Embed Size (px)
Citation preview

Crystal Structure of A Bifunctional Deaminaseand Reductase Involved in Riboflavin Biosynthesis
Bacterial RibG is an attractive candidate for development of antimi-crobial drugs because of its involvement in the riboflavin biosyn-thesis. The crystal structure of Bacillus subtilis RibG at 2.41-Å resolu-tion displayed a tetrameric ring-like structure with an extensiveinterface of ~2400 Å2 per monomer. The N-terminal deaminasedomain belongs to the cytidine deaminase superfamily. A structure-based sequence alignment of a variety of nucleotide deaminasesreveals not only the unique signatures in each family member forgene annotation, but also putative substrate-interacting residuesfor RNA-editing deaminases. The strong structural conservationbetween the C-terminal reductase domain and the pharmaceuti-cally important dihydrofolate reductase suggests that the tworeductases involved in the riboflavin and folate biosynthesesevolved from a single ancestral gene. Together with the binding ofthe essential cofactors, zinc ion and NADPH, the structural compari-son assists substrate modeling into the active-site cavities, allowingidentification of specific substrate recognition. Finally, the presentstructure reveals that the deaminase and the reductase are sepa-rate functional domains, and that domain fusion is crucial for theenzyme activities through formation of a stable tetrameric structure.
Flavin coenzymes are ubiquitous in all organisms because of theirinvolvements in central metabolic pathways. Plants and many microor-ganisms obtain the precursor riboflavin by biosynthesis, whereas ani-mals depend on nutritional sources. Numerous pathogenic microorgan-isms are unable to takeup flavins from the environment and hence areabsolutely dependent on their endogenous production. Therefore, theenzymes involved in riboflavin biosynthesis have the potential to be-come attractive candidates for the design of new defenses against anti-biotic-resistant pathogens.
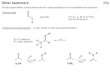
During riboflavin biosynthesis, GTP cyclohydrolase II first catalyzesthe hydrolytic C8 release of GTP to yield formate and pyrophosphate as
Biology
81
Beamline
BL17B2 Protein X-ray CrystallographybeamlineSP12B2 Protein X-ray Crystallographybeamline
Authors
S.-H. LiawNational Yang-Ming University,Taipei, Taiwan
Fig. 1: The deamination and reduction steps in the riboflavin biosynthesis.

side products. The product, 2, 5-diamino-6-ribo-sylamino-4(3H)-pyrimidinone 5’-phosphate (com-pound 1) is converted into 5-amino-6-ribitylamino-2,4 (1H, 3H)-pyrimidinedione 5’-phosphate (com-pound 4) by deamination of the pyrimidine ring andNAD(P)H-dependent reduction of the ribose (Fig. 1).The deamination and reduction steps have been
shown to proceed in the opposite order in yeast andE. coli. Most eubacteria contain a bifunctional pro-tein; for instance the Bacillus subtilis RibG (BsRibG) is composed of an N-terminal deaminase domain (D domain) and a C-terminal reductase domain (R domain). In contrast, in fungi, plants and mostarchaea, these two enzymes are separate. To gainstructural insights into the inhibitor design, substratespecificity and evolution, we have solved the BsRibGstructure at 2.41-Å resolution.
Fusion of Two Enzyme Domains in RibG
Analytical ultracentrifugation experiments clear-ly demonstrated that BsRibG exists as a tetramer insolution as well as in crystal form, where the enzymeforms a tetrameric ring-like structure (Fig. 2). Mole-cules A and B interact with each other through theirD domains, while molecule A makes extensive con-tacts with molecule C through their R domains. Thereare no contacts between molecules A and D in thetetramer, and the D and R domains make only a fewcontacts. There is no evidence for any dependence ofthe active sites within the RibG tetramer. Previousdeletion mutants have demonstrated that the N-ter-minal 147 residues and the C-terminal 248 residuesof BsRibG were sufficient for their respective enzymeactivities. However, these truncated proteins couldnot be isolated due to poor stability. Therefore, eventhough the two enzyme domains can fold indepen-dently, the domain fusion is crucial for the enzymeactivities through formation of a stable tetramericstructure.
Structural Conservation and Divergence in the CDASuperfamily
The D domain belongs to the cytidine deaminase(CDA) superfamily, which consists of the mononu-cleotide deaminases involved in nucleotide metabo-lism, and the RNA(DNA)-editing deaminases involvedin gene diversity and in anti-virus defense. Thesedeaminases catalyze the hydrolytic deamination ofcytosine, guanine, adenine moieties and several oftheir therapeutically useful analogues. Detailed struc-tural comparisons reveal a common three-layered α/β/α structure with the five β-strands (β1-β5)and three helices (αA -αC) (Fig. 3(A)). The active-sitearchitecture of the D domain resembles those of theCDA members, which share a similar zinc-assisteddeamination mechanism. The substrate was then mo-deled into the active site through superposition of
Biology
82
Fig. 2: Structure of BsRibG. Ribbon views of thetetramer (A) and monomer (B). The tightly boundzinc ion in the D domain is shown as a sphere(magenta) with the cofactor NADPH and themodeled substrates as ball-and-stick represen-tations. Molecules A-D in the tetramer and theinter-domain helices are colored in red, black,blue, green, and magenta, respectively. The D andR domains are made up of a central β-sheetflanked by α-helices, and have few contacts witheach other. The two enzyme domains can foldindependently, however, the domain fusion iscrucial for the enzyme activities through forma-tion of a stable tetrameric structure.

the nucleobase rings because of the highly con-served interaction networks surrounding the targetamino group.
In contrast, the C-terminal segment beyond theβ4 strand, is quite diverse and may make a majorcontribution to the structural plasticity and func-tional diversity among the CDA members (Fig. 3(A)).For instance, yeast cytosine deaminase (yCD) and thet-RNA specific adenosine deaminase from Aquifexaeolicus (AaTADA) unexpectedly superimpose verywell, with a rmsd of 1.08 Å for 115 Cα atoms with22% sequence identity. The major structural differ-ence around the active-site cavity is the C-terminalhelix. In yCD as well as Bacillus subtilis guanine de-aminase (BsGD), genetic changes to alter the substratespecificity is through an introduction of substrate-recognition residues at the C-terminal tail (Asp155 inyCD and Tyr156 in BsGD), which then forms a “flap”capping and hence narrowing the opening of theactive-site cavity upon substrate binding to limit thepocket size for the nucleobase. In contrast, the C-ter-minal tail in AaTADA as well as CDA, dCMP deami-nase, and RibG, swings away to enlarge the active-site cavity for their larger substrates.
In combination with a sequence-structure analy-sis, a structure-based sequence alignment of theCDA members was constructed, which reveals uni-que member signatures useful for gene annotationand an optimal alignment for comparative modeling.To date, apolipoprotein B mRNA-editing catalyticsubunit 1 (APOBEC1) and its sequence homologues,activation-induced deaminase (AID), APOBEC2 andthe tandem repeats APOBEC3A to 3H, are the onlyidentified C-to-U RNA (DNA)-editing deaminases inhumans. AID is an essential B cell-specific factorrequired for antibody maturation, while severalAPOBECs are involved in defense against a broadrange of retroviruses. We have modeled residues 1-160 of human AID into a CDA fold, which revealsthe possible effects of the AID loss-of-function pointmutants of AID in patients with hyper-IgM syndrometype 2.
Structural Conservation and Divergence betweenBsRibG and DHFRs
A structural homology search by DALI revealedthat the R domain of BsRibG displays significant struc-tural similarity to dihydrofolate reductase (DHFR)(Fig. 3(B)). DHFR catalyzes the NADPH-utilizing reduc-tion of DHF to tetrahydrofolate. The R-R structure ofBsRibG is similar to the highly stable dimeric Thermo-toga maritime DHFR (TmDHFR). Structural superposi-tion of BsRibG and TmDHFR reveals a rmsd of 1.5 Å
Biology
83
Fig. 3: (A) Structural superposition of BsRibG(red), yCD (blue), T4-bacteriophage dCMP deami-nase (green), and AaTADA (yellow). The zinc ion isdisplayed as a sphere (magenta) with the yCDinhibitor, 3,4-dihydrouracil (DHU) (cyan) as ball-and-stick representations. These deaminsesshare the conserved β-sheet and helices αA -αC, even a part of the αE helix. T4dCMPD con-tains a ~60-residue insertion, which folds intotwo helices (αB’ and αB’’) and flexible loops.The C-terminal tail of yCD folds backward to limitthe pocket size, whereas those of the remainingmembers swing away to enlarge the active-sitecavity. (B) Structural superposition of the Rdomain of BsRibG (red) and TmDHFR (green). TheR domain contains an extra ~25-residue inser-tion, which folds into the αD’ helix and the βD’strand.

for 120 Cα atoms with 25% sequence identity. Thestrong conservation of the tertiary structures sug-gests that these two reductases involved in the ribo-flavin and folate biosyntheses are descended from asingle ancestral gene and thereby define a new super-family.
The enzymatic mechanism of DHFR has beenchemically and structurally studied in detail. Simi-larly, RibG is expected to catalyze the reduction of acyclic ribosyl into an open ribityl group by hydridetransfer from the C4 atom of the nicotinamide ring ofNAD(P)H to the C1’ of the ribose with concomitantprotonation of O5’. The binding architecture of thenicotinamide ring is virtually identical in DHFR andRibG. Thus, based on the structural comparison, thesubstrate was modeled into the active site withsubsequent energy minimization. These substrate-binding residues are highly conserved in the reduc-tases and located in similar spatial positions as thosein DHFRs. Many DHFR inhibitors such as methotrex-ate, pyrimethamine and trimethoprim, have longbeen used clinically in the treatment of cancer, rheu-matoid arthritis, malaria, and bacterial and fungalinfection. Therefore, the R domain may become animportant target for new drug design.
Biology
84
Experimental Sation
Protein Crystallographic end station
Publications
.S. C. Chen, Y. C. Chang, C. H. Lin, C. H. Lin,and S. H. Liaw. J. Biol. Chem. 281, 7605(2006)..S. H. Liaw, Y. J. Chang, C. T. Lai, H. C. Chang,
and G. G. Chang. J. Biol. Chem. 279, 35479(2004)..T. P. Ko, J. J. Lin, C. Y. Hu, Y. H. Hsu,
A. H. J. Wang, and S. H. Liaw. J. Biol. Chem.278, 19111 (2003).
Contact e-mail