Embed Size (px)
Citation preview

Chem.-Biol. Interactions, 22 (1978) 309--327 © Elsevier/North-Holland Scientific Publishers Ltd.
309
COMPARATIVE EFFECT OF A FAMILY OF SUBSTITUTED THIOPSEUDOUREAS ON PROTEIN SYNTHESIS BY RAT LIVER AND WALKER CARCINOMA RIBOSOMES*
ANDRES CARMONA and NESTOR F. GONZALEZ-CADAVID
Departamento de Biologfa Celular, Facultad de Cieneias, Universidad Central de Venezuela, Apartado 10098, Caracas (Venezuela)
(Received June 1st, 1977) (Revision received January 25th, 1978) (Accepted April 5th, 1978)
SUMMARY
In order to assess the role played respectively by the pseudothiourea group and the alkylic chain in the inhibition of protein synthesis and tumour growth caused by compound AHR-1911, a series of eight related substances were studied.
The blockade of protein synthesis on liver and Walker carcinoma ribosomes and on suspensions of Walker carcinoma cells, depended es- sentially on the length of the alkylic chain and the substitution in C-1. The minimum chain was 9 carbons and a plateau in the activity was reached at 11 carbons. Replacement of the thiourea group in C-1 by an NH2 group did not change the pattern. A double bond in the distal section of the chain (AHR-1911) increased inhibition on intact cells with a parallel decrease in cytotoxici ty, and reduced the aggregation of ribosomes, protein synthesis factors and other proteins.
The ant i tumour effect depends on the pseudothiourea group and is not caused primarily by interference with protein synthesis.
Aminoacyl tRNA binding and transfer appeared to be targets of AHR-1911, but this did not affect significantly tRNA charge or nascent peptide release. Drug binding to ribosomes and their subsequent aggregation can be regulated by K + concentration and temperature. It is assumed that the inhibition of protein synthesis is caused by AHR-1911 effects on elongation factors, imp airing their interaction with ribosomes.
INTRODUCTION
The substituted thiopseudoureas are a group of pharmacological agents
*This work was supported by a grant from the Consejo de Desarrollo Cientffico y Humanlstico de la Universidad de Venezuela.

310
which include a few derivatives with ant i tumour activity. One of them, the S-(10-undecen-l-yl) isothiouronium iodide (AHR-1911) has been shown to decrease the growth of the Walker-256 carcinosarcoma and to inhibit protein synthesis in vitro by liver polyribosomes, as indicated by the interference with processes such as the incorporation of[~4C]leucine into protein, the poly(U)-stimulated formation of polyphenylalanine, and the release of nascent chains by puromycin [ 1]. In order to evaluate the respective roles of the pseudothiourea group and the alkylic chain, we selected a series of dif- ferent related compounds and compared their effects on protein synthesis in vitro with the degree of aggregation of ribosomes, soluble factors and other proteins. Our results suggest that the inhibition of protein synthesis exerted by compound AHR-1911 is due both to the presence of a long alkylic chain and substitution in C~, either by a pseudouthiourea or an amino group, but that the ant i tumour action is restricted to the thiourea derivative. We also investigated the mechanism of action of compound AHR-1911, showing that it inhibits the aminoacyl transfer reaction at two levels: the enzymic binding of aminoacyl tRNA to the ribosomes by pre- sumably interacting with elongation factors, and the subsequent stage of transpeptidation, without affecting the charge of tRNA. The ribosomal aggregation induced by the drug is shown to be a temperature dependent process reverted by high ionic strength and puromycin.
MATERIALS AND METHODS
Materials
Chemicals. All pseudothioureas tested were S-(alkyl-1) isothiouronium derivatives, the alkyl substituent being as follows (the counter anion indi- cated immediately after): hexanyl, bromide; heptanyl, bromide; nonanyl, iodide; undecanyl, bromide; 10-undecenyl, bromide; 10-undecenyl, iodide {compound AHR-1911): dodecanyl, iodide. They were kindly provided by Professor Nicola Ercoli of this Institute. Dodecylamine hydrochloride was purchased from Eastman Kodak Co., Rochester, N.Y. 14650, U.S.A. L- U-[14C]leucine (sp. radioactivity 473 uCi/mg) were obtained from the Commisariat a' l'Energie Atomique (C.E.A.), Gif-sur-Ivette, France. [methoxy-3H] puromycin (sp. radioactivity 3280 mCi/mmol) was from New England Nuclear, Boston, Mass., U.S.A. S-(10-undecen-l-yl)[14C] isothiouro- nium iodide ([14C]AHR-1911) was synthesized by Professor Ercoli from [ 14C] thiourea.
Anisomycin and sparsomycin were kindly given by Pfizer Central Research (U.S.A.) and Dr. Israel Algranatti, respectively. Dithiothreitol and Sephadex G-25 (25--80 pm) were purchased from Sigma Chemical Co., St. Louis, Mo., U.S.A. Phosphotungstic acid was from E. Merck, Darmstadt, Germany; glutaraldehyde (25% w/v) was from BDH Chemicals Ltd., Poole, Dorset, U.K. The membrane filters (HAW 304 FO, 0.45 ~m of pore size) were from Millipore Intertech Inc., Bedford, Mass., U.S.A. Trypsin 1 : 2 5 0 from hog

311
pancreas was from ICN Pharmaceuticals, Inc., Cleveland, Ohio, U.S.A. Earl&s balanced salt solution was from Grand Island Biological Co., New York 14072, U.S.A. Deoxyribonuclease 1 from beef pancreas was from Sigma Chemical Co., St. Louis, MO., U.S.A.
Animals. Male Sprague-Dawley rats (150-160 g body wt.) from a closed colony bred at the Instituto de Medicina Experimental, Universidad Central de Venezuela, Caracas, Venezuela, were used throughout. The rats were starved for 18-20 h before killing by decapitation. The Walker 256 carcino- sarcoma was transplantedsubcutaneusly on the fore-dorsal region as described [ 1,2] and the animals were killed nine days after inoculation.
Methods Incubation of ribsomes under conditions ofprotein synthesis. A detergent-
treated ribosomal fraction with a high polyribosome/monoribosome ratio (C-ribosomes) was prepared from the liver of one or several rats by the method of Wettstein et al. [ 31 as modified by Ragnotti et al. [4]. A poly- ribosomeenriched fraction containing ribosomes not bound to endoplasmic reticulum membranes (free ribosomes) was obtained from the Walker carci- noma as indicated for liver ribosomes, except that homogenization was carried out with 20 strokes of the Potter-Elvehjem pestle and that no deoxy- cholate was added to the post-mitochondrial fraction. The pH5 fraction was obtained as described previously [5] and the pH5 fraction supematant was brought to pH 7.8 as done by Moldave [ 61. In certain cases this last fraction was filtered through a 1 X 19 cm column of Sephadex G-25 equili- brated with medium B (0.15 M sucrose, 10 mM MgC12, 35 mM Tris-HCl buffer pH 7.8 at 2O”C, and 25 mM KCl). The preparation of ribosomal subunits was performed according to von der Decken et al. [ 71. All fractions unless stated, were stored at -60°C until use.
Incubations for protein synthesis were carried out for 30 min at 37°C as indicated in 100 ~1 volume containing rRNA (250 pg/ml), pH5 fraction (1 mg protein/ml), sucrose(90mM), Mg” (6 mM), Tris-HCl (pH 7.8 at 20°C 21 mM), K+ (25 mM), pyruvate kinase (50 fig/ml), ATP (2 mM), phosphoenol pyruvate (10 mM), GTP (0.25 mM) and [14C]leucine (0.25 pCi/ml). Radio- activity measurements were done in 0.1 ml samples by the glass-fibredisc method. The extent of ribosomal agglutination was measured as described previously [2], and in the case of soluble factors the precipitates were dissolved in N-NaOH and estimated by a modification of Lowry’s [8] procedure.
Incubation of carcinoma cells under conditions for protein synthesis. Pieces of tumour were trypsinized in the presence of 56 wg/ml of deoxyribo- nuclease [9], the cells harvested, washed with Earle’s medium containing DNAse and finally suspended in the same medium at lo6 viable cells/ml (trypan blue test), containing 0.05 MCi [“Cl leucine/ml. Aliquots of 1 ml were incubated in triplicate at 37°C for 2 h under aseptic conditions. Protein was precipitated and washed twice with 10% trichloroacetic acid, and the residues dissolved in 0.1 ml formic acid.

312
Charge o f tRNA with [3H]leucine. When the effect of AHR-1911 was assayed on this process, the incubation was performed as described for the estimation of protein synthesis except that no ribosomes were present and [3H]leucine (1 pCi/ml) was used. In certain instances the energy source (pyruvate kinase, ATP, GTP, and phosphoenol pyruvate) was omitted. After 30 re_in at 37°C the samples were chilled in an ice bath and 50 pl of a solution of L-leucine (10 mg/ml) and 5 ml of 10% trichloroacetic acid were added. The samples were then passed through Millipore filters and the precipitates washed three times with 5% trichloroacetic acid containing L-leucine (5 mg/ml). The filters were finally used for radioactivity deter- minations. Control values from three separate experiments performed in quadruplicate were 2600 {2500--2800) dpm/tube .
Preparation o f L-[~4C]leu - and [14C]aminoacyl-tRNA. The L-[14C] leu- tRNA was obtained by a modification of the procedure of Moldave [10], incubating in a final volume of 2.3 ml, 4 uCi of [~4C]leucine, 2 ml of con- centrated freshly prepared pH5 fraction (36 mg protein/ml) and 8.7 mM ATP. The [ '4C]aminoacyl- tRNA was prepared in a similar way with 4 pCi of [~4C]amino acid mixture. In the absence of an accurate estimation of the endogenous pool of L-leucine adsorbed in the pH5 fraction and the extent of actual charge of the tRNA (that is whether saturation was achieved or not), we prefer to express the specific radioactivity of the tRNA as dpm/pmol of total tRNA. The values obtained were 5--8 dpm/pmol for the L-[ '4C]teu-tRNA and 20 dpm/pmol for the [ ~4C] aminoacyl-tRNA.
Aminoacyl transfer from L-['4C] leu-tRNA to ribosomes. The incubation was performed as described for the estimation of protein synthesis except that[ '4C]leucine and pH5 fraction, and no energy source aside GTP was present. The incubation contained ribosomes (0.5 mgRNA/ml) , pH5 fraction supernatant (2--3 mg protein/ml), sucrose (90 mM), Mg 2÷ (6 mM), Tris--HC1 buffer (pH 7.8 at 20°C 21mM), K ÷ (25 mM), GTP (0.25 mM), ['4C] leu-tRNA (20 ~g RNA/ml) and compound AHR-1911 at the appropriate concentration (absent in controls), all in a total volume of 0.5 ml. In certain instances the pH5 fraction supernatant was ommitted, and in other experiments no GTP was added and the pH5 fraction supernatant was filtered through Sephadex G-25 to remove the endogenous soluble GTP. Some samples lacked both GTP and the pH5 fraction supernatant. The pH5 fraction supernatant corresponded to a single batch in order to avoid variations owing to a different GTP content. In all cases blanks wi thout r ibosomes were included. After 60 min at 37°C the samples were chilled in an ice bath and diluted with 3 ml of cold medium B, filtering through millipore filters, and then were washed three times with 3 ml of cold medium B and used for radio- activity determinations.
Considering the limitations in the estimation of the actual specific radio- activity of L-[~4C] leu-tRNA (see above), all results of binding are expressed as pmoles of total tRNA bound, assuming that the other tRNA species follow the behaviour of leu-tRNA.
Assessment o f aminoacyl tRNA binding and peptide bond formation in

313
the aminoacyl transfer reaction. A partially purified mixture of elongation factors EF-1 and EF-2 was used instead of the pH5 fraction supernatant. This mixture was prepared applying Moldave's [6] procedure until precipi- tation at 65% saturation with (NH4)2SO4 and subsequent gel filtration through Sephadex G-25 column and was named "S-65 fraction". The amino- acyl transfer reaction was carried out as above and samples were taken for measuring the total binding of [14C] aminoacyl tRNA. Other samples were submit ted to three different procedures as follows: (a) trichloroacetic acid was added to 10% w/v, left at 0°C for 10 rain and incubated at 90°C for 15 min, then passed through the millipore filter and washed 3 times with 3 ml portions of 5% trichloroacetic acid; (b) 1 M NaOH was added to 0.3 M, the samples were incubated overnight at 37°C, trichloroacetic acid was added to 10% w/v and filtered 10 rain after as above; (c) the same as in (b), except that a heating step in the 10% trichloroacetic acid used for precipitation was included as in (a).
Binding of [~4C] AHR-1911 to ribosomal particles and soluble factors. For the ribosomal particles the technique used was similar to that for the binding of L-[14C] leu-tRNA in the absence of soluble factors and GTP. The incubations contained ribosomes or ribosomal particles (0.06 mgRNA/ml) , sucrose (90 raM), Mg 2÷ (6 raM), Tris--HC1 buffer (pH 7.8 at 20°C, 21 mM), K ÷ (25 raM) and compound AHR-1911 at the appropriate concentration (absent in controls), all in a total volume of 0.5 ml. In all cases blanks wi thout r ibosomes were included. After 30 rain. at 37°C the samples were treated exactly as described for the binding of [ ~4C] leu-tRNA. For the soluble factors the procedure was similar except that pH5 fraction (0.8--1 mg protein/ml) or pH5 fraction supernatant (2--3 mg protein/ml) were substi- tu ted for the ribosomal particles.
Electron microscopy of aggregated ribosomes. Ribosomes were incubated at 0.25 mg RNA/ml in a medium containing 90 raM-sucrose, 21 mM-Tris- HC1 (pH 7.8 at 20°C), 25 mM-KC1 and 6 mM-MgCl2, in the presence of 0.42 mM-AHR-1911 at 0°C for 30 min. The aggregated ribosomes were sedimented at 2000 g for 5 min and the pellet resuspended with Vortex mixer in the same medium without the drug. The ribosomal concentration was adjusted to 0.1 rag RNA/ml and a drop was put on a Formvar~overed copper grid for fixation and negative staining [ 11]. The grids were examined in a Siemens Elmiscop 1 at 30000× magnification.
Pepdityl [3H]puromycin formation on ribosomes. Two techniques were followed differing mainly in the concentrat ion of puromycin and KC1. The first was based on the procedure of Pestka et al. [12], incubating ribosomes (0.2 mg RNA/ml) in a medium containing 50 mM Tris--HC1 (pH 7.2 at 20°C) 500 mM KCI, 6 mM MgCl2, 30 mM sucrose and 4 gM [3H] puromycin, with or without AHR-1f111 (0.42 raM) all in a total volume of 0.5 ml. After 30 rain at 37°C puromycin and trichloroacetic acid solutions were added to 40 gM and 10% w/v, respectively and left for 10 min at 0°C. The suspensions were filtered through millipore, the discs washed 2-times with 3 ml of 5% w/v trichloroacetic acid and 10-times with 3 ml of ethanol, and finally used for radioactivity determinations.

314
The other precedure was a modification [1] of the method of Cannon [13]. Ribosomes (1 mg RNA/ml) were incubated for 5 min at 37°C in medium B in the presence or absence of 0.42 mM AHR-1911 and [all]- puromycin was then added to 0.5 mM, continuing the incubation for 15 min more at 37°C. Cold medium B (0.25 ml) was added and the ribosomes sedimented at 165 000 g for I h. The pellets were gently washed with medium B, resuspended in 0.6 ml of medium B with a Vortex mixer and albumin was added to 0.2 mg/ml. Three samples of 0.1 ml each were layered on three glass fibre discs, dryed and treated with 10% w/v trichloroacetic acid. The discs were washed twice with 3 ml of 5% w/v trichloroacetic acid and 10 times with 3 ml of ethanol and finally used for radioactivity determinations. The supernatants from the centrifugations were treated in the same way to estimate the peptidyl-puromycin released to the medium.
Radioactivity measurements. All the samples on the glass fibre discs were counted for radioactivity in vials with 2 ml of a scintillation liquid containing 0.4% of PPO (2.5~liphenyloxazole) and 0.005% of POPOP (1.4-bis-(5- phenyloxazol-2-yl) benzene) in toluene. The millipore discs were dissolved in vials with 10 ml of a similar scintillation mixture in 60% toluene--40% 2-methoxyethanol. In other cased, 100 pl aliquots were directly mixed with this latter cocktail. The samples from the cell incubations dissolved in formic and were added to 4 ml of a scintillation fluid with Triton-× 100 instead of 2-methoxyethanol. Determinations of radioactivity were carried ou t in an automatic liquid scintillation spectrometer (Packard Tri-Carb Model 385). Values for efficiency for glass fibre discs were 78% (14C) and 20% (all) and for the other cases were 70% (14C) and 25% (all).
Assay of antitumour acivity. Three separate groups of three rats each, bearing Walker turnouts transplanted 24 h before, were injected twice daily for 7 days, intramuscularly in the legs, with 0.12 ml of propyleneglycol containing the drug. The rats were killed at the 8th day and the treated turnout wt. /control tumour wt. ratios (T/C ratios) were obtained by com- paring the values with the average of 3 control groups of two rats each. Other similar groups of rats shaved on the dorsal skin where the tumour was transplanted, were treated topically by rubbing 0.5 ml of the same solution until complete absorption. The application was repeated twice daily. T/C ratios were obtained as before.
The effect on L-1210 lymphocyt ic leukemia was assayed in groups of 10 treated and 10 control DBA/2 mice bearing the leukemia transplanted by intraperitoneai injection of 1 × 106 cells. The drugs, dissolved in propylen- glycol as described, were injected intramuscularly or intraperitoneally or applied topically in the ventral skin once daily until death occurred. The ratio between life span of treated and control groups was established.
RESULTS
Inhibition of protein synthesis and ribosomal aggregation The inhibitory activity of pseudothioureas and related compounds on
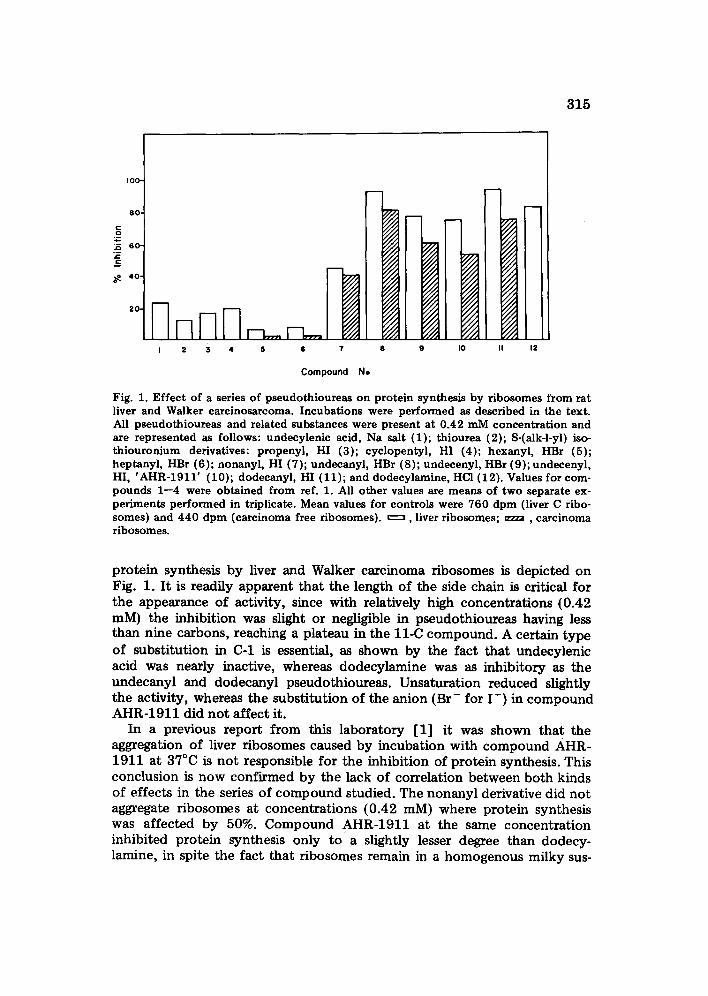
315
I 0 0 -
8 0 -
,~ 6o-
N 4o-
2 0 -
I 2 3 4 5 S ?
r11
S
I I0
I 12
Compound No
Fig. 1. Effect of a series of pseudothioureas on protein synthesis by ribosomes from rat liver and Walker carcinosarcoma. Incubations were performed as described in the text. All pseudothioureas and related substances were present at 0.42 mM concentration and are represented as follows: undecylenic acid, Na salt (1); thiourea (2); S-(alk-l-yl) iso- thiouronium derivatives: propenyl, HI (3); cyclopentyl, HI (4); hexanyl, HBr (5); heptanyl, HBr (6); nonanyl, HI (7); undecanyl, HBr (8); undecenyl, HBr (9); undecenyl, HI, 'AHR-1911' (10); dodecanyl, HI (11); and dodecylamine, HC1 (12). Values for com- pounds 1--4 were obtained from ref. 1. All other values are means of two separate ex- periments performed in triplicate. Mean values for controls were 760 dpm (liver C ribo- somes) and 440 dpm (carcinoma free ribosomes). ~ , liver ribosomes; . . . . , carcinoma ribosomes.
protein synthesis by liver and Walker carcinoma ribosomes is depicted on Fig. 1. It is readily apparent that the length of the side chain is critical for the appearance of activity, since with relatively high concentrations {0.42 mM) the inhibition was slight or negligible in pseudothioureas having less than nine carbons, reaching a plateau in the l l -C compound. A certain type of substitution in C-1 is essential, as shown by the fact that undecylenic acid was nearly inactive, whereas dodecylamine was as inhibitory as the undecanyl and dodecanyl pseudothioureas. Unsaturation reduced slightly the activity, whereas the substitution of the anion (Br- for I - ) in compound AHR-1911 did not affect it.
In a previous report from this laboratory [1] it was shown that the aggregation of liver ribosomes caused by incubation with compound AHR- 1911 at 37°C is not responsible for the inhibition of protein synthesis. This conclusion is now confirmed by the lack of correlation between both kinds of effects in the series of compound studied. The nonanyl derivative did not aggregate ribosomes at concentrations (0.42 mM) where protein synthesis was affected by 50%. Compound AHR-1911 at the same concentration inhibited protein synthesis only toga slightly lesser degree than dodecy- lamine, in spite the fact that ribosomes remain in a homogenous milky sus-

316
pension in the presence of the former drug whereas are completely precipi- tated by dodecylamine.
Interaction with soluble factors and other proteins The presence of a double bond in compound AHR-1911, or its bromide
derivative, causes a high degree of specificity in the protein precipitating action since AHR-1911 (0.42 mM) does not agglutinate proteins such as albumin, cytochrome c, chymotrypsinogen or concanavalin in 30 min incubations of 0.1 mg/ml solutions either at 0 ° or 37°C, and affects only slightly haemoglobin at 37°C. In contrast, the saturated long chain pseudo- thioureas (undecanyl and dodecanyl) and dodecylamine precipitate con- siderably all these proteins, even at 0°C. The pH 5 fraction is partially pre- cipitated (40--50%) by the whole series of compounds starting from the nonanyl derivative, when incubated at 37°C. In experiments where the pH 5 fraction (0.9 mg protein/ml) or its supernatant (3 mg protein/ml) were incubated with 0.42 mM AHR-1911 for 30 min at 0°C and the amount of sedimented protein estimated, the former was precipitated partially (24% protein), whereas the latter was not affected at all. At room temperature, the precipitation of pH 5 fraction was somewhat enhanced (34%) without still affecting the supematant . At 37°C, more than half the pH 5 fraction was precipitated against only 20% in the suI0ernatant. In the last cases the increase of K + concentrat ion of 0.5 M did not interfere with precipitation at 37 °, differing with what occurs with ribosomes [1] (see below) thus indicating that a different mechanism operates in both cases.
Inhibition o f protein synthesis in intact cells Carcinoma cells are easily isolated in suspension by submitting the solid
tumour to a trypsinization procedure which is simpler than the methods normally applied for yielding liver cells. Thus trypsinized cells are 90% viable and comparable to the more physiological system based on suspensions of the ascites form of this tumour [14], allowing evaluation of the ICs0 (micro- molar concentratkm of each drug that inhibits protein synthesis by 50%). Compound AHR-1911 and its bromide derivative were nearly twice more active than the saturated analogues or dodecylamine. The nonanyl pseudo- thiourea, which does not agglutinate liver or carcinoma ribosomes in vitro and affects only slightly the pH5 fraction at doses lower than 0.4 mM had approximately the same inhibitory capacity as dodecylamine, and the heptanyl derivative was totally inactive. The ICs0 of 17 pM (6 pg/ml) found for compound AHR-1911 is approx. 15-fold higher than that for emetine in the same system and no cell kill occurs with AHR-1911 at this concentration, although dodecylamine at the ICs0 is highly cytotoxic.
Role o f the thiourea group in antitumour action Compound AHR-1911 has been shown previously [1] to decrease con-
siderably the growth of the Walker carcinoma implanted subcutaneously in the dorsal region of the rat. The reduction in tumour weight was achieved
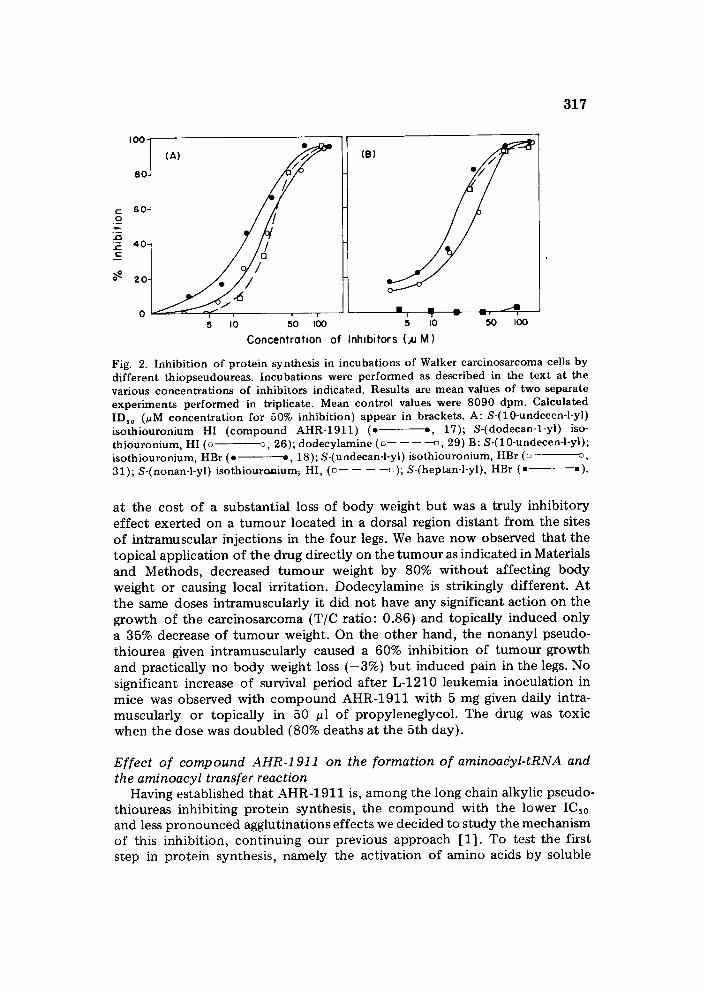
317
C o
- l . -
35 F ¢ -
'°°1 (A)
'°1 6 0 "
5 I 0 50 I00
(B)
n ~ = __, j~ll, 5 I0 50 I00
Concentration of Inhibitors (~ M)
Fig. 2. Inhibition of protein synthesis in incubations of Walker carcinosarcoma cells by different thiopseudoureas. Incubations were performed as described in the text at the various concentrations of inhibitors indicated. Results are mean values of two separate experiments performed in triplicate. Mean control values were 8090 dpm. Calculated IDho (~M concentration for 50% inhibition) appear in brackets. A: S-(10-undecen-l-yl) isothiouronium HI (compound AHR-1911) ( . o, 17); S-(dodecan-l-yi) iso- thiouronium, HI (o o, 26); dodecylamine (u---------~, 29) B: S-(10-undecen-l-yl); isothiouronium, HBr (o . , 18); S-(undecan-l-yl) isothiouronium, HBr (o o, 31); S-(nonan-l-yl) isothiouxcmium, HI, (u . . . . G ); S-(heptan-l-yl), HBr (• • ).
a t the cos t o f a subs tant ia l loss of b o d y weight bu t was a t ru ly inh ib i to ry e f f ec t exe r t ed on a t u m o u r loca ted in a dorsal region d i s t an t f r o m the sites o f i n t r amuscu la r in jec t ions in the fou r legs. We have n o w observed tha t the top ica l app l i ca t ion o f the d rug d i rec t ly on the t u m o u r as indica ted in Materials and Methods , decreased t u m o u r weight b y 80% w i t h o u t a f fec t ing b o d y weigh t or causing local i r r i ta t ion. D o d e c y l a m i n e is s tr ikingly d i f fe ren t . A t the same doses in t r amuscu la r ly it did no t have any signif icant ac t ion on the g rowth of the ca r c i nos a r com a (T/C ra t io : 0 .86) and topica l ly induced on ly a 35% decrease of t u m o u r weight . On the o the r hand , the n o n a n y l pseudo- t h iou rea given in t ramuscu la r ly caused a 60% inhib i t ion of t u m o u r g rowth and prac t ica l ly no b o d y weight loss ( - 3 % ) b u t induced pain in the legs. No signif icant increase of survival per iod a f t e r L-1210 l eukemia inocu la t ion in mice was observed wi th c o m p o u n d A H R - 1 9 1 1 wi th 5 mg given daily intra- muscu la r ly or top ica l ly in 50 ~l o f p ropy leneg lyco l . The drug was toxic w h e n the dose was d o u b l e d (80% dea ths a t the 5th day) .
Effect of compound AHR-1911 on the formation of aminoadyl-tRNA and the aminoacyl transfer reaction
Having es tabl ished t ha t A H R - 1 9 1 1 is, a m o n g the long chain alkylic pseudo- th ioureas inhibi t ing p ro te in synthesis , the c o m p o u n d with the lower ICs0 and less p r o n o u n c e d agglu t ina t ions e f fec t s we dec ided to s tudy the m e c h a n i s m o f this inhibi t ion, con t inu ing our previous a p p r o a c h [1] . To tes t the first s tep in p ro te in synthes is , namely the ac t iva t ion of amino acids by soluble

318
factors, the charge of tRNAs with [3H] leucine was studied upon incubation with the drug.
At 0.42 mM AHR-1911, a concentrat ion which caused a 78% inhibition of protein synthesis by liver polyribosomes [1], the slight inhibition (13%) detected on the formation of [3 H] leu-tRNA was too small to account for the overall effect on protein synthesis. This is evident considering the large excess of aminoacyl-tRNAs, in respect to their utilization by the ribosomes, occuring under the usual conditions of the in vitro systems for protein synthesis. At a higher concentration (0.56 mM) the effect becomes signi- ficant (46%) but it is probably due to the considerable precipitation of soluble factors from the pH5 fraction caused by this concentrat ion of AHR- 1911, and even so the inhibition is about half that observed when measuring total protein synthesis with C--ribosomes. These results suggest that the charge itself of tRNAs with amino acids is not a main factor in the inhibition of protein synthesis exerted by comparatively low concentrations of com- pound AHR-1911.
As rat liver polyribosomes do no t perform efficiently the initiation step we decided to investigate the next stage in the process, that is the aminoacyl transfer reaction in the ribosomes occuring during elongation. It involves aminoacyl tRNA binding and transpeptidation. There are two different kinds of binding reactions, one enzymic depends on elongation factor EF-1, GTP and aminoacyl-tRNA [15, 16] and the other, non-enzymic, takes place in the absence of EF-1 and GTP, becoming significant at high Mg 2+ concentrations [17]. The non-enzymic binding is negligible in the presence of soluble factors at 6 mM Mg 2+ and therefore the total aminoacyl tRNA bound under the conditions of our assay can be considered as representative of aminoacyl transfer depending on enzymic binding alone [16, 17].
Fig. 3a shows the aminoacyl transfer occuring after enzymic binding in the complete system in the presence of increasing concentrations of com- pound AHR-1911. The drug induces some retention of L-[14C]leu-tRNA in the filters in the absence of r ibosomes in a process rising steadily from negligible at 0.14 mM to about 6% of the tRNA input at 0.56 raM. Even at this high concentration this should not affect the availability of L-leu-tRNA for the reaction but it has to be considered for correcting the observed bindings. The retention of L-[ ~4C]leu-tRNA is probably due to an interaction of compound AHR-1911 with GTP-tRNA-EF-1 since this complex passes completely in our conditions as it is shown by incubations in the absence of the drug (see also refs 16 and 18).
The corrected enzymic binding of L-[~4C]leu-tRNA was slightly increased at low concentrations of the drug (0.14 raM) but decreased steadily at higher concentrations until 100% inhibition at 0.56 mM of compound AHR- 1911. At the concentration where a 78% block of protein synthesis occurs, that is 0.42 mM, we observed an 88% inhibition of aminoacyl transfer in ribosomes. This suggests that this process is one of the targets of AHR-1911 action.
The GTP concentrat ion can be decreased considerably by omitting the
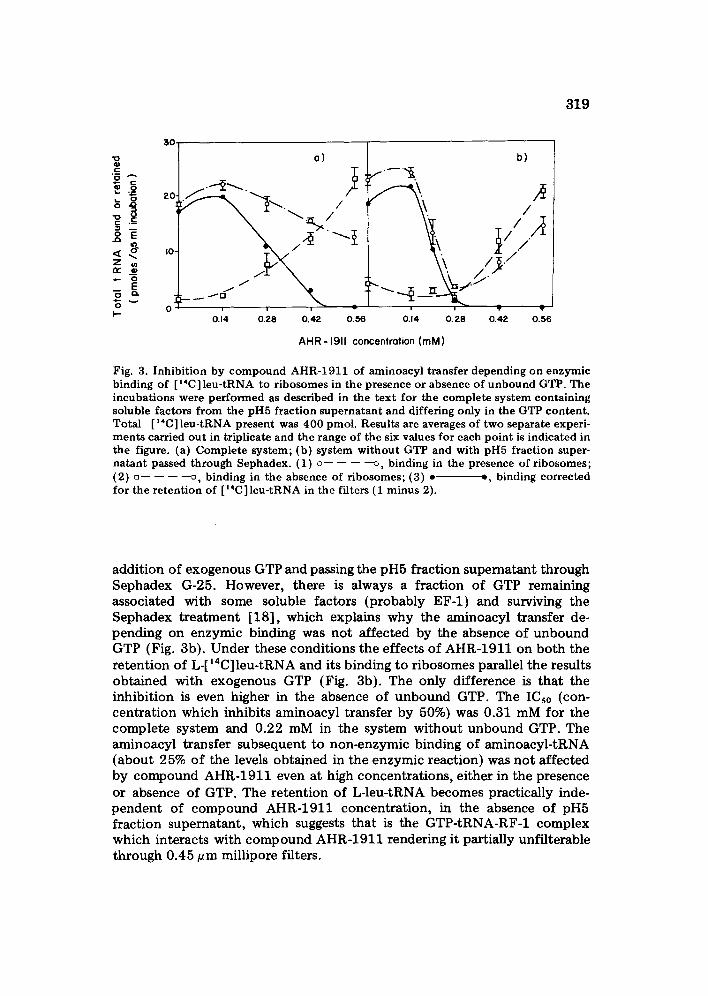
319
"I0
.E 0
OE g~
0 E
0 I.=
301 o)
2 0 .-~ " ~ /~
,o
i
0 .14
' "~ b)
= i - ! i T
0 . 2 8 0 . 4 2 0 . 5 6 0 .14 0 . 2 8 0 . 4 2 0 . 5 6
AHR-1911 concentration (mM)
Fig. 3. Inhibition by compound AHR-1911 of aminoacyl transfer depending on enzymic binding of [ ~4C] leu-tRNA to ribosomes in the presence or absence of unbound GTP. The incubations were performed as described in the text for the complete system containing soluble factors from the pH5 fraction supernatant and differing only in the GTP content. Total [ ~4C] leu-tRNA present was 400 pmol. Results are averages of two separate experi- ments carried out in triplicate and the range of the six values for each point is indicated in the figure. (a) Complete system; (b) system without GTP and with pH5 fraction super- natant passed through Sephadex. (1) o - - - - - - - - ~ , binding in the presence of ribosomes; (2) ~ . . . . D, binding in the absence of ribosomes; (3) • ~, binding corrected for the retention of [~4C ] leu-tRNA in the filters (1 minus 2).
addition of exogenous GTP and passing the pH5 fraction supernatant through Sephadex G-25. However, there is always a fraction of GTP remaining associated with some soluble factors (probably EF-1) and surviving the Sephadex treatment [18], which explains why the aminoacyl transfer de- pending on enzymic binding was not affected by the absence of unbound GTP (Fig. 3b). Under these conditions the effects of AHR-1911 on both the retention of L-[14C]leu-tRNA and its binding to ribosomes parallel the results obtained with exogenous GTP (Fig. 3b). The only difference is that the inhibition is even higher in the absence of unbound GTP. The ICs0 (con- centration which inhibits aminoacyl transfer by 50%) was 0.31 mM for the complete system and 0.22 mM in the system without unbound GTP. The aminoacyl transfer subsequent to non-enzymic binding of aminoacyl-tRNA (about 25% of the levels obtained in the enzymic reaction) was not affected by compound AHR-1911 even at high concentrations, either in the presence or absence of GTP. The retention of L-leu-tRNA becomes practically inde- pendent of compound AHR-1911 concentration, in the absence of pH5 fraction supernatant, which suggests that is the GTP-tRNA-RF-1 complex which interacts with compound AHR-1911 rendering it partially unfilterable through 0.45 gm millipore filters.
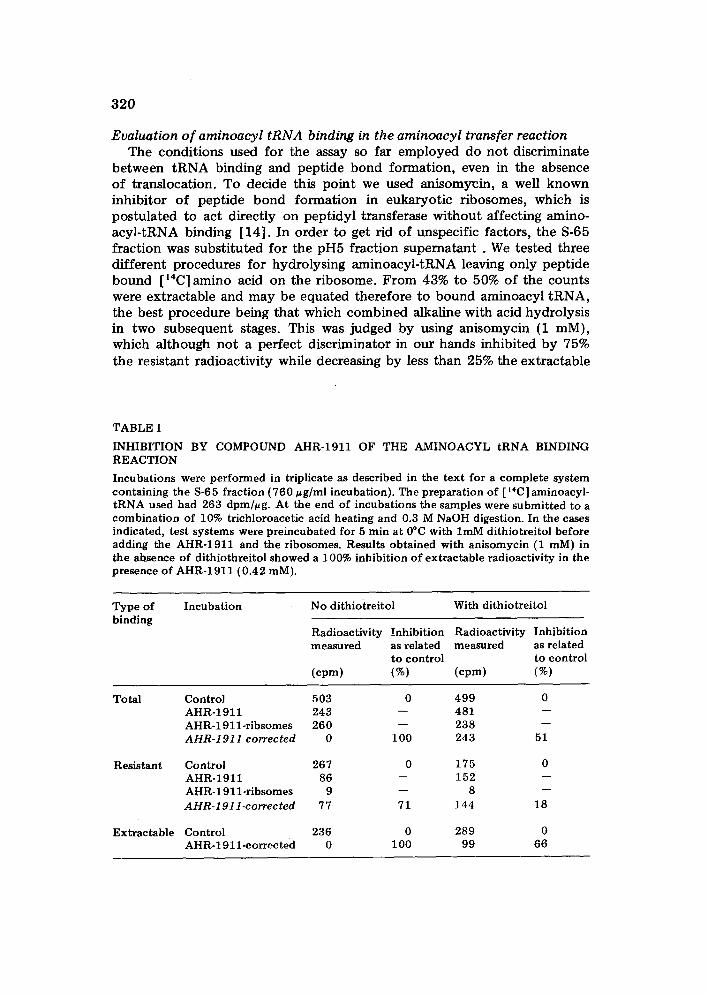
320
Evaluation o f aminoacyl tRNA binding in the aminoacyl transfer reaction The conditions used for the assay so far employed do not discriminate
between tRNA binding and peptide bond formation, even in the absence of translocation. To decide this point we used anisomycin, a well known inhibitor of peptide bond formation in eukaryotic ribosomes, which is postulated to act directly on peptidyl transferase without affecting amino- acyl-tRNA binding [14]. In order to get rid of unspecific factors, the S-65 fraction was substituted for the pH5 fraction supernatant. We tested three different procedures for hydrolysing aminoacyl-tRNA leaving only peptide bound ['4C] amino acid on the ribosome. From 43% to 50% of the counts were extractable and may be equated therefore to bound aminoacyl tRNA, the best procedure being that which combined alkaline with acid hydrolysis in two subsequent stages. This was judged by using anisomycin (1 mM), which although not a perfect discriminator in our hands inhibited by 75% the resistant radioactivity while decreasing by less than 25% the extractable
TABLE I
INHIBITION BY COMPOUND AHR-1911 OF THE AMINOACYL tRNA BINDING REACTION
Incubations were performed in triplicate as described in the text for a complete system containing the 8-6 5 fraction (760 u g/ml incubation). The preparation of [ '~C] aminoacyl- tRNA used had 263 dpm/ug. At the end of incubations the samples were submitted to a combination of 10% trichloroacetic acid heating and 0.3 M NaOH digestion. In the cases indicated, test systems were preincubated for 5 rain at 0°C with lmM dithiotreitol before adding the AHR-1911 and the ribosomes. Results obtained with anisomycin (1 raM) in the absence of dithiothreitol showed a 100% inhibition of extractable radioactivity in the presence of AHR-1911 (0.42 raM).
Type of Incubation No dithiotreitol With dithiotreitol binding
Radioactivity Inhibition Radioactivity measured as related measured
to control (cpm) (%) (cpm)
Inhibition as related to control (%)
Total
Resistant
Extractable
Control 503 AHR-1911 243 AHR-1911-ribsomes 260 A H R - 1 9 1 1 correc ted 0
Control 267 AHR-1911 86 AHR-] 911-ribsomes 9 A H R - 1 9 1 1 - c o r r e c t e d 77
Control 236 AHR-1911-corrected 0
0 499 0 - - 4 8 1 - -
- - 238 -- I00 243 51
0 175 0 - - 1 5 2 - -
- - 8 - -
71 144 18
0 289 0 100 99 66

321
counts, Sparsomycin (0.5 mM), although more active in the former step did also affect substantially the transfer reaction.
Therefore, for most of the experiments we preferred the indirect eva- luation of aminoacyl tRNA binding, by applying the two-stage hydrolysis in the absence of these inhibitors which would complicate interpretation. Table I shows that the extractable radioactivity was completely inhibited by AHR-1911 in a way compatible to an effect on enzymic aminoacyl-tRNA due to the postulated interaction of AHR-1911 and EF--1 gave high blanks for correction. Dithiotreitol decreased substantially the proportion of this interaction, as compared to the total aminoacyl transfer in the presence of ribosomes. Under these conditions, the effects on total aminoacyl transfer and on the presumable aminoacyl-tRNA binding were diminished but still remained significant.
Effect o f compound AHR-1911 on the transpeptidation reaction Gonzalez-Cadavid and Herrera [1] showed that the drug inhibited the
puromycin mediated release of peptide chains labelled in vitro from liver ribosomes and they inferred that transpeptidation itself was affected by compound AHR-1911. This would imply that inhibition of protein synthesis
TABLE II
E F F E C T OF AHR-1911 ON THE F O R M A T I O N OF PEPTIDYL-PUROMYCIN
In the first exper iment the incubat ion was pe r fo rmed by the procedure of Pestka [12] , at 4 t~M puromyc in (360 d p m / p m o l ) and 0.5 mM KCI. In the second exper iment the m e t h o d of Cannon [13] was fo l lowed, at 500 ~M puromyc in (36 d p m / p m o l ) and 25 mM KCI. In Expt . 3 the condi t ions were identical to the second one but the r ibosomes were pre incubated with pH5 fract ion for 5 min under condi t ions of pro te in synthesis. Com- pound AHR-1911 was used at 0.42 raM. In all cases [ 3 H ] pep t idy l -puromycin was measured as described in the text . Figures in brackets correspond to values above controls .
[ 3 H ] Puromycin Inhibi t ion incorpora t ion of incorpora t ion into pept ides as referred to in controls controls (dpm) (%)
A) No preincubation (1) High K ÷ -low pu romyc in Tota l 1166 (+5) (2) Low K ÷ -High pu romyc in Superna tan t 8276 40 Ribsosomes 3876 17 Tota l 12153 33
B) With preincubation ( 3 ) L o w K ÷ -high pu romyc in Superna tan t Ribosomes Tota l
34526 58 2666 (+65)
37197 49

322
is due to at least two targets of action: peptide formation and binding of aminoacyl-tRNA. Although this is possible because many inhibitors have two or even more sites in action, it was necessary to investigate whether the interference with chain release was in fact a process derived from the inter- action with transpeptidation (as suggested in the previous report) or a consequence of anchorage of the peptide chains in the ribosomes.
To decide be tween these possibilities the formation of peptidyl-[3H] puromycin was measured under three sets of conditions. The first one, at high K ÷ and low puromycin concentration, did not support the assumption of a direct effect on the pept idyl transferase site, since no inhibition of the formation of peptidyl-puromycin was observed even at 0.56 mM (Table II). At low K ÷ and high puromycin concentration, in a reaction similar to the one previously used for measuring the release of pulse-labelled chains from ribosome s, except that r ibosomes were no t preincubated under conditions of protein synthesis, the inhibition effect became evident. When ribosomes were no t preincubated in vitro, that is were not put in contact with soluble factors after the extensive washing occuring in the sucrose gradient, the
inhibition of total peptidyl-puromycin formation was slight (33%) and increased to 40% in the peptidyl-puromycin completely released from the ribosomes. But with r ibosomes preincubated in vitro with pH5 fraction under condit ions of protein synthesis, the inhibition of total peptidyl-puromycin formation reached 49% and the release to soluble peptidyl-puromycin was decreased by 58%. This means that there is a direct effect on transpeptidation coupled to only a slight retention of the synthesized peptides on the ribo- some s .
Interaction of compound AHR-1911 with ribosomal particles Since practically all of the antibiotics that inhibit the binding of aminoacyl-
tRNA to ribosomes interact with both the large and the small subunit (see ref. 14), it was important to s tudy this point concerning compound AHR-1911. The direct interaction with ribosomes which has been already described [ 1], leads to a particular kind of aggregation where the sedimented r ibosomes resuspended and incubated in the absence of free drug had still compound AHR-1911 bound to them aud~remained resedimentable at low speed. These r ibosomes were able to perform the puromycin reaction and the incorporation of [~4C]leucine into protein at nearly normal rate, thus suggesting that no relation exists between protein synthesis inhibition and ribosomal aggregation.
The morphological observation of these ribosomes shows that only some aggregates are detectable at 90.000X in spite of the fact that they were pellets from AHR-1911 incubations (Fig. 4 ) . The different sizes of the aggre- gates suggested that the original ones are broken down during specimen preparation for negative staining, indicating that the cross-links between ribosomes should be relatively labile. This, together with the fact that no gross structural alteration is visible, helps to understand why the ribosomes that have interacted with compound AHR-1911 are still able to function in protein synthesis.
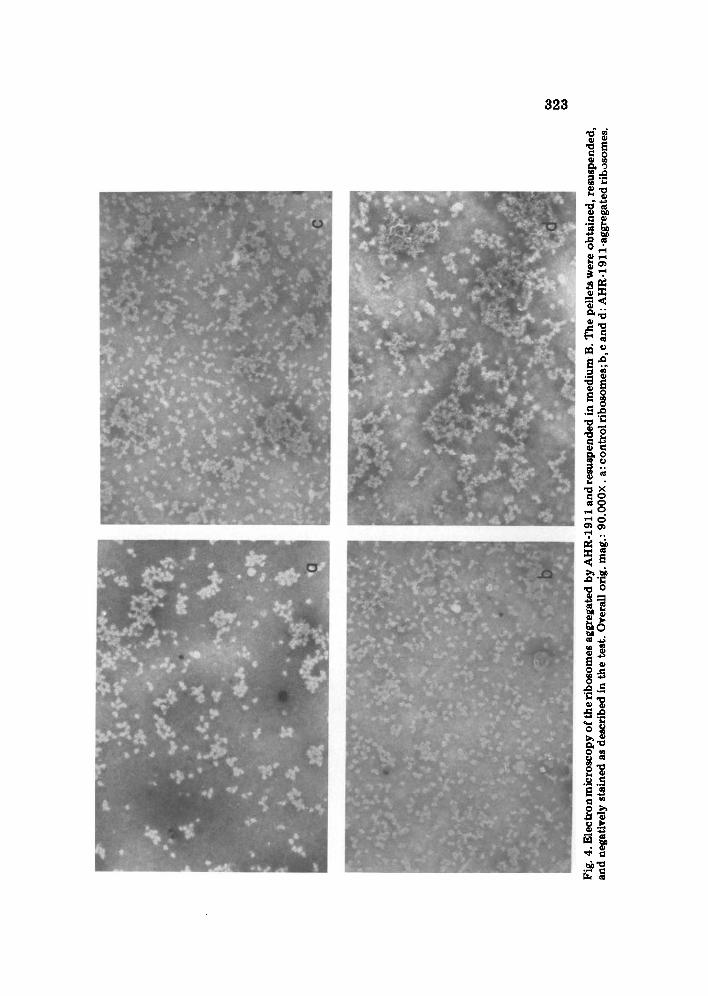
t~
c,o
Fig
. 4.
Ele
ctro
n m
icro
sco
py
of
the
rib
oso
mes
agg
rega
ted
by
AH
R-1
91
1 a
nd
res
usp
end
ed i
n m
ediu
m B
. T
he
pel
lets
wer
e o
bta
ined
, re
susp
end
ed,
and
neg
ativ
ely
stai
ned
as
desc
ribe
d in
th
e te
st.
Ove
rall
ori
g. m
ag.:
90
.00
0x
. a:
co
ntr
ol r
ibo
som
es;b
, c a
nd
d:
AH
R-1
91
1-a
gg
reg
ated
riL
osom
es.
324
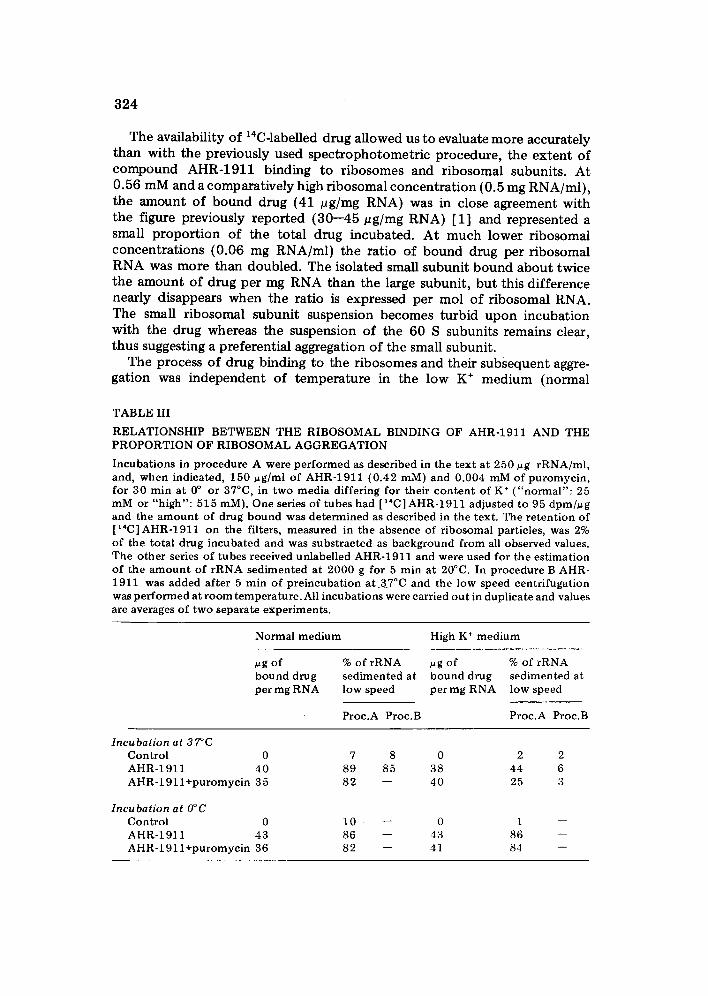
The availability of 14C-labelled drug allowed us to evaluate more accurately than with the previously used spectrophotometr ic procedure, the extent of compound AHR-1911 binding to ribosomes and r ibosomal subunits. At 0.56 mM and a comparatively high ribosomal concentrat ion (0.5 mg RNA/ml), the amount of bound drug (41 pg/mg RNA) was in close agreement with the figure previously reported (30--45 pg/mg RNA) [1] and represented a small proport ion of the total drug incubated. At much lower ribosomal concentrations (0.06 mg RNA/ml) the ratio of bound drug per ribosomal RNA was more than doubled. The isolated small subunit bound about twice the amount of drug per mg RNA than the large subunit, but this difference nearly disappears when the ratio is expressed per tool of ribosomal RNA. The small ribosomal subunit suspension becomes turbid upon incubation with the drug whereas the suspension of the 60 S subunits remains clear, thus suggesting a preferential aggregation of the small subunit.
The process of drug binding to the ribosomes and their subsequent aggre- gation was independent of temperature in the low K ÷ medium (normal
TABLE III
RELATIONSHIP BETWEEN THE RIBOSOMAL BINDING OF AHR-1911 AND THE P R O P O R T I O N OF RIBOSOMAL A G G R E G A T I O N
Incuba t ions in p rocedure A were p e r f o r m e d as descr ibed in the t ex t at 250 pg rRNA/mt , and, w h e n indicated, 150 pg/ml of AHR-1911 (0.42 mM) and 0.004 mM of pu romyc in , for 30 min at 0 ° or 37°C, in two media differ ing for their c o n t e n t o f K ÷ ( " n o r m a l " : 25 mM or "h igh" : 515 raM). One series of tubes had [ ~4C] AHR-1911 adjus ted to 95 d p m / u g and the a m o u n t of drug bound was de t e rmined as descr ibed in the text . The r e t en t ion of [ ~4C] AHR-1911 on the filters, measured in the absence of r ibosomal particles, was 2% of the total drug incuba ted and was subs t rac ted as background f rom all observed values. The o the r series of tubes received unlabel led AHR-1911 and were used for the es t imat ion of the a m o u n t o f r R N A sed imen ted at 2000 g for 5 min at 20°C. In p rocedure B AHR- 1911 was added af ter 5 min of p re incuba t ion a t 37°C and the tow speed cent r i fugat ion was pe r fo rmed at r o o m tempera ture . All incubat ions were carried ou t in dupl icate and values are averages of two separate exper iments .
Normal med ium High K ÷ medium
pg of % of rRNA pg of % of rRNA bound drug sed imen ted at b o u n d drug sed imen ted at per mg RNA low speed per mg RNA low speed
Proc.A Proc.B Proc .A Proc.B
Incubat ion at 37°C Cont ro l 0 AHR-1911 40 A H R - 1 9 1 1 + p u r o m y c i n 35
Incubat ion at O°C Contro l 0 AHR-1911 43 AHR-191 l + p u r o m y c i n 36
7 8 0 2 2 89 85 38 44 6 82 - - 40 25 3
1 0 - - 0 1 86 - - 43 86 82 -- 41 84 m

325
medium B) used initially for the incubations, but when the K ÷ was increased to 0.5 M two phenomena occurred {Table III). High K ÷ decreased the ribo- somal aggregation to about half, at 37°C, and the process became temperature- dependent since no protection against aggregation was noticeable at 0°C, thus suggesting either a ribosomal conformation change or an enzyme reaction. Most interestingly, the presence of very low concentrations of puromycin (4 uM) enhanced the protective effect of high K ÷, so that practically no ribosomes sedimented at low speed, and this was also temperature<lependent. In all cases, the level of drug binding did not change significantly, thus indicating that the aggregation occurs in a later stage after binding and can be affected by several conditions without decreasing the bound drug. The introduction of a preincubation at 37°C {Procedure B) enhanced the protective effect of both K ÷ and Na ÷ until it made negligible the ribosomal aggregation exerted by the drug. Similarly, experiments (not shown in Table III) where ribosomes were aggregated by incubation with the drug at 37°C and low K ÷, indicated that the effect can be reversed by addition of K ÷ to concentrations of 0.5 M. The K ÷ can be replaced by Na ÷ (0.5 M) with the same results.
DISCUSSION
The present study indicates that there is a direct correlation between the inhibitory effect on protein synthesis in vitro and the length of the alkylic chain, but essential for this effect is a subti tution in C1 either by a pseudo- thiourea or an NH2 group. Comparison between dodecanyl pseudothiorea and dodecylamine shows that there is not substantial difference on protein synthesis at the subcellular or the cellular level, between the insertion of a pseudouthiourea or an amino group. On the other hand the reduction of chain length by just one carbon (undecanyl pseudothiourea) does not change significantly the inhibitory activity, byt a further introduction of a distal double bond (AHR-1911) increases considerably this effect, decreases cytotoxici ty, and reduces the extent of agglutination of ribosome and soluble factors. This makes AHR-1911 a compound more suitable than the other available alkylic pseudothioureas, or dodecylamine itself, for dissecting the mechanism of action of these series of compounds on different steps in protein synthesis. The fact that dodecylamine is practically inactive against the growth of the Walker carcinoma, whereas AHR-1911 acts as carcino- static, shows that in contrast to the interference with protein synthesis the pseudothiourea group is important for turnout arrest. Therefore.the blockade of protein synthesis is not a main factor in this effect, which is more probably related to the observed inhibition of RNA and DNA synthesis [19].
AHR-1911 effects on protein synthesis are much more complex and differential than those which could arise from a mere detergent like action on the constituents of the in vitro system. The drug seems to affect at low concentration the interaction of leu-RNA with elongation factors and to inhibit its binding to ribosomes, but it does not affect the charge of tRNA,

326
or the translocation step [1]. Peptide bond formation is only significantly decreased when ribosomes are preincubated with soluble factors. It has been proposed [20, 21] that when tRNA has bound to the A site, the EF-1 factor must be released for transpeptidation to proceed. The interference with peptidyl-puromycin formation in preincubated ribosomes may be caused by stabilization of EF-1.
The ribosomal aggregation is another example of an interesting reversible interaction induced by AHR-1911, which has already been proved to be not directly linked to the inhibition of protein synthesis [1]. The fact that K ÷ counteracts agglutination in a temperature-dependent process could be interpreted as the result of conformational changes in the ribosomal structure making it refractory to aggregation. Puromycin probably potentiates the K ÷ effect by decreasing the ribosomal contact through the release of individual r ibosomes (monosomes) from the polyribosomes favoured by the dis- sociating action of high ionic strength.
All the evidence so far presented suggests therefore that the main func- tional effects of AHR-1911 on protein synthesis would be at the level of soluble factors involved in the elongation process, and their interaction with the ribosome itself (see also ref. 20). On the other hand, the aggregation of polyribosomes and its reversion by high ionic strength in a temperature- dependent process offers interesting similarities with the interaction between ribosomes and endoplasmic reticulum membranes [ 22] which should deserve further attention.
A C K N O W L E D G E M E N T S
The invaluable cooperation of Dr. N. Ercoli in providing both the un- labelled and the 14C-compound AHR-1911 is most gratefully acknowledged. Dr. Nathan Belcher from Pfizer Central Research and Dr. Israel Algranatti kindly provided the anisomycin and sparsomycin. Dr. Misuo Ogura helped in the electron microscopy. The rats were provided by the Insti tuto de Medicina Experimental (Universidad Central de Venezuela).
R E F E R E N C E S
1 N.F. Gonz~ilez-Cadavid and F. Herrera, Inhibi t ion of t ranslat ion in liver po lyr ibosomes by a new subst i tuted th iopseudourea with an t i tumour action, Biochem. J., 138 (1974) 129.
2 N.F. Gonz~ilez-Cadavid and J.L. P(irez, Indent i f ica t ion of the products of mi tochon- drial t ranscript ion in the Walker carcinosarcoma by the use of ac t inomycin D and e thidium bromide, Cancer Res., 36 (1976) 1754.
3 F.O. Wettstein, T. Staehel in and H. Noil, Ribosomal aggregate engaged in prote in synthesis : character izat ion o f the ergosome, Nature, 197 ( 1963 ) 430.
4 G. Ragnott i , G.R. Lawford and P.N. Campbell , Biosynthesis of microsomal nicoti- namide-adenine dinucleot ide phospha te -cy tochrome c reductase by membrane bound and free polysomes f rom rat liver, Biochem. J., 112 (1969) 139.
5 N.F. Gonz~tlez-Cadavid, J.P. Ortega and M. Gonz~ilez, The ceil-free synthesis of c y t o c h r o m e c by a microsomal fract ion f rom rat liver, Biochem. J., 124 (1971) 685.

327
6 K. Moldave, Aminoacyl transfer to ribosomal protein in the rat liver system, in L. Grossman and K. Moldave (Eds.), Methods in Enzymology, Vol. XII, Nucleic acids, part B, Academic Press, New York, 1968, pp. 721--725.
7 K. Norrby, F. Knutson and P.M. Ludin, On the single cell state in enzymatically produced tumor cell suspensions, Exp. Cell Res., 44 (1966) 421.
8 A. yon der Decken, P. Ashby, D. McIlreavy and P.N. Campbell, The reversible dis- sociation and activity of ribosomal subunits of liver and skeletal muscle from rats, Biochem. J., 120 (1970) 815.
9 O.H. Lowry, N.J. Rosebrough, A.L. Farr and R.J. Randall. Protein measurement with the Folin phenol reagent, J. Biol. Chem., 193 (1951) 265--275.
10 K. Moldave, The preparation of [~4C]aminoacyl soluble-RNA, in S.P. Coiowick and N.O. Kaplan (Eds.), Methods in Enzymology, Vol. VI, Academic Press, New York, 1963, pp. 757--761.
11 Th. W. O'Brien and G.F. Kalf, Ribosomes from rat liver mitochondria, II. Partial characterization, J. Biol. Chem., 242 (1967) 2180.
12 S. Pestka, R. Goorha, H. Rosenfeld, C. Neurath and H. Hintikka, Studies on transfer ribonucleic acid ribosome complexes, XX. Peptidylpuromycin synthesis on mammalian ribosomes, J. Biol. Chem., 247 (1972) 4258.
13 M. Cannon, The puromycin reaction and its inhibition by chloramphenicol, Eur. J. Biochem., 7 (1968) 137.
14 N.F. Gonzdlez-Cadavid, B. Dorta and A. Carmona, Inhibition of cytoplasmic protein synthesis by mitochondrial soluble factors in rat liver and Walker carcinosarcoma, in Th. Bucher et al. (Eds.), Genetics and Biogenesis of Chloroplasts and Mitochondra, Elsevier/North-Holland, Amsterdam, 1976, pp. 835--842.
15 D. V~zquez, Inhibitors of protein synthesis, FEBS. Lett., 40 (Suppl.) (1974) $63-$48. 16 F. Ibuki and K. Moldave, Evidence for the enzymatic binding of aminoacyl transfer
ribonucleic acid to rat liver ribosomes, J. Biol. Chem., 243 (1968) 791--798. 17 M.W. Nirenberg and P. Leder, RNA Code words and protein synthesis: The effect
of trinucleotides upon the binding of sRNA to ribosomes, Science, 145 (1964) 1339. 18 D. Richter, Formation of a ternary complex between yeast aminoacyl-tRNA binding-
factor, GTP and aminoacyl-tRNA, Biochem. Biophys. Res. Commun., 38 (1970) 864. 19 A. Cabrera, Efeeto del AHR-1911 sobre la sintesis del RNA eitoplasmatico en higado
y carcinosarcoma de Walker, Thesis, Facultad de Ciencias, U.C.V. Caracas, 1976. 20 H. Weissbach and N. Brot, The role of protein factors in the biosynthesis of proteins,
Cell, 2 (1974) 137. 21 H. Grasmuk, R. Nolan and J. Drews, Elongation factor 1 from ascites tumour cells:
interaction with ribosomes and elongation factor 2, Eur. J. Bioehem., 48 (1974) 485. 22 M.R. Adelman, D. Sabatini and G. Biobel. Ribosome membrane interaction: Non-
destructive disassembly of rat liver rough microsomes into ribosomal and mem- branous components, J. Cell. Biol., 56 (1973) 206.





























