Embed Size (px)
Citation preview

Thrombosis and Haemostasis 110.5/2013 © Schattauer 2013
910
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
Theme Issue Article
Crosstalk between platelets and the complement system in immune protection and diseaseAdmar Verschoor1; Harald F. Langer2
1Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Munich, Germany; 2Medizinische Klinik III, Klinik für Kardiologie und Kreislauf -erkrankungen, Eberhard Karls-Universität Tübingen, Tübingen, Germany
SummaryPlatelets have a central function in repairing vascular damage and stopping acute blood loss. They are equally central to thrombus formation in cardiovascular diseases such as myocardial infarction and ischaemic stroke. Beyond these classical prothrombotic diseases, immune mediated pathologies such as haemolytic uraemic syndrome (HUS) or paroxysmal nocturnal haemoglobinuria (PNH) also feature an increased tendency to form thrombi in various tissues. It has become increasingly clear that the complement system, part of the innate im-mune system, has an important role in the pathophysiology of these diseases. Not only does complement influence prothrombotic disease, it is equally involved in idiopathic thrombocytopenic purpura (ITP), an
autoimmune disease characterised by thrombocytopenia. Thus, there are complex interrelationships between the haemostatic and immune systems, and platelets and complement in particular. Not only does complement influence platelet diseases such as ITP, HUS and PNH, it also mediates interaction between microbes and platelets during sys-temic infection, influencing the course of infection and development of protective immunity. This review aims to provide an integrative overview of the mechanisms underlying the interactions between complement and platelets in health and disease.
KeywordsPlatelets, complement, inflammation, infectious disease, ITP, HUS, PNH
Correspondence to:Admar Verschoor, PhDInstitute for Medical Microbiology, Immunology and HygieneTechnische Universität München, Munich, GermanyE-mail: [email protected] toHarald F. Langer, MDDepartment of Cardiology and Cardiovascular MedicineUniversity Clinic of Tuebingen, Tuebingen, GermanyE-mail: [email protected]
Received: February 5, 2013 Accepted after major revision: July 15, 2013 Prepublished online: September 5, 2013
doi:10.1160/TH13-02-0102Thromb Haemost 2013; 110: 910–919
1. Introduction1.1 Platelet function in health and diseaseThe classical function of platelets is to cover and close endothelial and tissue wounds (1, 2). In addition to this temporary wound clo-sure, which is followed by stable thrombus formation, platelets contribute to long-term healing and regenerative mechanisms (3-5). However, if platelet activation occurs without proper regu-lation or at unfavourable locations, the intended platelet function to form a thrombus can result in life-threatening events such as myocardial infarction, stroke, or when vulnerable atherosclerotic plaques rupture, and arteries to the brain occlude (6-8).
The process of platelet thrombus formation is a rather well understood and characterised process with distinct and strictly regulated steps (9-11). It involves initial loose contact between the platelet and the injured vessel wall, firm adhesion, spreading and finally thrombus formation (12, 13). The initial loose contact of platelets with the subendothelial matrix is mediated by platelet gly-coprotein (GP) Ib-V-IX with von Willebrand Factor (vWF) (14, 15). Then, the glycoprotein receptor GPVI mediates interaction of
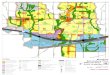
the platelet with collagen and, indeed, anti-GPVI monoclonal anti-bodies or blockade with soluble GPVI attenuates arterial thrombo-sis in vivo (16, 17). GPVI can directly promote adhesion of pla-telets to subendothelial collagen and activate further platelet ad-hesion receptors including GPIIb-IIIa (αIIbβ3) and GPIa-IIa (α2β1) (11). α2β1 represents a second collagen receptor while αIIbβ3 mediates irreversible adhesion to vWF (6). Subsequent in-side-out signalling mediates further platelet activation, and the re-sulting shape change and pseudopodia formation allow for effec-tive coverage of the injured vessel wall. Secondary mediators such as thromboxane A2 (TxA2) or adenosine diphosphate (ADP) pro-vide an amplification loop that initiates and reinforces further pla-telet aggregation (12). The latter is achieved through fibrinogen-mediated contacts between activated platelet receptors on individ-ual platelets (15, 18, 19). ▶ Figure 1 summarises the process of pla-telet thrombus formation.
During the last decades it has become increasingly evident that the relevance of platelets is not restricted to wound healing or thrombus formation, but that non-classical platelet functions con-tribute to inflammation and infection. Early insights into the pro-
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

© Schattauer 2013 Thrombosis and Haemostasis 110.5/2013
911Verschoor, Langer: Platelets and complement
motion of inflammation by platelets came from studies that recog-nised that atherosclerosis is in essence an inflammatory disease (20, 21). Platelet adhesion to endothelial cells was found to be cru-cial to the initiation of atherosclerosis, even before the actual plaque has formed (21). It was reported that platelets release che-mokines such as CCL5 or CXCL4 which contribute to athero-sclerosis, through a P-selectin-dependent process (20). A number of subsequent studies confirmed the early hypothesis that platelets contribute to vascular inflammation, e.g. by interacting with leu-kocytes (22-25). Moreover, interacting receptor / ligand pairs on platelets and leukocytes were identified (26-28). For instance, P-se-lectin glycoprotein ligand-1 (PSGL-1) on leukocytes mediates binding to platelet P-selectin (28), while leukocyte beta2-integrin Mac-1 (CD18/CD11b, CR3) promotes interaction with at least two platelet receptors, GPIb and the junctional adhesion molecule-C (23, 29-31). Platelet GPIIb/IIIa recruits leukocytes via formation of fibrinogen bridges with leukocyte Mac-1, an interaction triggered by platelet activating factor (32). For a schematic overview of leu-kocyte-platelet interactions, see the article by Langer and Chavakis in this Theme Issue (124). A further prominent mechanism by which activated platelets contribute to inflammation is the release of active biomolecules, such as interleukin (IL)-1beta, platelet fac-tor 4 or Rantes from platelet granules (33, 34). Recent research draws renewed attention to some of these processes, which were previously considered exclusively in the context of thrombus formation of classical cardiovascular diseases such as stroke or myocardial infarction, but are now being revisited in the context of infection (35), multiple sclerosis (36) or rheumathoid arthritis (37). Studies of prothrombotic or thrombocytopenic diseases such as idiopathic thrombocytopenic purpura (ITP), thrombotic thrombocytopenic purpura (TTP) or haemolytic uraemic syn-drome (HUS) likewise led to a deeper understanding of platelet
physiology, particularly their interaction with the complement sys-tem (38, 39).
1.2 The complement system
The complement system is a largely blood-borne cascade of pro-teins with its evolutionary roots in innate immune defence and homeostasis. Its activation is controlled by zymogens, proteolyti-cally activated enzyme precursors, which set in motion an expan-ding and self-sustaining array of effector molecules that have the potential to induce inflammation, tag (opsonise) targets for pha-gocytosis or directly lyse cells. The complement system’s ability to mark targets for destruction and removal plays not only a role in the combat of invading microbes, but also in maintaining homeos-tasis, for instance via the controlled clearance of dead and dying cells.
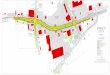
The biological effects of complement activation are broad, reaching into adaptive immunity, and are mediated in part via spe-cific receptors for complement activation products (complement receptors [CR]), and in part directly by the activation products themselves. Several complement activation pathways are recog-nised, differing in their modes of initiation, progression and mol-ecules involved. These are briefly reviewed and schematically rep-resented in ▶ Figure 2 to provide a frame of reference. For a further in depth description of the complement system, the reader is referred to (40).• The Alternative Pathway (AP) may be the most fundamental
and anciently preserved cascade of the complement system. It functions as a self-sustaining loop of activation, initiated by spontaneous conversion of the reactive internal thioesther within the central component C3. Generally, this reactivity is quickly neutralised in the aqueous environment of the blood,
Figure 1: Mechanisms of thrombus formation. Under physiological flow platelets do not adhere to the endothelial monolayer. Upon vascular injury pla-telets tether to the subendothelial space, become firmly adherent and finally form a thrombus by recruiting further platelets via activated GPIIbIIIa (αIIbβIII) and fibrinogen bridges. Involved receptor ligand pairs are depicted. vWF = von Willebrand factor. GP = glycoprotein. ECs = endothelial cells.
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

Thrombosis and Haemostasis 110.5/2013 © Schattauer 2013
912 Verschoor, Langer: Platelets and complement
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
resulting in an inactive form of C3. However, if a suitable hy-droxyl or amine acceptor group is present within close proxim-ity of the reactive thioesther, the resulting reaction can form a covalent bond that effectively tags the target with activated C3 (C3b). The self-sustaining nature of the AP stems from the fact that C3b becomes part of its own conversion complex (conver-tase) causing focussed activation of ever-more C3 molecules around the site of initial deposition. Obviously, hydroxyl or amine acceptor groups are not unique to foreign substances or microbes and equally occur in self molecules and on host cells. Therefore, the expression of complement regulatory molecules (CRM; also commonly referred to as complement regulatory proteins) limit activation on self-surfaces, while allowing unre-stricted complement activation on non-protected foreign or al-tered-self substances.
• The Classical Pathway (CP) forms a functional interface be-tween the complement system and antibodies of adaptive im-munity, thus providing the innate complement system with a mode of antigen specific activation. Once bound to its cognate antigen target, slight conformational changes in the antibody Fc-portion allow complement C1q to bind it. C1q can also bind selected targets directly or via intermediates including C-reac-tive protein (CRP), serum amyloid protein (SAP) or pentraxins without the need for antibody. Bound C1q proteolytically acti-vates serine proteases C1r and C1s which in turn (and anal-ogous to the activation of C3) convert C4 to C4b. Together with activated C2, C4b now forms the classical C3 convertase. Once C3 becomes activated and deposited by this convertase, it can feed into the AP, which further amplifies complement acti-vation.
Figure 2: Schematic overview of the complement system. Classical, Lectin and Alternative Pathways (CP, LP, and AP, respectively) commence from the left side of the diagram, leading to the converging point of C3-activation (top right). At each step of the cascade, new additions to the antigen (Ag) complex are shown in blue, their position taken within the complex shown in red, and released small proteolytic products are shown in green. With the convergence of the three initiation pathways at the level of C3 activation (top right), a C3 Amplification Loop is generated via the AP. Furthermore, at this point, also the Terminal Pathway is initiated with the formation of a C5
convertase, leading to the assembly of the membrane attack complex (MAC). The MAC ultimately interferes with the target cell’s structural integrity by penetrating its membrane (bottom right), while opsonisation with C3 acti-vation products provides complement receptor-expressing immune cells with a ligand to interact with the Ag. Several of the small proteolytic products (green) function as anaphylatoxins in the recruitment and activation of a broad range of immune defence mechanisms, including platelet activation. MBL (mannose binding lectin); MASP (MBL-associated serine proteases).
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

© Schattauer 2013 Thrombosis and Haemostasis 110.5/2013
913Verschoor, Langer: Platelets and complement
• The Lectin Pathway (LP) follows much the same principle of ac-tivation as the CP, short of its initiation, which in the LP is me-diated via a distinct set of zymogens which take the place of the C1 complex. To initiate the LP, mannose binding lectin (MBL) or ficollins must bind their target (typically specific sugar resi-dues) to activate MBL associated serine proteases (MASPs). Structurally, the LP initiation complex shows great similarity to the C1 complex of the CP, and also further complement acti-vation progresses via C4 and C2 as in the CP.
• The Terminal Pathway (TP). The aforementioned initiation pathways merge with the formation of their respective C3 con-vertases, and the pathways now progress into the formation of the membrane attack complex (MAC), a molecular structure that effectively forms a pore into the target membrane. The complex is initiated by the activation of C5 via the C5 conver-tase (a further modification of the C3 convertases), which re-cruits C6, C7, C8 and multiple copies of C9, which ultimately forms the pore, preventing the target cell to maintain osmotic pressure and functionality.
The unique modes of pathway initiation (i.e. microbe-specific antibody mediated or microbe-specific sugar residue-specific) provide the complement system with a degree of specificity to-wards its targets. Still, complement activation in the fluid phase creates potential for diffusion of activated products and non-inten-tional collateral damage to by-stander targets, such as neighboring self-cells. To limit such damage to the host, and to focus the des-tructive capacity of the complement system on the intended target, the system is tightly controlled via CRM which are present in the serum as well as on all host cells. CRM operate on various levels of complement activation, varying from destabilisation of the conver-tases to preventing the formation of a MAC on self-cells.
Apart from its functions in marking targets for phagocytosis via complement opsonisation and lysis via the MAC, the complement system alerts and supports other areas of the immune system. Pha-gocytic cells such as granulocytes, monocyte/macrophages and dendritic cells express high levels of phagocytic CR3 (CD11b) and CR4 (CD11c) with specificity for iC3b (the opsonising but inacti-vated form of C3b). Also the B cell co-receptor complex includes a CR, CR2 (CD21, with specificity for iC3b and its further proteo-lytic products C3d and C3dg), lowering the B cell activation threshold to antigen several orders of magnitude (41), with crucial effects during the response to infection (42, 43). Thus, opsoni-sation of a particular target with the proteolytic products of C3 en-ables a more effective antibody response than to the non-opso-nised target alone, in part also due to the expression of CR2 on fol-licular dendritic cells, which direct and orchestrate the B cell re-sponse (44). Activation of C3 or C5 generates small peptide-like split products, C3a and C5a respectively, also known as anaphylat-oxins. They have been shown to influence processes as diverse as angiogenesis (45) or asthma (46), histamine release or vasocon-striction (47), and generally mediate leukocyte activation and re-cruitment (48). C3a and C5a are recognised by C3aR and C5aR re-ceptors on granulocytes, mast cells and monocyte/macrophages.
1.3 Modes of interactions between the complement system and platelets
Complement and platelets share several levels of interaction. That after years of targeted research new points of interaction are regu-larly being discovered may in part be due to the fact that the sys-tems are typically addressed by separate groups of scientists and clinicians. The complexity of the cascades and their mechanisms of activation or regulation complicate the understanding further. However, it is clear that platelets do maintain a range of CR, as well as CRM, hinting at the importance of the complement system for their function and physiology. Moreover, it has been demonstrated that not only receptors, but also various complement factors can be detected on the platelet surface (49) and that thrombin acti-vated platelets initiate the complement cascade (50). Engagement of CR2 (receptor for iC3b C3d and C3dg) was reported to activate human platelets (51), while the iC3b receptor CR3 promotes arac-hadonic acid-induced platelet aggregation (52). Expression of iC3b receptor CR4 has also been reported (53), but its physiological role remains unclear. Not only C3-fragment receptors have been de-scribed on platelets, also C1q-receptor expression has been docu-mented, specifically gC1qR/p33 and cC1qR with affinity for the C1q globular heads and its collagenous tail, respectively, mediating platelet aggregating and activating effects (54-56). There are also strong indications that anaphylatoxin C3a and C5a receptors are present on human platelets, and C3a and its derivative C3adesArg induce platelet activation and aggregation in vitro, whereas in vivo data are not available to date (57, 58) Interaction between platelets and complement may also occur via receptors not typically re-garded as CR, such as P-selectin (57). Del Conde et al. could dem-onstrate that P-selectin binds C3b and this interaction resulted in both C3a generation and C5b-9 MAC complex formation (59). Formation of MAC-complexes further enhances the platelet and haemostatic response (60) and mice lacking C3 coincidentally show prolonged bleeding times (56). Combined, the general pic-ture emerges in which engagement of complement-receptors on platelets may have a platelet-activating function.
The pro-thrombotic effect of complement may obviously have advantages to the host in times of vascular damage and possibly associated infection, when both immune and haemostatic systems are simultaneously called upon and cross-talk enhances the re-sponse. A recent and novel example of interplay between comple-ment and platelets is reviewed in 4. Complement and platelets dur-ing systemic infection. This mechanism, involving complement C3 and platelet glycoprotein GPIb, was shown to have a strong en-hancing effect on the immune response to systemic bacterial infec-tion in vivo.
Nonetheless, pro-immune and pro-thrombotic interplay be-tween complement and platelets needs to remain focused on vas-cular damage, insult or infection and is to be avoided under nor-mal homeostatic conditions. Still, also under resting conditions platelets are continuously exposed to blood proteins, including complement, with the AP affording the complement system with a continuous, baseline-level of activation. This ever-present low-level complement activity requires a counter-balance on the pla-
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

Thrombosis and Haemostasis 110.5/2013 © Schattauer 2013
914 Verschoor, Langer: Platelets and complement
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
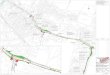
telet surface to avoid complement-mediated platelet activation, ag-gregation or even MAC mediated lysis. This complement restrain-ing action is provided by various CRM on human platelets, includ-ing C3b-inactivating and C3-convertase destabilising Factor H (bound via alphaIIbbeta3 [61] and Thrombospondin-1 [62]). Fur-thermore, the same functions are mediated on platelets by mem-brane co-factor (MCP or CD46, C3b inactivation) (63), decay-ac-celerating factor (DAF or CD55, interfering with C3 convertase) (64) and CD59, which destabilises the MAC (65). Similar roles have been reported for CRM on mouse platelets, where Crry (a CR1-related murine CRM), DAF and factor H protect platelets from complement destruction (66). In ▶ Figure 3 we provide a shematic representation that summarises several modes of cross-talk between complement compounds and the platelet surface. In the following, we will review the current understanding and mo-lecular underpinnings when an imbalance between the action of CRM-mediated protection and complement activation arises in persons with low expression of or mutations in CRM (see 2. Com-plement in diseases with a prothrombotic state), or in the presence of CP-activating autoantibodies to specific self-targets (see 3. Complement in immune thrombocytopenia).
2. Complement in diseases with a prothrombotic state
In HUS or PNH, involvement of the complement system and pla-telet activation have been firmly established.
Two forms of HUS are recognised: HUS and atypical HUS (aHUS). Whereas HUS is caused by infection with bacterial strains such as Escherichia coli which produce cytotoxins (67), atypical HUS (aHUS) is a genetic or acquired disorder which is connected to a dysregulated complement system (68, 69). Still, both consti-tute diseases featuring microvascular thrombosis with subsequent thrombocytopenia, haemolytic anaemia and dysfunction of af-fected organs, accompanied by complement activation (38). For reasons of simplicity, we summarise both diseases as HUS.
Platelet function in HUS is modulated by various synergistic ef-fects including endothelial cell activation, and complement acti-vation shifts the disease phenotype from a “thromboresistant” state (preventing thrombosis on the endothelial monolayer) to a “pro-thrombotic” state (see also 1.3. Modes of interactions between the complement system and platelets) (68). The prothrombotic phe-notype results from synergy between the release of P-selectin, vWF and Weibel-Pallade bodies (70) and the C3a or C5a-mediated up-regulation of adhesion receptors and tissue factor on endothelial cells (71). Distinct mutations uncovered in recent years offer a mechanistic basis for the prothrombotic state of HUS patients. The net effect of aHUS-associated mutations is an increased suscepti-bility to uncontrolled activation of the AP on self-cell surfaces, re-duced circulating C3 levels in a subset of patients, deposits of C3 and C5b-9 MAC on glomerular endothelium and on circulating platelets (38). When platelets are exposed to sera from aHUS pa-tients with Factor H mutations C3 and C9 deposition on platelets associated with CD40L and P-selectin expression - markers of pla-telet activation -, aggregate formation, and generation of tissue fac-
Figure 3: Modes of crosstalk between complement compo-nents and the platelet surface and potential relevance for disease. Under homeostatic condi-tions, complement regula-tory molecules (CRM) pre-vent excessive comple-ment deposition and acti-vation on the platelet sur-face. Under pathological conditions, uncontrolled complement activation on platelets or insufficient counterbalance by CRM results in platelet dam-age, thrombosis and dis-ease progression.
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

© Schattauer 2013 Thrombosis and Haemostasis 110.5/2013
915Verschoor, Langer: Platelets and complement
tor-expressing microparticles can be observed (72). In contrast, complement deposition and platelet activation were reduced when unmutated Factor H was preincubated with platelets and minimal when normal serum was used (72). Most of the mutations of Fac-tor H are located in the exons coding for the C-terminal region of the protein (73), which is responsible for binding to the cell surface or surface-associated C3b (74). Mutant Factor H (75) as well as factor H neutralised by HUS-associated autoantibodies (76, 77) cannot exert functions preventing complement activation. On pla-telets, C-terminal bound factor H prevents complement activation, as treatment of platelets with mutated factor H resulted in comple-ment propagation with subsequent platelet activation (72). Specifi-cally, for the FH-E1198 Stop mutation within short consensus re-peats of the C-terminal region a reduced binding to platelets and, thus, lack of complement inhibition on the cell surface could be demonstrated (72). Also mutations unrelated to the FH-E1198 Stop mutation but resulting in dysfunctional factor H or reduced factor H serum levels led to increased platelet activation (72). Be-sides factor H mutations, MCP mutations which affect extracellu-lar domain involved in complement regulation, can cause aHUS and result in reduced MCP expression or decreased binding to C3b and reduced cofactor activity (78). Moreover, mutations in complement factor I located in the protease domain are relevant for defective C3b inactivation (79). Gain-of-function mutations in C3 or complement factor B (80, 81) can produce hyperactive regu-lation-resistant C3-convertase leading to HUS. Also, mutants af-fecting the complement inhibitory molecule thrombomodulin have diminished capacity to inactivate C3b via complement factor H and complement factor I (82). The collective evidence points to-ward a model in which mutations in CRM that down-modulate complement activity or, vice versa, complement mutations that re-sist down-modulation lead to dysregulation of normal platelet ac-tivation, resulting in a hyperactive and pro-thrombotic state.
Similar to HUS, PNH is also characterised by a prothrombotic state. PNH is a rare, acquired stem cell disorder, which becomes clinically apparent with haemolytic anaemia, bone marrow failure and thrombophilia with high probability of thrombosis (39). The manifestations of thrombosis include visceral thrombosis (e.g. of hepatic veins and mesenteric veins), cerebrovascular thrombosis and pulmonary embolism (83). PNH is caused by a lack of several proteins on the blood cell surface (84-86) due to a mutation in the X-linked phosphatidylinositol glycan class A (PIGA) gene (87, 88), which is necessary for the biosynthesis of the glycosyl phosphati-dylinositol (GPI) anchor (89, 90). This mutation results in a lack of complement regulators DAF (91, 92) and CD59 (93, 94) anchoring to the platelet surface. Absence of these CRM is are also closely as-sociated with haemolytic anaemia (39). It was demonstrated that PNH platelets differ from normal platelets in their interaction with activated complement components (95). After alternative comple-ment activation, greater amounts of C3 are present on the platelet surface and, in contrast to wild type platelets, PNH platelets show release of serotonine after C3 fixation (95). Moreover, upon expo-sure to MAC, PNH platelets lacking CD59 have more expression of coagulation factor Va, present more catalytic surface for the pro-thrombinase complex (VaXa) and have increased prothrombinase
activity compared to healthy platelets (96). Such observations closely link to the observed increased thrombosis in many PNH patients. In contrast, some PNH patients present with reduced pla-telet counts and their platelets reveal reduced reactivity regarding central functions such as clot formation, adhesion and aggre-gation. Considering prothrombotic nature of PNH, it may be con-cluded that this platelet hyporeactivity may be due to reactive down-regulation of function in response to chronic hyperstimu-lation, a form of “platelet exhaustion” (97).
A study with 187 PNH patients treated with Eculizumab (Sol-iris, Alexion Pharmaceuticals), a humanised monoclonal antibody which inhibits cleavage of C5 into C5a and C5b and preventing MAC-mediated cell damage, revealed clearly reduced thrombosis. The study showed a 85% reduction in thrombosis incidence using Eculizumab (98), but could not establish at what level (C5a, C5b or MAC) Eculizumab exerts its effect on platelet activation and thrombosis (39). Future studies will have to clarify exactly how complement influences platelet function and activation in HUS and PNH.
Beside HUS and PNH there are other diseases where interplay between complement and thrombotic responses may be relevant, such as systemic lupus erythematodes (SLE) and TTP. Based on the increasing mechanistic understanding, there is ongoing dis-cussion if TTP and HUS should be combined under the term “thrombotic microangiopathies” (38). The same is true for ITP (see 3. Complement in immune thrombocytopenia), which shares mechanistic features with TTP and HUS, including complement-induced platelet alterations with consequences for blood homeos-tasis. ITP and TTP, however, also have features that are decidedly distinct from HUS: ITP and TTP are autoantibody mediated auto-immune diseases that coincide with other clinical constellation such as HIV-infection or systemic lupus erythematodes. Both are characterised by thrombocytopenia and both present with abrupt onset of manifestation (99). Still, whereas ITP is associated with platelet lysis, TTP links to functional platelet alterations. More-over, TTP patients have, in contrast to ITP patients, measurable activation of platelets and endothelial cells (99). Therefore, further research is needed to classify the diseases in detail and to provide new evidence-based avenues for targeted, differential and effective treatment. For reasons of brevity, we refer for further reading on SLE and TTP to (100, 101).
3. Complement in immune thrombocytopenia
ITP, also known as immune thrombocytopenia , is a condition characterised by low platelet counts in the presence of autoanti-bodies directed to platelet antigens. Patients present with a tenden-cy for bleedings, most obviously detected as bruises of the skin, known as purpura, due to spontaneously occurring intracutaneous bleeding (102). There are clear indications that complement acti-vation is involved in the disappearance of platelets from the circu-lation of patients with ITP (103). Under normal conditions in healthy persons, ever-present low level AP complement activation and deposition is controlled and sufficiently counteracted by CRM
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

Thrombosis and Haemostasis 110.5/2013 © Schattauer 2013
916 Verschoor, Langer: Platelets and complement
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
present in the circulation as well as associated with the cell mem-brane (see 1.3. Modes of interactions between the complement sys-tem and platelets) and indeed, CRM deregulation is associated with thrombotic diseases (see 2. Complement in diseases with a prothrombotic state). However, in the presence of autoantibodies (typically IgG isotypes) that specifically target platelet epitopes and can activate complement via the CP (see 1.2. The complement sys-tem) (104), it appears that these protective mechanisms become over-stretched. As a result, more complement is deposited on the platelet than under usual conditions, and the presence of comple-ment and antibody on the platelet surface promotes their uptake by phagocytic cells, especially those located in the reticuloen-dothelial system such as hepatic Kupffer cells and splenic macro-phages. Indeed, ITP patients with a high degree of complement fixation onto their platelets benefit significantly from splenectomy, and subsequently show increased numbers of circulating platelets (105). In addition to phagocytosis, also direct damage inflicted by the accumulation of MAC onto platelet and megakaryocyte mem-branes is thought to contribute to the low platelet numbers in ITP patients (106). Short of splenectomy, which is not considered a first line treatment, currently approved treatment options aim to replenish platelets via transfusion or stimulation of thrombopoie-sis (Eltrombopag, Romiplostim) (107), or to interfere with their consumption via immunomodulation therapies that utilise trans-fusion of pooled immunoglobulins (IVIG), general immune sup-pression via glucocorticoids, or interfere with the source of the anti-platelet antibodies by depletion of peripheral B cells (Rituxi-mab) (108). Combined clinical and laboratory data suggest that therapies, which target complement deposition and MAC formation (eculizumab), could be effective in ITP, but no clinical trial data are available to date.
4. Complement and platelets during systemic infection
Infections of the blood stream provide ample opportunity for in-teractions between microbes, platelets and the complement sys-tem. Systemic infections become particularly apparent when they result in acute clinical disease, for instance during disseminated intravascular coagulation (DIC) or bacterial sepsis (“blood poi-soning”). The vast majority of sepsis cases are associated with bac-terial infection (109). Especially people and patients with generally weakened defences to infection are vulnerable to developing this condition. Sepsis has remained notoriously difficult to control clinically, with mortality rates reaching 50% in cases of septic shock (109). Factors closely associated with a bad prognosis for septic patients are the occurrence of DIC (110) and high levels of complement activation (111). The mechanisms of sepsis are com-plex, and ultimately result in the deterioration of the normal bal-ance between initiation and resolution of haemostasis, a combi-nation of coagulation and platelet aggregation. To make matters worse, haemostasis factors may simultaneously contribute to ex-acerbation of sepsis via the generation of anaphylatoxins (112) from an already overheated complement system (111).
To prevent aforementioned conditions from arising, it is a prin-ciple task of our immune system to remove blood-borne bacteria from the circulation. Blood clearance is achieved through the ac-tive capture, uptake and killing of bacteria by professional phago-cytes. Large populations of these cells, particularly macrophages, reside in spleen and liver, organs with a great capacity for filtering and clearing the circulation (113, 114). The complement system plays not only a role in direct lysis of microbes via MAC, but also a central role in the acquisition and retention of bacterially derived “antigen”, as is evident from the expression of a broad palette of complement receptors (115) and/or ligands (42, 43, 116) by all phagocyte populations (see also 1.2 The complement system). In-deed, complement is vital for the proper function of the immune response to bacteria, as complement deficiencies are associated with recurrent bacterial infections, including bacteraemia and sep-sis (117, 118).
Various blood-borne systems, including those traditionally as-sociated with haemostasis, participate in (immune) protective mechanisms. Indeed, platelets contain a broad array of substances that are not primarily associated with haemostasis but rather with immune defence, in some species even compounds that are di-rectly bactericidal (119). Moreover, platelets induce neutrophils to extrude web-like structures, appropriately known as neutrophil extracellular traps (NETs) that can entrap bacteria (120). The close biochemical similarities between the complement, coagulation and fibrinolytic cascades, all principally blood-borne systems, cre-ate for more areas of interaction than previously realised (121).
While controlling the ongoing blood infection via phagocytic clearance is the first goal, the immune system is laid out to “learn” from this experience and to “remember” so it can protect more ef-ficiently against re-infection in the future. It has been shown that platelets may interact with dendritic cells mediated by adhesion molecules JAM-C on platelets and Mac-1 on dendritic cells (122). Recent work reveals a novel mechanism in which complement and platelets aid T-cell responses to bacteria in the blood stream (123). In this mechanism, interaction between bacteria and platelets via complement C3 is needed to efficiently shuttle the bacteria into antigen presenting DC. In turn, better targeting of bacteria to DC results in more efficient induction of T-cell immunity. The com-plement and haemostatic systems are linked via GPIb, a receptor on platelets for von vWF, and bacterial adhesion to platelets tip the clearance balance from macrophages towards DC, thus boosting the generation of effective anti-microbial T-cell responses. Impor-tantly, and in contrast to DC and macrophages, T cells “re-member” their encounters with microbes, and their ability to form “memory” improves their effectiveness during a reinfection. Thus, this novel mechanism of platelet- and complement-directed clear-ance provides a blueprint that can aid the rational design of novel strategies to exploit the immunity-boosting capacity of platelets and complement during systemic infection.
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

© Schattauer 2013 Thrombosis and Haemostasis 110.5/2013
917Verschoor, Langer: Platelets and complement
5. Summarisation
In conclusion, complement activation and platelet thrombus formation occur in close spatiotemporal proximity in various homeostatic and pathophysiological contexts, such as during the resolution of vascular wounds, infections of the blood stream, or several diseases with prothrombotic state. Taking both comple-ment and haemostatic systems into consideration, integrating them in research efforts as well as clinical practice, may foster new insights into mechanisms of disease and generate novel targets for drug development and diagnosis.
AcknowledgementsH. F. L. is supported by the Volkswagen Foundation (Lichtenberg program), the Tuebingen Platelet Investigative Consortium (TuePIC) and the Clinical research unit 274 supported by the Ger-man Research Foundation, and the Wilhelm Sander Foundation. A.V. is supported by the Collaborative Research Centre SFB 914 (Trafficking of Immune Cells in Inflammation, Development and Disease, Project B4) from the German Research Foundation.
Conflicts of interestNone declared.
References1. Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets.
Arterioscler Thromb Vasc Biol 2008; 28: 403-412.2. Ho-Tin-Noe B, Demers M, Wagner DD. How platelets safeguard vascular integ-
rity. J Thromb Haemost 2011; 9 (Suppl 1): 56-65.3. Langer H, May AE, Daub K, et al. Adherent platelets recruit and induce differ-
entiation of murine embryonic endothelial progenitor cells to mature endothe-lial cells in vitro. Circ Res 2006; 98: e2-10.
4. Langer HF, Gawaz M. Platelets in regenerative medicine. Basic Res Cardiol 2008; 103: 299-307.
5. Griffin XL, Wallace D, Parsons N, Costa ML. Platelet rich therapies for long bone healing in adults. Cochrane Database Syst Rev 2012; 7: CD009496.
6. Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res 2004; 61: 498-511.
7. Kleinschnitz C, Pozgajova M, Pham M, et al. Targeting platelets in acute experi-mental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007; 115: 2323-2330.
8. Stoll G, Kleinschnitz C, Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood 2008; 112: 3555-3562.
9. Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nat Med 2011; 17: 1423-1436.
10. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res 2007; 100: 1673-1685.
11. Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central re-ceptor? Blood 2003; 102: 449-461.
12. Langer H, Gawaz M. The role of platelets for the pathophysiology of acute cor-onary syndromes. Hamostaseologie 2006; 26: 114-118.
13. Langer H, Schonberger T, Bigalke B, Gawaz M. Where is the trace? Molecular imaging of vulnerable atherosclerotic plaques. Semin Thromb Hemost 2007; 33: 151-158.
14. Ruggeri ZM. Platelets in atherothrombosis. Nat Med 2002; 8: 1227-1234.15. Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto
fibrinogen or translocation on von Willebrand factor. Cell 1996; 84: 289-297.
16. Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for pla-telet recruitment to the injured arterial wall in vivo. J Exp Med 2003; 197: 41-49.
17. Massberg S, Konrad I, Bultmann A, et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J 2004; 18: 397-399.
18. Ware J, Russell S, Ruggeri ZM. Generation and rescue of a murine model of pla-telet dysfunction: the Bernard-Soulier syndrome. Proc Natl Acad Sci USA 2000; 97: 2803-2808.
19. Uff S, Clemetson JM, Harrison T, et al. Crystal structure of the platelet glycopro-tein Ib(alpha) N-terminal domain reveals an unmasking mechanism for recep-tor activation. J Biol Chem 2002; 277: 35657-35663.
20. Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 2003; 9: 61-67.
21. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med 2002; 196: 887-896.
22. Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med 2009; 13: 1211-1220.
23. Santoso S, Sachs UJ, Kroll H, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med 2002; 196: 679-691.
24. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflam-mation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007; 7: 678-689.
25. Schober A, Manka D, von Hundelshausen P, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002; 106: 1523-1529.
26. von Hundelshausen P, Koenen RR, Weber C. Platelet-mediated enhancement of leukocyte adhesion. Microcirculation 2009; 16: 84-96.
27. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflam-mation and cardiovascular disease. Circ Res 2007; 100: 27-40.
28. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood 2008; 111: 5271-5281.
29. Ehlers R, Ustinov V, Chen Z, et al. Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med 2003; 198: 1077-1088.
30. Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 2000; 192: 193-204.
31. Chavakis T, Santoso S, Clemetson KJ, et al. High molecular weight kininogen regulates platelet-leukocyte interactions by bridging Mac-1 and glycoprotein Ib. J Biol Chem 2003; 278: 45375-45381.
32. Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest 1997; 100: 2085-2093.
33. Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost 2008; 99: 480-486.
34. Patzelt J, Langer HF. Platelets in angiogenesis. Curr Vasc Pharmacol 2012; 10: 570-577.
35. Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with pla-telets. Nat Rev Microbiol 2006; 4: 445-457.
36. Langer HF, Choi EY, Zhou H, et al. Platelets Contribute to the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Circ Res 2012; 110: 1202-1210.
37. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME et al. Pla-telets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010; 327: 580-583.
38. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all dis-eases of complement activation. Nat Rev Nephrol 2012; 8: 622-633.
39. Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology 2012; 217: 1080-1087.
40. Verschoor A, Carroll MC. Complement and its receptors in infection. Am Soc Microbiol Press 2013; 219-240.
41. Dempsey PW, Allison ME, Akkaraju S, et al. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 1996; 271: 348-350.
42. Verschoor A, Brockman MA, Knipe DM, et al. Cutting edge: myeloid comple-ment C3 enhances the humoral response to peripheral viral infection. J Immu-nol 2001; 167: 2446-2451.
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

Thrombosis and Haemostasis 110.5/2013 © Schattauer 2013
918 Verschoor, Langer: Platelets and complement
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
43. Verschoor A, Brockman MA, Gadjeva M, et al. Myeloid C3 determines induc-tion of humoral responses to peripheral herpes simplex virus infection. J Immu-nol 2003; 171: 5363-5371.
44. Brockman MA, Verschoor A, Zhu J, et al. Optimal long-term humoral responses to replication-defective herpes simplex virus require CD21/CD35 complement receptor expression on stromal cells. J Virol 2006; 80: 7111-7117.
45. Langer HF, Chung KJ, Orlova VV, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010; 116: 4395-4403.
46. Zhang X, Kohl J. A complex role for complement in allergic asthma. Expert Rev Clin Immunol 2010; 6: 269-277.
47. Ali H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol Lett 2010; 128: 36-45.
48. Peng Q, Li K, Sacks SH, Zhou W. The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm Allergy Drug Tar-gets 2009; 8: 236-246.
49. Hamad OA, Nilsson PH, Wouters D, et al. Complement component C3 binds to activated normal platelets without preceding proteolytic activation and pro-motes binding to complement receptor 1. J Immunol 2010; 184: 2686-2692.
50. Hamad OA, Ekdahl KN, Nilsson PH, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost 2008; 6: 1413-1421.
51. Nunez D, Charriaut-Marlangue C, Barel M, et al. Activation of human platelets through gp140, the C3d/EBV receptor (CR2). Eur J Immunol 1987; 17: 515-520.
52. Cosgrove LJ, d'Apice AJ, Haddad A, et al. CR3 receptor on platelets and its role in the prostaglandin metabolic pathway. Immunol Cell Biol 1987; 65: 453-460.
53. Vik DP, Fearon DT. Cellular distribution of complement receptor type 4 (CR4): expression on human platelets. J Immunol 1987; 138: 254-258.
54. Peerschke EI, Ghebrehiwet B. Human blood platelet gC1qR/p33. Immunol Rev 2001; 180: 56-64.
55. Wautier JL, Souchon H, Reid KB, et al. Studies on the mode of reaction of the first component of complement with platelets: interaction between the collagen-like portion of C1q and platelets. Immunochemistry 1977; 14: 763-766.
56. Peerschke EI, Ghebrehiwet B. C1q augments platelet activation in response to aggregated Ig. J Immunol 1997; 159: 5594-5598.
57. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med 1983; 158: 603-615.
58. Martel C, Cointe S, Maurice P, et al. Requirements for membrane attack com-plex formation and anaphylatoxins binding to collagen-activated platelets. PLoS One 2011; 6: e18812.
59. Del C, I, Cruz MA, Zhang H, et al. Platelet activation leads to activation and propagation of the complement system. J Exp Med 2005; 201: 871-879.
60. Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today 1991; 12: 338-342.
61. Mnjoyan Z, Li J, Afshar-Kharghan V. Factor H binds to platelet integrin al-phaIIbbeta3. Platelets 2008; 19: 512-519.
62. Vaziri-Sani F, Hellwage J, Zipfel PF, et al. Factor H binds to washed human pla-telets. J Thromb Haemost 2005; 3(1):154-162.
63. Yu GH, Holers VM, Seya T, et al. Identification of a third component of comple-ment-binding glycoprotein of human platelets. J Clin Invest 1986; 78: 494-501.
64. Nicholson-Weller A, March JP, Rosen CE, et al. Surface membrane expression by human blood leukocytes and platelets of decay-accelerating factor, a regula-tory protein of the complement system. Blood 1985; 65: 1237-1244.
65. Morgan BP. Isolation and characterization of the complement-inhibiting pro-tein CD59 antigen from platelet membranes. Biochem J 1992; 282: 409-413.
66. Barata L, Miwa T, Sato S, et al. Deletion of Crry and DAF on murine platelets stimulates thrombopoiesis and increases factor H-dependent resistance of pe-ripheral platelets to complement attack. J Immunol 2013; 190: 2886-2895.
67. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005; 365: 1073-1086.
68. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009; 361: 1676-1687.
69. Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol 2010; 25: 2431-2442.
70. Morigi M, Galbusera M, Gastoldi S, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 2011; 187: 172-180.
71. Tedesco F, Pausa M, Nardon E, et al. The cytolytically inactive terminal comple-ment complex activates endothelial cells to express adhesion molecules and tis-sue factor procoagulant activity. J Exp Med 1997; 185: 1619-1627.
72. Stahl AL, Vaziri-Sani F, Heinen S, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008; 111: 5307-5315.
73. Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell rec-ognition. Am J Hum Genet 2001; 68: 485-490.
74. Ferreira VP, Herbert AP, Cortes C, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol 2009; 182: 7009-7018.
75. Manuelian T, Hellwage J, Meri S, et al. Mutations in factor H reduce binding af-finity to C3b and heparin and surface attachment to endothelial cells in hemo-lytic uremic syndrome. J Clin Invest 2003; 111: 1181-1190.
76. Strobel S, Hoyer PF, Mache CJ, et al. Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Neph-rol Dial Transplant 2010; 25: 136-144.
77. Jozsi M, Strobel S, Dahse HM, et al. Anti factor H autoantibodies block C-ter-minal recognition function of factor H in hemolytic uremic syndrome. Blood 2007; 110: 1516-1518.
78. Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2006; 17: 2017-2025.
79. Kavanagh D, Richards A, Noris M, et al. Characterization of mutations in com-plement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immu-nol 2008; 45: 95-105.
80. Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008; 112: 4948-4952.
81. Goicoechea dJ, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syn-drome. Proc Natl Acad Sci USA 2007; 104: 240-245.
82. Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypi-cal hemolytic-uremic syndrome. N Engl J Med 2009; 361: 345-357.
83. Ziakas PD, Poulou LS, Pomoni A. Thrombosis in paroxysmal nocturnal hemog-lobinuria at a glance: a clinical review. Curr Vasc Pharmacol 2008; 6: 347-353.
84. Kunstling TR, Rosse WF. Erythrocyte acetylcholinesterase deficiency in paroxy-smal nocturnal hemoglobinuria (PNH). A comparison of the complement-sen-sitive and insensitive populations. Blood 1969; 33: 607-616.
85. Nicholson-Weller A, March JP, Rosenfeld SI, et al. Affected erythrocytes of pa-tients with paroxysmal nocturnal hemoglobinuria are deficient in the comple-ment regulatory protein, decay accelerating factor. Proc Natl Acad Sci USA 1983; 80: 5066-5070.
86. Selvaraj P, Rosse WF, Silber R, et al. The major Fc receptor in blood has a phos-phatidylinositol anchor and is deficient in paroxysmal nocturnal haemoglobi-nuria. Nature 1988; 333: 565-567.
87. Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 1993; 73: 703-711.
88. Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science 1993; 259: 1318-1320.
89. Mahoney JF, Urakaze M, Hall S, et al. Defective glycosylphosphatidylinositol an-chor synthesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood 1992; 79: 1400-1403.
90. Takahashi M, Takeda J, Hirose S, et al. Deficient biosynthesis of N-acetylgluco-saminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidyli-nositol anchor biosynthesis, in cell lines established from patients with paroxy-smal nocturnal hemoglobinuria. J Exp Med 1993; 177: 517-521.
91. Nicholson-Weller A, Burge J, Fearon DT, et al. Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J Immunol 1982; 129: 184-189.
92. Nicholson-Weller A. Decay accelerating factor (CD55). Curr Top Microbiol Im-munol 1992; 178: 7-30.
93. Holguin MH, Fredrick LR, Bernshaw NJ, et al. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989; 84: 7-17.
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80

© Schattauer 2013 Thrombosis and Haemostasis 110.5/2013
919
94. Holguin MH, Wilcox LA, Bernshaw NJ, et al. Relationship between the mem-brane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989; 84: 1387-1394.
95. Dixon RH, Rosse WF. Mechanism of complement-mediated activation of human blood platelets in vitro: comparison of normal and paroxysmal noctur-nal hemoglobinuria platelets. J Clin Invest 1977; 59: 360-368.
96. Wiedmer T, Hall SE, Ortel TL, et al. Complement-induced vesiculation and ex-posure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood 1993; 82: 1192-1196.
97. Grunewald M, Grunewald A, Schmid A, et al. The platelet function defect of paroxysmal nocturnal haemoglobinuria. Platelets 2004; 15: 145-154.
98. Hillmen P, Muus P, Duhrsen U, et al. Effect of the complement inhibitor eculizu-mab on thromboembolism in patients with paroxysmal nocturnal hemoglobi-nuria. Blood 2007; 110: 4123-4128.
99. Kravitz MS, Shoenfeld Y. Thrombocytopenic conditions-autoimmunity and hy-percoagulability: commonalities and differences in ITP, TTP, HIT, and APS. Am J Hematol 2005; 80: 232-242.
100. Chen M, Daha MR, Kallenberg CG. The complement system in systemic auto-immune disease. J Autoimmun 2010; 34: J276-J286.
101. Zipfel PF, Wolf G, John U, et al. Novel developments in thrombotic microan-giopathies: is there a common link between hemolytic uremic syndrome and thrombotic thrombocytic purpura? Pediatr Nephrol 2011; 26: 1947-1956.
102. Cines DB, Bussel JB, Liebman HA, et al. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113: 6511-6521.
103. Najaoui A, Bakchoul T, Stoy J, et al. Autoantibody-mediated complement acti-vation on platelets is a common finding in patients with immune thrombocy-topenic purpura (ITP). Eur J Haematol 2012; 88: 167-174.
104. McMillan R. Autoantibodies and autoantigens in chronic immune thrombocy-topenic purpura. Semin Hematol 2000; 37: 239-248.
105. Bell WR, Jr. Role of splenectomy in immune (idiopathic) thrombocytopenic purpura. Blood Rev 2002; 16: 39-41.
106. Kurata Y, Curd JG, Tamerius JD, et al. Platelet-associated complement in chronic ITP. Br J Haematol 1985; 60: 723-733.
107. Basciano PA, Bussel JB. Thrombopoietin-receptor agonists. Curr Opin Hema-tol 2012; 19: 392-398.
108. Pels SG. Current therapies in primary immune thrombocytopenia. Semin Thromb Hemost 2011; 37: 621-630.
109. Alberti C, Brun-Buisson C, Burchardi H, et al. Epidemiology of sepsis and in-fection in ICU patients from an international multicentre cohort study. Inten-sive Care Med 2002; 28: 108-121.
110. Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999; 341: 586-592.
111. Nakae H, Endo S, Inada K, et al. Serum complement levels and severity of sep-sis. Res Commun Chem Pathol Pharmacol 1994; 84: 189-195.
112. Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol 2010; 185: 5628-5636.
113. Morris DH, Bullock FD. The importance of the spleen in resistance to infec-tion. Ann Surg 1919; 70: 513-521.
114. Parker JT, Franke E. The fate of typhoid bacilli injected intravenously into nor-mal and typhoid immune rabbits. J Med Res 1919; 39: 301-309.
115. van Lookeren CM, Wiesmann C, Brown EJ. Macrophage complement recep-tors and pathogen clearance. Cell Microbiol 2007; 9: 2095-2102.
116. Gadjeva M, Verschoor A, Brockman MA, et al. Macrophage-derived comple-ment component C4 can restore humoral immunity in C4-deficient mice. J Im-munol 2002; 169: 5489-5495.
117. Cunnion KM, Benjamin DK, Jr., Hester CG, et al. Role of complement recep-tors 1 and 2 (CD35 and CD21), C3, C4, and C5 in survival by mice of Staphy-lococcus aureus bacteremia. J Lab Clin Med 2004; 143: 358-365.
118. Figueroa JE, Densen P. Infectious diseases associated with complement defi-ciencies. Clin Microbiol Rev 1991; 4: 359-395.
119. Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011; 11: 264-274.
120. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extra-cellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13: 463-469.
121. Markiewski MM, Nilsson B, Ekdahl KN, et al. Complement and coagulation: strangers or partners in crime? Trends Immunol 2007; 28: 184-192.
122. Langer HF, Daub K, Braun G, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arte-rioscler Thromb Vasc Biol 2007; 27: 1463-1470.
123. Verschoor A, Neuenhahn M, Navarini AA, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol 2011; 12: 1194-1201.
124. Langer HF, Chavakis T. Platelets and neurovascular inflammation. Thromb Haemost 2013; 110: 888-893.
Verschoor, Langer: Platelets and complement
Plat
elet
s: b
asic
mec
hani
sms
and
tran
slat
iona
l im
plic
atio
ns
For personal or educational use only. No other uses without permission. All rights reserved.Downloaded from www.thrombosis-online.com on 2017-06-16 | IP: 54.191.40.80