Embed Size (px)
Citation preview

OLIGONUCLEOTIDES 16:323–335 (2006)© Mary Ann Liebert, Inc.
Comparison of Different Strategies to Select Aptamers Against a Transmembrane Protein Target
CARINE PESTOURIE,1 LAURA CERCHIA,2 KARINE GOMBERT,1 YOUSSEF AISSOUNI,3
JOCELYNE BOULAY,3 VITTORIO DE FRANCISCIS,2 DOMENICO LIBRI,3
BERTRAND TAVITIAN,1 and FRÉDÉRIC DUCONGÉ1
ABSTRACT
Binding of aptamers is dependent on their target conformation, which in turn is conditioned by thetarget’s environment. Therefore, selection of aptamers against the active forms of membrane pro-teins could require their correct membrane insertion in order to maintain their native conformation.Here, we compare different SELEX strategies to identify aptamers against the mutated form of themembrane receptor tyrosine kinase RETC634Y. (1) selections S1 and S2 against living cells trans-formed to express the protein yielded a minority of RET-targeted aptamers while the bulk of ap-tamers recognized more abundant membrane proteins, suggesting that a high level of expression ofthe target protein is crucial to allow the isolation of aptamers at cell surface; (2) selection S3 againstthe purified extracellular moiety of RET yielded aptamers unable to recognize RET expressed at thecell membrane; (3) crossover selections S4 and S5 alternating cells and recombinant RET enhancedthe enrichment of the aptamers directed against RET; however, these aptamers displayed a weakeraffinity for Ret than those obtained with S1 and S2. In our case, using transformed cell lines as thepartitioning matrix during SELEX appears to be essential in order to obtain aptamers able to recog-nize the RET receptor tyrosine kinase in its physiologic environment.
INTRODUCTION
APTAMERS are nucleic acid sequences selected by aniterative selection procedure to act as specific li-
gands for molecular targets. Among the myriad of three-dimensional structures that a large combinatorial pool ofsequences (1014–1015) may adopt, those who bind the de-sired target are favored by the selection procedure com-monly named SELEX (systematic evolution of ligandsby exponential enrichment) (Ellington and Szostak,1990; Osborne et al., 1997; Tuerk and Gold, 1990). Themost successful in vivo applications of aptamers havebeen described against biomedical extracellular markersthat are easier to access in vivo (for review see Pestourieet al. [2005]). Recently, the Food and Drug Administra-
tion has approved one aptamer (Macugen™), that in-hibits the human vascular endothelial growth factor 165(VEGF165), for the treatment of age-related macular de-generation and several aptamers are actually in clinicaltrials (Thiel, 2004). Remarkably, the majority of theseaptamers have been designed against soluble blood-cir-culating targets while few have been selected againstmembrane proteins (Chen et al., 2003; Cui et al., 2004;Daniels et al., 2002; Lupold et al., 2002; Mori et al.,2004) even though this class of proteins represent thevast majority of known drug targets.
Given the high specificity of the aptamers for their tar-get’s conformation, it is questionable whether classic SELEX using purified proteins represents a relevantstrategy for the selection of aptamers capable to bind to
1CEA, DSV, DRM, Service Hospitalier Frédéric Joliot, INSERM U803, Orsay, France.2Instituto per l’Endocrinologia e l’Oncologia Sperimentale “G. Salvatore” del CNR, Napoli, Italy.3Centre de Génétique Moléculaire, C.N.R.S., Gif sur Yvette, France.
323

TA
BL
E1.
DIF
FER
EN
TSE
LE
X S
TR
AT
EG
IES
AG
AIN
STR
ET
C63
4Y
Sele
ctio
nsS1
aS2
S3a
S4S5
Sele
ctio
n ty
peA
gain
st l
ivin
g ce
llsA
gain
st l
ivin
g ce
llsA
gain
st p
urif
ied
Cro
ssov
er s
elec
tion
Cro
ssov
er s
elec
tion
prot
ein
cells
/pro
tein
ste
p 1
cells
/pro
tein
ste
p 2
Star
ting
pool
1014
1014
1014
7thro
und
of S
114
seq
uenc
es f
rom
4th
roun
dof
S4
Tar
get
PC12
/ME
N2A
PC12
/ME
N2A
EC
-Ret
C63
4Y-P
AE
C-R
etC
634Y
-PA
PC12
/ME
N2A
Firs
t co
unte
rsel
ectio
nPC
12PC
12Ig
G b
eads
IgG
bea
ds—
Seco
nd c
ount
erse
lect
ion
PC12
/ME
N2B
PC12
——
—N
umbe
r of
rou
nds
1515
134
3
14 s
eque
nces
fro
m S
4 us
ed f
or S
5
a Sel
ectio
ns p
revi
ousl
y de
scri
bed
(Cer
chia
et a
l., 2
005)
.

membrane-inserted proteins in the context of live cells. Asan alternative, several studies have demonstrated the possi-bility to apply SELEX directly to complex biologic systems(viruses, parasites, cell membranes, cells) (Blank et al.,2001; Daniels et al., 2003; Hicke et al., 2001; Homann andGoringer, 1999; Morris et al., 1998; Ohuchi et al., 2006;Wang et al., 2000). In a previous work, we developed awhole-living cell SELEX protocol to target the transmem-brane receptor tyrosine kinase (RTK) RET (rearranged dur-ing transfection) in its natural environment (Cerchia et al.,2005). We used PC12 cells that express the human mutantdimerized receptor RETC634Y (PC12/MEN2A) as target forSELEX and performed counterselection against both PC12cells that do not express RET and PC12/MEN2B cells thatexpress the human monomeric mutant protein RETM918T.We found one aptamer, named D4, which bound specifi-cally to RET and blocked RET dimerization-dependent sig-naling pathways induced either by GDNF or by the C634Yactivating mutation. Interestingly, D4 did not bind to a re-combinant extracellular RET fragment, highlighting thestrength of our approach. However, this aptamer did notdistinguish the dimeric form of RET from the monomerform in spite of counterselection and represent a minorityof the selected sequences at the end of the SELEX (2 D4-related sequences of 67).
Therefore, the question remains open as to which oftwo SELEX strategies alternatives is the most advanta-geous: (1) SELEX against a recombinant protein, whichhas the advantage of a pure, defined target but the disad-vantage of lacking the natural membrane environmentwhich may be important for conformation or (2) SELEXagainst cells expressing the target protein, with the oppo-site advantages of a native target conformation but in amore complex environment.
Here, we address this issue by comparing differentstrategies to guide the SELEX towards RETC634Y-ap-tamers (Table 1). We performed a new SELEX againstwhole-living cells without counter-selection againstPC12/MEN2B, and a crossover SELEX alternatingRETC634Y expressing cells and the recombinant purifiedextracellular domain of the RetC634Y protein as targets.The crossover SELEX leads to a higher enrichment ofaptamers against RET. However, the selected aptamersusing pure whole-living cells SELEX display a better ap-parent Kd. This study brings new insights into the respec-tive advantages of each of these different methods for theselection of aptamers targeting membrane proteins.
MATERIALS AND METHODS
Cells, protein, and oligonucleotides
PC12 cells were grown in RPMI 1640 supplementedwith 10% horse serum and 5% heat-inactivated fetal calfserum. PC12/MEN2A and PC12/MEN2B cell lines,
expressing RETC634Y and RETM918T, respectively, weregrown in the same culture medium with the appropriate se-lection pressure, as reported previously (Califano et al.,1996). Purified recombinant EC-RetC634Y-PA fragments areexcreted by cells and recovered on immunoglobulin G(IgG) sepharose beads (Amersham Biosciences GE Health-care) as described (Cerchia et al., 2003). Oligonucleotideswere obtained from Eurogentec (Seraing, Belgium).
Whole-living cell SELEX
The first whole living cells SELEX S1 has been de-scribed (Cerchia et al., 2005). Our new cell SELEX S2has been performed in the same conditions except thecounterselection step against PC12/MEN2B that was re-placed by a second counter-selection against PC12 andminor modifications in the selection pressure summa-rized in Fig. 1B. This new cell SELEX was performedwith a new library named S2: 5�TAATACGACTCAC-T A T A G G G A G A C A A G A A T A A C ( N 5 0 ) T T C -GACAGGAGGCTCAC AACAGGA3�. (N50) indicateda random sequence of 50 nucleotides. Polymerase chainreaction (PCR) amplifications of this library was per-formed using a different set of primers to prevent cross-contamination between libraries (P10 and P30 or P10 and P20, respectively, for the S1 and S2). The primer sequences are: P30: 5�TCCTGTTGTGAGCCTCCTGT-CGTT3�; P20: 5�TCCTGTTGTGAGCCTCCTGTCG-AA3�; P10: 5�TAATACGACTCACTATAGGGAGA-CAAGAATAAACGCTCAA3�.
At the end of SELEX, sequences of the pools werecloned with TOPO-TA cloning kit (Invitrogen Life Tech-nologies) before sequencing. The sequences have beencompared by Clustal (Thompson et al., 1997) and struc-ture prediction were obtained by the Mfold software(Zuker, 2003).
SELEX on recombinant EC-RetC634Y-PA protein
Forty microliters of IgG sepharose beads or 3.6 pmol ofpurified recombinant EC-RetC634Y-PA protein immobi-lized on 40 �L of IgG sepharose beads as described (Cer-chia et al., 2003) were washed twice with NET 500 buffer(50 nM Tris-HCl pH 8.0, 500 nM NaCl, 0.05% NP40)and twice with RPMI-1640 before selection. Prior to use,the 2� fluoro-pyrimidine (2�F-Py)-RNA pool was heatedat 85°C for 5 minutes, cooled on ice for 5 minutes, and al-lowed to warm up at room temperature for 5 minutes todisrupt intermolecular interactions. Then all the selectionsteps were performed at 37°C. The 2�F-Py-RNA pool (1nmol) was incubated on the IgG sepharose beads in a finalvolume of 500 �L of RPMI-1640. After 15 minutes of in-cubation, sequences that were not retain to the beads wererecovered by centrifugation (1000g, 1 minute) and incu-bated 15 minutes with the EC-RetC634Y-PA first immobi-lized on IgG beads. Unbound and weakly associated 2�F-
APTAMER SELECTION STRATEGY 325
Py RNAs were washed off by two washes with RPMI-1640 and 2 washes with TEV protease buffer (50 mM TrisHCl pH8.0, 3 mM MgCl2, 0.5 mM ethylenediamine-tetraacetic acid [EDTA]). Then, the 2�F-Py RNA-RetC634Y
complexes were recovered by TEV protease cleavage andcentrifugation. The bound 2�F-Py RNAs were extractedby phenol-chloroform followed by ethanol precipitation.After a DNAse digestion step, the isolated 2�F-Py RNAswere reverse-transcribed using SuperscriptII (InvitrogenLife Technologies) in a final volume of 100 �L and thenamplified by PCR. The resulting double stranded DNAtemplates were in vitro transcribed into 2�F-Py RNA be-fore a subsequent round of selection.
Restriction fragment length polymorphism analysis
Ten microliters of PCR from different rounds of selec-tion were amplified by 8 rounds of PCR with [32P] 5�-end-labeled primer P10 (15.106 Bq/pmole). The radiolabeledPCR products (6.6.103 Bq for each sample) was digested at37°C during 1hour by a combination of RsaI, AluI, HaeIII,and HinPI endonuclease (10 units each) in REact I® buffer(Invitrogen Life Technologies). After an ethanol precipita-tion, the digested samples were subsequently analyzed us-ing electrophoresis on a denaturing polyacrylamide gel(20% acrylamide/bis acrylamide 19:1, 7 M urea, 1�TBE). After the electrophoresis, the dried gel was exposedon imaging plate and analyzed using PhosphorImager.
Binding experiments
2�F-Py RNAs were 5� end dephosphorylated usingbacterial alcaline phosphatase (Invitrogen Life Technolo-
gies) before [32P] 5�-end-labeled (3.103 Bq.pmole�1) us-ing T4 kinase (Invitrogen Life Technologies) accordingto the supplier’s instructions. 2.105 Cells were grown 48hours on 24-well collagen-treated plates (BD Falcon).Various concentrations of aptamers were incubated 10minutes at 37°C on cells in 200 �L of RPMI in the pres-ence of 100 �g � mL�1 of polyInosine (Sigma, St. Louis,MO) as nonspecific competitor. After 5 washes with 500�L of RPMI 1640, bound sequences were recovered in350 �L of sodium dodecyl sulfate (SDS) 0.6 % and esti-mated by Cerenkov counting. The amount of radioactiv-ity recovered from each well was normalized by measur-ing their protein content using the BCA™ Protein AssayKit (Pierce). Nonspecific binding of 2�F-Py RNAs oncells was assessed using a naive pool (i.e., the startingpool of the selection), and these background values weresubtracted from the values obtain from the aptamer at thesame concentration.
Apparent Kd and total target concentration (Cmax) foreach aptamers were determined by Scatchard analysis ac-cording to the equation:
[bound aptamer]/[aptamer] ��(1/Kd) � [bound aptamer] � [Cmax]/Kd
Competitive binding experiments were performed in thesame conditions except the addition of cold aptamer dur-ing the incubation period or time on cells.
UV cross-linking of the selected aptamers
Aptamers were uniformly [32P]-radiolabeled (50.103
Bq.pmole�1) by incorporation of GTP�32P during in
PESTOURIE ET AL.326
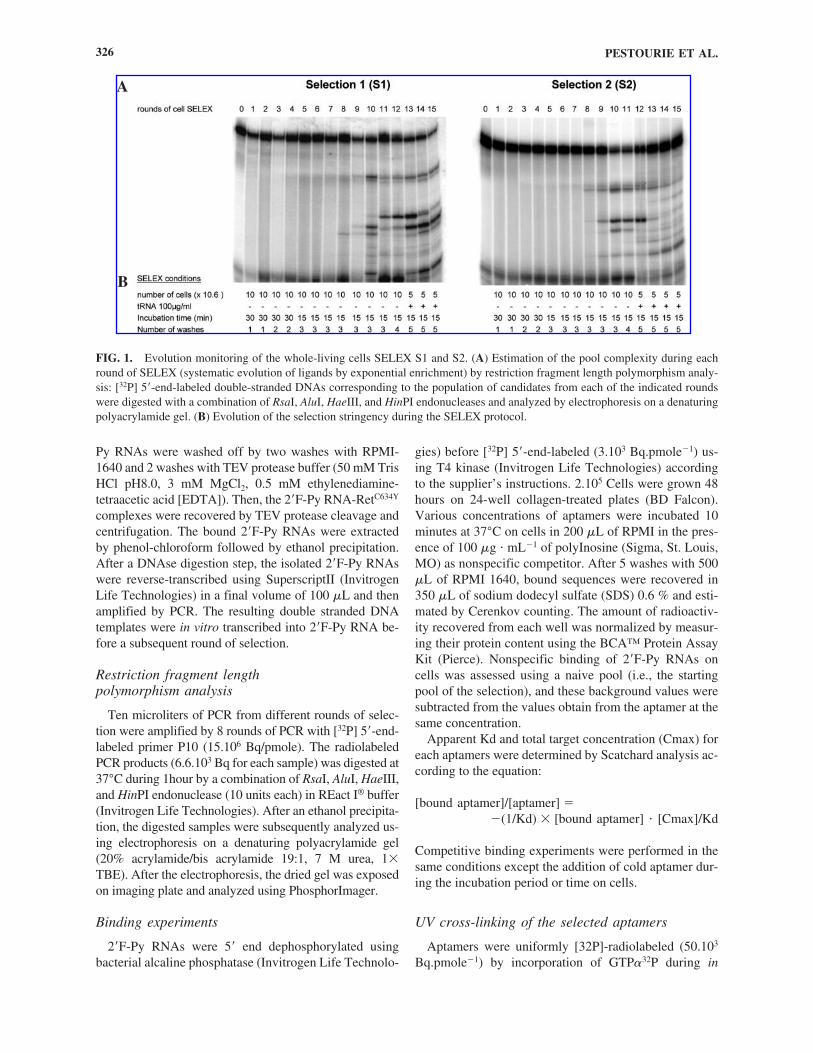
FIG. 1. Evolution monitoring of the whole-living cells SELEX S1 and S2. (A) Estimation of the pool complexity during eachround of SELEX (systematic evolution of ligands by exponential enrichment) by restriction fragment length polymorphism analy-sis: [32P] 5�-end-labeled double-stranded DNAs corresponding to the population of candidates from each of the indicated roundswere digested with a combination of RsaI, AluI, HaeIII, and HinPI endonucleases and analyzed by electrophoresis on a denaturingpolyacrylamide gel. (B) Evolution of the selection stringency during the SELEX protocol.
A
B

vitro transcription. Three hundred nanomoles of radiola-beled aptamers were incubated 15 minutes at 37°C on1.106 monolayer cells grown on 6 wells collagen-treatedplates (BD Falcon), in the presence of 250 �g � ml�1 oftotal RNA (Sigma) as nonspecific competitor. Unboundsequences were removed and 200 �L of fresh RPMI-1640 were added to the cells before UV irradiation of0.25 J.cm�2. After irradiation, UV-crosslinked 2�F-PyRNA-cells complexes were subjected to RNase digestion(30 minutes, 37°C, 10 �g.�L�1 RNase A and 50U.�L�1
RNase T1) in 300 �L of PBS before extraction withRIPA buffer and analyzed using 6% sodium dodecyl sul-fate-polyacrylamide gel electrophoresis (SDS-PAGE)and autoradiography.
RESULTS
Selection of aptamers on PC12/MEN2A cells
As a first approach to select aptamers against the mu-tant RETC634Y protein in its native conformation, we per-formed SELEX on whole-living PC12/MEN2A cells ex-pressing the membrane-inserted form of the protein(Califano et al., 1996). In our selection system, the stableexpression of constitutively activated RET leads to achange in the morphology of the PC12 cells that switchfrom a round morphology to an elongated shape withneurite-like prolongations (Califano et al., 1996). As amatter of rule, each round of SELEX is performed in twosequential steps: a first negative or counterselection stepagainst the capture system in the absence of the desiredtarget, followed by a positive selection on the same cap-ture system with the target. The members of the oligonu-cleotide library that bind to the capture system during thecounterselection are discarded while those that bind dur-ing the positive selection are kept and amplified. This en-sures the selection of aptamers directed against the targetbut not the other elements of the aptamer capturing moi-ety. We reasoned that counterselection is likely to beeven more important when selection is done on complextargets such as live cells and compared two selectionstrategies (S1 and S2) differing essentially in the counterselection steps.
In the S1 selection previously done (Cerchia et al.,2005), counterselection was performed first on round-shaped PC12 and then on PC12/MEN2B cells with theelongated morphology (similar to PC12/MEN2A), whilein the S2 selection, both rounds of counter selection weredone against PC12 (Table 1). The selections steps wereperformed as previously described (Cerchia et al., 2005).Briefly, the selection was started with a pool of approxi-mately 1014 sequences, containing a 50-base random re-gion flanked by two primer hybridization sites. We used2�F-Py-modified RNA, which are both nuclease-resistantand compatible with transcription (Cerchia et al., 2005).
The unbound sequences after the two counterselectionsteps were then incubated with the PC12/MEN2A cellsand, after washing, the bound sequences were recoveredby phenol-chloroform extraction amplified by reversetranscriptase-polymerase chain reaction (RT-PCR) andtranscribed into 2�F-Py-RNA. During the selection pro-cess, we progressively increased the selective pressure byincreasing the number of washes and decreasing the in-cubation time (Fig. 1B). We monitored the evolution ofthe pool during each round by restriction fragment lengthpolymorphism analysis (RFLP) with a combination ofRsaI, AluI, HaeIII, and HinPI endonucleases, all ofwhich recognize short restriction sites (Bartel andSzostak, 1993) (Fig. 1A). During the first rounds of theselection, the sequence composition of the pools was ex-tremely heterogeneous and lead to a smearing RFLP pro-file. After 8 rounds of selection, some sequences werepredominantly amplified and dominated in abundancethe aptamer pool, resulting in discrete restriction bandsfor both selections. During rounds 8 to 11 and 8 to 10, forS1 and S2, respectively, RFLP profiles changed at eachround, indicating an evolution of the sequence distribu-tion in the library. The RFLP profile then remained un-changed in the next rounds (rounds 11 to 12 and 10 to 11,for S1 and S2, respectively), suggesting that the popula-tion had stopped to evolve under the selection pressure.Hence, during the following rounds the selection strin-gency was increased both by addition of tRNA as com-petitor and by decreasing the number of target cells (Fig.1B). This led to the appearance of new RFLP profiles andto their stabilization for both strategies.
No increase in the total amount of aptamers bound tothe cells was observed during the selection (data notshown). A plausible explanation could be that given thehigh concentration of 2�F-Py-RNA (1 �M) used at eachround, the background of unspecific binding hid the en-richment of specific sequences.
Identification of high-affinity PC12/MEN2A cells aptamers
After 15 rounds, the resulting RNA pools from S1 andS2 selection were cloned and sequenced. Sequences re-sulting from S2 were highly different from those result-ing from S1 (Table 2; Cerchia et al., 2005). Only one S2sequence, B25, was identical to the S1 aptamer D24, ex-cept for the primer sequences that were made intention-ally distinct in order to prevent cross-contaminations be-tween selections. Interestingly, D24 displays the sameaffinity than D4 as well a common conserved stem-inter-nal loop-stem predictive structure that has been previ-ously described to be sufficient for the binding to theRET protein (Fig. 2) (Cerchia et al., 2005). As for S1,three predominant sequences (B26, B11, B23) repre-sented over 50% of the pool. Most of the sequences weretested in a low concentration (100 nM) for their binding
APTAMER SELECTION STRATEGY 327

TA
BL
E2.
AN
AL
YSI
SO
FSE
QU
EN
CE
SIN
TH
EPO
OL
SA
FTE
RFI
FTE
EN
RO
UN
DS
OF
WH
OL
E-L
IVIN
GC
EL
LS
SEL
EX
Nam
eF
requ
ency
Sequ
ence
sK
d nM
Cm
ax
Liv
ing
cells
SE
LE
X S
1 (c
ount
er-s
elec
tion
agai
nst
PC12
and
PC
12/M
EN
2B)
D14
23/6
7G
GC
CA
UA
GC
GC
AC
CA
CC
AA
GA
GC
AA
AU
CC
CU
AA
GC
GC
GA
CU
CG
AG
UG
AG
C—
D12
19//6
7G
GG
CU
UC
AU
AA
GC
UA
CA
CC
GG
CC
AA
CG
CA
GA
AA
UG
CC
UU
AA
GC
CC
GA
UU
57�
934
2�
94
D30
06/6
7A
GG
CG
AG
CC
CG
AC
CA
CG
UC
AG
UA
UG
CU
AG
AC
AA
CA
AC
GC
CC
GC
GU
GG
UA
C64
� 1
953
1�
241
D71
05/6
7G
GC
CC
UU
AA
CG
CA
AA
AA
CG
AA
GG
AU
CA
UC
GA
UU
GA
UC
GC
CU
UA
UG
GG
CU
51�
19
265
� 1
37D
5104
/67
GA
CC
CG
UA
UG
AA
GG
UG
GC
GC
AG
GA
CA
CG
AC
CG
UC
UG
CA
AU
GA
GC
GA
GC
—D
7702
/67
GG
GC
CA
AU
CG
AA
GC
CG
GU
AA
UU
CC
CA
AA
CU
AA
CG
UG
CA
AA
CU
GC
AC
CC
GC
—D
401
/67
GC
GC
GG
GA
AU
AG
UA
UG
GA
AG
GA
UA
CG
UA
UA
CC
GU
GC
AA
UC
CA
GG
GC
AA
CG
35�
315
8�
18
D24
01/6
7G
GU
AU
GU
AG
GG
AA
UA
GC
AC
UU
UU
UU
UG
CG
UA
UA
CC
UA
CA
CC
GC
AG
CG
35�
315
8�
18
Indi
vidu
als
08/6
7�
/ND
Liv
ing
cells
SE
LE
X S
2 (t
wo
coun
ter-
sele
ctio
n ag
ains
t PC
12)
B26
15/6
6C
AA
AG
CG
UG
UA
UU
CU
CG
UG
AG
CC
GA
CC
AU
CG
UU
GC
GA
AC
AU
CC
CC
GG
—B
1110
/66
GG
CA
AU
CG
AA
GA
UG
AA
AC
UU
GC
CG
UC
AC
UA
UC
CC
AG
AG
CC
CA
GA
GG
GA
C17
0�
46
1063
� 3
42B
2309
/66
CC
CC
AA
CC
CG
AG
CC
UG
CU
AC
GC
AG
CC
AG
UA
GC
AU
AC
GC
UU
GC
GA
AA
CG
39�
834
4�
113
B37
03/6
6G
UU
CG
GC
UG
GC
CA
CU
AG
CA
AA
AA
CU
AG
GU
UA
GG
CG
GC
GA
AG
UG
CC
GG
AG
C—
B58
03/6
6U
CC
GU
CA
CC
CC
GA
GU
AC
UG
AG
GA
AC
GC
AA
AC
UC
AG
UG
UC
CA
UU
GU
GA
AA
CN
DB
6103
/66
AG
CC
AA
CG
UG
AC
CA
UC
UC
UU
CU
CG
AA
CG
AA
CG
CG
UA
AU
CC
UC
GC
GC
CG
AN
DB
802
/66
CG
AG
AC
UG
GU
GC
GA
GC
CC
UA
GA
GC
AU
UU
AU
GC
UC
CA
UA
CG
AU
UG
GU
AC
GA
ND
B43
02/6
6C
CC
CG
CU
UU
UU
GA
CG
UG
AU
CG
GA
CG
CG
UA
UC
GU
AA
CG
UC
AG
CA
GU
CG
AG
CN
DB
1302
/66
GC
AA
CU
GC
GU
UU
AA
UC
CC
AG
AG
UC
GA
CA
UA
CG
AG
CG
CC
AU
GA
UC
UG
CC
GN
DB
8202
/66
CG
CC
CG
AA
GA
GU
CU
UA
UG
AC
AC
AC
AA
GG
UG
UG
UC
CC
AA
GC
UG
UA
GN
DB
2501
/66
GG
UA
UG
UA
GG
GA
AU
AG
CA
CU
UU
UU
UU
GC
GU
AU
AC
CU
AC
AC
CG
CA
GC
G35
� 3
158
� 1
8In
divi
dual
s15
/66
�/N
D
S1 a
nd S
2. S
eque
nce
of R
NA
-2�F
-Py
apta
mer
s (5
�to
3�
from
lef
t to
rig
ht)
obta
ined
fro
m t
he s
elec
tions
S1
and
S2. F
or s
impl
icity
, fix
ed-p
rim
er s
eque
nce
at 5
�an
d 3�
extr
emiti
es a
re r
emov
ed. T
he f
requ
ency
indi
cate
s th
e cl
one
abun
danc
e of
a s
eque
nce.
For
the
D4-
rela
ted
apta
mer
s (D
4, D
24, a
nd B
25),
gra
y se
quen
ces
indi
cate
a c
onse
n-su
s pr
edic
tive
inte
rnal
loop
fla
nked
by
stem
reg
ion
form
ed b
y un
derl
ined
seq
uenc
es. B
indi
ng e
xper
imen
ts o
n PC
12/M
EN
2A c
ells
hav
e be
en p
erfo
rmed
at 1
00 n
M. I
n th
eco
lum
n fo
r K
d, “
—”
indi
cate
s no
sig
nifi
cant
bin
ding
at t
his
conc
entr
atio
n in
com
pari
son
to th
e st
artin
g po
ol a
nd N
D in
dica
te n
ot te
sted
for
bin
ding
. For
thos
e se
quen
ces
whi
ch d
ispl
ay a
ffin
ity, b
indi
ng c
urve
s w
ere
perf
orm
ed a
s de
scri
bed
(Cer
chia
et
al.,
2005
), t
o es
timat
e th
eir
appa
rent
Kd
(nM
) an
d th
eir
appa
rent
Cm
ax (
pM).
Sta
ndar
d de
viat
ions
wer
e de
term
ined
fro
m a
t lea
st f
our
inde
pend
ent e
xper
imen
ts.

capacity to PC12/MEN2A cells (Table 2). Surprisingly,no binding could be demonstrated at this concentrationfor the two sequences predominant in both selections,D14 and B26 (Table 2). One explanation, as has beenshown in the case of SELEX performed against phospho-lipids bilayers (Vlassov et al., 2001), could be that thesesequences require the formation of a supramolecularstructure with another sequence in order to bind to cells.However, we decided to focus our work on aptamers thatbound cells when tested individually.
The five other sequences predominantly selected fromS1 (D30, D71, D12) and S2 (B11 and B23) displayedaffinity for the PC12/MEN2A cells, and so did the groupof sequences with a common predicted structure D4,D24-B25. From S1, we previously identified D4 as an in-hibitor of Ret (Cerchia et al., 2005). Aptamers from S2were also assayed for the ability to inhibit RET signaling.However, neither B11 nor B23 could block the phospho-rylation of RetC634Y and of its downstream effector, theextracellular signal-regulated kinase (ERK) (data notshown).
In order to determine the apparent Kd and maximumconcentration of targets (Cmax) more precisely, we per-
formed binding assays to PC12/MEN2A cells in at leastthree independent experiments for each aptamer (Table 2).Most of the aptamers presented an apparent Kd around 50nM, except for aptamer B11 that exhibited an apparent Kdof approximately 200 nM. Interestingly, while the affinityof B11 is roughly 4 times lower than that of the other ap-tamers, its higher Cmax indicates that B11 has a highernumber of binding sites, which could explain why it waspreserved during the selection process.
It should be noted that although the apparent Kd deter-minations were well reproducible between the experi-ments while the estimations of apparent Cmax were some-what variable, except for the aptamer D4 (standarddeviation � 10% in comparison to 30%–50%, respec-tively). This may result from the fact that aptamers such asD4 binds to RET whose cellular expression is constant,while other aptamers bind to proteins whose expressionmay be more variable. Furthermore, RET under the con-trol of MMLV promoter is known to be highly expressedin PC12/MEN2A (Califano et al., 1996). Hence, the higherapparent Cmax of the other aptamers (two to six timehigher, see table 2) suggests that they were selected againsthighly expressed targets present at the cell surface.
APTAMER SELECTION STRATEGY 329
FIG. 2. Predicted structures from the E38-related aptamers and the D4-related aptamers. (A) Comparison of a secondary struc-ture prediction for the E38 and B69 aptamers. Structures were predicted using MFOLD software version 3.1 (Zuker, 2003). Con-sensus motif of E38-related aptamers (E38 and B69) are in gray, primer sequences are in bold. (B) Consensus motif of E38-relatedaptamers (E38 and B69) and D4-related aptamers. The consensus motif of D4-related aptamers was previously identified by com-parison of D24 and D4 predicted structure (Cerchia et al., 2005). X-Y represents a base pair.

Analysis of aptamer targets by UV-cross-link
The apparent Cmax was also found to vary betweenaptamers in a range from 260 pM to 1 nM, suggestingthat they had been selected against different targets orbinding sites on the cell surface. To test this hypothesis,we cross-linked radiolabeled aptamers to PC12,PC12/MEN2B, or PC12/MEN2A using UV irradiation.The cross-linked aptamer-protein complexes were di-gested by RNase A and RNase T1 before an SDS-PAGEto determine more precisely the size of the radiolabeledproteins by autoradiography (note that even if 2�F-Py-RNA are nuclease resistant, they could be digested usinghigh concentration of RNases). Out of the six aptamerstested, only three could be efficiently cross-linked totheir targets with the UV irradiation conditions used (Fig.3). Different molecular weights were found for the pro-teins radiolabeled by the individual aptamers, confirmingthat they bound to different target proteins. Aptamer B23cross-linked to a protein of 150 kd on the three differentcell lines, demonstrating that this protein is not RET andis not specific of the PC12/MEN2A cells, in spite of thecounterselection step. In contrast, aptamer B11 and D12crosslinked proteins (one protein around 180 kd and twoproteins around 100 kd, respectively) on PC12/MEN2Aand PC12/MEN2B cells but not on PC12 cells, demon-strating that counterselection had been partially effectiveduring the selection of these aptamers. Moreover, wecould not immunoprecipitate any of the cross-linked pro-teins with a RET antibody, which suggests they are notinvolved in a complex with RET (data not shown).
Crossover SELEX
In our previous S3 selection, aptamers selected againstthis purified recombinant protein were unable to recog-
nize the PC12/MEN2A cells (Cerchia et al., 2005), al-though, this protein has been proven to be functional(Cerchia et al., 2003). Therefore, we hypothesized thatusing sequences preselected on cells as a starting pool forSELEX against the purified protein could lead to identifyaptamers recognizing a common domain between the pu-rified protein and the protein in its native environment.So, we tested a crossover SELEX procedure (S4 and S5)in order to enrich the selection for aptamers binding to theRET protein in the pool selected on PC12/MEN2A cells.To this end, we alternated SELEX on cells with SELEXon the recombinant extracellular domain of RetC634Y
fused to protein A (EC-RetC634Y-PA). We chose to startfrom the pool obtained after the first 7 rounds of the S1SELEX, which was the last pool before the appearance ofa RFLP profile (Fig. 1). We reasoned that this pool hadbeen enriched in sequences selected against cell surfacetargets while maintaining a significant level of sequencediversity as demonstrated by the absence of a specificRFLP profile.
During SELEX against EC-RetC634Y-PA, the counter-selection step was realized by incubating the pool withIgG beads in order to eliminate sequences with affinityfor the binding support. The pool of sequences were thenincubated with recombinant EC-RetC634Y-PA immobi-lized on IgG beads. This protein contains a domain be-tween protein A and EC-RetC634Y that is specificallycleavable by the TEV protease. In addition to the coun-terselection step, it allows to elute specifically the ap-tamer-RetC634Y complex after several washes and im-proves the SELEX protocol to prevent enrichment inaptamers against IgG beads.
RFLP analysis of the crossover SELEX showed a rapidevolution with the appearance of several intense bandsafter only 2 rounds, but no significant further evolution
PESTOURIE ET AL.330
FIG. 3. UV-cross linking analysis of aptamers. Autoradiography from the sodium dodecyl sulfate-polyacrylamide gel elec-trophoresis (SDS-PAGE) analysis of the UV-cross linking products formed between the [32P]-labeled B11, B23, and D12 aptamersand the PC12 (wt), PC12/MEN2A (2A) and PC12/MEN2B (2B) cells. The cross-linked protein-aptamer complexes were first di-gested by RNases to estimate more precisely the size of the protein.

TA
BL
E3.
AN
AL
YSI
SO
FSE
QU
EN
CE
SFR
OM
CR
OSS
OV
ER
SEL
EX
S4
Nam
eF
requ
ency
1Se
quen
ces
Kd
nMC
max
Fre
quen
cy 2
E8
18/5
1G
TC
GT
CC
TG
CC
TG
TA
AG
GC
CC
AC
AA
GA
AA
TG
TG
TG
TG
CG
TT
CG
AG
CC
—0
E15
5/51
CA
CG
AG
CC
CT
CG
CG
AC
AA
TA
TG
CG
AC
GA
AT
TC
TA
AG
CC
AT
GT
GA
GT
AC
C—
0E
374/
51G
TG
GT
GA
AC
CG
GC
AA
AC
AC
TG
CC
CT
CT
AC
CT
TT
AG
CG
CT
AG
AT
GC
CT
GG
C—
0E
433/
51C
CC
CC
TG
TA
TT
GT
GC
GC
TT
GA
TG
AA
CC
AA
GT
GT
CA
CG
TT
CG
AG
CC
CA
AN
D1/
41E
92/
51C
CG
CA
CG
GG
CC
AC
TG
AA
TC
GA
CC
AT
TA
TT
GG
TC
CC
TT
CG
CA
CT
AT
CG
GC
G—
0E
242/
51G
GA
CC
GG
CA
GA
GT
TG
CA
AT
GG
CA
AA
CC
AA
GT
CC
AG
GC
CC
TC
AG
TG
CG
C—
0E
352/
51G
TG
AG
AC
GA
GC
CC
TG
TG
AT
AG
CA
CA
AA
AT
GC
CC
TT
CG
AT
TC
CC
AA
CG
—0
E38
1/51
GC
AC
CA
TT
CA
TG
AG
CT
AC
CA
AC
TT
GA
AG
AG
TC
AA
CA
TA
CG
TT
CG
TA
CG
G10
4�
826
1�
51
36/4
1In
divi
dual
14/5
1�
/ND
2/41
(inc
ludi
ng
E71
)
Sequ
ence
of
RN
A-2
�F-P
y ap
tam
ers
(5�
to 3
�fr
om le
ft to
rig
ht)
obta
ined
aft
er e
ight
rou
nds
of S
1 an
d fo
ur r
ound
s of
SE
LE
X o
n th
e re
com
bina
nt e
xtra
cellu
lar
dom
ain
ofR
ET
C63
4Y. F
or s
impl
icity
, fix
ed-p
rim
er s
eque
nce
at 5
�an
d 3�
extr
emiti
es a
re r
emov
ed. T
he f
requ
ency
1 in
dica
tes
the
clon
e ab
unda
nce
of a
seq
uenc
e. B
indi
ng e
xper
imen
tson
PC
12/M
EN
2A c
ells
hav
e be
en p
erfo
rmed
at 1
00 n
M. I
n th
e co
lum
n fo
r K
d, —
indi
cate
no
sign
ific
ant b
indi
ng a
t thi
s co
ncen
trat
ion
in c
ompa
riso
n to
the
star
ting
pool
and
ND
indi
cate
not
test
ed f
or b
indi
ng. F
or th
e se
quen
ce w
hich
dis
play
s af
fini
ty, b
indi
ng c
urve
s w
ere
perf
orm
ed to
est
imat
e its
app
aren
t Kd/
(nM
) an
d its
app
aren
t Cm
ax(p
M).
Sta
ndar
d de
viat
ions
wer
e de
term
ined
fro
m a
t le
ast
four
ind
epen
dent
exp
erim
ents
. Fo
urte
en s
eque
nces
wer
e us
ed a
t eq
uim
olar
con
cent
ratio
n fo
r th
ree
roun
ds o
f SE
LE
X o
n PC
12/M
EN
2A c
ells
. The
fre
quen
cy 2
indi
cate
s th
e cl
one
abun
danc
e of
thos
e se
quen
ces
afte
r th
ese
thre
e n
ew r
ound
s.

of the population has been observed beyond the thirdround (data not shown). Sequences obtained from thecrossover SELEX S4 (7 rounds against cells followed by4 rounds against the recombinant protein) were totallydifferent from the ones obtained after S1, S2, and S3(Table 3). Except the sequence E71 that was nearly iden-tical to D4 (only one mutation in the minimum bindingsite of D4; Table 3), only one other sequence (E38) shareda predicted consensus structure with sequence B69 previ-ously identified after S2 (Fig. 3), but these sequenceswere found in low abundance after this crossover SELEXprocedures (one of 66 and 51, for S2 and S4, respec-tively). Most of the sequences identified from crossoverSELEX were tested for binding on PC12/MEN2A cellsat 100 nM (Table 3). Intriguingly, sequence E71 did notbind to the cells, demonstrating that the mutation of the Ainto U at the 34th position abolish the binding of D4.Only E38 and E43 show good affinity for thePC12/MEN2A cells. This suggests that sequences withlow affinity for cell membrane-inserted RET but highaffinity for the recombinant protein were predominantlyselected by the crossover-SELEX S4. If this is the case,alternating SELEX on living cells with SELEX on re-combinant protein should lead to an enrichment in se-quences recognizing the native, membrane-inserted pro-tein. To verify this hypothesis, we mixed in equimolarconcentrations the E38, E43 and E71 sequences with 11other sequences selected by crossover-SELEX and per-formed three rounds of SELEX on PC12/MEN2A cellswithout counter-selection steps (S5, Table 1). As ex-pected, E38 was predominantly amplified and repre-sented 90% of the final pool, while E43, which displays alower affinity was slightly amplified (approximately 2%;Table 3). This result emphasizes the advantageous capac-ity of a mixed (i.e., purified protein and cells) selectionprocedure to direct the selection of aptamers able to rec-ognize Ret in its native conditions.
Comparison of D4-related aptamers and E38-related aptamers
Binding experiments of aptamer E38 on PC12/MEN2A cells revealed an affinity twofold lower thanthat of aptamer D4 (apparent Kds around 100 nM and 40 nM, respectively). In addition, the conserved stem-internal loop-stem present in the predicted structure ofcell-selected aptamers D4, D24, and B25 was unable tobind the EC-RETC634Y recombinant protein, which ex-plain why these aptamers were not selected by the S4crossover-SELEX. Interestingly, an identical predictedsecondary structure could be identified between E38 andB69 (Fig. 2), even though the internal loop of these twoaptamers is so different that it is uncertain whether theywould adopt the same conformation for RET binding.However, D4 was able to compete for binding of E38 on
PC12/MEN2A (Fig. 4; Cerchia et al., 2005), as shown bythe reduction of 50% of E38 binding when both aptamerswhere added at equimolar concentration (100 nM). Incomparison, neither a naïve library nor the aptamer D30could inhibit the binding of E38. This result strongly sug-gests that D4 and E38 recognize an overlapping region ofRET. However, E38, in contrast to D4, was unable to in-hibit the RET auto-phosphorylation and subsequent sig-nalling (data not shown). Interestingly, in comparison toD4, the primer region of E38 seems to be important forits consensus structure sheared with B69 (see Figure 2)which suggest that this aptamer could be futher im-proved; for instance using randomized E38 as pool forSELEX.
DISCUSSION
The plasma membrane has been extensively targetedfor drug design and membrane proteins account for 70%of all known drug targets. Among them, RTKs have beendemonstrated to be excellent target for the treatment ofmany cancers (Gschwind et al., 2004; Krause and VanEtten, 2005). Two classes of drug are currently used:small molecules (PM � 1000 da) and humanized anti-bodies. Aptamers could provide a new strategy with sev-eral advantages. Specifically, they seem to lack immuno-genicity and can be chemically synthesized with severalmodifications to improve their stability against nucleasesor extend their functionality. However, aptamers arehighly specific for the conformation of the target against
PESTOURIE ET AL.332
1x D400
20
40
60
80
100
120
140
2x D4 1x S1S0 2x S1S0 1x D30 2x D30
FIG. 4. Competitive experiments for the binding of E38. [32P]5�-end-labeled E38 have been incubated at 100 nM onPC12/MEN2A cells in the absence or presence of 100 and 200nM (1� and 2�, respectively) of D4, naïve library (i.e., thestarting pool from S1) and D30 cold sequence as indicated. Af-ter several washes, sequences bound to cells were estimated.

which they have been selected. Several aptamers selectedagainst the recombinant extracellular domain of trans-membrane proteins recognize their targets on the cell sur-face (Chen et al., 2003; Lupold et al., 2002; Mi et al.,2005; Mori et al., 2004). In our case, however, SELEXS3 performed on the recombinant extracellular domain ofRETC634Y did not provide aptamers able to recognize theprotein in a cell surface environment (Cerchia et al.,2005), which most likely implies a different mode ofrecognition for the native and recombinant proteins.
Our data suggest that in the case of RET and possiblyother transmembrane proteins using living cells trans-formed to express a transmembrane protein of choice as atarget for SELEX is a valuable alternative approach todrive the identification of functional aptamers. As amodel system, we raised RNAse-resistant 2�-F pyrimi-dine aptamers directly against PC12 cells expressing theRETC634Y mutant form (PC12/MEN2A) which is consti-tutively dimerized and activated (Cerchia et al., 2005).Counterselection steps are usually performed during SE-LEX to prevent amplification of aptamers against shamtargets from the selection matrix (nitrocellulose, chro-matography medium, etc.). However, cells represent acomplex selection support and counterselection is diffi-cult to achieve. For instance, the presence of activatedRETC634Y in PC12 induce a burly differentiation of cellmorphology, i.e., a flat phenotype with elongated neuriticprocesses while PC12 cells are round-shaped (Califano etal., 1996). We compare two different cell SELEX (S1and S2) to investigate the impact of counterselection.During S1, counterselection was performed against PC12and PC12/MEN2B that express the RETM918T monomerform constitutively activated and displayed quite thesame phenotype than PC12/MEN2A (Cerchia et al.,2005), while S2 was performed in the same manner but without counterselection against PC12/MEN2B.Both selections identified D4-related aptamers selectivefor RET but unable to distinguish the dimeric form ofRET from the monomer in spite of counterselection onPC12/MEN2B for S1. It suggests that using a non-trans-formed cell line for counter-selection steps is sufficientto select aptamers against a transmembrane protein of in-terest, even if this target induces a strong change in thecellular phenotype upon transformation. However, bothSELEX predominantly enrich aptamers against other tar-gets than RET. Interestingly, all these aptamers displayan apparent Kd in the same range around 50 nM, exceptfor the aptamer B11 that displays a worse apparent Kd of170 nM but a much higher target number than the others.We could observe that all aptamers present quite thesame Cmax/Kd ratio. As expected, this ratio suggeststhat the evolution of the pool during cell SELEX is notonly managed from the affinity factor but also from thetarget concentration. In fact, B11 represent 10 of 66 se-quences after S2 whereas B25 (D4-related aptamer) only
represents 1 of 66 sequences which suggests that with thesame Cmax/Kd ratio, aptamers against target with higherCmax are predominantly amplified. However, D12 wastwo times more amplified from S1 in comparison to D30and D71 (19/67 in comparison to 6/67 and 5/67, respec-tively) whereas they present similar Cmax and Kd, sug-gesting that other factors affect the evolution of cells SELEX (for instance a better amplification during PCR).
These data are consistent with recent reports (Blank etal., 2001; Daniels et al., 2003; Hicke et al., 2001), de-scribing the predominant isolation of aptamers againstabundant proteins in whole-cell SELEX.
Counterselections are performed to reduce selection ofaptamers against uninteresting abundant targets ex-pressed at cell surface. However, we demonstrated thatcounterselections were partially inefficient in our case,particularly for PC12/MEN2B whose phenotype is closeto PC12/MEN2A. These counterselection steps could beimproved by increasing the number of cells or incubationtime. Recently, a post-negative selection have been de-scribed to be efficient (Ohuchi et al., 2006). It is particu-larly important when differential cell SELEX is used toidentify new markers of interest. Nevertheless, success ofcounterselection should be largely dependant of the rela-tive concentration of the desired target in comparison tothe other, a ratio that is difficult to assess. The lower isthis ratio, the more the counterselection proceduresshould be efficient.
Even if this last point remains a challenging task,whole-living cell SELEX conserves two major advan-tages: (1) selection without a prior purification of the tar-gets and (2) conservation of membrane proteins in theirnative conformation similar to the in vivo conditions.Moreover, crossover selection from cell SELEX to therecombinant protein could promote the identification ofaptamers against conserved epitopes between the nativeand the recombinant protein. Such crossover procedurehas already been used to select aptamers against theTenascin-C (Hicke et al., 2001). In our case, thecrossover strategy was partially ineffective, only two ap-tamers could be identified, they display a weaker affinityfor RET than those obtained from pure whole-living cellSELEX and did not exhibit any inhibition of RET activ-ity. Two main reasons could explain these different re-sults: (1) in comparison to our unsuccessful S3 selection,SELEX against the recombinant Tenascin-C identifiedaptamers able to recognize the target at cell surface sug-gesting that a structurally similar domain was targeted inthe recombinant and native forms of the protein and (2)after cell SELEX, aptamers against RET are present inlow abundance whereas they were predominantly ampli-fied against the Tenascin-C. Maybe alternating these se-lections (cells and protein) each round could be the betterchoice. Another possibility could be to use cells from dif-ferent lineages transformed to express a target of interest.
APTAMER SELECTION STRATEGY 333

In conclusion, SELEX against the recombinant extra-cellular part of transmenbrane proteins is technically lesschallenging than using transformed whole-living cellsduring SELEX. However, the latter approach opens anew avenue for holistic selection of aptamers when theformer approach is ineffective. This class of aptamers arepromising tools for many applications (diagnosis, imag-ing, therapeutic, etc.). For instance, an aptamer targetingthe PSMA, a cell-surface receptor overexpressed inprostate cancer cells, have recently been used to increasespecifically the delivery of siRNA in vitro (Chu et al.,2006) and in vivo (McNamara et al., 2006).
ACKNOWLEDGMENTS
This work was supported by the EEC contract QLG1-CT-2000-00562 (OLIM), the European Molecular Imag-ing Laboratory (EMIL) network EU contract LSH-2004-503569, la fondation de France, the CEA-CNRSImagerie du petit animal programme, a grant no. 3527from the Association pour la Recherche sur le Cancer,the CNRS, and the MIUR-FIRB grant RBNE0155LB.C.P. is supported by a doctoral fellowship from the CEA.F.D. was supported by a post-doctoral fellowship fromthe CEA. We wish to thank R. Souza for the gift of aT7Y639F RNA polymerase expressing plasmid.
REFERENCES
BARTEL, D.P., and SZOSTAK, J.W. (1993). Isolation of newribozymes from a large pool of random sequences [see com-ment]. Science 261, 1411–1418.
BLANK, M., WEINSCHENK, T., PRIEMER, M., andSCHLUESENER, H. (2001). Systematic evolution of a DNAaptamer binding to rat brain tumor microvessels. selectivetargeting of endothelial regulatory protein pigpen. J. Biol.Chem. 276, 16464–16468.
CALIFANO, D., D’ALESSIO, A., COLUCCI-D’AMATO,G.L., DE VITA, G., MONACO, C., SANTELLI, G., DIFIORE, P.P., VECCHIO, G., FUSCO, A., SANTORO, M.,and DE FRANCISCIS, V. (1996). A potential pathogeneticmechanism for multiple endocrine neoplasia type 2 syn-dromes involves ret-induced impairment of terminal differ-entiation of neuroepithelial cells. Proc. Natl. Acad. Sci. USA93, 7933–7937.
CERCHIA, L., DUCONGE, F., PESTOURIE, C., BOULAY,J., AISSOUNI, Y., GOMBERT, K., TAVITIAN, B., FRAN-CISCIS, V.D., and LIBRI, D. (2005). Neutralizing Aptamersfrom whole-cell SELEX inhibit the RET receptor tyrosine ki-nase. PLoS Biol. 3, e123.
CERCHIA, L., LIBRI, D., CARLOMAGNO, M.S., and DEFRANCISCIS, V. (2003). The soluble ectodomain ofRetC634Y inhibits both the wild-type and the constitutivelyactive Ret. Biochem. J. 372, 897–903.
CHEN, C.H., CHERNIS, G.A., HOANG, V.Q., and LAND-GRAF, R. (2003). Inhibition of heregulin signaling by an ap-tamer that preferentially binds to the oligomeric form of hu-man epidermal growth factor receptor-3. Proc. Natl. Acad.Sci. USA 100, 9226–9231.
CHU, T.C., TWU, K.Y., ELLINGTON, A.D., and LEVY, M.(2006). Aptamer mediated siRNA delivery. Nucleic AcidsRes. 34, e73.
CUI, Y., RAJASETHUPATHY, P., and HESS, G.P. (2004).Selection of stable RNA molecules that can regulate thechannel-opening equilibrium of the membrane-boundgamma-aminobutyric acid receptor. Biochemistry 43,16442–16449.
DANIELS, D.A., CHEN, H., HICKE, B.J., SWIDEREK, K.M.,and GOLD, L. (2003). A tenascin-C aptamer identified by tu-mor cell SELEX: systematic evolution of ligands by expo-nential enrichment. Proc. Natl. Acad. Sci. USA 100,15416–15421.
DANIELS, D.A., SOHAL, A.K., REES, S., and GRISSHAM-MER, R. (2002). Generation of RNA aptamers to the G-pro-tein-coupled receptor for neurotensin, NTS-1. Anal.Biochem. 305, 214–226.
ELLINGTON, A.D., and SZOSTAK, J.W. (1990). In vitro se-lection of RNA molecules that bind specific ligands. Nature346, 818–822.
GSCHWIND, A., FISCHER, O.M., and ULLRICH, A. (2004).The discovery of receptor tyrosine kinases: targets for cancertherapy. Nat. Rev. Cancer 4, 361–370.
HICKE, B.J., MARION, C., CHANG, Y.F., GOULD, T., LY-NOTT, C.K., PARMA, D., SCHMIDT, P.G., and WAR-REN, S. (2001). Tenascin-C aptamers are generated using tu-mor cells and purified protein. J. Biol. Chem. 276,48644–48654.
HOMANN, M., and GORINGER, H.U. (1999). Combinatorialselection of high affinity RNA ligands to live African try-panosomes. Nucleic Acids Res. 27, 2006–2014.
KRAUSE, D.S., and VAN ETTEN, R.A. (2005). Tyrosine ki-nases as targets for cancer therapy. N. Engl. J. Med. 353,172–187.
LUPOLD, S.E., HICKE, B.J., LIN, Y., and COFFEY, D.S.(2002). Identification and characterization of nuclease-stabi-lized RNA molecules that bind human prostate cancer cellsvia the prostate-specific membrane antigen. Cancer Res. 62,4029–4033.
MCNAMARA, J.O., 2ND, ANDRECHEK, E.R., WANG, Y.,VILES, K.D., REMPEL, R.E., GILBOA, E., SULLENGER,B.A., and GIANGRANDE, P.H. (2006). Cell type-specificdelivery of siRNAs with aptamer-siRNA chimeras. Nat.Biotechnol. 24, 1005–1015.
MI, J., ZHANG, X., GIANGRANDE, P.H., MCNAMARA,J.O., 2ND, NIMJEE, S.M., SARRAF-YAZDI, S., SUL-LENGER, B.A., and CLARY, B.M. (2005). Targeted inhibi-tion of alphavbeta3 integrin with an RNA aptamer impairsendothelial cell growth and survival. Biochem. Biophys. Res.Commun. 338, 956–963.
MORI, T., OGURO, A., OHTSU, T., and NAKAMURA, Y.(2004). RNA aptamers selected against the receptor activatorof NF-kappaB acquire general affinity to proteins of the tu-mor necrosis factor receptor family. Nucleic Acids Res. 32,6120–6128.
PESTOURIE ET AL.334

MORRIS, K.N., JENSEN, K.B., JULIN, C.M., WEIL, M., andGOLD, L. (1998). High affinity ligands from in vitro selec-tion: Complex targets. Proc. Natl. Acad. Sci. USA 95,2902–2907.
OHUCHI, S.P., OHTSU, T., and NAKAMURA, Y. (2006). Se-lection of RNA aptamers against recombinant transforminggrowth factor-beta type III receptor displayed on cell surface.Biochimie 88, 897–904.
OSBORNE, S.E., MATSUMURA, I., and ELLINGTON, A.D.(1997). Aptamers as therapeutic and diagnostic reagents:problems and prospects. Curr. Opin. Chem. Biol. 1, 5–9.
PESTOURIE, C., TAVITIAN, B., and DUCONGE, F. (2005).Aptamers against extracellular targets for in vivo applica-tions. Biochimie 87, 921–930.
THIEL, K. (2004). Oligo oligarchy-the surprisingly smallworld of aptamers. Nat. Biotechnol. 22, 649–651.
THOMPSON, J.D., GIBSON, T.J., PLEWNIAK, F., JEAN-MOUGIN, F., and HIGGINS, D.G. (1997). TheCLUSTAL_X windows interface: flexible strategies for mul-tiple sequence alignment aided by quality analysis tools. Nu-cleic Acids Res. 25, 4876–4882.
TUERK, C., and GOLD, L. (1990). Systematic evolution of li-gands by exponential enrichment: RNA ligands to bacterio-phage T4 DNA polymerase. Science 249, 505–510.
VLASSOV, A., KHVOROVA, A., and YARUS, M. (2001).Binding and disruption of phospholipid bilayers bysupramolecular RNA complexes. Proc. Natl. Acad. Sci. USA98, 7706–7711.
WANG, J., JIANG, H., and LIU, F. (2000). In vitro selection ofnovel RNA ligands that bind human cytomegalovirus andblock viral infection. RNA 6, 571–583.
ZUKER, M. (2003). Mfold web server for nucleic acid foldingand hybridization prediction. Nucleic Acids Res. 31,3406–3415.
Address reprint requests to:Dr. Frédéric Ducongé
CEA/DSV/DRM Service HospitalierFrédéric JoliotINSERM U803
4 Place du général Leclerc91401 Orsay
France
E-mail: [email protected]
Received July 26, 2006; accepted in revised formSeptember 13, 2006.
APTAMER SELECTION STRATEGY 335


This article has been cited by:
1. S. Ueno, S. Yoshida, A. Mondal, K. Nishina, M. Koyama, I. Sakata, K. Miura, Y. Hayashi, N. Nemoto, K. Nishigaki, T. Sakai.2012. In vitro selection of a peptide antagonist of growth hormone secretagogue receptor using cDNA display. Proceedingsof the National Academy of Sciences 109:28, 11121-11126. [CrossRef]
2. Christian A Bippes, Daniel J Muller. 2011. High-resolution atomic force microscopy and spectroscopy of native membraneproteins. Reports on Progress in Physics 74:8, 086601. [CrossRef]
3. Pooja Dua, Soyoun Kim, Dong-ki Lee. 2011. Nucleic acid aptamers targeting cell-surface proteins. Methods 54:2, 215-225.[CrossRef]
4. Elina Zueva, Laila Illán Rubio, Frédéric Ducongé, Bertrand Tavitian. 2011. Metastasis-focused cell-based SELEX generatesaptamers inhibiting cell migration and invasion. International Journal of Cancer 128:4, 797-804. [CrossRef]
5. Kristina W Thiel, Paloma H Giangrande. 2010. Intracellular delivery of RNA-based therapeutics using aptamers. TherapeuticDelivery 1:6, 849-861. [CrossRef]
6. Laura Cerchia, Vittorio de Franciscis. 2010. Targeting cancer cells with nucleic acid aptamers. Trends in Biotechnology 28:10,517-525. [CrossRef]
7. Hari P. Dwivedi, R. Derike Smiley, Lee-Ann Jaykus. 2010. Selection and characterization of DNA aptamers with bindingselectivity to Campylobacter jejuni using whole-cell SELEX. Applied Microbiology and Biotechnology 87:6, 2323-2334.[CrossRef]
8. Kristina W. Thiel, Paloma H. Giangrande. 2009. Therapeutic Applications of DNA and RNA Aptamers. Oligonucleotides19:3, 209-222. [Abstract] [Full Text PDF] [Full Text PDF with Links]
9. Claudia M. Dollins , Smita Nair , Bruce A. Sullenger . 2008. Aptamers in Immunotherapy. Human Gene Therapy 19:5,443-450. [Citation] [Full Text PDF] [Full Text PDF with Links]
10. Aniela Wochner, Marcus Menger, Martina Rimmele. 2007. Characterisation of aptamers for therapeutic studies. ExpertOpinion on Drug Discovery 2:9, 1205-1224. [CrossRef]





























