Embed Size (px)
Citation preview

Coronary Vasospastic Angina: Current
Understanding and the Role of Inflammation
Ming-Jui Hung and Wen-Jin Cherng
Coronary vasospastic angina (CVsA) plays an important role in myocardial ischemia including stable angina, acute
coronary syndromes, and sudden cardiac death. Inflammation status from either endothelium or adventitia can
cause endothelial dysfunction. Thereafter, the endothelial dysfunction further induces vascular smooth
muscle hypercontraction through the enhanced rho-kinase with the resultant clinical event. With better
understanding of the interactions between inflammation, endothelium, and smooth muscle cells, we and other
investigators have provided new insights into the basic pathophysiology of CVsA. Apart from calcium channel
blockers, nitrates, and the rho-kinase inhibitor fasudil, anti-inflammatory treatment is helpful in some patients
with refractory CVsA. Additional studies are needed to clarify the mechanisms of recurrent CVsA.
Key Words: Angina � Coronary vasospasm � Inflammation
HISTORY OF CORONARY VASOSPASM (CVsp)
In 1959, Prinzmetal and his colleagues described a
syndrome characterized by angina at rest with transient
ST-segment elevation in patients with diseased coronary
arteries, which is different from that seen in classic an-
gina.1
In these circumstances where the heart burden is
not increased, the angina must be due to a reduction in
coronary blood flow. Prinzmetal proposed the term “vari-
ant angina” and suggested that it was caused by spasm of
a major coronary artery, because it was relieved promptly
by administration of nitroglycerin. With the advent of
coronary angiography, it became apparent that variant
angina was caused by CVsp and it may occur at the site of
a coronary stenosis2
or in normal coronary arteries, the
so-called “variant of the variant”3
or “coronary vasospastic
angina (CVsA)”.4
Recently, many investigators found that
much of the underlying cause of CVsA is associated with
ST-segment depression rather than ST-segment elevation.5,6
Therefore, the term “variant angina” is usually denoted
as angina with transient ST-segment elevation.
CLINICAL CHARACTERISTICS OF CVsA
CVsA is different from typical atherosclerotic angina in
the pathophysiologies. CVsp, however, might be induced
by exercise, particularly in the morning in some patients
with variant angina,7
and might cause exercise-induced
angina with ST-segment depression in some patients with
stable effort angina5
(Figure 1). The authors postulated that
the spastic arteries are not normal, in that the spastic ar-
teries cannot dilate fully in response to exercise as in nor-
mal coronary arteries. There are daily, weekly, monthly,
and circadian variations in the incidence of CVsA.8
Circadian variation in the incidence of attacks in
patients with CVsA
CVsA occurs usually at rest, particularly from mid-
night to early morning.9
Yasue et al.10
compared coro-
1 Acta Cardiol Sin 2013;29:1�10
Review Article Acta Cardiol Sin 2013;29:1�10
Received: June 28, 2012 Accepted: September 19, 2012
Department of Cardiology and Medical Research Center, Chang
Gung Memorial Hospital, Keelung, Chang Gung University College of
Medicine, Taiwan.
Address correspondence and reprint requests to: Dr. Ming-Jui Hung,
Department of Cardiology, Chang Gung Memorial Hospital, No. 222,
Maijin Road, Keelung 20401, Taiwan. Tel: 886-2-2431-3131 ext.
3168; Fax: 886-2-2433-5342; E-mail: [email protected]
nary arteriograms recorded in the early morning with
those recorded in the afternoon in patients with variant
angina. In the early morning, the tone of the major cor-
onary artery was increased and its diameter was small.
Under such conditions, mild exercise could induce CVsp
resulting in attacks; the administration of nitroglycerin
dilated the artery markedly. In contrast, in the after-
noon, the major coronary artery was usually already di-
lated, and its tone was low on the control coronary ar-
teriograms. Under such conditions, exercise could in-
duce little coronary vasoconstriction and no attacks
usually occurred except in patients with severe organic
stenosis, in whom only a mild spasm could occlude the
artery and result in angina attacks. To record quantita-
tively the difference in the tone in the major coronary
arteries observed in the early morning and in the after-
noon, Yasue et al.10
measured the diameter of the major
coronary artery on both the control coronary arterio-
grams and the coronary arteriograms taken after nitro-
glycerin administration. The percentage increase in di-
ameter of the major coronary artery after nitroglycerin
administration was significantly greater in the early
morning than in the afternoon. This may be one of the
reasons that there is a circadian variation in the exercise
capacity of most patients with variant angina. A pathol-
ogy study illustrated the complexity of the local neural
events that modulate the tone of the coronary arter-
ies.11
The occurrence of CVsA in the early morning has
been noted to be associated with rapid eye move-
ment.12
Therefore, a rapid elevation of sympathetic ac-
tivity during augmented parasympathetic activity has
been suggested to be related to the occurrence of CVsA
in the early morning.
Acute coronary syndrome
There is general consensus in the medical commu-
Acta Cardiol Sin 2013;29:1�10 2
Ming-Jui Hung et al.
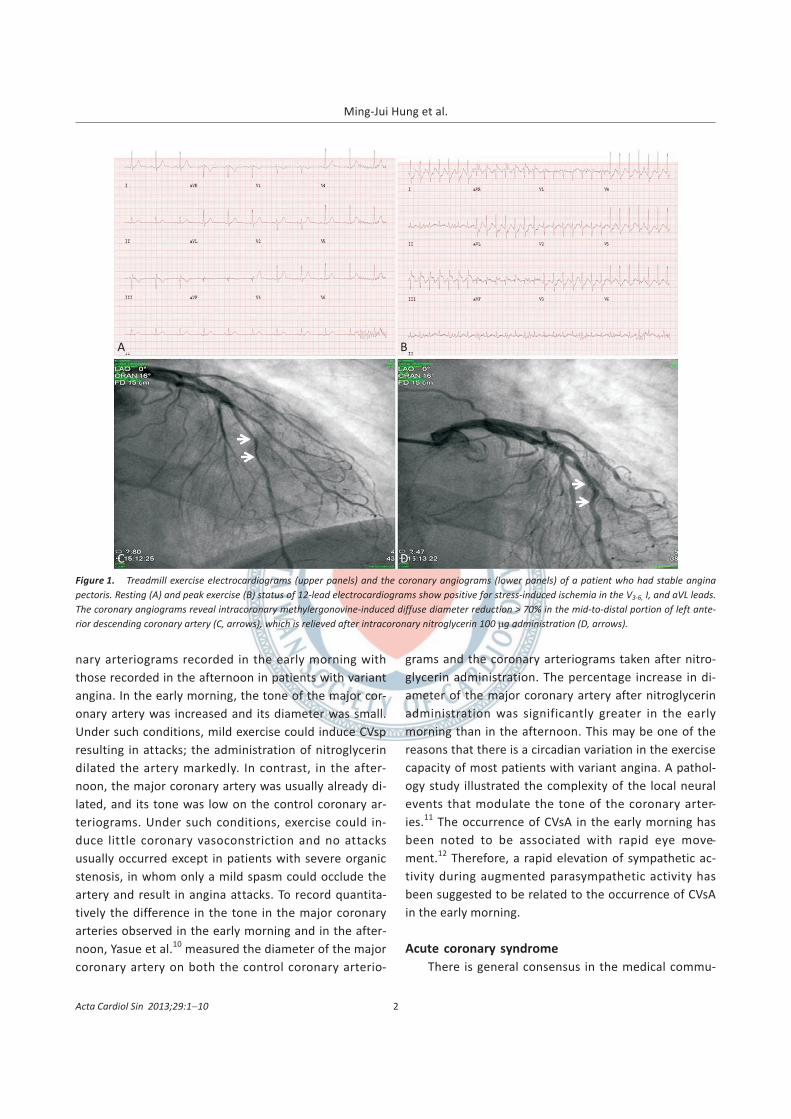
Figure 1. Treadmill exercise electrocardiograms (upper panels) and the coronary angiograms (lower panels) of a patient who had stable angina
pectoris. Resting (A) and peak exercise (B) status of 12-lead electrocardiograms show positive for stress-induced ischemia in the V3-6, I, and aVL leads.
The coronary angiograms reveal intracoronary methylergonovine-induced diffuse diameter reduction > 70% in the mid-to-distal portion of left ante-
rior descending coronary artery (C, arrows), which is relieved after intracoronary nitroglycerin 100 �g administration (D, arrows).
A B
C D

nity that intracoronary thrombus plays a major role in
the pathogenesis of acute myocardial infarction. Yasue
et al.13
found that CVsp was presumed to be responsible
for the acute myocardial infarction because the culprit
artery was patent without delay of visualization in
17.9% of patients in the early phase of acute myocardial
infarction. Our report also found that the responsible
coronary arteries were patent in 12% of patients with
acute myocardial infarction (Figure 2).14
There was in-
farct-related CVsp involvement in 95% in these patients.
This may be explained by the spontaneous resolution of
either spasm or thrombus, or both. Oshima et al.15
re-
ported that CVsp causes intracoronary thrombus forma-
tion, supporting the concept that CVsp is one of the pri-
mary factors contributing to acute myocardial infarc-
tion. Therefore, CVsp appears to play a role in the pro-
duction of acute myocardial infarction in these patients.
Most of the patients with CVsA present as angina
with ST-segment depression and/or T-wave inversion on
electrocardiogram, which is an acute coronary syn-
drome. If there is no cardiac enzyme elevation, we de-
fine the cardiac event as an unstable angina because
most of the CVsA occurs at rest. Angiographically normal
or near-normal coronary arteries occurs in 25% of pa-
tients with acute coronary syndrome irrespective of the
provocative agents.16,17
The CVsp can be induced in
50-60% of these patients. After initial management (i.e.
oxygen, aspirin, nitroglycerin, and/or morphine) for
acute coronary syndrome, follow-up electrocardiograms
are important to indicate the role of CVsp for the acute
coronary syndrome. If there is a normalized ST-segment
after the initial management, the CVsp may play a role
in acute coronary syndrome.
Intracoronary administration of methylergonovine
Provocative testing for CVsp is required to clarify its
role in the pathogenesis of angina pectoris, especially in
patients without significant obstructive coronary artery
disease (CAD). To ensure a valid provocative test, vasodi-
lators (calcium antagonists and nitrates) must be with-
drawn for at least 24 hours, except sublingual nitroglyc-
erin if necessary. The nitroglycerin solution must be well
prepared before starting intracoronary methylergo-
novine testing, in part to abolish intracoronary me-
thylergonovine-induced CVsp immediately through the
intracoronary route (50-1000 �g). There have been
several agents or procedures, including ergonovine
maleate, methylergonovine maleate, acetylcholine or
hyperventilation reported that induce CVsp in patients
with CVsA. Intracoronary ergonovine administration has
been a popular method to induce CVsp during angio-
graphic study because of its high sensitivity and specific-
ity.18
This test was administrated using a step-wise dose
of ergonovine (1, 5, 10, and 30 �g) every 3 minutes;
3 Acta Cardiol Sin 2013;29:1�10
Inflammation and Coronary Vasospasm
Figure 2. A serial 12-lead electrocardiograms and the coronary
angiograpms of a patient who had coronary vasospasm-induced acute
myocardial infarction. The patient had normal coronary angiograms 1
year before acute myocardial infarction. Initial resting 12-lead electro-
cardiograms at emergency department showed complete atrioven-
tricular block with ST-elevation in the II, III, aVF and V1-4 (A) and right
ventricular infarction (B). The emergency coronary angiogram revealed
total occlusion of right coronary artery since the orifice (C, arrow)
inititally. After intra-aortic nitroglycerin 200 �g administration, patent
proximal and mid-portion of right coronary artery with thrombi-oc-
cluded (D, arrow) in the distal portion of right coronary artery was
noted. A thrombuster-catheter (E, arrow) was used to aspirate thrombi.
After thrombi suction and further intracoronary 200 �g nitroglycerin ad-
ministration, the right coronary artery was patent (F). The 12-lead elec-
trocardiograms recorded at the day after coronary intervention (G) and
6 months later (H) showed evolutional electrocardiographic ST-segment
and T-wave changes of inferior myocardial infarction.
A B
C D
E F
G H

the drug was first introduced into the right coronary ar-
tery and subsequently into the left coronary artery.
CVsp was defined as a decrease of � 50%19
or 70%20
in
the diameter of an arterial lumen with concurrent chest
pain and/or ischemic ST-T changes during the provoca-
tion testing. Recently, the Japanese Circulation Society
published a guideline to define a positive indicator on a
provocative test as a decrease of � 90% in the diameter
of an arterial lumen, with concurrent chest pain and/or
ischemic ST-segment changes during the provocation
test.4
However, Yasue et al.8
suggested that there are no
limits on the degree of lumen diameter reduction re-
quired to diagnose CVsp since ischemia must accom-
pany the changes of vessel size in a period of time. Al-
though there are different criteria in vessel diameter,
the angina and/or ischemic electrocardiographic changes
during provocation testing are necessary in defining a
positive provocation test result. After a CVsp had been
diagnosed, the administration of intracoronary ergo-
novine was stopped and 50-1000 �g of intracoronary ni-
troglycerin was administered. The ergonovine maleate
and acetylcholine are not available in Taiwan. Therefore,
only methylergonovine maleate was used in the CVsp
provocative testing in our studies, with the same in-
tracoronary dose regimen as in ergonovine maleate. Al-
though it is useful in the diagnosis of CVsA, safety is still
a major concern. The contraindications for intraco-
ronary methylergonovine testing included pregnancy,
severe hypertension (systolic blood pressure > 180 mm
Hg), moderate to severe aortic stenosis and uncon-
trolled ventricular arrhythmia.19
Some studies have re-
ported complications of intravenous injection of er-
gonovine in patients who underwent cardiac cathe-
terization, including ventricular tachycardia, ventricular
fibrillation, and death.21,22
The importance of intra-
coronary nitroglycerin rather than intravenous or sub-
lingual route in relieving provocative CVsp is empha-
sized.22
Since Hackett et al.18
introduced the intra-
coronary route for provocation testing in 1987, the gen-
eral consensus has been that the intracoronary rather
than the intravenous route is safer because of negligible
drug recirculation and avoidance of effects on branches
with critical stenosis.19
Recently, a Japanese multicenter
study found that intracoronary ergonovine provocation
testing induces ventricular tachycardia/ventricular fibril-
lation in 0.8% of patients with CVsA,23
which is similar to
our prior study.24
They concluded that spasm provoca-
tion tests have an acceptable level of safety; the evalua-
tion of spasm type may provide useful information for
the risk stratification of CVsA patients. Consistent with
the Japanese study, there was no mortality reported in
our prior study.24
Because ventricular tachycardia or
ventricular fibrillation is a possible complication follow-
ing intracoronary methylergonovine administration, its
use outside the cardiac catheterization laboratory is not
recommended.
In theory, the diagnosis of CVsp must be made on
the coronary angiographic findings during the attack.
However, it is not practical to perform coronary angio-
graphy during an attack in every patient, and this step is
unnecessary. During coronary angiography, adequate
doses (50-1000 �g) of intracoronary nitroglycerin ad-
ministration help cardiologists differentiate spontane-
ous CVsp from fixed obstructive CAD. Angina pectoris
that is relieved promptly after nitroglycerin may be diag-
nosed as CVsA even without angiographic evidence, if
one of following characteristics is noticed:8
1) the attack
occurs at rest, particularly from midnight to early morn-
ing; 2) there is marked circadian variation of exercise ca-
pacity, the attack easily induced by exercise in the
morning but not by even vigorous exercise in the after-
noon; 3) the attack is associated with ST-segment eleva-
tion on the electrocardiogram; 4) the attack is induced
by hyperventilation; or 5) the attack is suppressed by
calcium antagonists, but not by beta-blockers.
Prevalence of CVsp
In the Japanese population, CVsp frequency appears
to be greater than that in Western populations,25
and
the diagnosis of variant angina (angina with transient
ST-segment elevation) is made in a high percentage
(10% to 70%) of patients with anginal symptoms re-
ferred to Japanese medical centers. Our prior investiga-
tion also found that 27% of angina patients who under-
went coronary angiography and had CVsA (angina with
and without transient ST-segment elevation).26
In pa-
tients who had angina and no significant obstructive
CAD, the prevalence of CVsp was around 50%.27
In pa-
tients who had acute coronary syndrome and no signifi-
cant obstructive CAD, the prevalence of CVsp was esti-
mated to be 57%,17
which is similar to data from Ger-
many.16
The diagnosis of CVsp depends on angiographic
Acta Cardiol Sin 2013;29:1�10 4
Ming-Jui Hung et al.

protocols and provocation tests, which varies from labo-
ratory to laboratory. Premedication with spasmolytic
drugs such as nitroglycerin or calcium antagonists,
avoidance of coronary constrictors, and daily or monthly
variation of disease activity may lead to a failure to diag-
nose CVsp. Therefore, an estimate of the prevalence of
CVsp in different populations remains to be defined. Al-
though the racial difference in coronary constrictor re-
sponse has been strongly suggested,25
the underestima-
tion of CVsp worldwide is an important issue in the cur-
rent percutaneous coronary intervention era.
Risk factors for the development of CVsA
Cigarette smoking was found to be an unequivocal
risk factor for CVsA in the literature (adjusted odds ratio
of 2.41).28
Similarly, our investigation29
has found a
multivariate-adjusted odds ratio of 2.58, which is similar
to other studies. In our recent study, synergistic interac-
tion was verified between smoking and high-sensitivity
C-reactive protein (hs-CRP) in the development of
CVsp.29
This interaction was linear and monotonic among
smokers. In nonsmokers, however, hs-CRP had a th-
reshold effect on CVsp development. A decreased odds
ratio to 2.12 was observed when hs-CRP was added into
the multivariate analysis between smoking and CVsp,
suggesting that hs-CRP was an important covariate of
CVsp. Further analysis showed that the relationship be-
tween hs-CRP and CVsp differs between men and wo-
men.30
The non-threshold model in men and a threshold
model in women provide evidence that more smokers in
men (life-style) and age (induction time) contribute to
the natural history of CVsp development. In addition,
we also found that hypertension is a negative predictor
for CVsp, which suggests that the pathogenesis of CVsp
differs from that of coronary atherosclerosis.
Treatment and prognosis of CVsA
Table 1 depicts the treatment strategies for CVsA
patients. Calcium antagonists play a cornerstone role in
the management of CVsA.8
Of importance, the long-act-
ing calcium antagonist should be given before going to
bed at night as CVsA occurs usually from midnight to the
early morning.8
Usually, a high-dose long-acting calcium
antagonist (e.g. nifedipine 80 mg/day, amlodipine 20
mg/day, diltiazem 360 mg/day, or verapamil 480 mg/
day) is started.31
It may require 2 calcium antagonists
(dihydropyridine and non-dihydropyridine) to relieve
CVsA. While long-acting nitrates are also effective in
preventing CVsA attack, the occurrence of nitrate toler-
ance may limit their use as a first-line approach.8,32
In
contrast, �-blockers do not suppress, but rather may ag-
gravate CVsp in patient with variant angina.32
Although
platelets may play a role in precipitating or aggravating
CVsp, intravenous prostacyclin or cyclooxygenase inhibi-
tors have thus far not been shown to have beneficial ef-
fects.32
Recent clinical research shows that magnesium,
antioxidants, rho-kinase (ROCK) inhibitor fasudil, and
fluvastatin are also beneficial to treat CVsA.8
Addition-
5 Acta Cardiol Sin 2013;29:1�10
Inflammation and Coronary Vasospasm
Table 1. Treatment strategies for CVsA
Strategy Recommendation
Cigarette cessation Obligatory
Long-acting calcium antagonist 1. Use before going to bed at night
2. Combination of dihydropyridine and non-dihydropyridine if recurrent CVsA
Long-acting nitrates Decreased potency by tolerance
Statin Fluvastatin
Magnesium Intravenous infusion
RhoA/ROCK inhibitor Intravenous or oral Fasudil
Vitamin C Intravenous infusion
Vitamin E Oral
Valsartan A case report
Prednisolone Case reports
Coronary artery bypass surgery Controversial
Coronary stenting Controversial
Implantable cardioverter defibrillator For life-threatening ventricular arrhythmias
CVsA, coronary vasospastic angina; ROCK, Rho-associated coiled-coil containing protein kinase.

ally, the aggravating factors for CVsA such as cigarette
smoking, catecholamines, muscarinic agonists, ergot al-
kaloids, prostaglandins, alcohol, emotional stress, and
propranolol must be avoided. Coronary intervention
plays a limited role in patients with CVsA and organic
stenosis.33
In the current coronary stenting era, there
are some reports of coronary stenting in the manage-
ment of drug-refractory CVsA.34-36
Gaspardone et al.34
reported that coronary stent placement is effective in
preventing angina attacks and methylergonovine-in-
duced CVsp in 6 of 9 patients 6 months after stenting.
They performed coronary stent placement in CVsA pa-
tients because of persistent angina attacks despite med-
ical treatment (up to 960 mg diltiazem or 100 mg
nifedipine and nitrates). Other reports also showed suc-
cessful coronary stenting in the management of CVsA.
However, their reports did not use the combination and
maximal doses of 2 calcium antagonists in terms of ag-
gressive medical therapy for CVsA.35,36
In theory, the cor-
onary stenting is considered to be effective to abolish
the CVsA in the absence of concomitant calcium antago-
nist treatment after coronary stenting. Calcium antago-
nists were still used after coronary stenting in these re-
ports indicating that single-vessel or multi-vessel CVsp
may develop at sites different from the initial stenosis.
Therefore, Yuksel et al.37
suggested that before coronary
stenting can be advocated as an established treatment
resistant to maximal medical therapy, a randomized trial
should be carried out. Nevertheless, approximately 20%
of CVsA patients do not respond to treatment with 2 cal-
cium antagonists plus a long-acting nitrates.31
Under the
circumstances, coronary stenting in combination with
adequate coronary vasodilators doses may be helpful in
patients CVsA and severe organic stenosis.4,31,32,38
On
the other hand, coronary interventions are contraindi-
cated in patients with CVsA without severe organic ste-
nosis.4,31,38
As coronary arteries treated with coronary
interventions show time-dependent loss of endothe-
lial-dependent and -independent vasomotor function,
with imbalanced contraction/dilation capacity,39
the
suggestion has been that cardiologists must keep CVsA
in mind before performing coronary intervention.8,34,37
An implantable cardioverter defibrillator with aggressive
medical therapy for CVsA was reported to be effective in
patients who had syncope, ventricular tachycardia, or
survived cardiac arrest outside the hospital.4
The prognosis among Japanese patients with variant
angina is better than that among western patients.40,41
The difference is probably due to the fact that the per-
centage of patients with multivessel disease or impaired
left ventricular function or both was smaller, and the
percentage of patients who received a calcium antago-
nist (diltiazem or nifedipine) as the initial treatment was
higher in the Japanese series.40
By multivariate analysis,
the intake of calcium antagonists, extent and severity of
coronary artery disease, and multivessel CVsp were
shown to be significantly independent predictors of sur-
vival without myocardial infarction. In Taiwanese pa-
tients who had CVsA and no significant obstructive CAD,
the long-term prognosis was found to be good despite
11% of recurrent CVsA.42
The most common factors for
recurrent CVsA are continued smoking and self-discon-
tinuation of calcium antagonists. The occurrence of an-
gina attacks is difficult to assess in individuals with CVsA
because their frequency tends to fluctuate spontane-
ously and the attacks are not necessarily accompanied
by symptoms. Based on the observation of recurrent an-
gina pectoris, it appears reasonable to suggest that
treatment should not be discontinued in cases of CVsA,
even when the patients are asymptomatic.42,43
INFLAMMATION AND CVsA
Determinants of CVsp: endothelium and smooth
muscle
There is no single mechanism that can explain the
entire process of CVsA. In 1980s, the autonomic nervous
system was found to play an important role in the
pathophysiology of CVsA.8
In the 1990s, oxidative stress,
deficiency of nitric oxide activity, respiratory alkalosis,
magnesium deficiency and insulin deficiency were iden-
tified as other possible mechanisms for CVsA.8
In the
late 1990s and early 2000s, mutation of the promoter in
endothelial nitric oxide synthase gene8
and polymor-
phism of paraoxonase gene44
were found to be associ-
ated with CVsA. In another large Japanese cohort
study,45
The nicotinamide adenine dinucleotide hydro-
gen/nicotinamide adenine dinucleotide phosphate hy-
drogen oxidase p22 phox gene is a susceptibility locus
for CVsp in men, and the stromelysin-1 and interleukin-
6 genes are susceptibility loci in women. In this study,
Acta Cardiol Sin 2013;29:1�10 6
Ming-Jui Hung et al.

the inflammation gene was firstly identified to be as-
sociated with CVsp. These findings indicate that dys-
functional endothelium secondary to down-regulation
of endothelial nitric oxide synthase and oxidative inacti-
vation of nitric oxide is one of the major mechanisms re-
sponsible for CVsA. In addition, inflammation is indi-
cated as a possible contributor in the development of
CVsA. However, a deficiency of endothelial nitric oxide
may not explain the complete mechanism of CVsA be-
cause all atherosclerotic coronary arteries are not nec-
essarily associated with CVsp in spite of the deficiency
of nitric oxide activity. In 2006, Kakket et al.46
found that
spontaneous CVsp occurs in KATP mutant mice, which
arise from a smooth muscle-extrinsic process. They pos-
tulated that endothelial dysfunction with loss of KATP
channels and decreased nitric oxide production and/or
bioavailability promotes smooth hypercontraction. Ano-
ther possibility includes the sympathetic neurons,
where opening of presynaptic KATP channels decreases
norepinephrine release, enhancing smooth muscle relax-
ation to dilate coronary arteries. A defect in these chan-
nels decreasing the threshold for norepinephrine re-
lease might be associated with CVsp. In 2011, Qipshidz
et al.47
found that folic acid treatment attenuates acetyl-
choline-induced coronary vasoconstriction in hyper-
homocysteinemic cystathionine beta synthase hyter-
zygote mice. They suggested that CVsp is related to the
regulation of endothelial nitric oxide synthase expres-
sion, nitric oxide availability, and tissue homocysteine.
In addition to deficient endothelial nitric oxide ac-
tivity, hyperreactivity of the coronary smooth muscle
seems to play an important role in the pathogenesis of
CVsA.48
Shimokawa and colleagues developed swine
models of CVsp and showed that ROCK activity is en-
hanced in coronary artery smooth muscle after wrap-
ping the coronary artery with interleukin-1 beads.48
They also demonstrated that ROCKs expression and ac-
tivity are enhanced at the inflammatory/arteriosclerotic
coronary lesions.49
Recently, we50
found that ROCK ac-
tivity in circulating neutrophils is a useful biomarker for
the diagnosis and disease activity assessment in patients
with CVsA. Some investigators found that decreased en-
dothelial nitric oxide synthase activity increase ROCK ac-
tivity in coronary arteries.51
These findings connect the
activity of ROCK to endothelial nitric oxide and are in
agreement with the clinical observations that spastic
arteries are inflammatory and are supersensitive to
both vasoconstrictor agonists and nitrates.
Role of inflammation
In 1978, Lewis et al.52
first reported a patient who
died of cardiogenic shock due to variant angina and lo-
calized pericarditis. They postulated that there was a
link between inflammation and CVsA. In the mid and
later 2000s, we showed that chronic inflammation was
associated with CVsA, as evidenced by elevated peri-
pheral leukocyte and monocyte counts, hs-CRP, inter-
leukin-6, and adhesion molecules.27
It was demon-
strated that the serum level of cortisol, one of the im-
portant stress hormones, causes coronary hyperre-
activity through activation of ROCK in pigs in vivo.53
Cigarette smoking, a major risk factor for CVsA, is as-
sociated with low-grade inflammation.54
These findings
suggest that there is increased inflammatory status in
patients with CVsA and the inflammation may con-
tribute to the occurrence of CVsp. An interaction be-
tween smoking and hs-CRP was recently reported by
our group and the relationship between hs-CRP and
CVsA differed between men and women.30
We also
have recently demonstrated that ROCK activity in pe-
ripheral leukocyte independently predicts the presence
and severity of CVsA and the level of ROCK activity cor-
relates with plasma interleukin-6.50
These findings sug-
gest that there is increased inflammatory status in pa-
tients with CVsA and the inflammation may cause endo-
thelial dysfunction, ROCK activity accentuation and fi-
nally contribute to the vascular smooth muscle hyper-
contraction, the occurrence of CVsp.
Some investigators found that inflammatory clinical
condition is associated with CVsA.8
Several reports have
suggested a possible link between allergic disease and
CVsA.55,56
The pathogenesis of bronchial asthma has
been recently attributed to hyperreactivity of the air-
way caused by inflammation, and corticosteroids are
considered to work by alleviating that inflammation.57
There is an analogy with the pathophysiology of CVsA
and bronchial asthma; that is CVsp may be induced by
arterial hyperreactivity caused by local inflammation in
the coronary arterial wall and corticosteroids suppress
the hyperreactivity by alleviating the inflammation in
the vessel wall. In fact, Forman et al.58
reported a pa-
tient with CVsA complicated by sudden death in whom
7 Acta Cardiol Sin 2013;29:1�10
Inflammation and Coronary Vasospasm

mast cell infiltration was found at the site of angio-
graphic documentation of CVsp, which indicates that
CVsp can also occur as the result of adventitial inflam-
mation in addition to endothelial inflammation. Some
other studies also reported that focal infiltration of in-
flammatory cells was seen in the adventitia or plaque of
the coronary artery in patients with CVsA.59
Even where
there is no evident narrowing during angiography, dif-
fuse intimal thickening or intimal bump by precursor
medial contraction is demonstrated from optical coher-
ence tomography.60,61
Nitric oxide availability is reduced
under the circumstances of low-grade inflammation of
atherosclerosis or diffuse intimal thickening.62
Endothe-
lial nitric oxide synthase has been reported to be quan-
titatively associated with caveolin-1 in endothelial cells.
The vascular relaxation in response to acetylcholine was
also much larger in aortic rings collected from caveolin-
1–/–
mice than in their wild-type littermates.63
Of note,
isolated aortic rings from caveolin-1–/–
mice were unable
to maintain a constant contractile tone, oscillating at 1
Hz frequency. These studies support the proposition
that, in the basal condition, endothelial nitric oxide
synthase becomes hyperactivated in the absence of
caveolin-1. Based on the above analyses, it appears rea-
sonable to speculate that CVsp is an early inflammatory
coronary artery condition because of the presence of
low-grade inflammation-related endothelial dysfunction
with resulting diffuse intimal thickening and impaired ni-
tric oxide production.64
Because the lack of normal nitric
oxide activity does impact the normal vessel, the vessel
tends to constrict in the presence of low-grade inflam-
mation such as cigarette smoking, stress, airway dis-
ease, and allergy.
CONCLUSION
Identification of CVsp is important in our daily
clinical practice because of a wide disparity between
treatment strategies for fixed obstruction versus vaso-
spasm of coronary arteries. Adequate doses of intra-
coronary nitroglycerin administration help cardiologists
differentiate spontaneous CVsp from fixed obstructive
CAD. However, cessation of cigarette smoking and
adequate dose and timing of calcium antagonists are
the cornerstone for CVsA therapy. Accumulating evi-
dence suggests that inflammation substantially con-
tributes to the development of CVsA. Clinical studies
with antiinflammtory therapies further support the role
of inflammation in CVsA. Release of inflammatory medi-
ators, such as interleukin-6, histamine, or serotonin
leads to nitric oxide availability reduction, endothelial
dysfunction, and precursor medial contraction. The final
common pathway in CVsp is vascular smooth muscle
contraction by increased ROCK activity. With the under-
standing of interactions between inflammation, endo-
thelium, and smooth muscle cells, we and other investi-
gators have provided some insights into the basic patho-
physiology of CVsp. More studies are needed to clarify
the mechanisms of recurrent CVsA.
REFERENCES
1. Prinzmetal M, Kennamer R, Merliss R, et al. Angina pectoris I. A
variant form of angina pectoris; preliminary report. Am J Med
1959;27:375-88.
2. MacAlpin RN, Kattus AA, Alvaro AB. Angina pectoris at rest with
preservation of exercise capacity: Prinzmetal’s variant angina.
Circulation 1973;47:946-58.
3. Cheng TO, Bashour T, Kelser GA Jr, et al. Variant angina of
Prinzmetal with normal coronary arteriograms. A variant of the
variant. Circulation 1973;47:476-85.
4. JCS Joint Working Group. Japanese Circulation Society of Guide-
lines for Diagnosis and Treatment of Patients with Vasospastic
Angina (Coronary Spastic Angina) (JCS 2008): digest version, Cir J
2010;74:1745-62.
5. Cheng CW, Yang NI, Lin KJ, et al. Role of coronary spasm for a pos-
itive noninvasive stress test result in angina pectoris patients
without hemodynamically significant coronary artery disease.
Am J Med Sci 2008;335:354-62.
6. Nakagawa H, Morikawa Y, Mizuno Y, et al. Coronary spasm
preferentially occurs at branch points: an angiographic com-
parison with atherosclerotic plaque. Circ Cardiovasc Interv
2009;2:97-104.
7. Hung MJ, Hung MY, Cheng CW, et al. Clinical characteristics of pa-
tients with exercise-induced ST-segment elevation without prior
myocardial infarction. Circ J 2006;70:254-61.
8. Yasue H, Nakagawa H, Itoh T, et al. Coronary artery spasm--
clinical features, diagnosis, pathogenesis, and treatment. J
Cardiol 2008;51:2-17.
9. Liu PC, Cheng CW, Hung MJ, et al. Variant angina with angio-
graphically normal or near-normal coronary arteries: a 10-year
experience. J Int Med Taiwan 2010;21:79-89.
10. Yasue H, Omote S, Takizawa A, et al. Coronary arterial spasm in
ischemic heart disease and its pathogenesis. A review. Circ Res
Acta Cardiol Sin 2013;29:1�10 8
Ming-Jui Hung et al.

1983;52:I147-52.
11. Jougasaki M, Yasue H, Takahashi K. Perivascular nerve lesion of
the coronary artery involved in spasm in a patient with variant
angina. Pathology 1989;21:304-7.
12. Otsuka K, Yanaga T, Watanabe H. Variant angina and REM sleep.
Am Heart J 1988;115:1343-6.
13. Yasue H, Ogawa H, Okumura K. Coronary artery spasm in the gen-
esis of myocardial ischemia. Am J Cardiol 1989;63:29E-32E.
14. Hung MJ, Cherng WJ. Comparison of white blood cell counts in
acute myocardial infarction patients with significant versus in-
significant coronary artery disease. Am J Cardiol 2003;91:
1339-42.
15. Oshima S, Ogawa H, Yasue H, et al. Increased plasma fi-
brinopeptide A levels during attacks induced by hyperventilation
in patients with coronary vasospastic angina. J Am Coll Cardiol
1989;14:150-4.
16. Ong P, Athanasiadis A, Hill S, et al. Coronary artery spasm as a fre-
quent cause of acute coronary syndrome: the CASPAR (Coronary
Artery Spasm in Patients with Acute Coronary Syndrome) Study. J
Am Coll Cardiol 2008;52:523-7.
17. Hung MJ, Cheng CW, Yang NI, et al. Coronary vasospasm-induced
acute coronary syndrome complicated by life-threatening car-
diac arrhythmias in patients without hemodynamically signifi-
cant coronary artery disease. Int J Cardiol 2007;117:37-44.
18. Hackett D, Larkin S, Chierchia S, et al. Induction of coronary
artery spasm by a direct local action of ergonovine. Circulation
1987;75:577-82.
19. Scanlon PJ, Faxon DP, Audet AM, et al. ACC/AHA guidelines for
coronary angiography. A report of the American College of Cardi-
ology/American Heart Association Task Force on practice guide-
lines (Committee on Coronary Angiography). Developed in col-
laboration with the Society for Cardiac Angiography and Inter-
ventions. J Am Coll Cardiol 1999;33:1756-824.
20. Baim DS. Coronary angiography. In: Baim DS, eds. Grossman’s
Cardiac Catheterization, Angiography, and Intervention. 7th
ed.
Philadelphia: Lippincott Williams & Wilkins; 2006:215.
21. Bertrand ME, LaBlanche JM, Tilmant PY, et al. Frequency of
provoked coronary arterial spasm in 1089 consecutive patients
undergoing coronary arteriography. Circulation 1982;65:1299-
306.
22. Buxton A, Goldberg S, Hirshfeld JW, et al. Refractory ergonovine-
induced coronary vasospasm: importance of intracoronary nitro-
glycerin. Am J Cardiol 1980;46:329-34.
23. Takagi Y, Yasuda S, Takahashi J, et al. Clinical implications of prov-
ocation tests for coronary artery spasm: safety, arrhythmic com-
plications, and prognostic impact: Multicentre Registry Study of
the Japanese Coronary Spasm Association. Eur Heart J 2012 July
10 [E-pub ahead of print], doi: 10.1093/eurheartj/ ehs199.
24. Hung MY, Hung MJ, Cheng CW, et al. Safety and predictors of a
positive result of intracoronary ergonovine testing in patients
with ischemic heart disease without hemodynamically signifi-
cant coronary artery stenosis in Taiwan. Acta Cardiol Sin 2007;
23:150-9.
25. Beltrame JF, Sasayama S, Maseri A. Racial heterogeneity in coro-
nary artery vasomotor reactivity: differences between Japanese
and Caucasian patients. J Am Coll Cardiol 1999;33:1442-52.
26. Hung MJ, Cherng WJ, Yang NI, et al. Relation of high-sensitivity
C-reactive protein level with coronary vasospastic angina pec-
toris in patients without hemodynamically significant coronary
artery disease. Am J Cardiol 2005;96:1484-90.
27. Hung MJ, Cherng WJ, Cheng CW, et al. Comparison of serum lev-
els of inflammatory markers in patients with coronary vaso-
spasm without significant fixed coronary artery disease versus
patients with stable angina pectoris and acute coronary syn-
dromes with significant fixed coronary artery disease. Am J
Cardiol 2006;97:1429-34.
28. Sugiishi M, Takatsu F. Cigarette smoking is a major risk factor for
coronary spasm. Circulation 1993;87:76-9.
29. Hung MY, Hsu KH, Hung MJ, et al. Interaction between cigarette
smoking and high-sensitivity C-reactive protein in the develop-
ment of coronary vasospasm in patients without hemodyna-
mically significant coronary artery disease. Am J Med Sci
2009;338:440-6.
30. Hung MY, Hsu KH, Hung MJ, et al. Interactions among gender,
age, hypertension and C-reactive protein in coronary
vasospasm. Eur J Clin Invest 2010;40:1094-103.
31. de Lemons JA, O’Rourke RA. Unstable angina and non-ST-seg-
ment elevation myocardial infarction. In: Fuster V, Eds. Hurst’s
the Heart. 12th
ed. New York: McGraw-Hill; 2008: 1371-2.
32. Pinto DS, Beltrame JF, Crea F. Variant angina. Available at: http://
www.uptodate.com/contents/variant-angina?source=search_
result&search=variant+angina&selectedT itle=1%7E103.
Accessed August 24, 2012.
33. Tanabe Y, Itoh E, Suzuki K, et al. Limited role of coronary
angioplasty and stenting in coronary spastic angina with organic
stenosis. J Am Coll Cardiol 2002;39:1120-6.
34. Gaspardone A, Tomai F, Versaci F, et al. Coronary artery stent
placement in patients with variant angina refractory to medical
treatment. Am J Cardiol 1999;84:96-8.
35. Chou HH, Lim KE, Ko YL. Treatment of spontaneous left main cor-
onary artery spasm with a drug-eluting stent. Acta Cardiol Sin
2009;25:43-6.
36. Chou MT, Huang TY. Coronary stenting for coronary vasospasm
complicated with refractory ventricular tachycardia and fibril-
lation. Acta Cardiol Sin 2012;28:145-7.
37. Yuksel UC, Celik T, Iyisoy A, et al. Polymorphic ventricular tachy-
cardia induced by coronary vasospasm: a malignant case of vari-
ant angina. Int J Cardiol 2007;121:210-2.
38. Cannon CP, Braunwald E. Unstable angina and non-ST elevation
myocardial infarction. In: Libby P, Eds. Braunwald’s Heart Dis-
ease: A Textbook of Cardiovascular Medicine. 8th
ed. Philadel-
phia: Saunders, 2008:1339.
39. Plass CA, Sabdyusheva-Litschauer I, Bernhart A, et al. Time
course of endothelium-dependent and -independent coronary
vasomotor response to coronary balloons and stents: compari-
son of plain and drug-eluting balloons and stents. J Am Coll
9 Acta Cardiol Sin 2013;29:1�10
Inflammation and Coronary Vasospasm

Cardiol Intv 2012;5:741-51.
40. Yasue H, Takizawa A, Nagao M, et al. Long-term prognosis for pa-
tients with variant angina and influential factors. Circulation
1988;78:1-9.
41. Walling A, Waters DD, Miller DD, et al. Long-term prognosis of
patients with variant angina. Circulation 1987;76:990-7.
42. Hung MJ, Hung MY, Cheng CW, et al. Comparison of clinical char-
acteristics and prognosis in Taiwanese patients with coronary
vasospastic angina pectoris without significant fixed coronary ar-
tery disease versus patients with significant fixed coronary artery
disease and either stable angina pectoris or acute coronary syn-
dromes. Am J Med Sci 2007;334:160-7.
43. Ueda O, Kohchi K, Kishi Y, et al. Long lasting spasticity in con-
trolled vasospastic angina. Heart 1999;81:528-32.
44. Ito T, Yasue H, Yoshimura M, et al. Paraoxonase gene Gln192Arg
(Q192R) polymorphism is associated with coronary artery
spasm. Hum Genet 2002;110:89-94.
45. Murase Y, Yamada Y, Hirashiki A, et al. Genetic risk and gene-
environment interaction in coronary artery spasm in Japanese
men and women. Eur Heart J 2004;25:970-7.
46. Kakkar R, Ye B, Stoller DA, et al. Spontaneous coronary vaso-
spasm in KATP mutant mice arises from a smooth muscle-
extrinsic process. Circ Res 2006;98:682-9.
47. Qipshidze N, Metreveli N, Lominadze D, et al. Folic acid improves
acetylcholine-induced vasoconstriction of coronary vessels iso-
lated from hyperhomocysteinemic mice: an implication to coro-
nary vasospasm. J Cell Physiol 2011;226:2712-20.
48. Kandabashi T, Shimokawa H, Miyata K, et al. Inhibition of myosin
phosphatase by upregulated rho-kinase plays a key role for coro-
nary artery spasm in a porcine model with interleukin-1 beta.
Circulation 2000;101:1319-23.
49. Kandabashi T, Shimokawa H, Miyata K, et al. Evidence for protein
kinase C-mediated activation of Rho-kinase in a porcine model of
coronary artery spasm. Arterioscler Thromb Vasc Biol 2003;
23:2209-14.
50. Hung MJ, Cherng WJ, Hung MY, et al. Increased leukocyte Rho-as-
sociated coiled-coil containing protein kinase activity predicts
the presence and severity of coronary vasospastic angina. Ath-
erosclerosis 2012;221:521-6.
51. Nohria A, Grunert ME, Rikitake Y, et al. Rho kinase inhibition im-
proves endothelial function in human subjects with coronary ar-
tery disease. Circ Res 2006;99:1426-32.
52. Lewis JR, Kisilevsky R, Armstrong PW. Prinzmetal’s angina, nor-
mal coronary arteries and pericarditis. Can Med Assoc J
1978;119:36-9.
53. Hizume T, Morikawa K, Takaki A, et al. Sustained elevation of se-
rum cortisol level causes sensitization of coronary vasocon-
stricting responses in pigs in vivo: a possible link between stress
and coronary vasospasm. Circ Res 2006;99:767-75.
54. Yasue H, Hirai N, Mizuno Y, et al. Low-grade inflammation,
thrombogenicity, and atherogenic lipid profile in cigarette
smokers. Circ J 2006;70:8-13.
55. Kounis NG, Zavras GM. Histamine-induced coronary artery
spasm: the concept of allergic angina. Br J Clin Pract 1991;
45:121-8.
56. Chang PH, Hung MJ, Yeh KY, et al. Oxaliplatin-induced coronary
vasospasm manifesting as Kounis syndrome: a case report. J Clin
Oncol 2011;29:e776-8.
57. Busse WW, Lemanske RF Jr. Asthma. N Engl J Med 2001;
344:350-62.
58. Forman MB, Oates JA, Robertson D, et al. Increased adventitial
mast cells in a patient with coronary spasm. N Engl J Med
1985;313:1138-41.
59. Kohchi K, Takebayashi S, Hiroki T, et al. Significance of adventitial
inflammation of the coronary artery in patients with unstable
angina: results at autopsy. Circulation 1985;71:709-16.
60. Morikawa Y, Uemura S, Ishigami K, et al. Morphological features
of coronary arteries in patients with coronary spastic angina: as-
sessment with intracoronary optical coherence tomography. Int
J Cardiol 2011;146:334-40.
61. Tanaka A, Shimada K, Tearney GJ, et al. Conformational change in
coronary artery structure assessed by optical coherence tomo-
graphy in patients with vasospastic angina. J Am Coll Cardiol
2011;58:1608-13.
62. Hung MJ, Cherng WJ, Hung MY, et al. Interleukin-6 inhibits
endothelial nitric oxide synthase activation and increases endo-
thelial nitric oxide synthase binding to stabilized caveolin-1 in
human vascular endothelial cells. J Hypertens 2010;28:940-51.
63. Drab M, Verkade P, Elger M, et al. Loss of caveolae, vascular dys-
function, and pulmonary defects in caveolin-1 gene-disrupted
mice. Science 2001;293:2449-52.
64. Noma K, Kihara Y, Higashi Y. Striking crosstalk of ROCK signaling
with endothelial function. J Cardiol 2012;60:1-6.
Acta Cardiol Sin 2013;29:1�10 10
Ming-Jui Hung et al.





























