also integrin activation status and intracellular MAPKsignalling.
Discussion
Themorbidity andmortality of RCC is strongly associatedwithits high propensity to metastasize. Recently acquired datasuggest that the CXCR4–CXCL12 axis may be a critical

Fig. 9 – FACS analysis of intracellular integrin expression in A498 cells. A498 cells were incubated with 500 ng/ml CXCL12 (SDF)for 4 h or remained untreated (control). Cells were then washed in blocking solution and stained with specific monoclonalantibodies as listed inmaterials andmethods. Amouse IgG1-PE or IgG2a-PE was used as the isotype control. Fluorescence wasanalyzed using a FACScan flow cytometer, and a histogram plot was generated to show PE-fluorescence. Alpha1, alpha2 andalpha4 level were not changed by CXCL12, histograms are therefore related to untreated cells. However, CXCL12 enhancedalpha3, alpha5, beta1 and beta3 integrins which is all indicated in the figure (control vs. SDF-treated). One from threeindependent experiments.
4061E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
determinant for metastatic potential. This may be important,since current therapeutic options for RCC are limited.
Information about CXCR4 in RCC is sparse and mainlybased on gene analysis [2,3]. This is the first report whichpresents evidence that CXCR4 not only drives chemotacticmovement towards a CXCL12 gradient but is also involved in
RCC–endothelial cell and RCC–matrix communication. Ourexperiments were carried out on three carcinoma cell lines,A498, Caki-I and KTC-26, which all express CXCR4 mRNA andCXCR4 protein. However, significant amounts of CXCR4surface receptors were only detected on A498 cells and,according to this observation, only A498 responded to a

Fig. 10 – CXCR4 engagement increases intracellular integrinproteins. A498 cells were incubated with 500 ng/ml CXCL12(SDF) for 10 min, 4 h or remained untreated (control). Celllysates were then analyzed by specific antibodies againstintegrin subtypes as listed inmaterials andmethods.β-Actinserved as the internal control. One representativeexperiment of three is shown.
Fig. 11 – Beta1 integrins are relevant for adhesion events. CXCL1function-blocking antibodies (clone 6S6) orwith IgG antibodies, anfor 60 min. Control cultures were not treated with monoclonal ansample, the remaining cells were fixed and counted in five diffeMean values were calculated from the five counts. Mean adhesiorepresentative of six experiments is shown. *Significant differen
4062 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
CXCL12 stimulus. Therefore, neither the CXCR4 gene nor theintracellular CXCR4 content, but rather the CXCR4 surfacelevel may determine the adhesion potential of (CXCR4-expressing) RCC cells.
The amount of CXCR4 appearing at the cell surface waslimited, and confocal analysis was necessary to clearlydemonstrate receptor localization along the surface mem-brane. Still, CXCR4 surface receptors on A498 cells werefunctionally active. Obviously, receptor saturation occurs ata very low level and, consequently, quantitative receptorenhancement beyond a specific threshold may not acceleratemigratory processes of the tumor cells. Since no data areavailable dealing with this issue, this assumption is purelyspeculative. However, a similar phenomenon has beenascribed to prostate carcinoma cells. In vitro assays demon-strated that traces of CXCR4 enabled these cells to migratetowards CXCL12 [6]. Prostate tumor cells also invadedextracellular matrix components in response to CXCL12, butat rates not corresponding to CXCR4 surface expression [7].
Presumably, CXCL12 release but not CXCR4 level is criticalfor cell migration, and cancerous cells expressing CXCR4 aremore likely to seed distant sites where high amounts ofCXCL12 are found. In fact, our dose–response analysisrevealed that RCC migration strongly depended on CXCL12concentration, and neutralization of CXCL12 in SCID miceabrogated metastasis of RCC cells to target organs which werecharacterized by high levels of CXCL12 [5].
2-treated A498 cells were preincubated with beta1d then added to immobilized collagen, laminin, or fibronectintibodies. Non-adherent tumor cells were washed off in each
rent fields (5×0.25 mm2) using a phase contrast microscope.n capacity is depicted as counted cells/mm2. Onece to controls.

Fig. 12 – CXCR4 engagement activates the intracellular signalling cascade. A498 cells were incubated with 500 ng/ml CXCL12(SDF) for 10 min, 4 h or remained untreated (control). Intracellular signalling was evaluated using the appropriate monoclonalantibodies recognizing ILK, FAK or phosphorylated FAK (pFAK; left part of the panel), total ERK1, JNK, p38, or thephosphorylated form of ERK (pERK1/2), JNK (pJNK), or p38 (pP38; right part of the panel). β-Actin served as the internal control.One representative experiment of three is shown.
4063E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
Chemotaxis towards a CXCL12 gradient may not exclu-sively explain the potential for neoplastic cells to migrate andinvade other tissues. Tumor cell contact with the vessel walland underlying matrix must occur to allow penetration andinitiation of secondary tumors. Our results indicate thatCXCR4–CXCL12 is an important mediator for the adherenceof RCC cells to endothelium and for the interaction withextracellular matrix proteins such as laminin, fibronectin andcollagen. Indeed, CXCL12-enhanced attachment of CXCR4-positive A498 cells (but not of CXCR4-negative Caki-I and KTC-26) to HUVEC, laminin, fibronectin and collagen could beantagonized by CXCR4-specific antibodies or CXCR4 knockdown. This intriguing finding argues for a novelmechanism ofCXCR4 and clearly expands the current knowledge of thisparticular chemokine. Nevertheless, laminin was derivedfrom the EHS mouse tumor and, due to interspecies differ-ences, the data should be interpreted with caution.
CXCR4 did not regulate adhesion itself, but served as asignalling element to modulate the expression pattern ofintegrin adhesion receptors. Our conclusion is based on threeobservations: (1) CXCR4 engagement specifically increasedbinding of A498 cells to extracellular matrix proteins whichmediate integrin-dependent attachment, but not to poly-D-lysine which mediates integrin-independent attachment. (2)CXCR4 engagement altered integrin expression and activity.(3) CXCR4 blockade subsequent to CXCL12 stimulation en-hanced tumor cell binding, whereas CXCR4 blockade prior toCXCL12 stimulation did not.
The cross talk between CXCR4 and integrins might be thekey event necessary to establish close contact of RCC cells to
the vascular wall and underlying matrix proteins. Severalreports have documented the pivotal role of integrins in RCCdissemination. In particular, improper localization of integrinsat the cell surface has been observed in mammalian cancercells and is believed to be closely related to their metastaticbehaviour [8]. There is also evidence that translocation ofintegrins into lipid rafts activates integrin signalling pathways[8]. Motility studies have shown that RCC cells mainly utilizeintegrin beta1-mediated mechanisms for cell adhesion andmigration [9,10]. According to our present and recent experi-ments, this indicates the involvement of beta1 receptors inRCC–extracellular matrix and RCC–HUVEC interaction [11].
In vitro data and xenotransplant models refer to beta3integrins as potential mediators of RCC malignancy [10,12].However, although beta3 integrins have been detected in renaltubular epithelial cells in human kidney tissues [13,14], andwere suggested to be involved in cell transdifferentiationprocesses [15], their role in RCC is still unclear. Meanwhile,several integrin antagonists have been designed for clinicaltrials [16–19].
Fluorescence analysis revealed that CXCR4 engagement byCXCL12 enhances intracellular alpha3 and alpha5 subtypeexpression. In close context, gene array analysis of tumorsgrown in kidneys of severe combined immunodeficient miceshowed that alpha3 integrin was in particular involved intumor metastasis [20], and Rissanen et al. observed increasedintegrin alpha3 accompanying increased RCC malignancy [9].Based on our assay, beta1 and beta3 became down-regulatedat the cell surface but concomitantly up-regulated in thecytoplasm. We assume that redistribution processes take

4064 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
place, which at least partially dislocate integrin receptors intothe inner compartment of the tumor cell. The relevance of thisphenomenon is not clear. Very novel findings suggest thatintegrin internalization may contribute to tumor metastasis.Winterwood et al. have shown that integrin internalization isa key mechanism, necessary to control adhesion and migra-tion dynamics of A431 epidermal carcinoma cells. In goodaccordance, steady state cell surface expression of beta1 andother integrins impairedmotile behaviour in the same culturemodel [21]. Consistent results are based on a CHO cell culturemodel which implicates integrin trafficking as an importantcomponent during the process of cell spreading [22]. Transmi-grating tumor cells represent a dynamic form of polarized cellsby forming a migrating front and retracting edges. Thus,integrin trafficking might allow rapid re-targeting to differentdomains where new migrating fronts are generated, and/ormodulation of the interaction with the actin cytoskeleton anddownstream effector components [23].
Beside quantitative integrin alterations, ILK and FAK (totaland phosphorylated) became enhanced in A498 cells byCXCL12, which may point to a specific activation of theintegrin receptors. Nevertheless, this is still speculative asmodification of ILK and FAK is an indirect link to integrinactivation and further experiments are necessary to demon-strate that CXCL12–CXCR4 unquestionably activates integrins(i.e., changes their conformation). This is the first report ofCXCL12-induced ILK and FAK alteration in RCC cells, however,it has been demonstrated on other types of cancer thatintegrin-mediated tumor adhesion strongly depends onintegrin-linked downstream signalling: Integrin beta1-medi-ated activation of ILK and FAK has been shown to promoteadhesion of colon cancer cells to a laminin matrix [24], totrigger invasion of glioma cells through Matrigel [25], and hasbeen linked tometastasis of prostate and colon cancer [26–28].We, therefore, postulate that CXCR4 is a pivotal element in themalignant progression of RCC, triggering integrin-dependenttumor invasion after occupation of CXCR4 by CXCL12 hadtaken place. Ongoing studies are now investigating whetherintegrins become activated by CXCR4-induced intracellularsignalling or whether CXCR4–integrin co-localization is re-quired to activate integrins.
Investigating the signalling components which may par-ticipate in CXCR4mediated cell adhesion, we found that ERK1/2 and JNK became strongly and p38 modestly activated. Sincenumerous functions have been ascribed to these proteins,interpretation is ambiguous. Integrin-driven adhesion ofovarian [29], colon [30], or pancreatic cancer cells [31] toextracellular matrix proteins has been demonstrated to beconnected to both FAK phosphorylation and ERK signalling.However, it is also postulated that the same integrin mayproduce distinct responses through specific signalling cas-cades. Notably, FAK activation may induce adhesion andoriented migration of tumor cells, whereas FAK combinedwith ERK signalling may support tumor growth and survival[32,33].
Pan et al. have provided evidence that neutralization ofCXCL12 in a SCID mice model abrogated metastasis of RCC totarget organs expressing high levels of CXCL12, withoutaltering tumor cell proliferation and apoptosis [5]. Based onthese results, it seems more likely that ILK/FAK/MAPK
signalling is tightly linked to enhanced RCC adhesion, asseen in our in vitro assay.
Very recently, an alternate membrane-associated recep-tor, CXCR7, has been characterized which binds with highaffinity to CXCL12 [34]. Therefore, it cannot be completelyruled out that some CXCL12 effects reported in the presentinvestigation may have been caused by CXCR7 engagement.Nevertheless, this seems unlikely since: (1) CXCR7 has beendetected in the breast tumor cell line MCF-7 and humancervical carcinoma HeLa cells, but not on RCC cell lines. (2)CXCR7-specific mRNA has been found in normal mousekidney tissues, but did not correlate with cell surface proteinexpression. (3) Binding of CXCL12 to CXCR7 did not elicit theCa2+ mobilization and MAP kinase signalling characteristic ofCXCR4-activation [34].
To summarize, a novel link between the CXCR4–CXCL12and integrin–ILK/FAK axis in RCC adhesion has been demon-strated. Further experiments should elucidate whether this isa unique or ubiquitous phenomenon. Similar mechanismshave been described in prostate carcinoma and small cell lungcancer cells [6,35]. If this model is also valid for other CXCR4expressing tumor entities, monoclonal antibodies directedagainst this receptor might become an effective tool intreating such malignancies. Small molecule antagonists toCXCR4, such as AMD3100, have already been developed [36],and the results presented here suggest that RNA interferencestrategies may prevent CXCR4-mediated tumor invasion.
Acknowledgments
We would like to thank Karen Nelson for critically reading themanuscript. This work was supported by the “Horst Müggen-burg-Stiftung”, “Jung-Stiftung”, “Ebert-Stiftung” and “Held-Hecker-Stiftung”.
R E F E R E N C E S
[1] A. Zlotnik, Chemokines and cancer, Int. J. Cancer 119 (2006)2026–2029.
[2] J. Jones, H. Otu, D. Spentzos, S. Kolia, M. Inan,W.D. Beecken, C.Fellbaum, X. Gu, M. Joseph, A.J. Pantuck, D. Jonas, T.A.Libermann, Gene signatures of progression and metastasis inrenal cell cancer, Clin. Cancer Res. 11 (2005) 5730–5739.
[3] A.J. Schrader, O. Lechner, M. Templin, K.E. Dittmar, S.Machtens, M. Mengel, M. Probst-Kepper, A. Franzke, T.Wollensak, P. Gatzlaff, J. Atzpodien, J. Buer, J. Lauber, CXCR4/CXCL12 expression and signalling in kidney cancer, Br. J.Cancer 86 (2002) 1250–1256.
[4] D. Zagzag, B. Krishnamachary, H. Yee, H. Okuyama, L.Chiriboga, M.A. Ali, J. Melamed, G.L. Semenza, Stromalcell-derived factor-1alpha and CXCR4 expression inhemangioblastoma and clear cell–renal cell carcinoma: vonHippel-Lindau loss-of-function induces expression of a ligandand its receptor, Cancer Res. 65 (2005) 6178–6188.
[5] J. Pan, J. Mestas, M.D. Burdick, R.J. Phillips, G.V. Thomas, K.Reckamp, J.A. Belperio, R.M. Strieter, Stromal derived factor-1(SDF-1/CXCL12) and CXCR4 in renal cell carcinomametastasis, Mol. Cancer 5 (2006) 56.
[6] T. Engl, B. Relja, D. Marian, C. Blumenberg, I. Muller, W.D.Beecken, J. Jones, E.M. Ringel, J. Bereiter-Hahn, D. Jonas, R.A.Blaheta, CXCR4 chemokine receptor mediates prostate tumor

4065E X P E R I M E N T A L C E L L R E S E A R C H 3 1 3 ( 2 0 0 7 ) 4 0 5 1 – 4 0 6 5
cell adhesion through alpha5 and beta3 integrins, Neoplasia8 (2006) 290–301.
[7] S. Singh, U.P. Singh, W.E. Grizzle, J.W. Lillard Jr.,CXCL12–CXCR4 interactions modulate prostate cancer cellmigration, metalloproteinase expression and invasion, Lab.Invest. 84 (2004) 1666–1676.
[8] Q. Huang, H.M. Shen, G. Shui, M.R. Wenk, C.N. Ong, Emodininhibits tumor cell adhesion through disruption of themembrane lipid Raft-associated integrin signaling pathway,Cancer Res. 66 (2006) 5807–5815.
[9] J. Rissanen, M. Korhonen, V.P. Lehto, I. Virtanen, Lamininalpha1 chain in human renal cell carcinomas andintegrin-mediated adhesion of renal cell carcinoma cells tohuman laminin isoforms, J. Pathol. 200 (2003) 157–167.
[10] W. Brenner, F. Benzing, J. Gudejko-Thiel, R. Fischer, G. Farber,J.G. Hengstler, B. Seliger, J.W. Thuroff, Regulation of beta1integrin expression by PKCepsilon in renal cancer cells, Int. J.Oncol. 25 (2004) 1157–1163.
[11] A.Oertl, B.Relja, J.Makarevic, E.Weich, S.Hofler, J. Jones,D. Jonas,H. Bratzke, P.C. Baer, R.A. Blaheta, Altered expression of beta1integrins in renal carcinoma cell lines exposed to thedifferentiation inducer valproic acid, Int. J. Mol. Med. 18 (2006)347–354.
[12] M. Korhonen, H. Sariola, V.E. Gould, L. Kangas, I. Virtanen,Integrins and laminins in human renal carcinoma cells andtumors grown in nude mice, Cancer Res. 54 (1994) 4532–4538.
[13] P. Roy-Chaudhury, G. Hillis, S. McDonald, J.G. Simpson, D.A.Power, Importance of the tubulointerstitium in humanglomerulonephritis: II. Distribution of integrin chains beta 1,alpha 1 to 6 and alpha V, Kidney Int. 52 (1997) 103–110.
[14] A. Baraldi, G. Zambruno, L. Furci, M. Ballestri, A. Tombesi, D.Ottani, L. Lucchi, E. Lusvarghi, Beta 1 and beta 3 integrinupregulation in rapidly progressive glomerulonephritis,Nephrol. Dial. Transplant. 10 (1995) 1155–1161.
[15] Y. Shi, Z. Tu, W. Wang, Q. Li, F. Ye, J. Wang, J. Qiu, L. Zhang, H.Bu, Y. Li, Homologous peptide of connective tissue growthfactor ameliorates epithelial to mesenchymal transition oftubular epithelial cells, Cytokine 36 (2006) 35–44.
[16] S.K. Kuwada, Drug evaluation: volociximab, anangiogenesis-inhibiting chimeric monoclonal antibody, Curr.Opin. Mol. Ther. 9 (2007) 92–98.
[17] D.G. McNeel, J. Eickhoff, F.T. Lee, D.M. King, D. Alberti, J.P.Thomas, A. Friedl, J. Kolesar, R. Marnocha, J. Volkman, J.Zhang, L. Hammershaimb, J.A. Zwiebel, G. Wilding, Phase Itrial of a monoclonal antibody specific for alphavbeta3integrin (MEDI-522) in patients with advanced malignancies,including an assessment of effect on tumor perfusion, Clin.Cancer Res. 11 (2005) 7851–7860.
[18] K.W. Beekman, A.D. Colevas, K. Cooney, R. Dipaola, R.L. Dunn,M. Gross, E.T. Keller, K.J. Pienta, C.J. Ryan, D. Smith, M.Hussain, Phase II evaluations of cilengitide in asymptomaticpatients with androgen-independent prostate cancer:scientific rationale and study design, Clin. Genitourin. Cancer4 (2006) 299–302.
[19] M.E. Cianfrocca, K.A. Kimmel, J. Gallo, T. Cardoso, M.M.Brown, G. Hudes, N. Lewis, L. Weiner, G.N. Lam, S.C. Brown,D.E. Shaw, A.P. Mazar, R.B. Cohen, Phase 1 trial of theantiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a betaintegrin antagonist, in patients with solid tumours, Br. J.Cancer 94 (2006) 1621–1626.
[20] M. Bockhorn, S. Roberge, C. Sousa, R.K. Jain, L.L. Munn,Differential gene expression in metastasizing cells shed fromkidney tumors, Cancer Res. 64 (2004) 2469–2473.
[21] N.E. Winterwood, A. Varzavand, M.N. Meland, L.K. Ashman,C.S. Stipp, A critical role for tetraspanin CD151 in alpha3beta1and alpha6beta4 integrin-dependent tumor cell functions onlaminin-5, Mol. Biol. Cell 17 (2006) 2707–2721.
[22] M. Skalski, M.G. Coppolino, SNARE-mediated traffickingof alpha5beta1 integrin is required for spreading inCHO cells, Biochem. Biophys. Res. Commun. 335 (2005)1199–1210.
[23] A.M. Powelka, J. Sun, J. Li, M. Gao, L.M. Shaw, A. Sonnenberg,V.W. Hsu, Stimulation-dependent recycling of integrin beta1regulated by ARF6 and Rab11, Traffic 5 (2004) 20–36.
[24] K. Kato, K. Shiga, K. Yamaguchi, K. Hata, T. Kobayashi, K.Miyazaki, S. Saijo, T. Miyagi, Plasma-membrane-associatedsialidase (NEU3) differentially regulates integrin-mediatedcell proliferation through laminin- and fibronectin-derivedsignalling, Biochem. J. 394 (2006) 647–656.
[25] Q. Shi, S. Bao, L. Song, Q. Wu, D.D. Bigner, A.B. Hjelmeland,J.N. Rich, Targeting SPARC expression decreases gliomacellular survival and invasion associated with reducedactivities of FAK and ILK kinases, Oncogene 26 (2007)4084–4094.
[26] J.R. Graff, J.A. Deddens, B.W. Konicek, B.M. Colligan, B.M.Hurst, H.W. Carter, J.H. Carter, Integrin-linked kinaseexpression increases with prostate tumor grade, Clin. CancerRes. 7 (2001) 1987–1991.
[27] Z.Z. Zeng, Y. Jia, N.J. Hahn, S.M. Markwart, K.F. Rockwood, D.L.Livant, Role of focal adhesion kinase andphosphatidylinositol3′-kinase in integrin fibronectin receptor-mediated, matrixmetalloproteinase-1-dependent invasion by metastatic prostatecancer cells, Cancer Res. 66 (2006) 8091–8099.
[28] V. Thamilselvan, D.H. Craig, M.D. Basson, FAK associationwithmultiple signal proteins mediates pressure-induced coloncancer cell adhesion via a Src-dependent PI3K/Akt pathway,FASEB J. 21 (2007) 1730–1741.
[29] S. Hapke, H. Kessler, B. Luber, A. Benge, P. Hutzler, H. Hofler,M. Schmitt, U. Reuning, Ovarian cancer cell proliferationand motility is induced by engagement of integrin alpha(v)beta3/Vitronectin interaction, Biol. Chem. 384 (2003)1073–1083.
[30] R.S. Sawhney, M.M. Cookson, Y. Omar, J. Hauser, M.G.Brattain, Integrin alpha2-mediated ERKandcalpain activationplay a critical role in cell adhesion and motility via focaladhesion kinase signaling: identification of a novel signalingpathway, J. Biol. Chem. 281 (2006) 8497–8510.
[31] H. Sawai, Y. Okada, H. Funahashi, Y. Matsuo, H. Takahashi, H.Takeyama, T. Manabe, Activation of focal adhesion kinaseenhances the adhesion and invasion of pancreatic cancercells via extracellular signal-regulated kinase-1/2 signalingpathway activation, Mol. Cancer 4 (2005) 37.
[32] N. Desban, J.C. Lissitzky, P. Rousselle, J.L. Duband,alpha1beta1-integrin engagement to distinct laminin-1domains orchestrates spreading, migration and survival ofneural crest cells through independent signaling pathways, J.Cell Sci. 119 (2006) 3206–3218.
[33] A. Manohar, S.G. Shome, J. Lamar, L. Stirling, V. Iyer, K.Pumiglia, C.M. DiPersio, Alpha 3 beta 1 integrin promoteskeratinocyte cell survival through activation of a MEK/ERKsignaling pathway, J. Cell Sci. 117 (2004) 4043–4054.
[34] J.M. Burns, B.C. Summers, Y.Wang, A.Melikian, R. Berahovich,Z. Miao, M.E. Penfold, M.J. Sunshine, D.R. Littman, C.J. Kuo, K.Wei, B.E. McMaster, K. Wright, M.C. Howard, T.J. Schall, Anovel chemokine receptor for SDF-1 and I-TAC involved in cellsurvival, cell adhesion, and tumor development, J. Exp. Med.203 (2006) 2201–2213.
[35] T.N. Hartmann, J.A. Burger, A. Glodek, N. Fujii, M. Burger,CXCR4 chemokine receptor and integrin signaling co-operatein mediating adhesion and chemoresistance in small celllung cancer (SCLC) cells, Oncogene 24 (2005) 4462–4471.
[36] A.F. Cashen, B. Nervi, J. DiPersio, AMD3100: CXCR4 antagonistand rapid stem cell-mobilizing agent, Future Oncol. 3 (2007)19–27.












![Moderate Restriction of Macrophage-Tropic Human ......9], and this observation led to the identification of the CCR5 and CXCR4 chemokine receptors as HIV-1 co-receptors for viral fusion](https://img.pdfslide.us/doc/110x75/606645ddad14062d597e7589/moderate-restriction-of-macrophage-tropic-human-9-and-this-observation.jpg)















