-
COMMENTARY
ls cancer a disease of self-seeding?Larry Norton & foan
Massagu6
The high cell density, rapid growth rate and large population
size of cancer are conventionally attributed to apathologically
high ratio of cell production to cell death. Yet these features
might also or instead result frominappropriate cell movement,
already understood to underlie invasion and metastasis. This
integrating concept couldinduce a broadening of our existing
anticancer pharmacopoeia, which, with mitosis as its predominant
target, is nowseldom curative.
Although epithelial cancers are diverse geno-tlpically and
phenoryaically, several featuresare universall-5.One of these is
the abnormalcapacity of cancer cells to migrate, which manifests in
two ways. The first is invasion: the cells'ability to become
dislodged and travel withinthe tissue of origin. The second,
metastasis,involves travel beyond the tissue of origin.Aberrant
cell mobility is thought to be distinctfrom (while sharing some
molecular pathwayswith) another universal feature: an
increasedratio ofproliferation to cell deathr,6,7. This lat-ter
paired abnormality, rather than cell mobil-ity, is thought to
underlie tumorigenesis, theexpanded generation of cancer cells that
pro-duces large, destructive masses. So, the currentview of cancer
incorporates a set of overlap-ping dichotomies: invasion versus
metastaticspread, both relating to cell mobility; tumori-genesis
versus metastatic behavior, the formerthought to result from cell
proliferation, thelatter from cell movement.
Here we seek to unifir these dichotomies byconsidering the
possibiliry that pathologic cellmobility, in addition to behg
crucial to tumorinvasion and metastatic dissemination, canalso
contribute significantly to primary tumorgrowth. Moreover, the
mechanism by whichcell mobility can increase tumor size can
alsoproduce high cell density and rapid grouth rate.
Lary \o'tor is in lre Depa'r'rerrs or Meoicire,Vemor al
Sloan-Kenerirg Carcer Ce.ter aro tl-eWeill College of Med cine of
Cornell University, Newvork, New Yor( 10021. USA ano Joa^
lvassagLes in the Cancer B ology and Genet cs Programand Howard
Hughes Medlca Institute, MemorialSloan-Kettering Cancer Center, New
York, New York1002i, usA.[-marl I [email protected]
We call this proposed mechanism'self-seeding',a term used in
botany to describe the abilityof plants to overgrow an ecosystem.
Considerhow a mass of weeds dominates a field: not bythe massive
increase in size of individual weedplants, but rather by the
continuous propaga-tion of new weeds both within the weed
bed(density) and at its periphery (invasion). Incancer, this
concept would translate to escapeecells that constantly re-seed a
tumor mass,making that mass a dense collection of con-tiguous small
growths that inflltrate and poten-tially destroy its host organ.
Furthermore, justas a self-seedingweed population might
spreadbeyond its field of origin, so might cancers seeddistant
metastases.
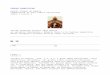
The logic of self-seedingMany of the faculties that permit
distant seed-ing could logically enable self-seeding (Fig. I
).These include separating from an anatomicmooring,lysing a
proteinaceous and carbohy-drate matrix, intravasating, circulating,
adher-ing to an endothelium, extravasating, attachingin a target
location, inducing angiogenesis andpropagating in the target
environmentl. Seedsmight depart from the primary site of
tumorformation or from a metastatic site. Returningto the site of
origin would constitute self-seed-ing or re-seeding, whereas
lodging in another(distant) site would constitute the process
ofmetastasis.
Cancer invasion is symbolized in Figure Iby pathways A and D.
Metastasis is syrnbolizedby pathway C. Here we hlpothesize the
exis-tence of pathways B and E, through which adislodged, wandering
cancer cell, having accessto the entire systemic circulation, has a
finiteprobability of returning to its site of birth. Tobe sure,
this return trip could well be faced with
an unfavorable circulation pattern, includingencounters with
intricate, tortuous capillarybeds that may retain the wandering
cells beforethey can return to their site of origin. Thosecells
that do manage the return trip, however,would find themselves in
thewelcoming micro-environment in which they first developed orin
which they took rootl-s. The co-evolution ofcancer cells and their
stroma that creates sucha microenvironment is often termed'field
can-cerization', and maybe reflected in the presenceof tumor and
stroma-specific gene expression
._ q l)Datrerns" -'.The molecular mediators of self-seeding
in the primary site (pathways A and/or B)and self-seeding in
metastatic sites (pathwaysD and/or E) might be partially
overlapping.Successful self-seeding might well require thecontin
ual gene ration of sel l-renewi n g p rogeny.Therefore, we envision
that some proportionof self-seeds may be acting the role of, or
besynonpnous with, cancer'stem' cells, moreaccurately called
tumor-initiating ce11s13,14.And, to the extent that normal stem
cells mayundergo tumorigenic mutations, their
intrinsicself-renewing and migratory properties mightturn such
cells into higtrly effective self-seeds.
An appealing aspect of the concept of self-seeding is that it
could explain diverse char-acteristics of cancer, extending the
analogy toa weed-strewn garden. Dysplasia could be theconsequence
of the tumor being a spatiallydisoriented conglomerate of
functionallyindependent smaller masses. As self-seededloci continue
to self-seed, the result wouldbe more and younger loci of new
growthand hence more histologic disorganizationas well as a
preponderance of immature cells.Furthermore, independent masses at
theperiphery of a cancer would be de facto points
NATURE MEDICINE VOLUME 12 NUMBER 8 AUCUST 2006
-
COMMENTARY
of invasion whether they arose by pathways Aor B in the primary
site, or pathways D or E ina metastatic site. Each new growing
compo-nent of the conglomerate would tend to attractits own
independentblood supply, promotinghypervascularityls. If self-seeds
return to theprimary tumor's organ of origin but do notattach to
the mass of the primary tumoq thiswould convey the appearance of
multifocal-ity. (Of course, true multifocality-multiplepoints of
primary carcinogenesis-may alsocoexist with this process.) As the
cells estab-lishing these noncontiguous seeded loci aresamples of
the heterogeneous collection ofcells already present, they may be
sufficientlydistinct that they convey the appearance
ofpolyclonality. Moreover, once established, sat-ellite lesions
could further evolve and inter-mingle, consistent with the
observation thatmultifocal lesions often seem to be
polyclonal.Self-seeding might also be relevant to thehotly debated
question of whether a primarytumor exhibits the expression
signature of itsmetastatic descendents: self-seeding wouldtend to
equalize the molecular profiles of anaggressive tumor and its
metastases, theirdegree of similarity increasing proportionalto the
degree to which self-seeding accountsfor the growing mass of the
primary tumor.
The mathematics of self-seedingSelf-seeding could explain
increased celldensity, high mitotic rate and large tumorsize by
reference to two related biomath-ematical concepts: fractal
geometry andGompertzian kinetics. Biological structuresare best
described mathematically by fractalgeometry rather than.the
Euclidian geom-etry of regular solid masses like spheresand
cubesl6'17. By Euclidian geometry thevolume of a biologic mass
increases by thecube of its diameter. In contrast, by
fractalgeometry the number of cells in that massincreases by a
power constant less thanthree. Hence, the ratio of cell number
tovolume (termed 'cell density') falls as thevolume increases.
Because of this, a fullydeveloped normal organ, or even a
largesection of that adult organ, would in mostcases have a lower
total cell density than thewhole organ in its primordial, smaller
state.In contrast, if-as self-seeding would sug-gest-a cancerous
mass arising from thatorgan is a conglomerate of small, incipi-ent,
component masses, each would have asmall volume and hence a high
cell density'creating a high cell density in the cumula-tive whole.
(An important exception to thisgeneral rule is discussed
below.)
The abnormally high cell density found inmost cancers is
labeled'hyperproliferation'
4{
A*Tff;t n"X
Metaslatictumor
Systemic circulalion
Figure I The self seeding concept of cancer growth and
metastasis. The primary tumor mass on thelower left is a composite
of three smaller sub tumors: the central, larger one is a
proliferating collectionof cells at the original site of
carcinogenesis; the other two sub-tumors on the lower left are the
progenyof self-seeds that spread from ihat original site. Because
each of ihe three is relatively small comparedwith the volume of
their sum, they are relatively both dense and rapidly growing (see
text). A self-seedto the primary tumor may follow pathway A,
comprised of dislodging (orange arrow) and proteolysisof the
extracellular matrix (yellow arrow) followed by reattachment in or
at the primary site, thenproliferation and angiogenesis. Or the
self-seed might follow pathway B, which includes
intravasation(green anow), circulation and extravasation (purple
arrow), then return to the primary tumor bed,proliferation and
angiogenesis. A cell following pathway C traces the same steps, but
relocates to ametastatic siie, where growth can occur by
proliferation, angiogenesis and self-seedtng at that site,either
directly (pathway D) or via circulatory flow (pathway E). The
expressed gene sets responsible forthese processes may be shared
completely or partially. For example, pathways B and C may require
thesame biochemical processes, and hence the same gene expression
pattern, with the final destinaiion(primary or metastatic sites)
determined stochastically. Or pathway C may use the same genes
aspathway B, but with some additional properties. The genes
responsible for paihways D or E may besignificantly overexpressed,
whereas the genes associated with pathways A or B may not, which
wouldexplain virulent growth in a metastatic but not the primary
site. Many oiher patterns of local and distantgrowth are consistent
with the model, as discussed in the text.
because its microscopic appearance-more to imperceptible further
growth (Fig' 2).cells per unit of space-is regarded properly
Gompertzian kinetics has proven useful
asasevidenceofmorecelldivisionsinconjunc- well as applicable in the
clinic, particularlytion with relatively fewer cell deaths. This in
the design of improved cancer treatmentdoes not, however, imply
that the regulation regimensls.of mitosis or apoptosis is
necessarilyabnor- If a malignant tumol is a conglomeratemal. An
alternative cause of high cell density of component Gompertzian
masses, eachcould be the cells' normal response to the with a
relatively high growth rate because it'start-up'nature of the small
tumor seedlings is relatively small, then the whole
conglom-comprising the malignant conglomerate. erate-being the sum
of its parts-would
One of the consequences of high cell den- have a high growth
rate as well. In addition, asity is a high density of proliferating
cells. conglomerate of small Gompertzian tumorsThis might be one
reason why small-vol- would grow large because it would sum theume
masses grow mole rapidly relative to many individual Gompertzian
plateaus oftheir sizes than masses of larger volume, as the
components. Hence, the concept of self-first described
mathematically by Benjamin seeding provides an explanation for both
theGompertzl8. This phenomenon results in a increased growth rate
and the increased totalsigmoid growth curve on an arithmetic plot
volume of malignancy without needing toof size versus time, with
population size even- hlpothesize deviant regulation of mitosis
ortually approaching a plateau phase of slow cell death at the
individual cellular level.
I AUCUST
-
The relationship between ptoliferationand self-seedingThe above
analysis does not ignore the obvi-ous facts that abnormalities in
mitotic regu-lation and an aberrant ability to survive suchstresses
as cell-cycle checkpoint alterations,hypoxia, glucose deprivation
or interstitialfluid pressure changes are commonly foundin cancer
cel1sl,6. We merely indicate thatabnormal cell mobility could
contribute tophenomena previously attributed entirely toother
processes. Furthermore, it is possiblethat hlperactive mitogenic
pathways and eva-sion of growth inhibitory constraints may
benecessary but not sufficient for overt carcino-genesis.
Hyperproliferation is also common inmany benign conditions such as
dermatosesand premalignant lesions. Furthermore, micegenetically
engineered to overexpress purelymitogenic oncogenes
characteristically demon-strate benign hlperplasias in many
organsT'1e.Tiue cancers arise from some, but only some, ofthe cells
in these genetically altered organs. Tobecome malignant, therefore,
l"umors requireadditional abilities, which maybe present fromthe
outset (for example, in normal tissue stemcells that undergo
transformation) or may beacquired later. We argue that one of these
abili-ties is the capacity to self-seed. In this regard,the
observation that some potent oncogenessimultaneously disrupt both
cell adhesion(part ofthe seeding process) and mitotic-apop-totic
regulation may explain their efficienry incausing virulent
cancetT'19. That the expres-sion of matrix metalloproteases (also
part ofthe seeding process) maybe sufficient to causemalignant
transformation in transgenic mice issimilarly supportive of our
concept2-1.
Increased proliferation is clearly an adverseprognostic marker
in many types of epithe-lial cancer2o. Although this is usually
thoughtto reflect the cancer cells' mitotic-apoptoticdysfunction,
we may wish to consider that italso could reflect the
aggressiveness of self-seeding. As more vigorous self-seeding
wouldproduce more loci of growthwithin the tumorconglomerate, it
would result in faster overallgrowth rate as well as larger
potential totaltumor volume. Furthermore, and perhapsmost
importantly, a self-seeding etiology ofhlperproliferation would
correlate well witha tumor's metastatic ability, which is, after
all,the prime cause of poor prognosis in clinicalcancer.
This line of reasoning may cast new light onone of the clinical
truisms of epithelial can-cer, that large primary tumor size is a
poorprognostic feature. The conventional view isthat cancers gain
metastatic ability throughan accumulation of mutations as they grow
tolarge size. Yet perhaps some tumors grow to be
large, and have a poor prognosis, because theyseed aggressively
to self and to distant sites.Cancers may not be bad because they
are big;they may be big because they are bad.
The molecular links between metastasisand self-seedingLending
credence to the mathematical reason-ing above is recent molecular
evidence that theability of a wandering cancer cell to re-seed
itsoriginal mass may be linked to the ability toseed distant sites.
Primary tumors frequentlyexpress genes whose products alter the
micro-environment, such as extracellular matrix pro-teases,
glycosylases, proangiogenesis factors,regulators of cell adhesion,
and mediators ofinflammation and angiogenesisl s'7'e-14'1e.Gene
expression signatures that include thesegenes predict metastatic
behavior and hencepoor prognosis in breast cancers9-12. Indeed,so
important is the abiliry of a cancer to alter itsenvi ron m ent
that poor-prognosis gene-expres-sion signatures are emerging which
largelyexclude genes that purely mediate proliferationor
survivalll'12.
It is commonly assumed that these environ-ment-altering genes
are associated with poorprognosis only because they are
responsiblefor metastatic behavior. However, althoughhuman breast
cancer xenografts with highmetastatic capacity in the laboratory do
dem-onstrate a gene expression profile associatedwith metastatic
behavior (and hence poorprognosis) in the clinic, so do cell clones
withless prominent metastatic behaviorl9'21'22.We therefore
consider that the genes in the'poor-prognosis' environment-altering
setmay underlie the self-seeding that permitsgrowth in the primary
site and also lay thefoundation for distant seeding. It should
beexpected, therefore, that genes in addition tothose in the
poor-prognosis set are neededto promote growth in metastatic sites,
andin fact there is evidence that this is the case.In highly
metastatic laboratory models ofhuman cancer, there are genes that
are bothhighly expressed and responsible for site-specific
metastases22. Some of these genes arealso found to be expressed in
human primarytumors that show a high risk of developingmetastasis
to the lung22.
Of particular importance in this regard isthe experimental
evidence that a subset oflungmetastasis-associated genes also
contribute totumor growth in the mammary gland22. Thisfinding may
be interpreted as follows: theseparticular genes may act not only
as metasta-sis-producing, distant-seeding genes (pathwaysC and
possibly D and/or E in Fig. 1), but alsoas tumor self-seeding genes
(pathways B andpossibly A). Furthermore, a different and dis-
COMMENTARY
8x
2x
01020304050Arbitrary time scale
Figure 2 Gompertzian growth curves, This patternof growth is
ubiquitous in nature. As the numberof cells increases with time the
growth rate relativeto the number of cells decreases,
eventuallyapproaching zero as the number of cells approachesa
limiting plateau size (1010 in this example). lf atumor is
comprised of many Gompertzian masses,each arising from a self-seed,
rather than one mass,as would occur in the absence of self-seeding,
theconglomerate's maximum size will be the sum ofmany plateaus,
which could be very large, possibleeven lethal. Self seeding might
cause Gompertziangrowth itself . Gompertz's original equation
isshown as the solid line18. The dashed line, whichis almost
indistinguishable from the solid line, is anew equation (below) in
which proliferation occursin an anatomic region of the mass that
has a lowerfractal dimension than the region where apoptosisoccurs.
Self-seeding would create such a patternsince the seeds,
approaching the mass from theoutside (pathway B in Fig. 1) or
migrating outward(pathway A), would congregate on the periphery
(orleading edge) of the growing mass rather ihan themass's volume.
d/V/df = h1 . /Vt(a/c) - h2 . /Vt(b/c).The constants are h1 > h2
> 0, and 3 >c > b > a >2. (ln this example, a = 2.8,
b = c = 2.9.)
tinct gene subset is associated with the growthof metastases in
the lung but not in the pri-marysite22. Perhaps some of these
genes,whichmight be associated with pathway C, are caus-ing
self-seeding (pathways D and/or E) in themetastatic masses but not
in the primarv site(pathways A and/or B).
Some natural history implications of self-seedingThe concept
that many genes may be involvedin these processes could explain
some of thecomplex and enigmatic phenotypes that areobserved in
nature. For example, one couldenvision a cancer that expresses
metastasis-permissive genes (pathway C in Fig. 1) butlacks
prominent capability to colonize meta-static sites (pathways D
and/or E). This wouldresult in the production of latent
metastasesrestricted in their volume expansion. Perhapsthis is why
some individuals with breast cancerwho have cells of epithelial
origin in their bonemarrow presumed to be breast cancer
metas-tases, never demonstrate clinically significantosseous
disease.
Another eccentric phenotlpe would bethat of a cancer with such
intense self-seedine
10 x
boE o'3 +'
NATURE MTDICINE VOLUME 12 I NUMBER 8 AUCUSI 2006
-
COMMENTARY
power (pathways A and B) that it would neverform a discrete
tumor mass in the primary site,but rather demonstrate a diffuse
infiltrationof the organ of origin. This would provide anexception
to the general rule cited above thatcancer cells form dense masses.
As the genesthat underlie such behavior maywell be associ-ated also
with pathways C, D and E, we shouldexpect to see virulent
metastases in many dis-tant organs. Many adenocarcinomas of
thepancreas may act in this way. In other cancert)?es, metastases
in organs with especiallycancer-supporting stroma may also
exhibitsuch behavior. If a cancer's pathways C andD and/or E are
most prominent, the diseasecould become widely metastatic while
neverdemonstrating a large or even a discernableprimary mass.
Indeed, on rare occasions wedo observe breast and lung cancers that
actlike this. Another possibility is that a primarycancer could
spin offa cell qpe with the capac-ify to strongly self-seed
(pathways D and/or E)onlyin one spot in one distant organ. In such
acase the individual would benefit considerablyfrom resection or
irradiation of that solitarymetastasis in addition to control of
the pri-mary tumor. Hence, variations on the themesdepicted in
Figure I could well encompassdiverse clinical presentations.
It must be emphasized, however, that all ofthe above examples
ofdiscrepancies in growthcharacteristics between primary and
metastaticsites are exceptions to the general ruie. In thevast
preponderance of cases of clinical cancerthere is such a consistent
association of largetumor size, anaplasia, rapid growth rate
andmetastatic behavior that it leads one to hypoth-esize. as we
have, that the molecular roots ofthese phenomena are likely to be
the same orvery closely related.
A therapeutic implication of self-seedingBecause of our
community's historic focus oncell proliferation as the core
aberrancy in can-cer, almost al1 anticancer drugs now in com-mon
use were developed as antiproliferativeinterventions. These drugs
have unquestion-ably proven usefi-rl because they shrink
tumors,which improves prognosis bypostponing deathor delaying
recurrence after resectioni8'20. Cureof established epithelial
cancers is uncommon,
however, and even the advances we have madeir the use
ofpostsurgical adjuvant drug therapymay reflect time delays (as
regrowth proceedsfrom residual cells) rather than eradicationof all
cells in some individuals with cancerts.Moreover, whatever benefits
are gained fromthe use of antimitotic drugs are often at thecost of
considerable toxicity because cellularproliferation is so intrinsic
to the viability andfunction of most normal tissues.
The fact that our current therapeutic strat-egies have been
largely disappointing to dateshould not surprise us if further
research con-firms that seeding as well as mitosis is at thecore of
malignanry. Only recently have clinicaloncologists started to
explore agents directedagainst targets other than those involved
incell proliferation. Antiangiogenesis is onesuch example. In these
clinical trials, however,our bias toward mitosis as a primary
targetis evident in our tendency to combine newtherapeutics with
antiproliferative agents,usually chemotherapy23 2s. This may notbe
an ideal long-term strategy. In addition,neoangiogenesis is only
one of many possibletargets along the paths to successful
self-seed-ing, and may not be the best, as it is far downthe
pathway. But as we develop antiseedingdrugs we must beware of new
toxicities, aswe might be disrupting certain crucial physi-ological
processes-wound healing and gam-ete production being examples.
ConclusionExperimental and theoretical results aresuggesting the
possibility that the processof self-seeding, a putative close
relative ofthe process of metastasis, may influence orunderlie many
important features of epithe-lial cancer. These include high ceil
density,invasion, multifocality, amplified growthrate, enlarged
cell-population size, and thecapacity to spread to and reproduce
thesefeatures in distant organ sites. Testing thevalidity of this
hypothesis will require acombination of approaches. For example,
itwould be of interest to see whether tumorsize is causally linked
to the effect of metas-tasis-gene expression signatures. We
alsoneed to determine whether metastatic tumormasses have the
ability to attract circulating
cells released from the same tumor, andwhether this self-seeding
ability, contribut-ing to tumor growth, depends on pro-meta-static
genes. Yet ifvalidated by these or othermeans, this new concept
would help us tobetter understand malignant diseases andperhaps to
discover how to manage themmore effectively.
ACKNOWLEDGMENTSThe authors express their appreciation to A.B.
Olshenfor his review of mathematical details, to T.J. Rileyfor
drawing the figures, and to C. Pearce for editingthe manuscript. We
also acknowledge with gratitudediscussions with our principal P0 1
-CA94060-04colleagues R. Benezra, M. Iasin, S. Larson,M. Moynahan,
N. Rosen and H. Varmus and withJ. Bromberg, G. Gupta, C. Hudis, R.
Kurtz,A. Minn,D. Spriggs and C. Van Poznak.
1. Hanahan, D. & Welnberg, R.A. Cel/ 100, 57-70(2000).
2. Rizki, A, & Bissell, M.J. Cancer Cell6,7-2 QAOq.3.
lvlott, .i.D. & Werb, Z. Cur. Opin. Cell Biol. t6, 55a
564 (2004).4. Lynch, C.C. & Matrisian, L.M. Differentiation
7O,
561-573 (2002).5. .Joyce, J.A, Cancer Cell 7, 513-520 (2005).6.
I\4assague, J. Nature432,29A 306(2004).7. t1,Y., Hively, WP
&Varmus, l,.l.)ncogene79,1002-
1009 (2000).8. Garcla, S.8., Park, H.S., Nove li, lvl, & Wr
ght, N.A.
J. PaLhol.187, 6l 8l (1999).9. van de Vijver, N4.J. et al. N.
Engl. J. Mled.347,7999
2049 QOOZJ.10. Chang, H.Y. et al. Proc. Natl. Acad. Sci. USA
7O2,
3734-3743 (2005).ll.Troester, M.A. et al. Cancer Res. 64, 4218
4226
(2004).12. Foekens, J.A. et al. J. Clin. oncol.24, 1665 167I
(2006).13. Al-Hajj, M. & Clarke, M.F. Ancogene23,7274
7242
QA04).14. Polyak, K. & Hahn, W.C. Nat. Med. 12, 296-300
(2006).15. Folkrnan, J. & Kallurl, R. Cancer Medicine. 6th
ed.
161-194 (BC Decker, Hamilton,0ntario, 2003).16. Norton, L. Prog.
Clin. Bial. Res, 354A, 109 132
( 1990).17. Schneider, 8.S., Hastings, H,lV, & lVlaytal, G.
Proc.
Biol. Sci.263,129 131 (1996).18. Norton, L. Oncologist.6 Suppl
3, 30 35 (2001).19. Hutchinson, J.N. & lvlul er,W,). Oncogene
19,6130
6137 (2000).20, Dang, C,, Gllewski, T.A., Surbone, A. &
Norton, L.
Cytokinetics. in Cancer lWedicine 6th edn 645 668(BC Decker,
Hamilton, Ontario, 2003).
27. Kang, Y. et al. Cancer Cell 3, 537 549 (2043).22. Minn,
A..). et al. Nature 436,5I8 524 (2045).23. Kabbinavar, F.F. etal.
J. Clin.4nco1.23,3706-3772
(2005).24. Eastern Cooperative 0ncoLogy Group. Clin. Lung
Cancer
6,276-278 (2005).25. Miller, K.D. et al. J. Clin. )ncal. 23,
792-799
(2005).
. ^* @ nature Publishing grouP
To order reprints, please contact:
Am eric as ; T el 21 2 1 26 927 8 ; F ax 212 6'7 9 08 43 ;
author-reprints@nature. comEurope/UK/ROW: Tel + 44 (0)20 7833 4000;
Fax + 44 (0)20 7843 4500; [email protected]
Japan & Korea: Te1 +81 3 3267 8'751L; Fax +8i 3 3267 8746;
[email protected]
878 VOLUME 12 I NUMBER 8 AUCUST 2006 NATURE MEDICINE