Embed Size (px)
Citation preview

CXCR1 AND CXCR2 ACTIVATION AND REGULATION: ROLE OF ASPARTATE 199 OF THE SECOND EXTRACELLULAR LOOP OF CXCR2 IN CXCL8-MEDIATED RAPID RECEPTOR INTERNALIZATION.
Mohd W. Nasser, Sandeep K. Raghuwanshi, Kimberly M. Malloy, Pavani Gangavarupu#, Joong-Youn Shim, Krishna Rajarathnam# and Ricardo M. Richardson
Julius L. Chambers Biomedical/Biotechnology Research Institute and the Department of Biology, North Carolina Central University, Durham, NC 27707
#Department of Biochemistry and Molecular Biology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555-1055
Running title: Regulation of CXCR1 and CXCR2 *To whom correspondence should be addressed: Ricardo M. Richardson, Ph.D.; Julius L. Chambers Biomedical/Biotechnology Research Institute, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707., Tel.: 919-530-6421; Fax: 919-530-7780, Email:[email protected] Interleukin-8 (IL-8 or CXCL8) interacts with two receptors CXCR1 and CXCR2 to activate leukocytes. Upon activation, CXCR2 internalizes very rapidly relative to CXCR1 (~90% vs ~10%, respectively, after 5 min). The carboxyl-terminus of the receptors were shown to be necessary for internalization but are not sufficient to explain the distinct kinetics of downregulation. To determine the structural determinant(s) that modulate receptor internalization, various chimeric and point mutant receptors were generated by progressively exchanging specific domains or amino acids between CXCR1 and CXCR2. The receptors were stably expressed in RBL-2H3 cells and characterized for receptor binding, intracellular Ca2+ mobilization, phosphoinositide hydrolysis, phosphorylation, internalization and, MAP kinase activation. The data herein indicate that the second extracellular loop (2ECL) of the receptors is critical for the distinct rate of internalization. Replacing the 2ECL of CXCR2 for that of CXCR1 (B2ECLA) or aspartate-199 for its valine counterpart (BD199VA) delayed CXCR2 internalization similar to that of CXCR1. Replacing aspartate-199 for asparagine (BD199N), restored CXCR2 rapid internalization. Structure modeling of the 2ECL of the receptors also suggests that aspartate-199 plays a critical role in stabilizing and modulating CXCR2 rapid internalization relative to CXCR1. BD199N internalized rapidly but migrated as a single phosphorylated form like CXCR1(~75 kDa)
whereas B2ECLA and BD199VA showed a slow and a fast migrating form like CXCR2 (~45 and ~65 kDa) but internalized like CXCR1. These data further undermine the role of receptor oligomerization in CXCL8 receptors internalization. Like CXCR1, BD199VA also induced sustained ERK activation and cross-desensitized Ca2+ mobilization to CCR5, relative to BD199N and CXCR2. Altogether, the data suggest that the 2ECL of the CXCL8 receptors are important in modulating their distinct rate of downregulation thereby signal length and post-internalization activities. Chemokines are a family of structurally related peptides that regulate inflammation through cell surface G-protein coupled receptors (GPCRs) on leukocytes. These peptides mediate diverse biological and biochemical activities including adhesion to endothelium, directed migration, and activation of cytotoxic activities such as the respiratory burst and exocytosis (1,2). Chemokines have been classified into four families; C, CC, CXC, and CX3C based on the number and positions of the N-terminal-conserved cysteine residues. Most chemokines activate more than one chemokine receptor and many chemokine receptors are activated by multiple chemokines (3). CXCL8 activates two receptors, CXCR1 and CXCR2. CXCR1 is specific for CXCL8 whereas CXCR2 also interacts with CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL7(4). Upon activation, both receptors couple to pertussis toxin (Ptx)-sensitive G protein to mediate
1
http://www.jbc.org/cgi/doi/10.1074/jbc.M610289200The latest version is at JBC Papers in Press. Published on January 4, 2007 as Manuscript M610289200
Copyright 2007 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

phosphoinositide (PI) hydrolysis, intracellular Ca2+ mobilization, chemotaxis, and exocytosis. CXCR1, but not CXCR2, activates phospholipase D (PLD) and mediates respiratory burst, suggesting that the two receptors may play different physiological roles (5,6). Like many GPCRs, both receptors become phosphorylated, desensitized and internalized upon exposure to CXCL8. Over 95% of CXCR2 internalizes in the first 2-5 min of activation as compared to ~10% of CXCR1 (7-10). CXCR2 also recovers more slowly (~35% after 90 min) to the cell surface than CXCR1 (~100% after 90 min) upon removal of CXCL8 (7,8,11-14). This difference in receptor trafficking appears to be an important factor in the distinct ability of CXCR1 and CXCR2 to mediate leukocyte activation and regulation in response to CXCL8 (9,10). To date, the molecular basis for the differential regulation of the CXCL8 receptors remains unclear. CXCR1 and CXCR2 are highly homologous (77%) (15,16). The most divergent regions are the amino terminus (N-terminus), the fourth transmembrane domain (TMD), the second extracellular loop (ECL), and the carboxyl terminal (C-terminal) (15-18). While both receptors internalize via a phosphorylation/arrestin/dynamin-dependent mechanism, CXCR2 was also shown to internalize via a phosphorylation-independent mechanism (19). This partial internalization of the receptors requires the interaction of the C-terminal dileucine motif LLKIL with scaffold proteins such as adaptor protein-2 (AP-2) (20). Previous studies using phosphorylation-deficient and C-terminal deletion mutants of CXCR1 and CXCR2 indicate that the cytoplasmic tails of the receptors are necessary for receptor phosphorylation and subsequent arrestin binding but not sufficient to account for the differences in receptor internalization and recycling (10). Recent studies in our laboratory and others (8,10,13,14,21,22) also demonstrated that chimeric receptors in which the C-terminal of CXCR1 was exchanged for that of CXCR2, and vice versa, mediated cellular responses and internalized as well as the wild-type (WT) receptors. This suggests that domains other than the C-terminals modulate the rates of CXCR1 and CXCR2 trafficking in response to CXCL8. In the present work, we sought to determine the structural determinants of CXCR2 involved in rapid receptor internalization relative to CXCR1. To that end, chimeric and point mutant receptors exchanging different regions or specific
amino acids of CXCR2 for their CXCR1 counterparts were generated and expressed in RBL-2H3 cells. The receptors were characterized for their pharmacological and physiological properties as well as their ability to undergo receptor internalization, phosphorylation and desensitization in response to CXCL8 occupancy. The data herein indicate that the 2ECL of the CXCR2 is critical for its rapid internalization relative to CXCR1. Point mutants and computational modeling studies further identified Asp199 of the 2ECL as a key modulator of CXCR2 rapid internalization. MATERIALS AND METHODS Materials: [32P]Orthophosphate (8500-9120 Ci/mmol), myo-[2-3H]inositol (24.4 Ci/mmol) and [125I]-CXCL8 were purchased from Perkin Elmer. CXCL8/IL-8 was prepared as described previously (23). GROα (CXCL1) was purchased from Peprotech. Geneticin (G418) and all tissue culture reagents were purchased from Life Technologies, Inc. Monoclonal 12CA5 antibody, protein G-agarose and protease inhibitors were purchased from Boehringer Manheim. Anti-Human IL-8RA (CXCR1) and IL-8RB (CXCR2) antibodies were purchased from Pharmagen. Phorbol 12-myristate 13-acetate (PMA) was purchased from Sigma. All other reagents are from commercial sources. cDNAs encoding the chimeric mutants AB1, BA1, ABA and BAB were generous gifts from Dr. Philip M. Murphy, NIH. A, refers to CXCR1 and B, CXCR2. To facilitate the interpretation of the data, A and B were also employed throughout this manuscript to designate the chimeric and point mutant receptors. The chimeras were excised from pCEP4 using NotI and XhoI and subcloned into the expression vector pcDNA3. Construction of chimeric and point mutants of CXCR1 and CXCR2. The CXCR1 and CXCR2 cDNAs possess a unique conserved BamHI restriction site located at the junction of the second extracellular loop (2ECL) and transmembrane domain 5 (TMD5). This site is also conserved in chimeric mutants ABA and BAB and was used to generate the chimeric receptors (17). A4TMDB and BB4TMDA were generated by polymerase chain reaction (PCR) using ABA and BAB as templates, T7 pcDNA3 as forward primer and two reverse primers corresponding to the 2ECL of
2
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

CXCR1 (A2ECL): 5′ GGGCAGGATCCGTAACACCATCCGCCATTTTGCTGTGTCGTTGCCCAGGACCTCATAGCAAACTGGGCTGGAATTGTTTGGATGGACGGTCCTTCG 3′, and CXCR2 (B2ECL): 5′AGGCAGGATCCGCAACAGCATCCGCCAGTTTGCTGTATTATTTCCCATGTCCTCATAGCAGGCTGGACTAACATTGGATGAGTAGTAAGCCTGGCG 3′. The Bam H1 site is depicted by underlined sequences. For the generation of A2ECLB and B2ECLA, the same reverse primers were employed using CXCR1 (B2ECL) and CXCR2 (A2ECL) as templates, respectively. The resulting PCR products were digested with Not I and BamHI and ligated into pcDNA3-CXCR1 (A4TMDB & A2ECLB) or pcDNA3-CXCR2 (B4TMDA & B2ECLA) digested with the same restriction enzymes. Point mutations were introduced into pcDNA3-CXCR1 or pcDNA3-CXCR2 constructs using the QuickChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Forward and reverse primers were designed with single base changes to incorporate amino acid point mutations. The identity of all chimeric and point mutant constructs and fidelity of all PCR based coding sequences were verified by sequencing. Cell culture and transfection: RBL-2H3 cells were maintained as monolayer cultures in Dulbacco’s modified Eagle’s medium (DMEM) supplemented with 15% heat inactivated fetal bovine serum, 2 mM glutamine, penicillin (100 units/ml), and streptomycin (100 mg/ml) (24). RBL-2H3 cells (1x107 cells) were transfected by electroporation with 20 μg of pcDNA3 containing the receptor cDNAs and geneticin-resistant cells were cloned into single cell by fluorescence-activated cell sorter (FACS) analysis. Levels of protein expression were monitored by fluorescence-activated cell sorting (FACS) analysis. FACS analysis: For flow cytometric analysis, RBL cells were detached by Versene treatment, washed with HEPES buffered Hanks’ balanced salt solution (HHBSS) and resuspended in the same medium. Cells (1-5x106 cells) were incubated with anti-CXCR1 or anti-CXCR2 antibodies (1 μg/ml) in a total volume of 400 μl of HHBSS for 60 min at 4 oC. The cells were then washed and incubated
with fluorescein (FITC)-antimouse IgG for 60 min at 4 oC. Cells were then washed and analyzed for cell surface expression of the receptor on a Beckton Dickenson FACScan cytometer (10). Radioligand binding assays and receptor internalization: Radioligand binding assays were carried out as decribed previously (10). Briefly, RBL-2H3 cells were subcultured overnight in 24-well plates (0.5x106 cells/well) in growth medium. Cells were then rinsed with Dulbecco’s modified Eagles medium supplemented with 20 mM Hepes, pH 7.4 and 10 mg/ml BSA and incubated on ice for 2-4 hours in the same medium (250 μl) containing [125I]-CXCL8 (0.1-1 nM). Reactions were stopped with 1 ml of ice-cold PBS containing 10 mg/ml BSA, and washed 3 times with the same buffer. Then cells were solubilized with RIPA buffer (200 μl), dried under vacuum and bound radioactivity was counted (25). Nonspecific radioactivity bound was determined in the presence of 500 nM unlabelled CXCL8. Kd and Bmax were determined using the GraphPad radioligand binding data analysis. For receptor internalization, cells were incubated with ligand for 0-60 min at 37 °C. Cells were then washed with ice-cold PBS and 125I-CXCL8 binding (0.1-1 nM) was carried out as described above. Phosphoinositide (PI) hydrolysis and calcium measurement: RBL-2H3 cells were subcultured overnight in 96-well culture plates (50,000 cells/well) in inositol-free medium supplemented with 10% dialyzed fetal bovine serum and 1 μCi/ml [3H]inositol. Cells were then washed with HEPES-buffered saline supplemented with 100 mM LiCl2 in the presence of 0.1% bovine serum albumin and preincubated with 50 μl of the same buffer for 10 min at 37 ºC. Cells were then stimulated with 100 nM CXCL8 or CXCL1. Reactions were terminated 10 min later by addition of 200 μl of chloroform: methanol: 4 N HCl (100:200:2), 75 μl of 0.1 N HCl, and 75 μl of chloroform. Total [3H]inositol phosphates in the aqueous phase were separated on columns of Dowex formate (24,26). For calcium mobilization, cells (5x106) were washed with HEPES buffered saline and loaded with 1 μM Indo I-AM in the presence of 1 μM pluronic acid for 30 minutes at room temperature. The cells were then washed with HEPES and resuspended in 1.5 ml of Siriganian buffer. Intracellular calcium increase in
3
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

the presence or absence of ligands was measured as described previously (27). Phosphorylation of receptors: Phosphorylation of receptors was performed as described previously (10,25,28). RBL-2H3 cells (5x106) expressing the receptors were incubated with [32P]orthophosphate (150 μCi/dish) for 90 min. The labeled cells were then stimulated with the indicated ligands for 5 minutes at 37 oC. Cells were then washed with ice-cold PBS and solubilized in 1 ml of radioimmunoassays buffer (RIPA) containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS. Cell lysates were immunoprecipitated with specific antibodies against either the N-terminus of CXCR1 or CXCR2 and analyzed by SDS electrophoresis and visualized by autoradiography. Extracellular regulated kinase (ERK) activity. For ERK activity, RBL-2H3 cells (5x106) expressing the receptors were washed three times with PBS, then resuspended in PBS containing CXCL8 (100 nM) for different periods of time at 37 oC. The reactions were stopped with ice-cold PBS, cells were collected by centrifugation and membranes were prepared and assayed for protein concentration as described previously (28). Membrane proteins (~50μg) were resolved in 10% SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against either ERK 1/2 or phospho-ERK 1/2 (28). Detection was carried out with HRP-conjugated sheep anti-mouse antibody and ECL. Homology modeling of human CXCR2. Multiple sequence alignment of the rhodopsin and human CXCR2, along with human CXCR1, was performed by CLUSTALW (http://align.genome.jp) using the BLOSUM matrix. To prohibit any gap within the TMD core, a gap-open penalty of 15.0 and a gap-extension penalty of 0.1 were used. The calculated sequence alignment was validated by examining alignment of the highly conserved amino acid residues, including the NLAxxD motif in TMD2, the D/ERY motif in TMD3, the CWxP motif in TMD6, and the NPxxY motif in TMD7 of GPCRs (29).
To construct the seven TM helices and the β3-β4 hairpin structure at the N-terminal region of 2ECL for human CXCR2, the Cα coordinates of the
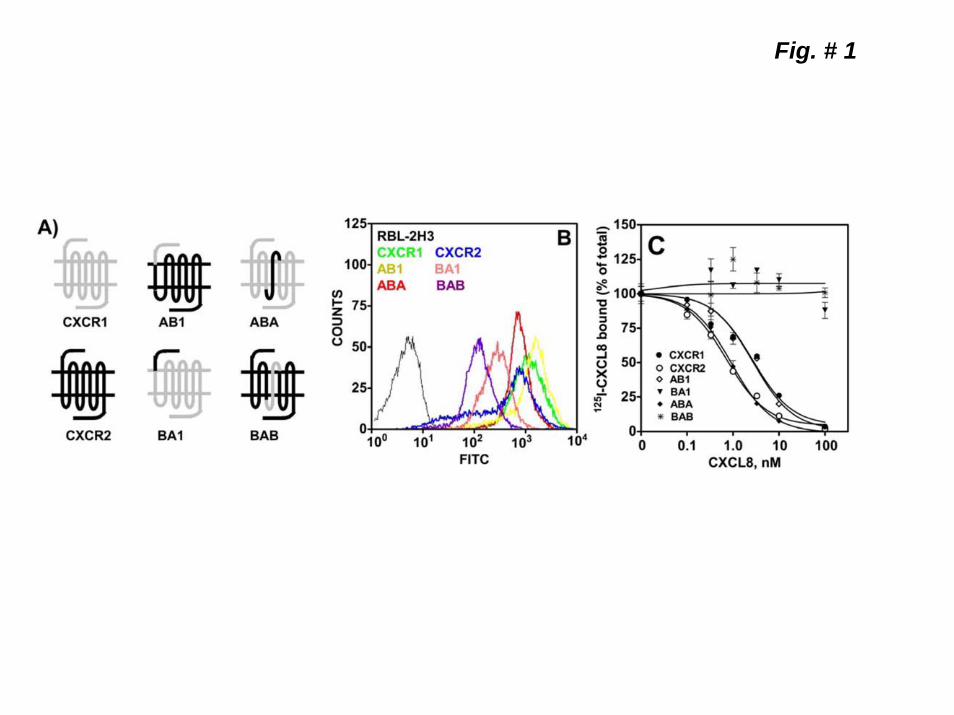
recently published x-ray crystal structure of bovine rhodopsin (PDB ID: 1U19) (30) were used as the template. The intracellular and extracellular loops (i.e., 2ECL C-terminal region, ICL3 and 3ECL) that are different in length from those of rhodopsin were constructed using the loop search tool implemented in InsightII homology module, utilizing the Cα-distance matrix from a selected dataset of Brookhaven Protein Databank (PDB). The typical disulfide linkage between TMD3 and 2ECL common to most GPCRs was inserted between C119 and C196. After a short minimization, the resulting structure was subjected to a 500 ps MD simulation at 300 K, during which the transmembrane backbone atoms and the H-bonds in the 2ECL β3-β4 hairpin structure were restrained. The receptor model was sampled by extracting a snapshot of the structure every 1 ps during the simulation. The averaged structure from the last fifty 50 ps was energy minimized, with the restraints mentioned above first, then with the transmembrane backbone constraint only. Overall 3 separate simulations were repeated for the wild-type CXCR1 and CXCR2 (i.e., AV190 and BD199), the mutated forms (i.e., BBD199VA, BD199N and AV190DB mutants). Energy minimization and MD simulations were carried out on a Silicon Graphics Origin 350 workstation using the Consistent Force Field (CFF) (31) implemented in InsightII (Version 2005, Accelrys Inc. San Diego, CA). The cell multipole method (32) with a distance-dependent dielectric constant (ε=ε r, with ε = 4.0) was used for summation of non-bonding interactions. RESULTS Expression of CXCR1 and CXCR2 chimeric mutants in RBL-2H3 cells. In order to determine the structural determinants involved in the rapid internalization of CXCR2 relative to CXCR1, four chimeric receptors exchanging the amino terminus (AB1 and BA1) or the region comprising the 4th transmembrane domain (4TMD) and the 2nd extracellular loop (2ECL) of CXCR1 for those of CXCR2 and vice versa (ABA and BAB) were stably expressed in RBL-2H3 (Fig 1, A & B). Competition binding of [125I]CXCL8 to the chimeric receptors in the presence of unlabeled CXCL8 and Scatchard analysis demonstrated that AB1 and ABA, but not BA1 and BAB, bound CXCL8 with affinities similar to CXCR1 and
4
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

CXCR2, respectively (Fig, 1-C & Table 1). All receptors, however, stimulated a rapid and transient increase in free intracellular Ca2+ mobilization and phosphoinositide (PI) hydrolysis in response to both CXCL8 and CXCL1 (Fig 2, A, B&C). Except for CXCR2, CXCL1-induced significantly lower PI hydrolysis relative to CXCL8 (Fig 2-C). No significant difference was found in CXCL8-induced PI hydrolysis to AB1, ABA, and BA1, relative to CXCR1 and CXCR2 (3.5-4 fold over basal) (Fig 2-C). BAB-mediated PI hydrolysis to CXCL8 was significantly lower (~2.5 fold over basal) than the WT receptors. Since BAB displayed no ligand binding properties (Table 1), it is difficult to determine whether this difference is due to a decrease affinity of the chimeric receptor for CXCL8 or impaired ability to couple to Gi to activate PLCβ. We next measured the rate of internalization of the chimeric receptors upon CXCL8 (100 nM) pretreatment. CXCL8 induced time-dependent internalization of all 4 receptors (Fig 2-D). AB1 and ABA internalized as rapidly as CXCR2 (~90% after 5-10 min) whereas BA1 and BAB (~35% after 60 min) internalized far slower than CXCR1 (~55% after 60 min).
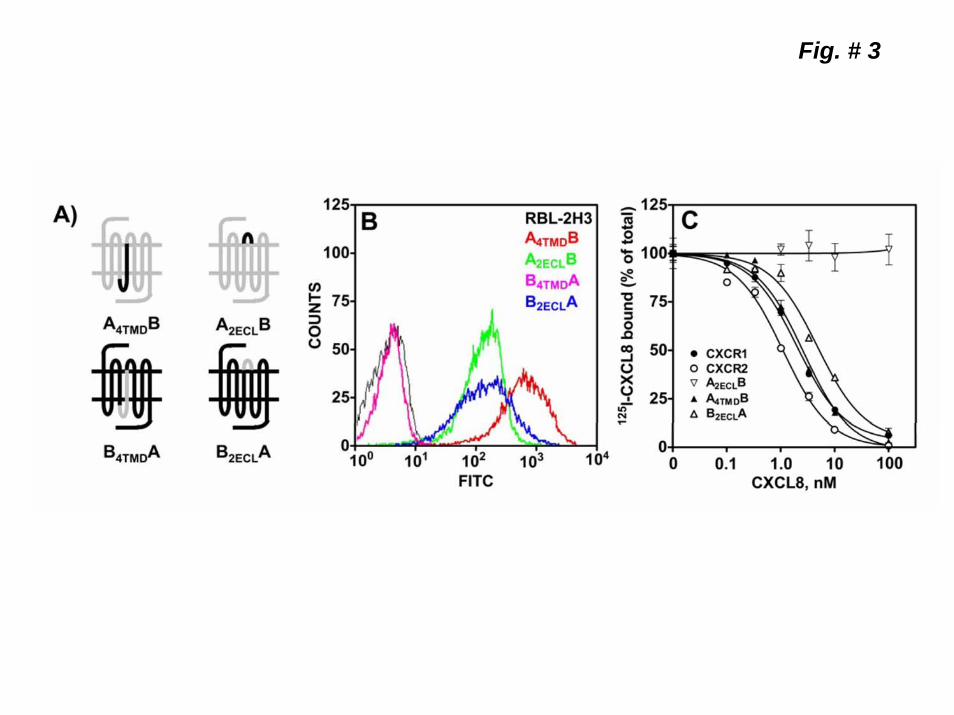
Role of the 4th transmembrane domain (4TMD) and the 2nd extracellular loop (2ECL) of CXCR1 and CXCR2 in receptor internalization. To further delineate the region(s) of CXCR1 and CXCR2 involved in receptor internalization, four additional chimeras were constructed (Fig 3-A) by exchanging either the 4TMD domains (A4TMDB and BB4TMDA) or the 2ECL (A2ECLB and B2ECLA). Except for B4TMDA, all chimeras were stably expressed in RBL-2H3 cells (Fig 3-B) and characterized. A4TMDB and B2ECLB A, but not A2ECLB, bound CXCL8 with affinities similar to the WT receptors (Fig 3-C & Table 1). A4TMDB and B2ECLA mediated intracellular Ca2+ mobilization and PI hydrolysis in response to both CXCL8 and CXCL1 (Fig 4, A & B). A4TMDB, like CXCR1, mediated greater responses to CXCL8 whereas B2ECLA, like CXCR2, responded to both ligands with similar potency. Upon CXCL8 pretreatment, A4TMDB internalized as well as CXCR1 (~55% after 60 min) (Fig 4-C). Interestingly, B2ECLA also showed a slower time course of internalization (~50% after 60 min, Fig 4-C).
Role of receptor glycosylation in CXCR2 internalization. CXCR1 and CXCR2 express two N-glycosylation motifs in their 2ECL (Fig 5-A, boxed motifs). Since the motifs are different between the two receptors, we constructed four chimeras in which the glycosylation sites of CXCR2 were converted to those of CXCR1 (BNNSSA and BN203DA), and vice versa (ASNVSB, AD194NB) (Fig 5-A). The mutants were stably expressed in RBL-2H3 cells and characterized. Except for ASNVSB, all receptor variants bound CXCL1 and CXCL8 to induce intracellular Ca2+ mobilization (Fig 5-B). Upon CXCL8 pretreatment, BBNNSSA and BN203DA internalized like CXCR2 whereas AD194NB internalized like CXCR1 (Fig 5-C). Identification of specific residue of the 2nd ECL involved in receptor internalization. In order to identify the specific residue(s) of CXCR2 involved in rapid receptor internalization relative to CXCR1, we exchanged all divergent amino acids (aa) in the 2nd ECL of CXCR2 for their CXCR1 counterparts and, vice versa (Table II). Except for BY188HA, all receptors were stably expressed in RBL-2H3 cells (Table II). The expressed receptors, except for AV190DB, bound CXCL8 to induce intracellular Ca2+ mobilization similar to the WT receptors. We next measured receptor internalization upon exposure to CXCL8 (60 min). As shown in Figure 6-A, the CXCR1 mutants AH179YB, AS183VB, AL191MB and AV201LB internalized like CXCR1 (~55% after 60 min) whereas AP180SB (~40%), AV186AB (~40%) and AK197NB (~50%) were slighty more resistant to CXCL8-mediated internalization than CXCR1. All the CXCR2 mutants internalized as rapidly as CXCR2, except for BD199VA which displayed delayed internalization similar to that of CXCR1 (~55% after 60 min, Fig 6-B).
To further determine the role of D199 in CXCR2 rapid internalization, we generated a CXCR2 mutant in which D199 was replaced for asparagine (N) (BD199N). BD199N was stably expressed in RBL-2H3 cells and was shown to bind CXCL8 as well as CXCR2 and BD199VA (Fig 7-A) to induce intracellular Ca2+ mobilization and internalization (Fig 7-B & data not shown). BD199N internalization was identical to that of CXCR2 whereas BD199VA internalized like CXCR1 (Fig 7-B).
5
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Role of receptor phosphorylation and dimerization in CXCL8-mediated receptor internalization. It was previously reported that phosphorylated CXCR1 migrated as a single monomeric band of ~75 kDa whereas CXCR2 showed two phosphorylated forms: a slow dimeric (~65 kDa) and a fast monomeric (~45 kDa) form (10,33-35). To assess the role of receptor phosphorylation and dimer formation in CXCL8-induced receptor internalization, 32P-labeled cells expressing CXCR1, CXCR2, ABA, BAB, A4TMDB, BB2ECLA, BD199VA and BD199N were stimulated with either CXCL8 (100 nM) or PMA (100 nM) for 5 min. The cell lysates were immunoprecipitated with specific antibodies directed against the N-terminus of either CXCR1 (CXCR1, ABA & A4TMDB; Fig 8-A) or CXCR2 (CXCR2, B2ECLB A, BBD199VA & BD199N; Fig 8-B). The receptors were homologously phosphorylated by CXCL8 (lanes 2) and heterologously by PMA (lanes 3). Phosphorylation by PMA (lanes 3) was less than that of CXCL8 (lanes 2). CXCR2, ABA, A4TMDB, B2ECLB A and BD199VA displayed two forms of phosphorylated receptors (Fig 8-A and B). The electrophoretic mobilities of the two forms of A4TMDB (~70 and ~50 kDa); B2ECLA and BD199VA (~70 and ~45 kDa) were slightly different than those of CXCR2 and ABA (~65 and ~45 kDa). BBD199VA also showed a greater phosphorylation of the dimeric form (~70 kDa) than the monomeric one (~45 kDa) (Fig 8-B). CXCR1 (Fig 8-A) and BD199NB (Fig 8-B) migrate as a single phosphorylated band (~75 kDa). CXCR1, CXCR2, BD199VA and BD199N mediated-ERK activation: CXCR1 and CXCR2 were shown to mediate mitogen-activated protein kinase (MAPK) activation to different extents, based on the cell lines (36-38). In order to assess the ability of BD199VA and BD199N to activate MAPK in RBL-2H3 cells, we measured ERK1/2 phosphorylation in response to CXCL8. As shown in Figure 9, CXCR1, CXCR2, BD199VA, and BBD199N induced time-dependent phosphorylation of ERK1/2 upon activation by CXCL8 (100 nM). Maximum response was obtained at ~5 min. CXCR1 and BD199VB A induced ERK1/2 phosphorylation to a greater extent relative to CXCR2 and BBD199N (Fig 9-B).
Desensitization of BD199VA and BD199N mediated intracellular Ca2+ mobilization. We next studied the ability of B BD199VA, BD199N to mediate and undergo desensitization by monitoring for CXCL8 and CXCL1-induced intracellular Ca mobilization. As shown in Table III, B
2+
D199VA, BD199N,B CXCR1 or CXCR2 mediated Ca2+ mobilization was desensitized by pretreatment of the cells with a first dose of CXCL8 or CXCL1 (100 nM). Homologous desensitization by CXCL8 was more potent (85-100%) than that of CXCL1 (47-84%). CXCR1, but not CXCR2, was shown to mediate receptor cross-desensitization in response to CXCL8 activation (14,25,27). In order to determine whether BD199VA and BBD199N mediate cross-regulatory signals, RBL-2H3 stably co-expressing the RANTES (CCL5) receptor CCR5 along with BD199VA (RBL-CCR5/BD199VA), BD199N (RBL-CCR5/BD199N), CXCR1 (RBL-CCR5/CXCR1) or CXCR2 (RBL-CCR5/CXCR2) were generated. CXCL8 and CCL5 mediated cross-desensitization of Ca mobilization was measured. As shown in Table IV, pretreatment of RBL-CCR5/CXCR1 or RBL-CCR5/B
2+
D199VA cells with a first dose of CXCL8 (100 nM) cross-desensitized CCL5 (100 nM)-induced Ca mobilization by 56% and 41%, respectively. CCL5-mediated Ca was resistant to cross-desensitization by CXCL8 in RBL-CCR5/CXCR2 (2%) and RBL-CCR5/B
2+
2+
D199N (0%) cells. CCL5 activation of CCR5 cross-desensitized CXCL8-induced intracellular Ca in all four cell lines (Table IV).
2+
Computational Modeling of the 2ECLs of CXCR1, CXCR2, B BD199VA and BD199N. Multiple sequence alignment and homology models of CXCR1 and CXCR2 were constructed based on the recent x-ray structure of rhodopsin (30). Molecular Dynamics (MD) simulations of the extracellular loop region of the WT CXCR1 and CXCR2 and the mutant forms BD199VA, BD199N and AV190DB were performed. The molecular interaction involving D199 of CXCR2 (BD199) was measured by the H-bond formation with the neighboring residues (Table V). As shown in Figure 10-B, D199 of CXCR2 forms H-bonds with R185, T186 and R208. The H-bonding network is similar to the one between the equivalent D190 of rhodopsin and R177, Y192 and T193 (Fig 10-A). When D199 was replaced for V (BD199VA), due to the highly
6
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

hydrophobic nature of V, the neighboring polar or charged residues (R185, T186, Y188, M200, N202, and R208) could not interact with it to form the H-bonding network (Fig 10-C). In contrast, when D199 was replaced with an N, BD199NB , H-bond could be formed with T186 and R185, but not Arg208 (Fig 10-D). Apparently, the replacement was sufficient to regenerate the H-bonding network and stabilize the receptor in a CXCR2-like conformation thereby restoring rapid receptor internalization. DISCUSSION CXCL8 and its receptors CXCR1 and CXCR2 play a critical role in the pathophysiology of several inflammatory diseases including multiple sclerosis, rheumatoid arthritis and asthma. Upon activation, these receptors mediate similar as well as different cellular functions. This distinction correlates with the receptors differing kinetics of internalization and recycling upon activation by CXCL8 (10). CXCR1 internalizes slower (50-60% after 60 min) and mediates leukocyte cytotoxicity (i.e. respiratory burst) and receptor cross-regulatory signals (6,25). In contrast, CXCR2 internalizes rapidly (90-95% after 2-5 min) but mediates greater post-internalization signals [i.e. mitogen activated protein (MAP) kinase and protein kinase B (AKT) activation] (37). Attempts to determine the structural determinants responsible for the different kinetics of receptor downregulation have been focusing predominantly on the intracellular domains of the receptor (13,22). Recent studies using tail deleted and chimeric receptors in which the tail of CXCR1 was exchanged for that of CXCR2 and, vice versa, have shown that the C-terminals are important for receptor phosphorylation, desensitization, adaptor protein binding and downregulation but not sufficient to explain the distinct kinetics of internalization of the two receptors in response to CXCL8 interaction (10). In this study we employed a set of new (A4TMDB, B4TMDA, A2ECLB & B2ECLA) as well previously published (ABA, BAB, AB1 & BA1; (17) chimeras and point mutant receptors to further analyze domains upstream of the C-terminal of the two receptors which may modulate receptor downregulation. The data herein indicate that the second extracellular loop (2ECL) modulates CXCR2 rapid internalization relative to CXCR1. This contention is based on the following observations. First, the CXCR1 derived chimera
ABA which expressed the 2ECL and fourth transmembrane domain (4TMD) of CXCR2 internalized like CXCR2, ~90% in the first 5 min of activation (Fig 2-D). Second, the CXCR2 chimera B2ECLA which expressed the 2nd extracellular loop of CXCR1 internalized like CXCR1 (~50% after 60 min) (Fig 4-C). Third, the point mutant receptor BBD199VA in which aspartate 199 of the 2ECL of CXCR2 was exchanged for its valine (V) counterpart of CXCR1, showed decrease CXCL8 mediated internalization as compared to CXCR2 (~45% versus 95%, respectively, after 60 min; Fig 6-B and 7-B).
Interestingly, when D199 was exchanged for asparagine (N) instead of valine (BD199N), rapid internalization of CXCR2 (~90% after 5 min) was restored (Fig 7-B). These results further suggest that D199 of the 2ECL of CXCR2 plays a critical role in stabilizing the receptor in the conformation necessary for its rapid internalization upon binding to CXCL8. Homology modeling of the 2ECL of CXCR2 based on the crystal structure of rhodopsin also supports this contention. As shown in Figure 10-B, D199 may interact with threonine 186 (T186), arginine 185 (R185) and R208, to form an H-bonding network similar to the one formed between D190 and R177, tyrosine 192 (Y192) and T193 of rhodopsin (Fig 10-A). When D199 was replaced for the corresponding hydrophobic valine of CXCR1, BD199VA, (Fig 10-C) no H-bond could be formed causing a conformational change in the 2ECL similar to that of CXCR1 (Fig 10-D). In contrast, the exchange of D199 for N, BBD199N, (Fig 10-E) partially restored the H-bounding network with T186 and R185, thereby, the rapid kinetic of receptor internalization (Fig 10-D). Despite the differences in H-bonding patterns between CXCR1 and CXCR2, it cannot be ruled out, however, that other factors may contribute to the differences in the structure and rate of internalization. These receptors interact with several adaptor proteins (i.e. arrestins, AP-2, Src, HIP) to mediate signal transduction (10,19,20). It could be that conformational changes induced by D199 altered receptor/adaptor proteins complex formation and, thereby, receptor downregulation (39).
A question addressed in this study was the role of receptor glycosylation in the distinct kinetics of CXCR1 and CXCR2 downregulation. Both receptors express two glycosylation sites with different sequence motifs within their 2ECL (Fig 5) (16,17). It was previously shown that
7
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

glycosylation of the CXCR2 does not affect ligand binding, signaling, or trafficking abilities (40). Since exchanging the 2ECL of CXCR2 for that of CXCR1 (B2ECLA) changed the rate of internalization (Fig 4-C), this raised the possibility that CXCR1-like glycosylation could influence a change in the kinetic of internalization. However, when the glycosylation motifs of the 2ECL of CXCR2 were exchanged for those of CXCR1 (BNNSSA and BN203DA), or vice versa (AD194NB), no significant differences were found in CXCL8 binding to both receptors or in the rates of receptor internalization, relative to the WT receptors (Fig 5-C and data not shown). These observations suggests that receptor glycosylation plays no role in receptor internalization and further indicate that Asp199 is solely responsible for the rapid internalization of CXCR2.
Receptor oligomerization has been shown to play an important role in the activation and regulation of several chemokine receptors including CCR2, CCR5, CXCR4 (41-44). To date, contrasting results exist regarding the dimerization of CXCR1 and CXCR2 and their roles in receptor-mediated cellular functions. Previous studies from our group and others using native and epitope-tagged receptors have shown two migrating forms of CXCR2 (~40 and ~65 kDa) but one form of CXCR1 (~75 kDa) (9,10,33,34). Trettel et al (35), recently reported that CXCR2, but not CXCR1, functions as a dimer and that could affect the kinetic of receptor downregulation. Wilson et al (45), however, demonstrated that both CXCR1 (~45 and ~75 kDa) and CXCR2 (~40 and ~65 kDa) are capable of forming dimers. In this study, since both CXCR1 and CXCR2 internalize in a phosphorylation-dependent fashion, we used CXCL8-induced receptor phosphorylation to assess the role of receptor dimerization in receptor internalization. The data herein, clearly showed one form of CXCR1 and further suggested that receptor internalization is independent of receptor dimerization (Figs 4, 6-8). First, the chimeras BB2ECLA and A4TMDB internalized like CXCR1 but displayed a slow and a fast migrating form like CXCR2 (Fig 4-C & Fig 8-B). Second, BD199N migrated as a single slow form (~75 kDa) similar to that of CXCR1 but internalized as rapidly as CXCR2 (~90% after 5 min) (Fig 7-B & Fig 8-B). Third, the related mutant BD199VB A showed two forms (~75 and ~45 kDa) but internalized like CXCR1(~45% after 60 min) (Fig 7-B & Fig 8-B).
The contrast with the results reported by Wilson et al (45) is unclear. One explanation could be that native CXCR1 predominantly exists as a dimer, and CXCR2 as an equal mixture of both dimer and monomer. Thus, since dimerization of the receptor occurs in the endoplasmic reticulum (ER), it could be that overexpression of the receptors in human embryonic kidney (HEK293) cells affected the rate of dimerization resulting in two forms of CXCR1 (~75 and ~45 kDa). Against this hypothesis is that Tretell et al (35) also used the HEK293 expression system in their study.
CXCR1 but not CXCR2 was shown to mediate receptor cross-regulatory signals (27). This distinction was attributed to the length of signaling which for CXCR2 was shortened by rapid receptor internalization (14). Indeed, the mutant BBD199VA which displayed delayed CXCL8-mediated receptor internalization, cross-desensitized CCR5-mediated intracellular Ca mobilization to CCL5 (Table IV). In contrast, BD199N
2+
B which internalized as rapidly as CXCR2 failed to cross-desensitize responses to CCL5. BD199VA, like CXCR1, also induced a sustained ERK1/2 activation as compared to BD199N and CXCR2 (fig 9). Sai et al (46) have recently reported that delayed internalization of the CXCR2 mutant IL/AA-CXCR2 in HL60 cells correlated with decreased ERK1/2 activation. These results are in contrast to the ones presented in this work and may well reflect differences between cell lines. Supporting this contention is that CXCR2 expressed in mouse embryonic fibroblast (MEF) cells deficient in βarrestin 1/2-/- expression also displayed decreased receptor internalization and sustained ERK1/2, stress kinases p38 and c-Jun N-terminal protein kinase activation, relative to wild-type MEF (38). Furthermore, several phosphorylation and internalization resistant mutants of CCR5 and the follicle-stimulating hormone receptor (FSH-R) expressed in RBL-2H3 and HEK293 cells, respectively, were also shown to induce sustained ERK1/2 activation relative to the WT receptors (47,48)
In all, the data herein provide new insights into the role of extracellular domains of the receptors in modulating signal length and ability to mediate a distinct set of cellular responses. A drawback, however, is that key chimera or point mutant such as A2ECLB and AV190DB expressed but failed to bind or respond to ligand (Fig 3-B, Fig 4-
8
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

A and Table II). These are common limitations in studies using cellular and genetic approaches to elucidate the complexities of receptor activation and regulation. Despite these limitations, however, these studies pointed out the importance of the 2nd extracellular loop, and more specifically Asp199, in the distinct ability of CXCR1 and CXCR2 to mediate and regulate inflammatory responses to
CXCL8. As chimeras such as BAB and BA1, failed to show ligand binding, but mediated functional responses (i.e. Ca2+ mobilization, PI hydrolysis) and underwent CXCL8-dependent internalization, the data further support the notion that distinct domains of the receptors may be involved in ligand interaction and receptor activation/regulation (17).
ACKNOWLEDGEMENTS
We are grateful to Dr. Philip M. Murphy for the generous gift of plasmid cDNAs expressing receptor chimeric mutants. This work was supported by National Institutes of Health Grants AI-38910 (to R. M. R.) and AI-069152 (to K. R.) We thank Ernest Johnson and James Crisp for their technical support.
REFERENCES 1. Baggiolini, M., Dewald, B., and Moser, B. (1997) Annu Rev Immunol 15, 675-705 2. Locati, M., and Murphy, P. M. (1999) Annu Rev Med 50, 425-440 3. Murphy, P. M., Baggiolini, M., Charo, I. F., Hebert, C. A., Horuk, R., Matsushima, K., Miller, L. H.,
Oppenheim, J. J., and Power, C. A. (2000) Pharmacol Rev 52(1), 145-176 4. Baggiolini, M. (2000) Immunol Rev 177, 5-7 5. L'Heureux, G. P., Bourgoin, S., Jean, N., McColl, S. R., and Naccache, P. H. (1995) Blood 85(2),
522-531 6. Jones, S. A., Wolf, M., Qin, S., Mackay, C. R., and Baggiolini, M. (1996) Proc Natl Acad Sci U S A
93(13), 6682-6686 7. Barlic, J., Khandaker, M. H., Mahon, E., Andrews, J., DeVries, M. E., Mitchell, G. B., Rahimpour,
R., Tan, C. M., Ferguson, S. S., and Kelvin, D. J. (1999) J Biol Chem 274(23), 16287-16294 8. Feniger-Barish, R., Ran, M., Zaslaver, A., and Ben-Baruch, A. (1999) Cytokine 11(12), 996-1009 9. Richardson, R. M., DuBose, R. A., Ali, H., Tomhave, E. D., Haribabu, B., and Snyderman, R.
(1995) Biochemistry 34(43), 14193-14201 10. Richardson, R. M., Marjoram, R. J., Barak, L. S., and Snyderman, R. (2003) J Immunol 170(6),
2904-2911 11. Barlic, J., Andrews, J. D., Kelvin, A. A., Bosinger, S. E., DeVries, M. E., Xu, L., Dobransky, T.,
Feldman, R. D., Ferguson, S. S., and Kelvin, D. J. (2000) Nat Immunol 1(3), 227-233 12. Chuntharapai, A., and Kim, K. J. (1995) J Immunol 155(5), 2587-2594 13. Prado, G. N., Suzuki, H., Wilkinson, N., Cousins, B., and Navarro, J. (1996) J Biol Chem 271(32),
19186-19190 14. Richardson, R. M., Pridgen, B. C., Haribabu, B., Ali, H., and Snyderman, R. (1998) J Biol Chem
273(37), 23830-23836 15. Holmes, W. E., Lee, J., Kuang, W. J., Rice, G. C., and Wood, W. I. (1991) Science 253(5025), 1278-
1280 16. Murphy, P. M., and Tiffany, H. L. (1991) Science 253(5025), 1280-1283 17. Ahuja, S. K., Lee, J. C., and Murphy, P. M. (1996) J Biol Chem 271(1), 225-232 18. Catusse, J., Liotard, A., Loillier, B., Pruneau, D., and Paquet, J. L. (2003) Biochem Pharmacol
65(5), 813-821 19. Fan, G. H., Yang, W., Sai, J., and Richmond, A. (2001) J Biol Chem 276(20), 16960-16968 20. Fan, G. H., Yang, W., Wang, X. J., Qian, Q., and Richmond, A. (2001) Biochemistry 40(3), 791-800 21. Ben-Baruch, A., Bengali, K. M., Biragyn, A., Johnston, J. J., Wang, J. M., Kim, J., Chuntharapai, A.,
Michiel, D. F., Oppenheim, J. J., and Kelvin, D. J. (1995) J Biol Chem 270(16), 9121-9128
9
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

22. Matityahu, E., Feniger-Barish, R., Meshel, T., Zaslaver, A., and Ben-Baruch, A. (2002) Eur J Immunol 32(12), 3525-3535
23. Rajagopalan, L., and Rajarathnam, K. (2004) J Biol Chem 279(29), 30000-30008 24. Ali, H., Richardson, R. M., Tomhave, E. D., Didsbury, J. R., and Snyderman, R. (1993) J Biol Chem
268(32), 24247-24254 25. Richardson, R. M., Pridgen, B. C., Haribabu, B., and Snyderman, R. (2000) J Biol Chem 275(13),
9201-9208 26. Ali, H., Tomhave, E. D., Richardson, R. M., Haribabu, B., and Snyderman, R. (1996) J Biol Chem
271(6), 3200-3206 27. Nasser, M. W., Marjoram, R. J., Brown, S. L., and Richardson, R. M. (2005) J Immunol 174(11),
6927-6933 28. Brown, S. L., Jala, V. R., Raghuwanshi, S. K., Nasser, M. W., Haribabu, B., and Richardson, R. M.
(2006) J Immunol 177(5), 3242-3249 29. Baldwin, J. M., Schertler, G. F., and Unger, V. M. (1997) J Mol Biol 272(1), 144-164 30. Okada, T., Sugihara, M., Bondar, A. N., Elstner, M., Entel, P., and Buss, V. (2004) J Mol Biol
342(2), 571-583 31. Maple, J., Hwang, M.J., Stockfisch, T. P., and Hagler, A. T. (1994) Israel J. of Chem. 34, 195 32. Ding, H. Q. K., N.; Goddard, W. A., III. (1992) J Chem Phys 97, 4309-4315 33. Mueller, S. G., Schraw, W. P., and Richmond, A. (1994) J Biol Chem 269(3), 1973-1980 34. Mueller, S. G., White, J. R., Schraw, W. P., Lam, V., and Richmond, A. (1997) J Biol Chem
272(13), 8207-8214 35. Trettel, F., Di Bartolomeo, S., Lauro, C., Catalano, M., Ciotti, M. T., and Limatola, C. (2003) J Biol
Chem 278(42), 40980-40988 36. Jones, S. A., Moser, B., and Thelen, M. (1995) FEBS Lett 364(2), 211-214 37. Shyamala, V., and Khoja, H. (1998) Biochemistry 37(45), 15918-15924 38. Zhao, M., Wimmer, A., Trieu, K., Discipio, R. G., and Schraufstatter, I. U. (2004) J Biol Chem
279(47), 49259-49267 39. Vaidehi, N., Schlyer, S., Trabanino, R. J., Floriano, W. B., Abrol, R., Sharma, S., Kochanny, M.,
Koovakat, S., Dunning, L., Liang, M., Fox, J. M., de Mendonca, F. L., Pease, J. E., Goddard, W. A., 3rd, and Horuk, R. (2006) J Biol Chem 281(37), 27613-27620
40. Ludwig, A., Ehlert, J. E., Flad, H. D., and Brandt, E. (2000) J Immunol 165(2), 1044-1052 41. Babcock, G. J., Farzan, M., and Sodroski, J. (2003) J Biol Chem 278(5), 3378-3385 42. Issafras, H., Angers, S., Bulenger, S., Blanpain, C., Parmentier, M., Labbe-Jullie, C., Bouvier, M.,
and Marullo, S. (2002) J Biol Chem 277(38), 34666-34673 43. Rodriguez-Frade, J. M., Vila-Coro, A. J., de Ana, A. M., Albar, J. P., Martinez, A. C., and Mellado,
M. (1999) Proc Natl Acad Sci U S A 96(7), 3628-3633 44. Vila-Coro, A. J., Mellado, M., Martin de Ana, A., Lucas, P., del Real, G., Martinez, A. C., and
Rodriguez-Frade, J. M. (2000) Proc Natl Acad Sci U S A 97(7), 3388-3393 45. Wilson, S., Wilkinson, G., and Milligan, G. (2005) J Biol Chem 280(31), 28663-28674 46. Sai, J., Walker, G., Wikswo, J., and Richmond, A. (2006) J Biol Chem 281(47), 35931-35941 47. Kara, E., Crepieux, P., Gauthier, C., Martinat, N., Piketty, V., Guillou, F., and Reiter, E. (2006) Mol
Endocrinol 20(11), 3014-3026 48. Kraft, K., Olbrich, H., Majoul, I., Mack, M., Proudfoot, A., and Oppermann, M. (2001) J Biol Chem
276(37), 34408-34418
FOOTNOTES
1The abbreviations used are: IL-8 or CXCL8, interleukin-8; CXCR1, IL-8 receptor A; CXCR2, IL-8 receptor B; RBL-2H3, rat basophilic leukemia-2H3; PMA, phorbol 12-myristate 13-acetate; IP, inositol phosphate; G protein, GTP-regulatory protein; FITC, fluorescein isothiocyanate, βarr, β-arrestin; ECL, extracellular loop; TMD, transmembrane domain; WT, wild-type; RIPA, radio-immunoprecipitation assay.
10
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

FIGURE LEGENDS
Figure 1. Schematic representation and expression of wild-type CXCR1 and CXCR2 and the chimeric receptor mutants AB1, BA1 ABA and BAB. A) Gray traces represent CXCR1 and black traces CXCR2. B) FACS analysis of surface expression of wild-type and chimeric receptors in RBL-2H3 cells after staining with CXCR1 (CXCR1, AB1 and ABA) and CXCR2 (CXCR2, BA1and BAB) specific antibodies. C) Competition binding of [125I]CXCL8 to CXCR1, CXCR2 AB1, BA1, ABA, and BAB in the presence of different concentrations of CXCL8 (0-100 nM). Representative experiments are from three independent determinations. Figure 2. Functional characterization of the wild-type and chimeric receptors. For intracellular Ca2+ mobilization, RBL-2H3 cells (5 x 106) stably expressing CXCR1, AB1 or ABA (A) and CXCR2, BA1 or BAB (B) were loaded with indo-1 and CXCL8 and CXCL1 (100 nM) mediated Ca2+ mobilization were measured. C) For the generation of inositol phosphates ([3H]IPs), RBL-2H3 cells (50,000 cells/ well) were cultured overnight in the presence of myo[2-3H]inositol (1μCi/ml). Cells were preincubated (10 min, 37 0C) with HEPES-buffered saline containing 10 mM LiCl in a total volume of 200 μl and stimulated with CXCL8 or CXCL1 (100 nM) for 10 min. Supernatant was used to determine the release of ([3H]IPs). Data are represented as the fold stimulation over the basal. The experiment was repeated three times with similar results. D) For CXCL8-mediated internalization, cells were treated with CXCL8 (100 nM) at different times, washed and assayed for [125I]CXCL8 (0.1nM) binding. The values are presented as a percentage of totals, which is defined as the total amount of [125I]CXCL8 bound to control (untreated) cells. The experiment was repeated five times with similar results. Figure 3. Schematic illustration and surface expression of 2ECL (A2ECLB and B2ECLA) and 4TMD (A4TMDB and B4TMDA) chimeric mutants of CXCR1 and CXCR2. A) Gray traces represent CXCR1 and black traces CXCR2. B) FACS analysis of surface expression of the chimeric receptors in RBL-2H3 cells after staining with CXCR1 (A4TMDB and A2ECLB) or CXCR2 (B4TMDA and B2ECLA) specific antibodies. C) Competition binding of [125I]CXCL8 to A2ECLB, A4TMDB, and B2ECLA in the presence of different concentrations of CXCL8 (0-100 nM). Data shown are representative of one of three experiments performed in triplicate. Figure 4. Functional characterization and internalization of A2ECLB, A4TMDB and B2ECLA. CXCL8 and CXCL1-induced intracellular Ca2+ mobilization (A) phosphoinositide hydrolysis (B) and receptor internalization (C) were measured as described in the legend of Figure 2. The experiments were repeated three times with similar results. Figure 5. Mutation and characterization of the glycosylation motifs of the 2ECL of CXCR1 and CXCR2. A) Amino acid sequences of the 2ECL of CXCR1 and CXCR2 and the chimeric mutants exchanging the glycosylation motif of CXCR1 for those of CXCR2 (ASNVSB and AD194NB), and vice versa (BNNSSA and BN203DA). Boxed are the glycosylation motifs. CXCL8 and CXCL1-induced intracellular Ca2+ mobilization (B) and receptor internalization (C) were measured as described in the legend of Figure 2. The experiments were repeated three times. Figure 6. CXCL8-mediated internalization of point mutants of the 2ECL of CXCR1 and CXCR2. RBL-2H3 cells (5 x 105 cells /well) stably expressing CXCR1, CXCR2 or mutant receptors exchanging divergent amino acids of the 2ECL of CXCR1 for their CXCR2 counterpart (A), and vice versa (B) were treated with CXCL8 (100 nM) for 60 min, washed and assayed for [125I]-CXCL8 binding. The values are presented as a percentage of totals, which is defined as the total amount of The Kd values bound to control (untreated) cells. Data shown are representative of one of three experiments performed in triplicate.
11
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 7. Expression and characterization of BD199VA and BD199N mutants. A) FACS analysis of surface expression of CXCR2, BD199VA and BD199N in RBL-2H3 cells after staining with CXCR2 specific antibodies. Inset is the Kd values for BD199VA and BD199N. B) Time-course of CXCL8-induced receptor internalization was determined as described in the Figure 2 legend. The experiment was repeated four times with similar results. Figure 8. Phosphorylation of CXCR1, CXCR2, ABA, A4TMDB, B2ECLA, BD199VA and BD199N. 32P-labeled RBL-2H3 cells (5 x 106 cells / 60 mm plate) expressing CXCR1, ABA or A4TMDB (A) and CXCR2, BB2ECLA, BD199VA or BD199N (B) were incubated for 5 min with or without stimulants as shown. Cells were lysed, immunoprecipitated with specific antibodies against CXCR1 (A) or CXCR2 (B) and then analyzed by SDS-PAGE and autoradiography. The results are from a representative experiment that was repeated three times. Figure 9. CXCR1, CXCR2, BD199VA and BD199N-induced ERK1/2 phosphorylation. RBL-2H3 cells stably expressing CXCR1 (A), CXCR2 (B), BD199VA (C) and BD199N (D) were stimulated with CXCL8 (100 nM) for 0-20 min. ERK 1/2 phosphorylation and total ERK were determined by western blot using a phospho-ERK 1/2 (pERK) and total ERK 1/2 (tERK) specific antibodies. Figure 10. Computational modeling of 2ECL of rhodopsin, CXCR2, CXCR1, and the receptor mutants BD199VA, BD199N and AV190DB. Receptor interactions involving D190 of rhodopsin (A) and its corresponding residue D199 of the 2ECL of CXCR2 (B), V190 of CXCR1 (C) V199 of BD199VA (D) N199 of B BD199N (E), and D190 of AV190DB (F). The protein secondary structural elements are displayed as follow: α-helix, white; β-sheet, yellow; turn, cyan; and random coil, purple. Color coding of receptor residue atoms: green, C; red, O; and blue, N, respectively. H-bond forming residues (in stick depiction) in association with (A) D190 in rhodopsin (Y192, T193 and R177), (B) D199 in CXCR2 (T186, R208 and R185), (E) N199 in BD199N (T186 and R185), and (F) D190 in AV190DB (R199) are shown in white dotted line. No H-bond was formed in CXCR1 (C) and BD199VA (D). The residues within 3.0 Å of V190 (CXCR1) or V199 (BD199VA) are represented.
12
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Table I Ligand binding affinity of wild type and chimeric CXCR1 and CXCR2 expressed in RBL-2H3 cells. 125I-CXCL8 binding sites per cell and the apparent affinity binding (Kd) values for wild type and mutant receptors
Kd (nM) BBmax (receptors/ cell)
CXCR1 4.70 ± 0.095 13564 ± 679 AB1 4.09 ± 0.049 17682 ± 1820 ABA 1.95 ± 0.01 14974 ± 1109 CXCR2 2.48 ± 0.13 14092 ± 527 BA1 ND ND BAB ND ND
A4TMDB 2.3 ± 0.074 13608 ± 387 A2ECLB ND ND BB4TMDA ND ND BB2ECLA 5.0 ± 0.102 8995 ± 461
1
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Table II Expression and functional characterization of the 2ECL mutants of CXCR1 and CXCR2 expressed in RBL-2H3 cells. a Surface expression of wild type and 2ECL mutants of CXCR1 and CXCR2 were determined by FACS after staining with CXCR1 (A) or CXCR2 (B) specific antibodies. +, indicated the level of expression was that of wild type CXCR1 or CXCR2 and ND, not detectable. b Intracellular Ca2+ mobilization was measured upon stimulation with 100 nM CXCL8. +++, indicated Ca2+flux was similar to that of the wild type receptors (348±2.8 and 305±0.7 nM for CXCR1 and CXCR2 respectively); ++ (~200 nM) and ND, not detectable. The experiments were repeated twice with similar results. Receptors Expressiona Ca2+ Fluxb Receptors Expressiona Ca2+ Fluxb
CXCR1 + +++ CXCR2 + +++ AH179YB + +++ BBY188HA ND ND AP189SB + +++ BBS189PA + +++ AS183VB + +++ BBV192SA + +++ AV186AB + ++ BBA195VA + +++ AV190DB + ND BBD199VA + +++ AL191MB + +++ BBM200LA + +++ AK197NB + ++ BBN206KA + +++ AV201LB + +++ BBL201VA + +++
2
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Table III
Homologous desensitization of Ca2+ mobilization elicited by a first dose of CXCL8. RBL-2H3 cells (5 x 106 cells/assay) expressing wild type or mutants CXCR1 and CXCR2 were loaded with indo-1 and stimulated with CXCL8 or CXCL1 (100 nM). Cells were rechallenged 3 min later with a second dose of CXCL8 (100 nM) and peak intracellular Ca2+ mobilization was determined. Data are the means ± S.E. of three different experiments. _______________________________________________________________________
Cells/Treatment Peak Ca2+ Mobilization (nM) % Desensitization _______________________1st dose_______2nd dose___________________________ RBL-CXCR1 CXCL8 348 ± 2.8 17 ± 1.7 95 CXCL1 88 ± 13 47 ± 7.7 47 RBL-CXCR2 CXCL8 305 ± 0.7 0.0 ± 0.0 100 CXCL1 284 ± 23 104 ± 11 63 RBL-BD199VA CXCL8 299 ± 35 44 ± 0.7 85 CXCL1 200 ± 0.7 55 ± 8 73 RBL-BD199NCXCL8 225 ± 2.5 0.0 ± 0.0 100 CXCL1 212 ± 15 33 ± 4.9 84 _______________________________________________________________________
3
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Table IV
BBD199VA and BD199N mediated cross-desensitization of intracellular Ca mobilization elicited by CCR5. RBL-2H3 cells (5 x 10 cells/assay) co-expressing CCR5 along with CXCR1 (RBL-CXCR1-CCR5), CXCR2 (RBL-CXCR2-CCR5), B
2+
6
D199VA (RBL-BD199VA-CCR5) or BD199N (RBL-BD199N-CCR5) were loaded with Indo-1 AM and stimulated with a first dose of CXCL8 or CCL5 (100 nM). Cells were rechallenged 3 min later with a second dose of either CXCL8 or CCL5 (100 nM) and peak intracellular Ca mobilization was determined. Values for cross-desensitization are represented as percentage of inhibition of the Ca response elicited in the absence (first dose) of pretreatment. Data are the means ± SE of three different experiments.
2+
2+
Cells/Treatment Peak Ca2+ Mobilization (nM) % Cross-desensitization _______________________________________________________________________ RBL-CXCR1-CCR5 CXCL8→ CCL5 376 ± 15 → 149 ± 13 56 CCL5 → CXCL8 331 ± 9 → 210 ± 10 44 RBL-CXCR2-CCR5 CXCL8→ CCL5 296 ± 33 → 316 ± 29 2 CCL5 → CXCL8 322 ± 22 → 108 ± 17 65 RBL-BD199VA-CCR5 CXCL8→ CCL5 343 ± 19 → 186± 17 41 CCL5 → CXCL8 281 ± 27 → 202 ± 25 33 RBL-BD199N-CCR5 CXCL8→ CCL5 305 ± 24 → 283 ± 11 0 CCL5 → CXCL8 285 ± 16 → 117 ± 9 59
4
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Table V. Comparison of hydrogen bonding interactions for rhodopsin (D190), CXCR2 (D199), the mutant BD199N (N199), and the mutant AV190DB (D190).a
H-bond acceptor H-bond donor Rhodopsinb D190(Oδ2) R177(HNε) D190(Oδ2) R177(HNη2) D190(Oδ1) Y192(HN) D190(Oδ1) T193(HN) CXCR2c D199(O) R185(HN η2) N199(Nδ2) T186(HN) D199(Oδ1) T186(HOγ) D199(Oδ2) R208(HNε) BD199N N199(O) R185(HN η2) N199(Nδ2) T186(HN) T186(Oγ1) N199(HNδ) N199(Oδ1) T186(HOγ) AV190DB D190(Oδ2) R199(HNε) a H-bond acceptor and donor atoms are indicated by their positions from the Cα position. For example, D190 (Oδ2) indicates the second oxygen atom of the side chain carboxylate in the D190. b Two additional H-bonds as part of the H-bonding network are T193(HOγ)-R177(Nη2) and R177(HNη2)-T193(Oγ1). c Two additional H-bonds within H-bonding network are T186(Oγ)-R208(HNε) and T186(Oγ)-R208(HN η2).
5
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from
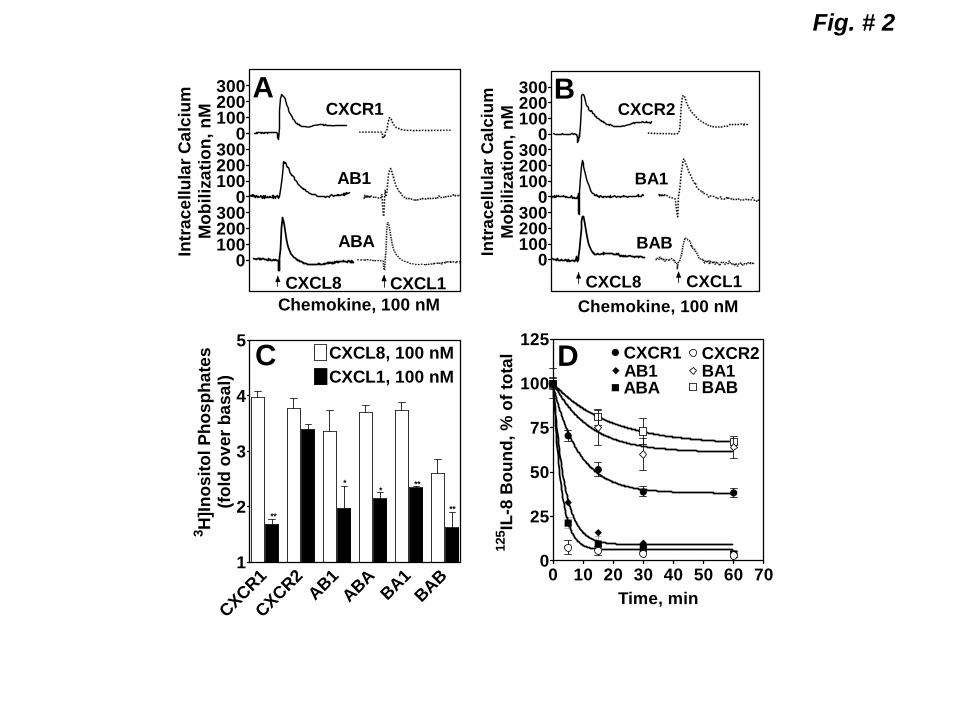
Fig. # 2
0100200300
0100200300
0100200300
A
CXCL8 CXCL1
CXCR1
AB1
ABA
Chemokine, 100 nM
Intr
acel
lula
r Cal
cium
Mob
iliza
tion,
nM
0100200300
0100200300
0100200300 B
CXCL8 CXCL1
CXCR2
BA1
BAB
Chemokine, 100 nM
Intr
acel
lula
r Cal
cium
Mob
iliza
tion,
nM
CXCR1
CXCR2
AB1ABABA1BAB
1
2
3
4
5CXCL8, 100 nMCXCL1, 100 nM
C
3 H]In
osito
l Pho
spha
tes
(fold
ove
r bas
al)
**
**
**
**
0 10 20 30 40 50 60 700
25
50
75
100
125CXCR1 CXCR2AB1ABA
BA1BAB
125 IL
-8 B
ound
, % o
f tot
al D
Time, min
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from
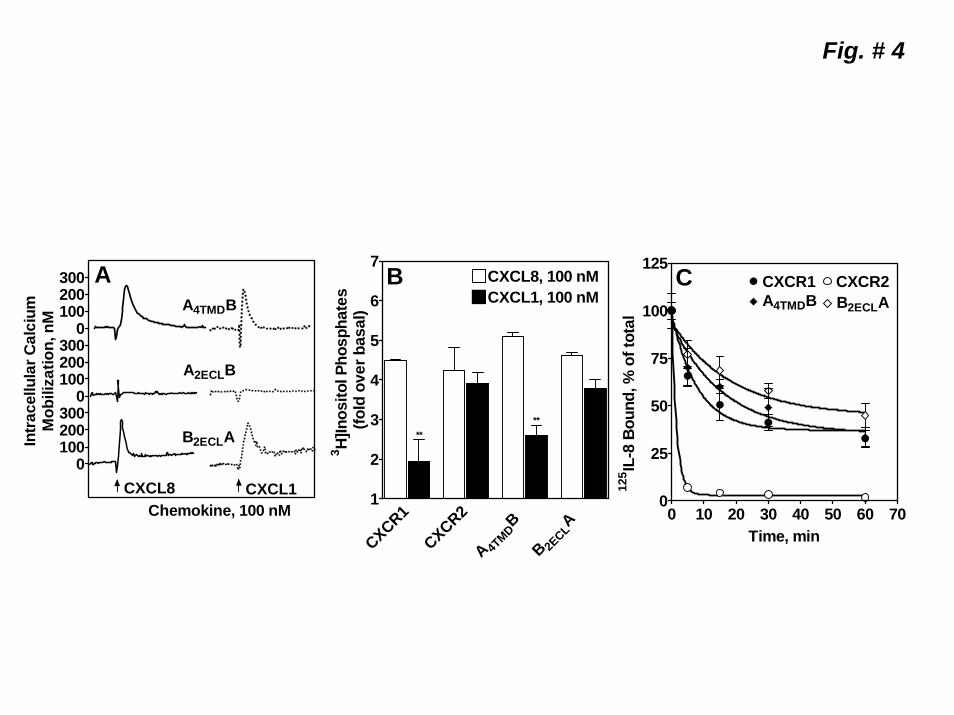
Fig. # 4
0100200300
0100200300
0100200300 A
CXCL8 CXCL1
A4TMDB
A2ECLB
B2ECLA
Chemokine, 100 nM
Intr
acel
lula
r Cal
cium
Mob
iliza
tion,
nM
CXCR1CXCR2A 4T
MDB
B 2ECLA
1
2
3
4
5
6
7
CXCL1, 100 nMCXCL8, 100 nMB
3 H]In
osito
l Pho
spha
tes
(fol
d ov
er b
asal
)
****
0 10 20 30 40 50 60 700
25
50
75
100
125
125 IL
-8 B
ound
, % o
f tot
al
C CXCR1 CXCR2A4TMDB B2ECLA
Time, min
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from

A). CXCR1 178 HPNNSSPVCYEVLGNDTAKWRMVLR 204 CXCR2 187 YSSNVSPACYEDMGNNTANWRMLLR 213ASNVSB 178 HPSNVSPVCYEVLGNDTAKWRMVLR 204AD194NB 178 HPNNSSPVCYEVLGNNTAKWRMVLR 204BNNSSA 187 YSNNSSPACYEDMGNNTANWRMLLR 213BN203DA 187 YSSNVSPACYEDMGNDTANWRMLLR 213
0100200300
0100200300
0100200300
0100200300
ASNVSB
AD194NB
BN203DA
BNNSSA
CXCL1CXCL8
Chemokine, 100 nM
B
Intr
acel
lula
r C
a2+ M
obili
zatio
n, n
M
0 10 20 30 40 50 60 700
25
50
75
100
125CXCR1CXCR2
BNNSSABN203DA
AD194NB
125 IL
-8 B
ound
, % o
f tot
al
C
Time, min
Fig. # 5
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from
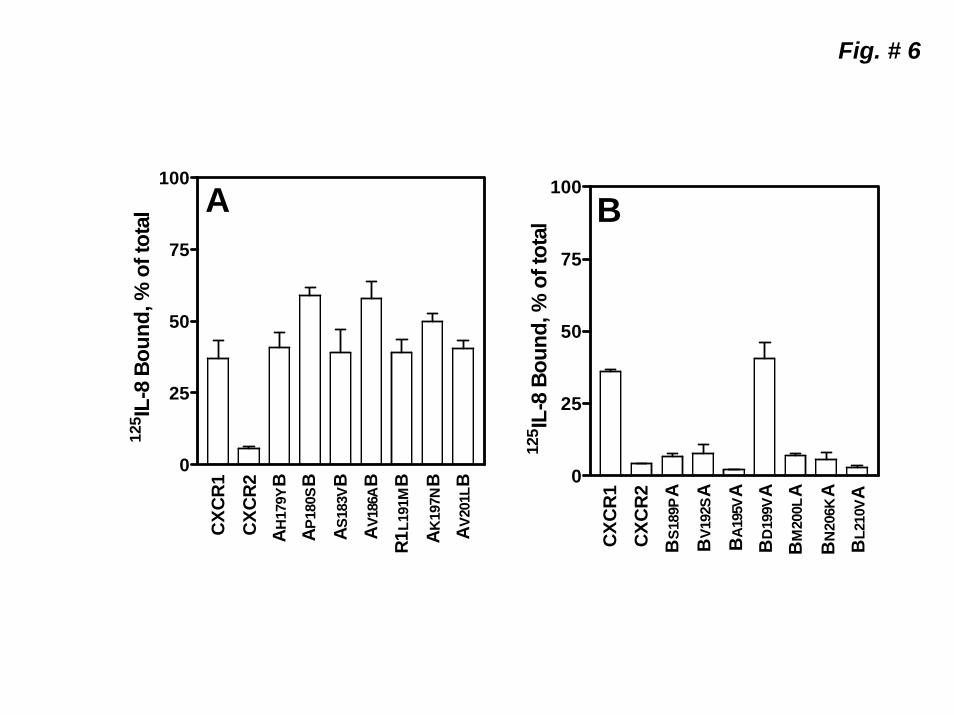
Fig. # 6
0
25
50
75
100
125 IL
-8 B
ound
, % o
f tot
al
B
CXC
R1
CXC
R2
BS1
89P
AB
V192
SA
BA1
95V
AB
D19
9VA
BM
200L
A
BN
206K
AB
L210
VA
0
25
50
75
100
125 IL
-8 B
ound
, % o
f tot
al
A
AH17
9YB
CXC
R1
CXC
R2
A P18
0SB
A V20
1LB
A S18
3VB
A V18
6AB
R1L1
91M
BA K
197N
B
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from

Fig. # 7
0
25
50
75
100
125
FITC
CO
UN
TS
RBL-2H3CXCR2BD199VABD199N
A
100 101 102 103 104
Cell Kd (nM) BD199VA 0.68 ± 0.29BD199N 2.33 ± 0.23
0 10 20 30 40 50 60 700
25
50
75
100
125CXCR1CXCR2BD199VABD199N
125 I
L-8
Boun
d, %
of t
otal
B
Time, min
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from

CXCR1 CXCR2 BD199VA
116→
52→
37→
kDa
PMA (100 nM) - - + - - + - - + - - + - - + - - + - - +
ABA
93→
BD199N
1 2 3 1 2 3 1 2 3A4TMDB B2ECLA
1 2 3
Fig. # 8
A) B)
1 2 31 2 31 2 3
CXCL8 (100 nM) - + - - + - - + - - + - - + - - + - - + -
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from
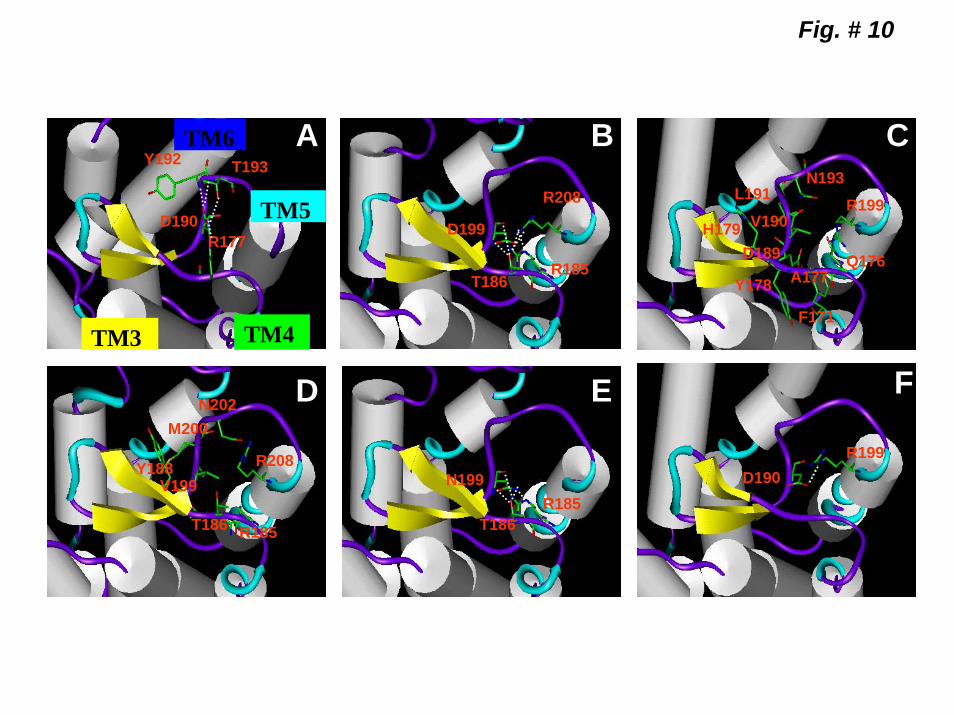
Fig. # 10
Y192
D190
T193
R177D199
R208
R185T186
N193
V190L191
D189H179
Y178 A177
F171
R199
Q176
V199Y188
M200N202
T186 R185
R208N199
T186R185
D190R199
TM6
TM5
TM4TM3
A B C
D E F
by guest on July 3, 2018 http://www.jbc.org/ Downloaded from

Joong-Youn Shim, Krishna Rajarathnam and Ricardo M. RichardsonMohd W. Nasser, Sandeep K. Raghuwanshi, Kimberly M. Malloy, Pavani Gangavarupu,
extracellular loop of CXCR2 in CXCL8-mediated rapid receptor internalizationCXCR1 and CXCR2 activation and regulation: Role of aspartate 199 of the second
published online January 4, 2007J. Biol. Chem.
10.1074/jbc.M610289200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from