Embed Size (px)
Citation preview

Biochimica et Biophysica Acta, 308 (1973) 1-8 Elsevier Scientific Publishing Company, Amsterdam - Printed in The Netherlands
BBA 97630
CONFORMATIONS OF N6-MONOSUBSTITUTED ADENINE DERIVATIVES
CRYSTAL STRUCTURE OF N6-METHYLADENINE
HELENE STERNGLANZ and CHARLES E. BUGG
Institute of Dental Research and Laboratory of Molecular Biology, University of Alabama in Bir- mingham University, Station, Birmingham, Ala. 35294 (U.S.A.)
(Received November 13th, 1972)
SUMMARY
Three-dimensional X-ray diffraction data were used to determine the crystal structure of N6-methyladenine, a base that occurs in modified DNAs and in the anti- cod on loop of Escherichia coli tRNA TM. Crystals of N6-methyladenine are monoclinic, space group P2z/c, with a=9.911(1), b=5.850(1), c = l 1.680(4)fit and fl=92.49(2) °. Intensity data were collected with an automated diffractometer. The structure was solved by direct methods and refined by least-squares to R=0.033. The conformation of the base is such that the methyl group is nearly coplanar with the purine ring and is pointing away from the imidazole moiety. This conformation is similar to that found for adenine derivatives with bulky substituents at the N(6)position. When in this conformation, N6-methyladenine could not form normal, complementary base pairs with thymine or uracil, and would therefore disrupt double-helical regions of nucleic acids. Such an effect might account for the biological roles of N6-methyladenine in modification and restriction processes, and in the anticodon loop of E. coli tRNA TM.
INTRODUCTION
Adenine derivatives that /lave substituents on the N(6)amino group occur as minor components of many nucleic acids z. Those derivatives with a single substi- tuent at the N(6) position would be expected to assume one of two conformations that are related to each other by a rotation of 180 ° around the C(6)-N(6)bond (Fig. 1). To maintain partial double-bond character in the C(6)-N(6) bond, the amino group must be approximately coplanar with the purine moiety, and, therefore, the substituent must point either toward (Fig. la) or away from (Fig. lb) the imidazole ring of the base. Since the position of the substituent will have major effects on the hydrogen- bonding capabilities of the base, the physical properties of nucleic acids that contain N(6) monosubstituted adenine bases will be greatly influenced by the relative stabili- ties of these two conformations. If the substituent were directed toward the imidazole
Abbreviations: m6Ade, N6-methyladenine; i~Ade, N6-(A2-isopentenyl)adenine; i6ms2Ade, N 6- (A 2-isopentenyl)-2-methylthioadenine.

2 I-L S q ' E R N G L A N Z , C. E. B U G G
H R R H
N N
H H
H H
H H
(Q) (b)
Fig. 1. Two feasible conformations for adenine derivatives with a single substituent, R, at the N(6) position, a, R pointing toward the imidazole ring; b, R pointing away from the imidazole ring.
ring, as shown in Fig. la, the base would be able to form Watson-Crick hydrogen- bonded pairs with uracil and thymine. However, adenine derivatives in the alternate configuration (Fig. lb) could not form normal Watson-Crick base pairs, because the R group would block the amino group from participating in complementary hydro- gen-bonding. Therefore, it is important to understand the influence of various substi- tuents upon the relative stability of these two conformational states.
Recent crystallographic studies have shown that the conformation depicted in Fig. lb is preferred by adenine derivatives that have bulky substituents at the N(6) position 2-4. In this paper, we describe the crystal structure and conformation of N6-methyladenine (m6Ade), a base found in some phage and bacterial DNAs s- 7, as well as in the anticodon loop of Escherichia eoli tRNA va~ (refs 8, 9). Our results in- dicate that even small aliphatic substituents at the N(6) position of adenine force the base to assume the conformation shown in Fig. lb. This finding suggests that the biological roles of m6Ade may be related to the effect of the methyl group on base pairing within nucleic acids.
EXPERIMENTAL
Clear, lath-shaped crystals of N6-methyladenine were obtained by slowly evaporating an acetonitrile-n-butylether solution of the base. Weissenberg and oscil- lation photographs showed that the crystals are monoclinic; the space group is P2t/c, as indicated by the systematic absence of reflections h 0 1 with I odd and 0 k 0 with k odd. A crystal fragment with approximate dimensions of 0.4, 0.4 and 0.2 mm was mounted on a Picker FACS-1 diffractomoter with its b axis slightly inclined to the axis of the diffractometer. Approximate cell parameters for use in collection of intensity data were calculated by aleast-squares analysis of the angular settings for ten medium- angle reflections (CuK~, 2----- 1.5418 A).
Intensity data were collected with the diffractomoter, by use of a scintillation counter, nickd-filtered copper radiation, and a 0-20 scanning technique. The scanning speed was l°/min, and a 20-s background measurement was performed at each termi- nus of the scans. Measurements were made for all 1170 unique reflections with 20~< 128 ° .
Immediately after data collection, accurate values for the cell parameters were determined by a least-squares analysis of 20 values for 12 high-angle reflections

CRYSTAL STRUCTURE OF N6-METHYLADENINE 3
(CuK~, 2=1.54051 A). These cell parameters were not significantly different from those obtained prior to the collection of intensity data. Crystal data are listed in Table I.
TABLE I
CRYSTAL DATA CuK0t = 1.5418 A
The reported standard deviations are double those obtained from the least-squares analysis. The density was measured by flotation in a mixture of benzene and carbon tetrachloride.
Data Value
Stoichiometry C6N sl--[7 Z 4 Space group P2Jc a 9.911(1) A b 5.850(1) A c 11.680(4) A fl 92.49 (2) p (calculated) 1.462 g- cm-a p (observed) 1.46 g. cm-3
8:4 cm -1
The intensitities were assigned variances, tr2(I), according to the statistics of the scan and background counts plus a correctional term (0.03 S) 2, S being the scan counts. The intensities and their variances were corrected for Lorentz and polarization factors, and absorption corrections were applied by using the program ORABS 1°. The data were then scaled by means of a Wilson H plot.
Trial coordinates for the 11 nonhydrogen atoms were easily determined by direct methods with the computer program MULTAN ~2. A modified version of the full-matrix least-squares program ORFLS :3 was used to refine the trial structure. The quantity minimized was Zw(Fo2--Fc2/k2) 2, with. k as a scale factor and the weight w equal to 1/tr2(Fo2). Scattering factors for the nonhydrogen atoms were from the International Tables for X-Ray Crystallography14; anomalous dispersion correction factors for these atoms were from Cromer and Liberman I s, and hydrogen- atom scattering factors were from Stewart et aL 16. All hydrogen atoms were located in difference Fourier maps that were calculated during the latter stages of refinement. Final cycles of refinement included all positional parameters, anisotropic temperature factors for the heavy atoms, isotropic temperature factors for the hydrogen atoms, and Zachariasen's ~7 isotropic extinction parameter g (as formulated by Coppens and HamiltonlS). The final R index (ZI I f o l - IFcl I/ZIFol) is 0.033, and the goodness-of-fit {[(~,w(Fo2--Fc2/k2)~/(m--s)]l/~, where m is the number of reflections used and s is the number of parameters refined}, is 2.19. During the last cycle of refinement, no parameter shifted more than 0.05 of its standard deviation. A final three-dimensional electron-density difference map showed no peaks or troughs that exceeded 0.19 e/A_ 3 in magnitude.

4 H. STERNGLANZ, C. E. BUGG
RESULTS
The molecular conformation and the heavy-atom thermal ellipsoids are shown in Fig. 2. The molecule is in the conformation shown in Fig. lb, with the methyl substituent directed away from the imidazole ring. Tables of the final heavy-atom parameters and their estimated standard deviations and of the hydrogen atom para- meters and their estimated standard deviations have been deposited together with a table of the observed and calculated structure factors and a table of atomic deviations from a least-squares plane throughthe purine ring*. The estimated standard deviations in positional coordinates are 0.001-0.002 A for the carbon and nitrogen atoms and about 0.02 _~ for the hydrogen atoms. Bond lengths and angles are listed in Tables II and IIi, respectively; these values are in agreement with those found for N6-(A 2-iso- pentenyl)adenine 3. As in most crystal structures of purines, the purine moiety is slightly nonplanar, but none of the atoms in the purine ring deviate from the purine plane by more than 0.015 A. The amino group is slightly tilted, with atoms N(6) and C(methyl) each displaced by about 0.05 A from the purine plane.
H(2MET) ~H(N6) H ( I M E T ) ~ / ~
C (MET)~:~:~/~ N(6) ~ N(?')
H I 3 M E T ) ~ c ( 5 ) \ ~C(SI
Fig. 2. Conformation of N6-methylMenine. The nonhydrogen atoms are represented by thermal ellipsoids defined by the principal axes of thermal vibralion and scaled to include 50 ~ probability. The hydrogen atoms are represented by spheres of 0.1-A. radius (this drawing was prepared by using the computer program ORTEP21).
The hydrogen-bonding and crystal-packing schemes are depicted in Fig. 3. The pattern of hydrogen bonding between bases is the same as that in the crystal structures of N6-(A2-isopentenyl)adenine (i6Ade) and N6-(A2-isopentenyl)-2-methyl - thioadenine (i6ms2Ade) 2' 3. The bases are hydrogen bonded to form continuous rib- bons, wherein adjacent bases are linked across crystallographic inversion centers by N(9)-H---N(3) and N(6)-H---N(7) hydrogen bonds. Atom N(1) does not participate in hydrogen bonding. The ribbons of hydrogen-bonded bases are stacked in the b direction, and separated by an interplanar spacing of 3.42 A. Fig, 4 shows the base- stacking pattern. As in numerous other crystal structures of adenine derivatives, and
* These tables can be obtained on request from the Elsevier Publishing Company, BBA Data Deposition, P.O. Box 330 Amsterdam, The Netherlands. Reference should be made to Number BBA/DD/003/97630/308 0973) 1-8.
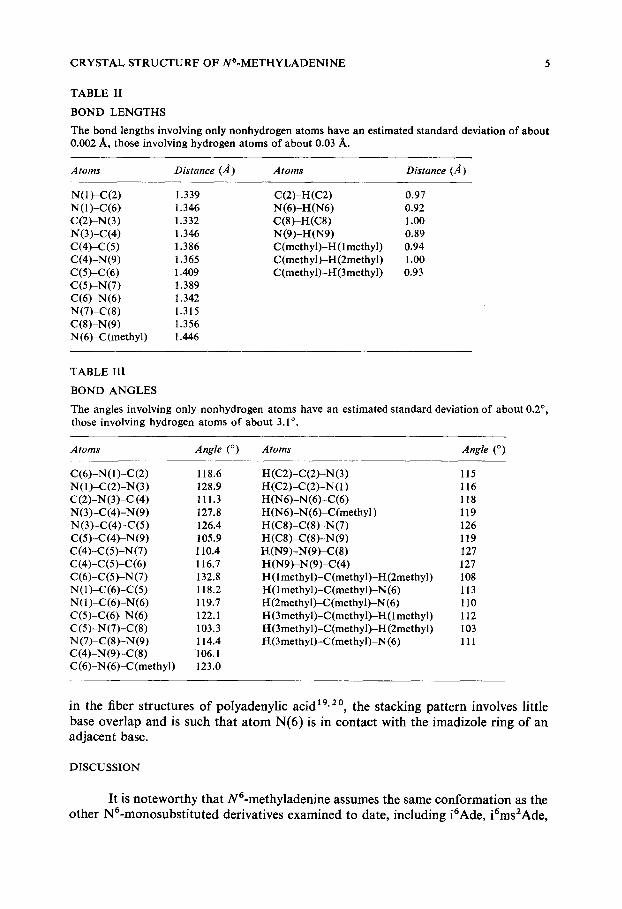
CRYSTAL STRUCTURF OF N6-METHYLADENINE 5
TABLE 1I
BOND LENGTHS
The bond lengths involving only nonhydrogen atoms have an estimated standard deviation of about 0.002 A, those involving hydrogen atoms of about 0.03 A.
A toms Distance (,4) A toms Distance (A)
N(1)-C(2) 1.339 N (1)-C(6) 1.346 C(2)-N(3) 1.332 N(3)-C(4) 1.346 C(4)-C(5) 1.386 C(4)-N(9) 1.365 C(5)-C(6) 1.409 C(5)-N(7) 1.389 C(6)-N(6) 1.342 N(7)-C(8) 1.315 C(8)-N(9) 1.356 N (6)-C(methyl) 1.446
C(2)-H(C2) 0.97 N(6)-H(N6) 0.92 C(8)-H(C8) 1.oo N(9)-H(N9) 0.89 C(methyl)-H(lmethyl) 0.94 C(methyl}-H(2methyl) 1.00 C (methyl)-I-[(3 methyl) 0.93
TABLE III
BOND ANGLES
The angles involving only nonhydrogen atoms have an estimated standard deviation of about 0.2 °, those involving hydrogen atoms of about 3.1°.
Atoms Angle (o) Atoms Angle (o)
C(6)-N(1)-C(2) 118.6 N(I)-C(2)-N(3) 128.9 C(2)-N(3)-C (4) 111.3 N(3)-C(4)-N(9) 127.8 N (3)-C(4)-C(5) 126.4 C(5)-C(4)-N(9) 105.9 C0)-C(5)-N(7) 110.4 c(4)-c(5)-c(6) 116.7 C(6)-C(5)-N (7) 132.8 N(1)-C(6)-C(5) 118.2 N(1)~(6)-N(6) 119.7 C(5)-C(6)-N(6) 122.1 C(5)-N(7)-C(8) 103.3 N (7)-C(8)-N(9) 114.4 C(4)-N(9)-C(8) 106.1 C(6)-N(6)-C(methyl) 123.0
H(C2)-C(2)-N(3) 115 H(C2)-C(2)-N(1) 116 H (N6)-N(6)-C(6) 118 H (N6)-N(6)-C(methyl) 119 H(C8)-C(8)-N(7) 126 H(C8)-C(8)-N(9) 119 H(N9)-N(9)-C(8) 127 H(N9)-N(9)-C(4) 127 H(lmethyl)-C (methyl)-H(2methyl) 108 ~(1 methyl)-C(methyl)-N (6) 113 l-[(2methyl)-C(methyl)-N (6) 110 H (3methyl)-C(methyl)-H(1 methyl) 112 H(3methyl)-C(methyl)-H (2methyl) 103 H(3methyl)-C (methyl)-N (6) 111
in the fiber s t ructures o f po lyadenyl ic acid 19,2°, the s tacking pa t t e rn involves l i t t le base ove r l ap and is such tha t a t o m N(6) is in contac t wi th the imadizole r ing o f an ad jacen t base.
DISCUSSION
It is no tewor thy tha t N6-methyladenine assumes the same confo rma t ion as the o ther N6-monosubs t i t u t ed derivat ives examined to date, inc luding i6Ade, i6ms2Ade,

6 H. STERNGLANZ, C. E. BUGG
Fig. 3. The crystal packing scheme as viewed down the b axis. Hydrogen-bond distances and angles are shown (this drawing was prepared by using the computer program ORTEP2~).
J N
N
Fig. 4. Base stacking pattern as viewed perpendicular to the plane of the base. The bases are par- allel and are separated by an interplanar spacing of 3.42 A.
and N ~- [2-(4-imidazolyl)-ethyl]adenine 2- 4. This finding indicates that even relatively small aliphatic groups at the N(6) position tend to point away from the imidazole ring. Examination of space-filling models suggests that if the molecules were in the conformation depicted in Fig. la, the methyl-hydrogen atoms would form close con- tacts with a tom N(7); however, the methyl group does not suffer crowding when directed away from the imidazole ring. Since the observed conformation has now been found in four different crystal structures, it is unlikely to be simply a consequence of solid-state forces. Therefore, it appears that the most stable conformation of N 6- monosubstituted adenine derivatives is the one depicted in Fig. 1 b.

CRYSTAL STRUCTURE OF N6-METI-IYLADENINE 7
Assuming that the conformation of m6Ade is the same in polynucleotides as in this crystal structure, one would expect that this base might have a considerable effect on the secondary and tertiary structures of nucleic acids. In the observed confor- mation, the methyl group would block the formation of normal Watson-Crick base pairs between adenine and either thymine or uracil. Thus methylation of adenine residues would tend to disrupt the regularity of double helical regions. Such effects might account for the biological roles of m6Ade within nucleic acids.
For example, m6Ade plays a major part in DNA modification and restriction processes. Bacterial modification enzymes are methylases that can convert certain adenine residues of DNA to m6Ade. The modification enzymes methylate only those adenine bases which occur at DNA sites that are normally recognized by bacterial restriction enzymes. The restriction enzymes are endonucleases that can cleave unmodified DNA molecules. After modification, however, the specific sites that contain m6Ade are no longer recognized by restriction enzymes. Our results indicate that m6Ade residues might have a major effect on the secondary structure of DNA at restriction sites. When in the conformation found in this crystal structure, m6Ade bases would interfere with base-pairing at these sites, thereby causing considerable distortions in the regions that are normally cleaved by restriction enzymes. Such distortions might be sufficient to inhibit the binding of restriction enzymes, thus accounting for the effect of m6Ade.
The minor base, m6Ade, is also found in the anticodon loop of E. coli tRNA TM, where it occupies the position adjacent to the 3' end of the anticodon triplet 9. In several other tRNAs this position is occupied by i6Ade, which also assumes a confor- mation that would inhibit normal Watson-Crick hydrogen bonding. It has been sug- gested that by disrupting complementary pairing within tRNA, i6Ade helps to main- tain the anticodon loop in a single stranded conformation that enhances codon-anti- codon interactions. Conceivably, m6Ade could influence the properties of tRNA vai through similar effects.
ACKNOWLEDGEMENT
We are indebted to Miss Catherine Sims for assistance with the preparation of this manuscript. This work was supported by N.I.H. Grants CA-12159, DE-02670 and RR-145.
REFERENCES
1 Hall, R. H. (1971) The Modified Nucleosides in Nucleic Acids, pp. 257-280, Columbia University Press, New York
2 McMullan, R. K. and Sundaralingam, M. (1971) J. Am. Chem. Soc. 93, 7050-7054 3 Bugg, C. E. and Thewalt, U. (1972) Biochem. Biophys. Res. Commun. 46, 779-784 4 Thewalt, U. and Bugg, C. E. (1972) Acta Crystallogr. B28, 1767-1773 5 Arber, W. and Lirm, S. (1969) Annu. Rev. Biochem. 38, 467-500 6 Smith, J. D., Arber, W. and Kiihnlein, U. (1972) J. MoL Biol. 63, 1-8 7 Meselson, M., Yvan, R. and Heywood, J. (1972) Annu. Rev. Biochem. 41,447-466 8 Yaniv, M. and Barrell, B. G. (1969) Nature 222, 278-279 9 Kimura, F., Harada, F. and Nishimura, S. (1971) Biochemistry 10, 3277-3283
10 Wehe, D. J., Busing, W. R. and Levy, H. A. (1962) ORABS, A Fortran Prooramfor Calculating Sinole Crystal Absorption Corrections, Report ORNL-TM-229, Oak Ridge National Laboratory, Tennessee

8 H. STERNGLANZ, C. E. BUGG
11 Wilson, A. J. C. (1942) Nature 150, 151-152 12 Germain, G., Main, P. and Woolfson, M. M. (1971) Acta Crystallogr. A27, 368-376 13 Busing, W. R., Martin, K. O. and Levy, H. A. (1962) ORFLS, A Fortran Crystalloyraphic
Least-Squares Pro#ram, Report ORNL-TM-305, Oak Ridge National Laboratory, Tennessee 14 International Tables for X-Ray Crystallo#raphy (1962) Vol. III, pp. 202-205, Kynoch Press,
Birmingham 15 Cromer, D. T. and Liberman, D. (1970) J. Chem. Phys. 53, 1891-1898 16 Stewart, R. F., Davidson, E. R. and Simpson, W. T. (1965) J. Chem. Phys. 42, 3175-3187 17 Zachariasen, W. I-L (1963) Acta Crystallogr. 16, 1139-1144 18 Coppens, P. and Hamilton, W. C. (1970) Acta Crystallogr. A26, 71-83 19 Bugg, C. E., Thomas, J. M., Sundaralingam, M. and Rao, S. T. (1971) Biopolymers 10, 175-219 20 Bugg, C. E. (1972) in The Jerusalem Symposia on Quantum Chemistry and Biochemistry, Vol. 1V,
The Purines-Theory and Experiment (Bergmann, E. D. and Pullman, B., eds), pp. 178-204, The Israel Academy of Sciences and Humanities, Jerusalem
21 Johnson, C. K. (1965) ORTEP, A Fortran Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Report ORNL-3794, Oak Ridge National Laboratory, Oak Ridge, Tennessee





























